

Abstract
Bronchopulmonary dysplasia (BPD) is a chronic lung disease of premature human infants, which may persist through adulthood. Airway inflammation has been firmly established in the pathogenesis of BPD. Previous studies to reduce airway inflammation with high-dose dexamethasone demonstrated adverse neurological outcomes, despite lower incidences of BPD. Instillation of budesonide and surfactant can facilitate early extubation and reduce the incidence of BPD and death among very low birth weight infants. However, the pharmacokinetics of budesonide and its distribution into the lung and brain are unknown. Therefore, 5 premature lambs were administered 0.25 mg/kg budesonide, with surfactant as the vehicle. Plasma and tissue samples were taken from the lambs for measurement of budesonide, 16α-hydroxy prednisolone, and budesonide palmitate using LC/MS/MS. Peak plasma budesonide concentrations were inversely correlated with the oxygenation index (correlation coefficient of −0.75). Plasma budesonide concentrations were extremely low (~10% of expected) for two lambs that had high oxygenation indices and were excluded from further analyses. For the remaining 5 premature lambs, a non-compartmental analysis demonstrated an AUCinf of 148.77 ± 28.16 h*μg/L, half-life of 4.76 ± 1.79 h, and Cmax of 46.17 ± 17.71 μg/L. Using population pharmacokinetic methods, a one-compartment model with exponential residual error and first-order absorption adequately described the data. The apparent clearance and apparent volume of distribution of budesonide were estimated at 6.29 L/h (1.99 L/h/kg) and 29.1 L (9.2 L/kg), respectively. Budesonide and budesonide palmitate, but not 16α-hydroxy prednisolone, were detected in lung tissue. In this study, budesonide and its metabolites were not detected in the brain, which suggests that intratracheal instillation suggests that after local pulmonary deposition, there is no evidence of budesonide accumulation in the central nervous system. Overall, these results show that peak plasma budesonide concentrations are inversely correlated with the oxygenation index and that lung-specific delivery of budesonide avoids accumulation of budesonide in the brain.
Keywords: Budesonide, infants, lambs, pharmacokinetics, premature, surfactant
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is characterized by pathologic changes in alveolar architecture, including: reduced alveolar number, thickened septa, and decreased capillary growth. These effects result in poor respiratory gas exchange, which can persist throughout adulthood, leading to chronic lung disease (CLD) [1–3]. Risk factors for BPD include extremely low gestational age and need for supplemental oxygen and positive pressure ventilation [4–6]. The pathophysiology of BPD is incompletely understood, but inflammation has been firmly established in the pathogenesis [7]. Studies have identified high neutrophil counts in tracheal aspirates shortly after birth, possibly triggered by chorioamnionitis, in addition to elevations in pro-inflammatory cytokines such as tumor necrosis factor-alpha, interleukin-8, and intercellular adhesion molecule-1 among neonates with BPD [8, 9]. Due to the limited number of effective therapeutic options available for the prevention of BPD and the role of inflammation in BPD, anti-inflammatory agents may be warranted for premature infants at high-risk of developing BPD.
Glucocorticoids are potent anti-inflammatory drugs that are commonly used in the neonatal intensive care unit setting. Several studies have investigated dexamethasone therapy for the treatment of BPD [10–19]. These studies, however, have reported conflicting results. Yeh et al. administered dexamethasone IV within 12 hours of birth, which was continued for 4 weeks, with tapering dosages the last 3 weeks. Their results demonstrated that fewer premature infants treated with dexamethasone had neonatal CLD compared to their placebo counterparts [10]. However, long-term follow-up of the dexamethasone-treated premature infants demonstrated more disabilities associated with significant neuromotor and cognitive deficits, less somatic growth in length, and smaller head circumference in school-aged children [20]. They concluded that the systemic administration of high-dose dexamethasone could not be recommended due to its adverse safety profile. Subsequently, Yeh et al. conducted a pilot study to reduce BPD in very low birth weight premature infants with severe respiratory distress via topical administration of budesonide into the lung, using surfactant as a vehicle [21]. Intratracheal instillation of budesonide in surfactant was associated with less risk of the composite outcome of death or BPD. However, the pharmacokinetics of budesonide in this group of premature infants were not determined. Additionally, the extent to which budesonide distributes into the brain, which could lead to neuromotor and cognitive deficits later in life, has not been assessed. Therefore, our study evaluated the pharmacokinetics of budesonide administered with surfactant as a vehicle in premature lambs and characterized the deposition of budesonide as well as its major metabolite and fatty acid derivative in lung and brain tissue 24 hours after a single 0.25 mg/kg dose of budesonide.
MATERIALS AND METHODS
Materials
Budesonide was purchased from IVAX Pharmaceuticals (Sellersville, PA). Surfactant was supplied by ONY, INC. (Amherst, New York). Budesonide and prednisolone (internal standard) for analytical purposes were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO) and 16α-hydroxy prednisolone (major budesonide metabolite) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Budesonide palmitate (a major fatty acid ester derivative of budesonide) was synthesized by reacting 31 mg budesonide with palmitoyl chloride (1:1.3 molar ratio) for 12 h at room temperature in 3 mL dry pyridine with constant stirring. Reactions and reagent handling were performed using dried glass containers and syringes to maximize product formation. The organic layer was then washed with H2O and concentrated under reduced pressure to obtain the ester. Final purification was achieved by passage through silica gel using cyclohexane:ethyl acetate (5:3). The product was a mixture of two products derived from the racemic acetal center at the CH2OH position. The products had a mass of 668.5 (m/z 669.5) which were confirmed by 1H NMR, 1H-1H COSY and HSQC NMR spectra. All other chemicals were HPLC grade and purchased from Sigma-Aldrich.
Management of Premature Lambs
All protocols adhered to APS/NIH guidelines for animal research and were approved by the Institution Animal Care and Use Committees (IACUC) at the University of Utah and University at Buffalo, State University of New York. Detailed methods have been published previously by Reyburn et al. and Null et al. [22, 23]. Briefly, pregnant ewes that carried single or twin fetuses at 131–133 days (term 150 days) were given an intramuscular injection of dexamethasone phosphate (6 mg; Vedco, Inc., St. Joseph, MO) 24–36 hours before operative delivery. On the morning of delivery, ewes received intramuscular ketamine hydrochloride (10–20 mg/kg; Fort Dodge Laboratories, Fort Dodge, IA) followed by inhalation of 1% isoflurane (Abbot Laboratories, North Chicago, IL). Fetuses were exposed by midline hysterotomy and catheters were placed in the common carotid artery and external jugular vein. Lambs were intubated with a cuffed (inflated with 2 mL of air) endotracheal tube. Ten mL of lung liquid was aspirated and replaced with surfactant mixed with budesonide, administered at 0.25 mg/kg. The endotracheal tube was then clamped and the lamb was delivered.
Sample Collection and Extraction
Plasma samples were collected from lambs during the time range of 15 min to 24 hours. Plasma extractions were adapted from Wang et al. [24] to include a liquid-liquid extraction. Plasma samples were thawed at room temperature and 50 μL (200 μM) of internal standard working solution was added to 1 mL of plasma. If 1 mL of plasma was not available, blank ovine plasma was added to account for matrix effects. Fifty μL of 4% formic acid in water was added to acidify the plasma. Samples were vortexed followed by the addition of 1 mL of 30% ethanol after which the sample was incubated at 4°C for 15 min. Samples were spun at 3500 rpm for 20 minutes to remove protein precipitate and supernatants were transferred to a new tube containing 2 mL methyl tert-butyl ether. Samples were shaken for 20 min at 4°C then spun at 3500 rpm for 20 minutes. The organic layer was transferred to a fresh tube and dried under air until completion. Samples were reconstituted in 40 μL of 30:70 methanol (MeOH) and H2O, and clarified by centrifugation immediately prior to LC/MS/MS analysis.
Tissue extractions were based on Miller-Larsson et al. [25] and modified to incorporate a liquid-liquid extraction rather than a solid phase extraction. Approximately 30 mg of tissue was disrupted in 5 mL of ethanol (EtOH) using a Sonics Vibra Cell sonicator operated at 30% amplitude for 25 seconds at 4°C. This mixture was then shaken vigorously for 5 hours at 4°C. Samples were clarified by centrifugation and the EtOH extract was then transferred to a new tube and dried under air at room temperature. Samples were reconstituted in 40 μL 30:70 MeOH:H2O immediately prior to liquid chromatography-tandem mass spectrometry (LC/MS/MS) analysis.
Analyte Measurements
LC/MS/MS analysis was conducted using either a Thermo LCQ Advantage Max ion trap instrument equipped with a Finnigan Surveyor LC pump, Surveyor Autosampler, and universal Ion Max source, or a Thermo-Finnigan TSQ Quantum AM triple-quadrupole mass spectrometer system with Perkin Elmer Series 200 Autosampler and Micropumps. Both systems were operated with Thermo Xcalibur software version 2.0 (Thermo Fisher Scientific, Waltham, MA). Positive electrospray ionization (ESI) was used and the instrument detection systems were optimized using budesonide. Analytes were resolved on a 50 cm × 2 mm i.d. Luna 5μm C18 reverse-phase HPLC column (Phenomenex Inc., Torrance, CA) and eluted with a linear gradient of 30–100% MeOH over 13 minutes followed by isocratic elution with 100% MeOH for 6 minutes. The aqueous solvent was 0.1% formic acid, the flow rate was 0.250 mL/min, and the column temperature was 40°C. Specific analytes were identified by retention time using the following precursor-to-product ion transitions, determined using analytical standards: m/z 431 → 413 (budesonide), m/z 377 → 323 (16α-hydroxy prednisolone), m/z 669 → 323 (budesonide-palmitate), and m/z 361 → 343 (prednisolone). Quantitation was based on the analyte-to-internal standard (prednisolone) peak area ratios using the Thermo Xcalibur 2.0 software. Standard curves for plasma ranged from 0.5 ng/mL to 100 ng/mL. The lower limit of quantification was 0.5 ng/mL for budesonide palmitate and 16α-hydroxy prednisolone, and 2.0 ng/mL for budesonide. Calibration curves for tissue ranged from 14 pg/mg to 3 μg/mg. The lower limit of quantitation for all analytes in tissue was 14 pg/mg.
Non-Compartmental Pharmacokinetic Modeling
A non-compartmental approach using Phoenix WinNonlin, version 5.1 (Pharsight, Mountain View, CA) was used to estimate the area under the time-concentration curve extrapolated to infinity (AUCinf) using the log-linear trapezoidal method. The terminal elimination half-life, and the maximum concentration (Cmax) after the single dose of budesonide administered with surfactant were also determined using Phoenix WinNonlin.
Population Pharmacokinetic Modeling
The absorption constant (Ka), apparent clearance (CL/F), and apparent volume of distribution (Vd/F) were estimated using a nonlinear mixed-effects model that was implemented in Monolix 4.3.1 (Lixoft, Orsay, France) using the stochastic approximation expectation maximization (SAEM) algorithm [26] combined with a Markov Chain Monte Carlo (MCMC) procedure. The number of MCMC chains was fixed to four. One- and two-compartment structural models were evaluated and selected based on their goodness of fit. Model stability was assessed by altering the initial estimates for budesonide Ka, CL/F, and Vd/F. Unstable models and those that produced erroneous results (e.g. negative parameter estimates) were disregarded. Diagnostic plots were used to assess the model’s fit. Population weighted residuals were plotted versus time and population predictions. Models were compared by assessing the biological plausibility of the parameter estimates, the variability of the parameter estimates, and the -2 log likelihood or the objective function value (OFV). Model variability and random effects were classified as one of two types of error: between subject variability (BSV) and residual unexplained variability (RUV). BSV was assumed to be log-normally distributed according to an exponential equation:
| (1) |
where Pi is the pharmacokinetic parameter of the ith individual, θpop is the population mean for P, and η represents the normally-distributed between subject random effect with a mean of zero and a variance of ω2. Additive, proportional, combined additive and proportional, and exponential RUV error models were evaluated. The final model used an exponential model of the form:
| (2) |
where Yij is the observed concentration for the ith individual at time j, Ŷij is the individual predicted concentration, and ε represents the normally-distributed error term with a mean of zero and variance of σ2.
Covariate Analysis
Several characteristics of the lambs were tested for their influence on budesonide pharmacokinetic parameters. The covariates that were tested included birth weight, gestational age, gender, multiple gestation, and study site. Power, exponential, and linear models were evaluated for each of the covariates. In addition, each covariate was evaluated after it was centered on the population mean. Forward addition was utilized to determine significant covariates. A decrease in the OFV ≥ 3.84 was considered significant for one degree of freedom at p = 0.05 based on the χ 2 distribution. Backward elimination was used to remove covariates from the model with an increase in the OFV ≥ 6.63 corresponding to 1 degree of freedom at p = 0.01.
Model Evaluation
Base and final models were evaluated using goodness-of-fit plots. Observed drug concentrations were inspected for their correlation with predicted concentrations. The −2 and +2 region criterion was used to assess the population weighted residual (PWRES) plots that were constructed. Uncertainty in pharmacokinetic parameter estimates was quantitatively assessed by calculating standard errors and 95% confidence intervals for all pharmacokinetic parameter estimates. Additionally, normalized prediction distribution errors (NPDE) were plotted against time and population-predicted budesonide concentrations to assess for model misspecification. A prediction-corrected visual predictive check (VPC) was also performed by simulating 500 budesonide concentrations at each time point [27].
RESULTS
Pharmacokinetics
Seven premature lambs were used for this study, for which 33 plasma concentration measurements are included in this study. Characteristics of each lamb are listed in Table 1. Two lambs at 1 hour had budesonide concentrations more than 3 standard deviations below the mean of the group, which were determined to be outliers and excluded from the analysis (p < 0.05, Grubbs’ test for outliers). Further investigation into these lambs revealed that their oxygenation status was different. These two lambs required higher concentrations of inspired oxygen to meet the prospective physiological target for oxygenation (arterial partial pressure of oxygen of 60 to 90 mmHg) than the other lambs (Table 1). We used the oxygenation index as a standard clinical expression of oxygenation status. Plotting oxygenation index for the first 3 hours of life versus budesonide concentrations at 1 hour of life demonstrated that the two lambs also had very low concentration of budesonide. We interpret these results to mean that the lung parenchyma was not recruited during resuscitation and subsequent hours of life, resulting in decreased absorption of budesonide from the lungs into the systemic circulation. Decreased absorption would result in decreased plasma concentration of budesonide, which led us to exclude these animals from further analyses. For the five lambs that remained, 25 plasma concentrations were obtained. All samples after 9 hours were below the LLOQ of 2 ng/mL for budesonide and were excluded from the analysis. In addition, neither 16α-hydroxy prednisolone nor budesonide-palmitate were detected in the plasma samples (Fig. 1).
Table 1.
Premature lamb characteristics.
| Lamb | Weight (kg) | Gestational Age (days) | Sex | Oxygenation Index (Mean for First 3h of Life) | Site | Multiple Gestation |
|---|---|---|---|---|---|---|
| 1 | 3.6 | 139 | Male | 2.83 | New York | Yes |
| 2 | 2.0 | 133 | Male | 2.67 | New York | Yes |
| 3 | 3.1 | 133 | Female | 3.76 | Utah | Yes |
| 4 | 3.7 | 131 | Female | 4.66 | Utah | No |
| 5 | 3.5 | 132 | Female | 5.01 | Utah | No |
| 6* | 4.2 | 132 | Female | 12.63 | Utah | Yes |
| 7* | 3.6 | 132 | Female | 6.07 | Utah | Yes |
Excluded from the study because concentrations were more than 3 standard deviations below the mean of the group; Oxygenation index = (MAP*FiO2)/PaO2
Fig. (1).
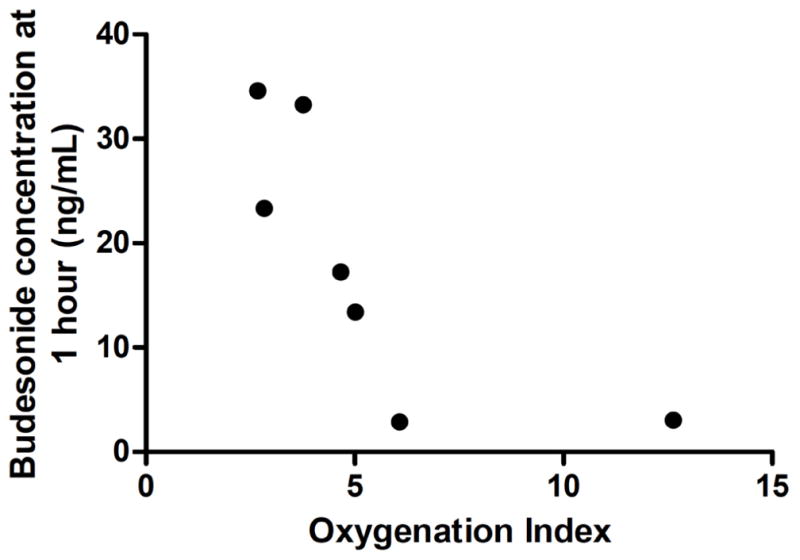
Correlation plot of oxygenation index (mean for the first 3h of life) vs budesonide concentrations at 1 h for all lambs.
A non-compartmental analysis was used to describe the AUCinf, half-life, and Cmax for the lambs (Table 2). For the population pharmacokinetic analysis, the data were well described by a one-compartment model with first order-absorption and first-order elimination (Table 3). Estimation of Ka was initially attempted; however, Ka could not be reliably parameterized and the absorption rate constant was fixed to 10 h−1 as done previously by Wu et al. [28] Covariate analyses identified no significant covariates, which was not unexpected due to the limited sample size and the premature lamb model system. Diagnostic plots were visually inspected to confirm the selection of the final model (Fig. 2A and 2B). PWRES and NPDE plots were used to further evaluate the model (Fig. 3A, 3B, 3C and 3D). The data were equally distributed around zero and concentrated between the −2 and +2 range. Furthermore, data were simulated to produce a visual predictive check (Fig. 4). All of the simulations were within the 10th, 50th, and 90th percentiles, although the 90th and 50th percentiles overlapped at time zero. Overall, the VPC demonstrated reasonable agreement between observed and simulated concentrations.
Table 2.
Non-compartmental pharmacokinetic parameter estimates.
| Parameter | Mean ± Standard Deviation |
|---|---|
| AUCinf (h*μg/L) | 148.77 ± 28.16 |
| Half-life (h) | 4.76 ± 1.79 |
| Cmax (μg/L) | 46.17 ± 17.71 |
AUCinf – area under the concentration-time curve extrapolated to infinity; Cmax – maximum concentration.
Table 3.
One compartment model population pharmacokinetic parameter estimates.
| Parameter | Estimate | R.S.E. (%) |
|---|---|---|
| Ka (h−1) | 10.0 (fixed) | n.a. |
| Vd/F (L) | 29.1 | 15 |
| CL/F (L/h) | 6.29 | 9 |
| ω-BSV | ||
| Vd/F | 0.225 | 70 |
| CL/F | 0.0478 | 565 |
| σ-RUV | 0.380 | 17 |
Ka – Absorption rate constant; Vd/F – apparent volume of distribution; CL/F – apparent clearance; BSV- between subject variability; RUV- residual unexplained variability; R.S.E. – relative standard error.
Fig. (2). Observed budesonide concentrations versus population predicted and individual predicted budesonide concentrations in 5 premature lambs.

Population predicted concentrations are presented in A and individual predicted concentrations are presented in B for the final model. The solid line represents the line of reference and the dotted line represents the locally-weighted spline of the model.
Fig. (3). The population weighted residuals (PWRES) and normalized prediction distribution errors (NPDE) versus time and population predicted budesonide concentrations.

PWRES are presented in panels A and C and NPDE are presented in panels B and D. The solid line is the reference line at zero.
Fig. (4). Visual predictive check of the final ovine budesonide model.

The dark gray shading represents the 10th and 90th percentiles. The light gray shading represents the 50th percentile.
Tissue Analysis
Lung, as well as brain gray matter and white matter, were analyzed in 2 of the premature lambs 24 hours after budesonide administration. Budesonide was detected in the lung of one lamb and was quantitated at 34 pg/mg of lung tissue (no budesonide was detected at 24 hours in the plasma). The other lamb did not have detectable budesonide in the lung. Budesonide-palmitate in the lung of both lambs was detected, although it was below the LLOQ of 14 pg/mg. No budesonide metabolite was detected. Budesonide was not detected in either the gray or the white matter in either of the lambs. Additionally, all brain tissue samples were below the LLOQ (14 pg/mg) for 16α-hydroxy prednisolone and budesonide-palmitate.
DISCUSSION
This is the first study to describe the pharmacokinetics of budesonide given with surfactant as a vehicle in premature lambs. In addition, this is the first study to characterize the metabolism of budesonide in lambs. Our results demonstrate that the pharmacokinetic profile of budesonide is similar to that seen in premature human infants given the same dose of budesonide. Although the Cmax was higher in lambs than that reported in premature human infants (approximately 20 ng/mL), the half-life is around 4 hours, similar to that documented by Yeh et al. [21] In addition, our study demonstrates that the pharmacokinetics of budesonide in premature lambs is adequately described by a one-compartment model, with a population clearance equal to 6.29 L/h (1.99 L/h/kg) and volume of distribution equal to 29.1 L (9.2 L/kg), after fixing Ka to 10 h−1. Covariate analyses did not identify any significant covariates, although this was not surprising due to the study’s limited sample size and use of a premature lamb model.
The residual variability of the population model was moderately high (38%) and could not be reduced by incorporating any measured factors related to the lambs included in this study. In particular, the samples taken right after surfactant instillation demonstrated the greatest amount of variability (Fig. 2A). We speculate that this pharmacokinetic variability is attributable to variability in initial intrapulmonary distribution of budesonide, which was given by mixing budesonide with surfactant and then instilling the combined product through a tube passed through the endotracheal tube to reach the lungs. However, this variability would likely have been even greater if instillation had been given with just drug alone. Other studies using surfactant as a vehicle for instillation of antibiotics [29–31] and another glucocorticoid, dexamethasone, [32] in animals also demonstrated more consistent lung absorption when administered with surfactant compared to administration of the drug alone, supporting use of surfactant as a drug-delivery vehicle into the lung.
Previous studies with glucocorticoids administered systemically as a treatment for BPD demonstrated poor long-term neurological outcomes [10, 12, 20]. This is likely due to high concentrations of glucocorticoids that crossed the blood brain barrier and adversely affected neurologic development or caused abnormal cerebral blood flow and ischemia. Consequently, one of the goals of our study was to measure the concentrations of budesonide, 16α-hydroxy prednisolone, and budesonide-palmitate in the gray and white matter of the brain to assess the degree to which intratracheal instillation of budesonide in surfactant has the potential to penetrate the brain and result in adverse neurological outcomes similar to those seen with high-dose dexamethasone [33]. Our study revealed no detectable budesonide, 16α-hydroxy prednisolone, or budesonide-palmitate in the gray or white matter of the brain in this premature lamb model study, which may be evidence of a favorable safety profile, although the sample size is small. Furthermore, budesonide is extensively metabolized by cytochrome P450 (CYP) 3A enzymes in vitro [34]. However, cytochrome P450 3A4 activity level is low in premature newborns that progressively increases to adult activity [35]. The activity in human newborns is adequate to metabolize budesonide because the major metabolite, 16α-hydroxy prednisolone, has been measured in neonates in plasma at comparable concentrations to budesonide which was administered via surfactant as well [21]. In the premature lambs, however, no metabolite was detected in the plasma (LLOQ 0.5 ng/mL) or the tissue (LLOQ 14 pg/mg), which suggests that lamb CYP3A enzymes do not metabolize budesonide as readily as human neonatal CYP3A enzymes. Theoretically, this could result in more bioavailable budesonide, with the potential to cross the blood brain barrier in these premature lambs, compared to premature human infants. Support for this may be found in the higher Cmax observed in premature lambs compared to that reported previously in premature human infants [21]. Furthermore, the plasma Cmax was around 1000-fold higher than 50% of the maximal dose required to achieve biological effects with budesonide [36], even though no budesonide was detected in the brain. In aggregate, not detecting the budesonide metabolite in the plasma, coupled with the lack of detectable concentrations of budesonide in both gray and white matter, may be cautiously interpreted as evidence supporting future clinical trials of this therapy in premature human infants.
This study presents the first pharmacokinetic description of budesonide administered in surfactant as a vehicle in premature lambs, which will be used to inform future neonatal clinical trials. In this study, the half-life of budesonide in premature lambs was similar to that observed in premature human infants. Our study identified undetectable concentrations in brain tissue, which suggests that intratracheal instillation of budesonide, using surfactant as the vehicle, may feature a favorable neurological safety profile. Based on our pharmacokinetic parameter estimates and tissue measurements, future studies of budesonide administered in surfactant are warranted in premature human infants at-risk for BPD.
Acknowledgments
JKR designed the research study, processed plasma samples, performed LC/MS/MS analysis, performed modeling analysis, and wrote the manuscript. CS performed modeling analysis, wrote the manuscript and critically reviewed the manuscript. MJD performed the lamb studies. KHA performed the lamb studies and critically reviewed the manuscript. EE designed the research study, provided surfactant for the studies, and critically reviewed the manuscript. ZL performed the fatty acid synthesis and validation. CAR performed the LC/MS/MS analysis and critically reviewed the manuscript. PLB designed the research study and critically reviewed the manuscript. RAB designed the research study and critically reviewed the manuscript. RMW designed the research study and critically reviewed the manuscript.
Footnotes
CONFLICT OF INTEREST
This work was supported by an unrestricted research grant from ONY, INC as well as the National Institutes of Health Eunice Kennedy Shriver National Institute of Child Health and Human development [Grant HD060559]. JKR was supported by the Pharmacotherapy Subspecialty Award from the Primary Children’s Hospital Foundation. CS was supported by the American Foundation for Pharmaceutical Education’s Clinical Pharmaceutical Sciences Fellowship. KHA is supported by NIH grant HL110002. EE is part owner of ONY, INC.
References
- 1.Northway WH, Jr, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. 1967;276(7):357–68. doi: 10.1056/NEJM196702162760701. [DOI] [PubMed] [Google Scholar]
- 2.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. 2007;357(19):1946–55. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- 3.Wong PM, Lees AN, Louw J, et al. Emphysema in young adult survivors of moderate-to-severe bronchopulmonary dysplasia. Eur Respir J. 2008;32(2):321–8. doi: 10.1183/09031936.00127107. [DOI] [PubMed] [Google Scholar]
- 4.Sinkin RA, Cox C, Phelps DL. Predicting risk for bronchopulmonary dysplasia: selection criteria for clinical trials. Pediatrics. 1990;86(5):728–36. [PubMed] [Google Scholar]
- 5.Coalson JJ, Winter V, deLemos RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 1995;152(2):640–6. doi: 10.1164/ajrccm.152.2.7633720. [DOI] [PubMed] [Google Scholar]
- 6.Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med. 1999;160(4):1333–46. doi: 10.1164/ajrccm.160.4.9810071. [DOI] [PubMed] [Google Scholar]
- 7.Wright CJ, Kirpalani H. Targeting inflammation to prevent bronchopulmonary dysplasia: can new insights be translated into therapies? Pediatrics. 2011;128(1):111–26. doi: 10.1542/peds.2010-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speer CP. Inflammation and bronchopulmonary dysplasia. Semin Neonatol. 2003;8(1):29–38. doi: 10.1016/s1084-2756(02)00190-2. [DOI] [PubMed] [Google Scholar]
- 9.Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics. 1996;97(2):210–5. [PubMed] [Google Scholar]
- 10.Yeh TF, Lin YJ, Hsieh WS, et al. Early postnatal dexamethasone therapy for the prevention of chronic lung disease in preterm infants with respiratory distress syndrome: a multicenter clinical trial. Pediatrics. 1997;100(4):E3. doi: 10.1542/peds.100.4.e3. [DOI] [PubMed] [Google Scholar]
- 11.Wang JY, Yeh TF, Lin YJ, Chen WY, Lin CH. Early postnatal dexamethasone therapy may lessen lung inflammation in premature infants with respiratory distress syndrome on mechanical ventilation. Pediatr Pulmonol. 1997;23(3):193–7. doi: 10.1002/(sici)1099-0496(199703)23:3<193::aid-ppul4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 12.Yeh TF. Prevention of chronic lung disease (CLD) in premature infants with early dexamethasone therapy. Pediatr Pulmonol Suppl. 1997;16:35–6. doi: 10.1002/ppul.1950230819. [DOI] [PubMed] [Google Scholar]
- 13.Mammel MC, Green TP, Johnson DE, Thompson TR. Controlled trial of dexamethasone therapy in infants with bronchopulmonary dysplasia. Lancet. 1983;1(8338):1356–8. doi: 10.1016/s0140-6736(83)92139-6. [DOI] [PubMed] [Google Scholar]
- 14.Avery GB, Fletcher AB, Kaplan M, Brudno DS. Controlled trial of dexamethasone in respirator-dependent infants with bronchopulmonary dysplasia. Pediatrics. 1985;75(1):106–11. [PubMed] [Google Scholar]
- 15.Garland JS, Alex CP, Pauly TH, et al. A three-day course of dexamethasone therapy to prevent chronic lung disease in ventilated neonates: a randomized trial. Pediatrics. 1999;104(1 Pt 1):91–9. doi: 10.1542/peds.104.1.91. [DOI] [PubMed] [Google Scholar]
- 16.Rastogi A, Akintorin SM, Bez ML, Morales P, Pildes RS. A controlled trial of dexamethasone to prevent bronchopulmonary dysplasia in surfactant-treated infants. Pediatrics. 1996;98(2 Pt 1):204–10. [PubMed] [Google Scholar]
- 17.Cummings JJ, D’Eugenio DB, Gross SJ. A controlled trial of dexamethasone in preterm infants at high risk for bronchopulmonary dysplasia. N Engl J Med. 1989;320(23):1505–10. doi: 10.1056/NEJM198906083202301. [DOI] [PubMed] [Google Scholar]
- 18.Early postnatal dexamethasone therapy for the prevention of chronic lung disease. Pediatrics. 2001;108(3):741–8. doi: 10.1542/peds.108.3.741. [DOI] [PubMed] [Google Scholar]
- 19.Dexamethasone therapy in neonatal chronic lung disease: an international placebo-controlled trial. Collaborative Dexamethasone Trial Group. Pediatrics. 1991;88(3):421–7. [PubMed] [Google Scholar]
- 20.Yeh TF, Lin YJ, Lin HC, et al. Outcomes at school age after postnatal dexamethasone therapy for lung disease of prematurity. N Engl J Med. 2004;350(13):1304–13. doi: 10.1056/NEJMoa032089. [DOI] [PubMed] [Google Scholar]
- 21.Yeh TF, Lin HC, Chang CH, et al. Early intratracheal instillation of budesonide using surfactant as a vehicle to prevent chronic lung disease in preterm infants: a pilot study. Pediatrics. 2008;121(5):e1310–8. doi: 10.1542/peds.2007-1973. [DOI] [PubMed] [Google Scholar]
- 22.Reyburn B, Li M, Metcalfe DB, et al. Nasal ventilation alters mesenchymal cell turnover and improves alveolarization in preterm lambs. Am J Respir Crit Care Med. 2008;178(4):407–18. doi: 10.1164/rccm.200802-359OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Null DM, Alvord J, Leavitt W, et al. High-frequency nasal ventilation for 21 d maintains gas exchange with lower respiratory pressures and promotes alveolarization in preterm lambs. Pediatr Res. 2014;75(4):507–16. doi: 10.1038/pr.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Tang Y, Moellmann H, Hochhaus G. Simultaneous quantification of budesonide and its two metabolites, 6beta-hydroxybudesonide and 16alpha-hydroxyprednisolone, in human plasma by liquid chromatography negative electrospray ionization tandem mass spectrometry. Biomed Chromatogr. 2003;17(2–3):158–64. doi: 10.1002/bmc.233. [DOI] [PubMed] [Google Scholar]
- 25.Miller-Larsson A, Mattsson H, Hjertberg E, Dahlback M, Tunek A, Brattsand R. Reversible fatty acid conjugation of budesonide. Novel mechanism for prolonged retention of topically applied steroid in airway tissue. Drug Metab Dispos. 1998;26(7):623–30. [PubMed] [Google Scholar]
- 26.Kuhn E, Lavielle M. Maximum likelihood estimation in nonlinear mixed effects models. Comput Stat Data Anal. 2005;49:1020–38. [Google Scholar]
- 27.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. Aaps J. 2011;13(2):143–51. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu K, Goyal N, Stark JG, Hochhaus G. Evaluation of the administration time effect on the cumulative cortisol suppression and cumulative lymphocytes suppression for once-daily inhaled corticosteroids: a population modeling/simulation approach. J Clin Pharmacol. 2008;48(9):1069–80. doi: 10.1177/0091270008320607. [DOI] [PubMed] [Google Scholar]
- 29.van’t Veen A, Mouton JW, Gommers D, Lachmann B. Pulmonary surfactant as vehicle for intratracheally instilled tobramycin in mice infected with Klebsiella pneumoniae. Br J Pharmacol. 1996;119(6):1145–8. doi: 10.1111/j.1476-5381.1996.tb16016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van’t Veen A, Gommers D, Mouton JW, Kluytmans JA, Krijt EJ, Lachmann B. Exogenous pulmonary surfactant as a drug delivering agent: influence of antibiotics on surfactant activity. Br J Pharmacol. 1996;118(3):593–8. doi: 10.1111/j.1476-5381.1996.tb15442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kharasch VS, Sweeney TD, Fredberg J, et al. Pulmonary surfactant as a vehicle for intratracheal delivery of technetium sulfur colloid and pentamidine in hamster lungs. Am Rev Respir Dis. 1991;144(4):909–13. doi: 10.1164/ajrccm/144.4.909. [DOI] [PubMed] [Google Scholar]
- 32.Nimmo AJ, Carstairs JR, Patole SK, Whitehall J, Davidson K, Vink R. Intratracheal administration of glucocorticoids using surfactant as a vehicle. Clin Exp Pharmacol Physiol. 2002;29(8):661–5. doi: 10.1046/j.1440-1681.2002.03712.x. [DOI] [PubMed] [Google Scholar]
- 33.Halliday HL, Ehrenkranz RA, Doyle LW. Early postnatal (<96 hours) corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database Syst Rev. 2003;(1):CD001146. doi: 10.1002/14651858.CD001146. [DOI] [PubMed] [Google Scholar]
- 34.Moore CD, Roberts JK, Orton CR, et al. Metabolic pathways of inhaled glucocorticoids by the CYP3A enzymes. Drug Metab Dispos. 2013;41(2):379–89. doi: 10.1124/dmd.112.046318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver--evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247(2):625–34. doi: 10.1111/j.1432-1033.1997.00625.x. [DOI] [PubMed] [Google Scholar]
- 36.Jaffuel D, Demoly P, Gougat C, et al. Transcriptional potencies of inhaled glucocorticoids. Am J Respir Crit Care Med. 2000;162(1):57–63. doi: 10.1164/ajrccm.162.1.9901006. [DOI] [PubMed] [Google Scholar]