

Abstract
Nucleic acid aptamers (NAAs) are short synthetic DNA or RNA molecules that specifically fold into distinct three-dimensional structures able to specifically recognize a target. While NAAs show unprecedented promise in a variety of applications, including sensing, therapeutics and diagnostics, one major limitation involves the lack of stability towards omnipresent nucleases. Therefore, we herein report a systematic truncation and incorporation of 2′-O-methyl bases to a DNA aptamer, which results in increased stability without affecting affinity. One of the newly designed analogues is stable up to 24 hours, demonstrating that 2′-O-methyl RNA is an attractive modification to DNA aptamers, especially when therapeutic applications are intended.
Keywords: Aptamer, stability, 2′-O-methyl RNA, DNA, serum
TOC image
The systematic truncation and incorporation of 2′-O-methyl bases to a DNA aptamer resulted in increased stability without affecting affinity.
Nucleic acid aptamers (NAAs) are short synthetic DNA or RNA molecules capable of folding into distinct three-dimensional structures able to specifically recognize a target.[1] NAAs acquire their unique structures from the intrinsic nature of self-assembly via intra-molecular hydrogen bonding, which, in turn, leads to distinct three-dimensional shapes.[2] NAAs typically adopt structures based on Watson-Crick base pairing, as well as non-canonical base pairing, with the most favorable three-dimensional arrangement suitable for the target recognition. The synthetic nature of NAAs is attractive in a wide range of biomedical applications, such as biosensors, diagnostic molecules, drug-delivery agents and immunomodulators.[3] Another attractive feature arises from easily introducible chemical functionalities, giving aptamers the versatility required for a wide spectrum of applications.[4] While NAAs show unprecedented promise for both therapeutic and imaging applications, a major limitation arises from their susceptibility to nuclease degradation.[5] Therefore, post-SELEX (Systematic Evolution of Ligands by Exponential enrichment) structure-activity relationship (SAR) studies of NAAs typically focus on improving the most favorable fold by truncating NAAs, followed by modification of NAA segments to enhance stability against nucleases.[6]
More specifically, following aptamer selection via SELEX that uses natural DNA/RNA, SAR studies are performed to develop functional NAAs. This includes systematic truncation and synthesis of aptamer analogues, followed by investigation of the affinity of each analogue to determine the shortest length of aptamer with the highest affinity. Truncated aptamers with the highest affinity are subsequently subjected to systematic substitution of modified nuclease-resistant nucleic acid analogues in the 3′- and 5′- end to prevent exonuclease attack. However, such substitution in the “motifs” of NAA structure has typically led to loss of aptamer-target binding; therefore, substitution of “motif” sequences by unnatural nucleic acid analogues is usually avoided.[7] Moreover, it has been widely argued that endonuclease activity is greatly minimized when aptamers are bound to their targets, thus eliminating the extensive need for modification of “motif” structures.
The challenges with nuclease stability have resulted in the notable development of nuclease-resistant RNA aptamers through utilization of DNA and/or RNA with modified nucleic acids in SELEX experiments.[8] However, a number of challenges still exist in using modified RNA bases during the SELEX process.[9] For example, the use of modified nucleic acids during aptamer selection relies on engineered polymerase variants capable of accepting modified nucleic acids followed by efficient amplification. However, not all engineered polymerases are successful in efficiently incorporating modified nucleic acids during polymerase chain reactions (PCRs). On the other hand, ready availability of polymerases capable of efficient amplification to enrich and evolve DNA/RNA SELEX libraries is more suitable in identifying successful aptamers. Nonetheless, post-SELEX modification of DNA aptamers still requires improvement in stability. Thus far, post-SELEX modification of DNA aptamers has been accomplished by systematic substitution of locked nucleic acid (LNA) monomers in defined segments of DNA aptamers.[6a, 7] This approach has been widely investigated as a potential improvement in structural and nuclease stability without compromising aptamer affinity. However, apart from LNA monomers, the use of 2′-modified RNA bases as a modification for DNA aptamers to stabilize duplex regions and increase nuclease resistance remains largely uninvestigated. RNA bases have two analogues with chemical modifications introduced at the 2′-position of the furanose ring: 2′Fluoro-RNA and 2′O-methyl RNA. In particular, 2′O-methyl RNA bases are an excellent alternative to LNA owing to their resistance to ribonucleases, their low manufacturing cost and accessibility.[10] In terms of nuclease resistance, it has been shown that 2′-O-methyl RNA bases are resistant to both 3′exonuclease and S1 nuclease, which is an endonuclease, suggesting that 2′O-methyl RNA bases can also prevent some endonuclease degradation.[11] Both 2′-F-RNA and natural DNA bases were shown to be more susceptible to degradation from 3′ exonuclease present in calf serum.[12]
To test the hypothesis that systematic truncation and incorporation of 2′-O-methyl bases to a DNA aptamer result in increased stability without affecting affinity, we used an aptamer selected against myeloid leukemia, which was recently introduced by Sefah et al., to design new analogues.[13] In particular, aptamer KH1C12 is reported to show high affinity and specificity towards myeloid leukemia cell line HL60 and can specifically recognize a subset of myeloid leukemia.[13] However, this aptamer consists of natural DNA that is susceptible to nuclease degradation, and its long length hinders large-scale chemical synthesis. Therefore, to develop a second-generation aptamer based on original aptamer KH1C12, we herein report a systematic truncation of KH1C12, followed by systematic modification with 2′-O-methyl ribonucleic acid (2′O-CH3 RNA) analogues to design highly stable second-generation variants.
Truncation
The reported full-length KH1C12 aptamer selected against myeloid leukemia cells was 76 bases. This length, which is typical for an NAA, results from the fact that the fixed primer regions, which are required for polymerase chain reaction during SELEX, normally contribute approximately 50% of the aptamer’s length. Typically, a randomized region of an nucleic acid library employed in SELEX ranges from 35–50 bases to allow formation of adequate secondary structures to generate a structurally diverse library. The randomized region of an aptamer mainly contributes to this structural diversity and often plays a significant role in the recognition of the desired target. Thus, the primer region may not be needed for aptamer binding. However, the complete removal of fixed primer regions, as a strategy to generate minimized versions of a functional aptamer, has not always been successful. Therefore, to engineer NAA with a minimum length, it is imperative to systematically truncate nucleic bases from the 3′- and 5′-end. Truncation is done to reduce the length of an aptamer and thereby increase the most favorable fold with the highest affinity. Furthermore, short aptamers are better suited to the design of multivalent scaffolds, essentially because shorter lengths facilitate higher yields during solid-state synthesis, thus lowering the cost of synthesis. The lower cost is particularly important in generating therapeutic multimeric aptamers.
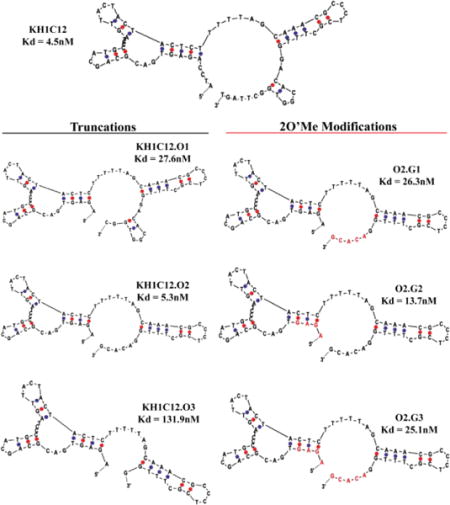
The predicted most favorable secondary structure of KH1C12 using m-fold is shown in Scheme 1. We utilized the predicted structure to design three analogues of KH1C12 aptamer by systematically removing bases from the 5′- or 3′− end. First, we truncated 4 bases from the 5′-end (5′A1T2C3C4) and 5 bases from the 3′-end, i.e., 3′T76G75T74T73C72, from the fixed primer regions of KH1C12 to design the first analogue, KH1C12.O1 (Scheme 1). Next, we truncated 5 more bases (C67G66G65T64G63) from the 3′-end of KH1C12.O1 to design a second analogue, KH1C12.O2 (Scheme 1). Finally, we truncated 5 more bases (G61C60A59C58A57) from the 3′-end of KH1C12.O2 to design the third analogue, KH1C12.O3. Each truncated aptamer was synthesized using standard phosphoramidite chemistry with fluorescein-dT at the 3′-end. The synthesized aptamers were purified using reversed-phase HPLC, followed by post-synthesis work-up and characterization by UV/Vis spectroscopy (see Methods in Supporting Information (SI)). We first evaluated the binding affinities of each truncated aptamer analogue to determine if these truncations affected the most favorable fold of the aptamer. In doing so, different concentrations of each aptamer against HL60 target cells were incubated using appropriate buffers on ice for 45 minutes, followed by washing to remove the unbound sequences. Aptamer binding to HL60 cells was evaluated using flow cytometry. In parallel, a variable concentration of randomized sequence was used as a control to monitor background sticking. Based on the affinity analysis, we did observe slight difference in the affinity for KH1C12.O1 (27.5±13.4nM nM) or KH1C12.O2 (5.25±1.40 nM) (Fig. S1a, b). The KH1C12.O2 analogue showed an affinity similar to that reported for the original full-length aptamer at 4°C (Scheme 1). However, analogue KH1C12.O3 did not show any saturation with increasing concentration (Fig. S1c), suggesting that 1) the removal of additional 5 bases at the 3′-end played a significant role in destabilizing the most optimized fold or 2) the truncated bases were within the binding motif of the aptamer. Therefore, affinity analyses suggest that bases could only be removed from the 3′ end of KH1C12, as reflected in the first two designs and that the functional fold of the aptamer remained intact in these designs.
Scheme 1. Systematic truncation of aptamer KH1C12 and modification with 2’-O-methyl RNA bases.
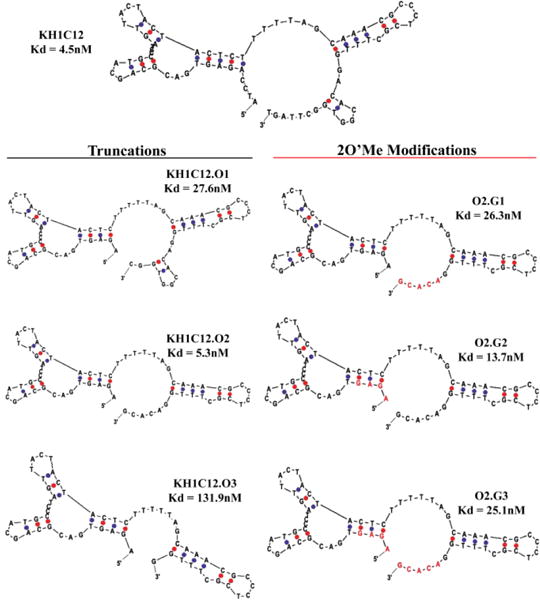
Affinity was determined 4°C with each construct calculated using the method described in ref[14]. Corresponding binding curves are listed in Figure S1. Red letters indicate 2’-O-methyl bases.
2′-O-Methyl RNA incorporation
To increase nuclease stability, we next investigated the possibility of modifying KH1C12.O2 with the unnatural nucleic acid derivative. We only modified KH1C12.O2 because its functional fold remained intact after optimization with affinity comparable to the original full-length aptamer. It has been shown that unnatural analogues of nucleic acid base modifications introduced at the 2′ position could enhance both duplex stability and resistance towards nucleases. LNA incorporation of a DNA aptamer has been shown to improve nuclease stability and melting temperature. Also, owing to economical synthesis and high nuclease stability, 2’-O-methyl RNA has been shown to be attractive in the post-SELEX step for RNA aptamers. Importantly, however, no studies have previously reported that systematic introduction of 2′-O-methyl RNA bases into DNA aptamers could enhance nuclease stability. Therefore, we next investigated whether the bio-stability of a DNA aptamer could be enhanced without compromising its affinity by systematically substituting 2′-O-methyl RNA in defined segments of the KH1C12.O2 analogue. To accomplish this, we again designed three different analogues, but this time, we substituted purines and pyrimidines of truncated aptamer KH1C12.O2 by 2′-O-methyl RNA, as shown in Scheme 1. First, we substituted 2′-O-methyl RNA bases to the double-stranded region (G61C60A59C58A57) of the 3′-end of KH1C12.O2 (Scheme 1: O2.G1). Second, we substituted 2′-O-methyl RNA bases to the 5′- end (A1G1A1G1) of KH1C12.O2 analogue (Scheme 1: O2.G2). Finally, we introduced 2′-O-methyl RNA to both 3′- and 5′- ends of the KH1C12.O2 (G61C60A59C58A57 and A1G1A1G1) analogue (Scheme 1: O2.G3). These three analogues of KH1C12.O2 reflected the best possible scenarios with which to evaluate the effect of 2′-O-methyl RNA on the structure of a DNA aptamer. All newly designed analogues modified with 2′-O-methyl RNA were synthesized using standard phosphoramidite chemistry and were purified using reversed-phase HPLC. We also incorporated 3′fluorescein-dT to facilitate the detection of aptamer binding to HL60 using flow cytometry. Each construct was evaluated for affinity against target HL60 cells similar to the method used above for assessing the affinity of truncated analogues (see Methods in SI). Interestingly, when 2′-O-methyl RNA was incorporated into the 3′-end of the aptamer (O2.G1 analogue), the affinity of the aptamer was reduced by 6-fold compared to the unmodified version at 4°C (26.3±4.9nM, Fig. S2A). However, O2.G2 with 2′-O-methyl RNA substituted at the 5′-end showed only a 3-fold decrease in affinity compared to the unmodified analogue (13.7±2.3nM, Fig. S2B). Finally, O2.G3 showed 6-fold decrease in affinity (25.1±7.3nM, Fig S2C) similar to that of O2.G1 compared to the unmodified variant. This observation suggests that the bases at the 5′-end of A1G2A3G4 are not directly associated with target binding. It should be noted that the segment modified with 2′-O-methyl RNA leads to DNA:RNA heteroduplexes in which sugars have been shown to adopt C3′-endo conformation, shifting the conformational equilibrium of the sugar moieties. This feature also occurs in 2′O-methyl-modified RNA analogues. These types of conformational shifts are also shown to increase Tm of duplexes, which, in turn, increases thermo-stability.
Therefore, incorporation of 2′-O-methyl bases at the 5′-end of O2.G1 might favor the adoption of most favorable conformation, resulting in only minimal effect on affinity. We observed that minimization at the truncation step to generate analogue KH1C12.O3 led to complete loss of aptamer binding. Similarly, we observed that the substitution of 2′-O-methyl RNA at the 3-end spanning the same 5 nucleic acid bases lowered the affinity by about 6-fold. Complete substitution of DNA bases with 2′-O-methyl RNA at both 3′ and 5′ ends diminished affinity by about 6-fold. Taken together, these observations suggest that bases at the 3′-end might play a significant role in aptamer-target binding. We next evaluated the binding of modified O2.G1, O2.G2 and O2.G3 with HL60 using unmodified KH1C12 as a control at physiological temperature. We observed that all three variants bound to HL60 cells at 1μM concentration at physiological temperature. Binding of 2′-O-methyl analogues was similar to that of unmodified KH1C12, suggesting that affinity at physiological temperatures was retained and was comparable with binding affinity of the original aptamer (Fig. 1A and B).
Figure 1.
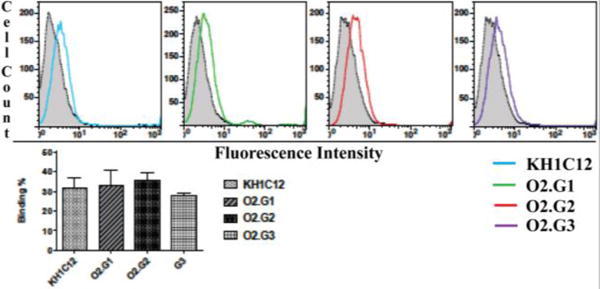
Flow cytometric histograms (top) of aptamer analogues binding to HL60 cells and a bar diagram (bottom) representing overall conclusion from three independent experiments at physiological temperature. Binding was calculated as [(control−aptamer)/aptamer] × 100 and quantified using Graph Pad Prism software.
Specificity and stability of truncated and modified aptamers
We next evaluated the specificity of aptamer variants O2.G1, O2.G2 and O2.G3 against Daudi cells, a Burkitt′s lymphoma cell line, and Jurkat cells, a T-Acute Leukemia cell line. Based on previous reports, full-length KH1C12 did not bind these cells; therefore, they were appropriate for specificity analysis. We employed the HL60 cell line as the positive control. We followed a method similar to that described in Sefah et al., but with 100,000 HL60, Daudi or Jurkat cells. The specificity of all analogues was retained and did not show any nonspecific sticking to Jurkat or Daudi cells, suggesting that truncation and incorporation of 2′-O-methyl RNA bases do not interfere with aptamer specificity (Fig. 2).
Figure 2.
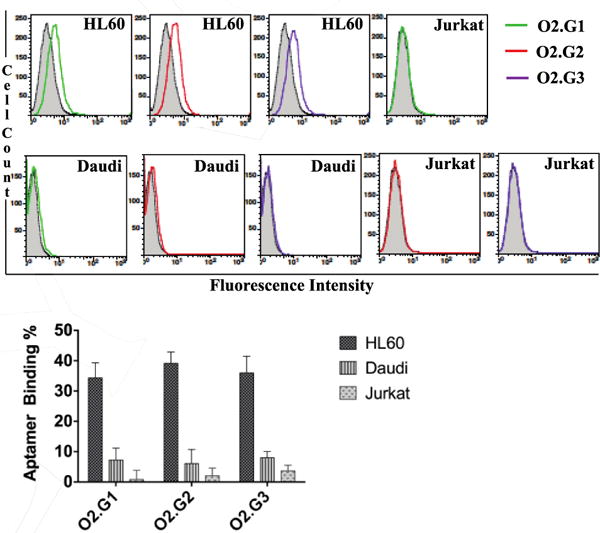
Analysis of specificity of 2′-O-methyl RNA-substituted O2.G analogues against three different cell lines. Bar diagrams represents overall conclusion from three independent specificity assays. The specificity of the aptamer sequences was evaluated by incubating 1.0 × 105 HL60 cells, Jurkat (T cells) and Daudi (B cells) with concentrations of 500nM of Fluorescein-dT-labeled aptamer separately in 200μL of binding buffer on ice for 45min. Cells were then washed once with 2.0mL of wash buffer and reconstituted in 250 μL of wash buffer. The binding of the constructs was determined using the equation [(control-aptamer)/aptamer] × 100 and quantified using Graph Pad Prism software.
The stability of all three analogues was evaluated in human serum using 50 pmoles of aptamer in 20 μL in human serum or PBS buffer. Degradation was compared at 9 different time points. Stability was evaluated up to 24 hours with intervals of 0.25, 0.5, 1, 2, 4, 3, 6, 12 and 24 hours. Interestingly, O2.G1 with 2′-O-methyl RNA bases at the 3′- end digested only 19.0±68% at the 24-hour time point (Fig. 3, top panel-right). The highest rate of digestion was observed for O2.G2 with 74.4±26.7% at 24 hours (Fig. 3, top panel-middle), suggesting that 3′-end modification showed higher stability than that of 5′-end modification. Finally, O2.G3 with modification at both 3′ and 5′ ends showed a trend similar to that of O2.G1 with 30.9±9.8% digestion at the 24-hour time point (Figure 3, top panel-left). No degradation of any modified analogue was noted when PBS was used in place of human serum (Fig. 3, bottom panel). Differences in the stability of the modified versions suggest that the 3′-end modification of aptamer using 2′-O-methyl RNA bases is more suitable in terms of increasing the nuclease stability of a DNA aptamer.
Figure 3.
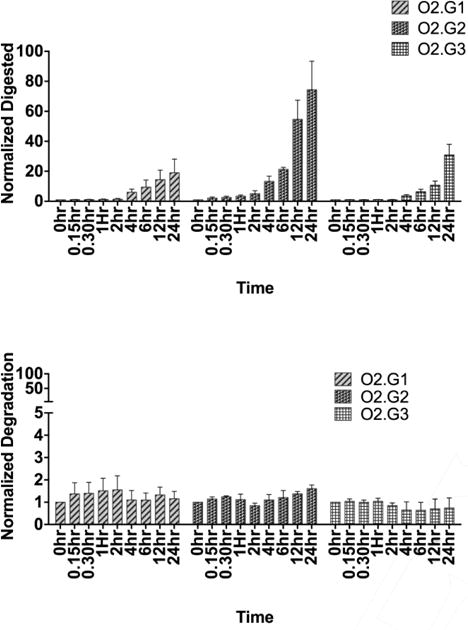
Analysis of stability of 2′-O-methyl RNA-substituted O2.G analogues in human serum (upper panel) and in PBS (lower panel). The digestion by nuclease was less than 10% up to 2 hours, with gradual increase in digestion noted after 2h. The aptamer analogues in PBS did not degrade more than 1% up to 24 hours.
NAAs are attractive synthetic molecules with which to develop therapeutics and diagnostics. Introduction of SELEX and the first aptamers occurred nearly 26 years ago.[15] Since then, significant progress has been made in developing new SELEX methods and introducing extended alphabet with chemically modified nucleic acids into aptamers, SOMAmers and spiegelmers.[16] The use of DNA aptamers during SELEX shows significant advantages over RNA aptamers, predominantly by the lack of 2′ hydroxyl group in the furanose ring, making DNA aptamers less reactive and relatively stable. However, endo- and exonuclease activity towards DNA aptamers is a major challenge in transforming naked DNA aptamers into therapeutics. One way of circumventing nuclease instability involves the use of chemically modified and stable 2′-O-X-analogues at the 3′ and 5′ ends, resulting in second-generation functional DNA aptamer analogues. Herein, we demonstrated a systematic truncation of an aptamer developed against human myeloid leukemia cells to increase stability, but without compromising affinity. In the present work, systematic introduction of 2′-O-methyl RNA at the 3′-end shows higher stability when compared to 5′-end modification. We observed stability up to 24 hours, yet only 19.0±6.70% of aptamer analogue O2.G1 had been digested by nucleases present in human serum when 2′-O-methyl RNA was added to the 3′-end of the DNA aptamer, suggesting that 3′-end modification of DNA aptamer using 2′-O-methyl ribonucleic acids is an attractive avenue to pursue for enhancement of nuclease resistance.
Supplementary Material
Acknowledgments
Authors are grateful for funding of this work NIGMS (grant 5SC3GM105578-04) grant Lauri Strauss Leukemia Foundation and NSF CBET (grant 40F68-02).
Footnotes
Supporting information for this article is given via a link at the end of the document
Experimental Section
Please see Supporting Information (SI) for detailed experimental methods.
References
- 1.Gold L, Polisky B, Uhlenbeck O, Yarus M. Annu Rev Biochem. 1995;64:763–797. doi: 10.1146/annurev.bi.64.070195.003555. [DOI] [PubMed] [Google Scholar]
- 2.Eaton BE, Gold L, Zichi DA. Chem Biol. 1995;2:633–638. doi: 10.1016/1074-5521(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 3.a Bunka DH, Stockley PG. Nat Rev Microbiol. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]; b Chu TC, Twu KY, Ellington AD, Levy M. Nucleic Acids Res. 2006;34:e73. doi: 10.1093/nar/gkl388. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Famulok M, Mayer G. Chem Biol. 2014;21:1055–1058. doi: 10.1016/j.chembiol.2014.08.003. [DOI] [PubMed] [Google Scholar]; d Gold L. Harvey Lect. 1995;91:47–57. [PubMed] [Google Scholar]; e Keefe AD, Pai S, Ellington A. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lee JF, Stovall GM, Ellington AD. Curr Opin Chem Biol. 2006;10:282–289. doi: 10.1016/j.cbpa.2006.03.015. [DOI] [PubMed] [Google Scholar]; g Ye M, Hu J, Peng M, Liu J, Liu J, Liu H, Zhao X, Tan W. Int J Mol Sci. 2012;13:3341–3353. doi: 10.3390/ijms13033341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verma S, Eckstein F. Annu Rev Biochem. 1998;67:99–134. doi: 10.1146/annurev.biochem.67.1.99. [DOI] [PubMed] [Google Scholar]
- 5.Lakhin AV, Tarantul VZ, Gening LV. Acta Naturae. 2013;5:34–43. [PMC free article] [PubMed] [Google Scholar]
- 6.a Shangguan D, Tang Z, Mallikaratchy P, Xiao Z, Tan W. Chembiochem. 2007;8:603–606. doi: 10.1002/cbic.200600532. [DOI] [PubMed] [Google Scholar]; b Eaton BE, Gold L, Hicke BJ, Janjic N, Jucker FM, Sebesta DP, Tarasow TM, Willis MC, Zichi DA. Bioorg Med Chem. 1997;5:1087–1096. doi: 10.1016/s0968-0896(97)00044-8. [DOI] [PubMed] [Google Scholar]
- 7.Mallikaratchy PR, Ruggiero A, Gardner JR, Kuryavyi V, Maguire WF, Heaney ML, McDevitt MR, Patel DJ, Scheinberg DA. Nucleic Acids Res. 2011;39:2458–2469. doi: 10.1093/nar/gkq996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Burmeister PE, Lewis SD, Silva RF, Preiss JR, Horwitz LR, Pendergrast PS, McCauley TG, Kurz JC, Epstein DM, Wilson C, Keefe AD. Chem Biol. 2005;12:25–33. doi: 10.1016/j.chembiol.2004.10.017. [DOI] [PubMed] [Google Scholar]; b Sefah K, Yang Z, Bradley KM, Hoshika S, Jimenez E, Zhang L, Zhu G, Shanker S, Yu F, Turek D, Tan W, Benner SA. Proc Natl Acad Sci U S A. 2014;111:1449–1454. doi: 10.1073/pnas.1311778111. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Thiel WH, Bair T, Peek AS, Liu X, Dassie J, Stockdale KR, Behlke MA, Miller FJ, Jr, Giangrande PH. PLoS One. 2012;7:e43836. doi: 10.1371/journal.pone.0043836. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pinheiro VB, Holliger P. Curr Opin Chem Biol. 2012;16:245–252. doi: 10.1016/j.cbpa.2012.05.198. [DOI] [PubMed] [Google Scholar]; e Kusser W. J Biotechnol. 2000;74:27–38. doi: 10.1016/s1389-0352(99)00002-1. [DOI] [PubMed] [Google Scholar]
- 9.Crouzier L, Dubois C, Edwards SL, Lauridsen LH, Wengel J, Veedu RN. PLoS One. 2012;7:e35990. doi: 10.1371/journal.pone.0035990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue H, Hayase Y, Imura A, Iwai S, Miura K, Ohtsuka E. Nucleic Acids Res. 1987;15:6131–6148. doi: 10.1093/nar/15.15.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cummins LL, Owens SR, Risen LM, Lesnik EA, Freier SM, McGee D, Guinosso CJ, Cook PD. Nucleic Acids Res. 1995;23:2019–2024. doi: 10.1093/nar/23.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi M, Minakawa N, Matsuda A. Nucleic Acids Res. 2009;37:1353–1362. doi: 10.1093/nar/gkn1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sefah K, Tang ZW, Shangguan DH, Chen H, Lopez-Colon D, Li Y, Parekh P, Martin J, Meng L, Phillips JA, Kim YM, Tan WH. Leukemia. 2009;23:235–244. doi: 10.1038/leu.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a Zumrut HE, Ara MN, Fraile M, Maio G, Mallikaratchy P. Nucleic Acid Ther. 2016;26:190–198. doi: 10.1089/nat.2016.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zumrut HE, Ara MN, Maio GE, Van NA, Batool S, Mallikaratchy PR. Anal Biochem. 2016;512:1–7. doi: 10.1016/j.ab.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]; b Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 16.a He W, Elizondo-Riojas MA, Li X, Lokesh GL, Somasunderam A, Thiviyanathan V, Volk DE, Durland RH, Englehardt J, Cavasotto CN, Gorenstein DG. Biochemistry. 2012;51:8321–8323. doi: 10.1021/bi300471d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kraemer S, Vaught JD, Bock C, Gold L, Katilius E, Keeney TR, Kim N, Saccomano NA, Wilcox SK, Zichi D, Sanders GM. PLoS One. 2011;6:e26332. doi: 10.1371/journal.pone.0026332. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Oberthur D, Achenbach J, Gabdulkhakov A, Buchner K, Maasch C, Falke S, Rehders D, Klussmann S, Betzel C. Nat Commun. 2015;6:6923. doi: 10.1038/ncomms7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.