

Abstract
Oxidative damage resulting from increased lipid peroxidation (LPO) is considered an important factor in the development of late onset/age-related Alzheimer’s disease (AD). Deuterium-reinforced polyunsaturated fatty acids (D-PUFAs) are more resistant to the reactive oxygen species-initiated chain reaction of LPO than regular hydrogenated (H-) PUFAs. We investigated the effect of D-PUFA treatment on LPO and cognitive performance in aldehyde dehydrogenase 2 (Aldh2) null mice, an established model of oxidative stress-related cognitive impairment that exhibits AD-like pathologies. Mice were fed a Western-type diet containing either D- or H-PUFAs for 18 weeks. D-PUFA treatment markedly decreased cortex and hippocampus F2-isoprostanes by approximately 55% and prostaglandin F2α by 20–25% as compared to H-PUFA treatment. D-PUFAs consistently improved performance in cognitive/memory tests, essentially resetting performance of the D-PUFA-fed Aldh2−/− mice to that of wildtype mice fed a typical laboratory diet. D-PUFAs therefore represent a promising new strategy to broadly reduce rates of LPO, and combat cognitive decline in AD.
Keywords: lipid peroxidation, cognition, Alzheimer’s disease, polyunsaturated fatty acid, deuterium-based therapeutics
Graphical Abstract
Lipid peroxidation (LPO) is an important factor in Alzheimer’s disease (AD) pathogenesis. Deuterated polyunsaturated fatty acids (D-PUFAs) are resistant to ROS-initiated LPO because of the isotope effect on the rate-limiting step. A diet containing D-PUFAs instead of H-PUFAs in Aldh2−/− AD mice dramatically improves cognition and memory by decreasing LPO, thus representing a promising new strategy to reduce LPO, and combat cognitive decline in AD.

Introduction
Oxidative damage due to increased oxidative stress is considered an important factor in the pathogenesis of Alzheimer’s disease (AD) and other neurodegenerative diseases including retinal diseases, Parkinson’s disease (PD) and Huntington’s disease. High brain levels of polyunsaturated fatty acids (PUFAs) and transition metals, coupled with high oxygen utilization and modest antioxidant defense, creates an environment especially vulnerable to oxidative damage [1]. Oxidative damage is present in the very early stages of AD [2] suggesting it is a primary driving force in AD pathogenesis [3–5].
Reactive oxygen species (ROS)-initiated non-enzymatic lipid peroxidation (LPO) of PUFAs such as linoleic (LA), linolenic (ALA), arachidonic (ARA), eicosapentaenoic (EPA) and docosahexaenoic (DHA) acids is the major contributor to oxidative damage, resulting in altered membrane fluidity and changes in membrane-bound enzymes and receptors [6]. LPO of PUFAs by reactive oxygen species (ROS) is initiated by abstraction of bis-allylic hydrogens, generating resonance-stabilized free radicals which subsequently react with molecular oxygen to form lipid peroxyls. These newly formed species abstract a hydrogen atom off an adjacent PUFA molecule thus propagating the chain reaction. Propagation is eventually terminated by a chain-terminating antioxidant such as vitamin E or by homologous recombination. LPO results in the formation of two broad categories of secondary byproducts derived through either endoperoxide or hydroperoxide intermediates. Examples of the former are F2-isoprostanes (F2-IsoPs) derived from ARA, EPA-derived F3 isoprostanes, and F4-neuroprostanes (F4-NeuroPs) derived from DHA. The chemical stability of these metabolites has led to their use as reliable biomarkers for monitoring LPO. Decomposition of hydroperoxide intermediates gives rise to a number of reactive aldehydes such as malondialdehyde, acrolein, 4-hydroxy-2-nonenal (HNE), 4-hydroxy-2-hexenal (HHE) and many others [7–9].
Despite epidemiological studies indicating that antioxidants reduce AD risk, prospective clinical trials of antioxidants for the treatment or prevention of AD have been disappointing [5,10–12]. With respect to the prevention of LPO-induced oxidative damage, chain terminating antioxidants such as vitamin E are limited in their effectiveness by the low ratio of vitamin E in membranes relative to PUFA content (vitamin E/PUFA = 1/2000) [13]. Another strategy to limit LPO is to decrease the rate-limiting initiation step of PUFA autoxidation: abstraction of bis-allylic hydrogen atoms. This has been accomplished by substitution of hydrogen with deuterium (Fig. 1) at bis-allylic sites of PUFAs [14–17]. This substitution (D-PUFA versus H-PUFA) exhibits an isotope effect [18] with respect to the ease of abstraction at the bis-allylic site, resulting in a decrease in the rate of PUFA peroxidation. This strategy has been used to demonstrate partial protection of the nigrostriatal dopaminergic pathway in the MPTP mouse model of Parkinson’s disease [19], and to improve cholesterol handling and reduce atherosclerosis development in a hyperlipidemic mouse model of atherosclerosis [20].
Fig 1.

11,11-D2-LA (a) an omega-6 D-linoleic acid used in this study, is enzymatically converted into 13,13- D2-Arachidonic acid (b). 11,11,14,14-D4-ALA (c), an omega-3 D-linolenic acid used in this study, is enzymatically converted into 13,13,16,16-D4-EPA (d) and 15,15,18,18-D4-DHA (e) [31].
We have developed an oxidative stress-based mouse model of age-related cognitive impairment with AD-like biochemical and structural pathologies, based on gene deletion of aldehyde dehydrogenase 2 (ALDH2) [21]. This enzyme is important for the detoxification of LPO-derived aldehydes and reactive carbonyls [13,22,23] and both free HNE and HNE protein adducts accumulate in the brains of AD patients [24–29], and are associated with AD pathogenesis and altered amyloid-β disposition. These mice exhibit increases in HNE protein adducts, and deficits in both working and spatial memory as assessed by the novel object recognition (NOR), Y-maze, and Morris Water Maze (MWM) tasks [21], and also exhibit anxiety-like behaviours [30].
In the current study, we evaluated whether a diet enriched in D-PUFAs decreases LPO, thus providing protection against the cognitive impairment present in our mouse model of oxidative stress. We used the deuterated versions of the essential fatty acids LA and ALA, since they are also converted in situ to other PUFAs such as ARA and DHA (Fig. 1), thus allowing deuterium incorporation into a broad range of membrane lipids. The Aldh2−/− mouse is an excellent model with which to assess the effects of a D-PUFA-enriched diet, since the basis of the model is increased lipid peroxidative damage because of the inability to catabolize toxic reactive carbonyl compounds that are derived from PUFAs.
Results
D-PUFAs efficiently incorporate into tissues
Aldh2−/− mice were treated with diets containing either deuterium-reinforced D-PUFAs or control H-PUFAs (see Table 1 for composition of the diets). At the end of the study, 18 weeks after Western-type diet feeding, efficient D-PUFA incorporation was confirmed by measuring deuterium content in brain sections of the mice. The difference between the D-PUFA (33342±3223‰) and the H-PUFA (−140.7±12.5‰; p<0.001) groups corresponds to an approximate 35% D-PUFA incorporation (i.e. D-PUFA fraction of total PUFA) [20, 31]. This level of D-PUFA substitution is biologically relevant as approximately 10–20% is sufficient to terminate LPO [16].
Table 1.
Composition of H- and D-PUFA-containing diets. AIN-93M rodent-based diets were modified to contain 12% fat with 1.2% D-linoleic:D-linolenic(1:1 weight ratio), or 1.2% H-linoleic:H-linolenic (1:1 weight ratio). gm%, grams of each component per 100 grams of diet.
| H-diet | D-diet | |||
|---|---|---|---|---|
| gm% | kcal% | gm% | kcal% | |
| D-Linoleic acid, ethyl ester | - | - | 0.6 | 1.2 |
| D-Linolenic acid, ethyl ester | - | - | 0.6 | 1.2 |
| H-Linoleic acid, ethyl ester | 0.6 | 1.2 | - | - |
| H-Linolenic acid, ethyl ester | 0.6 | 1.2 | - | - |
| Oleate, ethyl | 2.9 | 6.2 | 2.9 | 6.2 |
| Coconut oil, 101 (hydrogenated) | 7.5 | 16.0 | 7.5 | 16.0 |
| Casein | 13.5 | 12.8 | 13.5 | 12.8 |
| L-cystine | 0.0 | 0.2 | 0.0 | 0.2 |
| Corn starch | 43.2 | 40.9 | 43.2 | 40.9 |
| Maltodextrin 10 | 12.1 | 11.4 | 12.1 | 11.4 |
| Sucrose | 9.7 | 9.1 | 9.7 | 9.1 |
| Cellulose | 4.8 | 0.0 | 4.8 | 0.0 |
| t-Butylhydroquinone | 0.0 | 0.0 | 0.0 | 0.0 |
| Mineral Mix S10022M | 3.4 | 0.0 | 3.4 | 0.0 |
| Vitamin Mix V10037 | 1.0 | 0.9 | 1.0 | 0.9 |
| Choline bitartrate | 0.2 | 0.0 | 0.2 | 0.0 |
D-PUFAs reduce lipid peroxidation products
Since oxidative stress is considered an important factor in the development of AD, and since D-PUFAs have been shown to reduce LPO products in other models [14–17,19,20,32,33], we investigated whether D-PUFA treatment also reduces LPO in the Aldh2−/− mouse model of sporadic AD. D-PUFA treatment markedly decreased esterified F2-IsoPs by approximately 55% in both cortex and hippocampus, and prostaglandin F2α (PGF2α) levels by 20–25% (Fig. 2), indicating that D-PUFAs effectively reduce brain LPO products in Aldh2−/− mice.
Fig. 2.

Decrease in F2-IsoPs and PGF2α in cortex and hippocampus from Aldh2−/− mice fed the D-PUFA diet. 18 weeks after initiation of either the D-PUFA or H-PUFA diet, homogenates of cortex or hippocampus were analyzed for bound F2-IsoPs or PGF2α. Data are presented as means ± s.d. (n=6–8) and were analyzed by Student’s t-test for unpaired data. *, significant difference from H-PUFA as indicated (*p<0.05 ***p<0.001).
D-PUFAs prevent cognitive impairment and anxiety-like behaviour in Aldh2−/− mice
We used three widely used and accepted tests of spatial and working memory to assess the effects of the D-PUFA diet on memory deficits in Aldh2−/− mice. The Morris water maze (MWM) task assesses spatial reference memory, spontaneous alternations in the Y-maze is a test for spatial working memory, and the open field novel object recognition (NOR) task assesses working memory in the absence of a reference memory component. The version of the MWM task used in this study consisted of 3 days of cued platform training, in which mice swim to a visible platform, followed by 5 days of testing in which the platform was hidden. There were no differences in latency times between the two diets for the cued platform training (Fig. 3A, 3B, 3C), as we have found when comparing wildtype and Aldh2−/− mice [21]. For the hidden platform trials, after 2 weeks on the D-PUFA or H-PUFA diet, there was a significant decrease in escape latency times for D-PUFA diet mice compared to H-PUFA diet mice on the fourth and fifth days of testing (blocks 7 and 8 in Fig. 3A). Differences in escape latencies became more marked after 10 or 18 weeks on the two diets (Fig. 3B and 3C), and were significantly different on days 2, 3, 4, and 5 of testing (blocks 5–8). In the probe trial, mice fed the D-PUFA diet spent more time in the target quadrant (Fig 3D) and had a greater number of platform crosses (Fig. 3E) compared to H-PUFA diet mice after either 2, 10, or 18 weeks on the two diets. We also evaluated changes in task performance over time in animals on each of the diets. For mice on the H-PUFA diet, there were no differences in performance in any of the trial blocks over the 18 week study period. For animals on the D-PUFA diet, the only difference was a decrease in latency time in the first trial block (block 4) after 18 weeks on diet compared to that of mice after either 2 or 10 weeks on diet. In the probe trials, time in the target quadrant and number of platform crosses did not change over time for either of the groups. Thus memory deficits in mice fed the H-PUFA diet, and task performance in mice fed the D-PUFA diet, remained stable for the duration of the study. Similarly, task performance in wildtype and Aldh2−/− mice fed a standard diet (LabDiet® 5015 Mouse diet) remains stable in animals between 3 and 12 months of age (data not shown).
Fig. 3.

Superior performance in the Morris Water Maze task in Aldh2−/− mice fed the D-PUFA diet. 2 weeks (A) 10 weeks (B) or 18 weeks (C) after the initiation of the D-PUFA or H-PUFA diet, escape latency (time to reach the hidden platform) was determined in a 3 day cued trial block (4 trials per day) followed by a 5 day hidden trial block (6 trials per day). Day 9 was a probe trial, in which the time spent in the target quadrant (D) and number of platform crosses (E) were determined (total time of the trial was 60 s). Performance in the MWM task in a separate cohort of wildtype animals was compared to Aldh2−/− mice fed the D-PUFA diet for 2 weeks (F). In (F), no significant differences were found at any trial block. In (A, B, C, and F), data are expressed as the mean ± s.e.m. of the average scores in each trial block (n=16 for D-PUFA, n=15 for H-PUFA, and n=20 for wildtype). In D and E, data are expressed as the mean ± s.d. (n=16 for D-PUFA and n=15 for H-PUFA). In D, the dotted line represents the expected time that would be spent in any quadrant by chance alone. Data were analyzed by 2-way ANOVA and a Bonferroni post-hoc test in (A, B, C, and F) and by Student’s t-test for unpaired data in (D and E), *, significant difference from D-PUFA as indicated (*p<0.05, ** p<0.01, ***p<0.001).
In the open field Novel object recognition (NOR) task (Fig. 4) and spontaneous alternations in the Y-maze (Fig. 5), Aldh2−/− mice fed the H-PUFA diet showed a progressive decrease in performance in both memory tasks, compared to a progressive increase in performance in mice on the D-PUFA diet. Performance in the NOR task was significantly different between the two groups after 10 and 18 weeks on diet, and in the Y-maze task after 18 weeks on diet.
Fig. 4.

Superior performance in the in the Novel Object Recognition (NOR) task in Aldh2−/− mice fed the D-PUFA diet. Male and female mice were subjected to the NOR task 2, 10, and 18 weeks after initiation of either the D-PUFA or H-PUFA diet, and the frequency of visits to the objects and the time spent exploring each object was recorded. (A) Ratio of time spent with the novel object in relation to the familiar object. (B) Discrimination index (difference in time exploring the novel and familiar object, divided by total exploration time). Data are presented as the mean ± s.d. (n=16) and were analyzed by Student’s t-test for unpaired data. ***Significant difference from D-PUFA (p<0.001).
Fig. 5.
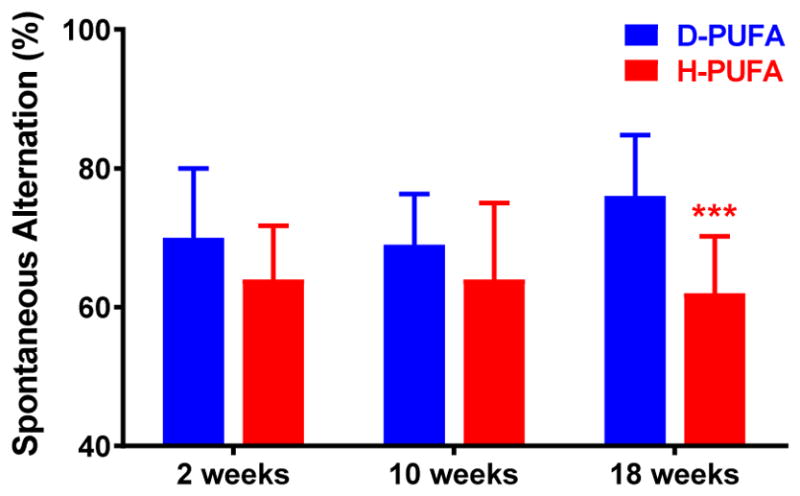
Superior performance in the Y-maze task by Aldh2−/− mice fed the D-PUFA diet. The spontaneous alternation rate in the Y-maze task was assessed in male and female mice 2, 10, and 18 weeks after initiation of either the D-PUFA or H-PUFA diet. Data are presented as means ± s.d. (n=16) and were analyzed by Student’s t-test for unpaired data. *** Significant difference from D-PUFA (p<0.001).
The open field test was used to assess spontaneous locomotor activity during free exploration in a dimly lit environment, and revealed no differences between groups in total time moving (D-PUFA, 161±29 s; H-PUFA, 163±17 s. p> 0.05, Student’s t-test for unpaired data) or total distance travelled (D-PUFA, 83±20 m; H-PUFA, 83±18 m. p> 0.05, Student’s t-test for unpaired data). Similarly, exploratory behaviour, as measured by time spent rearing, revealed no difference between groups (D-PUFA, 33±16 s; H-PUFA, 29±11 s. p> 0.05, Student’s t-test for unpaired data). This suggests that there is no difference in pattern of movement or exploratory behaviour in an open field, which is perhaps not surprising, since wildtype and Aldh2−/− mice do not exhibit differences in these parameters [30].
The Light/dark box test was used to assess aversion to bright illumination as a measure of anxiety-like behaviour. Although both groups showed a preference for the dark chamber, in mice fed the D-PUFA diet for 10 weeks there was a significant increase in both time spent in the light side and total number of crosses between the light and dark sides, compared to mice on the H-PUFA diet (Fig. 6). The differences observed here between D-PUFA- and H-PUFA-fed mice were similar to those between wildtype and Aldh2−/− mice of a comparable age [30].
Fig. 6.
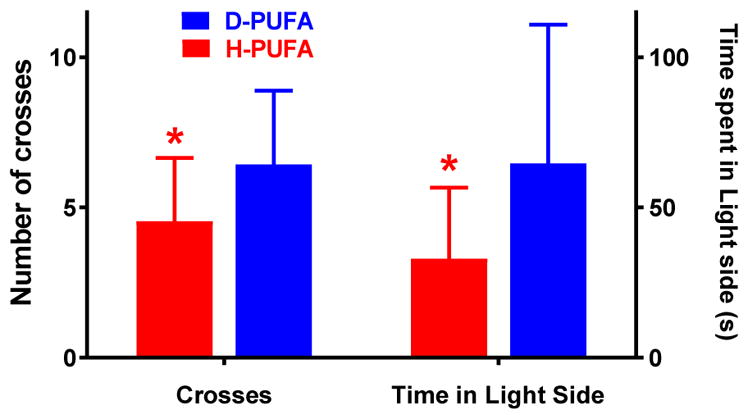
Increased time spent in, and number of crosses into, the illuminated chamber of the Light/Dark Box by Aldh2−/− mice fed the D-PUFA diet for 10 weeks. Mice were placed into the dark side of the light/dark box chamber and allowed to explore for a 5 min period. The number of crosses between the light and dark sides of the chamber, and the total time spent in the light side were recorded. Data are presented as means ± s.d. (n=16 for D-PUFA and n=13 for H-PUFA) and were analyzed by Student’s t-test for unpaired data. *, significant difference from D-PUFA (*p<0.05).
Discussion
Deuterium-reinforced D-PUFAs are resistant to non-enzymatic LPO and have been shown to reduce oxidative stress in several experimental models [14–17,19,20,32,33]. In the present study, we wished to evaluate whether alteration of lipid metabolism by a D-PUFA-enriched diet resulted in improvement of cognitive deficits in our model of oxidative stress-induced cognitive impairment that exhibits AD-like pathological changes. We found that oral administration of the ethyl esters of 11,11-D2-LA and 11,11,14,14-D4-ALA markedly reduced LPO products in Aldh2−/− mice. This was associated with improved performance in three different memory tasks that assess both working and spatial memory, and also with a reduction in anxiety-like behaviour.
Our protocol for the MWM, in which a 3-day cued trial is followed by a 5-day hidden platform trial, provides a thorough assessment of spatial reference memory. The lack of differences between D-PUFA and H-PUFA fed mice during the 3-day cued trial suggests that eyesight, swim speed, basic strategies, and motivation were the same in each treatment group. Also, we did not observe nonspecific behavioural changes such as floating or thygmotaxis in either group. Mice fed the D-PUFA diet performed significantly better in all measures of the hidden platform portion of MWM task after only 2 weeks on diet, with differences becoming more marked after 10 and 18 weeks on diet. In both the Y-maze NOR tasks, Aldh2−/− mice fed the H-PUFA diet showed progressive decreases in performance compared to progressive increases in performance in mice on the D-PUFA diet. In the Light/dark box test, mice fed the D-PUFA diet exhibited a reduction in the anxiety-like behaviour that is seen in this model (aversion to bright illumination). Thus reduction of non-enzymatic LPO by D-PUFAs has cognition-restoring properties in Aldh2−/− mice, and suggests that there are therapeutic opportunities afforded by oral dosing with D-PUFAs for the treatment of neurodegenerative diseases such as AD.
Although wildtype animals fed the D-PUFA or H-PUFA diets were not used in this study, when the performance of mice fed the D-PUFA diet for 2 weeks is compared to that of wildtype animals of a similar age, but fed LabDiet® 5015 Mouse diet, no significant differences in escape latency were found at any trial block (Fig. 3F). Both diets contained 12% fat, but had differing amounts and ratios of linoleic and linolenic acids. Thus the D-PUFA diet prevented the loss in spacial reference memory performance that would otherwise occur in the Aldh2−/− mice. Furthermore, the differences in performance in all three memory tasks between mice fed the D-PUFA and H-PUFA diets for 18 weeks, were very similar to the differences between wildtype and Aldh2−/− mice of a comparable age [21], indicating that D-PUFAs essentially reset the performance of Aldh2−/− mice to that of wildtype mice.
Isotope ratio mass spectrometry indicated incorporation of deuterium into brain lipids, to an extent similar to that found in previous studies [31]. This level of incorporation was sufficient to markedly reduce LPO products, as measured by the decrease in esterified F2-IsoPs, consistent with previous studies in yeast [16] and mice [20]. F2-IsoPs are derived from non-enzymatic oxidation of ARA. ARA can also undergo enzymatic oxidation to yield PGF2α as well as numerous other products catalyzed by cyclooxygenases and lipoxygenases. Comparison of F2-IsoPs and PGF2α levels in the D-PUFA fed animals shows that there was a substantially greater reduction in non-enzymatic LPO than enzymatic lipid oxidation. (Fig. 2). That D-PUFAs also had an effect on enzymatic oxidation can be explained by the fact that 11,11-D2-LA is efficiently converted to 13,13-D2-ARA [31]. The presence of deuterium at carbon 13 on ARA would be expected to influence both non-enzymatic and enzymatic oxidation since this is the site of action of the cyclooxygenase enzymes. Non-enzymatic oxidation is also influenced by the presence of other D-PUFAs in lipid membranes, slowing down the free radical chain reaction and resulting in a greatly decreased level of F2-IsoPs.
F2-IsoPs are recognized as reliable biomarkers of endogenous LPO by virtue of their ubiquitous nature, their chemical stability in biological fluids and tissues, and their amenability to quantitative GC/MS analysis [34], and were the measure used in the current study for assessment of non-enzymatic LPO, rather than the more semi-quantitative assessment afforded by immunoblot analysis of carbonyl protein adducts. In the context of AD progression, increased levels of F2-IsoPs and F4-NeuroPs were found in the frontal, parietal, and occipital lobes of subjects with amnesic mild cognitive impairment (MCI) relative to age-matched normal control subjects [35]. These levels were comparable to those found in subjects with late stage AD, suggesting that LPO of ARA and DHA is an early event that continues throughout the progression of AD. This is consistent with other studies of individuals with MCI, early stage AD, and late stage AD which examined the formation of protein adducts of LPO-derived reactive aldehydes such as HNE [27–29,36,37] and HHE [38]. Although much of the literature has focussed on the identification and potential role of HNE protein adducts in AD pathogenesis, protein adducts of HHE, an omega-3 oxidation product, have received much less attention [39]. Given that HHE and HNE exhibit similar toxicities in cultured neurons [40] and that DHA, the predominant omega-3 PUFA in many brain regions, is 30–100% more abundant than ARA, the predominant, omega-6 PUFA [41,42], raises the possibility that HHE protein adducts could contribute to reactive aldehyde-induced neuronal damage.
Previous experimental studies show that antioxidant strategies [43,44] protect neurons in AD models, but almost all clinical trials with antioxidants for the treatment or prevention of AD have been consistently negative [5,10–12, but see 45 and related Editorial]. This does not necessarily refute the importance of oxidative stress and LPO in the pathogenesis of AD, or the clinical potential of D-PUFA treatment, since studies claiming the ineffectiveness of antioxidant interventions suffer from variations in treatment regimens and trial duration, lack of therapeutic drug monitoring, lack of monitoring oxidative stress reduction caused by drug treatment, and choice and dosage of antioxidants [5]. For example, under some conditions the lipophilic chain-terminating antioxidant, Vitamin E, can also act as a pro-oxidant, thus serving to increase LPO [46]. Also unresolved is the issue of the relative importance of enzymatic versus non-enzymatic, as well as two-electron versus one-electron processes in LPO [13]. Based on data presented here and in other studies [19,20], we propose that specific inhibition of non-enzymatic LPO by D-PUFAs may prove beneficial in a number of disease states involving LPO-induced damage, and that this mechanism for LPO reduction is likely superior to general antioxidant inhibition or enzymatic inhibition of LPO. It is noteworthy that D-PUFAs are not antioxidants per se: they do not quench reactive oxygen species (ROS), nor do they affect cellular redox status or the antioxidant-ROS ratio, thus allowing normal ROS-mediated signalling pathways to remain intact.
The relevance and importance of ALDH2 to antioxidant defense is suggested by a number of studies in which inhibition or lack of ALDH2 increases the vulnerability to HNE- and reactive aldehyde-induced damage [47–49], whereas increased expression or activation of ALDH2 confers protective effects [48–53]. ALDH2 is expressed in numerous brain regions [54] and its expression/activity is increased in the temporal cortex and putamen in AD brains [55] and also in the putamen in PD brains [56], suggesting a protective response to AD- and PD-associated increases in LPO. In addition, some epidemiological studies have reported an increased risk for AD in individuals possessing the Glu504Lys loss of function mutation of ALDH2 [57,58]. The Glu504Lys ALDH2 polymorphism is also associated with an increased incidence of certain cancers [59], with an increased risk for developing schizophrenia [60], and for developing PD in some studies [61,62] but not others [63]. Interestingly, increased striatal concentrations of the toxic dopamine metabolite, 3,4-dihydroxyphenylacetaldehyde (DOPAL) are seen in Aldh1A1/Aldh2 double knockout mice [64]. These mice exhibit age-related impairments in motor performance, and have been suggested as an animal model of PD, which is also associated with increased LPO and HNE protein adduct formation [65].
The above considerations provided the rationale for the development of Aldh2−/− mice as an alternative and complementary model to the numerous transgenic mouse models of AD that are dependent on overexpression of mutant human genes linked to early-onset familial AD. Indeed, in a recent study [66] of NMZ (4-methyl-5-(2-(nitrooxy)ethyl)thiazol-3-ium chloride), a neuroprotective agent that targets synaptic failure by a combination of GABA-mimetic activity and increased NO/cGMP/CREB signalling, memory was improved in Aldh2−/− mice as well as in APP/PS1 and 3xTg transgenic mice. The Aldh2−/− mice are characterized by age-related decreases in the performance of several hippocampus-dependent memory tasks, together with a number of AD-like pathological changes, including increases in amyloid β (Aβ), phosphorylated tau protein, defective CREB signalling, synaptic loss, and several vascular pathologies [21]. Because of the variety of biochemical changes exhibited by Aldh2−/− mice, and a lack of certainty as to the relative importance of these changes in mediating the observed cognitive deficits, we chose to use the changes in the behavioural responses as our index of efficacy of the D-PUFA-enriched diet. Of relevance to the improved cognitive performance exhibited by D-PUFAs is a recent study using quench-assisted MRI which demonstrated increased free radical production (and by extension increased LPO) in the dorsal CA1 region of the hippocampus of Aldh2−/− mice relative to wildtype mice [67]. In rodents, this hippocampal subfield is associated with the encoding of spatial memory (which can be assessed by behavioural tests such as the MWM task), and is therefore consistent with the idea of a role for LPO in the impaired spatial reference memory exhibited by Aldh2−/− mice, and further, its amelioration by D-PUFA treatment.
A limitation of the current study stems from a relatively small number of mice studied. Due to limitations in the amount of available brain tissue, this precluded the analysis of F3-IsoPs and F4-NeuroPs, more relevant proxies for monitoring the omega-3 damage than F2-IsoPs and PGF2α. Given a potentially greater role for omega-3 derived LPO products in neuronal diseases than the corresponding omega-6 LPO products [39], we are currently planning a larger study to address this issue, as well as various PK/PD studies, studies using either 11,11-D2-LA or 11,11,14,14-D4-ALA alone or in different ratios, and studies using other D-PUFAs such as D-ARA, D-EPA and D-DHA. We also plan to use in vivo quench-assisted MRI [67] to see if the inhibition of LPO by D-PUFAs affects the neuronal redox status.
D-PUFA treatment as a viable therapeutic strategy to mitigate LPO-induced oxidative damage has been examined in the clinical setting (September 2016) in a first-in-human, Phase 1/2 clinical trial for Friedreich’s ataxia, a mitochondrial disease characterized by rampant lipid peroxidation. This 28-day trial of Retrotope Inc.’s lead compound, RT001 (bideuterated linoleic acid ethyl ester), in 18 Friedreich’s ataxia patients met all of its primary safety, tolerability and pharmacodynamics goals (manuscript in preparation). The results of the current study show that D-PUFAs dramatically improve cognitive performance in an animal model of oxidative stress-induced cognitive impairment by reducing LPO, and provide a rationale for future studies to determine whether D-PUFA treatment also reduces LPO, cognitive/memory decline and MCI/AD risk in humans.
Materials and methods
Animal model and diet
All procedures for animal experimentation were undertaken in accordance with the principles and guidelines of the Canadian Council on Animal Care and were approved by the Queen’s University Animal Care Committee. Animals were maintained under a 12 h light/dark cycle, with free access to food and water. The Aldh2−/− mouse colony was generated as described [21]. The cohort of Aldh2−/− mice used in the study was generated by mating heterozygotes, and the progeny genotyped by PCR analysis of genomic DNA extracted from ear punches using the primers as reported [68].
Mice were fed Mouse Diet AIN 93M (Research Diets, New Brunswick, NJ, USA), containing defined saturated and monounsaturated fatty acids (Table 1). The PUFA component of the diet was 1.2 g (1.2%) LA and ALA per 100 g diet in a 1:1 ratio. The experimental diet contained 11,11-D2 LA ethyl ester and 11,11,14,14-D4 α-ALA ethyl ester [14, Fig. 1], whereas the control diet had the same ratios of non-deuterated, H-PUFA. The total combined fat composition of the diet was 12.0% (saturates, monounsaturates and PUFAs). The control and experimental diet were supplied by Retrotope Inc. Male and female Aldh2−/− mice were fed either D- or H-PUFA-supplemented diet for 18 weeks, beginning at two months of age. Behavioural testing using the novel object recognition (NOR) task, spontaneous alternation in the Y-maze, and Morris water maze task (MWM) were performed after the animals had been on the D- or H-PUFA diet for 2, 10 or 18 weeks. The open field test and light/dark box tests were performed once, after animals had been on the diet for 10 weeks. At the end of the study, samples of cortex and hippocampus were obtained for determination of deuterium incorporation and for assessment of PGF2α and esterified F2-IsoP.
Deuterium incorporation
At the end of the study, mice were anaesthetised with isoflurane and the brain perfused with ice-cold PBS by trans-cardiac perfusion. The brain was removed and frozen in liquid nitrogen until analysis. Deuterium incorporation was determined in the brain as described previously [19,20]. In short, samples of cortex and hippocampi were sonicated and aliquots of tissue samples were lyophilized for deuterium evaluation using isotope ratio mass spectrometry (ir-MS). Samples were placed into silver capsules using CCl4 to avoid contamination by hydrogen. Subsequently, samples were loaded into a Costech Zero Blank Auto-sampler connected to a Thermo/Finnigan thermochemical elemental analyzer (Thermo Fisher, Bremen, Germany) and analyzed in triplicate as described. Each run was standardized through the use of at least two standards of different isotopic values. Data are presented as delta D/H (in permille, ‰), where delta D/H = (Rsample/Rstandard) x 1000; with R being the ratio between the abundances of deuterium and hydrogen (R = D/H).
Lipid peroxidation products
The lipid peroxidation markers (esterified) F2-IsoPs and PGF2α were determined in cortex and hippocampus at the Eicosanoid Core Laboratory at Vanderbilt University Medical Center. Samples were pre-treated and analysed for (esterified) F2-IsoPs and PGF2α using GC/negative ion chemical ionization MS as described in detail previously [69–71].
Behavioral Analyses
Open-field NOR task, the Y-maze task and the MWM task were performed as described [21]. The NOR task consisted of three consecutive days of testing per trial; habituation, training with two identical objects, and testing with one familiar and one novel object. On the testing day, animals were allowed to explore the objects until they accumulated 30 seconds of total object exploration time. Two measures of behaviour were assessed: frequency of visits to the objects, and the time spent exploring each object. From the latter measure the discrimination index (difference in time exploring the novel and familiar object, divided by total exploration time) and the ratio of time spent with the novel object in relation to the familiar object were calculated.
For the Y-maze task, mice were placed in the center of the maze and allowed to explore the three maze arms freely for 10 min. The maze was surrounded by distinct spatial cues so that the mice could distinguish between arms. The spontaneous alternation rate was calculated as the total triads containing entries into each of the three arms without repeated entry into a previously visited arm, divided by the total number of arm entries.
For the MWM task, the maze consisted of a circular pool (1.2m in diameter) filled with water (approximately 23°C) made opaque by non-toxic white paint. A circular platform (15cm in diameter) was submerged approximately 1cm below the surface in the northeast quadrant of the maze. The escape latency (time to reach the hidden platform) was determined in in a 3-day cued trial block (4 trials per day) followed by a 5-day hidden trial block (6 trials per day) [72]. For cued platform training, a black-and-white striped pole (2.2 cm diameter and 15 cm height) was attached to the center of the platform. Each mouse was allowed 60 seconds to locate the platform per trial. Mice that could not find the platform within 60 seconds were gently guided towards it. Day 9 was a 60 second probe trial, in which the time spent in each quadrant and number of platform crosses were recorded.
The open field test was used to assess spontaneous locomotor activity during free exploration, and was performed as described previously [73]. The open field test was conducted using a square Plexiglass® open field apparatus (45cm x 45cm x 25cm). Each mouse was placed individually in the centre and allowed to explore freely for 5 minutes. The test was performed under dim light conditions and minimal background noise. Spontaneous locomotion was assessed by a grid of infrared beams using a software data acquisition system (TSE Actimot, Scientific Products and Equipment, Concord, ON, Canada). The total time spent moving, total distance travelled, and time spent rearing were recorded.
The Light/dark box test was used to assess aversion to bright illumination as a measure of anxiety-like behaviour. The opaque plastic arena (50cm x 25cm x 18cm) was arranged as described [74]. It was divided unequally such that two-thirds of the space was considered the “light” side and one-third was the “dark” side. The compartments were divided by an opaque barrier with a hole (6cm x 6cm) along the bottom to allow free movement between sides. The dark side was enclosed by opaque plastic sheets to block light and the light side was illuminated by a 60W bulb suspended 20 cm above the arena. Animals were placed in the dark side and allowed to explore freely for 6 minutes. An entry was considered when the centre of mass of the animal crossed the division between sides. The latency to enter the light side and total time spent in the light and dark sides were recorded.
Data analysis
Data is expressed as the mean ± s.d or s.e.m. and was analyzed by two way analysis of variance with Bonferroni’s post-hoc test, or a two-tailed Student’s t test for unpaired data, as indicated. A p-value of 0.05 or less was considered statistically significant.
Acknowledgments
The authors wish to thank Mrs. Diane Anderson, Lihua Xue and Chloe Lowry for technical assistance. Aldh2−/− mice were kindly provided by Dr. T. Kawamoto, University of Occupational and Environmental Health, Kitakyushu, Japan. Financial support and D-PUFA reagents were provided by Retrotope, Inc. BMB is supported by The Alzheimer’s Society Research Program grant 365170 and by CIHR Canadian Vascular Network grant 395023. GLM is supported by NIH grant DK020593.
Abbreviations
- AD
Alzheimer’s disease
- ALA
linolenic acid
- ALDH2
Aldehyde dehydrogenase 2
- ARA
arachidonic acid
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- F2-IsoP
F2-isoprostanes
- HHE
4-hydroxy-2-hexenal
- HNE
4-hydroxy-2-nonenal
- LA
linoleic acid
- LPO
lipid peroxidation
- MCI
mild cognitive impairment
- MWM
Morris water maze
- NOR
novel object recognition
- PD
Parkinson’s disease
- PGF2α
prostaglandin F2α
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
Footnotes
Authors’ Contributions
AE, BMB and MSS planned the experiments and wrote the manuscript. AE, NMC and MG performed the behavioural testing, and AE, NMC, GLM and EP performed data analysis. All authors read and approved the final manuscript.
Conflict of Interest: MSS is Chief Science Officer and a shareholder of Retrotope Inc.
References
- 1.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 2.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurology. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 3.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA. β-Amyloid-associated free radical oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Chem Res Toxicol. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 5.Praticò D. Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Di Domenico F, Tramutola A, Butterfield DA. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.10.490. doi.org/10.1016/j.freeradbiomed.2016.10.490. [DOI] [PubMed]
- 7.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 8.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 9.Kawai Y, Takeda S, Terao J. Lipidomic analysis for lipid peroxidation-derived aldehydes using gas chromatography-mass spectrometry. Chem Res Toxicol. 2007;20:99–107. doi: 10.1021/tx060199e. [DOI] [PubMed] [Google Scholar]
- 10.Galasko DR, Peskind E, Clark CM, Quinn JF, Ringman JM, Jicha GA, Cotman C, Cottrell B, Montine TJ, Thomas RG, Aisen P. Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol. 2012;69:836–841. doi: 10.1001/archneurol.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vellas B, Coley N, Ousset P-J, Berrut G, Dartigues JF, Dubois B, Grandjean H, Pasquier F, Piette F, Robert P, Touchon J, Garnier P, Mathiex-Fortunet H, Andrieu S. GuidAge Study Group. Long-term use of standardised Ginkgo biloba extract for the prevention of Alzheimer’s disease (GuidAge): a randomised placebo-controlled trial. Lancet Neurol. 2012;11:851–859. doi: 10.1016/S1474-4422(12)70206-5. [DOI] [PubMed] [Google Scholar]
- 12.Kryscio RJ, Abner EL, Caban-Holt A, Lovell M, Goodman P, Darke AK, Yee M, Crowley J, Schmitt FA. Association of antioxidant supplement use and dementia in the prevention of Alzheimer’s disease by vitamin E and selenium trial (PREADViSE) JAMA Neurol. 2017 doi: 10.1001/jamaneurol.2016.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4. Oxford University Press; Oxford: 2007. [Google Scholar]
- 14.Hill S, Hirano K, Shmanai VV, Marbois BN, Vidovic D, Bekish AV, Kay B, Tse V, Fine J, Clarke CF, Shchepinov MS. Isotope-reinforced polyunsaturated fatty acids protect yeast cells from oxidative stress. Free Radic Biol Med. 2011;50:130–138. doi: 10.1016/j.freeradbiomed.2010.10.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shchepinov MS. Reactive Oxygen Species, Isotope Effect, Essential Nutrients, and Enhanced Longevity. Rejuvenation Res. 2007;10:47–59. doi: 10.1089/rej.2006.0506. [DOI] [PubMed] [Google Scholar]
- 16.Hill S, Lamberson CR, Xu L, To R, Tsui HS, Shmanai VV, Bekish AV, Awad AM, Marbois BN, Cantor CR, Porter NA, Clarke CF, Shchepinov MS. Small amounts of isotope-reinforced polyunsaturated fatty acids suppress lipid autoxidation. Free Radic Biol Med. 2012;53:893–906. doi: 10.1016/j.freeradbiomed.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andreyev AY, Tsui HS, Milne GL, Shmanai VV, Bekish AV, Fomich MA, Pham MN, Nong Y, Murphy AN, Clarke CF, Shchepinov MS. Isotope-reinforced polyunsaturated fatty acids protect mitochondria from oxidative stress. Free Radic Biol Med. 2015;82:63–72. doi: 10.1016/j.freeradbiomed.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 18.Westheimer FH. The magnitude of the primary kinetic isotope effect for compounds of hydrogen and deuterium. Chem Rev. 1961;61:265–273. [Google Scholar]
- 19.Shchepinov MS, Chou VP, Pollock E, Langston JW, Cantor CR, Molinari RJ, Manning-Boğ AB. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson’s disease. Toxicol Lett. 2011;207:97–103. doi: 10.1016/j.toxlet.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 20.Berbée JFP, Mol IM, Milne GL, Pollock E, Hoeke G, Lütjohann D, Monaco C, Rensen PCN, van der Ploeg LHT, Shchepinov MS. Deuterium-reinforced polyunsaturated fatty acids protect against atherosclerosis by lowering lipid peroxidation and hypercholesterolemia. Atherosclerosis. 2017 doi: 10.1016/j.atherosclerosis.2017.06.916. doi.org/10.1016/j.atherosclerosis.2017.06. [DOI] [PubMed]
- 21.D’Souza Y, Elharram A, Soon-Shiong R, Andrew RD, Bennett BM. Characterization of Aldh2−/− mice as an age-related model of cognitive impairment and Alzheimer’s Disease. Mol Brain. 2015;8:27. doi: 10.1186/s13041-015-0117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picklo MJ, Olson SJ, Markesbery WR, Montine TJ. Expression and activities of aldo-keto oxidoreductases in Alzheimer disease. J Neuropath Exp Neurol. 2001;60:686–695. doi: 10.1093/jnen/60.7.686. [DOI] [PubMed] [Google Scholar]
- 23.Yoval-Sánchez B, Rodríguez-Zavala JS. Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem Res Toxicol. 2102;25:722–729. doi: 10.1021/tx2005184. [DOI] [PubMed] [Google Scholar]
- 24.Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 25.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 26.McGrath LT, McGleenon BM, Brennan S, McColl D, McIlroy S, Passmore AP. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. Q J Med. 2001;94:485–490. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 27.Reed TT, Pierce WM, Markesbery WR, Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis. 2008;30:107–120. doi: 10.1016/j.nbd.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Perluigi M, Sultana R, Cenini G, Domenico FD, Memo M, Pierce WM, Coccia R, Butterfield DA. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics Clin Appl. 2009;3:682–693. doi: 10.1002/prca.200800161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czegledy NM, Bennett BM. Anxiety- and depression-like behaviours in a mouse model of oxidative stress-induced cognitive impairment. Canadian Association for Neuroscience; Montreal: 2017. May, [Google Scholar]
- 31.Shchepinov MS, Roginsky VA, Brenna JT, Molinari RJ, To R, Tsui H, Clarke CF, Manning-Bog AB. Deuterium protection of polyunsaturated fatty acids against lipid peroxidation: a novel approach to mitigating mitochondrial neurological diseases. In: Watson RS, De Meester F, editors. Omega 3 Fatty Acids in Brain and Neurological Health. Elsevier Academic Press; Amsterdam: 2014. pp. 373–383. [Google Scholar]
- 32.Lamberson CR, Xu L, Muchalski H, Montenegro-Burke JR, Shmanai VV, Bekish AV, McLean JA, Clarke CF, Shchepinov MS, Porter NA. Unusual kinetic isotope effects of deuterium reinforced polyunsaturated fatty acids in tocopherol-mediated free radical chain oxidations. J Amer Chem Soc. 2014;136:838–841. doi: 10.1021/ja410569g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cotticelli MG, Crabbe AM, Wilson RB, Shchepinov MS. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013;1:398–404. doi: 10.1016/j.redox.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milne GL, Dai Q, Roberts LJ. The isoprostanes—25 years later. Biochim Biophys Acta. 2015;1851:433–445. doi: 10.1016/j.bbalip.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58:730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 36.Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer Disease brain. Free Radic Biol Med. 2013;62:157–169. doi: 10.1016/j.freeradbiomed.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Domenico F, Tramutola A, Butterfield DA. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.10.490. [DOI] [PubMed] [Google Scholar]
- 38.Bradley MA, Xiong-Fister S, Markesbery WR, Lovell MA. Elevated 4-hydroxyhexenal in Alzheimer’s disease (AD) progression. Neurobiol Aging. 2012;33:1034–1044. doi: 10.1016/j.neurobiolaging.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long EK, Picklo MJ., Sr Trans-4-hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: make some room HNE. Free Radic Biol Med. 2014;49:1–8. doi: 10.1016/j.freeradbiomed.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 40.Long EK, Murphy TC, Leiphon LJ, Watt J, Morrow JD, Milne GL, Howard JR, Picklo MJ., Sr Trans-4-hydroxy-2-hexenal is a neurotoxic product of docosahexaenoic (22:6; n-3) acid oxidation. J Neurochem. 2008;105:714–724. doi: 10.1111/j.1471-4159.2007.05175.x. [DOI] [PubMed] [Google Scholar]
- 41.Pawlosky RJ1, Bacher J, Salem N., Jr Ethanol consumption alters electroretinograms and depletes neural tissues of docosahexaenoic acid in rhesus monkeys: nutritional consequences of a low n-3 fatty acid diet. Alcohol Clin Exp Res. 2001;25:1758–1765. [PubMed] [Google Scholar]
- 42.Lim SY, Doherty JD, Salem N., Jr Lead exposure and (n-3) fatty acid deficiency during rat neonatal development alter liver, plasma, and brain polyunsaturated fatty acid composition. J Nutr. 2005;135:1027–1033. doi: 10.1093/jn/135.5.1027. [DOI] [PubMed] [Google Scholar]
- 43.Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20(Suppl 2):S609–631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012 doi: 10.1155/2012/472932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. Editorial commentary (1997) N Engl J Med 336, 1245–7. [DOI] [PubMed] [Google Scholar]
- 46.Bowry VW, Ingold KU, Stocker R. Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem J. 1992;288:341–344. doi: 10.1042/bj2880341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohsawa I, Nishimaki K, Yasuda C, Kamino K, Ohta S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J Neurochem. 2003;84:1110–1117. doi: 10.1046/j.1471-4159.2003.01619.x. [DOI] [PubMed] [Google Scholar]
- 48.Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 49.Duan Y, Gao Y, Zhang J, Chen Y, Jiang Y, Ji J, Zhang J, Chen X, Yang Q, Su L, Zhang J, Liu B, Zhu Z, Wang L, Yu Y. Mitochondrial aldehyde dehydrogenase 2 protects gastric mucosa cells against DNA damage caused by oxidative stress. Free Radic Biol Med. 2016;93:165–176. doi: 10.1016/j.freeradbiomed.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo JM, Liu AJ, Zang P, Dong WZ, Ying L, Wang W, Xu P, Song XR, Cai J, Zhang SQ, Duan JL, Mehta JL, Su DF. ALDH2 protects against stroke by clearing 4-HNE. Cell Res. 2013;23:915–930. doi: 10.1038/cr.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu SH, Zhang HF, Yang ZB, Li TB, Liu B, Lou Z, Ma QL, Luo XJ, Peng J. Alda-1 reduces cerebral ischemia/reperfusion injury in rat through clearance of reactive aldehydes. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:87–94. doi: 10.1007/s00210-013-0922-8. [DOI] [PubMed] [Google Scholar]
- 53.Ding J, Zhang Q, Luo Q, Ying Y, Liu Y, Li Y, Wei W, Yan F, Zhang H. Alda-1 attenuates lung ischemia-reperfusion injury by reducing 4-hy- droxy-2-nonenal in alveolar epithelial cells. Crit Care Med. 2016;44:e544–e552. doi: 10.1097/CCM.0000000000001563. [DOI] [PubMed] [Google Scholar]
- 54.Picklo MJ, Olson SJ, Markesbery WR, Montine TJ. Expression and activities of aldo-keto oxidoreductases in Alzheimer disease. J Neuropath Exp Neurol. 2001;60:686–695. doi: 10.1093/jnen/60.7.686. [DOI] [PubMed] [Google Scholar]
- 55.Michel TM, Gsell W, Käsbauer L, Tatschner T, Sheldrick AJ, Neuner I, Schneider F, Grünblatt E, Riederer P. Increased activity of mitochondrial aldehyde dehydrogenase (ALDH) in the putamen of individuals with Alzheimer’s disease: a human postmortem study. J Alzheimers Dis. 2010;19:1295–1301. [Google Scholar]
- 56.Michel TM, Käsbauer L, Gsell W, Jecel J, Sheldrick AJ, Cortese M, Nickl-Jockschat T, Grünblatt E, Riederer P. Aldehyde dehydrogenase 2 in sporadic Parkinson’s disease. Parkinsonism Relat Disord. 2014;20(Suppl 1):S68–72. doi: 10.1016/S1353-8020(13)70018-X. [DOI] [PubMed] [Google Scholar]
- 57.Kamino K, Nagasaka K, Imagawa M, Yamamoto H, Yoneda H, Ueki A, Kitamura S, Namekata K, Miki T, Ohta S. Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimer’s disease in the Japanese population. Biochem Biophysical Res Commun. 2000;273:192–196. doi: 10.1006/bbrc.2000.2923. [DOI] [PubMed] [Google Scholar]
- 58.Wang B, Wang J, Zhou S, Tan S, He X, Yang Z, Xie Y-C, Li S, Zheng C, Ma X. The association of mitochondrial aldehyde dehydrogenase gene (ALDH2) polymorphism with susceptibility to late-onset Alzheimer’s disease in Chinese. J Neurolog Sci. 2008;268:172–175. doi: 10.1016/j.jns.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 59.Seitz HK, Meier P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl Res. 2007;149:293–297. doi: 10.1016/j.trsl.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 60.Zheng F, Yan H, Liu B, Yue W, Fan L, Liao J, Cui Y, Lu T, Jiang T, Zhang D. ALDH2 Glu504Lys Confers Susceptibility to Schizophrenia and Impacts Hippocampal-Prefrontal Functional Connectivity. Cereb Cortex. 2017;27:2034–2040. doi: 10.1093/cercor/bhw056. [DOI] [PubMed] [Google Scholar]
- 61.Liu j, Sui S, Xu l, Lin K, Zong J, Yang Y, Wang H. Association of ADH2 and ALDH2 genetic polymorphisms with susceptibility to Parkinson’s disease in a Chinese population. Int J Clin Exp Pathol. 2016;9:9436–9442. [Google Scholar]
- 62.Zhao CC, Cai HB, Wang H, Pan SY. Role of ADH2 and ALDH2 gene polymorphisms in the development of Parkinson’s disease in a Chinese population. Genet Mol Res. 2016:15. doi: 10.4238/gmr.15038606. [DOI] [PubMed] [Google Scholar]
- 63.Zhang X, Ye YL, Wang YN, Liu FF, Liu XX, Hu BL, Zou M, Zhu JH. Aldehyde dehydrogenase 2 genetic variations may increase susceptibility to Parkinson’s disease in Han Chinese population. Neurobiol Aging. 2015;36:2660, e9–13. doi: 10.1016/j.neurobiolaging.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 64.Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, Mash DC, Kopin IJ, Sharabi Y. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem. 2013;126:591–603. doi: 10.1111/jnc.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7:e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo J, Lee SH, VandeVrede L, Qin Z, Ben Aissa M, Larson J, Teich AF, Arancio O, D’Souza Y, Elharram A, Koster K, Tai LM, LaDu MJ, Bennett BM, Thatcher GR. A multifunctional therapeutic approach to disease modification in multiple familial mouse models and a novel sporadic model of Alzheimer’s disease. Mol Neurodegener. 2016;11:35. doi: 10.1186/s13024-016-0103-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berkowitz BA, Lenning J, Khetarpal N, Tran C, Wu JW, Berri AM, Dernay K, Haacke EM, Shafie-Khorassani F, Podolsky RH, Gant JC, Maimaiti S, Thibaut O, Murphy GG, Bennett BM, Roberts R. In vivo imaging of prodromal hippocampus CA1 subfield oxidative stress in models of Alzheimer’s Disease and Angelman Syndrome. FASEB J. 2017;31:4179–4186. doi: 10.1096/fj.201700229R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Isse T, Oyama T, Kitagawa K, Matsuno K, Matsumoto A, Yoshida A, Nakayama K, Nakayama K, Kawamoto T. Diminished alcohol preference in transgenic mice lacking aldehyde dehydrogenase activity. Pharmacogenetics. 2002;12:621–626. doi: 10.1097/00008571-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 69.Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- 70.Milne GL, Gao B, Terry ES, Zackert WE, Sanchez SC. Measurement of F2-isoprostanes and isofurans using gas chromatography-mass spectrometry. Free Radic Biol Med. 2013;59:36–44. doi: 10.1016/j.freeradbiomed.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Menon R, Fortunato SJ, Milne GL, Brou L, Carnevale C, Sanchez SC, Hubbard L, Lappas M, Drobek CO, Taylor RN. Amniotic fluid eicosanoids in preterm and term births: effects of risk factors for spontaneous preterm labor. Obstet Gynecol. 2011;118:121–134. doi: 10.1097/AOG.0b013e3182204eaa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Delpolyi AR, Fang S, Palop JJ, Yu G, Wang X, Mucke L. Altered navigational strategy use and visuospatial deficits in hAPP transgenic mice. Neurobiol Aging. 2006;29:253–266. doi: 10.1016/j.neurobiolaging.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 73.Gould TD, Dai DT, Kovacsics CE. The open field test. Mood and anxiety related phenotypes in mice. Neuromethods. 2009;42:1–20. [Google Scholar]
- 74.Bourin M, Hascoet M. The mouse light/dark box test. Eur J Pharmacol. 2003;463:55–65. doi: 10.1016/s0014-2999(03)01274-3. [DOI] [PubMed] [Google Scholar]