

Abstract
Although the majority of SUMO substrates are localized in the nucleus, SUMOylation is not limited to nuclear proteins and can be also detected in extra-nuclear proteins. In this review, we will highlight and discuss how SUMOylation in different cellular compartments regulate biological processes. First, we will discuss the key role of SUMOylation of proteins in the extra-nuclear compartment in cardiomyocytes, which is overwhelmingly cardio-protective. On the other hand, SUMOylation of nuclear proteins is generally detrimental to the cardiac function mainly because of the trans-repressive nature of SUMOylation on many transcription factors. We will also discuss the potential role of SUMOylation in epigenetic regulation. In this review, we will propose a new concept that shuttling of SUMO proteases between the nuclear and extra-nuclear compartments without changing their enzymatic activity regulates the extent of SUMOylation in these compartments and determines the response and fate of cardiomyocytes after cardiac insults. Approaches focused specifically to inhibit this shuttling in cardiomyocytes will be necessary to understand the whole picture of SUMOylation and its pathophysiological consequences in the heart, especially after cardiac insults. This article is part of a Special Issue entitled: Genetic and epigenetic control of heart failure - edited by Jun Ren & Megan Yingmei Zhang.
Keywords: SUMOylation, p90RSK, SENP2, Potassium channel, SERCA2a, DRP-1, NEMO, PKCα, AMPK, Ubc9, XBP-1, PPARs, ERK5, HDACs
1. Introduction
SUMOylation is an important post-translational modification in which one or more small-ubiquitin like modifier (SUMO) peptides are conjugated to a protein and contributes to the complexity of eukaryotic proteomes. SUMOylation is a dynamic and reversible process that requires conjugation and de-conjugation enzymes.
Reversibility of protein SUMOylation is achieved by de-SUMOylation enzymes called sentrin/SUMO-specific proteases (SENPs; SENP1-7). Certain SENPs, especially SENP1 and 2, contain both nuclear localization and export signal domains, and shuttling of SENPs from one compartment of the cell to another has an effect on altering SUMOylation levels in different sub-cellular regions. Here, we will review the role of SUMOylation in both extra-nuclear and nuclear compartments of cardiomyocytes and discuss the potential impact of de-SUMOylation enzyme’s shuttling on the control of the response and fate of cardiomyocyte upon cardiac insults.
1.1. SUMOyation (SUMO conjugation)
SUMOylation is a lysine-targeted post-translational modification (PTM). SUMO is a 10-kDa polypeptide [1,2] and is covalently conjugated to targeted molecules (substrates) at a lysine (K) residue within the specific consensus motif Ψ-K-x-D/E, in which Ψ is a hydrophobic residue, x is any amino acid, and D/E represent negatively charged aspartic acid or glutamic acid residue. There are four SUMO isoforms (SUMO 1–4) in human, and SUMO1 shares ~50% sequence identity with SUMO2, while SUMO2 and 3 differ by only three NH2-terminus residues (97% identity). Therefore, SUMO2 and 3 together are considered to form a subfamily, which is distinct from SUMO1. SUMO1–3 are ubiquitously expressed whereas SUMO4 is limited to kidney, lymph node, and spleen [2,3]. The functional role of SUMO4 remains controversial, because the C-terminus proline residue unique to SUMO4 inhibits the maturation of SUMO4 into the conjugatable form (or modifier) [4].
As shown in Fig. 1, SUMOylation utilizes an enzymatic machinery similar to that of ubiquitination, which has been reviewed extensively [3]. In brief, inactive precursor SUMO is maturated by SUMO proteases sentrin/SUMO-specific proteases (SENPs) to expose the di-glycine C-terminus motif and then activated by an E1 activating enzyme that is a heterodimer containing SAE1/SAE2 subunits. Next, activated SUMO binds to the E2 conjugating enzyme (Ubc9) via the transesterification reaction to facilitate SUMO protein attachment to lysine (K) residues on target proteins in conjunction with the E3 ligating enzyme. SUMO E3 ligase acts as an adaptor between Ubc9-SUMO and the substrate protein. The efficient and targeted SUMO modification of substrates can be accomplished by SUMO E3 ligase [2]. Several SUMO E3 ligases have been identified including the protein inhibitor of activated STAT (PIAS) family (PIAS1, PIAS2(x), PIAS3, PIAS4(y)), Polycomb-2 protein (Pc2), and RanBP2/Nup358 [5]. It is thought that SUMO E3 ligases determine the substrate specificity.
Fig. 1.
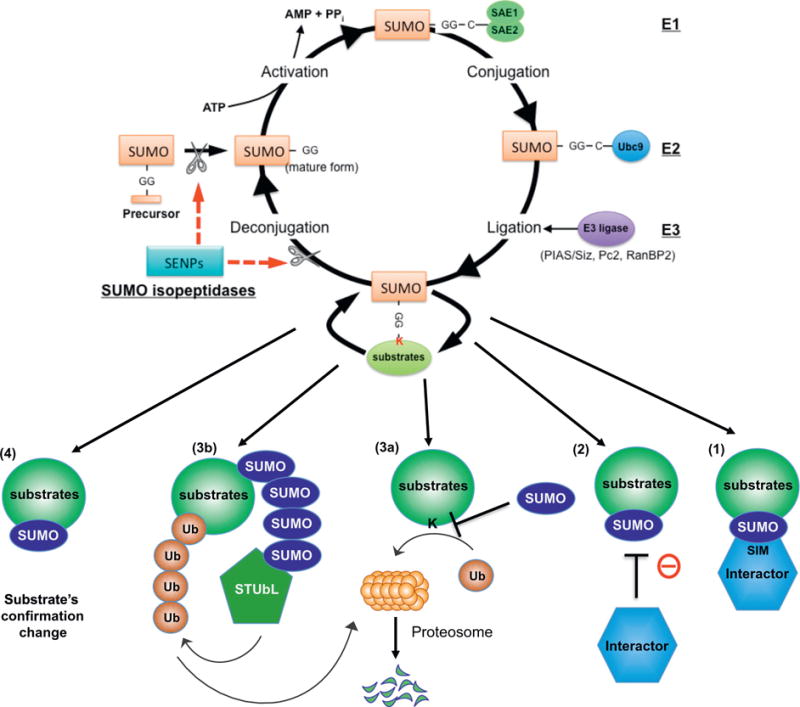
SUMOylation system and its regulatory mechanism. The SUMOylation system is a dynamic process of protein modification achieved by two enzyme systems; one which consisting of conjugates SUMO to substrates and one which de-conjugates. [176–179] SUMO proteins are covalently attached to certain residues of specific target substrates and change the function of these substrates. Prior to conjugation, the E1-activating enzyme, SAE1–SAE2 heterodimer, activates the mature form of SUMO [180]. SUMO is then transferred to Ubc9, an E2 conjugase, forming a thioester bond between Ubc9 and SUMO [181]. Lastly, SUMO E3 ligases, including a family of protein inhibitors such as activated STAT (PIAS1-4), transfer SUMO to the target substrate containing the free е-amino group of a lysine residue [182]. De-SUMOylation enzymes are also involved in the process of SUMOylation. Sentrin/SUMO-specific proteases (SENPs; SENP1–7) catalyze both de-conjugation of SUMOylated substrates and editing of the SUMO precursor into the matured form which now have a pair of glycine residues at the C terminus [183,184]. Adapted, reprinted, and modified from Heo et al. [22] with permission from ANTIOXIDANTS AND REDOX SIGNALING, April 2016, published by Mary Ann Liebert, Inc., New Rochelle, NY.
The majority of SUMO substrates are localized in the nucleus. Such nuclear substrates include proteins that regulate cell cycle progression, DNA repair and replication, and gene transcription. However, it is also becoming evident that many SUMOylated molecules are localized in extra-nuclear compartments, and they regulate protein functions including intracellular trafficking, apoptosis, protein stability, and enzyme activity [1,3,6]. SUMO-modification of substrates is known to play a major role in the following molecular events and processes: 1) SUMOylation provides a platform to recruit molecules that non-covalently bind to SUMO via SUMO-interacting motif (SIM), 2) SUMOylation can promote or block the association of molecules that interact with SUMOylated substrates, 3) SUMOylation can regulate substrate stability by competing for the lysine site with ubiquitination or degradation by recruiting the SUMO-targeted ubiquitin ligase (STUbL) family of proteins to the SUMOylated substrates, and 4) SUMOylation induces conformational changes in proteins so that their interaction with other molecules can be regulated [7,8] (Fig. 1).
1.2. De-SUMOylation (SUMO deconjugation)
SUMOylation is a dynamic and reversible modification of substrates and allows transient changes in signal transduction (Fig. 1). It is important to note that not only SUMOylation but also de-SUMOylation can play a key role in regulating cellular signaling pathways. SENPs are a family of enzymes with isopeptidase activity. In addition to cleaving the C-terminus of pro-SUMO to expose the d-glycine motif required for conjugation of mature SUMO as explained above, SENPs can catalyze de-conjugation of SUMO from target proteins. Six isoforms of SENP (1–3, 5–7) exist in humans and they exhibit preference toward distinct SUMO forms. SENP1 and 2 are involved in both maturation and de-conjugation of both SUMO1 and 2, although SENP2 has a lower processing (maturation) activity but a higher de-conjugating activity [9–12]. SENP3 and 5 favor SUMO2 and 3 over SUMO1 for both maturation and de-conjugation [13–15]. SENP6 and 7 cannot effectively remove monomeric SUMOs but are able to cleave polymeric chains of SUMO2/3 and edit lysine-linked polySUMO2/3 chains [11,16]. In addition to the SENP family, de-SUMOylating isopeptidase 1 and 2 (DeSI1 and 2) and ubiquitin-specific protease-like 1 (USPL1) can also function as SUMO-protease. So far, BTB-ZF is the only molecule identified as a target of DeSI1 [17], and USPL1 acts preferentially on SUMO2/3 and edits polySUMO2/3 chains with a limited capacity for the general pro-SUMO processing [18]. Since most SENPs have both pro-SUMO processing (maturation) and de-conjugation functions and since maturation can promote SUMO conjugation, the level of SUMOylation of each substrate may depend on the combined effects of SUMO ligases and SENPs. Further investigations will be necessary.
Different SENPs appear to have certain overlapping substrates, but they can also have a high degree of substrate specificity. SUMOylation of hypoxia-inducible factor 1α (HIF1α) decreases its stability leading to its degradation in response to hypoxia. SENP1 enhances HIF1α stability through de-SUMOylation. Indeed, HIF1α in SENP1−/− mouse embryos shows increased levels of SUMOylation and reduced levels of stability. In contrast, HIF1α stability is not affected in SENP2−/− embryos, indicating the specificity of SENP1 and 2 in regulating HIF1α SUMOylation [19,20].
It is interesting that both SENP1 and SENP2 are self-regulated through transcriptional feedback loops. For example, under conditions of genotoxic stress, there is an early increase in SENP2 mRNA followed by de-SUMOlyation of NEMO (NF-κB Essential Modulator)/IKK (inhibitor of κB kinase) γ by SENP2. NEMO then decreases SENP2 transcription thereby creating a negative feedback loop to prevent the survival of damaged cells [21].
1.3. SENPs sub-cellular localization
SENPs are mainly localized in the nucleus, but certain SENPs, especially SENP1 and 2, contain both nuclear localization and export signal (NLS and NES) domains and are able to shuttle between the nucleus and the cytoplasm. Such changes in the localization of SENPs within a cell have profound effects on SUMOylation levels in different cellular compartments (Fig. 2). Recently, we have reported the crucial role of SENP2 phosphorylation in regulating SENP2 nuclear export [22]. The NLS is located near the N-terminus of SENP2 and the leucine-rich, CRM1-dependent NES sequence in the central region. Because we found the involvement of both p90RSK kinase activity and SENP2 in disturbed flow (d-flow)–initiated p53 SUMOylation and subsequent apoptosis, we investigated whether p90RSK could directly phosphorylate SENP2. We subjected phosphorylated SENP2 to proteolytic digestion and liquid chromatography–tandem mass spectrometry and identified T368 as a SENP2 phosphorylation site by p90RSK. SENP2-T368A mutant inhibited p53 SUMOylation by d-flow, suggesting that SENP2-T368 phosphorylation is a key regulator of p53 SUMOylation and plays a critical role in EC apoptosis. Furthermore, we found that d-flow increased SENP2 nuclear export, which was significantly inhibited by the T368A mutation. In addition, although it was very low in the steady laminar flow area, a robust increase in anti-phospho-SENP2-T368 staining was detected in the cytoplasm of ECs in the d-flow areas of the mouse aorta. Taken together, these data suggest the crucial role of SENP2 T368 phosphorylation in regulating SENP2 nuclear export, which subsequently upregulates nuclear p53 SUMOylation (Fig. 2). The extra-nuclear localization of SENP1 [23], DeSI1, DeSI2 [17], and USPL1 [18] was also reported, but the regulatory mechanisms and functional consequences of different sub-cellular localization of these SUMO proteases are unclear. The role of SUMOylation in the nucleus and the cytoplasm can be very different. In the following two sections, we will review the role of SUMOylation in the extra-nuclear and the nuclear compartments and discuss how such compartmentalization can be achieved and what consequences it may have on the regulation of cellular functions.
Fig. 2.
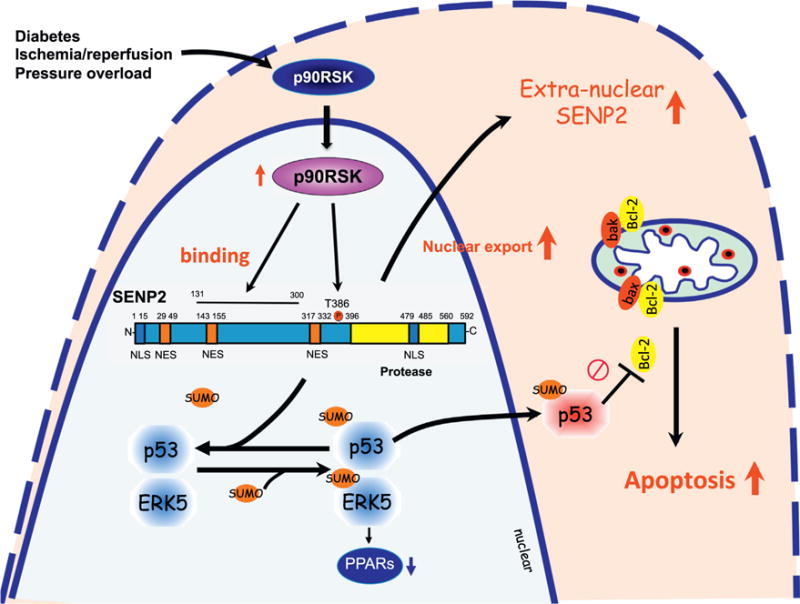
SENP2 shuttling between the nuclear and extra-nuclear compartments: SENP2 contains a bipartite nuclear localization signal (NLS) sequence at the N terminus domain and a leucine-rich, CRM1-dependent nuclear export signal (NES) sequence in the central region. These NLS and NES sequences are involved in SENP2 shuttling between the nucleus and the cytoplasm, which regulates levels of SUMOylation of proteins in these compartments. Activated p90RSK associates with SENP2, and phosphorylates T368 site, leading to SENP2 nuclear export. This nuclear export diminishes the nuclear SENP2 de-SUMOylation function in the nucleus, and consequently up-regulates SUMOylation of p53 and ERK5in the nucleus. p53 SUMOylation increases p53 nuclear export and binds with Bcl-2 in the cytoplasm, which inhibits Bcl-2 anti-apoptotic effects, and induces apoptosis. ERK5 SUMOylation inhibits transcriptional activity of PPARs, which can be detrimental to regulating the cardiac function and remodeling after cardiac insult.
1.4. SUMOylation events in extra-nuclear compartments
In this section, we will discuss the functional consequences of various SUMOylated substrates and events localized in (a) the plasma membrane; K+ channel, (b) the sarcoplasmic reticulum (SR) membrane; SR Ca2+ ATPase 2a (SERCA2a), (c) the mitochondrial membrane; DRP1, and (d) the cytosol; NF-κB essential modulator (NEMO)/inhibitor of κB kinase (IKK)γ, protein kinase C α (PKCα), adenosine monophosphate-activated protein kinase (AMPK), and ubiquitin-proteasome system (UPS) (Fig. 3).
Fig. 3.
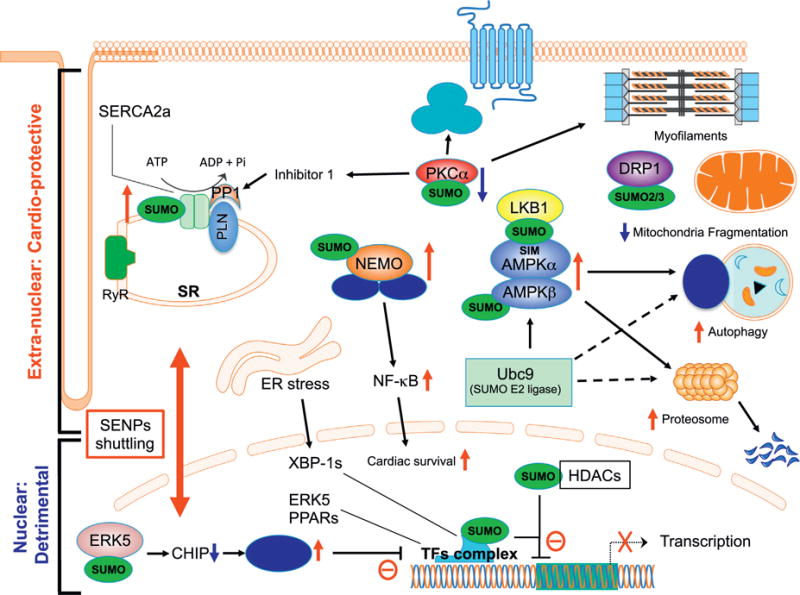
Diagram of signaling in a cardiomyocyte showing how SUMO modification differentially controls extra-nuclear and nuclear events, which can be coordinately regulated by the shuttling of SENPs between these two compartments. In the cytoplasm, up-regulation of SUMO modification on SERCA2a, NEMO, LKB1, and AMPKs up-regulates their functions, while SUMOylation of PKCa and DRP1 inhibits PKCa kinase activation and DRP1 function for inducing mitochondrial fragmentation. In the nucleus SUMOylation of proteins in the tissue factor complex including XBP-1s, ERK5, and PPARs inhibits their transcriptional activity. ERK5 SUMOylation inhibits ERK5-mediated ICER reduction by inhibiting CHIP E3 ligase activity, which can also inhibit TFs activity. SUMOylation of HDACs can augment their function, leading to accelerated inhibition of transcription. These data suggest that SUMOylation events in the extra-nuclear compartments in cardiomyocytes are cardio-protective, while the nuclear SUMOylation events are detrimental to these cells. We propose the shuttling of SENPs between the two compartments plays an important role in the response to various forms of cardiac insults and decides the fate of the cardiomyocytes.
1.4.1. Plasma membrane; potassium channels
1.4.1.1. Kv1.5 (potassium voltage-gated channel subfamily A member 5, KCNA5)
Kv1.5 is responsible for the IKur repolarizing current in atrial myocytes and also regulates vascular tone in peripheral vascular beds. It has been reported that de-SUMOylaion of Kv1.5 mediated by SENP2 leads to a substantial hyperpolarizing shift in the voltage dependence of steady-state inactivation [24]. Since significant V50 shift of Kv1.5 in the depolarizing direction could not be detected by overexpressing SUMO3 and Ubc9, neither Ubc9 nor SUMO3 appears to regulate Kv1.5 function. However, the inhibition of Kv1.5 SUMOylation induced by the cytoplasm-targeted and constitutively active SENP2 with de-SUMOylation activity caused a significant hyperpolarization shift in the voltage dependence of inactivation without altering the total current density or voltage dependence of Kv1.5 activation, suggesting that changes in localization of SENP2 regulate Kv1.5 function. Kv1.5 is widely expressed in the cardiovascular system [25], and its role in the familial form of atrial fibrillation [26] has been reported [27]. Further investigation is necessary to determine the exact molecular events that lead to Kv1.5 SUMOylation and the role of SENP2 in this process.
1.4.1.2. Potassium voltage-gated channel subfamily Q member (KCNQ)
Five KCNQ genes (KCNQ1 to KCNQ5) codify a family of 5 different voltage-gated potassium ion channels (KV7.1 to KV7.5), which are mainly expressed in the nervous and cardiac systems [28]. Yeh’s group has elucidated SENP2’s role in Kv7.2 SUMOylation using mice homozygous for the floxed SENP2 allele with a neomycin insert (SENP2fxN/fxN) which induced a significant reduction in SENP2 transcription and protein expression. These SENP2fxN/fxN mice appeared healthy at birth, but developed convulsive seizures followed by sudden death at 6–8 weeks of age. Reduced SENP2 levels in these mice created a hyper-SUMOylation environment and led to accumulation of SUMO-1 and SUMO-2/3 proteins in the brain and heart [29]. It is worth noting that the expression of SENP2 is abundant in the hippocampal region, which is a highly relevant area of the brain for seizure. Yeh’s group also found that SUMOylation of Kv7.2 was significantly enhanced in hippocampal neurons. Hyper-SUMOylation of this potassium channel protein diminished the M-current, which is conducted by Kv7, leading to a more positive resting membrane potential and increased excitability of hippocampal neurons. These data suggest the pathophysiological role of SENP2 in epilepsy via regulating the plasma membrane Kv7.2 function. The possible role of Kv7.2 SUMOylation in regulating cardiac arrhythmia is under investigation.
1.4.1.3. Potassium channel subfamily K member 1 (KCNK1)
KCNK1 is expressed in the heart and the central nervous system and regulates the sinus rhythm [30] and background leak currents stabilizing neuronal excitability [31]. KCNK1 can be SUMOylated at K274, and the point mutation of this lysine to glutamic acid (KCNK1 K274E) has been shown to increase KCNK1 current [32], suggesting that SUMOylation is inhibitory for this channel. Since overexpression of SENP1 inhibited KCNK1 SUMOylation and also increased KCNK1 current [33], SENP1 can be crucial for the regulation of KCNK1 SUMOylation and subsequent generation of K current. At present, however, the regulatory mechanism of KCNK1 SUMOylation in response to various stimuli remains unclear.
In summary, SENP1 and 2 are de-SUMOylation enzymes and play a major role in regulating the SUMOylation state of certain potassium channels and consequently their channel activities. However, it is not yet fully understood how the action of these de-SUMOylating enzymes is controlled in the cell. Obvious mechanisms would be to regulate expression levels of these enzymes, to regulate their enzymatic activity, or to control both of these aspects. Another mechanism, which is highlighted in this review, is to regulate intracellular localization of these SENPs without changing their levels of expression and enzymatic activity. SENPs are generally considered to function in the nucleus, but once they are exported from the nucleus, they can function in the extra-nuclear compartment including the plasma membrane. Such a mechanism may indeed play a role in the regulation of certain potassium channels by SENP1 and 2.
1.4.2. SR membrane; SERCA2a SUMOylation - SUMO1 vs. SUMO2/3
SERCA2a is a transporter found in the SR membrane of cardiomyocytes and transports Ca2+ into and out of the SR, and its dysfunction results in various types of heart failure. Hajjar’s group has reported the role of SERCA2a SUMOylation by SUMO1 in the process of heart failure [34] (Fig. 3). They found that SERCA2a was SUMOylated at two sites, K480 and K585, and that mutation of these lysine residues to arginine significantly decreased the ATP-binding affinity and ATPase activity of SERCA2a. In addition, they found that SUMOylation stabilized SERCA2a in the cell by inhibiting ubiquitination and subsequent proteosomal degradation. These data suggest that SERCA2a SUMOylation can improve cardiac function via up-regulating its activity and expression. To support these findings the authors also performed SUMO1 gene delivery experiments in both mice [34] and pigs[35] and used a small molecule activator of SERCA2a SUMOylation (N106) [34, 36] and found that these interventions which promoted SERCA2a SUMOylation improved cardiac function. Moreover, they showed that the improved cardiac function by elevated SUMO1 levels and that this improvement was diminished by the depletion of SERCA2a. These data appear to suggest a possible role of SERCA2a in the SUMO1-induced improvement of cardiac function. However, since it is well known that the depletion of SERCA2a inhibits the whole process of excitation-contraction coupling. Therefore, these experiments cannot exclude the involvement of other SUMOylated substrates, which can improve cardiac function independent on SERCA2a.
The role of SUMO1 in cardiac function was tested in cardiac muscle specific Sumo1-overexpressing transgenic mice, and the result showed that pressure overload-induced cardiac hypertrophy and dysfunction were inhibited by SUMO1 overexpression, suggesting the cardio-protective effect of SUMO1 [34]. In contrast, cardiac specific Sumo2 overexpressing mice showed dose-dependent cardiac hypertrophy and dysfunction [37]. In this study, the contribution of calpain 2 and calpastatin SUMOylation to cardiomyocyte apoptosis has been suggested. SUMO2-modified calpastatin was shown to be degraded more efficiently, which subsequently increased calpain 2 expression. SUMOylation of calpain 2 by SUMO2 increased its enzymatic activity. These results indicate that SUMO2 modification up-regulates the function of the calpastatin-calpain proteolytic system and consequently accelerates apoptosis. The SERCA2 SUMOylation in SUMO2 transgenic mice was not tested in this study. Since Kho et al. have only tested the level of co-immunoprecitation of SUMO2 with SERCA2a in human heart samples with no positive control [34], the contribution of SUMO2/3 in SERCA2a SUMOylation cannot be excluded. It is possible that SUMOylation of distinct sets of different substrates targeted by either SUMO1 or SUMO2/3 determines the beneficial and detrimental effects of SUMO1 and SUMO2, respectively, on cardiac function. However, as we will explain later, dynamin-related protein 1 (DRP1) SUMOylation by SUMO2/3 can also be protective of cardiac function. Therefore, we suggest that the existing contradictory data on the regulation of cardiac function by SUMOylation is not due so much to the difference between SUMO1 and SUMO2/3 but to the gene induction methods, which may force the expression of different SUMOs in different sub-cellular compartments. Since both SUMO1 and SUMO2 knock out mice are embryonic lethal [38,39], both are critical, at least, for embryonic development. Inducible cardiac specific SUMO1 and SUMO2 knock out mice may be necessary to clarify the role of SUMO isoforms in adult heart after cardiac insults.
1.4.3. Mitochondrial membrane; DRP1 SUMOylation - SUMO1 vs. SUMO2/3
Mitochondria are dynamic organelles whose overall morphology changes in response to cellular activity [40]. Such morphological changes include fission and fusion (i.e. elongation), which are under the tight regulation dictated by the physiological state of the cell [41,42]. Mitochondrial fusion is achieved by integrating outer and inner membranes of one mitochondrium with those of another mitochondrium and fission by pinching off of a mitochondrium in a manner similar to cytokinesis of animal cells. GTPase proteins from the dyamin family including mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy-1 (Opa1) are involved in the process of mitochondrial fusion [43,44], and ubiquitin ligases such as membrane-associated RING finger 5 (MARCH5) regulate the overall process [45]. Fission of mitochondria occurs in a sequential manner. First, constriction of mitochondrial tubules takes place. Next, the GTPase called DRP1 is mobilized from the cytosol to the outer membrane of mitochondria via several receptor proteins [46]. Upon reaching the outer membrane, DRP1 assembles into a scission complex by forming a spiral that surrounds the tubule. Then, in a GTP dependent manner, the DRP1 complex constricts the tubule to cause scission [46,47]. Lastly, the complex is disassembled.
It has been reported that SUMOylation of DRP1 regulates this ATPase and thus, also regulates mitochondrial fission (Fig. 3). Mitochondria-associated protein ligase (MAPL) is a 40 kDa protein located on the outer mitochondrial membrane[48]. Although MAPL can participate in the process of both ubiquitination and SUMOylation, under physiologic conditions MAPL preferentially functions as a SUMO E3 ligase for DRP1 SUMOylation (SUMO1) [49]. MAPL-mediated DRP1 SUMOylation (SUMO1) increases mitochondrial fission and hyper-fragmentation [50–52], leading to apoptosis in Cos7 and HeLa cells [48,49,53]. Cytochrome c functions as the terminal trigger for apoptotic cell death and is located in the intermembrane space [54]. Release of cytochrome c has been shown to depend on MAPL-mediated SUMOylation (SUMO1) of DRP1 [55]. The level of DRP1 SUMOylation can be reduced by de-SUMOylation enzymes including SENP5, SENP3, and SENP2 [56,57]. SENP5 is involved in mitochondrial fission via its interaction with DRP1 [52,58] and removes SUMO1, SUMO2, or SUMO3 [13,52]. DRP1 can be SUMOylated not only by SUMO1 but also by SUMO2 and 3. Interestingly, DRP1 modification by different SUMO isoforms has different functional consequences [56]. Depletion of SENP5 in COS7 and HeLa cells increases DRP1 SUMO1 modification by MAPL, leading to DRP1 association with mitochondria and increasing mitochondrial fragmentation and cellular apoptosis [50–52]. In contrast, cardiomyocyte-specific overexpression of SENP5 inhibits DRP1 SUMO2/3 modification and induces apoptosis via promoting the association of DRP1 with mitochondria [59]. These data suggest that DRP1 SUMO2/3 modification inhibits DRP1 recruitment to mitochondria whereas DRP1 SUMO1 modification promotes DRP1 binding to mitochondria and consequently up-regulates apoptosis [56]. Further studies are necessary to determine specific roles for the DRP1-SUMO1 and DRP1-SUMO2/3 conjugates in the process of mitochondrial fission.
Although SENP5 is localized primarily in the nucleus, a substantial amount of this enzyme is also found in the cytoplasm or the extra-nuclear compartment of the cell [51]. Zunino et al. have reported SENP5 translocation from the nucleus to the mitochondria during the G2/M transition [52]. Although the regulatory mechanism for this SENP5 translocation is unknown, this is another example that translocation of SUMO proteases between different intracellular compartments can occur, and such movements of de-SUMOyation enzyme may be able to alter SUMOylation states of various proteins located in different parts of the cell and regulate various cellular functions.
1.4.4. Cytosolic molecules; NEMO/IKKγ, protein kinase C α (PKCα), adenosine monophosphate-activated protein kinase (AMPK), and ubiquitin-proteasome system (UPS)
1.4.4.1. NEMO/IKKγ
NF-κB is a master regulator of cell survival and inflammation and plays an important role in various cardiac pathogenesis including heart failure [60], ischemic preconditioning [61], and apoptosis [62]. Under resting conditions, NF-κB is in an inactive state within the cytoplasm bound to IκB (Inhibitor of κB). When cells are stimulated by agonists such as TNF, IKK (IκB kinase) is activated and phosphorylates IκB, which then leads to degradation of IκB via ubiquitination. This not only frees NF-κB, but also unmasks its nuclear localization signal. NF-κB then translocates into the nucleus and transactivates pro-inflammatory genes [63]. SUMOylation has been reported to be involved at different levels of NF-κB regulation. IκBα can be modified by SUMO1 to protect it from ubiquitination and degradation, limiting NF-κB activation [64]. In contrast, modification of IκBα by SUMO2/3 confers the opposite effect and dissociates IκBα from NF-κB leading to NF-κB activation [65]. However, the exact pathophysiological role of IκBα SUMOylation remains unclear.
The IκB kinase (IKK) complex is an important regulatory kinase complex in regulating NF-κB signaling [66,67]. The complex consists of two kinases (IKKα and IKKβ) and a regulator subunit NEMO, also called IKKγ and is mainly expressed in the cytoplasm. NEMO/IKKγ does not possess catalytic activity but is required for IKK activation and its subsequent phosphorylation of IκB [68]. It has been reported that depletion of NEMO in cardiomyocytes promotes apoptosis and subsequent cardiac dysfunction via inhibiting the expression of anti-oxidant genes such as superoxide dismutase 2 and ferritin heavy chain. These data suggest that activation of NEMO/IKKγ-NF-κB signaling can be cardio-protective by inhibiting apoptosis [69]. Miyamoto’s group has reported that SUMOylation of NEMO during genotoxic stress leads to increased IKK activation and thus, NF-κB activation [70–76] (Fig. 3). Interestingly, a de-SUMOylation enzyme SENP2 is a downstream transcriptional target of NF-κB thereby creating a negative feedback mechanism for NF-κB activation through NEMO de-SUMOylation [21]. SENP6 attenuates Toll-like receptor-triggered inflammation in the endotoxin-induced murine sepsis model via de-SUMOylation of NEMO at K277 [77]. However these pathways are not well-characterized in the heart and await further investigation.
1.4.4.2. Protein kinase Cα
PKC contains multiple putative SUMOylation sites and modification by SUMO can affect its activity. Inactive PKCα is SUMOylated at the Lys465 site which can be de-SUMOylated by SENP1. Using rodent spinal cord neuronal cells, it was shown that PKCα activation by calcium was achieved only after de-SUMOylation by SENP1 [78]. This shows that SUMOylation can play an inhibitory role in PKCα kinase function. PKCα is known to negatively regulate cardiomyocyte contraction for which three specific extra-nuclear PKCα substrates play roles [79]. First, PKCα phosphorylates inhibitor 1 (I-1) at Ser67, which up-regulates protein phosphatase 1 activity, leading to greater phospholamban (PLN) de-phosphorylation and reduced activity of the SR Ca2+ ATPase (SERCA2) pump [80]. Second, PKCα activation increases G protein-coupled receptor kinase 2 (GRK2) phosphorylation and activity, and impairs β-agonist-stimulated ventricular function via abolishing cyclase activity [81]. Lastly, PKCα can phosphorylate cardiac troponin I (cTnI), cTnT, titin, and myosin binding protein C, the effect of which is to decrease the Ca2+ sensitivity and contractility of cardiomyocytes [79,82–85] (Fig. 3). These data suggest that inhibition of PKCα kinase activity by SUMOylation can be cardio-protective.
1.4.4.3. Adenosine monophosphate-activated protein kinase (AMPK) and ubiquitin-proteasome system (UPS)
AMPK is a stress-activated kinase, which can orchestrate the cellular response to a variety of stresses in the heart by regulating metabolism, protein synthesis, degradation, autophagy, and apoptosis [86]. AMPK is a complex of three subunits: a catalytic subunit (α) containing a serine-threonine kinase domain (KD) with a Thr172 phosphorylation site which is the target of liver kinase B1 (LKB1) and calcium-calmodulin-activated protein kinase kinase-β (CAMKKβ) and two regulatory subunits (β and γ) [86]. In unstressed cells, AMPK is mainly localized in the cytoplasm but can translocate to the nucleus after its activation [86]. Most studies have shown that endogenous AMPK activation is protective against cardiac insults including ischemia/reperfusion and pressure overload [86–90]. For example, depletion of AMPKα2 decreased glycolysis, lowered ATP levels, and impaired cardiac function after ischemia [91,92]. In addition, depletion of AMPKα2 resulted in greater cardiac hypertrophy and contractile dysfunction after pressure overload [93]. Rubio et al. have reported that AMPK SUMOylation (SUMO2) stimulates AMPK activation and inhibits its ubiquitin-dependent degradation [94] (Fig. 3). In addition, Yeh’s group has recently shown that LKB1 K178 SUMO1 modification promotes LKB1 association with AMPKα SIM, and accelerates AMPK activation [95] (Fig. 3). Taken together, these data suggest that the LKB1-AMPK SUMOylation can be cardio-protective.
It has been well established that accumulation of misfolded proteins contributes to the pathogenesis of heart failure. The ubiquitin-proteosome system (UPS) and selective autophagy are the two major mechanisms responsible for the removal of misfolded proteins, and both processes mainly occur in the cytoplasm. Robbins and his colleagues showed that depletion of Ubc9, the SUMO E2 conjugating enzyme, in cardiomyocytes caused accumulation of protein aggregates inside these cells and impaired cardiac function [96]. In addition, they also showed that Ubc9-mediated SUMOylation increased autophagy, which led to reduction of protein aggregate formation, fibrosis, and hypertrophy while at the same time, improving cardiac function and survival [97]. These studies were done using mice expressing a mutant α-B-crystallin in a cardiomyocyte specific manner (Fig. 3). It is unclear how Ubc9 regulates UPS and autophagy, but it is tempting to speculate the involvement of AMPK in this process, because AMPK SUMOylation increases AMPK activation which can upregulate UPS and autophagy.
In summary, most of the cardiac extra-nuclear SUMOylation events, except for a potassium channel and the calpastatin-calpain proteolytic system, are cardio-protective against cardiac insults (Table 1). It is important to state here that Wykoff et al. have reported that by utilizing a collection of epitope-tagged yeast strains, they found 82 proteins associated with SUMO [98]. In addition, by using immobilized metal affinity chromatography, Vertegaal et al. have identified 53 SUMO-conjugated proteins including 44 novel SUMO targets in HeLa cells [99]. These data suggest the possible existence of unstudied SUMOylated proteins in the heart. Of note, they also showed that SUMOylation was strongly related to transcription because nearly one-third of the identified target proteins are putative transcriptional regulators [99]. Therefore, to understand the whole picture of how SUMOylation regulates cardiac function, it is crucial to know the functional role of SUMOylation in regulating nuclear molecules including transcriptional factors. We will discuss the pathophysiological role of SUMOylation events in the nucleus in the next section.
Table 1.
Summary of transgenic models, phenotypes, and effects of SUMOylation (extra-nuclear events).
| Molecule | Transgenic model | Model characteristics | Phenotypes and the effect of SUMOylation |
|---|---|---|---|
| KCNA5 (Kv1.5) |
Kcna5 Replace mKv1.5 with 4-aminopyridine (4-AP)-insensitive channel rat Kv1.1 (SWAP mice) [162] |
|
|
| KCNK1 | Kcnk1−/− [163] |
|
|
| SERCA2a | Cardiac specific-Serca2a−/− [164] |
|
|
| DRP1 | Cardiac specific-inducible Drp1−/− |
|
|
| NEMO |
Nemo−/− Cardiac specific Nemo−/− [69] |
|
|
| PKCα |
PKCα−/− Cardiac specific PKCα−/− [80] |
|
|
| AMPK |
Ampkα1−/− [168,169] Ampkα2−/− [168,170] Ampkβ1β2−/− Skeletal and heart specific (under the control of muscle creatine kinase promoter) Ampkβ1β2−/− [171] Kinase dead Ampkα2 transgenic mice with muscle creatine kinase promoter (skeletal and cardiac expression) [87]. |
|
|
| LKB1 | Lkb1−/− [172] |
|
|
| UBC9 | Cardiac specific Ubc9 overexpression [97] |
|
|
1.5. SUMOylation in the nuclear compartment
In this section, we will discuss functional consequences of various SUMOylated substrates in the nucleus including (a) endoplasmic reticulum (ER) stress-mediated transcription factors, XBP-1s (spliced X-Box Binding Protein 1), (b) peroxisome proliferator-activated receptors (PPARs), (c) ERK5 (extracellular-signal regulated kinase 5)-CHIP (carboxyl terminus of HSP70-interacting protein)-ICER (inducible cAMP early repressor) complex, and (d) HDACs (histone deacetylases). Notable effects of SUMOylation in the nucleus are due to its trans-repression activity against a variety of transcriptional factors and SUMO modification of transcription factors and cofactors, which results in transcription repression [3,100] (Fig. 3). One of the possible inhibitory mechanisms is that covalent attachment of SUMO provides a new interaction interface that mediates recruitment of transcriptional corepressors [101]. For example, SUMOylation of transcriptional factor Elk-1 increases its affinity to HDAC2, which then induces histone de-acetylation and transcriptional repression of the c-fos promoter [102]. Another possible mechanism may involve REST corepressor 1 (RCOR1). RCOR1 plays a role as a corepressor by recruiting the RCOR1/KDM1 (histone lysine-specific demethylase)/HDAC1 and 2 complex to transcription factors and by increasing de-acetylation and de-methylation of histone tails to generate a repressive chromatin structure [103]. RCOR1 binds directly to SUMO2 with its non-consensus SUMO-interaction motif (SIM) [103]. This association is crucial for recruiting the RCOR1-KDM1-HDAC1/2 complex to various covalently SUMO-modified transcription factors, altering the acetylation and methylation status of histone and leading to transcriptional repression. As we will explain in this section, SUMOylation also has significant effects on the function of HDACs, which adds more complexity to the mechanism of SUMOylation-mediated trans-repression and its functional consequence.
1.5.1. Endoplasmic reticulum (ER) stress-mediated transcription factors; XBP-1s
ER stress is one of the endogenous sources of cellular stress, which occurs following the accumulation of misfolded proteins in the ER. To counterbalance the ER stress, cells utilize unfolded protein response (UPR), a three-pronged signaling pathway to restore the proper ER function. This entails inhibiting general protein synthesis but promoting ER processing in order to reduce misfolded protein aggregation in ER. Depending on the degree of stress, UPR can be either pro-survival or pro-apoptotic [8]. The three-pronged UPR pathway is mediated by three distinct ER-localized trans-membrane proteins: inositol-requiring kinase 1 (IRE1), PRKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [104]. It has been reported that IRE1 and PERK pathways are regulated by SUMOylation [105]. IRE1 possesses both kinase and endonuclease activities. Unfolded proteins in ER activate IRE1 and increase its auto-phosphorylation, which up-regulates IRE1 endoribonuclease activity and excises a 26 base pair fragment from unspliced XBP1 mRNA. Following religation by a putative tRNA ligase, active forms of spliced XBP1 mRNA are generated and XBP1 protein is synthesized. It has been reported that SUMOylation of XBP1s (by both SUMO1 and SUMO2/3) by PIAS2, a SUMO E3 ligase, inhibits transcriptional activity of XBP1s and subsequent UPR target gene expression [106] (Fig. 3). In addition, SENP1 has been suggested to play a role in maintaining ER-stress-mediated XBP1 activity by de-SUMOylating it [107]. XBP1 knock out mice are embryonic lethal [108]. In addition, it has been reported that overexpression of dominant negative XBP1 increases apoptosis in isolated cardiomyocytes in response to ischemia/reperfusion [109], suggesting that activation of ER stress may be cardio-protective. It appears, therefore, that inhibition of transcriptional activity of XBP1s by SUMOylation in the nucleus is detrimental after cardiac insult. Future study is necessary to verify this prediction.
1.5.2. Peroxisome proliferator-activated receptor isoforms (PPARs)
Cardiac energy substrate utilization is critically controlled by the PPAR family of transcription factors including PPARα, PPARβ/δ, and PPARγ, and all three isoforms are exclusively localized in the nucleus. Systemic PPARα null mice display decreased cardiac fatty acid oxidation rates while glucose oxidation rates and reliance on glucose metabolism for ATP production are increased [110,111], the condition of which induces cardiac dysfunction [112]. Cardiomyocyte specific deletion of PPAR-δ in mice showed reduced expression of key fatty acid oxidation genes and also reduced basal myocardial fatty acid oxidation, leading to progressive myocardial lipid accumulation, cardiac hypertrophy, and congestive heart failure with reduced survival [113]. Cardiomyocyte specific PPARγ null mice exhibited progressive cardiac hypertrophy with mitochondrial oxidative damage, and most mice died from dilated cardiomyopathy [114]. All three PPAR isoforms can be SUMOylated, and SUMO modification of the ligand binding domain of PPARα and γ inhibits their transcriptional activity [115,116] (Fig. 3). Although the specific SUMOylation sites of PPARβ/δ were not determined, SUMOylation of PPARβ/δ promoted their degradation and inhibited their transcriptional activity [117]. Although the precise role of SUMOylated PPARs in the heart remains unclear, these data suggest that SUMOylation of PPAR isoforms may be detrimental to maintaining cardiac function after various cardiac insults.
1.5.3. ERK5-PPARs
Although the localization of overexpressed ERK5 was cytosolic in resting cells and shifted to the nucleus after activation [118], Raviv et al. reported that endogenous inactive ERK5 was also exclusively expressed in the nucleus [119], which we have independently confirmed (data not shown). ERK5 plays a critical role in regulating the apoptotic pathway of the cardiomyocyte. ERK5 is a unique kinase that also possesses transcriptional activity and functions as a transcriptional co-activator for PPARs and myocyte enhancer factor-2 (MEF2), down-regulating the expression of proteins involved in apoptosis in endothelial cells and cardiomtocytes (Fig. 4). Mice with cardiomyocyte-specific ERK5 knockouts exhibit accelerated dysfunction and apoptosis of cardiac muscle cells after thoracic aorta constriction [120]. We have reported that SUMOylation at the N-terminus region (K6 and K22) of ERK5 significantly inhibits the C-terminus ERK5 transcriptional activity, which is independent on its kinase activation [121]. We found that deletion of PIAS1 significantly inhibited cardiomyocyte apoptosis induced by reactive oxygen species (ROS) and that overexpression of ERK5 K6R/K22R SUMOylation mutants also reduced ROS-induced apoptosis. These data suggest that ERK5 SUMOylation plays a major role in regulating cardiomyocyte apoptosis [122] (Fig. 4). Diabetic mediators (H2O2 and AGEs) and ischemia under the diabetic condition also increase ERK5 SUMOylation and promote apoptosis in cardiomyocytes in both in vitro and in vivo [121–123]. The contribution of PPARs to the ERK5 transactivation-mediated anti-inflammatory effect has been established [121,124], but their role in cardiomyocyte apoptosis mediated by SUMOylated ERK5 needs further investigation.
Fig. 4.

The primary structure of ERK5 and its regulation. ERK5, also called a big MAP kinase, is twice the size of other MAPKs and hence the largest kinase within its group. It possesses a catalytic N-terminus domain including the MAPK-conserved threonine/glutamic acid/tyrosine (TEY) motif in the activation loop with 50% homology with ERK1/2, and a unique C-terminus tail with transactivation domains. The activation of ERK5 occurs via interaction with and dual phosphorylation in its TEY motif by MEK. On the other hand, inflammatory stimuli or athero-prone flow (d-flow) leads to ERK5 deactivation via phosphorylation of Ser486 or Ser496, respectively. SUMO modification of the N-terminus K6 and K22 sites inhibit its own transactivation. B. Insulin growth factor-1 (IGF-1) or pre-conditioning activates ERK5 kinase activity in cardiomyocytes, leading to phosphorylation of the TEY motif and de-SUMOylation of the two sites which then fully activates ERK5 transcriptional activity. In contrast, ischemia under the diabetic condition (DM + MI), reactive oxygen species (ROS), and advanced glycation endo-products (AGE) increase ERK5 SUMOylation and ERK5 Ser496 phosphorylation, inhibiting ERK5 transcriptional activity and promoting ERK5 degradation via ERK5 Ser486 phosphorylation. CHIP; carboxyl terminus of HSP70-interacting protein, p90RSK: p90 ribosomal S6 kinase; PKCζ, protein kinase C-ζ; and PPARs, peroxisome proliferator-activated receptors. Adapted, reprinted, and modified from Heo et al. [22] with permission from Antioxidants and Redox Signaling, April 2016, published by Mary Ann Liebert, Inc., New Rochelle, NY.
1.5.4. ERK5-CHIP-ICER
Previously, our group has reported reduced expression of cAMP hydrolyzing enzymes including phosphodiesterase 3A (PDE3A) and increased expression of inducible cAMP early repressor (ICER) in failing hearts [125,126]. ICER is a pro-apoptotic transcriptional repressor, which inhibits transactivation of cAMP response element binding protein (CREB), and thus downregulates Bcl2. Furthermore, ICER represses PDE3A gene transcription, leading to increased cAMP availability and upregulation of PKA signaling, forming an autoregulatory positive feedback loop. Angiotensin II and isoproterenol (β-adrenergic receptor agonist) activate this mechanism by downregulating PDE3A and upregulating ICER, providing a mechanism for the observation that activation of neurohormonal systems affects myocyte apoptosis [126,127]. Interestingly, ERK5 activation induced by insulin growth factor 1 decreased ICER protein stability through ubiquitin-mediated degradation [128], and we found an important contribution of CHIP (carboxyl terminus of HSP70-interacting protein), an E3 ubiquitin (Ub) ligase, to this process [129] (Figs. 3, 4). It has been reported that CHIP has an important cardioprotective function as it reduces myocardial injury from ischemia/reperfusion after MI by inhibiting cardiac muscle cell apoptosis [130]. Indeed, depletion of CHIP increases infarct sizes and decreases survival [131]. Our group has reported that ICER is a CHIP in mice substrate and that in order for CHIP to ligate Ub to ICER, CHIP must form a complex with de-SUMOylated, hence activated, ERK5 as disruption of ERK5-CHIP binding by a small peptide fragment completely inhibits CHIP Ub ligase activity and consequently up-regulates ICER expression [132] (Fig. 3, 4). The precise mechanism of the de-SUMOylation dependent ERK5-CHIP association needs further investigation, but our data may suggest that ERK5 SUMOylation can be detrimental to the cell via inhibiting the association of ERK5 and CHIP.
1.5.5. Histone deacetylase (HDAC) 1 and 2
Histone acetylation is controlled by two types of enzymes, histone acetyltransferases (HATs) and HDACs [133–136]. HATs catalyze the addition of an acetyl group to specific lysine residues of histone, whereas HDACs do the opposite, catalyzing the removal of acetyl groups. In most cases, histone acetylation is associated with transcription of genes [133,137,138]. It has been reported that inhibitors of Class I HDACs (especially 1 and 2) can inhibit cardiac hypertrophy and preserve cardiac function [139]. For example, depletion of HDAC2 inhibits cardiac hypertrophy after pressure overload [140], while cardiomyocyte specific overexpression of HDAC2 promotes cardiac hypertrophy by reducing the expression of INPP5F (Inositol Polyphosphate-5-Phosphatase F), the gene encoding phosphatidylinositide phosphatase SAC2, which is a negative regulator of the Akt/GSK-3β pathway [140]. In addition, an inhibitor for HDAC1 and 2, 3-(4-substituted phenyl)-N-hydroxy-2-propenamide (SK-7041), [141] completely inhibits the hypertrophic response of the heart after pressure overload [142]. Not only against cardiac hypertrophy, it has also been reported that trichostatin A, pan-HDAC inhibitor, reduces infarct size and improves cardiac function after ischemia/reperfusion [143]. These data suggest that HDAC1 and 2 may have an exacerbating effect on cardiac hypertrophy and dysfunction after cardiac insults.
It has been reported that both HDAC1 and 2 can be SUMOylated [144,145]. Especially, HDAC1 SUMOylation with SUMO1, but not SUMO2, at K444 and K476 promotes HDAC1 stability and up-regulates its expression and activity [145]. Given that HDAC1 has a negative pathological effect, these data suggest that HDAC1 SUMOylation can be detrimental (Fig. 3). HDAC2 SUMOylation can increase p53 de-acetylation and promote NF-κB activation [144,146], which is anti-apoptotic. Of note, the involvement of p90RSK in SUMOylated HDAC2-mediated NF-κB activation has been suggested [146]. p90RSK increases NF-κB signaling via modulating cytosolic events such as IκB or NR-κB p65 phosphorylation [147]. It is possible that HDAC2 SUMOylation discussed here may occur in the extra-nuclear compartment (i.e. in the cytoplasm). Further investigation is necessary to clarify these issues.
As we have described in this section, SUMOylation can inhibit most of the transcriptional factor activity, and subsequently be detrimental after cardiac insult (Table 2). Therefore, although SERCA2 SUMOylation and overexpression of Ubc9 are cardio-protective, these events may not reflect the nature of SUMOylation in the whole heart. It is important to emphasize here that the SUMOylation process can be regulated by the localization changes of de-SUMOylation enzymes including SENP2. As we have explained above, since both SENP1 and 2 contain multiple NLS and NES domains, changes of sub-cellular localization of de-SUMOytion enzymes can be one of the important ways to regulate the “function” of these enzymes to coordinately control the cardiomyocytes responses to various cardiac insults in nuclear and extra-nuclear events.
Table 2.
Summary of transgenic models, phenotypes, and effects of SUMOylation (nuclear events)
| Molecule | Transgenic model | Model characteristics | Phenotypes and the effect of SUMOylation |
|---|---|---|---|
| XBP-1 | Xbp-1−/− [108,173] |
|
|
| PPARs |
Pparα−/− [110–112] Cardiac specific Pparγ−/− [114] Cardiac specific Pparδ−/− [113] |
|
|
| ERK5 |
Erk5−/− Cardiac-specific Erk5−/− |
|
|
| CHIP | Chip−/− |
|
|
| HDAC1 and 2 |
Hdac1−/− [175] Hdac2−/− [175] |
|
|
2. SUMOylation vs. ubiquitination
Although SUMOylation and ubiquitination uses a similar set of E1-E3 enzyme cascade, they use different set of E1–E3 enzymes. In addition, SUMO only shares ~18% homology with ubiquitin. SUMO is approximately 11 kDa in size, compared to the 8 kDa ubiquitin (Ub)molecule [148]. Previously, it has been suggested that SUMOylation acts as an antagonist of Ub, but it also promotes degradation by recruiting the SUMO-targeted Ub ligase as discussed above. It is important to state here that Ub can regulate protein functions by both degradation-dependent and independent mechanisms. When proteins bind to the lysine-48 (K48)-linked polyubiquitin chain, the protein is targeted to the proteasome for degradation. In addition to K48, the K63-linked poly-ubiquitin chain (K63-Ub chain) regulates proteins through a degradation-independent mechanism. For example, the IKK complex is activated when linked to the K63-Ub chain [149–152] (Fig. 5). We would like to briefly discuss here the functional role of Ub chains in regulating NF-kB activation, and its relationship to SUMOylation.
Fig. 5.
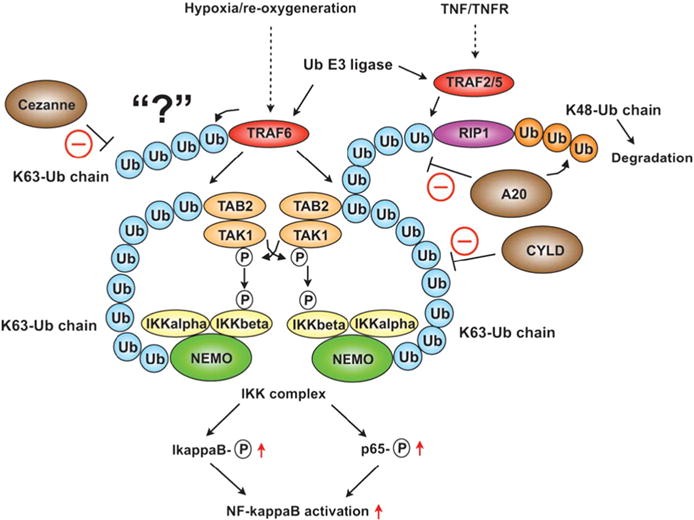
Ubiquitin-mediated TAK1 and IKK complex activation. Hypoxia/reperfusion activates the ubiquitin E3 ligase TRAF6, and TNF/TNFR activates TRAF2 and 5. TRAF6 increases K63-linked polyubiquitin chains, which associate with the TAB2 subunit of TAK1 kinase and activate TAK1 kinase. The K63-Ub chains also bind NEMO to recruit the IKK complex, thereby accelerating the phosphorylation of IKK Adapted, reprinted, and modified from Abe et al. [185] with permission from Circ Res, Jun 7, 2013, published by Wolters Kluwer Health, Inc.
Under Toll-like receptor (TLR) activation, Interleukin 1 receptor associated kinase (IRAK) 1 is phosphorylated by IRAK4 and then associates with and activates the ubiquitin E3 ligase TNF receptor associated factor 6 (TRAF6). TRAF6 complexes with the ubiquitin E2 complex composed of Ub conjugating enzyme (Ubc) 13 and Ub-conjugating enzyme variant (Uev) 1A [152,153] catalyzes the formation of K63-linked poly-ubiquitin chains on itself and other protein (Fig. 5). TRAF6 forms K63 Ub-chains, which promote the binding of the TAB2 (TGF-β-activated kinase 1 and MAP3K7-binding protein 2) subunit of the TAK1 (TGF-β-activated kinase 1) kinase complex and NEMO, leading to TAK1 activation [152,154]. Next, the K63-linked poly-Ub chain binds NEMO to recruit the IKK complex, activate IKK, phosphorylate IκB, and cause its degradation, and consequently activate NF-κB [155]. In addition to the K63-Ub chain formation, SUMO-1 modification of NEMO is required for NF-κB activation in response to genotoxic stress inducers [76]. First, DNA damage accelerates PIAS4 interaction with NEMO and preferentially stimulates site-selective modification of NEMO by SUMO1. DNA damage also activates ATM kinase and phosphorylates NEMO. Both SUMOylation and phosphorylation of NEMO promote its translocation to the cytoplasm, which is subsequently incorporated into the IKK complex. At the same time, K63-linked ubiquitination of BIRC2 (Baculoviral IAP repeat-containing protein 2) promotes NEMO mono-ubiquitination at K285, which is crucial for IKK complex activation [156]. These data suggest the importance of a cross-talk between ubiquitination and SUMOylation in NF-κB signaling [157].
The possible role of the SUMO chain has also been suggested [158]. SUMO can interact with substrates by covalently linking to specific lysine residues of substrate proteins or non-covalently associating with substrates via SUMO-interacting motifs (SIMs) [159]. These interactions are analogous to the ubiquitin system, in which a ubiquitin chain forms on the covalently attached ubiquitin and recruits ubiquitin-binding molecules, and such a molecular complex is able to efficiently activate signaling [158]. The molecules recruited to the ubiquitin chain are non-covalently bound to ubiquitins. Similarly, SUMO can form SUMO chains, to which molecules with SIMs are able to attach non-covalently, forming a SUMO chain–mediated molecular complex [159–161]. A growing number of proteins have been identified for which covalent SUMO association with substrate is regulated by non-covalent interaction of SUMO via SIM [158]. These data suggest that multiple SUMO-interacting molecules can be coordinately regulated by SUMO chain formation, but the exact regulatory mechanism and role of SUMO chain formation remain less clear than those of Ub chain formation. Further investigation will be necessary to determine the relationship between Ub chain and SUMO chain formation.
3. Conclusion
In this review, we have highlighted the role of SUMOylation in different sub-cellular locations particularly in heart. Available data show that SUMOylation events in the extra-nuclear compartment are overwhelmingly cardio-protective, while many SUMOylation events in the nucleus are detrimental to cardiac function, mainly because of the transrepressive nature of SUMOylation on transcription factors. As we also discussed, SUMOylation is an important and dynamic posttranslational protein modification occurring at different sub-cellular compartments, which is tightly regulated by the localization of SUMOylation and de-SUMOylation enzymes. In particular, SUMO proteases including SENP1 and 2 containing both NLS and NES and can shuttle between the cytoplasm and the nucleus. It is likely that shuttling of de-SUMOylation enzymes between the two compartments regulates SUMOylation events after cardiac insults (Fig. 3). Although conventional cardiac knockout or overexpression and gene transfer methods are powerful tools to determine the role of each SUMO-related molecule in cardiac dysfunction and remodeling, these methods are less useful for determining a dynamic regulation of SUMOylation mediated by shuttling of de-SUMOylation enzymes between the nuclear and extra-nuclear compartments. To determine the exact role of SUMOylation in the heart, it is necessary to clarify the precise molecular mechanism of de-SUMOylation enzyme shuttling. Experimental methods that can only inhibit the shuttling of de-SUMOylation enzyme but not SUMO ligase must be developed. Using such methods, we will be able to obtain more precise data on how SUMOylation within different compartments of cells is regulated by differential localization of SUMOylation and de-SUMOylation enzymes, which will aid us to determine the response and fate of cardiomyocytes by SUMOylation after cardiac insults.
Acknowledgments
Funding
The research activities of the authors are supported by grant from the National Institute of Health to Dr. Abe (HL-130193, HL-123346, HL-118462, HL-108551), and from American Heart Association to Dr. Le (AHA 13SDG14500033).
Abbreviations
- AMPK
adenosine monophosphate-activated protein kinase
- CAMKK
calcium-calmodulin-activated protein kinase kinase
- CHIP
carboxyl terminus of HSP70-interacting protein
- CVD
cardiovascular disease
- cTnI
cardiac troponin I
- d-flow
disturbed flow
- DeSI
de-SUMOylating isopeptidase
- DRP1
dynamin-related protein 1
- ER
endoplasmic reticulum
- ERK5
extracellular-signal regulated kinase 5
- GRK2
G protein-coupled receptor kinase
- HAT
histone acetyltransferases
- HDAC
histone deacetylase
- HIF
hypoxia-inducible factor
- ICER
inducible cAMP early repressor
- IKK
inhibitor of κB kinase
- KCNK1
potassium channel subfamily K member 1
- KDM1
histone lysine-specific demethylase
- LKB1
liver kinase B1
- MAPL
mitochondria-associated protein ligase
- NEMO
NF-κB Essential MOdulator
- NF-κB
nuclear factor kappa B
- NES
nuclear export signal
- NLS
nuclear localization signal
- p90RSK
p90 ribosomal S6 kinase
- PDE3A
phosphodiesterase 3A
- PKC
protein kinase C
- PIAS
protein inhibitor of activated STAT
- PLN
phospholamban
- PPAR
peroxisome proliferator-activated receptor
- PTM
post-translational modification
- RCOR1
REST corepressor 1
- ROS
reactive oxygen species
- SENP
sentrin/SUMO-specific protease
- SERCA2a
sarcoplasmic reticulum Ca2+ ATPase 2a
- SIM
SUMO-interacting motif
- SR
sarcoplasmic reticulum
- STUbL
SUMO-targeted ubiquitin ligase
- SUMO
Small Ubiquitin-related Modifier
- Ub
ubiquitin
- UPS
ubiquitin-proteasome system
- USPL
ubiquitin-specific protease-like
- XBP-1s
spliced X-Box Binding Protein 1
Footnotes
Competing financial interests
The authors have no competing financial interests.
Transparency document
The Transparency document associated with this article can be found, in online version.
This article is part of a Special Issue entitled: Genetic and epigenetic control of heart failure - edited by Jun Ren & Megan Yingmei Zhang.
References
- 1.Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Guo B, Yang SH, Witty J, Sharrocks AD. Signalling pathways and the regulation of SUMO modification. Biochem Soc Trans. 2007;35:1414–1418. doi: 10.1042/BST0351414. [DOI] [PubMed] [Google Scholar]
- 3.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 4.Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem Biophys Res Commun. 2005;337:517–520. doi: 10.1016/j.bbrc.2005.09.090. [DOI] [PubMed] [Google Scholar]
- 5.Gao C, Huang W, Kanasaki K, Xu Y. The role of ubiquitination and sumoylation in diabetic nephropathy. Biomed Res Int. 2014;2014:160692. doi: 10.1155/2014/160692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang E, Heo KS, Woo CH, Lee H, Le NT, Thomas TN, Fujiwara K, Abe J. MK2 SUMOylation regulates actin filament remodeling and subsequent migration in endothelial cells by inhibiting MK2 kinase and HSP27 phosphorylation. Blood. 2011;117:2527–2537. doi: 10.1182/blood-2010-08-302281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henley JM, Craig TJ, Wilkinson KA. Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiol Rev. 2014;94:1249–1285. doi: 10.1152/physrev.00008.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo C, Henley JM. Wrestling with stress: roles of protein SUMOylation and deSUMOylation in cell stress response. IUBMB Life. 2014;66:71–77. doi: 10.1002/iub.1244. [DOI] [PubMed] [Google Scholar]
- 9.Reverter D, Lima CD. Structural basis for SENP2 protease interactions with SUMO precursors and conjugated substrates. Nat Struct Mol Biol. 2006;13:1060–1068. doi: 10.1038/nsmb1168. [DOI] [PubMed] [Google Scholar]
- 10.Gong L, Millas S, Maul GG, Yeh ET. Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J Biol Chem. 2000;275:3355–3359. doi: 10.1074/jbc.275.5.3355. [DOI] [PubMed] [Google Scholar]
- 11.Bekes M, Prudden J, Srikumar T, Raught B, Boddy MN, Salvesen GS. The dynamics and mechanism of SUMO chain deconjugation by SUMO-specific proteases. J Biol Chem. 2011;286:10238–10247. doi: 10.1074/jbc.M110.205153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mikolajczyk J, Drag M, Bekes M, Cao JT, Ronai Z, Salvesen GS. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem. 2007;282:26217–26224. doi: 10.1074/jbc.M702444200. [DOI] [PubMed] [Google Scholar]
- 13.Gong L, Yeh ET. Characterization of a family of nucleolar SUMO-specific proteases with preference for SUMO-2 or SUMO-3. J Biol Chem. 2006;281:15869–15877. doi: 10.1074/jbc.M511658200. [DOI] [PubMed] [Google Scholar]
- 14.Kolli N, Mikolajczyk J, Drag M, Mukhopadhyay D, Moffatt N, Dasso M, Salvesen G, Wilkinson KD. Distribution and paralogue specificity of mammalian deSUMOylating enzymes. Biochem J. 2010;430:335–344. doi: 10.1042/BJ20100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay D, Ayaydin F, Kolli N, Tan SH, Anan T, Kametaka A, Azuma Y, Wilkinson KD, Dasso M. SUSP1 antagonizes formation of highly SUMO2/3-conjugated species. J Cell Biol. 2006;174:939–949. doi: 10.1083/jcb.200510103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen LN, Geoffroy MC, Jaffray EG, Hay RT. Characterization of SENP7, a SUMO-2/3-specific isopeptidase. Biochem J. 2009;421:223–230. doi: 10.1042/BJ20090246. [DOI] [PubMed] [Google Scholar]
- 17.Shin EJ, Shin HM, Nam E, Kim WS, Kim JH, Oh BH, Yun Y. DeSUMOylating isopeptidase: a second class of SUMO protease. EMBO Rep. 2012;13:339–346. doi: 10.1038/embor.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulz S, Chachami G, Kozaczkiewicz L, Winter U, Stankovic-Valentin N, Haas P, Hofmann K, Urlaub H, Ovaa H, Wittbrodt J, Meulmeester E, Melchior F. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012;13:930–938. doi: 10.1038/embor.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Luo Y, Yu L, Lin Y, Luo D, Zhang H, He Y, Kim YO, Kim Y, Tang S, Min W. SENP1 mediates TNF-induced desumoylation and cytoplasmic translocation of HIPK1 to enhance ASK1-dependent apoptosis. Cell Death Differ. 2008;15:739–750. doi: 10.1038/sj.cdd.4402303. [DOI] [PubMed] [Google Scholar]
- 20.Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584–595. doi: 10.1016/j.cell.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyamoto S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–130. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heo KS, Berk B, Abe JI. Disturbed flow-induced endothelial pro-atherogenic signaling via regulating post-translational modifications and epigenetic events. Antioxid Redox Signal. 2015 doi: 10.1089/ars.2015.6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benson MD, Li QJ, Kieckhafer K, Dudek D, Whorton MR, Sunahara RK, Iniguez-Lluhi JA, Martens JR. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci U S A. 2007;104:1805–1810. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overturf KE, Russell SN, Carl A, Vogalis F, Hart PJ, Hume JR, Sanders KM, Horowitz B. Cloning and characterization of a Kv1.5 delayed rectifier K+ channel from vascular and visceral smooth muscles. Am J Physiol. 1994;267:C1231–C1238. doi: 10.1152/ajpcell.1994.267.5.C1231. [DOI] [PubMed] [Google Scholar]
- 26.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 27.Hong Z, Smith AJ, Archer SL, Wu XC, Nelson DP, Peterson D, Johnson G, Weir EK. Pergolide is an inhibitor of voltage-gated potassium channels, including Kv1.5, and causes pulmonary vasoconstriction. Circulation. 2005;112:1494–1499. doi: 10.1161/CIRCULATIONAHA.105.556704. [DOI] [PubMed] [Google Scholar]
- 28.Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi Y, Wang J, Bomben VC, Li DP, Chen SR, Sun H, Xi Y, Reed JG, Cheng J, Pan HL, Noebels JL, Yeh ET. Hyper-SUMOylation of the Kv7 potassium channel diminishes the M-current leading to seizures and sudden death. Neuron. 2014;83:1159–1171. doi: 10.1016/j.neuron.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christensen AH, Chatelain FC, Huttner IG, Olesen MS, Soka M, Feliciangeli S, Horvat C, Santiago CF, Vandenberg JI, Schmitt N, Olesen SP, Lesage F, Fatkin D. The two-pore domain potassium channel, TWIK-1, has a role in the regulation of heart rate and atrial size. J Mol Cell Cardiol. 2016;97:24–35. doi: 10.1016/j.yjmcc.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Silveirinha V, Stephens GJ, Cimarosti H. Molecular targets underlying SUMO-mediated neuroprotection in brain ischemia. J Neurochem. 2013;127:580–591. doi: 10.1111/jnc.12347. [DOI] [PubMed] [Google Scholar]
- 32.Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell. 2005;121:37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Plant LD, Dementieva IS, Kollewe A, Olikara S, Marks JD, Goldstein SA. One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc Natl Acad Sci U S A. 2010;107:10743–10748. doi: 10.1073/pnas.1004712107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C, Kohlbrenner E, Fish KM, Kho C, Hajjar RJ. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci Transl Med. 2013;5:211ra159. doi: 10.1126/scitranslmed.3006487. [DOI] [PubMed] [Google Scholar]
- 36.Kho C, Lee A, Jeong D, Oh JG, Gorski PA, Fish K, Sanchez R, DeVita RJ, Christensen G, Dahl R, Hajjar RJ. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat Commun. 2015;6:7229. doi: 10.1038/ncomms8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim EY, Zhang Y, Ye B, Segura AM, Beketaev I, Xi Y, Yu W, Chang J, Li F, Wang J. Involvement of activated SUMO-2 conjugation in cardiomyopathy. Biochim Biophys Acta. 2015;1852:1388–1399. doi: 10.1016/j.bbadis.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Chen L, Wen S, Zhu H, Yu W, Moskowitz IP, Shaw GM, Finnell RH, Schwartz RJ. Defective sumoylation pathway directs congenital heart disease. Birth Defects Res A Clin Mol Teratol. 2011;91:468–476. doi: 10.1002/bdra.20816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Wansleeben C, Zhao S, Miao P, Paschen W, Yang W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014;15:878–885. doi: 10.15252/embr.201438534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jayashankar V, Mueller IA, Rafelski SM. Shaping the multi-scale architecture of mitochondria. Curr Opin Cell Biol. 2016;38:45–51. doi: 10.1016/j.ceb.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra P. Interfaces between mitochondrial dynamics and disease. Cell Calcium. 2016 doi: 10.1016/j.ceca.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 44.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem. 2014;155:273–279. doi: 10.1093/jb/mvu016. [DOI] [PubMed] [Google Scholar]
- 46.Ong SB, Kalkhoran SB, Cabrera-Fuentes HA, Hausenloy DJ. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol. 2015;763:104–114. doi: 10.1016/j.ejphar.2015.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zungu M, Schisler J, Willis MS. All the little pieces. Regulation of mitochondrial fusion and fission by ubiquitin and small ubiquitin-like modifier and their potential relevance in the heart. Circ J. 2011;75:2513–2521. doi: 10.1253/circj.cj-11-0967. [DOI] [PubMed] [Google Scholar]
- 49.Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zunino R, Schauss A, Rippstein P, Andrade-Navarro M, McBride HM. The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J Cell Sci. 2007;120:1178–1188. doi: 10.1242/jcs.03418. [DOI] [PubMed] [Google Scholar]
- 52.Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008;18:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 54.Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 55.Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell. 2015;59:941–955. doi: 10.1016/j.molcel.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 56.Mendler L, Braun T, Muller S. The ubiquitin-like SUMO system and heart function: from development to disease. Circ Res. 2016;118:132–144. doi: 10.1161/CIRCRESAHA.115.307730. [DOI] [PubMed] [Google Scholar]
- 57.Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Di Bacco A, Ouyang J, Lee HY, Catic A, Ploegh H, Gill G. The SUMO-specific protease SENP5 is required for cell division. Mol Cell Biol. 2006;26:4489–4498. doi: 10.1128/MCB.02301-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim EY, Zhang Y, Beketaev I, Segura AM, Yu W, Xi Y, Chang J, Wang J. SENP5, a SUMO isopeptidase, induces apoptosis and cardiomyopathy. J Mol Cell Cardiol. 2015;78:154–164. doi: 10.1016/j.yjmcc.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 60.Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98:100–103. doi: 10.1161/01.cir.98.2.100. [DOI] [PubMed] [Google Scholar]
- 61.Jancso G, Lantos J, Borsiczky B, Szanto Z, Roth E. Dynamism of NF-kappaB and AP-1 activation in the signal transduction of ischaemic myocardial preconditioning. Eur Surg Res. 2004;36:129–135. doi: 10.1159/000077253. [DOI] [PubMed] [Google Scholar]
- 62.de Moissac D, Mustapha S, Greenberg AH, Kirshenbaum LA. Bcl-2 activates the transcription factor NFkappaB through the degradation of the cytoplasmic inhibitor IkappaBalpha. J Biol Chem. 1998;273:23946–23951. doi: 10.1074/jbc.273.37.23946. [DOI] [PubMed] [Google Scholar]
- 63.Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2014;306:H317–H325. doi: 10.1152/ajpheart.00182.2013. [DOI] [PubMed] [Google Scholar]
- 64.Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- 65.Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S. Mechanism of hypoxia-induced NF-kappaB. Mol Cell Biol. 2010;30:4901–4921. doi: 10.1128/MCB.00409-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–2920. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
- 67.Mohan S, Konopinski R, Yan B, Centonze VE, Natarajan M. High glucose-induced IKK-Hsp-90 interaction contributes to endothelial dysfunction. Am J Physiol Cell Physiol. 2009;296:C182–C192. doi: 10.1152/ajpcell.00575.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kratsios P, Huth M, Temmerman L, Salimova E, Al Banchaabouchi M, Sgoifo A, Manghi M, Suzuki K, Rosenthal N, Mourkioti F. Antioxidant amelioration of dilated cardiomyopathy caused by conditional deletion of NEMO/IKKgamma in cardiomyocytes. Circ Res. 2010;106:133–144. doi: 10.1161/CIRCRESAHA.109.202200. [DOI] [PubMed] [Google Scholar]
- 70.Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell. 2011;43:180–191. doi: 10.1016/j.molcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol. 2011;31:2774–2786. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. 2006;8:986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 73.Wuerzberger-Davis SM, Nakamura Y, Seufzer BJ, Miyamoto S. NF-kappaB activation by combinations of NEMO SUMOylation and ATM activation stresses in the absence of DNA damage. Oncogene. 2007;26:641–651. doi: 10.1038/sj.onc.1209815. [DOI] [PubMed] [Google Scholar]
- 74.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 75.Wu ZH, Mabb A, Miyamoto S. PIDD: a switch hitter. Cell. 2005;123:980–982. doi: 10.1016/j.cell.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 76.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565–576. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 77.Liu X, Chen W, Wang Q, Li L, Wang C. Negative regulation of TLR inflammatory signaling by the SUMO-deconjugating enzyme SENP6. PLoS Pathog. 2013;9:e1003480. doi: 10.1371/journal.ppat.1003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun H, Lu L, Zuo Y, Wang Y, Jiao Y, Zeng WZ, Huang C, Zhu MX, Zamponi GW, Zhou T, Xu TL, Cheng J, Li Y. Kainate receptor activation induces glycine receptor endocytosis through PKC deSUMOylation. Nat Commun. 2014;5:4980. doi: 10.1038/ncomms5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Q, Molkentin JD. Protein kinase Calpha as a heart failure therapeutic target. J Mol Cell Cardiol. 2011;51:474–478. doi: 10.1016/j.yjmcc.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;15:15. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 81.Malhotra R, D’Souza KM, Staron ML, Birukov KG, Bodi I, Akhter SA. G alpha(q)-mediated activation of GRK2 by mechanical stretch in cardiac myocytes: the role of protein kinase C. J Biol Chem. 2010;285:13748–13760. doi: 10.1074/jbc.M110.109272. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82.Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res. 2007;101:195–204. doi: 10.1161/CIRCRESAHA.107.148288. [DOI] [PubMed] [Google Scholar]
- 83.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 84.Kooij V, Boontje N, Zaremba R, Jaquet K, dos Remedios C, Stienen GJ, van der Velden J. Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol. 2010;105:289–300. doi: 10.1007/s00395-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–638. doi: 10.1161/CIRCRESAHA.109.198465. 617 pp. following 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 89.Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003;278:28372–28377. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 90.Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, Bolli R. Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation. 2007;116:2809–2817. doi: 10.1161/CIRCULATIONAHA.107.725697. [DOI] [PubMed] [Google Scholar]
- 91.Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, Horman S, Vaulont S, Hoerter J, Viollet B, Hue L, Vanoverschelde JL, Bertrand L. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. 2006;291:H2875–H2883. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- 92.Carvajal K, Zarrinpashneh E, Szarszoi O, Joubert F, Athea Y, Mateo P, Gillet B, Vaulont S, Viollet B, Bigard X, Bertrand L, Ventura-Clapier R, Hoerter JA. Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and re-perfusion. Am J Physiol Heart Circ Physiol. 2007;292:H3136–H3147. doi: 10.1152/ajpheart.00683.2006. [DOI] [PubMed] [Google Scholar]
- 93.Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, Xu W, Wiczer B, Bernlohr DA, Bache RJ, Chen Y. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rubio T, Vernia S, Sanz P. Sumoylation of AMPKbeta2 subunit enhances AMP-activated protein kinase activity. Mol Biol Cell. 2013;24:1801–1811. doi: 10.1091/mbc.E12-11-0806. S1801–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ritho J, Arold ST, Yeh ET. A critical SUMO1 modification of LKB1 regulates AMPK activity during energy stress. Cell Rep. 2015;12:734–742. doi: 10.1016/j.celrep.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 96.Gupta MK, Gulick J, Liu R, Wang X, Molkentin JD, Robbins J. Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ Res. 2014;115:721–729. doi: 10.1161/CIRCRESAHA.115.304760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gupta MK, McLendon PM, Gulick J, James J, Khalili K, Robbins J. UBC9-mediated sumoylation favorably impacts cardiac function in compromised hearts. Circ Res. 2016;118:1894–1905. doi: 10.1161/CIRCRESAHA.115.308268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wykoff DD, O’Shea EK. Identification of sumoylated proteins by systematic immunoprecipitation of the budding yeast proteome. Mol Cell Proteomics. 2005;4:73–83. doi: 10.1074/mcp.M400166-MCP200. [DOI] [PubMed] [Google Scholar]
- 99.Vertegaal AC, Andersen JS, Ogg SC, Hay RT, Mann M, Lamond AI. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol Cell Proteomics. 2006;5:2298–2310. doi: 10.1074/mcp.M600212-MCP200. [DOI] [PubMed] [Google Scholar]
- 100.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 101.Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ., 3rd PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell. 2007;28:823–837. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang SH, Sharrocks AD. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell. 2004;13:611–617. doi: 10.1016/s1097-2765(04)00060-7. [DOI] [PubMed] [Google Scholar]
- 103.Ouyang J, Shi Y, Valin A, Xuan Y, Gill G. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol Cell. 2009;34:145–154. doi: 10.1016/j.molcel.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 105.Golebiowski F, Matic I, Tatham MH, Cole C, Yin Y, Nakamura A, Cox J, Barton GJ, Mann M, Hay RT. System-wide changes to SUMO modifications in response to heat shock. Sci Signal. 2009;2:ra24. doi: 10.1126/scisignal.2000282. [DOI] [PubMed] [Google Scholar]
- 106.Chen H, Qi L. SUMO modification regulates the transcriptional activity of XBP1. Biochem J. 2010;429:95–102. doi: 10.1042/BJ20100193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang Z, Fan Q, Zhang Z, Zou Y, Cai R, Wang Q, Zuo Y, Cheng J. SENP1 deficiency promotes ER stress-induced apoptosis by increasing XBP1 SUMOylation. Cell Cycle. 2012;11:1118–1122. doi: 10.4161/cc.11.6.19529. [DOI] [PubMed] [Google Scholar]
- 108.Masaki T, Yoshida M, Noguchi S. Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem Biophys Res Commun. 1999;261:350–356. doi: 10.1006/bbrc.1999.0972. [DOI] [PubMed] [Google Scholar]
- 109.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 110.Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JR, Belke DD, Severson DL, Kelly DP, Lopaschuk GD. A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem. 2002;277:4098–4103. doi: 10.1074/jbc.M106054200. [DOI] [PubMed] [Google Scholar]
- 111.Panagia M, Gibbons GF, Radda GK, Clarke K. PPAR-alpha activation required for decreased glucose uptake and increased susceptibility to injury during ischemia. Am J Physiol Heart Circ Physiol. 2005;288:H2677–H2683. doi: 10.1152/ajpheart.00200.2004. [DOI] [PubMed] [Google Scholar]
- 112.Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation. 2005;112:2339–2346. doi: 10.1161/CIRCULATIONAHA.105.534594. [DOI] [PubMed] [Google Scholar]
- 113.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 114.Ding G, Fu M, Qin Q, Lewis W, Kim HW, Fukai T, Bacanamwo M, Chen YE, Schneider MD, Mangelsdorf DJ, Evans RM, Yang Q. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc Res. 2007;76:269–279. doi: 10.1016/j.cardiores.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 115.Pourcet B, Pineda-Torra I, Derudas B, Staels B, Glineur C. SUMOylation of human peroxisome proliferator-activated receptor alpha inhibits its trans-activity through the recruitment of the nuclear corepressor NCoR. J Biol Chem. 2010;285:5983–5992. doi: 10.1074/jbc.M109.078311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shimizu M, Yamashita D, Yamaguchi T, Hirose F, Osumi T. Aspects of the regulatory mechanisms of PPAR functions: analysis of a bidirectional response element and regulation by sumoylation. Mol Cell Biochem. 2006;286:33–42. doi: 10.1007/s11010-005-9052-z. [DOI] [PubMed] [Google Scholar]
- 117.Wadosky KM, Willis MS. The story so far: post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am J Physiol Heart Circ Physiol. 2012;302:H515–H526. doi: 10.1152/ajpheart.00703.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Raviv Z, Kalie E, Seger R. MEK5 and ERK5 are localized in the nuclei of resting as well as stimulated cells, while MEKK2 translocates from the cytosol to the nucleus upon stimulation. J Cell Sci. 2004;117:1773–1784. doi: 10.1242/jcs.01040. [DOI] [PubMed] [Google Scholar]
- 120.Kimura TE, Jin J, Zi M, Prehar S, Liu W, Oceandy D, Abe J, Neyses L, Weston AH, Cartwright EJ, Wang X. Targeted deletion of the extracellular signal-regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload-induced apoptosis in the heart. Circ Res. 2010;106:961–970. doi: 10.1161/CIRCRESAHA.109.209320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- 122.Shishido T, Woo CH, Ding B, McClain C, Molina CA, Yan C, Yang J, Abe J. Effects of MEK5/ERK5 association on small ubiquitin-related modification of ERK5: implications for diabetic ventricular dysfunction after myocardial infarction. Circ Res. 2008;102:1416–1425. doi: 10.1161/CIRCRESAHA.107.168138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Heo KS, Chang E, Le NT, Cushman H, Yeh ET, Fujiwara K, Abe J. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013;112:911–923. doi: 10.1161/CIRCRESAHA.111.300179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Akaike M, Che W, Marmarosh NL, Ohta S, Osawa M, Ding B, Berk BC, Yan C, Abe J. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol Cell Biol. 2004;24:8691–8704. doi: 10.1128/MCB.24.19.8691-8704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ding B, Abe J, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, Yan C. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding B, Abe J, Wei H, Xu H, Che W, Aizawa T, Liu W, Molina CA, Sadoshima J, Blaxall BC, Berk BC, Yan C. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005;102:14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ding B, Price RL, Goldsmith EC, Borg TK, Yan X, Douglas PS, Weinberg EO, Bartunek J, Thielen T, Didenko VV, Lorell BH. Left ventricular hypertrophy in ascending aortic stenosis mice: anoikis and the progression to early failure. Circulation. 2000;101:2854–2862. doi: 10.1161/01.cir.101.24.2854. [DOI] [PubMed] [Google Scholar]
- 128.Yan C, Ding B, Shishido T, Woo CH, Itoh S, Jeon KI, Liu W, Xu H, McClain C, Molina CA, Blaxall BC, Abe J. Activation of extracellular signal-regulated kinase 5 reduces cardiac apoptosis and dysfunction via inhibition of a phosphodiesterase 3A/inducible cAMP early repressor feedback loop. Circ Res. 2007;100:510–519. doi: 10.1161/01.RES.0000259045.49371.9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Woo CH, Le NT, Shishido T, Chang E, Lee H, Heo KS, Mickelsen DM, Lu Y, McClain C, Spangenberg T, Yan C, Molina CA, Yang J, Patterson C, Abe J. Novel role of C terminus of Hsc70-interacting protein (CHIP) ubiquitin ligase on inhibiting cardiac apoptosis and dysfunction via regulating ERK5-mediated degradation of inducible cAMP early repressor. FASEB J. 2010;24:4917–4928. doi: 10.1096/fj.10-162636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Naito AT, Okada S, Minamino T, Iwanaga K, Liu ML, Sumida T, Nomura S, Sahara N, Mizoroki T, Takashima A, Akazawa H, Nagai T, Shiojima I, Komuro I. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ Res. 2010;106:1692–1702. doi: 10.1161/CIRCRESAHA.109.214346. [DOI] [PubMed] [Google Scholar]
- 131.Zhang C, Xu Z, He XR, Michael LH, Patterson C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2005;288:H2836–H2842. doi: 10.1152/ajpheart.01122.2004. [DOI] [PubMed] [Google Scholar]
- 132.Le NT, Takei Y, Shishido T, Woo CH, Chang E, Heo KS, Lee H, Lu Y, Morrell C, Oikawa M, McClain C, Wang X, Tournier C, Molina CA, Taunton J, Yan C, Fujiwara K, Patterson C, Yang J, Abe J. p90RSK targets the ERK5-CHIP ubiquitin E3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ Res. 2012;110:536–550. doi: 10.1161/CIRCRESAHA.111.254730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Illi B, Nanni S, Scopece A, Farsetti A, Biglioli P, Capogrossi MC, Gaetano C. Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ Res. 2003;93:155–161. doi: 10.1161/01.RES.0000080933.82105.29. [DOI] [PubMed] [Google Scholar]
- 134.Thangjam GS, Dimitropoulou C, Joshi AD, Barabutis N, Shaw MC, Kovalenkov Y, Wallace CM, Fulton DJ, Patel V, Catravas JD. Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am J Respir Cell Mol Biol. 2014;50:942–952. doi: 10.1165/rcmb.2013-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Iordache F, Buzila C, Constantinescu A, Andrei E, Maniu H. Histone deacetylase (HDAC) inhibitors down-regulate endothelial lineage commitment of umbilical cord blood derived endothelial progenitor cells. Int J Mol Sci. 2012;13:15074–15085. doi: 10.3390/ijms131115074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kumar A, Kim CS, Hoffman TA, Naqvi A, Dericco J, Jung SB, Lin Z, Jain MK, Irani K. p53 impairs endothelial function by transcriptionally repressing Kruppel-Like Factor 2. Arterioscler Thromb Vasc Biol. 2011;31:133–141. doi: 10.1161/ATVBAHA.110.215061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E, Tjwa M, Rossig L, Seto E, Augustin HG, Zeiher AM, Dimmeler S, Urbich C. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011;30:4142–4156. doi: 10.1038/emboj.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hellebrekers DM, Melotte V, Vire E, Langenkamp E, Molema G, Fuks F, Herman JG, Van Criekinge W, Griffioen AW, van Engeland M. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007;67:4138–4148. doi: 10.1158/0008-5472.CAN-06-3032. [DOI] [PubMed] [Google Scholar]
- 139.Abend A, Kehat I. Histone deacetylases as therapeutic targets—from cancer to cardiac disease. Pharmacol Ther. 2015;147:55–62. doi: 10.1016/j.pharmthera.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 140.Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- 141.Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Kim TY, Bang YJ. Class I histone deacetylase-selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin Cancer Res. 2004;10:5271–5281. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 142.Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, Zhou R, Ferrari V, Gruber P, Epstein JA. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest. 2003;112:863–871. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. 2007;76:473–481. doi: 10.1016/j.cardiores.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 144.Brandl A, Wagner T, Uhlig KM, Knauer SK, Stauber RH, Melchior F, Schneider G, Heinzel T, Kramer OH. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J Mol Cell Biol. 2012;4:284–293. doi: 10.1093/jmcb/mjs013. [DOI] [PubMed] [Google Scholar]
- 145.Citro S, Jaffray E, Hay RT, Seiser C, Chiocca S. A role for paralog-specific sumoylation in histone deacetylase 1 stability. J Mol Cell Biol. 2013;5:416–427. doi: 10.1093/jmcb/mjt032. [DOI] [PubMed] [Google Scholar]
- 146.Wagner T, Kiweler N, Wolff K, Knauer SK, Brandl A, Hemmerich P, Dannenberg JH, Heinzel T, Schneider G, Kramer OH. Sumoylation of HDAC2 promotes NF-kappaB-dependent gene expression. Oncotarget. 2015;6:7123–7135. doi: 10.18632/oncotarget.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 148.Muller S, Hoege C, Pyrowolakis G, Jentsch S. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol. 2001;2:202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 149.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 151.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 152.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 153.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–653. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 154.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 155.Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol Cell. 2010;40:63–74. doi: 10.1016/j.molcel.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 157.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. 2006;8:986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 158.Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. 2010;428:133–145. doi: 10.1042/BJ20100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kerscher O. SUMO junction-what’s your function? New insights through SUMO-interacting motifs. EMBO Rep. 2007;8:550–555. doi: 10.1038/sj.embor.7400980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pellegrino S, Altmeyer M. Interplay between ubiquitin, SUMO, and poly(ADP-ribose) in the cellular response to genotoxic stress. Front Genet. 2016;7:63. doi: 10.3389/fgene.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Vertegaal AC. Small ubiquitin-related modifiers in chains. Biochem Soc Trans. 2007;35:1422–1423. doi: 10.1042/BST0351422. [DOI] [PubMed] [Google Scholar]
- 162.London B, Guo W, Pan X, Lee JS, Shusterman V, Rocco CJ, Logothetis DA, Nerbonne JM, Hill JA. Targeted replacement of KV1.5 in the mouse leads to loss of the 4-aminopyridine-sensitive component of I(K,slow) and resistance to drug-induced qt prolongation. Circ Res. 2001;88:940–946. doi: 10.1161/hh0901.090929. [DOI] [PubMed] [Google Scholar]
- 163.Nie X, Arrighi I, Kaissling B, Pfaff I, Mann J, Barhanin J, Vallon V. Expression and insights on function of potassium channel TWIK-1 in mouse kidney. Pflugers Arch. 2005;451:479–488. doi: 10.1007/s00424-005-1480-9. [DOI] [PubMed] [Google Scholar]
- 164.Ver Heyen M, Heymans S, Antoons G, Reed T, Periasamy M, Awede B, Lebacq J, Vangheluwe P, Dewerchin M, Collen D, Sipido K, Carmeliet P, Wuytack F. Replacement of the muscle-specific sarcoplasmic reticulum Ca(2+)-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ Res. 2001;89:838–846. doi: 10.1161/hh2101.098466. [DOI] [PubMed] [Google Scholar]
- 165.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000;14:854–862. [PMC free article] [PubMed] [Google Scholar]
- 166.Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M. Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000;5:969–979. doi: 10.1016/s1097-2765(00)80262-2. [DOI] [PubMed] [Google Scholar]
- 167.Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV. Knockout of PKC alpha enhances insulin signaling through PI3K. Mol Endocrinol. 2002;16:847–858. doi: 10.1210/mend.16.4.0809. [DOI] [PubMed] [Google Scholar]
- 168.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 169.Foller M, Sopjani M, Koka S, Gu S, Mahmud H, Wang K, Floride E, Schleicher E, Schulz E, Munzel T, Lang F. Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J. 2009;23:1072–1080. doi: 10.1096/fj.08-121772. [DOI] [PubMed] [Google Scholar]
- 170.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ylikorkala A, Rossi DJ, Korsisaari N, Luukko K, Alitalo K, Henkemeyer M, Makela TP. Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science. 2001;293:1323–1326. doi: 10.1126/science.1062074. [DOI] [PubMed] [Google Scholar]
- 173.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- 174.Sohn SJ, Sarvis BK, Cado D, Winoto A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J Biol Chem. 2002;277:43344–43351. doi: 10.1074/jbc.M207573200. [DOI] [PubMed] [Google Scholar]
- 175.Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K, Abe J. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest. 2015;125:1299–1310. doi: 10.1172/JCI76453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, McClain C, Reinhart-King CA, King MR, Berk BC, Fujiwara K, Woo CH, Abe J. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011;193:867–884. doi: 10.1083/jcb.201010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K, Abe J. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013;127:486–499. doi: 10.1161/CIRCULATIONAHA.112.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nigro P, Abe J, Woo CH, Satoh K, McClain C, O’Dell MR, Lee H, Lim JH, Li JD, Heo KS, Fujiwara K, Berk BC. PKCzeta decreases eNOS protein stability via inhibitory phosphorylation of ERK5. Blood. 2010;116:1971–1979. doi: 10.1182/blood-2010-02-269134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997;16:5509–5519. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Johnson ES, Blobel G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J Biol Chem. 1997;272:26799–26802. doi: 10.1074/jbc.272.43.26799. [DOI] [PubMed] [Google Scholar]
- 182.Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 183.Li SJ, Hochstrasser M. A new protease required for cell-cycle progression in yeast. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 184.Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem. 2009;284:8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Abe J, Berk BC. Cezanne paints inflammation by regulating ubiquitination. Circ Res. 2013;112:1526–1528. doi: 10.1161/CIRCRESAHA.113.301518. [DOI] [PMC free article] [PubMed] [Google Scholar]