

Abstract
Background
FUN14 domain containing 1 (FUNDC1) is a highly conserved outer mitochondrial membrane protein. The aim of this study is to examine if FUNDC1 modulates the mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs), mitochondrial morphology, and function in cardiomyocytes and in intact hearts.
Methods
The impacts of FUNDC1 on MAMs formation and cardiac functions were studied in mouse neonatal cardiomyocytes, in mice with cardiomyocyte-specific Fundc1 gene knockout (Fundc1f/Y/CreαMyHC+/−), and in the cardiac tissues of the patients with heart failure.
Results
In mouse neonatal cardiomyocytes and intact hearts, FUNDC1 was localized in MAMs by binding to ER-resided inositol 1,4,5-trisphosphate type 2 receptor (IP3R2). Fundc1 ablation disrupted MAMs, reduced the levels of IP3R2 and Ca2+ in both mitochondria and cytosol whereas overexpression of Fundc1 increased the levels of IP3R2 and Ca2+ in both mitochondria and cytosol. Consistently, Fundc1 ablation increased Ca2+ levels in ER whereas Fundc1 overexpression lowered ER Ca2+ levels. Further, Fundc1 ablation in cardiomyocytes elongated mitochondria, and compromised mitochondrial functions. Mechanistically, we found that Fundc1 ablation-induced reduction of intracellular Ca2+ levels suppressed mitochondrial fission 1 protein (Fis1) expression and mitochondrial fission by reducing the binding of the cAMP response element binding protein (CREB) in the Fis1 promoter. Fundc1f/Y/CreαMyHC+/− mice but not their littermate control mice (Fundc1wt/Y/CreαMyHC+/−) exhibited cardiac dysfunction. The ligation of the left ventricle artery of Fundc1f/Y/CreαMyHC+/− mice caused more severe cardiac dysfunction than those in sham-treated Fundc1f/Y/CreαMyHC+/− mice. Finally, we found that the FUNDC1/MAMs/CREB/Fis1 signaling axis was significantly suppressed in the patients with heart failure.
Conclusions
We conclude that FUNDC1 binds to IP3R2 to modulate ER Ca2+ release into mitochondria and cytosol and that a disruption of FUNDC1 and IP3R2 interaction lowers the levels of Ca2+ in mitochondria and cytosol, both of which instigate aberrant mitochondrial fission, mitochondrial dysfunction, cardiac dysfunction, and heart failure.
Keywords: FUNDC1, IP3R2, MAMs, Cardiomyopathy, Heart failure
Introduction
Mitochondria and endoplasmic reticulum (ER) are interconnected organelles, both of which form an endomembrane network.1 These contact points where ER communicates with mitochondria are referred to as mitochondria-associated ER membranes (MAMs).1 These physical interactions between both organelles depend on complementary membrane proteins, which tether the two organelles together at specific sites. For example, the voltage-dependent anion channel 1 (VDAC1) of the outer mitochondrial membrane interacts with the inositol 1,4,5-triphosphate type 1 receptor (IP3R1) on the ER through the molecular chaperone glucose regulated protein 75 (Grp75), allowing Ca2+ transfer from the ER to mitochondria.2 The sigma-1 receptor (Sig1R) stabilizes MAMs by interacting with IP3R1 and VDAC1.3 Mitofusin 2 (Mfn2) has been proposed to be an additional physical tether.4, 5 Although numerous proteins have been identified in MAMs fractions, factors that control the formation and/or functions of MAMs are still poorly understood. Increasing evidence suggests that MAMs functions as a center for intermembrane transport of phospholipids, and MAMs are crucial to shape intracellular Ca2+ signaling and regulate mitochondrial bioenergetics.2, 6 As a result, dysfunctional MAMs is involved in the pathogenesis of neuronal disorders, such as Alzheimer’s7 and Parkinson’s8 disease. However, the roles of MAMs in cardiac physiology and/or pathologies remain poorly characterized.
Mitochondria are critical integrators of energy production, reactive oxygen species generation, signal transduction, and apoptosis.9 As the myocardium is a high-energy-demand tissue, mitochondria play a central role in maintaining optimal cardiac performance.10–12 Although mitochondrial fission and fusion are most evident in neonatal cardiomyocytes due to the features of perinatal cardiac metabolic maturation13–15, there is strong evidence that these processes are important in the adult cardiac tissue16–19. Indeed, growing evidence suggests deregulated mitochondrial dynamics plays a causative role in cardiovascular diseases.20–26 To maintain normal structure and function, mitochondria continuously join by the process of fusion and divide by the process of fission, so called mitochondrial dynamics.27 The execution of mitochondrial dynamics in mammalian cells requires the participation of a large number of proteins, including mitofusin 1/2 (Mfn1/2) and optic atrophy 1 (OPA1) as fusion proteins and dynamin-related protein 1 (DRP1), mitochondrial fission factor (MFF), and mitochondrial fission 1 protein (Fis1) as fission proteins. Recently, MAMs has been highlighted to play an important role in mitochondrial fission process by marking the division sites.
FUN14 domain containing 1 (FUNDC1), an outer mitochondrial membrane protein with 155 amino acids, is highly conserved from Drosophila to humans.28 FUNDC1 contains a hydrophobic transmembrane domain consisting of three α-helical stretches with its amino terminus exposing to the cytosol, and its carboxyl terminus stretching in the intermembrane space.28 Recent studies report that FUNDC1 localizes in MAMs of HeLa cells.29, 30 However, whether or not FUNDC1 regulates MAMs formation and influences MAMs function is unknown. Furthermore, the expression of Fundc1 in cardiac tissues, as well as its functions remain poorly studied. Here we report that FUNDC1 binds to inositol 1,4,5-trisphosphate type 2 receptor (IP3R2), a key player who is responsible for Ca2+ release from the ER into mitochondria and cytosol. As a result, cardiac-specific Fundc1 ablation instigates aberrant mitochondrial network, mitochondrial dysfunction, cardiac dysfunction, and heart failure.
Methods
Generation of the Cardiomyocyte-Specific Fundc1 Knockout Mice
The Fundc1 gene was located in Chromosome X. The floxed-Fundc1 mice were developed by Mouse Biology Program (MBP) (University of California, Davis, CA). The CreαMyHC+/− mice were purchased from Jackson Laboratory (Bar Harbor, ME) with the cardiac-specific alpha myosin-heavy chain (αMyHC) promoter driving expression of Cre specifically in cardiomyocytes. The cardiomyocyte-specific male Fundc1 knockout (Fundc1f/Y/CreαMyHC+/−) mice were generated by crossing female Fundc1wt/f/CreαMyHC+/− with male Fundc1wt/Y/CreαMyHC+/− mice. Littermate male Fundc1wt/Y/CreαMyHC+/− mice were considered as control mice. All mice (male) were C57BL/6J background and housed in temperature-controlled cages with a 12-h light-dark cycle and given free access to water and food.
Statistics
All the data were expressed as means ± standard deviation (SD). All experiments were performed at least in triplicates unless otherwise stated. Homogeneity of the variance was assessed via F test (two groups) or Brown-Forsythe test (≥ three groups). Normality of the data was assessed via Shapiro-Wilk test. When reporting results with two groups, we used standard Student’s t test when the assumptions (equal variance and normal distribution) are satisfied. Otherwise, the non-parametric Mann-Whitney U test was applied to analyze the difference between two groups. In the results with more than two groups, ANVOA or appropriate non-parametric tests were applied to analyze the difference. When one factor was involved, we used one-way ANOVA followed by Bonferroni post-hoc test when the assumptions (equal variance and normal distribution) are satisfied. Otherwise, we used the non-parametric test Kruskal-Wallis test followed by the Dunn’s post-hot test to correct for multiple comparisons. In results with two factors, two-way ANOVA followed by Bonferroni post-hoc test was applied for multiple comparisons when the assumptions (equal variances and normal distribution) are stratified. Otherwise, we used the non-parametric test Scheirer-Ray-Hare test (an extension of the Kruskal-Wallis test) followed by the Dunn’s post-hot test to correct for multiple comparisons. The P values were adjusted for multiple comparisons where appropriate. P values of less than 0.05 were considered statistically significant. All statistical analyses were carried out using SPSS Version 21 software.
Study Approval
The animal protocol in this study was approved by the Institutional Animal Care and Use Committee (IACUC) at Georgia State University. All human studies were approved by the human ethics committee of Wuhan Union Hospital of Huazhong University of Science and Technology (2017-S10005) and the subjects gave informed consent.
Results
Cardiomyocyte-Specific Deletion of Fundc1 Induces Mitochondrial Elongation and Heart Failure in Vivo
We first examined the distribution of FUNDC1 in different organs of mouse. As shown in Supplemental Figure 1A, mouse heart expressed higher levels of Fundc1. In cardiac tissue, FUNDC1 appeared to be exclusively localized within the mitochondria (Supplemental Figure 1B). To determine the contributions of FUNDC1 in maintaining normal cardiac structures and functions, we generated cardiomyocyte-specific Fundc1 knockout mice (Fundc1f/Y/CreαMyHC+/−, Supplemental Figure 2A). The knockout efficiency of Fundc1 gene was demonstrated by polymerase chain reaction, western blot, immunohistochemistry, and immunofluorescence (Supplemental Figure 2B through 2F). Cardiac functions of Fundc1f/Y/CreαMyHC+/− mice and their control littermates (Fundc1wt/Y/CreαMyHC+/−) were monitored using echocardiography at 10 weeks of age. Compared with Fundc1wt/Y/CreαMyHC+/− mice, the ratios of the early (E) to late (A) ventricular filling velocities (E/A) in Fundc1f/Y/CreαMyHC+/− mice were markedly reduced (Figure 1A and 1B), which was concurrent with an increase in the deceleration time of the E-wave (DT) (Figure 1A and 1C). Fundc1f/Y/CreαMyHC+/− mice also exhibited prolonged left ventricular isovolumic relaxation time (IVRT) (Figure 1A and 1D). These data collectively demonstrate that Fundc1f/Y/CreαMyHC+/− mice exhibit typical diastolic dysfunction. Moreover, the ejection fraction (EF) (Figure 1E and 1F; Supplemental Figure 3A), fractional shortening (FS) (Figure 1E and 1G; Supplemental Figure 3B), and cardiac output (Figure 1H) in Fundc1f/Y/CreαMyHC+/− mice were largely decreased when compared with those in Fundc1wt/Y/CreαMyHC+/− mice, indicating that Fundc1f/Y/CreαMyHC+/− mice also exhibited impaired systolic functions. As presented in Figure 1I, the main pathologic changes of heart from Fundc1f/Y/CreαMyHC+/− mice were the interstitial fibrosis of myocardium when compared with hearts from Fundc1wt/Y/CreαMyHC+/− mice. Furthermore, we detected increased positive signals of terminal deoxynucleotidyl transferase dUTP nick-end labeling staining in heart tissues from Fundc1f/Y/CreαMyHC+/− mice (Figure 1J and 1K), reflecting that Fundc1 cardiac-deficiency can cause apoptosis in cardiac tissues. Cardiac injury was also evidenced by cardiac stress gene expression profile, including the increase of β-myosin heavy chain (β-Mhc), atrial natriuretic peptide (Anp), and brain natriuretic peptide (Bnp) in Fundc1f/Y/CreαMyHC+/− hearts (Supplemental Figure 3C through 3E). These data indicate that cardiomyocyte-specific deletion of Fundc1 leads to heart failure.
Figure 1.
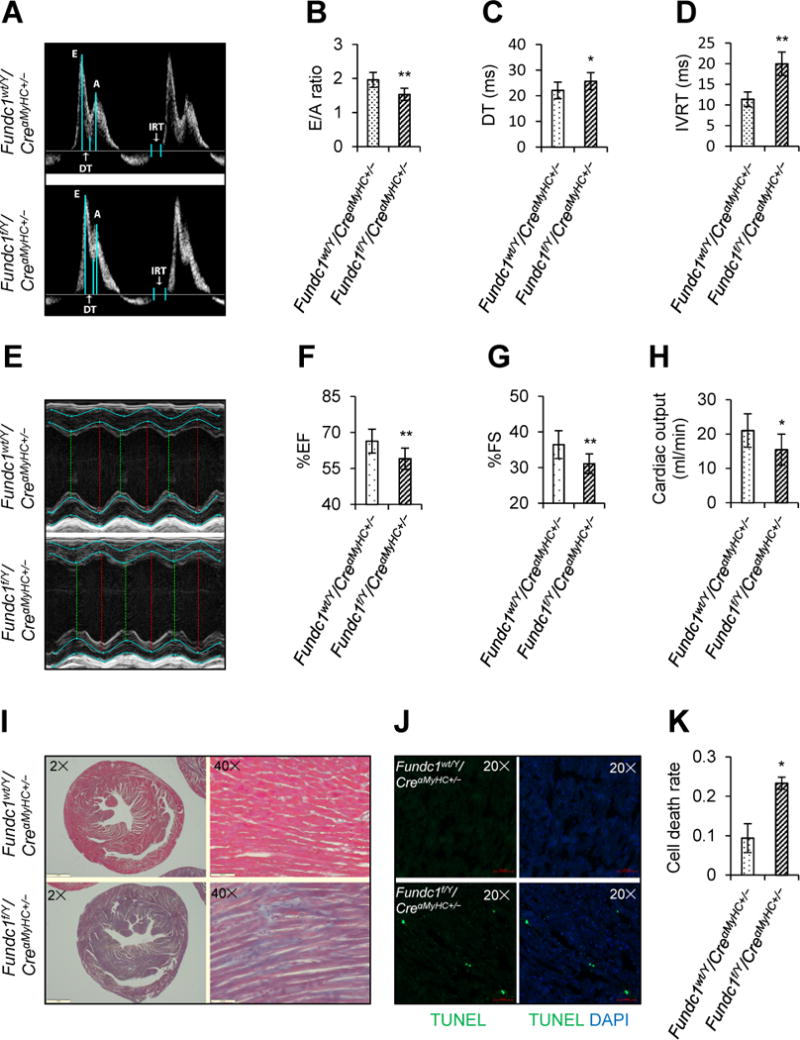
Effects of cardiac Fundc1 deletion on cardiac structure and function. A-H, Echocardiographic assessment was performed in 10-week-old male Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− mice. A, Representative Doppler flow measurements of mitral inflow. B, Ratios of the early (E) to late (A) ventricular filling velocities. C, Deceleration times (DT). D, Isovolumic relaxation times (IVRT). E, Representative images of M-mode echocardiography. F, Ejection fraction (EF). G, Fractional shortening (FS). H, Cardiac output. (Mean ± SD, n = 8 mice per group; *P < 0.05, **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/−.) I, Cardiac fibrosis was revealed by Masson’s trichrome staining of heart paraffin sections. Representative staining is shown (n = 8 mice per group). Scale bars, 1 mm (2×) and 500 μm (40×). J, Representative terminal-deoxynucleoitidyl transferase mediated nick end labeling (TUNEL) staining of heart frozen sections revealed cardiomyocyte apoptosis. Scale bars, 50 μm. K, Statistical analysis of TUNEL staining in J (mean ± SD, n = 8 mice per group; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−).
Since FUNDC1 is a mitochondrial protein, we further examined the effects of Fundc1 knockout on mitochondrial structure and function. Compared with the ovoid structure of mitochondria in Fundc1wt/Y/CreαMyHC+/− hearts, mitochondria in Fundc1f/Y/CreαMyHC+/− hearts were larger and more elongated (Figure 2A and 2B). This result was further evidenced by measuring the size of mitochondria isolated from hearts, showing that Fundc1-deficient mitochondria has ~2.5-fold increase of size than control mitochondria (Figure 2C and 2D). Furthermore, Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes displayed significantly lower basal and maximal respiratory capacity than Fundc1wt/Y/CreαMyHC+/− cardiomyocytes (Figure 2E and 2F). The mitochondrial membrane potential also decreased in the mitochondria isolated from Fundc1f/Y/CreαMyHC+/− hearts, compared with that from control hearts (Figure 2G and 2H). The levels of mitochondrial proteins, mitochondrial import inner membrane translocase subunit (TIM23) and VDAC1, in Fundc1f/Y/CreαMyHC+/− hearts were higher than those in Fundc1wt/Y/CreαMyHC+/− hearts (Figure 2I and 2J), indicating accumulation of compromised mitochondria. Taken together, cardiomyocyte-specific deletion of Fundc1 impairs cardiac functions, associated with mitochondrial elongation and accumulation of dysfunctional mitochondria.
Figure 2.
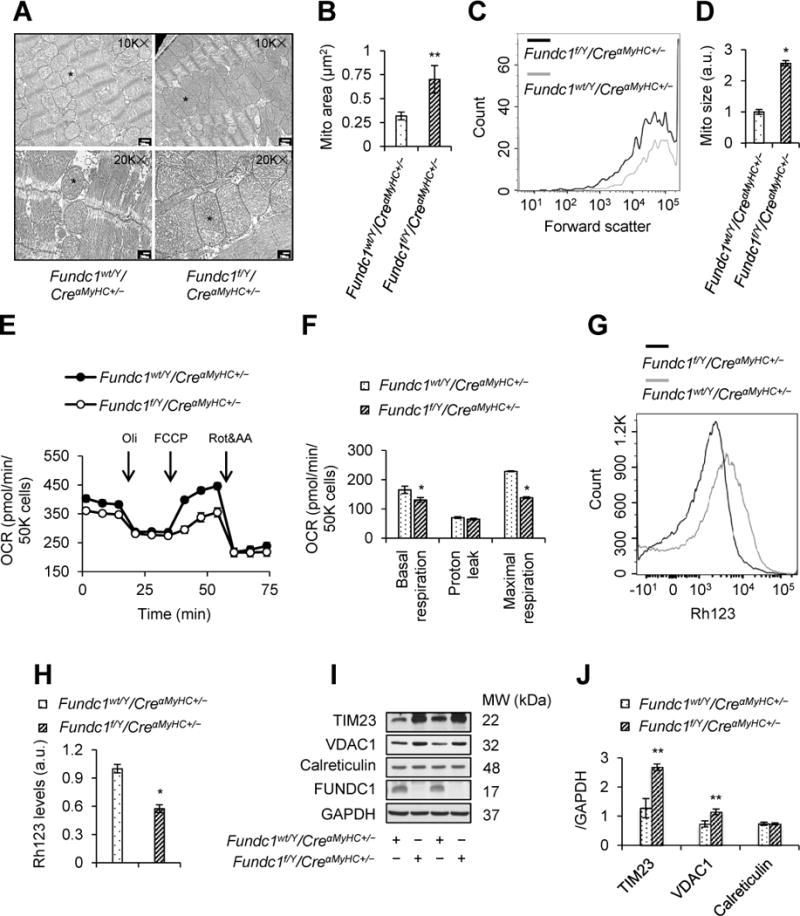
Effects of cardiac Fundc1 deletion on mitochondrial structure and function. A, Representative transmission electron microscopy (TEM) images of mitochondrial morphology in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− hearts. Asterisks indicate mitochondria with typical morphology. Scale bars, 500 nm (10K×) and 200 nm (20K×). B, Mitochondrial area (mean ± SD, n = 8 mice per group; **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/−). C, Flow cytometry (FCM) analysis of mitochondrial size (forward scatter) in adult mouse heart mitochondria. Representative size distribution curves are compared to paired controls; quantitative data are shown in D (mean ± SD, n = 8 mice per group; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). E, Oxygen consumption rate (OCR) was determined in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes using a Seahorse extracellular flux analyzer by sequential injection of 1 μM oligomycin (Oli), 2 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 1 μM antimycin A (AA) & rotenone (Rot). F, Basal respiration, proton leak, and maximal respiration were determined (mean ± SD, n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). G and H, Mitochondrial membrane potential (Δψm) in adult mouse heart mitochondria was determined by FCM using Rhodamine123 probe (Rh123). G, Representative FCM blot. H, Histogram of the intensities of Rh123-stained mitochondria in the indicated groups (mean ± SD, n = 8 mice per group; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). I, Western blot analysis of mitochondrial import inner membrane translocase subunit 23 (TIM23), voltage-dependent anion channel 1 (VDAC1), calreticulin, and FUNDC1 protein levels in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− hearts. J, Densitometric analysis of the protein levels in I (mean ± SD, n = 5 mice per group; **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/−).
Fis1 is Essential for FUNDC1-Induced Mitochondrial Fission in Cardiomyocytes
To establish the role of FUNDC1 in mitochondrial dynamics, we used time-lapse confocal imaging to measure mitochondrial fusion and fission in live neonatal cardiomyocytes. Interestingly, we observed that mitochondrial fission occurred at a lower frequency in Fundc1f/Y/CreαMyHC+/− cardiomyocytes relative to control Fundc1wt/Y/CreαMyHC+/− cardiomyocytes (26 ± 4 vs. 12 ± 1 events/cell in 10 min, Figure 3A and 3B). The proportion of fission events [assay as the ratio of fission/(fission + fusion) events] also decreased in Fundc1f/Y/CreαMyHC+/− cardiomyocytes (Figure 3C). These data suggested that Fundc1 knockout resulted in decreased mitochondria fission.
Figure 3.
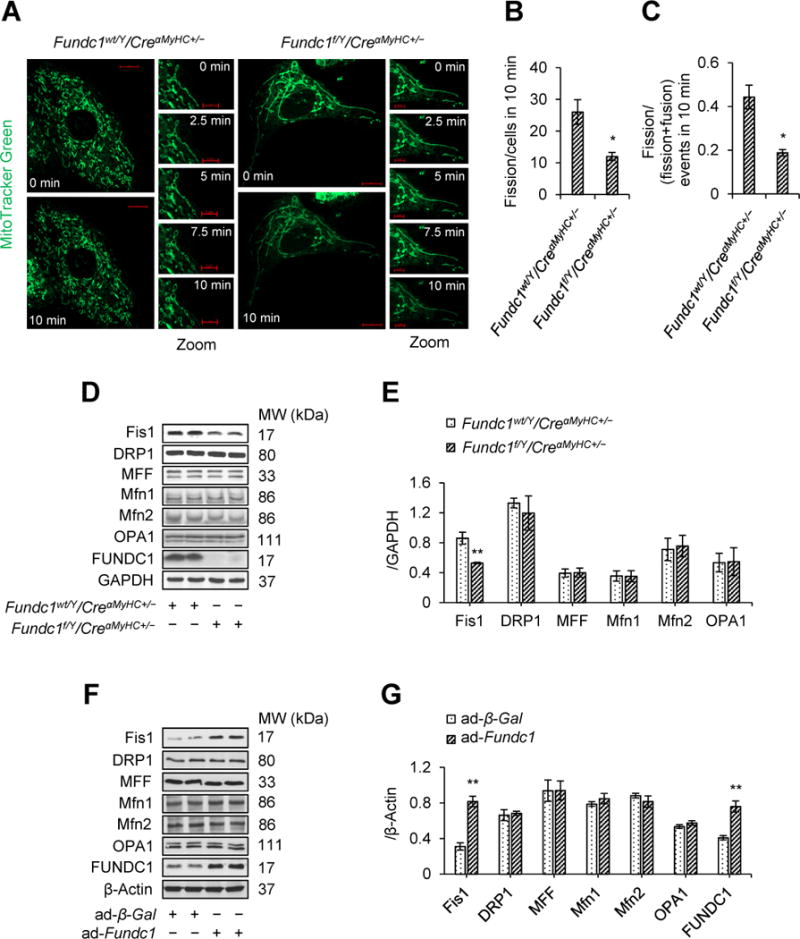
Deletion of Fundc1 decreases mitochondrial fission 1 protein (Fis1) protein level and inhibits mitochondrial fission. A-C, Mitochondrial morphology in live Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes stained with MitoTracker Red was captured using time-lapse confocal microscopy. Images were collected at 2.5 min intervals for 10 min. A, Representative time-lapse confocal images. Scale bars, 10 μm (whole cell images) and 5 μm (Zoomed images). Quantification of mitochondrial fission (B) and the ratio of fission and fission-plus-fusion events (C) in cells (mean ± SD, n = 5 independent experiments; 20 cells were quantified per group; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). D, Western blot analysis of Fis1, dynamin-related protein 1 (DRP1), mitochondrial fission factor (MFF), Optic atrophy 1 (OPA1), mitofusin 1/2 (Mfn1/2), and FUNDC1 protein levels in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− hearts. E, Densitometric analysis of the protein levels in D (mean ± SD, n = 5 mice per group, **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/−). F, The neonatal cardiomyocytes were infected with adenovirus encoding beta-galactosidase (ad-β-Gal) or FUNDC1 (ad-Fundc1) for 48 h. Protein levels of Fis1, DRP1, MFF, OPA1, Mfn1/2, and FUNDC1 were measured by western blot. G, Densitometric analysis of the protein levels in F (mean ± SD, n = 5 independent experiments; **P < 0.01 versus ad-β-Gal).
To gain insights into how FUNDC1 regulates mitochondrial dynamics, we investigated if FUNDC1 altered the expression of mitochondrial dynamics-related proteins in cardiomyocytes. Fundc1 knockout reduced Fis1 protein levels in the heart (Figure 3D and 3E). In contrast, adenoviral overexpression of Fundc1 in mouse neonatal cardiomyocytes increased Fis1 abundance (Figure 3F and 3G). Unexpectedly, neither overexpression nor ablation of Fundc1 affected the protein levels of DRP1, MFF, Mfn1/2, and OPA1 (Figure 3D through 3G). Similar results were also observed in cultured rat cardiac myoblasts (H9c2) with Fundc1 silencing (Supplemental Figure 4). These results suggest that FUNDC1 selectively regulates Fis1 in cardiomyocytes.
We next determined the contributions of Fis1 in FUNDC1-induced mitochondrial fission in H9c2 myoblasts. As depicted in Figure 4A and 4B, Fundc1 overexpression (GFP-FUNDC1) significantly increased mitochondrial fragmentation when compared to those transfected with green fluorescent protein (GFP) vectors. As expected, transfection with Fis1 siRNA but not control siRNA, markedly increased mitochondrial lengths and network in GFP-transfected cells, and prevented mitochondrial fragmentation caused by Fundc1 overexpression (Figure 4A and 4B). Furthermore, overexpression of Fis1 (GFP-Fis1) increased mitochondrial fragmentation in control siRNA-transfected cells and prevented mitochondrial elongation in Fundc1 siRNA-transfected cells (Figure 4C and 4D). Taken together, these data indicate that Fis1 mediates FUNDC1-induced mitochondrial fission.
Figure 4.
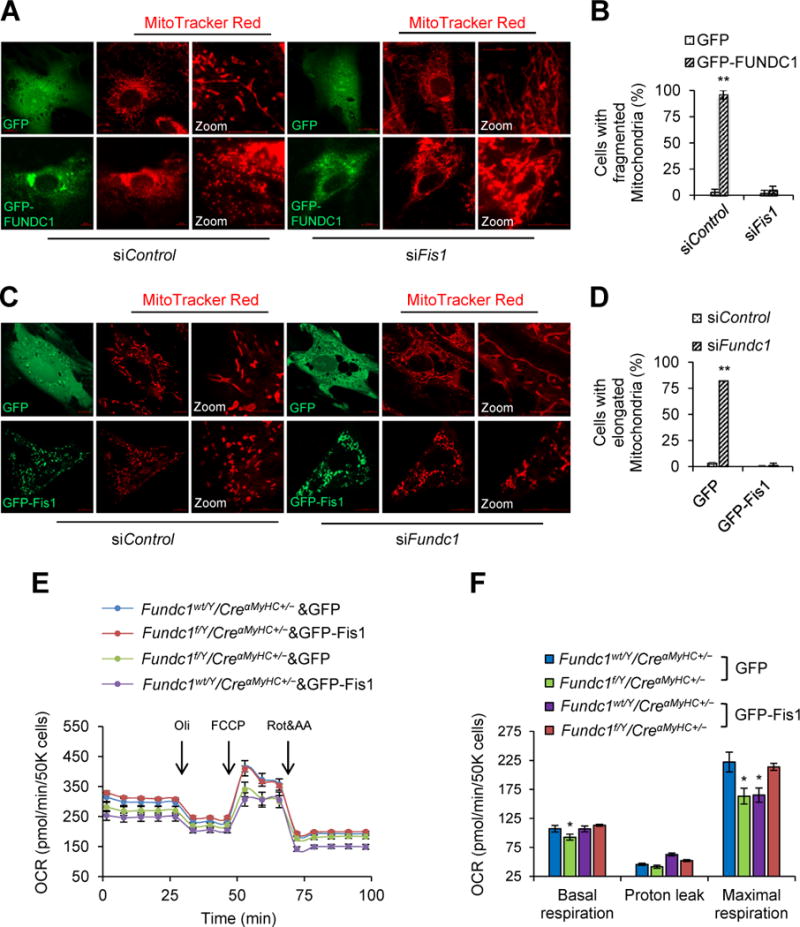
Mitochondrial fission 1 protein (Fis1) is essential for the induction of mitochondrial fission by FUNDC1. A, Representative confocal images of mitochondria in H9c2 myoblasts that were co-transfected with GFP-FUNDC1 plasmid and Fis1 siRNA (siFis1) for 48 h. A scramble siRNA (siControl) was used as gene silence control. Green fluorescent protein (GFP) vector plasmid was used as gene overexpression control. Mitochondria were labeled with MitoTracker Red. Scale bars, 10 μm. B, Quantification of the cells with fragmented mitochondria (mean ± SD, n = 5 independent experiments; 20 cells were quantified per group; **P < 0.01 versus GFP). C, Representative confocal images to show the mitochondrial network in Fundc1-silenced (siFundc1) H9c2 myoblasts with GFP or GFP-Fis1 overexpression. Mitochondria were labeled with MitoTracker Red. Scale bars, 10 μm. D, Quantification of the cells with elongated mitochondria (mean ± SD, n = 5 independent experiments; 20 cells were quantified per group; **P < 0.01 versus siControl). E, Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes were transfected with GFP-Fis1 or control GFP plasmid. Oxygen consumption rate (OCR) was determined using a Seahorse flux analyzer. F, Basal respiration, proton leak and maximal respiration were determined (mean ± SD, n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/− & GFP).
Since mitochondrial dynamics highly affects mitochondrial function, we sought to know whether or not Fis1 was involved in the impact of mitochondrial functional capacity by FUNDC1 in cardiomyocytes. As expected, overexpression of Fis1 (GFP-Fis1) (The transfection efficiency was shown in Supplemental Figure 5), but not GFP, in Fundc1f/Y/CreαMyHC+/− cells normalized oxygen consumption rate (OCR) (Figure 4E and 4F), suggesting that Fis1 mediates Fundc1 knockout-induced mitochondrial dysfunction.
Fis1 has been reported to recruit DRP1 into mitochondria, inducing mitochondrial fragmentation. To determine if DRP1 mediates FUNDC1-regulated mitochondrial dynamics, we first determined whether or not FUNDC1 physically interacts with DRP1. As depicted in Supplemental Figure 6A, FUNDC1 and DRP1 did not co-immunoprecipitate in H9c2 myoblasts. Similarly, FUNDC1 was absent in the anti-Fis1 immunoprecipitate (Supplemental Figure 6B). Further, there was no detectable interaction between DRP1 and Fis1 in Fundc1-overexpressing H9c2 myoblasts (Supplemental Figure 6C). Taken together, these data indicate that no physical interaction exists between FUNDC1, Fis1 and DRP1. DRP1 phosphorylation is important in its cytosol to mitochondrial translocation.31 To this end, we tested potential influence of FUNDC1 on DRP1 phosphorylation. Interestingly, Fundc1 overexpression failed to alter DRP1 phosphorylation at both serine 616 and serine 637 (Supplemental Figure 6D and 6E), two phosphorylation sites important for DRP1-mediated mitochondrial fission31. Consistently, Fundc1 overexpression did not alter the distributions of DRP1 in cytosolic and mitochondrial fractions (Supplemental Figure 6F).
Fis1 is Essential for FUNDC1-Induced Mitophagy in Cardiomyocytes
Since mitochondrial fission is essential for cells to undergo mitophagy32, 33 and FUNDC1 promoted mitochondrial fission, we next examined whether FUNDC1 mediates mitophagy in cardiomyocytes by assaying mt-Keima, a ratiometric pH-sensitive fluorescent protein that is targeted into the mitochondrial matrix34. The method was validated by showing that carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) enhanced mitophagy in cardiomyocytes (Supplemental Figure 7A and 7B). As described in Supplemental Figure 7C and 7D, adenoviral overexpression of Fundc1 but not β-Gal significantly increased the incidence of red mt-Keima puncta (lysosome-localized mitochondria), indicating that FUNDC1 promoted mitophagy. Importantly, Fis1 knockdown abolished enhanced mitophagy mediated by Fundc1 overexpression (Supplemental Figure 7C and 7D). Notably, FUNDC1-induced mitophagy could not be reduced by knockdown of Drp1 (Supplemental Figure 7E and 7F). To confirm this finding, we determined the co-localization of mitochondria and lysosomes. This approach was validated by showing that FCCP increased the co-localization of mitochondria and lysosomes (Supplemental Figure 8A and 8B). Consistently, Fundc1 overexpression increased co-localization of the two organelles while Fis1 silencing resulted in reduced co-localization, supporting that FUNDC1 promoted mitophagy and the mitophagy was abolished by silencing Fis1 (Supplemental Figure 8C and 8D). In addition, overexpression of Fundc1 but not β-Gal lowered the levels of mitochondrial proteins, TIM23 and VDAC1, without any effect on calreticulin (ER protein) levels in cardiomyocytes (Supplemental Figure 8E and 8F). Furthermore, silencing Fis1 prevented the reduction of mitochondrial proteins in Fundc1 overexpressing cardiomyocytes (Supplemental Figure 8E and 8F). Finally, in microtubule-associated protein 1A/1B-light chain 3 (Lc3)-deficient cells, Fundc1 overexpression could increase both Fis1 expression in cardiomyocytes (Supplemental Figure 9A and 9B) and increased mitochondrial fragmentation in H9c2 myoblasts (Supplemental Figure 9C and 9D). Taken together, these data indicate that Fis1 is required for FUNDC1-induced mitophagy.
FUNDC1 Regulates Fis1 at the Transcriptional Level by Activating CREB in a Ca2+ Dependent Manner
To elucidate how FUNDC1 regulates Fis1, we first examined if the ubiquitin-proteasome pathway was involved in regulating Fis1 protein turnover. To this end, mouse neonatal cardiomyocytes and H9c2 myoblasts were treated with MG132, a potent proteasome inhibitor. As shown in Supplemental Figure 10A and 10B, MG132 had no effect on Fis1 protein levels in cells. In addition, MG132 did not ablate the Fis1 reduction in Fundc1f/Y/CreαMyHC+/− cardiomyocytes (Supplemental Figure 10C) and Fundc1-silenced H9c2 myoblasts (Supplemental Figure 10D), indicating decreased level of Fis1 in Fundc1-deficient cells was not caused by increased degradation of Fis1. We next examined if FUNDC1 altered the levels of Fis1 messenger RNA (mRNA). As depicted in Supplemental Figure 11A, quantitative RT-PCR analysis showed Fis1 mRNA levels reduced significantly in Fundc1f/Y/CreαMyHC+/− hearts when compared to Fundc1wt/Y/CreαMyHC+/− hearts. Fundc1 ablation did not alter mRNA levels of Drp1, Mff, Mfn1/2, and Opa1 in heart (Supplemental Figure 11A). This indicates that FUNDC1 selectively regulates Fis1 expression at the transcriptional levels.
CREB is a ubiquitous transcription factor that can promote the expression of mitochondrial genes.35 Therefore, we investigated whether CREB was involved in FUNDC1-induced Fis1 expression. First, we examined whether FUNDC1 regulates CREB phosphorylation in heart, and found that Fundc1 deletion resulted in decreased phosphorylation of CREB at Ser133 (Supplemental Figure 11B and 11C). Conversely, the cardiomyocytes overexpressing Fundc1 (GFP-FUNDC1) exhibited higher level of phosphorylated CREB in the nucleus when compared to the control cells (GFP) (Supplemental Figure 11D and 11E), indicating that FUNDC1 promotes nuclear activation of CREB. It was necessary to further determine whether CREB is required for FUNDC1-induced Fis1 expression. As depicted in Supplemental Figure 11F through 11H, silencing of Creb reduced both Fis1 protein levels (Supplemental Figure 11F and 11G) and Fis1 mRNA levels (Supplemental Figure 11H) in cardiomyocytes. Furthermore, overexpressing of Fundc1 (ad-Fundc1) increased both Fis1 protein (Supplemental Figure 12A and 12B) and Fis1 mRNA levels (Supplemental Figure 12C) in the cardiomyocytes transfected with control siRNA but not in Creb siRNA transfected cells. Taken together, these data indicate that CREB is required for FUNDC1-induced Fis1 gene expression.
CREB activation is associated with the changes in intracellular Ca2+ levels [Ca2+]i. We therefore reasoned that FUNDC1 regulated CREB by alternating [Ca2+]i. To test this hypothesis, we pharmacologically modulated [Ca2+]i in Fundc1-ablated and Fundc1-overexpressed neonatal cardiomyocytes. BAPTA, which reduced [Ca2+]i levels, ablated Fundc1 overexpression-induced CREB phosphorylation and Fis1 expression (Supplemental Figure 12D and 12E). Conversely, increase of [Ca2+]i levels by thapsigargin (TG) prevented Fundc1 ablation-reduced phosphorylation of CREB and Fis1 expression (Supplemental Figure 12F and 12G). Neither TG nor BAPTA alone had effects on phosphorylated CREB levels and Fis1 levels in Fundc1wt/Y/CreαMyHC+/− cardiomyocytes. Overall, these results strongly indicated that FUNDC1 via the Ca2+/CREB pathway regulates Fis1 gene expression.
Next, we assayed how FUNDC1 promotes the activation of CREB. CREB activation is generally associated with the activation of protein kinase A (PKA). To test the role of PKA on the regulation of CREB by FUNDC1, we pharmacologically inhibited PKA activity in Fundc1-overexpressing cardiomyocytes with a PKA inhibitor H89. As depicted in Supplemental Figure 13, H89 did not affect CREB phosphorylation and Fis1 expression, suggesting a PKA-independent CREB activation.
Given that CREB is required by FUNDC1-mediated Fis1 gene expression, it was important to assay whether CREB directly induced Fis1 gene transcription. Analysis of the conserved motifs within promoters of Fis1 indicates that Fis1 promoter contains a putative CREB binding site at -477bp (Supplemental Figure 14A). We first assayed if overexpression of Creb enhances Fis1 expression in HEK293T cells. Western blot analysis showed that transfecting the cells with GFP-CREB plasmid increased Fis1 expression (Supplemental Figure 14B and 14C). Further, chromatin immunoprecipitation (ChIP) analysis showed that CREB bound to the Fis1 gene promoter in HEK293T cells (Supplemental Figure 14D and 14E). Importantly, these binding was significantly enhanced in cells overexpressing either GFP-CREB (Supplemental Figure 14D) or GFP-FUNDC1 (Supplemental Figure 14E). In contrast, silencing Fundc1 decreased the binding between CREB and Fis1 promoter (Supplemental Figure 14F).
To verify the specific binding location of CREB within Fis1 promoter, a 749-bp region from human Fis1 promoter that includes CREB binding site was cloned into the luciferase vector pGL-3, and then a serial titration of luciferase plasmids with different length of the promoter of human Fis1 was transfected into HEK293T cells (Supplemental Figure 14G). Two luciferase plasmids with -477bp site deletion were also generated, resulting in pGL–Fis1-749Del and pGL–Fis1-558Del. As shown in Supplemental Figure 14H, increased luciferase activity was evident in the cells transfected with the vectors containing putative binding site of CREB but not in the cells transfected with GFP. Furthermore, deletion of this site completely blocked the enhanced luciferase activity by Creb overexpression (Supplemental Figure 14H). Taken together, these results indicated that Ca2+-activated CREB binds to Fis1 promoter to initiate its transcription.
FUNDC1 Maintains MAMs, and Promotes Ca2+ Release from ER into Mitochondria
To look into how is the impact of FUNDC1 in intracellular calcium homeostasis, we evaluated the Ca2+ levels in mitochondria [Ca2+]m, cytosol [Ca2+]i, and ER [Ca2+]ER in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− cardiomyocytes. [Ca2+]m (Figure 5A and 5B) as well as [Ca2+]i (Figure 5C and 5D) were markedly lower in Fundc1f/Y/CreαMyHC+/− cardiomyocytes than those in Fundc1wt/Y/CreαMyHC+/− cells. In contrast, Fundc1f/Y/CreαMyHC+/− cells showed increased ER storage Ca2+ levels (Figure 5E and 5F). Next, we evaluated the levels of Ca2+ in Fundc1 overexpressing cells. As we expected, Fundc1 overexpression caused an increase of [Ca2+]m (Figure 5G and 5H), as well as [Ca2+]i (Figure 5I and 5J) in cardiomyocytes. We therefore hypothesized that Fundc1 ablation inhibited the Ca2+ release from the ER to mitochondria and cytosol.
Figure 5.
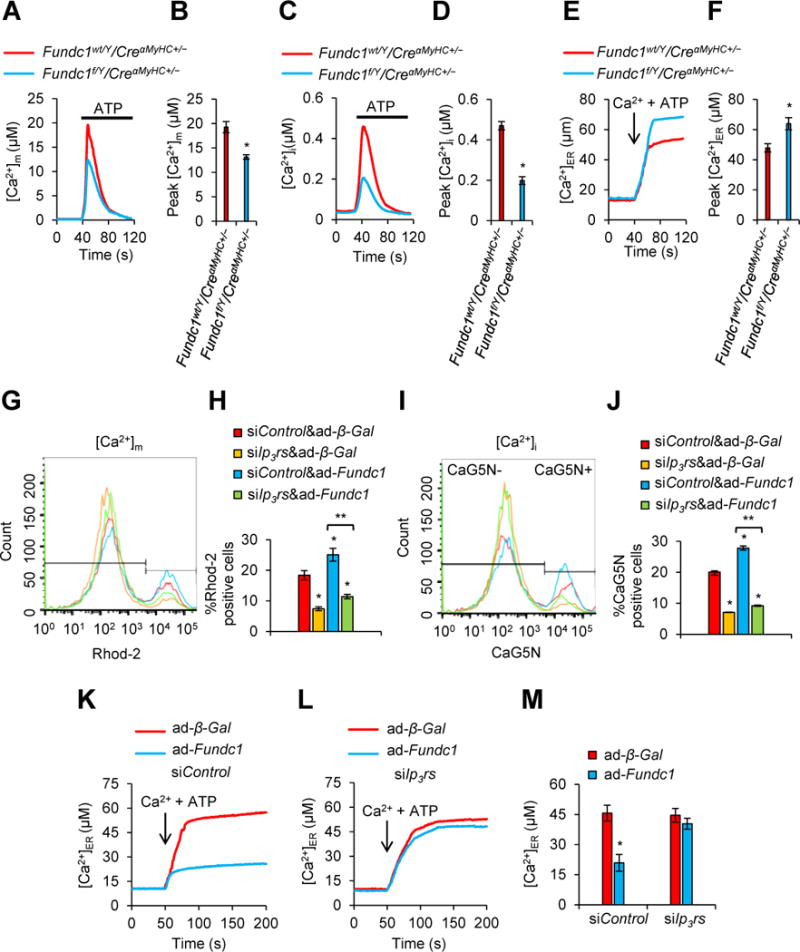
FUNDC1 promotes endoplasmic reticulum (ER) Ca2+ transfer to mitochondria and cytosol. A, Rhod-2 measurements of [Ca2+]m in response to ATP (0.1 mM) in Fundc1wt/Y/CreαMyHC+/− or Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes. B, Mean ± SD of [Ca2+]m from A (n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). C, Fura-2 measurements of cytosolic [Ca2+]i in response to ATP (0.1 mM) in Fundc1wt/Y/CreαMyHC+/− or Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes. D, Mean ± SD of [Ca2+]i from C (n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). E, Representative recordings of ER Ca2+ levels [Ca2+]ER during store filling initiated by addition of 100 nM Ca2+ and 1.5 mM ATP in Fundc1wt/Y/CreαMyHC+/− or Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes. F, Mean ± SD of [Ca2+]ER from E (n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). G-J, The neonatal cardiomyocytes overexpressing Fundc1 by infected with FUNDC1 adenovirus (ad-Fundc1) were transfected with three types of inositol 1,4,5-trisphosphate receptors (IP3Rs) siRNAs (siIp3r1, siIp3r2, and siIp3r3) for 48 h. A scramble siRNA (siControl) was used as gene silence control. Beta-galactosidase adenovirus (ad-β-Gal) was used as gene overexpression control. Flow cytometry (FCM) analysis were applied to detect resting [Ca2+]m and [Ca2+]i levels labeled with a florescence probe Rhod-2 and CaG5N, respectively in the cells. Representative FCM blots (G) and histogram (H) of the percentage of positive population of Rhod-2 cells in the indicated groups (mean ± SD, n = 5 independent experiments; *P < 0.05, **P < 0.01 versus siControl & ad-β-Gal). Representative FCM blots (I) and histogram (J) of the percentage of positive population of CaG5N cells in the indicated groups (mean ± SD, n = 5 independent experiments; *P < 0.05, **P < 0.01 versus siControl & ad-β-Gal). K-M, Effects of FUNDC1 on [Ca2+]ER. Representative recordings of [Ca2+]ER during store filling initiated by addition of 100 nM Ca2+ and 1.5 mM ATP in permeabilized cardiomyocytes infected with ad-β-Gal or ad-Fundc1 in the presence (K) or absence (L) of IP3Rs. Each trace represents mean of 15–20 cells in a single image field. M, Summary data showing the steady-state [Ca2+]ER after store filling (mean ± SD) for 40–100 cells pooled from at least 3 independent trials (mean ± SD, n = 5 independent experiments; *P < 0.05 versus siControl & ad-β-Gal).
Since IP3Rs are important ER Ca2+ release channels6, we assayed whether knockdown of Ip3rs affected FUNDC1-altered calcium homeostasis. As depicted in Figure 5G through 5J, in Fundc1 overexpressing cardiomyocytes, silencing of Ip3rs prevented the increase of both [Ca2+]m and [Ca2+]i, suggesting that Fundc1 overexpression enhanced [Ca2+]m and [Ca2+]i by promoting Ca2+ release from ER.
To further confirm the role of FUNDC1 on ER store Ca2+ release, we measured ER luminal Ca2+ levels in Ip3rs silencing cells. As shown in Figure 5K and 5M, [Ca2+]ER was markedly lower in Fundc1-overexpressing cardiomyocytes than those in control cardiomyocytes. Importantly, silencing of Ip3rs prevented Fundc1 overexpression-induced reduction of [Ca2+]ER in cardiomyocytes (Figure 5L and 5M). Taken together, these results indicate that FUNDC1 regulates Ca2+ release from ER to mitochondria and cytosol.
The regulation of calcium flux between the ER and mitochondria via IP3Rs is a major function of MAMs.36 Since FUNDC1’s localization in MAMs have been reported in HeLa cells29, 30, several steps were taken to determine if FUNDC1’s effects on intracellular calcium homeostasis in cardiomyocytes are dependent on MAMs formation. First, we detected FUNDC1 subcellular localization. As illustrated in Figure 6A and 6B, and Supplemental Figure 15, FUNDC1 co-localized with both mitochondria and ER, indicating that FUNDC1 is associated with MAMs, which was further verified by subcellular fractionation (Figure 6C). To test whether FUNDC1 can affect MAMs integrity, we examined the ER-mitochondrial contacts in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− cardiomyocytes. In Fundc1wt/Y/CreαMyHC+/− cardiomyocytes, the ER appeared as an interconnected network of cisternae spanning the entire cellular volume (Figure 6D; Supplemental Figure 16). In Fundc1f/Y/CreαMyHC+/− cardiomyocytes, the ER became aggregated and swollen, whereas mitochondria became longer and more interconnected (Figure 6D; Supplemental Figure 16). Notably, Fundc1f/Y/CreαMyHC+/− cardiomyocytes clearly showed a very low level of co-localization between ER and mitochondria when compared to Fundc1wt/Y/CreαMyHC+/− cardiomyocytes, as revealed by significantly decreased Pearson’s correlation coefficient between organelle-targeted fluorescent markers (Figure 6E). Consistently, Fundc1 silencing reduced co-localization of ER and mitochondria in H9c2 myoblasts (Supplemental Figure 17). We also used transmission electron microscopy (TEM) to determine the alterations in MAMs formation. We observed that Fundc1 deficiency reduced the association between ER and mitochondria in heart (Figure 6F). Quantitative analysis showed that the proportion of the ER adjacent to mitochondria was significantly reduced in Fundc1f/Y/CreαMyHC+/− heart (Figure 6G). Additionally, there is no changes on ER stress markers expression in Fundc1f/Y/CreαMyHC+/− hearts, although Fundc1 ablation strongly affects ER structure (Supplemental Figure 18). Since the changes in the MAMs related protein levels such as IP3Rs and phosphofurin acidic cluster sorting protein 2 (PACS-2) in both whole cell lysates and MAMs fractions are commonly used as an indication of enhanced or suppressed MAMs formation37, 38, we next determined if Fundc1 alteration affected IP3R2 and PACS2 in cardiomyocytes. Fundc1 deletion markedly reduced the abundance of IP3R2 and PACS2, without any effect on other ER proteins protein disulfide isomerase (PDI) and Sig1R, in both whole cell lysates (Supplemental Figure 19A and 19B) and MAMs fraction (Figure 6H and 6I). We did not find any change in the mRNA levels of Ip3rs and Pacs2 (Supplemental Figure 19C). Finally, to find out the molecular basis of how FUNDC1 maintains MAMs integrity, we found that FUNDC1 was co-immunoprecipitated with IP3R2 in Fundc1wt/Y/CreαMyHC+/− hearts and the interaction was abolished in Fundc1f/Y/CreαMyHC+/− hearts (Figure 6J), implying that FUNDC1 may form a protein bridge with IP3R2 to tether ER and mitochondria. There is no association between FUNDC1 and PACS-2 in hearts (Supplemental Figure 19D). Taken together, FUNDC1 regulates MAMs formation and function in cardiomyocytes.
Figure 6.
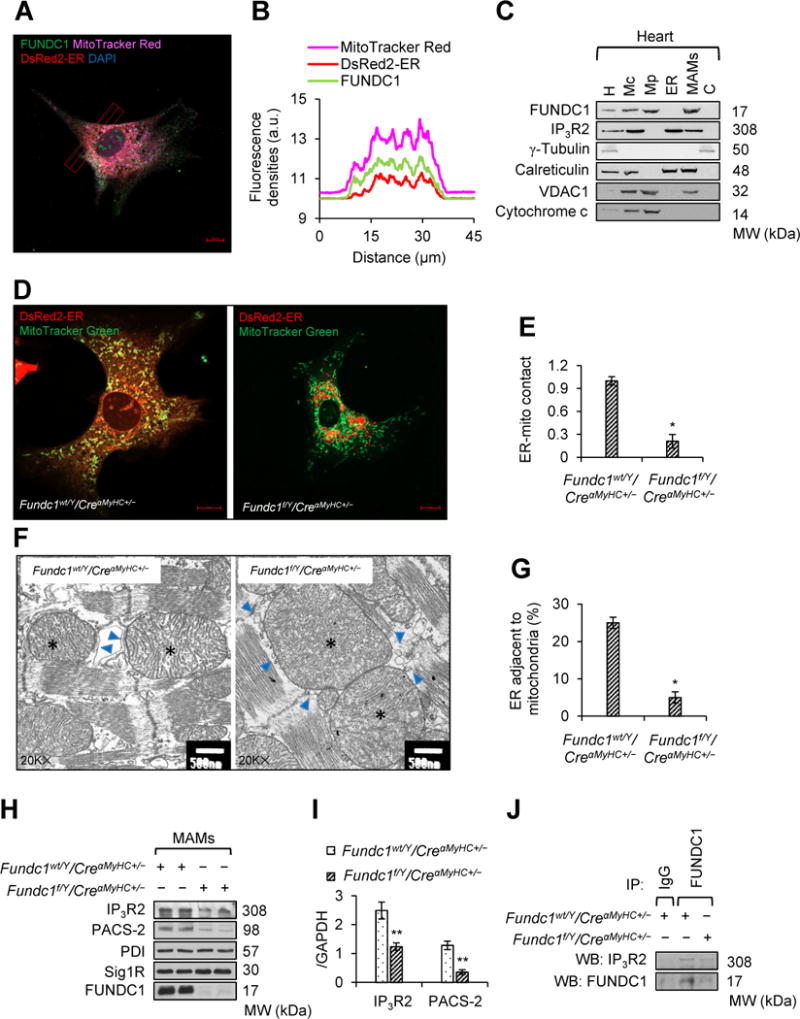
FUNDC1 mediates mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) formation. A, Co-localization of FUNDC1 with mitochondria and ER. Mitochondria was labeled with MitoTracker Red; ER was labeled with pDsRed2-ER; FUNDC1 was indicated by anti-FUNDC1; Nucleus was stained by DAPI. Representative confocal microscopy image is shown. The images of each single channel were given in Supplemental Figure 15. Scale bar, 10 μm. B, Traces of fluorescence intensity spatial profiles through the red line shown in A for MitoTracker Red, DsRed2-ER, and FUNDC1 fluorescence. C, Western blot analysis of the protein levels of FUNDC1 in homogenates (H), as well as crude mitochondria (Mc), pure mitochondria (Mp), MAMs, ER, and cytosolic (C) fractions from mouse hearts. Calreticulin was used as an ER marker, voltage dependent anion channel 1 (VDAC1) was used as a mitochondrial marker, and inositol 1,4,5-trisphosphate type 2 receptor (IP3R2) was used as a MAMs component (n = 5 mice per group). D, Representative confocal images showing the association between ER (DsRed2-ER) and mitochondria (MitoTracker Red) in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− neonatal cardiomyocytes (Scale bars, 10 μm; n = 5 mice per group). The images of each single channel were given in Supplemental Figure 16. E, Pearson’s coefficient, calculated by ZEN software, indicates the contact of ER and mitochondria (mean ± SD, n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). F, Representative transmission electron microscopy (TEM) images indicating the ER and mitochondrial contacts in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− hearts. Asterisks indicate mitochondria; Arrows indicate ER. Scale bars, 500 nm. G, Quantification of the ER adjacent to mitochondria in the heart was normalized by total ER length (mean ± SD, n = 5 independent experiments; *P < 0.05 versus Fundc1wt/Y/CreαMyHC+/−). H, Cardiac MAMs fractions were prepared from Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− mice. Protein levels of IP3R2, phosphofurin acidic cluster sorting protein 2 (PACS-2), protein disulfide isomerase (PDI), and sigma-1 receptor (Sig1R) in the homogenates were assayed by western blot. I, Densitometric analysis of the protein levels in H (mean ± SD, n = 5 independent experiments; **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/−). J, The interaction between FUNDC1 and IP3R2 in Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− hearts was determined by immunoprecipitation (IP) and western blot (WB) (n = 5 mice per group).
Acute Myocardial Infarction Exacerbates Heart Failure Phenotype in Cardiac Fundc1 Knockout Mice
Since we found that the heart failure phenotype by Fundc1 ablation became apparent in 10-week-old mice, and this phenotype became more obvious with the age increased. However, these mice at the age of 8 weeks appeared to have normal cardiac functions, as shown by echocardiography (Supplemental Table 1). We therefore sought to know whether heart attack challenge could exacerbate the heart failure phenotype in Fundc1-ablated mice. To this end, we subjected 8-week-old Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− mice mice to acute myocardial infarction (MI) induced by left anterior descending coronary artery ligation. Four weeks after MI, cardiac function, cardiac fibrosis, and survival rate were examined in each group. Echocardiography revealed that after MI, Fundc1f/Y/CreαMyHC+/− mice had significant increase in left ventricular internal diameters (LVID) at end-diastole and end-systole compared with Fundc1f/Y/CreαMyHC+/− mice received sham treatment (Figure 7A; Supplemental Table 2), indicating that left ventricular dilatation was severer in Fundc1f/Y/CreαMyHC+/− after MI. Moreover, the EF (Figure 7B) and FS (Figure 7C) were significantly lower in Fundc1f/Y/CreαMHC+/− mice treated with MI than those treated with sham. In addition, the fibrosis area, as determined by Masson’s trichrome staining (Figure 7D and 7E), was significantly higher in MI-treated Fundc1f/Y/CreαMyHC+/− mice than in sham-treated Fundc1f/Y/CreαMyHC+/− mice, indicative of an aggravation in myocardial scarring by MI in Fundc1 deficient mice. Additionally, the Fundc1f/Y/CreαMyHC+/− mice died at day 4 after MI challenge, and the mortality was up to 62.5% 4 weeks after MI (Figure 7F). Overall, our results indicate that heart attack challenge exacerbates the heart failure phenotype in Fundc1-ablated mice.
Figure 7.
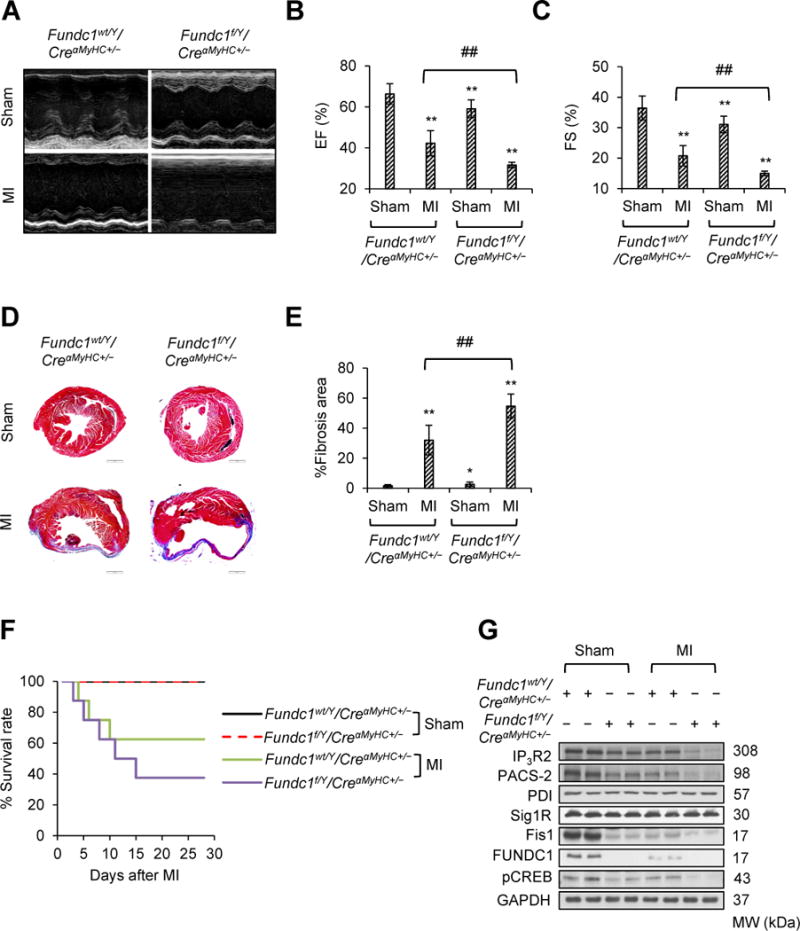
Acute myocardial infarction (MI) exacerbates cardiac dysfunction in cardiac Fundc1 knockout mice. Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− mice were subjected to left anterior descending coronary artery ligation (MI) or sham surgery. Four weeks after surgery, cardiac structure and function were evaluated. A, Representative images of M-mode echocardiograms of Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− mice subjected to MI or sham operation. The ejection fraction (EF) (B) and fractional shortening (FS) (C) are shown for each group (n = 8 mice per group, **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/− & Sham; ##P < 0.01 versus indicated group). D, Representative images show Masson’s trichrome staining of the cardiac tissues. Scale bars, 1 mm. E, Quantitative analyses of the left ventricular fibrotic area in each group (n = 8 mice per group, *P < 0.05, **P < 0.01 versus Fundc1wt/Y/CreαMyHC+/− & Sham; ##P < 0.01 versus indicated group). F, Kaplan-Meier curves of Fundc1wt/Y/CreαMyHC+/− and Fundc1f/Y/CreαMyHC+/− mice subjected to MI or sham operation (n = 8 mice per group). G, Protein levels of FUNDC1, phosphorylated cAMP response element binding protein (pCREB), inositol 1,4,5-trisphosphate type 2 receptor (IP3R2), phosphofurin acidic cluster sorting protein 2 (PACS-2), protein disulfide isomerase (PDI), and sigma-1 receptor (Sig1R), and mitochondrial fission 1 protein (Fis1) in hearts were assayed by western blot.
We next explored whether the exacerbation of heart failure was correlated with the further downregulation of FUNDC1/MAMs/CREB/Fis1 signaling by MI treatment. To this end, we examined the levels of FUNDC1/MAMs/CREB/Fis1 signaling in each group. The MI reduced FUNDC1 protein levels, concurrent with a reduction in MAMs-related proteins (IP3R2 and PACS-2), phosphorylated CREB, and Fis1 (Figure 7G). In Fundc1f/Y/CreαMyHC+/− mice, like we demonstrated above, there was a clear reduction in MAMs-related proteins (IP3R2 and PACS-2), phosphorylated CREB, and Fis1 (Figure 7G). More importantly, MI induced a further decrease in protein levels of IP3R2, PACS-2, pCREB, and Fis1 in Fundc1f/Y/CreαMyHC+/− mice (Figure 7G). Overall, our results indicate that MI exacerbates heart failure phenotype in cardiac Fundc1-ablated mice likely via its suppression of MAMs formation.
Downregulation of FUNDC1/MAMs/CREB/Fis1 Signaling in Human Heart Failure Patients
To further establish if FUNDC1 reduction occurred in human hearts as well as its contributions in heart failure in patients, four heart specimens from heart failure patients and four from control donors were examined. As depicted in Figure 8A and 8B, concurrent with a reduction in MAMs-related proteins including IP3R1 and inositol 1,4,5-trisphosphate type 3 receptor (IP3R3), the levels of FUNDC1, phosphorylated CREB, and Fis1 were significantly reduced in patients with heart failure compared to control donors. As expected, there was a reduced association of ER and mitochondria in failed hearts (Figure 8C). The proportion of the ER adjacent to mitochondria was significantly reduced in heart failure patients (Figure 8D). Compared with the ovoid structure of mitochondria in donor hearts, mitochondria in heart failure hearts were more elongated (Figure 8C and 8E). Taken together, reduced FUNDC1 is linked to reduce IP3Rs, decreased CREB/Fis1 signaling, elongated mitochondria, and reduced contact between ER and mitochondria.
Figure 8.
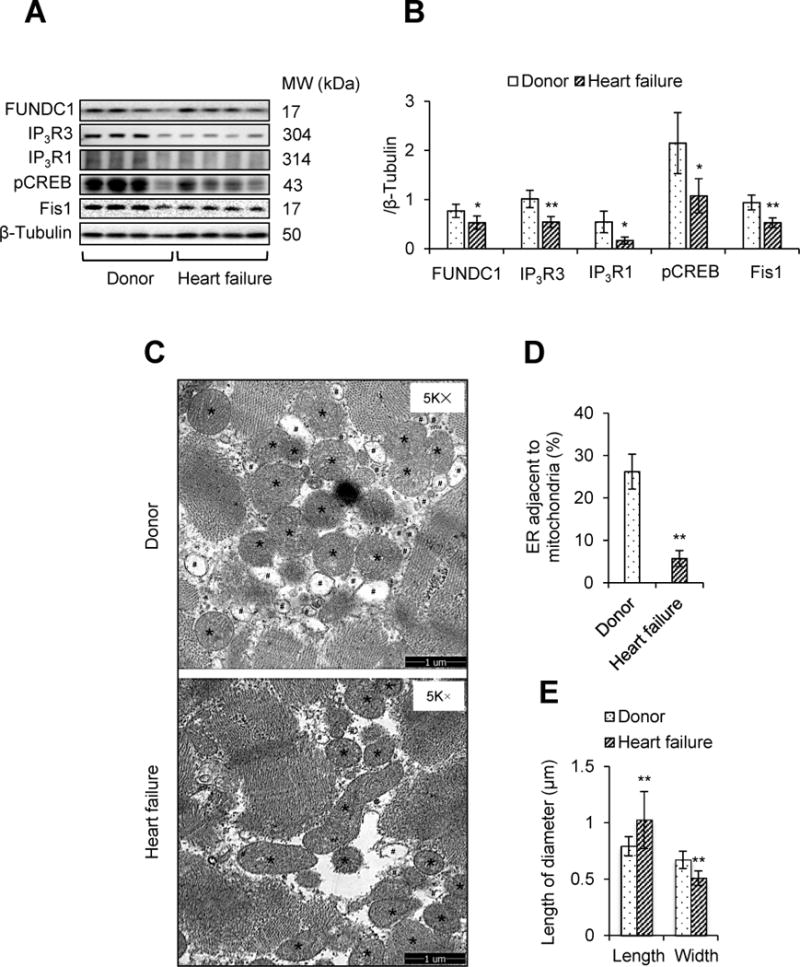
Downregulation of FUNDC1/MAMs/CREB/Fis1 signaling in human heart failure patients. A, The protein levels of FUNDC1, inositol 1, 4, 5-trisphosphate type 1 receptor (IP3R1), inositol 1, 4, 5-trisphosphate type 3 receptor (IP3R3), phosphorylated cAMP response element binding protein (pCREB), and mitochondrial fission 1 protein (Fis1) in human heart specimens from donor or heart failure patients were determined by western blot. B, Densitometric analysis of the protein levels in A (mean ± SD, n = 4; *P < 0.05, **P < 0.01 versus donor). C, Representative transmission electron microscopy (TEM) images indicating the endoplasmic reticulum (ER) and mitochondrial contacts in the human hearts of donors and heart failure patients. Asterisks indicate mitochondria; Pound signs indicate ER. Scale bars, 1 μm D, Quantification of the ER adjacent to mitochondria in the heart was normalized by total ER perimeter (mean ± SD, n = 4; **P < 0.01 versus donor). E, Mitochondrial length and width (mean ± SD, n = 4; **P < 0.01 versus donor).
Discussion
In the present study, we have for the first time found that MAMs-localized FUNDC1 binds to IP3R2 to mediate IP3Rs-dependent Ca2+ release from ER into both mitochondria and cytosol in cardiomyocytes. This reduction of intracellular Ca2+ levels in Fundc1-deficient cells suppresses the Ca2+-sensitive CREB-mediated Fis1 expression, disrupts mitochondrial fission, and causes mitochondrial dysfunction, all of which instigate cardiac dysfunction and heart failure.
MAMs is reported to be important in maintain calcium homeostasis but how MAMs regulates calcium homeostasis remains poorly characterized.37 In this study, we have identified that MAMs-localized FUNDC1 binds to IP3R2 to modulate Ca2+ release capacity of IP3R2 in cardiomyocytes and a disruption of FUNDC1-IP3R2 in MAMs promotes heart failure. The evidence can be summarized as follows: First, FUNDC1 is required for MAMs formation. Ablation of Fundc1 results in decrease of MAMs proteins abundance, along with dissociation between mitochondria and ER. Second, FUNDC1 physically interacts with IP3R2 in MAMs. Further, the interaction between FUNDC1 and IP3R2 provides a protein-protein bridge, which not only tether ER and mitochondria but also regulate IP3R2 stability. Third, FUNDC1 via IP3R2 controls MAMs-related calcium distribution/functions. Manipulation of Fundc1 expression by genetic manipulation shows that Fundc1 expression is strongly associated with mitochondrial Ca2+ flux and mitochondrial functional capacity. Consistently, we found that ER store Ca2+ levels increased in Fundc1 knockout cells while it decreased in Fundc1 overexpressed cells. Further, the decrease in ER store Ca2+ levels by Fundc1 overexpression were prevented by Ip3rs knockdown, indicating that the regulation of ER store Ca2+ by FUNDC1 is dependent of IP3Rs. Fourth, cardiomyocyte-specific deletion of Fundc1 impairs cardiac functions, associated with mitochondrial elongation and accumulation of dysfunctional mitochondria; Fifth, acute MI reduced Fundc1 expression and exacerbated the heart failure phenotype in Fundc1 knockout mice. Finally, Fundc1 expression was decreased in hearts from the patients who were diagnosed with dilated cardiomyopathy. Concomitantly, the contact between ER and mitochondria was largely reduced, along with the reduction in MAMs proteins, and the CREB/Fis1 signaling. Taken together, our results support a key role of FUNDC1 in maintaining normal mitochondrial function and morphology in cardiomyocytes and a reduction of FUNDC1 instigates cardiac dysfunction and heart failure.
Mitochondrial Ca2+ signaling plays an important role in controlling cell metabolism.39 For example, transient fluctuations in mitochondrial Ca2+ activate three matrix dehydrogenases and oxidative phosphorylation, stimulating ATP production, suggesting that maintaining a proper mitochondrial Ca2+ concentration is essential for mitochondrial and metabolic homeostasis. Our findings of Fundc1 deletion inhibits mitochondrial Ca2+ uptake and impairs mitochondrial respiration was consistent with earlier reports. For example, mitochondrial calcium uniporter regulator 1 (MCUR1) is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism.40 In both HeLa and HEK293T cells, stable or transient knockdown of Mcur1 reduced basal OCR, reflecting diminished oxidative phosphorylation.40 Further, deletion of all three isoforms of IP3Rs diminishes mitochondrial Ca2+ uptake, leading to compromised mitochondrial function.41 Of note, basal OCR were lower by 60% in IP3Rs-knockout cells compared with WT cells. Maximal OCR was also highly reduced in Fundc1-deficient cells.41 However, our result is not consistent with the report in Mfn2 (a MAMs structural protein) knockout cells showing inhibited mitochondrial Ca2+ uptake and but increased intracellular Ca2+ levels.4 In Mfn2 knockout cells4, the mitochondrial Ca2+ uptake was inhibited due to the disassociation of ER and mitochondria, but without any effect on ER Ca2+ release capacity, and therefore intracellular Ca2+ levels was increased. The precise molecular mechanism of how FUNDC1 regulates IP3Rs Ca2+ release channel activity warrants further investigation.
Another important finding of the present study is that we have for the first time uncovered a novel pathway by which increased MAMs activates a Ca2+-sensitive CREB transcription factor to directly bind the promoter of Fis1 resulting in its gene transcription. In our context, Ca2+ serves as a mediator to facilitate FUNDC1 to induce fission and mitophagy processes, suggesting a remarkable pathway that occurs in Ca2+-sensitized organs, particularly in cardiac muscle. The changes in intracellular Ca2+ concentration serve as a retrograde signal that activates a nuclear transcription factor CREB, which can directly regulate Fis1 transcription. Our evidence was summarized as follows: First, we observed that Fis1 expression was positively regulated by FUNDC1, without any effect on the protein levels of other mitochondrial dynamic machinery proteins. Second, genetic studies indicate that FUNDC1 and Fis1 work on the same pathway to induce mitochondrial fission, and FUNDC1 acts upstream of Fis1 because overexpression of Fis1 in Fundc1-deficient cells rescued the Fundc1 mutant phenotype while overexpression of Fundc1 failed to do so in Fis1-deficient cells. Finally, we have found that Fundc1 overexpression could not increase Fis1 expression in CREB-deficient cells. Our findings strongly support the notion that MAMs serve as an important signaling hub regulating mitochondrial dynamics, not only by providing potential fission site that has been previously reported38, 42, but also regulating dynamics-related gene expression at the transcriptional levels. Another important finding of our study is that FUNDC1-upregulated Fis1 facilitates mitophagy. Indeed, overexpression of Fundc1 increased mitochondrial fragmentation, accompanied by increased mitophagy. Inhibition of mitochondrial fission by gene silencing of Fis1 abolished the mitophagy in the cells overexpressing Fundc1. Our findings suggest that FUNDC1 induces mitophagy not only by acting as mitophagy receptor but also by enhancing mitochondrial fragmentation.
Several studies suggested CREB as an important regulator of gene expression involved in the pathophysiology of heart failure.43–46 However, CREB in cardiomyocyte mitochondrial dynamics has not been studied yet. We here reported that the phosphorylation levels of CREB at the site of Ser133 were significantly decreased in hearts of cardiac-specific Fundc1 knockout mice, along with elongated mitochondria and typical cardiac dysfunction. CHIP assay further indicates that CREB binds to the promoter sequence of Fis1, the expression of which is important for mitochondrial dynamics in cardiomyocytes47. With a luciferase assay method, we further observed that CREB increased Fis1 promoter activity. These results implicate that CREB was involved in the pathophysiology of disordered mitochondrial dynamics-associated cardiomyopathy by regulating mitochondrial dynamic machinery gene Fis1 expression. The finding of CREB as a transcription factor for Fis1, which largely extends the knowledge on how Fis1 expression is regulated. Wang et al. showed that Fis1 protein level was negatively regulated by miR-484, which could be trans-activated by forkhead box O3 (FoxO3a).47 In our experimental system, we used a silencer to decrease FoxO3a, but we could not see any change in Fis1 expression (data not shown). Another work identified Fis1’s proteasomal degradation by DJ-1 signaling in neuron by activating a RNF5 E3-ligase.48 However, we could not observe any change in Fis1 expression with different doses of MG132, a specific proteasome inhibitor, even after extended incubation time of 24 hours.
Recent study in HeLa cells shows that Ca2+ controls mitochondrial dynamics by regulating DRP1 phosphorylation and mitochondrial translocation via activating calcium/calmodulin-dependent kinase II α (CaMKIα).49 However, DRP1 appears not to be required for FUNDC1-induced mitochondrial fission in cardiomyocytes. First, there is no physical interaction between FUNDC1 and DRP1 in cardiomyocytes. Second, overexpression of Fundc1 failed to alter neither DRP1 protein expression nor DRP1 phosphorylation. Although Fundc1 overexpression dramatically increased Fis1 protein levels, it failed to promote the mitochondrial translocation of DRP1 as well as the interaction between DRP1 and Fis1. These results suggest that Fis1 works on mitochondrial dynamics in a DRP1-independent manner in cardiomyocyte. Consistent with our findings, there is no detectable interaction between purified DRP1 and hFis1.50, 51 Increased or reduced levels of hFis1 do not alter mitochondrial translocation of DRP1 in HeLa cells.52 Since Fis1 is not a GTPase and cannot directly drive mitochondrial fission, there should be some additional protein(s) work together with Fis1 to accomplish mitochondrial fission process. A recent study showed that mammalian Fis1 recruits the GTPase regulator domain-containing protein TBC1D15 and functions in mitochondrial morphology regulation independent of DRP1.53 However, we did not find any association between Fis1 and TBC1D15 in cardiomyocyte. Dynamin II is a fundamental component of the mitochondria which is recently defined to recruit to mitochondrial fission sites to facilitate fission process54, it is likely that Fis1 may function as the receptor of dynamin II. The molecular function of Fis1 in mammalian mitochondrial morphology regulation warrants further investigation.
In summary, in this study we found that Fundc1 reduction inhibits the integrity of MAMs and the MAMs-localized FUNDC1 regulates calcium concentrations in mitochondria and cytosol by regulating the levels and functions of IP3R2. The reduction of FUNDC1 in MAMs results in lower [Ca2+]m and [Ca2+]i, which, in turn, leads to inhibition of the CREB/Fis1 pathway, elongation of mitochondria, impaired mitochondrial function, and consequent cardiac dysfunction in the heart (Supplemental Figure 20). We conclude that FUNDC1 is essential in maintaining functional mitochondria and normal cardiac function. Deregulated FUNDC1 in cardiomyocytes via abnormal MAMs plays a causative role in the development of heart disease.
Supplementary Material
Clinical Perspective.
- What is new?
- For the first time, we demonstrate that a reduction of FUNDC1, a mitochondria-localized protein, instigates cardiac dysfunction and exacerbates cardiac dysfunction and remodeling in ischemic insults.
- In cardiomyocytes, FUNDC1 binds to IP3R2 to form MAMs, which modulates Ca2+ release from ER into mitochondria and cytosols. Fundc1 deletion lowers the levels of IP3R2 and Ca2+ in both mitochondria and cytosols.
- A reduction of intracellular Ca2+ results in mitochondrial fusion, mitochondrial dysfunction, cardiac dysfunction and heart failure by suppressing Ca2+-sensitive CREB-mediated Fis1 expression.
- What are the clinical implications?
- The FUNDC1-mediated MAMs formation was significantly suppressed in the patients with heart failure, supporting that FUNDC1 and MAMs actively participate in the development of heart failure.
- Restoring proper MAMs function may be a novel target for treating heart failure.
Acknowledgments
Sources of Funding
This study was supported by funding from the following agencies: National Heart Lung and Blood Institute (HL079584, HL080499, HL089920, HL110488, HL128014, HL132500, and HL137371), National Cancer Institute (CA213022), National Institute on Aging (AG047776), and American Heart Association (AHA) (16GRANT29590003). Dr. Zou is the Eminent Scholar in Molecular and Translational Medicine of Georgia Research Alliance. SW was supported by American Heart Association (AHA) postdoctoral fellowship award (16POST29960011). Dr. Kai Huang is supported in part by the Ministry of Science and Technology of China grant 2016 YFA0101100.
Abbreviations
- AA
Antimycin A
- ANP
Atrial natriuretic peptide
- BNP
Brain natriuretic peptide
- CaG5N
Calcium green-5N
- CaMKIα
Calcium/calmodulin-dependent kinase II α
- ChIP
Chromatin immunoprecipitation assay
- CREB
The cAMP response element binding protein
- DRP1
Dynamin-related protein 1
- DT
Deceleration time
- EF
Ejection fraction
- ER
Endoplasmic reticulum
- FCCP
Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- FCM
Flow cytometry
- Fis1
Mitochondrial fission 1 protein
- FoxO3a
Forkhead box O3
- FS
Fractional shortening
- FUNDC1
FUN14 domain containing 1
- GFP
Green fluorescent protein
- Grp75
Glucose regulated protein 75
- IP3R1
Inositol 1,4,5-triphosphate type 1 receptor
- IP3R2
Inositol 1,4,5-trisphosphate type 2 receptor
- IP3R3
Inositol 1,4,5-trisphosphate type 3 receptor
- IP3Rs
Inositol 1,4,5-trisphosphate receptors
- IVRT
Isovolumic relaxation times
- LAD
Left anterior descending coronary artery
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- LVID
Left ventricle internal diameter
- MAMs
Mitochondria-associated endoplasmic reticulum (ER) membranes
- MCUR1
Mitochondrial calcium uniporter regulator 1
- MFF
Mitochondrial fission factor
- Mfn1/2
Mitofusin 1/2
- MI
Myocardial infarction
- OCR
Oxygen consumption rate
- Oli
Oligomycin
- OPA1
Optic atrophy 1
- PACS-2
Phosphofurin acidic cluster sorting protein 2
- PDI
Protein disulfide isomerase
- PKA
Protein kinase A
- Rh123
Rhodamine123
- Rot
Rotenone
- SD
Standard deviation
- Sig1R
Sigma-1 receptor
- TEM
Transmission electron microscopy
- TG
Thapsigargin
- TIM23
Mitochondrial import inner membrane translocase subunit 23
- TUNEL
Terminal-deoxynucleoitidyl transferase mediated nick end labeling
- VDAC1
The voltage-dependent anion channel 1
- β-Gal
Beta-galactosidase
- β-MyHC
Beta myosin-heavy chain
Footnotes
Author contributions
MHZ and ZX conceived the project. MHZ, ZX, and SW designed the study and wrote manuscript. SW conducted confocal microscopy imaging, oxygen consumption rate measurement, flow cytometry analysis, immunoprecipitation and western blot. QL conducted cardiac function measurement and ChIP assay, and established myocardial infarction animal model. YD and ZM performed immunochemistry staining. XM and KH performed human studies. All authors have approved the manuscript.
Disclosures
None.
References
- 1.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marriott KS, Prasad M, Thapliyal V, Bose HS. Sigma-1 receptor at the mitochondrial-associated endoplasmic reticulum membrane is responsible for mitochondrial metabolic regulation. J Pharmacol Exp Ther. 2012;343:578–586. doi: 10.1124/jpet.112.198168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 5.Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A. 2015;112:E2174–2181. doi: 10.1073/pnas.1504880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 7.Fonseca AC, Moreira PI, Oliveira CR, Cardoso SM, Pinton P, Pereira CF. Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol Neurobiol. 2015;51:610–622. doi: 10.1007/s12035-014-8740-7. [DOI] [PubMed] [Google Scholar]
- 8.Cali T, Ottolini D, Negro A, Brini M. Alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287:17914–17929. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol. 2014;30:357–391. doi: 10.1146/annurev-cellbio-101011-155756. [DOI] [PubMed] [Google Scholar]
- 10.Lemieux H, Hoppel CL. Mitochondria in the human heart. J Bioenerg Biomembr. 2009;41:99–106. doi: 10.1007/s10863-009-9211-0. [DOI] [PubMed] [Google Scholar]
- 11.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–555. doi: 10.1172/JCI24405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 13.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW., 2nd Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012;111:1012–1026. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parra V, Verdejo H, del Campo A, Pennanen C, Kuzmicic J, Iglewski M, Hill JA, Rothermel BA, Lavandero S. The complex interplay between mitochondrial dynamics and cardiac metabolism. J Bioenerg Biomembr. 2011;43:47–51. doi: 10.1007/s10863-011-9332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuznetsov AV, Hermann M, Saks V, Hengster P, Margreiter R. The cell-type specificity of mitochondrial dynamics. Int J Biochem Cell Biol. 2009;41:1928–1939. doi: 10.1016/j.biocel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Archer SL. Mitochondrial dynamics — mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 18.Dorn GW., 2nd Mitochondrial dynamics in heart disease. Biochim Biophys Acta. 2013;1833:233–241. doi: 10.1016/j.bbamcr.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ong SB, Hall AR, Hausenloy DJ. Mitochondrial dynamics in cardiovascular health and disease. Antioxid Redox Signal. 2013;19:400–414. doi: 10.1089/ars.2012.4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. 2009;41:1846–1854. doi: 10.1016/j.biocel.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pennanen C, Parra V, Lopez-Crisosto C, Morales PE, Del Campo A, Gutierrez T, Rivera-Mejias P, Kuzmicic J, Chiong M, Zorzano A, Rothermel BA, Lavandero S. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J Cell Sci. 2014;127:2659–2671. doi: 10.1242/jcs.139394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lackner LL. Shaping the dynamic mitochondrial network. BMC Biol. 2014;12:35. doi: 10.1186/1741-7007-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 29.Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12:689–702. doi: 10.1080/15548627.2016.1151580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, Zhuang H, Zhang X, Chen H, Li S, Yang Y, Lu Y, Wang J, Zhu R, Zhang L, Sui S, Tan N, Zhao B, Zhang J, Li L, Feng D. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35:1368–1384. doi: 10.15252/embj.201593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang CR, Blackstone C. Drp1 phosphorylation and mitochondrial regulation. EMBO Rep. 2007;8:1088–1089. doi: 10.1038/sj.embor.7401118. author reply 1089–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. 2013;26:9–18. doi: 10.1016/j.devcel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi: 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi: 10.1016/j.chembiol.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 35.Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, Baba Y, Zhou Y, Kaufman BA, Cheung JY, Kurosaki T, Gill DL, Madesh M. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci Signal. 2015;8:ra23. doi: 10.1126/scisignal.2005673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal. 2015;22:995–1019. doi: 10.1089/ars.2014.6223. [DOI] [PubMed] [Google Scholar]
- 38.Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol. 2011;21:709–717. doi: 10.1016/j.tcb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 40.Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest. 1998;101:2415–2426. doi: 10.1172/JCI2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller FU, Boknik P, Horst A, Knapp J, Linck B, Schmitz W, Vahlensieck U, Bohm M, Deng MC, Scheld HH. cAMP response element binding protein is expressed and phosphorylated in the human heart. Circulation. 1995;92:2041–2043. doi: 10.1161/01.CIR.92.8.2041. [DOI] [PubMed] [Google Scholar]
- 45.Muller FU, Boknik P, Knapp J, Luss H, Neumann J, Vahlensieck U, Bohm M, Deng MC, Scheld HH, Schmitz W. Quantification of the cAMP response element binding protein in ventricular nuclear protein from failing and nonfailing human hearts. Biochem Biophys Res Commun. 1997;236:351–354. doi: 10.1006/bbrc.1997.6971. [DOI] [PubMed] [Google Scholar]
- 46.Muller FU, Neumann J, Schmitz W. Transcriptional regulation by cAMP in the heart. Mol Cell Biochem. 2000;212:11–17. doi: 10.1023/A:1007176030884. [DOI] [PubMed] [Google Scholar]
- 47.Wang K, Long B, Jiao JQ, Wang JX, Liu JP, Li Q, Li PF. miR-484 regulates mitochondrial network through targeting Fis1. Nat Commun. 2012;3:781. doi: 10.1038/ncomms1770. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q, Wu J, Wu R, Ma J, Du G, Jiao R, Tian Y, Zheng Z, Yuan Z. DJ-1 promotes the proteasomal degradation of Fis1: implications of DJ-1 in neuronal protection. Biochem J. 2012;447:261–269. doi: 10.1042/BJ20120598. [DOI] [PubMed] [Google Scholar]
- 49.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117:1201–1210. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- 51.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onoue K, Jofuku A, Ban-Ishihara R, Ishihara T, Maeda M, Koshiba T, Itoh T, Fukuda M, Otera H, Oka T, Takano H, Mizushima N, Mihara K, Ishihara N. Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci. 2013;126:176–185. doi: 10.1242/jcs.111211. [DOI] [PubMed] [Google Scholar]
- 54.Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. 2016;540:139–143. doi: 10.1038/nature20555. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.