

Abstract
Patients with hypertrophic cardiomyopathy (HCM), a disease associated with sarcomeric protein mutations, often suffer from sudden cardiac death (SCD) resulting from arrhythmia. In order to advance SCD prevention strategies, our understanding of how sarcomeric mutations in HCM patients contribute to enhanced arrhythmogenesis needs to be improved. Early afterdepolarizations (EADs) are an important mechanism underlying arrhythmias associated with HCM-SCD. Although the ionic mechanisms underlying EADs have been studied in general, whether myofilament protein dynamics mechanisms also underlie EADs remains unknown. Thus, our goals were to investigate if myofilament protein dynamics mechanisms underlie EADs and to uncover how those mechanisms are affected by pacing rate, sarcomere length (SL), and different levels of HCM-induced myofilament remodeling. To achieve this, a mechanistically-based bidirectionally coupled human electrophysiology-force myocyte model under the conditions of HCM was constructed. HCM ionic remodeling included a reduced repolarization reserve, while HCM myofilament modeling involved altered thin filament activation. We found that the mechanoelectric feedback (MEF) on calcium dynamics in the bidirectionally coupled model, via Troponin C buffering of cytoplasmic Ca2+, was the myofilament mechanism underlying EADs. Incorporating MEF diminished the degree of repolarization reserve reduction necessary for EADs to emerge and increased the frequency of EAD occurrence, especially at faster pacing rates. Longer SLs and enhanced thin filament activation diminished the effects of MEF on EADs. Together these findings demonstrate that myofilament protein dynamics mechanisms play an important role in EAD formation.
Keywords: Early afterdepolarizations, mechanoelectric feedback, hypertrophic cardiomyopathy, arrhythmia, computer modeling
1. Introduction
Hypertrophic cardiomyopathy (HCM), defined by unexplained ventricular hypertrophy, is the most commonly inherited cardiac disease, with a reported prevalence of 1 in 500 worldwide (Maron et al., 1995; Maron and Maron, 2013). Although the annual mortality from HCM is low (0.5 – 2%) (Elliott et al., 2014; Maron et al., 2014), certain patient subsets, such as young athletes, have enhanced risk of sudden cardiac death (SCD) (Maron et al., 2014; Maron et al., 2007). Thus far, the only treatment proven to prolong life and prevent SCD in HCM patients is implantation of a cardioverter defibrillator (ICD) (Gersh et al., 2011; Maron and Maron, 2013; Maron et al., 2003; Maron et al., 2000; Maron et al., 2013; Maron et al., 2007; Schinkel et al., 2012; Vriesendorp et al., 2013). The main risk markers used to identify those patients who will benefit most from an ICD include family history of HCM SCD, unexplained syncope, multiple-repetitive non-sustained ventricular tachycardia (NSVT) episodes, abnormal exercise blood pressure response, late gadolinium enhancement ≥15% of left ventricular mass, and massive left ventricular hypertrophy ≥ 30 mm (Gersh et al., 2011; Maron, 2002; Maron, 2010; Maron and Maron, 2013; Maron et al., 2003; Spirito et al., 2014; Spirito et al., 2009; Spirito et al., 2000). However, HCM patients who lack all risk factors are still not immune to SCD (Pastore et al., 1999; Spirito et al., 2014). In addition, the risk stratification models used to identify high risk adult patients are not as effective at identifying children with high risk (Maron et al., 2013). Furthermore, since SCD risk in HCM patients is highest in younger patients (<30 years old) (Maron et al., 2014), ICDs must be implanted early in life, increasing the likelihood that those individuals will experience device-related complications (Maron et al., 2013). Despite the challenges of SCD risk stratification in young HCM patients and their increased probability of device complications, other treatment options are limited. In fact despite wide prevalence, many consider HCM an orphan condition because it lacks disease-specific pharmacological treatment (Spoladore et al., 2012), which may be a consequence of the wide genetic and phenotypic heterogeneity among HCM patients. In particular, more than 1500 mutations identified on at least 11 genes, encoding thick and thin myofilament protein components, have been found to underlie HCM (Bos et al., 2009; Ingles et al., 2013; Maron et al., 2012; Niimura et al., 1998; Seidman and Seidman, 2011), though the causality and contribution to disease progression for each is not fully understood (Bezzina et al., 2015). To complicate matters, a new HCM subgroup, described as ‘genotype positive-phenotype negative,’ has been discovered in which patients have one or more pathogenic mutations but no disease phenotype (Gersh et al., 2011; Gray et al., 2011; Maron and Semsarian, 2010; Maron et al., 2011). The lack of disease-specific pharmacological treatment, the challenges of accurately stratifying HCM SCD patients, and the incomplete understanding of the relationship between HCM genotype, phenotype and outcome (Charron et al., 2010) underscore the need to improve SCD prevention strategies for HCM patients by improving our understanding of how sarcomeric mutations in HCM patients contribute to the enhanced arrhythmogenesis underlying SCD (Coppini et al., 2013; Maron et al., 2000).
Patients with HCM often have cardiac arrhythmias, including atrial fibrillation, NSVT, and sustained ventricular tachycardia, as well as prolonged QTc (Coppini et al., 2013; Coppini et al., 2014; Maron et al., 2000). Early afterdepolarizations (EADs), defined as a slowing or reversal of normal repolarization during the plateau or rapid depolarization phases, have been implicated as the primary mechanism underlying many arrhythmias associated with HCM (Weiss et al., 2010; Yan et al., 2001) and have also been found in patients with HCM (Coppini et al., 2013). Many studies have investigated the ionic mechanisms of EADs in lethal ventricular arrhythmias and have found that EADs occur when repolarization reserve is reduced due to enhanced inward current, diminished outward current, or both. In addition, there must also be a regenerative increase in net inward current that has the ability to overcome and reverse repolarization. The ion channels typically implicated in this positive feedback are the L-type Ca2+ current (ICaL) and the Na+-Ca2+ exchange current (INaCa) (Weiss et al., 2010). New studies have also begun to explore the ionic mechanisms of EADs in human HCM tissue, finding that ICaL and INaCa play an important role along with the rapid delayed rectifier current (IKr), the late Na+ current (INaL), and Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Coppini et al., 2013).
Although the ionic mechanisms of EADs have been studied, whether there are also myofilament protein dynamics mechanisms at play is poorly understood. Since mechanoelectric feedback (MEF) is known to be essential for normal function of the heart and is believed to be important in disease (Quinn, 2014; Taggart and Sutton, 1999; Tardiff et al., 2015; Zile and Trayanova, 2016) and since HCM is a disease caused by sarcomeric mutations, it is important to explore whether myofilament protein dynamics mechanisms could also contribute to EADs in human HCM. Thus, our goal was to utilize the capability of mechanistic computer simulations to investigate whether myofilament protein dynamics mechanisms modulate EAD formation in HCM for varying degrees of reduced repolarization reserve, and to uncover how these mechanisms are affected by pacing rate, sarcomere length, and the degree of HCM-induced myofilament remodeling.
2. Methods
2.1. Human Electrophysiology-Force Myocyte Model
To investigate whether myofilament protein dynamics mechanisms modulate EAD formation under the conditions of human HCM, a mechanistically-based human electrophysiology-force myocyte model was used. The electrophysiology-force model combined the human endocardial ventricular membrane kinetics model by Vandersickel et al (Vandersickel et al., 2014) and the myofilament protein dynamics model by Rice et al (Rice et al., 2008). The Vandersickel et al model, an extension of the ten Tusscher et al model (ten Tusscher and Panfilov, 2006), which incorporates updated L-type Ca2+ channel kinetics, was used because it is capable of generating EADs during reduced repolarization reserve. The Rice et al model, which describes the activation of the thin filament by intracellular calcium binding to Troponin C as well as thin filament binding to thick filament crossbridges (XBs) using a 6 state Markov model, was chosen for its computational efficiency and incorporation of important biophysical detail and cooperativity mechanisms. Since the Rice et al myofilament model was developed based on rabbit data, we adjusted it to match human force data as described elsewhere (Zile and Trayanova, 2016).
The ionic and myofilament models were bidirectionally coupled by incorporating MEF on calcium dynamics (Figure 1); this was done by incorporating a dynamic term for troponin buffering of intracellular calcium ([Ca]Troponin) using our previously developed approach (Zile and Trayanova, 2016) where we refer to this as “strong coupling.” In summary, the following equation was used to update the intracellular calcium concentration in the ionic model, using the [Ca]Troponin term calculated by the myofilament model, at each time step:
| (1) |
where [Ca]Total is the total calcium in the cytoplasm, [Ca]i is the free calcium in the cytoplasm, [Ca]Calmodulin is the total calcium buffered by calmodulin in the cytoplasm, and [Ca]Troponin is the total calcium bound to Troponin C and incorporates the cooperativity of calcium-troponin binding due to strongly bound nearby XBs. Bidirectionally coupling the models with a dynamic representation of [Ca]Troponin was important and necessary, because it has been shown to be crucial for accurately reproducing contractile experiment data in myocyte simulations (Ji et al., 2015).
Figure 1.
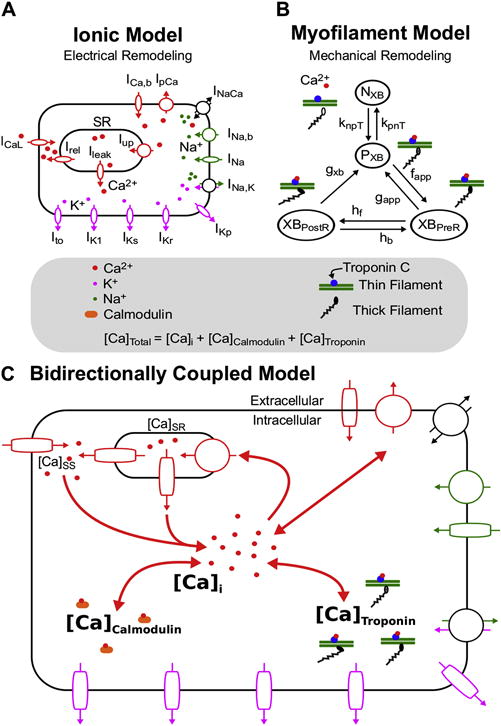
Ionic (A) and Myofilament (B) models combined to form the Bidirectionally Coupled Human Electrophysiology-Force Myocyte Model (C). A modified version of the ten Tusscher ionic model from Vandersickel et al (Vandersickel et al., 2014) was used for the ionic model. A modified version of the Markov state diagram of the myofilament model from Rice et al, which describes thin filament activation via free intracellular calcium ([Ca]i) binding to Troponin C as well as thin filament binding to thick filaments to form crossbridges (XBs), was used for the myofilament model. The transition rates (knpT and kpnT) between the thin filament states where XB formation is inhibited (NXB) and where weakly bound XB formation is possible (PXB) are both functions of perm50, kon, koffH, and koffL. The rate knpT is also dependent on kn_p, and kpnT is additionally dependent on kp_n. The XBPreR and XBPostR states represent a thin filament with a strongly bound XB that do not and do, respectively, have rotated myosin heads which induced strain (Rice et al., 2008). Bidirectional coupling was obtained by incorporating MEF on calcium dynamics via Equation 1, where the total cytoplasmic calcium ([Ca]Total) is equal to the sum of [Ca]i, the total calcium bound to calmodulin ([Ca]Calmodulin), and the total calcium bound to troponin C ([Ca]Troponin).
A unidirectionally coupled version of the model (with no MEF and referred to as “weakly coupled” in our previously developed approach (Zile and Trayanova, 2016)) was created by removing the [Ca]Troponin term from Equation 1. The sole purpose of this model was to aid in examining how MEF altered EAD emergence and the frequency of EAD occurrence, by comparing its simulations to those of the bidirectionally coupled model.
2.2. Incorporating HCM-Induced Remodeling
We simulated human HCM in our single cell electrophysiology-force models by incorporating ionic and myofilament remodeling. Ionic remodeling was represented by reducing repolarization reserve. Specifically, we increased the maximal conductance of the L-type Ca2+ current (GCaL) 2.5–5 fold (0.05 increments) and decreased the maximal conductance of the rapid delayed rectifier current (GKr) to 10–100% of its baseline value (in increments of 0.1%) in the ten Tusscher et al model, similar to Vandersickel et al (Vandersickel et al., 2014) and Zimik et al (Zimik et al., 2015) to represent increased ICaL density, CaV1.2 protein and CaCNA1.2, and decreased IKr density and HERG1b observed in human HCM (Coppini et al., 2013). These specific features of HCM remodeling were incorporated because they have been shown to be important to the development of EADs in previous studies of human HCM (Coppini et al., 2013). A range of values was used because HCM remodeling has been shown to be progressive over time (Gersh et al., 2011; Semsarian et al., 1997).
Myofilament remodeling was incorporated to simulate altered thin filament activation and myofilament Ca2+ sensitivity found in human HCM. Changes in thin filament activation have been linked to altered phosphorylation of cardiac Troponin I (cTnI) (Messer et al., 2007), mutations in cardiac troponin T (cTnT) (Miller et al., 2001) and mutations in cardiac myosin binding protein C (cMyBP-C) (Carrier et al., 2015; Kampourakis et al., 2014; Mun et al., 2014; Previs et al., 2015; Sequeira et al., 2015; Witayavanitkul et al., 2014), which occur in HCM in humans (Carrier et al., 2015; Sequeira et al., 2015) and animals (Miller et al., 2001; Warren et al., 2015). Although there are more sarcomeric mutations that cause HCM, such as in cardiac actin (ACTC), cardiac troponin C (TNNC1), and α-tropomyosin (TPM1) (Olivotto et al., 2015), less is known about how these affect thin filament activation, if at all. Furthermore, myocardial Ca2+ sensitivity, defined as the amount of Ca2+ necessary to produce half maximal force, is a function of thin filament activation and XB cycling rates and has been shown to be altered in HCM. Myocardial Ca2+ sensitivity has been found to increase by 1–7% in human HCM (Coppini et al., 2013; Robinson et al., 2007; Warren et al., 2015) and by 3–10% in animal models of HCM (Miller et al., 2001).
Despite the lingering uncertainty in the mechanisms by which these sarcomeric mutations lead to the altered myofilament properties outlined above and the uncertainty of the exact amount by which the myofilament properties are altered in HCM, these studies indicate that myofilament parameters involved in thin filament activation and Ca2+ sensitivity are important components of the disease manifestation and therefore may contribute to the emergence of EADs. To elucidate if and how changes to myofilament parameters in human HCM promote the emergence of EADs or modulate the frequency of EAD occurrence, we incorporated HCM remodeling into the Rice et al myofilament model. Specifically, we altered thin filament activation, embodied in the Rice et al model by these 3 parameters: perm50, koffH, and kon. The parameters knpT and kpnT are nonlinear transition rates that are functions of these 3 parameters and represent nearby regulatory unit (RU)-based and calcium-based activation of the thin filament, which is shown in Figure 1 as the transition of the thin filament from the NXB state (XB formation is inhibited) to the PXB state (weakly bound XB formation is possible). The parameter perm50 is the half activation constant for the shift of a thin filament RU from NXB to PXB, koffH is the rate constant for Ca2+ unbinding from the high affinity binding site of Troponin C, and kon is rate constant for Ca2+ binding to Troponin C. Due to the uncertainty in the literature regarding the exact amount that these parameters change in HCM, as described above, we explored HCM —induced remodeling of these 3 myofilament parameters within the range of 85% to 115% of their baseline values (increments of 5%) (Table 1).
Table 1.
Baseline and HCM values for important model parameters. GCaL and GKr are from the ten Vandersickel et al ionic model (Vandersickel et al., 2014). perm50, kon, and koffH are from the Rice et al myofilament model (Rice et al., 2008).
| Parameter | Baseline Value (BV) | HCM Value | Effect of HCM Remodeling on EADs | |
|---|---|---|---|---|
|
| ||||
| GCaL | 7.96*10 cm/(ms*μF) | Range of Values (Increments of 0.1): | Increase necessary for EAD Emergence | |
| BV*2.5 to BV*5.0 | ||||
|
| ||||
| GKr | 0.153 nS/pF | Range of Values (Increments of 0.1%): | Decrease necessary for EAD Emergence | |
| BV*10% to BV*100% | ||||
|
| ||||
| perm50
|
0.5 (unitless)
|
Range of Values (Increments of 5%): |
Increase
|
Enhances MEF effects on EAD emergence & frequency of occurrence |
| koffH
|
25 1/s
|
Increase
|
||
| kon | 47.5 1/μMs | BV*85% to BV*115% | Decrease | |
2.3. EAD Protocol
Studies of cardiomyocytes from HCM patients have shown that EADs arise at pacing frequencies ranging from 0.25–1 Hz (Coppini et al., 2013), consistent with clinical studies in which arrhythmias arose in HCM patients who were sedentary, asleep, or performing normal daily activities (Maron et al., 1994; Maron et al., 2009); EADs have also been induced, at these rates, in computational electrophysiological models of human ventricular arrhythmias associated with HCM (Vandersickel et al., 2014; Zimik et al., 2015). To induce EADs in the bidirectionally and unidirectionally coupled HCM electrophysiology-force myocyte models, we used a pacing protocol similar to those in Zimik et al (Zimik et al., 2015) and Vandersickel et al (Vandersickel et al., 2014). We isometrically paced the myocyte models at one of three pacing cycle lengths (CLs) (1000 ms; 1Hz, 2000 ms; 0.5 Hz, or 4000 ms; 0.25 Hz) for 100 beats. After the first 50 beats, all of our simulations had reached steady state, similar to previous studies (Zile and Trayanova, 2016). The final 50 beats were used to analyze the emergence of EADs and the frequency of their occurrence. Since sarcomere length (SL) changes during the cardiac cycle (ranging from 1.70 – 2.40 μm) (Trayanova and Rice, 2011) and since it is known to modulate MEF (Zile and Trayanova, 2016), isometric simulations for each pacing rate were run three times, once at a constant SL of 1.70, 1.90 or 2.40 μm.
To determine if MEF modifies EAD formation for varying degrees of HCM-induced reduced repolarization reserve in HCM myocytes, we compared EAD formation in simulations with the bidirectionally vs unidirectionally coupled HCM myocyte models using the protocol above. Since there is a wide spectrum of HCM-induced myofilament remodeling (including no myofilament remodeling at all) (Gersh et al., 2011; Gray et al., 2011; Maron and Semsarian, 2010; Maron et al., 2011), we first compared simulations with and without feedback that incorporated only HCM-induced ionic remodeling.
Then, to elucidate how the severity of HCM-induced myofilament remodeling alters the effects of MEF on EAD formation, we compared bidirectionally coupled simulations with only HCM-induced ionic remodeling (described above) to those with HCM-induced ionic remodeling and different amounts of HCM-induced myofilament remodeling (described below) using the aforementioned protocol. To incorporate different amounts of HCM-induced myofilament remodeling in the simulations, one of the 3 myofilament parameters (perm50, koffH, kon) was assigned a value 5–15% below or above its baseline value (as described in the previous section) for a given simulation.
2.4. Analysis of EADs
An EAD was defined as a slowing or reversal of normal repolarization during the plateau or rapid depolarization phases (phases 2 or 3) of an action potential (AP) (Vandersickel et al., 2014; Weiss et al., 2010). We narrowed this definition by requiring that the spontaneous depolarization(s) in phase 2 or 3 be larger than 1 mV. Therefore, an EAD was said to be elicited in a simulation if at least 1 of the final 50 APs had at least one spontaneous depolarization(s) in phase 2 or 3 that was larger than 1 mV.
The frequency of EAD occurrence was calculated as the number of beats that elicited an EAD. Specifically,
| (2) |
where the total number of beats were defined as the final 50 beats of each simulation.
3. Results
3.1. MEF Alters EAD Emergence and Frequency of Occurrence
Incorporating HCM-induced repolarization reserve reduction in the bidirectionally and unidirectionally coupled cellular electrophysiology-force models without HCM-induced myofilament remodeling elicited EADs similar to those shown previously (Vandersickel et al., 2014; Zimik et al., 2015), which resulted in an elongated active force transient, as expected (Coppini et al., 2013). However, there needed to be a sufficient reduction in repolarization reserve for EADs to emerge (Vandersickel et al., 2014; Zimik et al., 2015). An example of unaffected APs (without EADs) when repolarization reserve reduction is insufficient (GKr 50% of baseline, GCaL 3.8 fold above baseline) in the bidirectionally (light blue) and unidirectionally (red) coupled models is shown in Figure 2A, where transmembrane voltage (Vm) is plotted as a function of time for pacing at a CL of 2000 ms (0.5 Hz) and a SL of 1.90 μm. The corresponding active force traces, as a function of time (Figure 2C), were also unchanged. There were no differences in the APs or normalized active force traces between the bidirectionally and unidirectionally coupled models, despite a slight difference (32 μM vs 40 μM) in peak [Ca]Troponin (Figure 2B), the term that represents the feedback from the myofilament model to the ionic model.
Figure 2.
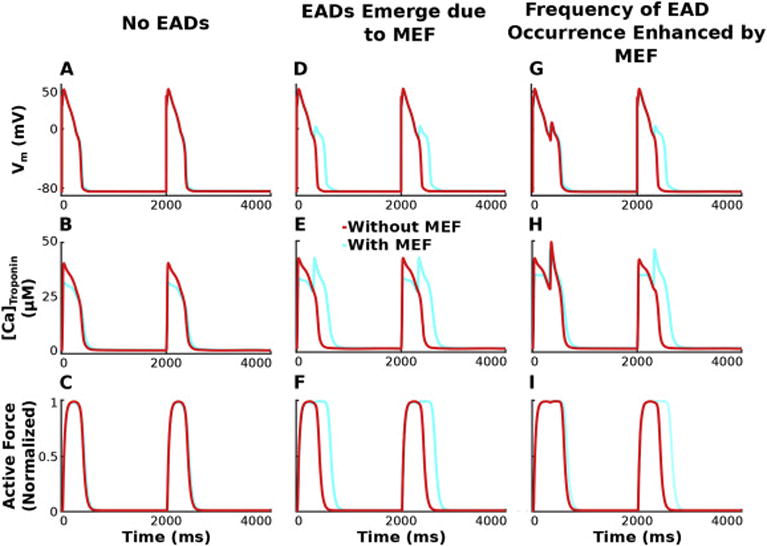
Vm (row 1), [Ca]Troponin (row 2), and active force (row 3) are plotted over time for simulations incorporating HCM-induced ionic remodeling in the absence of myofilament remodeling for a pacing CL of 2000 ms, SL=1.90 μm, and GKr 50% of baseline. Columns 1–3 have progressively enhanced GCaL (3.8, 4.0, 4.2 fold above baseline), illustrating that the degree of repolarization reserve reduction affects the emergence of EADs and the frequency of their occurrence differently for simulations with the bidirectionally coupled model (with MEF; light blue) vs the unidirectionally coupled model (without feedback; red).
An example of APs with EADs (sufficient reduction in repolarization reserve ensured by GCaL 4.0 fold vs 3.8 fold above baseline, other parameters identical to those in Fig. 2A–C) in the bidirectionally coupled model (light blue) is shown in Figures 2D and 3A, and the corresponding prolonged active force trace in Figures 2F and 3B. If MEF is removed, the resulting unidirectionally coupled model does not exhibit EADs or prolongation of the active force trace, despite the significant reduction in repolarization reserve. In the bidirectionally coupled model, Troponin C binds free intracellular Ca2+ and decreases the pool of free Ca2+ ions available in the cytoplasm ([Ca]i) as shown in Figure 3C. During the early AP, the feedback via the [Ca]Troponin term (Equation 1) shown in Figures 2E and 3D in the bidirectionally coupled model, resulted in smaller peak [Ca]i than that in the unidirectionally coupled model. This smaller [Ca]i in the bidirectionally coupled model causes the sodium-calcium exchanger current (INaCa) in Figure 3E to be larger while functioning in its reverse mode (since it is a function of the Ca2+ gradient across the membrane), allowing more Ca2+ ions into the cell without dramatically increasing [Ca]i (since many of these Ca2+ ions then bind to Troponin C). As the AP progresses, the remaining Ca2+ ions in the cytoplasm are available to be extruded from the cell via INaCa during its forward mode or via the sarcolemmal calcium pump (IpCa) shown in Figure 3F, or pumped into the sarcoplasmic reticulum (SR; Figure 3G) via the sarcoplasmic reticulum Ca2+-ATPase (Iup; Figure 3H). However, since INaCa is a function of the Ca2+ gradient across the membrane and since [Ca]i is smaller due to buffering by Troponin C in the bidirectionally coupled model, less Ca2+ ions are extruded from the cell via INaCa thereby leaving more to be pumped into the SR via Iup. Larger [Ca]SR in simulations with the bidirectionally coupled model increases the opening rate and decreases the closing rate of the ryanodine receptor (RyR), resulting in larger Ca2+ release from the SR (Irel; Figure 3I) and increased Ca2+ in the subspace ([Ca]ss; Figure 3J). If [Ca]ss is sufficiently large (as it is for the bidirectionally but not unidirectionally coupled model), spontaneous calcium-induced calcium release (CICR) occurs. This further increases [Ca]ss, which then via increased diffusion (Ixfer; Figure3K), dramatically increases [Ca]i during early repolarization resulting in increased INaCa in forward mode. If the inward current from INaCa in forward mode delays repolarization long enough (as it does with the bidirectionally coupled model), ICaL, as shown in Figure 3L, is able to recover from inactivation and increase enough to overcome the outward current through the rapid and slow delayed rectifier channels (IKr and IKs; Figures 3M–N), causing an EAD to form. This exemplifies how MEF can elicit EADs in bidirectionally coupled electrophysiology-force cell models under conditions of significant repolarization reserve, when EADs are not elicited in comparable simulations with the unidirectionally coupled model. Therefore, MEF diminishes the degree of repolarization reserve reduction necessary for EADs to emerge in simulations with the bidirectionally coupled model compared to those with the unidirectionally coupled model.
Figure 3.
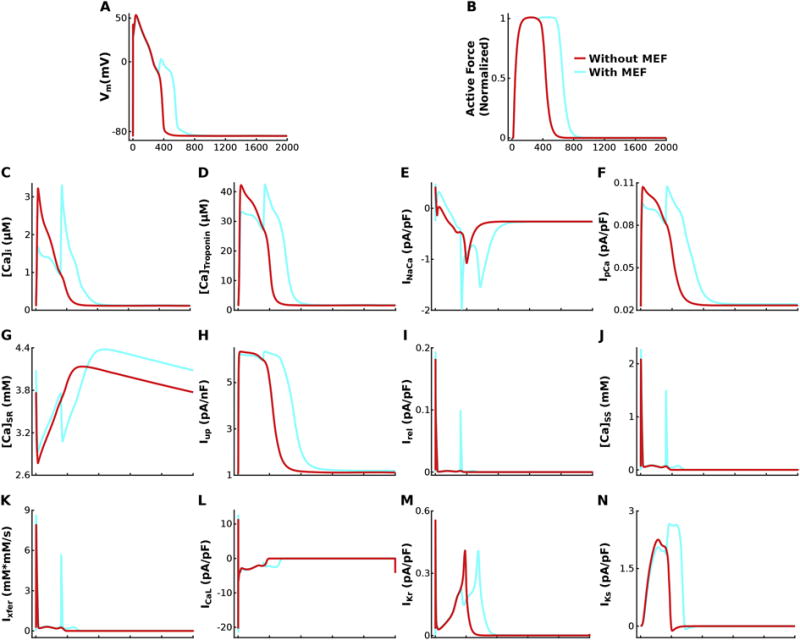
Vm (A), active force (B), [Ca]i, (C), [Ca]Troponin (D), INaCa (E), IpCa (F), [Ca]SR (G), Iup (H), Irel (I), [Ca]SS (J), Ixfer (K), ICaL (L), IKr (M), and IKs (N) are plotted over time for simulations incorporating HCM-induced ionic remodeling in the absence of myofilament remodeling for a pacing CL of 2000 ms, SL=1.90 μm, GKr 50% of baseline, and GCaL 4.0 fold above baseline, highlighting the mechanism underlying the effects of MEF on EAD emergence and frequency by comparing the bidirectionally coupled model (with MEF; light blue) to the unidirectionally coupled model (without MEF; red).
When repolarization reserve is reduced further (GCaL increased to 4.2 fold above baseline), both the bidirectionally and unidirectionally coupled models exhibit at least one AP with an EAD (Figure 2G) and one corresponding force trace that is of increased duration (Figure 2I). However, due to the difference in buffering of Ca2+ as a result of MEF (Figure 2H), the frequency of EAD occurrence is increased in the bidirectionally coupled model relative to the unidirectionally coupled one (2 APs with an EAD vs 1). After an EAD occurs in beat one, there is an excess of Ca2+ ions in the cytoplasm, due to Ca2+ release from the SR and from reactivated ICaL, for simulations with both the bidirectionally and unidirectionally coupled models. In the bidirectionally coupled model, some of those Ca2+ ions bind to Troponin C, reducing [Ca]i during the 2nd repolarization relative to that in the unidirectionally coupled model. The remaining free Ca2+ ions in the cytoplasm are then pumped into the SR by Iup, or extruded from the cell via INaCa, and IpCa. However, the decreased [Ca]i reduces forward mode INaCa and enhances the amount of Ca2+ stored in the SR for bidirectionally vs unidirectionally coupled models. If sufficiently large (as it is in this example for the bidirectionally but not unidirectionally coupled simulations), this [Ca]SR load is enough to cause increased Irel and elevated [Ca]ss in the second beat triggering spontaneous CICR and an EAD, thus increasing the frequency of EAD occurrence in bidirectionally vs unidirectionally coupled simulations due to MEF.
3.2. Faster Pacing Enhances Effects of MEF on EAD Emergence and Frequency
The stability diagrams of GKr and GCaL in Figure 4A–C illustrate that EADs arise when HCM-induced repolarization reserve is sufficiently reduced (smaller GKr and larger GCaL) in the absence of HCM-induced myofilament remodeling, most prominently at faster pacing rates; the light blue line divides simulations with the bidirectionally coupled model that elicited no EADs (on the left) from those that had at least one EAD (on the right) for CLs of 4000 ms (Figure 4A), 2000 ms (Figure 4B) and 1000 ms (Figure 4C), with SL=1.90 μm. The red lines denote the same division for simulation results with the unidirectionally coupled model.
Figure 4.
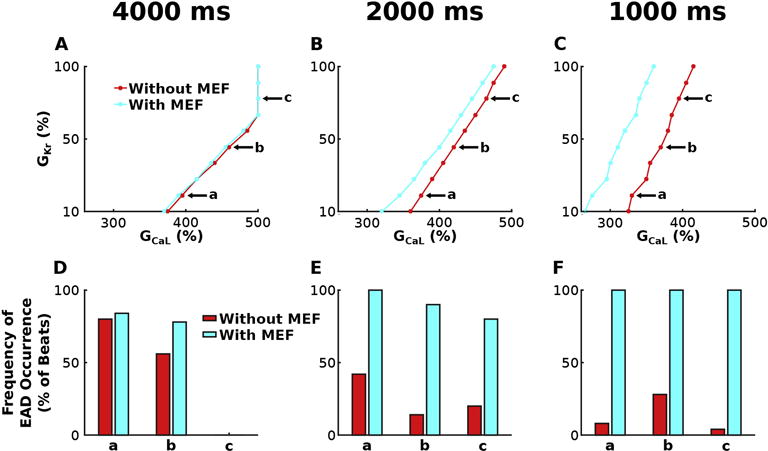
Stability diagrams of GKr and GCaL for simulations with the bidirectionally (light blue) and unidirectionally (red) coupled model incorporating HCM-induced ionic remodeling in the absence of myofilament remodeling for SL=1.90 μm at pacing CL=4000 ms (A) 2000 ms (B) and 1000 ms (C). The light blue (and red) line divides results of simulations with the bidirectionally coupled (unidirectionally coupled) model that elicited no EADs (on the left) from those that had at least one EAD (on the right). Simulations represented by the points on the lines all contained at least one EAD, except for 3 simulations at CL=4000 ms, with GCaL=5*baseline and GKr = 80, 90, 100% of baseline, respectively. Frequency of EAD occurrence is plotted in (D)–(F) for simulations at three representative points labeled a, b, and c in the corresponding stability diagrams in (A)–(C). Simulations at a, b, and c have GKr=20, 50 and 80% of baseline and the following GCaL values: 3.95, 4.60 and 5.00 times larger than baseline at CL=4000 ms; 3.75, 4.20 and 4.65 times larger at CL=2000 ms; and 3.30, 3.70, 3.95 times larger at CL=1000 ms.
Faster pacing enhanced the effects of MEF on the emergence of EADs, which is evidenced by the progressive separation of the red and light blue lines in Figure 4A–C. For pacing at CL=4000 ms, the degree of repolarization reserve reduction necessary to elicit EADs was similar for the bidirectionally and unidirectionally coupled models as shown in Figure 4A (minimal separation between lines); the minimum amount of repolarization reserve reduction necessary to elicit EADs was decreased on average by 1.0% for simulations with vs without MEF. At CL=2000 ms and CL=1000 ms, this effect was amplified; the minimum amount of repolarization reserve reduction necessary to elicit EADs was decreased on average by 4.6% at CL=2000 ms and 15.1% at CL=1000 ms for simulations with vs without MEF. This enhanced effect of MEF on the emergence of EADs with faster pacing was due to the progressively elevated [Ca]i during the early AP, which occurred due to a buildup of diastolic [Ca]i as a result of the progressively shorter time between beats needed for the Iup, INaCa, and IpCa to restore [Ca]i to normal. In the bidirectionally coupled model, this elevation in [Ca]i was lower than it was in the unidirectionally coupled model because some of the excess Ca2+ ions were able to bind to Troponin C instead of remaining unbound in the cytosol. Since Troponin C did not become saturated with Ca2+ even at the fastest pacing rates, the difference between [Ca]i in simulations with the unidirectionally vs bidirectionally coupled models was progressively enhanced as pacing rates were increased (CLs decreased). This difference in [Ca]i, as described in Results section 3.1 (via altered INaCa behavior, greater [Ca]SR, enhanced Irel, enlarged [Ca]ss, and CICR), reduced the degree of repolarization reserve reduction necessary for EADs to emerge in simulations with the bidirectionally coupled model. Since the difference in [Ca]i between the unidirectionally and bidirectionally coupled models progressively increased with increased pacing rate, the difference in the degree of repolarization reserve reduction necessary for EADs to emerge also became progressively larger.
Faster pacing also heightened the effects of MEF on the frequency of EAD occurrence. In Figure 4D–F, the frequency of EAD occurrence (defined in Methods 2.4) was plotted at three representative points labeled a, b, and c in the corresponding stability diagrams in Figure 4A–C. At these points, MEF increased the frequency of EAD occurrence in bidirectionally vs unidirectionally coupled simulations on average by 8.7% for CL=4000 ms (Figure 4D), 64.7% for CL=2000 ms, and 86.7% at CL=1000 ms. This progressive increase in EAD frequency with faster pacing, is due to progressively elevated [Ca]i during the early AP resulting in increasingly elevated [Ca]SR in the bidirectionally vs unidirectionally coupled models, as described in the preceding paragraph. Via the mechanism described in Results section 3.1, this progressive elevation in [Ca]SR in the subsequent beat progressively increased the probability of spontaneous CICR and thus the frequency of EAD occurrence.
3.3. Longer Sarcomere Length Diminishes MEF Effects on EAD Emergence and Frequency
Stability diagrams of GKr and GCaL, similar to those in Figure 4, illustrate how SL affects the degree of HCM-induced repolarization reserve reduction necessary for EAD emergence in simulations with the bidirectionally coupled electrophysiology-force myocyte model in the absence of HCM-induced myofilament remodeling for pacing at CL=2000 ms (Figure 5A) and CL=1000 ms (Figure 5B) for SL=1.70 μm (dark blue), 1.90 μm (light blue), and 2.40 μm (green). Since pacing at CL=4000 ms elicited little difference in the amount of repolarization reserve necessary for EAD formation or the frequency of EADs between the bidirectional and unidirectional models, we did not evaluate the effects of SL for pacing at CL=4000 ms. Pacing at CL=2000 ms, contracted myocytes (SL=1.70 μm) compared to those at resting length (SL=1.90 μm) required on average 2.4% less reduction in repolarization reserve for EAD emergence (dark blue line left shifted relative to light blue line). Stretched myocytes (SL=2.40 μm) required on average 2.1% more repolarization reserve reduction for EAD emergence compared to those at resting length (green line right shifted relative to light blue line). These examples show that shorter SLs enhance EAD formation. By definition, increasingly shorter SLs have a progressively decreased fraction of single-overlap thin filament opposing the thick filament. Since this portion of the thin filament has a higher binding affinity to calcium, a progressively smaller fraction of it causes Ca2+ to dissociate increasingly faster from Troponin C during early repolarization in simulations with shorter vs longer SLs. This reduction of Ca2+ buffering by Troponin C enhances [Ca]i during early repolarization resulting in enhanced forward mode INaCa. Since forward mode INaCa produces an inward current, increasingly larger INaCa for shorter SLs resulted in longer delays in AP repolarization. These increased delays provided ICaL with progressively longer times to recover from voltage-gated inactivation and to self-amplify sufficiently to become greater than the sum of outward currents (including IKr). Since ICaL is a function of GCaL, cases with diminished GCaL will require more time for self-amplification. Similarly enhanced GKr will result in larger outward currents requiring that ICaL be larger in order to overcome them, and thus requiring a longer period of delayed repolarization to allow greater self-amplification of ICaL. Since shorter SLs at CL=2000 ms result in increased delays in AP repolarization, EADs are elicited in simulations with increasingly large repolarization reserve (progressively larger GKr and smaller GCaL). However, SL does not affect the degree of repolarization reserve reduction necessary for EAD emergence at CL=1000 ms (Figure 5B), since ICaL had sufficient time to self-amplify in all cases due to the large [Ca]i at CL=1000ms.
Figure 5.
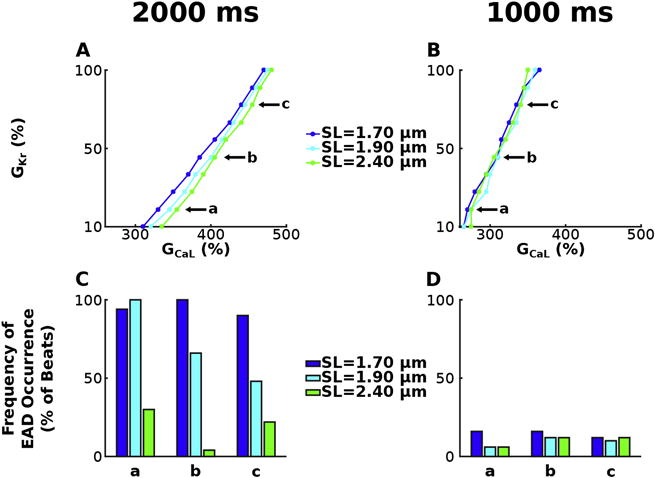
Stability diagrams of GKr and GCaL, similar to those in Figure 4, for simulations with the bidirectionally coupled electrophysiology-force myocyte model without myofilament remodeling for pacing at CL=2000 ms (A) and CL=1000 ms (B) for SL=1.70 μm (dark blue), 1.90 μm (light blue), and 2.40 μm (green). Frequency of EAD occurrence is plotted in (C) – (D) for simulations at three representative points labeled a, b, and c in the corresponding stability diagrams in (A) – (B). Simulations at a, b, and c have GKr=20, 50 and 80% of baseline and the following GCaL values: 3.55, 4.05 and 4.55 times larger than baseline at CL=2000 ms, and 2.75, 3.10 and 3.40 times larger at CL=1000 ms.
Longer SLs also diminished the effect of MEF on the frequency of EAD occurrence. In Figure 5C–D, the frequency of EAD occurrence was plotted for both models at three representative points labeled a, b, and c in the corresponding stability diagram in Figure 5A–B. The frequency of EAD occurrence at these points illustrates longer SLs result in fewer EADs. At SL=2.40 μm there are 52.7% less EADs than at SL=1.90 μm and 76.0% less than at SL=1.70 μm for CL=2000 ms (Figure 5C). For CL=1000 ms (Figure 5D), SL=2.40 μm had 0.7% less EADs than SL=1.90 μm and 4.7% less than SL=1.70 μm. Similarly, SL=1.90 μm resulted in 23.3% less EADs than SL=1.70 μm at CL=2000 ms, and 5.3% less than SL=1.70 μm at CL=1000 ms. This decrease in the frequency of EAD occurrence for longer SLs is due to the enhanced binding affinity of Troponin C for Ca2+. Via the mechanisms described in the preceding paragraph, this results in shorter delays in AP repolarization at longer SLs. When an equal degree of repolarization reserve reduction is incorporated in simulations, the cases with shorter delays in AP repolarization (due to diminished INaCa in forward mode) are less likely to have ICaL recover from inactivation and are thus less likely to exhibit EADs. Therefore, longer SLs are associated with a diminished frequency of EAD occurrence.
3.4. Increased Severity of HCM-Induced Myofilament Remodeling Alters Effects of MEF on EAD Emergence and Frequency
Stability diagrams of GKr and GCaL, similar to those in Figures 3 and 4, illustrate how the severity of HCM-induced myofilament remodeling affects the degree of HCM-induced repolarization reserve reduction necessary for EAD emergence in simulations with the bidirectionally coupled electrophysiology-force myocyte model. Figure 6 presents results regarding HCM-induced myofilament remodeling of parameters perm50 (Figure 6A), koffH (Figure 6B), and kon (Figure 6C) for pacing at CL=2000 ms and SL=1.90 μm. Simulations for the bidirectionally coupled model with both HCM-induced ionic and myofilament remodeling with CL=4000 ms and 1000 ms, and for CL=2000 ms with SL=1.70 and 2.40 μm are not shown since the effects of myofilament remodeling severity on MEF effects on EAD formation are similar to those shown in Figure 6. A 15% increase in perm50, a 15% increase in koffH, or a 15% decrease in kon, decreases the degree of repolarization reserve reduction required for EAD emergence (left shifted lines in Figures 5A–C respectively) on average by 3.0%, 1.0%, and 1.4% respectively compared to no remodeling. The reverse is true for a 15% decrease in perm50, a 15% decrease in koffH, or a 15% increase in kon; the degree of repolarization reserve reduction required for EAD emergence is increased on average by 1.2%, 1.2%, and 1.4% respectively compared to no remodeling. By definition, increasing perm50 decreases nearest-neighbor cooperativity, resulting in decreased binding affinity of Troponin C on nearby RUs to Ca2+, thus decreasing RU-based thin filament activation. By definition, increasing koffH (and decreasing kon) increases the unbinding rate (decreases the binding rate) of Ca to Troponin C, also resulting in decreased binding affinity of Troponin C to Ca2+, thus decreasing Ca-based thin filament activation. These examples show that diminishing TroponinC-Ca2+ binding affinity via HCM-induced remodeling increases the likelihood for EAD formation and results in EADs in simulations under conditions of greater repolarization reserve, via the same mechanism described in Results section 3.3. Therefore, diminishing TroponinC-Ca2+ binding affinity via HCM-induced remodeling enhances the effects of MEF on EAD emergence.
Figure 6.
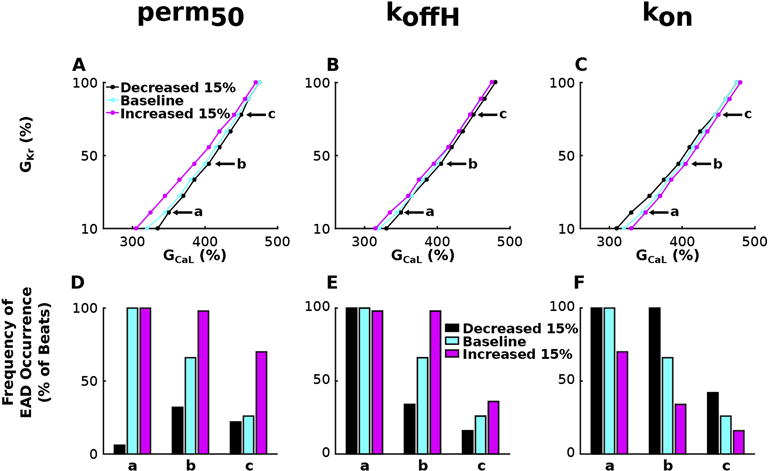
Stability diagrams of GKr and GCaL, similar to those in Figures 3 and 4, for simulations with the bidirectionally coupled electrophysiology-force myocyte model with HCM myofilament remodeling in parameters perm50 (A), koffH (B), and kon (C) for pacing at CL=2000 ms and SL=1.90 μm. Frequency of EAD occurrence is plotted in (D) – (F) for simulations at three representative points labeled a, b, and c in the corresponding stability diagrams in (A)–(C). Simulations at a, b, and c have GKr=20, 50 and 80% of baseline and GCaL values 3.50, 4.05, and 4.50 times larger than baseline.
HCM-induced myofilament remodeling also affected the frequency of EAD occurrence. In Figure 6 D–F, the frequency of EAD occurrence was plotted for simulations with the bidirectionally coupled electrophysiology-force myocyte model at three representative points labeled a, b, and c in the corresponding stability diagram in Figure 6A–C. The graphs illustrate that HCM-induced myofilament remodeling that decrease the binding affinity of Troponin C to Ca2+ (increased perm50, increased koffH, decreased kon) results in a larger number of EADs than in cases without remodeling. Myofilament remodeling that increases TroponinC-Ca2+ binding affinity results in fewer EADs. Simulations with myofilament remodeling that decrease binding affinity (15% increase in perm50, 15% increase in koffH, and a 15% decrease in kon) have an average of 25.3%, 13.3%, and 16.7% more EADs than respective simulations without myofilament remodeling at CL=2000 ms. Conversely, simulations with myofilament remodeling that increase binding affinity (15% decrease in perm50, 15% decrease in koffH, and a 15% increase in kon) have an average of 44.0%, 14.0% and 24.0% less EADs than respective simulations without myofilament remodeling at the same pacing rate.
4. Discussion
The present study examines whether myofilament protein dynamics mechanisms in HCM affect the incidence of EAD formation, which have been implicated as the primary mechanism underlying many arrhythmias associated with HCM. The goal of our study was to utilize the capability of mechanistic computer simulations to investigate whether myofilament protein dynamics mechanisms modulate EAD formation for varying degrees of reduced repolarization reserve in HCM, and to uncover how these mechanisms are affected by pacing rate, sarcomere length, and the severity of HCM-induced myofilament remodeling. Using bidirectionally and unidirectionally coupled human electrophysiology-force ventricular myocyte models, we showed that MEF on calcium dynamics in the bidirectionally coupled model, via Troponin C buffering of cytoplasmic Ca2+, was the myofilament protein dynamics mechanism underlying EADs. We showed that incorporating MEF diminished the degree of repolarization reserve reduction necessary for EADs to emerge and increased the frequency of EAD occurrence, especially at faster pacing rates. We also found that longer sarcomere lengths and enhanced thin filament activation diminished the effects of MEF on EADs. Together these findings demonstrate that myofilament protein dynamics mechanisms plays an important role in EAD formation and suggest that targeting MEF may be an alternative treatment approach for prevention of arrhythmia and thus SCD in HCM patients.
4.1. Modulation of EAD Incidence Occurs via MEF
SCD and arrhythmias have been shown to occur in patients with HCM, an inherited sarcomeric mutation-based disease (Coppini et al., 2013; Coppini et al., 2014; Maron et al., 2000). In addition, HCM patients with more than one sarcomeric mutation have a higher risk of SCD and a worse phenotype, independent of other risk factors (Van Driest et al., 2004). An important mechanism underlying many lethal arrhythmias found in HCM patients is EADs (Weiss et al., 2010; Yan et al., 2001), which also occur in patients with HCM (Coppini et al., 2013). In this HCM EAD study, the sarcomeric proteins cardiac myosin binding protein C and myosin heavy chain were mutated and evidence of altered [Ca]i transient kinetics was found. Furthermore, additional sarcomere mutations such as Troponin I (Messer et al., 2007), T (Miller et al., 2001), and C (Olivotto et al., 2015) and tropomyosin (Olivotto et al., 2015) have been found in HCM, all of which are sarcomeric proteins integral to Ca2+-Troponin C binding (Tardiff et al., 2015). Taken together, the results from these previous studies suggest that MEF, via altered Troponin C-Ca2+ buffering, is a likely mechanism by which sarcomeric mutations can modulate the incidence of EADs. Our results presented here provide proof that sarcomeric mutations, via altered Troponin C buffering of cytosolic Ca2+ (resulting from MEF), can indeed alter the degree of repolarization reserve reduction necessary for EADs to emerge and the frequency of EAD occurrence.
4.2. Effects of Altered Ca2+ Sensitivity on EADs and Arrhythmia in HCM
Ca2+ sensitivity is defined as the amount of Ca2+ necessary to produce half maximal force. When Ca2+ sensitivity is increased, less Ca2+ is required to produce half maximal force. Several studies have shown that increased Ca2+ sensitivity is associated with increased arrhythmogenicity (Baudenbacher et al., 2008; Huke and Knollmann, 2010). One animal study showed that the myosin inhibitor blebbistatin (which decreases Ca2+ sensitivity) reduced arrhythmia propensity in mice who had been hypersensitized to Ca2+ due to troponin mutations or Ca2+ sensitizing agents (Baudenbacher et al., 2008). Another showed that transgenic mice with different troponin T and I mutations had an increased incidence of premature ventricular complexes when injected with isoproterenol and an increased occurrence of ventricular tachycardia (Baudenbacher et al., 2008). However, none of these studies were conducted on human tissue or models and none incorporated the ionic remodeling changes that have been shown to occur in human HCM (Coppini et al., 2013). Neither did they investigate the myofilament protein dynamics mechanisms linking increased Ca2+ sensitivity to EADs or arrhythmia.
Human studies of HCM have shown that Ca2+ sensitivity is increased and that ventricular arrhythmias and SCD occur in HCM patients (Tardiff et al., 2015). However, ventricular arrhythmias and SCD also occur in dilated cardiomyopathy (DCM), which is characterized by a decrease in Ca2+ sensitivity (Sen-Chowdhry and McKenna, 2012). Therefore, to understand how altered Ca2+ sensitivity may potentially promote arrhythmogenicity in these sarcomeric mutation-based cardiac diseases, studies investigating the effects of Ca2+ sensitivity on the mechanisms underlying arrhythmia are needed.
This study investigated the effects of increased Ca2+ sensitivity on EADs in a bidirectionally coupled human HCM myocyte model. Our results show that sarcomeric mutations, which disrupt the regulation of the thin filament (decreased perm50, decreased koffH, increased kon) in a way that has a downstream effect of increasing Ca2+ sensitivity, increase the degree of repolarization reserve reduction necessary for EADs to emerge and decreases the frequency of EAD occurrence. Combined with the observations from the previous studies, this suggests that the enhanced arrhythmogenicity seen in HCM is not due to altered Ca2+ sensitivity, but rather due to HCM-induced ionic remodeling as suggested by Coppini et al (Coppini et al., 2013) or from secondary effects of myofilament remodeling at the tissue level, such as hypertrophy and fibrosis (Chan et al., 2014; Nucifora et al., 2015).
4.3 Limitations
A limitation of this study is that we do not incorporate sympathetic and parasympathetic stimulation, which is present during the cardiac cycle and could influence calcium cycling. Additionally, the conclusions of this study are in the context of a single myocyte; full understanding of the effects of myofilament dynamics on triggered activity (the manifestation of EADs at the organ level) will require tissue and whole heart simulations.
5. Conclusions
MEF, via Troponin C buffering of cytoplasmic Ca2+, was the myofilament protein dynamics mechanism modulating EAD formation in a bidirectionally coupled human electrophysiology-force myocyte model of HCM. Our results demonstrate that accounting for MEF on calcium dynamics diminished the degree of repolarization reserve reduction necessary for EADs to emerge and increased the frequency of EAD occurrence, especially at faster pacing rates. Longer sarcomere lengths and enhanced thin filament activation diminished the effects of MEF on EADs. These findings demonstrate that myofilament protein dynamics mechanisms play an important role in EAD formation.
Acknowledgments
This research was supported by National Institute of Health grants R01 HL12802 and PD1 HL123271 to Dr. Trayanova and by the David C. Gakenheimer Fellowship to Melanie Zile.
Abbreviations
- [Ca]Calmodulin
total calcium buffered by calmodulin in the cytoplasm
- [Ca]i
free intracellular calcium concentration
- [Ca]Total
total calcium in the cytoplasm
- [Ca]Troponin
total calcium bound to Troponin C
- CL
cycle length
- EAD
early afterdepolarization
- GCaL
maximal conductance of the L-type Ca2+ current
- GKr
maximal conductance of the rapid delayed rectifier current
- HCM
hypertrophic cardiomyopathy
- MEF
mechanoelectric feedback
- RU
regulatory unit
- SL
sarcomere length
- XB
thick filament crossbridge
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–903. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116:1919–36. doi: 10.1161/CIRCRESAHA.116.304030. [DOI] [PubMed] [Google Scholar]
- Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–11. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015;573:188–97. doi: 10.1016/j.gene.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RH, Maron BJ, Olivotto I, Pencina MJ, Assenza GE, Haas T, Lesser JR, Gruner C, Crean AM, Rakowski H, Udelson JE, Rowin E, Lombardi M, Cecchi F, Tomberli B, Spirito P, Formisano F, Biagini E, Rapezzi C, De Cecco CN, Autore C, Cook EF, Hong SN, Gibson CM, Manning WJ, Appelbaum E, Maron MS. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130:484–95. doi: 10.1161/CIRCULATIONAHA.113.007094. [DOI] [PubMed] [Google Scholar]
- Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L, Pankuweit S, Perrot A, Rapezzi C, Ristic A, Seggewiss H, van Langen I, Tavazzi L. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715–26. doi: 10.1093/eurheartj/ehq271. [DOI] [PubMed] [Google Scholar]
- Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–84. doi: 10.1161/CIRCULATIONAHA.112.134932. [DOI] [PubMed] [Google Scholar]
- Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, Mugelli A, Poggesi C, Tardiff J, Olivotto I. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol. 2014;64:2589–600. doi: 10.1016/j.jacc.2014.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) Eur Heart J. 2014;35:2733–79. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58:e212–60. doi: 10.1016/j.jacc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Gray B, Ingles J, Semsarian C. Natural history of genotype positive-phenotype negative patients with hypertrophic cardiomyopathy. Int J Cardiol. 2011;152:258–9. doi: 10.1016/j.ijcard.2011.07.095. [DOI] [PubMed] [Google Scholar]
- Huke S, Knollmann BC. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol. 2010;48:824–33. doi: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingles J, Sarina T, Yeates L, Hunt L, Macciocca I, McCormack L, Winship I, McGaughran J, Atherton J, Semsarian C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15:972–7. doi: 10.1038/gim.2013.44. [DOI] [PubMed] [Google Scholar]
- Ji YC, Gray RA, Fenton FH. Implementation of Contraction to Electrophysiological Ventricular Myocyte Models, and Their Quantitative Characterization via Post-Extrasystolic Potentiation. PLoS One. 2015;10:e0135699. doi: 10.1371/journal.pone.0135699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampourakis T, Yan Z, Gautel M, Sun YB, Irving M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc Natl Acad Sci U S A. 2014;111:18763–8. doi: 10.1073/pnas.1413922112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. Jama. 2002;287:1308–20. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010;121:445–56. doi: 10.1161/CIRCULATIONAHA.109.878579. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Kogan J, Proschan MA, Hecht GM, Roberts WC. Circadian variability in the occurrence of sudden cardiac death in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1994;23:1405–9. doi: 10.1016/0735-1097(94)90384-0. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–55. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–15. doi: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, 3rd, Spirito P, Ten Cate FJ, Wigle ED. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. 2014;64:83–99. doi: 10.1016/j.jacc.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Semsarian C. Emergence of gene mutation carriers and the expanding disease spectrum of hypertrophic cardiomyopathy. Eur Heart J. 2010;31:1551–3. doi: 10.1093/eurheartj/ehq111. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Semsarian C, Shen WK, Link MS, Epstein AE, Estes NA, 3rd, Almquist A, Giudici MC, Haas TS, Hodges JS, Spirito P. Circadian patterns in the occurrence of malignant ventricular tachyarrhythmias triggering defibrillator interventions in patients with hypertrophic cardiomyopathy. Heart Rhythm. 2009;6:599–602. doi: 10.1016/j.hrthm.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, Bardy GH, Favale S, Rea RF, Boriani G, Estes NA, 3rd, Spirito P. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342:365–73. doi: 10.1056/NEJM200002103420601. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, Estes NA, 3rd, Shannon KM, Ashley EA, Day SM, Pacileo G, Formisano F, Devoto E, Anastasakis A, Bos JM, Woo A, Autore C, Pass RH, Boriani G, Garberich RF, Almquist AK, Russell MW, Boni L, Berger S, Maron MS, Link MS. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61:1527–35. doi: 10.1016/j.jacc.2013.01.037. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, Epstein AE, Almquist AK, Daubert JP, Lawrenz T, Boriani G, Estes NA, 3rd, Favale S, Piccininno M, Winters SL, Santini M, Betocchi S, Arribas F, Sherrid MV, Buja G, Semsarian C, Bruzzi P. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. Jama. 2007;298:405–12. doi: 10.1001/jama.298.4.405. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Yeates L, Semsarian C. Clinical challenges of genotype positive (+)-phenotype negative (−) family members in hypertrophic cardiomyopathy. Am J Cardiol. 2011;107:604–8. doi: 10.1016/j.amjcard.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–59. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Miller T, Szczesna D, Housmans PR, Zhao J, de Freitas F, Gomes AV, Culbreath L, McCue J, Wang Y, Xu Y, Kerrick WG, Potter JD. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J Biol Chem. 2001;276:3743–55. doi: 10.1074/jbc.M006746200. [DOI] [PubMed] [Google Scholar]
- Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, Robbins J, Warshaw DM, Craig R. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci U S A. 2014;111:2170–5. doi: 10.1073/pnas.1316001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–57. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- Nucifora G, Muser D, Gianfagna P, Morocutti G, Proclemer A. Systolic and diastolic myocardial mechanics in hypertrophic cardiomyopathy and their link to the extent of hypertrophy, replacement fibrosis and interstitial fibrosis. Int J Cardiovasc Imaging. 2015;31:1603–10. doi: 10.1007/s10554-015-0720-0. [DOI] [PubMed] [Google Scholar]
- Olivotto I, d’Amati G, Basso C, Van Rossum A, Patten M, Emdin M, Pinto Y, Tomberli B, Camici PG, Michels M. Defining phenotypes and disease progression in sarcomeric cardiomyopathies: contemporary role of clinical investigations. Cardiovasc Res. 2015;105:409–23. doi: 10.1093/cvr/cvv024. [DOI] [PubMed] [Google Scholar]
- Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–94. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- Previs MJ, Prosser BL, Mun JY, Previs SB, Gulick J, Lee K, Robbins J, Craig R, Lederer WJ, Warshaw DM. Myosin-binding protein C corrects an intrinsic inhomogeneity in cardiac excitation-contraction coupling. Sci Adv. 2015;1 doi: 10.1126/sciadv.1400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn TA. The importance of non-uniformities in mechano-electric coupling for ventricular arrhythmias. J Interv Card Electrophysiol. 2014;39:25–35. doi: 10.1007/s10840-013-9852-0. [DOI] [PubMed] [Google Scholar]
- Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J. 2008;95:2368–90. doi: 10.1529/biophysj.107.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–73. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- Schinkel AF, Vriesendorp PA, Sijbrands EJ, Jordaens LJ, ten Cate FJ, Michels M. Outcome and complications after implantable cardioverter defibrillator therapy in hypertrophic cardiomyopathy: systematic review and meta-analysis. Circ Heart Fail. 2012;5:552–9. doi: 10.1161/CIRCHEARTFAILURE.112.969626. [DOI] [PubMed] [Google Scholar]
- Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743–50. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semsarian C, French J, Trent RJ, Richmond DR, Jeremy RW. The natural history of left ventricular wall thickening in hypertrophic cardiomyopathy. Aust N Z J Med. 1997;27:51–8. doi: 10.1111/j.1445-5994.1997.tb00914.x. [DOI] [PubMed] [Google Scholar]
- Sen-Chowdhry S, McKenna WJ. Sudden death from genetic and acquired cardiomyopathies. Circulation. 2012;125:1563–76. doi: 10.1161/CIRCULATIONAHA.111.025528. [DOI] [PubMed] [Google Scholar]
- Sequeira V, Najafi A, Wijnker PJ, Dos Remedios CG, Michels M, Kuster DW, van der Velden J. ADP-stimulated contraction: A predictor of thin-filament activation in cardiac disease. Proc Natl Acad Sci U S A. 2015;112:E7003–12. doi: 10.1073/pnas.1513843112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirito P, Autore C, Formisano F, Assenza GE, Biagini E, Haas TS, Bongioanni S, Semsarian C, Devoto E, Musumeci B, Lai F, Yeates L, Conte MR, Rapezzi C, Boni L, Maron BJ. Risk of sudden death and outcome in patients with hypertrophic cardiomyopathy with benign presentation and without risk factors. Am J Cardiol. 2014;113:1550–5. doi: 10.1016/j.amjcard.2014.01.435. [DOI] [PubMed] [Google Scholar]
- Spirito P, Autore C, Rapezzi C, Bernabo P, Badagliacca R, Maron MS, Bongioanni S, Coccolo F, Estes NA, Barilla CS, Biagini E, Quarta G, Conte MR, Bruzzi P, Maron BJ. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703–10. doi: 10.1161/CIRCULATIONAHA.108.798314. [DOI] [PubMed] [Google Scholar]
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–85. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J. 2012;33:1724–33. doi: 10.1093/eurheartj/ehs150. [DOI] [PubMed] [Google Scholar]
- Taggart P, Sutton PM. Cardiac mechano-electric feedback in man: clinical relevance. Prog Biophys Mol Biol. 1999;71:139–54. doi: 10.1016/s0079-6107(98)00039-x. [DOI] [PubMed] [Google Scholar]
- Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S, van der Velden J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:457–70. doi: 10.1093/cvr/cvv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Tusscher KH, Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol. 2006;291:H1088–100. doi: 10.1152/ajpheart.00109.2006. [DOI] [PubMed] [Google Scholar]
- Trayanova NA, Rice JJ. Cardiac electromechanical models: from cell to organ. Front Physiol. 2011;2:43. doi: 10.3389/fphys.2011.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–10. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- Vandersickel N, Kazbanov IV, Nuitermans A, Weise LD, Pandit R, Panfilov AV. A study of early afterdepolarizations in a model for human ventricular tissue. PLoS One. 2014;9:e84595. doi: 10.1371/journal.pone.0084595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriesendorp PA, Schinkel AF, Van Cleemput J, Willems R, Jordaens LJ, Theuns DA, van Slegtenhorst MA, de Ravel TJ, ten Cate FJ, Michels M. Implantable cardioverter-defibrillators in hypertrophic cardiomyopathy: patient outcomes, rate of appropriate and inappropriate interventions, and complications. Am Heart J. 2013;166:496–502. doi: 10.1016/j.ahj.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Warren CM, Karam CN, Wolska BM, Kobayashi T, de Tombe PP, Arteaga GM, Bos JM, Ackerman MJ, Solaro RJ. Green Tea Catechin Normalizes the Enhanced Ca2+ Sensitivity of Myofilaments Regulated by a Hypertrophic Cardiomyopathy-Associated Mutation in Human Cardiac Troponin I (K206I) Circ Cardiovasc Genet. 2015;8:765–73. doi: 10.1161/CIRCGENETICS.115.001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–9. doi: 10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witayavanitkul N, Ait Mou Y, Kuster DW, Khairallah RJ, Sarkey J, Govindan S, Chen X, Ge Y, Rajan S, Wieczorek DF, Irving T, Westfall MV, de Tombe PP, Sadayappan S. Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J Biol Chem. 2014;289:8818–27. doi: 10.1074/jbc.M113.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan GX, Wu Y, Liu T, Wang J, Marinchak RA, Kowey PR. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome : direct evidence from intracellular recordings in the intact left ventricular wall. Circulation. 2001;103:2851–6. doi: 10.1161/01.cir.103.23.2851. [DOI] [PubMed] [Google Scholar]
- Zile MA, Trayanova NA. Rate-dependent force, intracellular calcium, and action potential voltage alternans are modulated by sarcomere length and heart failure induced-remodeling of thin filament regulation in human heart failure: A myocyte modeling study. Prog Biophys Mol Biol. 2016;120:270–80. doi: 10.1016/j.pbiomolbio.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimik S, Vandersickel N, Nayak AR, Panfilov AV, Pandit R. A Comparative Study of Early Afterdepolarization-Mediated Fibrillation in Two Mathematical Models for Human Ventricular Cells. PLoS One. 2015;10:e0130632. doi: 10.1371/journal.pone.0130632. [DOI] [PMC free article] [PubMed] [Google Scholar]