

Abstract
A general approach to access sulfamate esters through preparation of sulfamic acid salts, subsequent activation with triphenylphosphonium ditriflate, and nucleophilic trapping is disclosed. The method proceeds in modest to excellent yields to incorporate nucleophiles derived from aliphatic alcohols and phenols. This approach can be employed to furnish differentially substituted sulfamides.
Graphical abstract
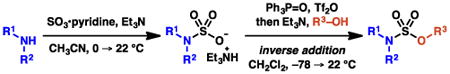
The sulfamate ester functional group has found broad utility, including use as nitrogen sources for amination and aziridination reactions,1 as electrophiles in cross-coupling reactions,2 and as an alcohol-masking moiety to modulate the bioactivity and bioavailability of pharmacologically relevant compounds (Scheme 1).3 One classical approach to access sulfamate esters relies on the use of sulfuryl chloride to furnish sulfamoyl or sulfonyl chloride intermediates. These procedures are inefficient or ineffective when the involved nucleophiles are sterically hindered, or electron deficient (Scheme 2, eq 3).4, 5 Although several alternative methods have been reported to access sulfamate esters (eqs 4–6),6, 7, 8 there are no operationally straightforward, efficient, general methods to prepare acyclic O-alkyl sulfamate esters incorporating primary or secondary alkyl substituents on the nitrogen. To identify a protocol that would provide access to these types of sulfamate esters with varied electronic and steric properties, we anticipated that initial sulfamation9 of an amine with a sulfur trioxide complex would furnish a sulfamic acid salt. We hypothesized that use of sulfamic acid salts would allow us to investigate an array of esterification strategies to install the sulfamate ester S–O bond. Based on this approach, described herein is a broadly effective protocol to prepare sulfamate esters (Scheme 3).
Scheme 1. Utility of sulfamate esters.

Scheme 2. Methods to prepare sulfamate esters.

Scheme 3. Disclosed strategy.

As anticipated, reaction of amines with sulfur trioxide sources provided facile access to diverse sulfamic acid salts (Table 1). Initially, treatment of 2,2,2-trifluoroethylamine with sulfur trioxide pyridine complex and triethylamine in acetonitrile furnished sulfamic acid salt 2a in quantitative yield as an oil without need for purification. Solid trimethylammonium salt 2b could be prepared through the reaction of 2,2,2-trifluoroethylamine with sulfur trioxide trimethylamine complex, and then recrystalized to purity. While the analogous sodium salt could be prepared from chlorosulfonic acid, incorporation of the ammonium cation simplified isolation and offered a better solubility profile in subsequent reactions. Using this strategy, sulfamic acid salts have been prepared efficiently from anilines, primary and secondary amines, enantioenriched amines, and amines with pendant heteroaromatic functionality.
Table 1. Preparation of sulfamic acid saltsa.

|
General reaction conditions: 1.0 equiv amine 1, 1.0 equiv sulfur trioxide pyridine complex (SO3•pyr),0.33 M acetonitrile, 1.5 equiv Et3N, 30 min, 0 → 22 °C.
1 equiv SO3•NMe3in place of SO3•pyr and Et3N.
>90% purity.
These readily accessible salts enabled us to interrogate a variety of tactics for esterification of 2,2,2-trifluoroethyl sulfamic acid salt 2a with n-pentanol (3a) to install a sulfamate ester S–O bond.While sulfamoyl chlorides have been used for efficient access of unsubstituted sulfamate esters,1, 2 activation by in situ generation of a sulfamoyl chloride furnished, at best, modest yields of N-(2,2,2-trifluoroethyl)sulfamate ester 4a (Table 2, entries 1–5). Anticipating that the reaction might be driven forward by formation of a strong phosphorous–oxygen double bond, DIAD and PPh3 were used to activate the salt, and furnish desired sulfamate ester 4a in moderate yield (entry 6). As an extension to this approach, the Hendrickson reagent10 furnished desired sulfamate ester 4a in slightly increased yield (entry 7). Under the optimal conditions, 1.5 equivalents of triethylammonium sulfamate 2a were activated by addition to a solution of triphenylphosphine ditriflate, which was generated in situ from 1.5 equivalents of Tf2O and 1.65 equivalents of Ph3PO (entry 10). Subsequent treatment with 3 equivalents of triethylamine and 1 equivalent of n-pentanol (3a) at −78 °C furnished sulfamate ester 4a in 95% isolated yield. Trimethylammonium sulfamate salt 2b reacted with similar efficiency under the optimal conditions (entry 11).
Table 2. Optimization of Sulfamate Ester Preparation.

| ||
|---|---|---|
|
| ||
| entrya | activating agent | yield (%)b4a |
| 1 | PCl5 (2.0 equiv) | 41 |
| 2 | POCl3 (2.0 equiv) | 44 |
| 3 | SOCl2 (2.0 equiv) | <5 |
| 4 | (COCl)2 (10.0 equiv) | ndc |
| 5 | trichlorotriazine (1.0 equiv) | <5 |
| 6d | DIAD, PPh3 | 50 |
| 7 | Tf2O (1.0 equiv), Ph3PO (2.1 equiv) | 56 |
| 8e | Tf2O (1.5 equiv), Ph3PO (3.15 equiv) | 71 |
| 9e | Tf2O (1.5 equiv), Ph3PO (1.65 equiv) | 78 |
| 10e, f | Tf2O (1.5 equiv), Ph3PO (1.65 equiv) | 95 |
| 11f, g | Tf2O (1.5 equiv), Ph3PO (1.65 equiv) | 94 |
General reaction conditions: 1.0 equiv n-pentanol (3a), 1.0 equiv sulfamate 2a, CH2Cl2, 2.0 equiv Et3N, Tf2O (1.5 equiv), Ph3PO (1.65 equiv), 18 h, −78 → 22°C.
Isolated yield.
Not detected.
No Et3N.
1.5 equiv triethylammonium sulfamate 2a.
3.0 equiv Et3N.
1.5 equiv trimethylammonium sulfamate 2b.
Under the optimized conditions, a range of N-substituted salts 2 can be converted to sulfamate esters 4 in modest to excellent yields (Table 3). While aryl and electron-deficient N-alkyl substituents are well-tolerated in the transformation (4a–d), more electron-rich N-alkyl substituents result in modest yields of sulfamate esters 4e–g. Of these, N-tert-butylsulfamate esters can be prepared in similar yield using tert-butanol and chlorosulfonyl isocyanate to generate N-tert-butylsulfamoyl chloride (Scheme 2, eq 4);6 however, N-methyl- and N-ethylsulfamate esters cannot be accessed via a similar strategy. In principle, these N-alkyl sulfamates could be prepared in a two-step approach featuring the mono-alkylation of O-pentyl sulfamate (Scheme 2, eq 5).7 Neither of these methods would be appropriate to access enantioenriched sulfamate ester 4i, which is generated without any stereochemical erosion using the disclosed approach.
Table 3. N- and O-substitutent variationsa.

|
General reaction conditions: 1.0 equiv alcohol 3 or amine 5, 1.5 equiv sulfamate 2, CH2Cl2, 3.0 equiv Et3N, Tf2O (1.5 equiv), Ph3PO (1.65 equiv), 18 h, −78→ 22°C.
1.0 equiv sodium pentoxide, 0 → 22°C.Alcohol and Et3N were omitted.
The optimized protocol is less effective at transforming salts that have been generated from secondary amines or that incorporate heteroaromatic substituents. When triethylammonium N,N-diethyl sulfamate (2h) is employed, diethylsulfamate ester 4h forms in 17% yield (see Supporting Information). Fortunately, this reaction proceeds in 68% yield if sulfamate 2h is treated with 1 equivalent of sodium pentoxide as the nucleophile. Notably, sulfamic acid salts incorporating nitrogen-containing heterocycle substituents are converted to sulfamate esters 4j–k. These products are not detected when utilizing PCl5 to activate the sulfamic acid salt via sulfamoyl chloride intermediates.
Under the optimized conditions, a variety of alcohols serve as effective nucleophiles to generate sulfamate esters in modest to excellent yields (Table 3, 4l–4w). Primary and secondary aliphatic alcohols, and phenols, including electron-deficient p-hydroxybenzonitrile (3o), are converted to sulfamate esters in high yield. In principle phenol-derived 4p could be generated from an activated sulfonyl imidazolium species (Scheme 2, eq 6).8 However, when sulfonyl imidazolium reagents are used for the synthesis of sulfamate esters, the alcohol portion must be installed first and the approach does not tolerate electron rich or neutral aliphatic alcohols. The disclosed reaction tolerates benzyl or silyl ether groups in alcohols 3r and 3s, respectively, and a phthalimide moiety in alcohol 3t, providing potential strategies for site-specific sulfamoylation of polylols and amino alcohols. As expected, these conditions efficiently incorporate more elaborate hydrocarbon scaffolds, such as those of tetrahydrogeraniol and 5α-cholestan-3β-ol, into sulfamate esters 4u–4w.
In addition to alcohols, nitrogen nucleophiles can be incorporated into sulfamides under the reaction conditions to furnish unsymmetrically substituted sulfamides 6a–6c. Sulfamides are valuable components of some bioactive small molecules, with some nonsymmetrically substituted sulfamides demonstrating higher bioactivity than symmetrically substituted analogues. 11 Nevertheless, few methods12 enable efficient preparation of unsymmetrically substituted sulfamides. Using this approach, differentially substituted sulfamides are accessible from primary, secondary, or tertiary amines, including sterically encumbered tert-butylamine (i.e, 5c→6c).
To conclude, the disclosed method employs inexpensive and readily available sulfur trioxide sources, primary and secondary alkyl amines, and aliphatic or aromatic alcohols to prepare sulfamate esters, many of which are not efficiently accessible through other known methods. Furthermore, the intermediate salts can be employed to generate differentially substituted sulfamides.This new approach provides ready access to a powerful pharmacologically relevant and synthetically versatile motif.
Supplementary Material
Acknowledgments
This project was funded by the American Chemical Society Petroleum Research Fund Doctoral New Investigator Program (PRF# 54824-DNI1) and by Duke University. JMB was supported by the National Institute of General Medical Sciences (T32GM007105-41) and the Burroughs Wellcome Fellowship. MAS was supported by a NSF predoctoral fellowship (NSF DGF 1106401).TDM was supported by the Williams College Alumni Fund. HRMS data was obtained by George Dubay and Matias Horst of Duke University. Characterization data were obtained on instrumentation secured by funding from the NSF (CHE-0923097, ESI-MS, George Dubay, the Duke Dept. of Chemistry Instrument Center), or the NSF, the NIH, HHMI, the North Carolina Biotechnology Center and Duke (Duke Magnetic Resonance Spectroscopy Center).
Footnotes
Author Contributions: The manuscript was written through contributions of all authors. / All authors have given approval to the final version of the manuscript.
Notes: The authors declare no competing financial interest.
References
- 1.(a) Espino CG, Wehn PM, Chow J, Du Bois J. J Am Chem Soc. 2001;123:6935–6936. [Google Scholar]; (b) Duran F, Leman L, Ghini A, Burton G, Dauban P, Dodd RH. Org Lett. 2002;4:2481–2483. doi: 10.1021/ol0200899. [DOI] [PubMed] [Google Scholar]; (c) Milczek E, Boudet N, Blakey S. Angew Chem Int Ed. 2008;47:6825–6828. doi: 10.1002/anie.200801445. [DOI] [PubMed] [Google Scholar]; (d) Liu Y, Guan X, Lai-Ming Wong E, Liu P, Huang JS, Che CM. J Am Chem Soc. 2013;135:7194–7204. doi: 10.1021/ja3122526. [DOI] [PubMed] [Google Scholar]; (e) Alderson JM, Phelps AM, Scamp RJ, Dolan NS, Schomaker JM. J Am Chem Soc. 2014;136:16720–16723. doi: 10.1021/ja5094309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Subbarayan V, Jin LM, Cui X, Zhang XP. Tetrahedron Lett. 2015;56:3431–3434. doi: 10.1016/j.tetlet.2015.01.186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Paradine SM, Griffin JR, Zhao J, Petronico AL, Miller SM, White MC. Nature Chem. 2015;7:987–994. doi: 10.1038/nchem.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Quasdorf KW, Antoft-Finch A, Liu P, Silbserstein AL, Komaromi A, Blackburn T, Ramgren SD, Houk KN, Snieckus V, Garg NK. J Am Chem Soc. 2011;133:6352–6363. doi: 10.1021/ja200398c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Agrawal T, Cook SP. Org Lett. 2014;16:5080–5083. doi: 10.1021/ol5024344. [DOI] [PubMed] [Google Scholar]; (c) Agrawal T, Cook SP. Org Lett. 2013;15:96–99. doi: 10.1021/ol303130j. [DOI] [PubMed] [Google Scholar]; (d) Quasdorf KW, Riener M, Petrova KV, Garg NK. J Am Chem Soc. 2009;131:17747–17749. doi: 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wehn PM, Du Bois J. Org Lett. 2005;7:4685–4688. doi: 10.1021/ol051896l. [DOI] [PubMed] [Google Scholar]
- 3.For a review, see: Winum JY, Scozzafava A, Montero JL, Supuran CT. Med Res Rev. 2005;25:186–228. doi: 10.1002/med.20021.
- 4.Binkley WW, Degering EF. J Am Chem Soc. 1939;61:3250–3251. [Google Scholar]
- 5.Spillane W. Chem Rev. 2014;114:2507–2586. doi: 10.1021/cr400230c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Timberlake JW, Alender J, Garner AW, Hodges ML, Özmeral C, Szilagyi S. J Org Chem. 1981;46:2082–2089. [Google Scholar]; (b) Hedayatullah M, Beji M. Bull Soc Chim Belg. 1988;97:219–225. [Google Scholar]
- 7.Abdoli M, Saeidian H. J Sulfur Chem. 2015;36:463–470. [Google Scholar]
- 8.For seminal reports, see: O'Connell JF, Rapoport H. J Org Chem. 1992;57:4775–4777.Ingram LI, Taylor SD. Angew Chem Int Ed. 2006;45:3503–3506. doi: 10.1002/anie.200600153.. For select recent advances, see: Ingram LJ, Desoky A, Ali AM, Taylor SD. J Org Chem. 2009;74:6479–6485. doi: 10.1021/jo9014112.Desoky AY, Hendel J, Ingram L, Taylor SD. Tetrahedron. 2011;67:1281–1287.. For a review, see: Abdoli M, Saeidian H. J Sulfur Chem. 2015;36:556–582.
- 9.For reviews, see: Gilbert EE. Chem Rev. 1962;62:549–589.Al-Horani RA, Desai UR. Tetrahedron. 2010;66:2907–2918. doi: 10.1016/j.tet.2010.02.015.
- 10.For a seminal report, see: Hendrickson JB, Schwartzman SM. Tetrahedron Lett. 1975;16:277.. For sulfonamide preparation using the Hendrickson reagent, see: Caddick S, Wilden JD, Judd DB. J Am Chem Soc. 2004;126:1024–1025. doi: 10.1021/ja0397658.
- 11.Schaal W, Karlsson A, Ahlsén G, Lindberg J, Andersson HO, Danielson UH, Classon B, Unge T, Samuelsson B, Hultén J, Hallberg A, Karlén A. J Med Chem. 2001;44:155–169. doi: 10.1021/jm001024j. [DOI] [PubMed] [Google Scholar]
- 12.For a recently disclosed method to access acyclic unsymmetrical sulfamides, see: Pantaine L, Richard F, Marrot J, Moreau X, Coeffard V, Greck C. Adv Synth Catal. 2016;358:2012–2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.