

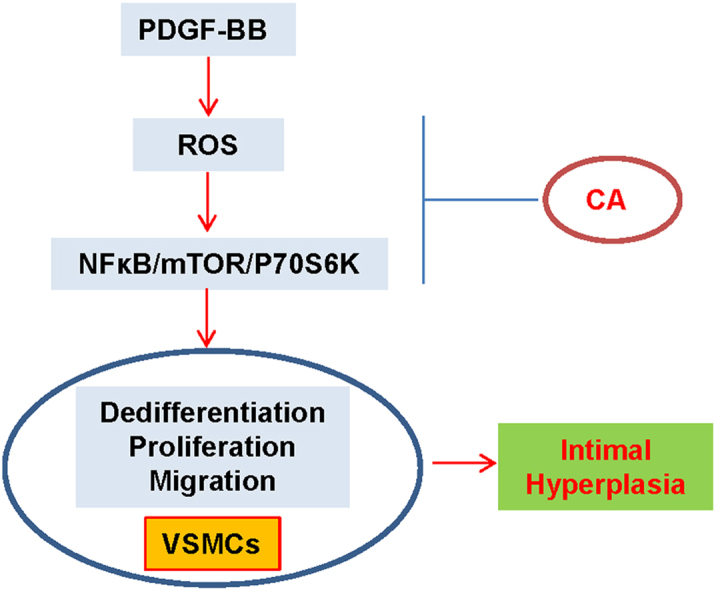
Abstract
Phenotypic switch of vascular smooth muscle cells (VSMCs) is characterized by increased expressions of VSMC synthetic markers and decreased levels of VSMC contractile markers, which is an important step for VSMC proliferation and migration during the development and progression of cardiovascular diseases including atherosclerosis. Chicoric acid (CA) is identified to exert powerful cardiovascular protective effects. However, little is known about the effects of CA on VSMC biology. Herein, in cultured VSMCs, we showed that pretreatment with CA dose-dependently suppressed platelet-derived growth factor type BB (PDGF-BB)-induced VSMC phenotypic alteration, proliferation and migration. Mechanistically, PDGF-BB-treated VSMCs exhibited higher mammalian target of rapamycin (mTOR) and P70S6K phosphorylation, which was attenuated by CA pretreatment, diphenyleneiodonium chloride (DPI), reactive oxygen species (ROS) scavenger N-acetyl-l-cysteine (NAC) and nuclear factor-κB (NFκB) inhibitor Bay117082. PDGF-BB-triggered ROS production and p65-NFκB activation were inhibited by CA. In addition, both NAC and DPI abolished PDGF-BB-evoked p65-NFκB nuclear translocation, phosphorylation and degradation of Inhibitor κBα (IκBα). Of note, blockade of ROS/NFκB/mTOR/P70S6K signaling cascade prevented PDGF-BB-evoked VSMC phenotypic transformation, proliferation and migration. CA treatment prevented intimal hyperplasia and vascular remodeling in rat models of carotid artery ligation in vivo. These results suggest that CA impedes PDGF-BB-induced VSMC phenotypic switching, proliferation, migration and neointima formation via inhibition of ROS/NFκB/mTOR/P70S6K signaling cascade.
Keywords: VSMCs, Chicoric acid, PDGF-BB, Proliferation, Migration
Graphical abstract
Highlights
-
•
Chicoric acid attenuated PDGF-BB-evoked VSMC phenotypic transformation, proliferation and migration.
-
•
Chicoric acid antagonized the activated ROS/NFκB/mTOR/P70S6K signaling pathway in VSMCs.
-
•
Chicoric acid treatment prevented intimal hyperplasia in rat models of carotid artery ligation.
1. Introduction
In mature and normal blood vessels, vascular smooth muscle cells (VSMCs) are characterized to be a highly quiescent and contractile phenotype associated with elevated levels of contractile markers proteins such as α-smooth muscle actin (α-SMA), SM22α and smooth muscle myosin heavy chain (SMMHC) [1], [2]. In atherosclerosis and arterial restenosis, VSMCs can switch to be a dedifferentiated, proliferative, and migratory phenotype by downregulating gene expressions of VSMC contractile markers and upregulating synthetic protein expressions of osteopontin (OPN) [3], [4]. Accumulating evidence indicates that VSMC phenotypic switching is widely observed in atherosclerosis, intimal hyperplasia, hypertension and postangioplasty restenosis [5], [6]. Unraveling the potential mechanisms of VSMC phenotypic switching may provide novel therapeutic target for the prevention and treatment of these diseases.
The aberrant VSMC proliferation and migration are core events in the pathophysiology of many cardiovascular diseases including atherosclerosis and restenosis after angioplasty [5]. The VSMC phenotypic switching is closely linked with excessive proliferation and migration of VSMCs, which is believed to a common vascular pathological condition in atherosclerosis, restenosis and vein bypass graft failure [3]. Therapeutic strategies against VSMC phenotypic switching, proliferation and migration may be beneficial for VSMC-related pathological conditions. In response to vascular injury, the activated inflammatory cells, platelets and VSMCs release the growth factors, especially platelet-derived growth factor (PDGF), thereby leading to a switch of VSMCs from a contractile phenotype to a synthetic phenotype [7], [8]. PDGF family is composed of five proteins including PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC and PDGF-DD [9], among which, PDGF-BB is described to be one of the most potent stimulants for VSMC proliferation and migration [10]. Therefore, PDGF-BB was utilized to induce VSMC dedifferentiation in this study.
Chicoric acid (CA) is isolated and purified from plant and vegetables, which is reported to possess antioxidant and anti-inflammatory activities [11], [12]. CA is established to be a new potential antidiabetic agent by stimulating insulin secretion [13]. CA functions as a regulator of cellular apoptosis, growth, differentiation, and immune response [14], [15], [16]. A recent study demonstrates that CA is a potent anti-atherosclerotic ingredient via attenuating oxidized low-density lipoprotein (oxLDL)-facilitated endothelial dysfunction [11]. However, no investigations were conducted concerning the effects of CA on the phenotypic switch, proliferation and migration of VSMCs. Therefore, this study was designed to explore the roles and molecular mechanisms of CA in the regulation of VSMC physiology.
2. Material and methods
2.1. Chemicals
Dulbecco's modified Eagle’s medium (DMEM), trypsin-EDTA, and fetal bovine serum (FBS) were obtained from Gibco BRL (Carlsbad, CA, USA). Recombinant human PDGF-BB was purchased from R&D Systems (Minneapolis, MN, USA). Chicoric acid (CA), rapamycin, diphenyleneiodonium chloride (DPI) and dhydroethidium (DHE) were bought from Sigma (St. Louis, MO, USA). Cell counting kit-8 (CCK-8) kits, NFκB inhibitor BAY 11-7082 and N-acetyl-lcysteine (NAC) were obtained from Beyotime Institute of Biotechnology (Shanghai, China). 5-Ethynyl-2′-deoxyuridine (EdU) Apollo kits were purchased from RiboBio (Guangzhou, China). Mitochondria-targeted antioxidant mitoquinone was purchased from Suzhou Vosun Chemical (Jiangsu, China) [17]. The transwell system was obtained from Corning (Corning, Inc., Cypress, CA). Antibodies against α-SMA, SM22α, PCNA, cyclin D1, P27 and horseradish peroxidase conjugated secondary antibodies were purchased from Proteintech Group, Inc (Wuhan, China). Antibodies against p65-NFκB, IκBα, and phosphor-IκBα were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against total or phosphorylated mTOR and P70S6K, SMMHC, Lamin B1, NOX2, p22phox, p47phox and goat anti-rabbit IgG H&L (Alexa Fluor® 488) were purchased from Abcam (Cambridge, MA, USA). The specific primers were synthesized by SangonBiotech Co.,Ltd. (Shanghai, China). The dose of inhibitors was selected according to previous studies and our preliminary studies [11], [14], [16], [18], [19], [20], [21], [22], [23], [24].
2.2. Animals
Male Sprague-Dawley rats (Vital River Biological, Beijing, China) were used in our experiments. All experiments were conformed to the rules and regulations of the Experimental Animal Care and Use Committee of Jiangnan University. All procedures were complied with the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH publication, 8th edition, 2011). The rats were housed on 12:12-h light–dark cycle in a temperature-controlled and humidity-controlled room, with free access to standard chow and tap water.
2.3. Cell culture
Human aortic VSMCs (FuDan IBS Cell Center, Shanghai, China) were in DMEM with 10% FBS and 1% penicillin/streptomycin in humidified air containing 5% CO2 under a condition at 37 °C. Cells grown to 80–90% confluence were passaged at a ratio of 1:3. The cells used in all experiments were passaged three to five times.
2.4. CCK-8 assay and EdU assay
The proliferation of VSMCs was detected by using a CCK-8 kit or EdU incorporation assay as our previous reports [25], [26], [27], [28]. For CCK-8 assay, the optical density was read by a microplate reader (SYNERGY H4, BioTek,Winooski, VT, USA) at a wavelength of 490 nm. For EdU analysis, the nuclear DNA was counterstained using Hoechst 33342, and the EdU positive images were captured by a fluorescence microscopy (80i, Nikon, Japan).
2.5. VSMC migration analysis
The migration assay was performed using a transwell chamber. Briefly, 8-μm pore size chambers with transparent polyester were placed into 24-well plates. After 24 h of incubation, non-migrated VSMCs were removed by cotton swabs. Migrated cells were fixed and stained with crystal violet (0.1%). The cells on bottom side of the membrane were counted under a phase-contrast microscope (80i, Nikon, Japan).
2.6. Western Blot
The total or nuclear proteins were obtained with the aid of commercially available kits (Beyotime Biotechnology, Shanghai, China) in accordance to the manufacturer’s protocol [20], [29]. Equal amount of proteins were electrophoresed, blotted, and then incubated with required primary antibodies at 4 °C overnight. The blots were then incubated with appropriate secondary horseradish peroxidase (HRP)-conjugated antibodies, the immunoreactive proteins were visualized by enhanced chemiluminescence (Millipore Darmstadt, Germany) [20].
2.7. Real-time PCR
Total RNA was extracted using Trizol reagent according to the manufacturer’s instructions. The equal RNA was used to generate cDNA using HiScriptQ RT SuperMix for qPCR (Vazyme, Nanjing, China). The real-time quantitative PCR was performed in triplicates by using ChamQTM SYBR® qPCR Master Mix (Vazyme, Nanjing, China). The average cycle thresholds (Ct) were employed to quantify fold-change. The 2-△△CT method was reported to calculate relative gene expression levels. The primer sequences for α-SMA: 5′-TTGAGAAGAGTTACGAGTTG-3′ (Forward), 5′-AGGACATTGTTAGCATAGAG-3′ (Reverse); SM22α: 5′-ACCCACCCTCCATGGTCTTC-3′ (Forward), 5′-CTTATGCTCCTGCGCTTTCT-3′ (Reverse); SMMHC: 5′-AAGAAACACAGACCAGGCGT-3′ (Forward), 5′-GGGTGAGTATCCAGCGGAAA-3′ (Reverse); OPN: 5′-CTGCCAGCAACCGAAGTTT-3′ (Forward), 5′-ACCATTCAACTCCTCGCTTTC 3-3′ (Reverse); GAPDH: 5′-CCACATCGCTCAGACACCAT-3′ (Forward), 5′-CCAGGCGCCCAATACG-3′ (Reverse).
2.8. Immunofluorescence staining
After fixed with 4% paraformaldehyde for 30 min, the stimulated VSMCs were permeabilized with 0.1% Triton X-100 in PBS for 15 min. Cells were blocked with 5% BSA for 1 h at room temperature, and then incubated with indicated primary antibodies at 4 °C overnight. After three washes with PBS, cells were detected with goat anti-rabbit IgG H&L Alexa Fluor® 488. Finally, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. Images were photographed by a fluorescence microscope (80i, Nikon, Tokyo, Japan).
2.9. Intracellular ROS measurement
The collected VSMCs were fixed and incubated with DHE (10 μM) 20 min in a dark and humidified container at 37 °C. The fluorescence signals were captured and quantified with the IMAGE-PRO PLUS 6.0 (Version 6.0, Media Cybernetics, Bethesda, MD, USA) by using the same parameters [20], [26].
2.10. Measurement of superoxide dismutase (SOD) and catalase (CAT) activities
The activities of SOD and CAT were determined by a Total Superoxide Dismutase Assay Kit with NBT (Beyotime Biotechnology, Shanghai, China) and a Catalase Assay Kit (Beyotime Biotechnology, Shanghai, China) following the manufacturer's instructions as previous reports [30], [31]. The activity was found as U/mg protein and the results were expressed as percentage of control.
2.11. Determination of malondialdehyde (MDA)
The levels of MDA were measured using commercial assay kits (Beyotime Biotechnology, Shanghai, China) according to the instructions provided by the manufacturer as previously described [32], [33].
2.12. Animal model of vascular injury
The unilateral carotid artery ligation was applied to mimic vascular injury in 8-week-old rats as previous reports [20], [34]. In short, the left common carotid arteries were ligated with a 6.0 silk suture that was proximal to the carotid bifurcation for 8 weeks, and the uninjured right carotid arteries served as control. Three days after vascular injury, CA (50 mg/kg/day) was administered gastric gavage to rats for a total of 8 weeks as previously described [35], [36], [37], [38]. At the end of experiments, the rats were sacrificed by overdose of pentobarbitalsodium (150 mg/kg, iv), and carotid arteries were harvested for hematoxylin and eosin (HE) staining and molecular biological analyses [39], [40].
2.13. Statistical analysis
All results were defined as mean ± S.E. Comparisons within two groups were made by Student′s t-test. Statistical analysis was performed by ANOVA/Dunnet t-test for multiple group comparisons. The criterion for statistical significance was set at p < 0.05.
3. Results
3.1. CA abrogated PDGF-BB-induced VSMC dedifferentiation
To test whether CA affected PDGF-BB-induced VSMC dedifferentiation, VSMCs were pretreated with different doses of CA for 6 h followed by stimulation with PDGF-BB for 24 h. The results showed that PDGF-BB promoted the VSMC phenotypic alteration from differentiated to dedifferentiated cells, as evidenced by increased VSMC synthetic gene OPN along with decreased VSMC contractile genes α-SMA, SMMHC, and SM22α at both protein (Fig. 1A and B) and mRNA levels (Fig. 1C). Pretreatment with CA antagonized VSMC dedifferentiation response to PDGF-BB in a dose-dependent manner (Fig. 1).
Fig. 1.

CA abrogated PDGF-BB-induced VSMC dedifferentiation. VSMCs were pretreated with various concentrations (10, 50 and 100 μM) of CA for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) Western blot was employed to quantitate the expression levels of contractile protein α-SMA, SMMHC, SM22α and synthetic proteins OPN. (B) Bar graph showing the relative protein level of α-SMA, SMMHC, SM22α and OPN. (C) Bar graph showing the relative mRNA level ofα-SMA, SMMHC, SM22α and OPN. Values are mean±SE. * P < 0.05 vs. Control, † P < 0.05 vs. PDGF-BB. n = 6 for each group.
3.2. CA abated PDGF-BB-induced VSMC proliferation and migration
Since VSMC dedifferentiation plays an essential role in proliferation and migration of VSMCs [4], we next decided to examine whether CA abrogated VSMC proliferation and migration in response to PDGF-BB. Treatment of VSMCs with PDGF-BB resulted in the increased proliferation and migration of VSMCs. However, CA pretreatment dose-dependently counteracted PDGF-BB-induced proliferation and migration of VSMCs, as evidenced by EdU incorporation assay (Fig. 2A and C), transwell migration assay (Fig. 2B and D) and absorbance assay (Fig. S1A). The cellular proliferation markers such as PCNA, cyclin D1 and P27 are considered to be involved in VSMC proliferation and migration [41], [42]. Consistently, pretreatment with CA concentration-relatedly mitigated the upregulated PCNA and cyclin D1 protein levels, as well as the downregulated P27 protein expression in PDGF-BB-incubated VSMCs (Fig. 2E and F).
Fig. 2.

CA abated PDGF-BB-induced VSMC proliferation and migration. VSMCs were pretreated with various concentrations (10, 50 and 100 μM) of CA for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) DNA synthesis in VSMCs determined with EdU incorporation assay. Blue fluorescence (Hoechst 33342) shows cell nuclei and green fluorescence (Edu) stands for cells with DNA synthesis. (B) Transwell assay was performed to determine the migration of VSMCs. (C) The ratio of EdU-positive cells to total cells. (D) Bar graph showing the number of migrated VSMCs. (E) Represented images showing the protein expressions of PCNA, cyclin D1 and P27. (F) Bar graph showing the relative protein level of PCNA, cyclin D1 and P27. Values are mean±SE. * P < 0.05 vs. Control, † P < 0.05 vs. PDGF-BB. n = 6 for each group.
3.3. CA inhibited PDGF-BB-induced VSMC dedifferentiation, proliferation and migration via suppressing mTOR/P70S6K signaling
Activation of mTOR/P70S6K signaling pathway participates in VSMC dedifferentiation, proliferation and migration [21]. EdU incorporation assay (Fig. 3A and C), CCK-8 assay (Fig. S1B) and transwell migration assay (Fig. 3B and D) results showed that blockade of mTOR/P70S6K with rapamycin markedly attenuated PDGF-BB-induced VSMC proliferation and migration. Meanwhile, rapamycin pretreatment obviously reversed PDGF-BB-triggered VSMC phenotypic alteration from differentiated to dedifferentiated cells by upregulating α-SMA protein level (Fig. 3E and F) and downregulating OPN protein expression (Fig. 3E and G). Furthermore, the abnormal changes in cellular proliferation markers including PCNA (Fig. 3E and H), cyclin D1 (Fig. 3E and I) and P27 (Fig. 3E and J) were all rectified by rapamycin pretreatment in VSMCs response to PDGF-BB. More importantly, western blot analysis illustrated that the increased P-mTOR and P-P70S6K protein levels in PDGF-BB-induced VSMCs were prevented by CA pretreatment (Fig. 3K). These data suggested that CA may inhibit mTOR/P70S6K signaling pathway to ameliorate PDGF-BB-evoked VSMC dedifferentiation, proliferation and migration.
Fig. 3.

CA inhibited PDGF-BB-induced VSMC dedifferentiation, proliferation and migration via suppressing mTOR/P70S6K signaling. VSMCs were pretreated with rapamycin (100 nM) for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) EdU assay. (B) Transwell assay was performed to determine the migration of VSMCs. (C) The ratio of EdU-positive cells to total cells. (D) Bar graph showing the number of migrated VSMCs. (E) Represented images showing the protein expressions of α-SMA, OPN, PCNA, cyclin D1 and P27. The relative protein expressions of α-SMA (F), OPN (G), PCNA (H), cyclin D1 (I) and P27 (J) were quantified. (K) VSMCs were pretreated with various concentrations (10, 50 and 100 μM) of CA for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. The phosphorylated and total mTOR and P70S6K protein levels were measure by western blot. Values are mean±SE. * P < 0.05 vs. Control + Vehicle (Veh) or Control, † P < 0.05 vs. PDGF-BB + Vehicle (Veh) or PDGF-BB. n = 6 for each group.
3.4. CA retarded PDGF-BB-induced VSMC dedifferentiation, proliferation and migration via suppressing NFκB signaling
NFκB is a critical signaling in VSMC dedifferentiation, proliferation and migration [20], [43]. Our results exhibited that pretreatment with Bay 11-7082, an inhibitor of NFκB, attenuated the increased EdU positive cells (Fig. 4A and C), migrated cells (Fig. 4B and D) and absorbance (Fig. S1C) in PDGF-BB-treated VSMCs. The decreased α-SMA protein level (Fig. 4E and F) and increased OPN protein expression (Fig. 4E and G) in PDGF-BB-treated VSMCs were normalized by Bay 11-7082 pretreatment. Furthermore, the proliferating markers including PCNA (Fig. 4E and H), cyclin D1 (Fig. 4E and I) and P27 (Fig. 4E and J) were restored by Bay 11-7082 pretreatment in PDGF-BB-incubated VSMCs. These results further verified that NFκB signaling extremely involved in PDGF-BB-induced VSMC biological effects. Therefore, we hypothesized that CA may impede NFκB signaling to restrain PDGF-BB-induced VSMC dedifferentiation, proliferation and migration. Our results showed that PDGF-BB promoted the translocation of p65-NFκB from cytoplasm to nucleus (Fig. 5A–D), accompanied with phosphorylation and degradation of IκBα in VSMCs (Fig. 5E and F), which were all antagonized by CA pretreatment. Immunofluorescence results further revealed that CA eliminated PDGF-BB-induced translocation of p65 of NFκB (Fig. 5G). Interestingly enough, inhibition of NFκB with Bay 11-7082 compromised activated mTOR/P70S6K signaling in VSMCs response to PDGF-BB (Fig. 5H). These data disclosed that NFκB-mediated mTOR/P70S6K signaling partially contributed to PDGF-BB-evoked VSMC dedifferentiation, proliferation and migration. Pretreatment with CA abated NFκB/mTOR/P70S6K signaling to abolish PDGF-BB-induced VSMC biological effects.
Fig. 4.

Bay117082 retarded PDGF-BB-induced VSMC dedifferentiation, proliferation and migration. VSMCs were pretreated with Bay117082 (10 μM) for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) EdU assay. (B) Transwell assay was performed to determine the migration of VSMCs. (C) The ratio of EdU-positive cells to total cells. (D) Bar graph showing the number of migrated VSMCs. (E) Represented images showing the protein expressions of α-SMA, OPN, PCNA, cyclin D1 and P27. The relative protein expressions of α-SMA (F), OPN (G), PCNA (H), cyclin D1 (I) and P27 (J) were quantified. Values are mean ± SE. * P < 0.05 vs. Control + Vehicle (Veh), † P < 0.05 vs. PDGF-BB + Vehicle (Veh). n = 6 for each group.
Fig. 5.

CA retarded PDGF-BB-induced NFκB signaling activation. VSMCs were pretreated with various concentrations (10, 50 and 100 μM) of CA for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) Represented images showing the protein expressions of p65-NFκB in nucleus. (B) Represented images showing the protein expressions of p65-NFκB in cytoplasm. (C) Bar graph showing the relative protein expressions of p65-NFκB in nucleus. (D) Bar graph showing the relative protein expressions of p65-NFκB in cytoplasm. (E) Represented images showing the protein expressions of IκBα and phosphorylated IκBα. (F) Bar graph showing the relative protein expressions of IκBα and phosphorylated IκBα. (G) The translocation of p65-NFκB from cytoplasm to nucleus was measure by immunofluorescence, white arrow showing the nuclear localization of p65-NFκB. (H) VSMCs were pretreated with Bay117082 (10 μM) for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. The phosphorylated and total mTOR and P70S6K protein levels were measure by western blot. Values are mean ± SE. * P < 0.05 vs. Control or Control + Vehicle (Veh), † P < 0.05 vs. PDGF-BB or PDGF-BB + Vehicle (Veh). n = 6 for each group.
3.5. CA prevented PDGF-BB-stimulated activation of ROS/NFκB/mTOR/P70S6K signaling cascade
Incubation of VSMCs with PDGF-BB increased the production of superoxide anions (Fig. 6A and B), upregulated NAD(P)H oxidase subunits p22phox (Fig. 6C and D), p47phox (Fig. 6C and E), NOX-2 (Fig. 6C and F) protein levels, and these changes were prevented by CA pretreatment (Fig. 6). It is particularly worthy noting that NAC (ROS scavenger, Fig. S2) and DPI (an inhibitor of flavincontaining enzyme including NADPH oxidase, Fig. S3) obviously blocked PDGF-BB-induced p65-NFκB nuclear translocation, phosphorylation and degradation of IκBα, as well as mTOR/P70S6K signaling activation in VSMCs. Moreover, compared with control cells, the SOD and CAT activities were obviously decreased, whereas the content of MDA was markedly increased in PDGF-BB-challenged VSMCs, and these changes were largely rectified by CA pretreatment (Fig. S4).
Fig. 6.

CA prevented PDGF-BB-induced ROS production in VSMCs. VSMCs were pretreated with various concentrations (10, 50 and 100 μM) of CA for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) ROS generation was evaluated by DHE fluorescence. (B) Relative ROS fluorescence intensity. (C) Represented images showing the protein expressions of NAD(P)H oxidase subunits p22phox, p47phox, NOX-2. The relative protein expressions of NAD(P)H oxidase subunits p22phox (D), p47phox (E), NOX-2 (F) were quantified. Values are mean ± SE. * P < 0.05 vs. Control, † P < 0.05 vs. PDGF-BB. n = 6 for each group.
DPI is taken as an unspecific inhibitor of flavoprotein-containing ROS producing enzymes, including NADPH oxidases or complexes I, III, and IV in the mitochondrial respiratory chain, which substantially blunts the intracellular ROS deposition [44], [45]. However, whether NAD(P)H oxidases or mitochondria contributed to PDGF-BB-stimulated ROS load in VSMCs remains uncertain in the present study. In the next step, we delineated the endogenous ROS source, which is crucial for PDGF-BB-induced VSMC biological function. Mitoquinone is a mitochondria-targeted antioxidant derived from ubiquinone by covalent attachment to a lipophilic triphenylphosphonium cation [46]. Our results showed that pharmacological inhibition of mitochondria ROS formation with mitochondrion-specific antioxidant mitoquinone also showed efficient inhibition on the ROS production in VSMCs induced PDGF-BB. Interestingly, the combined treatment of DPI and mitoquinone can not evoke a further reduction in ROS generation (Fig. S5), suggesting that both NADPH oxidase and mitochondria are vital involved players in PDGF-BB-triggered ROS production in VSMCs.
3.6. ROS were responsible for PDGF-BB-induced VSMC dedifferentiation, proliferation and migration
NFκB is taken as a redox-sensitive transcription factor, and intracellular ROS accumulation is critical for NFκB activation [20]. Our results showed that pretreatment with ROS scavenger NAC inhibited PDGF-BB-induced increased EdU positive cells (Fig. 7A and C), migrated cells (Fig. 7B and D) and absorbance (Fig. S1D) in VSMCs. The phenotypic changes from differentiated to dedifferentiated cells, as evidenced by decreased VSMC contractile genes α-SMA (Fig. 7E and F) and increased VSMC synthetic gene OPN (Fig. 7E and G) in PDGF-BB-treated VSMCs were alleviated by NAC pretreatment. In addition, the proliferating markers including PCNA (Fig. 7E and H), cyclin D1 (Fig. 7E and I) and P27 (Fig. 7E and J) were recovered by NAC pretreatment in PDGF-BB-treated VSMCs. Similar to the results from NAC, pretreatment with DPI also diminished PDGF-BB-evoked VSMC dedifferentiation, proliferation and migration (Fig. 8, Fig S1E). These results hinted that ROS are requisite factors for PDGF-BB-induced VSMC dedifferentiation, proliferation and migration.
Fig. 7.

NAC retarded PDGF-BB-induced VSMC dedifferentiation, proliferation and migration. VSMCs were pretreated with NAC (1 mM) for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) EdU assay. (B) Transwell assay was performed to determine the migration of VSMCs. (C) The ratio of EdU-positive cells to total cells. (D) Bar graph showing the number of migrated VSMCs. (E) Represented images showing the protein expressions of α-SMA, OPN, PCNA, cyclin D1 and P27. The relative protein expressions of α-SMA (F), OPN (G), PCNA (H), cyclin D1 (I) and P27 (J) were quantified. Values are mean ± SE. * P < 0.05 vs. Control + Vehicle (Veh), † P < 0.05 vs. PDGF-BB + Vehicle (Veh). n = 6 for each group.
Fig. 8.

DPI retarded PDGF-BB-induced VSMC dedifferentiation, proliferation and migration. VSMCs were pretreated with DPI (10 μM) for 6 h followed by stimulation with PDGF-BB (20 ng/mL) for 24 h. (A) EdU assay. (B) Transwell assay was performed to determine the migration of VSMCs. (C) The ratio of EdU-positive cells to total cells. (D) Bar graph showing the number of migrated VSMCs. (E) Represented images showing the protein expressions of α-SMA, OPN, PCNA, cyclin D1 and P27. The relative protein expressions of α-SMA (F), OPN (G), PCNA (H), cyclin D1 (I) and P27 (J) were quantified. Values are mean ± SE. * P < 0.05 vs. Control + Vehicle (Veh), † P < 0.05 vs. PDGF-BB + Vehicle (Veh). n = 6 for each group.
3.7. CA repressed intimal hyperplasia and suppressed ROS/NFκB/mTOR/P70S6K signaling cascade in vivo
In comparison with Sham rats, intimal hyperplasia was observed 8 weeks after injury (Fig. 9A). Administration of CA obviously reduced neointimal area and the ratio of neointimal area to media area in injured carotid arteries (Fig. 9A). CA prevented injury-evoked p65-NFκB nuclear translocation (Fig. 9B and C), phosphorylation and degradation of IκBα, as well as mTOR/P70S6K signaling activation (Fig. 9D and E). In addition, the upregulated NAD(P)H oxidase subunits p22phox, p47phox, NOX-2 protein expressions in injured carotid arteries were also mitigated by CA treatment (Fig. 9F and G). These results demonstrated a protective role of CA in the formation of hyperplastic neointima.
Fig. 9.

CA repressed intimal hyperplasia and suppressed ROS/NFκB/mTOR/P70S6K signaling cascade in vivo. Three days after vascular injury, CA (50 mg/kg/day) was administered gastric gavage to rats for a total of 8 weeks. (A) Representative HE staining of carotid arteries in sham and injury rats. (B) Represented blots showing the protein expressions of p65-NFκB in nucleus or in cytoplasm. (C) Bar graph showing the relative protein expressions of p65-NFκB in nucleus, p65-NFκB in cytoplasm and IκBα. (D) Represented images showing the protein expressions of IκBα, phosphorylated IκBα, phosphorylated and total mTOR and P70S6K. (E) Bar graph showing the relative protein expressions of phosphorylated IκBα, mTOR and P70S6K. (F) Represented images showing the protein expressions of NAD(P)H oxidase subunits p22phox, p47phox, NOX-2. (G) Bar graph showing the relative protein expressions of NAD(P)H oxidase subunits p22phox, p47phox, NOX-2. Values are mean ± SE. * P < 0.05 vs. Sham, † P < 0.05 vs. Veh. n = 6 for each group.
4. Discussion
The dysregulated VSMCs play a central role in vascular restenosis, hypertension and atherosclerosis [4]. Unlike cardiac or skeletal muscle cells, VSMCs undergo phenotypic changes from contractile phenotype to highly proliferative and migratory synthetic phenotype in response to injured stimuli [6]. The phenotypic switching of VSMCs is a fundamental step for proliferation and migration of VSMCs, and increased VSMC proliferation and migration are indispensable in atherosclerosis and restenosis after angioplasty [8]. The primary novel findings in this study are that CA pretreatment attenuated PDGF-BB-induced VSMC differentiation, proliferation and migration via suppression of ROS/NFκB/mTOR/P70S6K signaling cascade. These results provided the molecular mechanisms by which CA exerted a protective role in VSMC biology.
The normal and mature VSMCs exhibit higher contractile proteins including α-SMA, SM22α, calponin, SMMHC and myosin light chain kinase (MLCK) and lower synthetic proteins such as OPN, which play a prominent role in blood vessel tone, blood flow and blood pressure [21]. VSMCs have unique remarkable plasticity without terminally differentiate properties [47]. They can modulate their phenotypes response to the environmental stimuli from a differentiated and contractile state to synthetic state, and this process is also called dedifferentiation of VSMCs [47]. The phenotypic switch of VSMCs is one of the key cellular events in the development of atherosclerosis [48], [49]. Therefore, the new drugs can be designed to treat atherosclerosis, postangioplasty restenosis and hypertension on the basis of potential mechanisms involved in VSMC phenotypic switching. A recent study establishes that CA mitigates ox-LDL-induced adhesion molecules expressions in endothelial cells to mimic a potent anti-atherosclerotic ingredient [11]. However, it is unknown whether CA abrogated PDGF-BB-evoked VSMC dedifferentiation. In the present study, we showed that CA pretreatment prevented PDGF-BB induced VSMC differentiation, as reflected by decreased VSMC synthetic gene OPN along with increased VSMC contractile genes α-SMA, SMMHC, and SM22α. These results indicated that CA served as a therapeutic agent for inhibition of VSMC dedifferentiation.
The synthetic VSMCs exhibit accelerated proliferative and migratory abilities, which are critical for the development of vascular diseases including restenosis after angioplasty or bypass and atherosclerosis [4]. Therefore, it is urgent to elucidate the possible molecular mechanisms underlying the proliferation and migration of VSMCs. PCNA and cyclin D1 are suggested to be pro-proliferation genes in VSMCs, but P27 is supposed to exert anti-proliferative effect in VSMCs [1], [50]. In addition, cyclin D1 and P27 are cell cycle progression regulators, and they have opposite effect on VSMC proliferation [41]. Our results displayed that CA pretreatment reduced EdU-positive cells and migrated cells in response to PDGF-BB. Furthermore, CA mitigated the upregulated PCNA and cyclin D1 protein levels, as well as the downregulated P27 protein expression in PDGF-BB-incubated VSMCs. These results revealed that CA diminished PDGF-BB-induced VSMC proliferation and migration, and the protective effects of CA on PDGF-BB-stimulated VSMCs were ascribed to arrest of the cell cycle by activation of P27 and inhibition of cyclin D1 expression.
Mammalian target of rapamycin (mTOR) modulates a variety of cellular processes such as cell growth, migration and differentiation [51]. Specifically, mTOR plays a regulatory role in VSMC proliferation and migration via its downstream target P70S6K signaling [9], [21]. Activation of mTOR/P70S6K is responsible for VSMC dedifferentiation and proliferation [52]. Shear stress activates mTOR signaling to induce phenotypic modulation of VSMCs [53]. Exogenous miR-761 delivery inhibits angiotensin II-induced VSMC proliferation by targeting mTOR [54]. Sulforaphane is reported to diminish PDGF-induced VSMC proliferation in association with mTOR/P70S6K signaling inhibition [10]. CA is able to extend lifespan in Caenorhabditis elegans at least via inhibiting mTOR signaling [54]. These existing evidence drives us to explore whether mTOR/P70S6K signaling was implicated in CA-mediated protective effects on PDGF-BB-induced VSMC biological changes. Our results showed that blockade of mTOR/P70S6K alleviated PDGF-BB-induced VSMC dedifferentiation, proliferation and migration, which were consistent with the previous results [9], [21], [54]. The increased phosphorylated mTOR and P70S6K in VSMCs response to PDGF-BB were attenuated by CA pretreatment. In vivo study showed that employment of CA attenuated the activation of mTOR and P70S6K signaling in injured carotid arteries. These results hinted that CA-induced inhibition of mTOR/P70S6K signaling contributed to its antagonistic effects on PDGF-BB-evoked VSMC dedifferentiation, proliferation and migration.
Nuclear factor-κB (NFκB) is a transcription factor that is abundantly expressed in VSMCs, macrophages, and endothelial cells from human atherosclerotic lesions [55]. NFκB activation is a pivotal stimulator for VSMC dedifferentiation, proliferation and migration [20], [43], [56], [57]. Inhibitor κBα (IκBα) is an inhibitor of NFκB, which is bound to NFκB in the cytoplasm in unstimulated cells, the phosphorylated and degraded IκBα triggers nucleus translocation of NFκB and transcription expressions of NFκB-related genes [20], [58], [59]. It is recently established that CA significantly reversed lipopolysaccharide-elicited activation of NFκB in BV-2 microglia [16]. CA is shown to prevent systemic inflammation-induced memory impairment via inhibition of NFκB [18]. CA plays a prominent role in alleviating lipopolysaccharide-induced inflammatory response via inactivation of NFκB pathway [60]. Our data showed that NFκB inhibitor Bay 11-7082 pretreatment counteracted VSMC phenotypic change, proliferation and migration in response to PDGF-BB. CA pretreatment abolished PDGF-BB-induced translocation of p65-NFκB, phosphorylation and degradation of IκBα in VSMCs, suggesting that NFκB signaling inhibition was partially involved in the protective role of CA in VSMCs. Moreover, inhibition of NFκB with Bay 11-7082 suppressed the mTOR/P70S6K signaling in PDGF-BB-treated VSMCs. These results implied that PDGF-BB stimulated NFκB/mTOR/P70S6K signaling pathway to trigger dedifferentiation, proliferation and migration of VSMCs, which were substantially impeded by CA.
Oxidative stress plays a central role in NFκB activation, and excessive intracellular ROS contribute to dedifferentiation, proliferation and migration of VSMCs [20], [61], [62]. A major source for ROS in the cardiovascular system is NAD(P)H oxidases [63], [64]. In the present study, our results showed that CA pretreatment reversed the decreased SOD and CAT activities and ameliorated the upregulated MDA level in PDGF-BB-challenged VSMCs. These results hinted that CA may be a possible antioxidant to exert protective actions in VSMCs. Furthermore, we found that both NAC and DPI inhibited PDGF-BB-induced VSMC dedifferentiation, proliferation and migration. It is well established that both NAD(P)H oxidases and mitochondria are important producers of ROS in VSMCs [22], [65]. Although the antioxidant capacity of DPI is often attributed to the inhibition of NAD(P)H oxidases, several studies have identified that DPI is a potent inhibitor of mitochondrial ROS generation [66], [67], [68]. Our additional results displayed that both DPI and mitochondrion-targeted antioxidant mitoquinone suppressed ROS production in VSMCs induced by PDGF-BB. Interestingly, the combined treatment of DPI and mitoquinone did not evoke a further reduction in ROS generation, indicating that both NADPH oxidase and mitochondria participated in PDGF-BB-triggered ROS production in VSMCs.
In addition, the increased production of superoxide anions and upregulated NAD(P)H oxidase subunits p22phox, p47phox, NOX-2 protein levels in PDGF-BB-challenged VSMCs were prevented by CA pretreatment. These results revealed that CA could abrogate excessive ROS production to eliminate PDGF-BB-induced VSMC dedifferentiation, proliferation and migration. It is worthy to note that blockade of ROS generation diminished PDGF-BB-elicited p65-NFκB nuclear translocation, phosphorylation and degradation of IκBα, as well as mTOR/P70S6K signaling activation in VSMCs. The findings were further supported by the findings that the enhanced NAD(P)H oxidases and activated p65-NFκB/mTOR/P70S6K signaling cascade in injured carotid arteries were depressed by CA in rats. These results unveiled that ROS/NFκB/mTOR/P70S6K formed a signaling cascade, thereby contributing to PDGF-BB-induced VSMC dedifferentiation, proliferation and migration.
Taken together, our results disclose for the first time that CA antagonizes PDGF-BB-induced VSMC phenotypic alteration, proliferation and migration through regulating ROS/NFκB/mTOR/P70S6K signaling pathway. The activated ROS/NFκB/mTOR/P70S6K signaling axis was interrupted by CA, which provided a basis for possible clinical use of CA in atherosclerosis, intimal hyperplasia, hypertension and postangioplasty restenosis.
Acknowledgements
We thank the general support of experimental public platform, Wuxi School of Medicine, Jiangnan University. This work was supported by grants from the Young Scientists Fund of the National Natural Science Foundation of China (81700364), Jiangsu Natural Science Foundation (BK20170179), Fundamental Research Funds for the Central Universities (JUSRP11745), Project funded by China Postdoctoral Science Foundation (2017M611688) and Project funded by Jiangsu Postdoctoral Science Foundation (1701062C).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.11.012
Contributor Information
Xiang Liao, Email: aahuigh@sina.com.
Hai-Jian Sun, Email: haijsunjiangnan@jiangnan.edu.cn.
Appendix A. Supplementary material
Supplementary material
References
- 1.Liao X.H., Wang N., Zhao D.W., Zheng D.L., Zheng L., Xing W.J., Ma W.J., Bao L.Y., Dong J., Zhang T.C. STAT3 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J. Biol. Chem. 2015;290:19641–19652. doi: 10.1074/jbc.M114.630111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao T., Zhang L., Yao L.L., Zheng F., Wang L., Yang J.Y., Guo L.Y., Li X.Y., Yan Y.W., Pan Y.M., Jiang M., Chen L., Tang J.M., Chen S.Y., Jia N. S100B promotes injury-induced vascular remodeling through modulating smooth muscle phenotype. Biochim. Biophys. Acta. 2017 doi: 10.1016/j.bbadis.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Zhu L.H., Huang L., Zhang X., Zhang P., Zhang S.M., Guan H., Zhang Y., Zhu X.Y., Tian S., Deng K., Li H. Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin. Sci. 2015;129:129–145. doi: 10.1042/CS20140679. [DOI] [PubMed] [Google Scholar]
- 4.Bennett M.R., Sinha S., Owens G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015;214:33–50. doi: 10.1111/apha.12466. [DOI] [PubMed] [Google Scholar]
- 6.Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 7.Heusch G., Libby P., Gersh B., Yellon D., Bohm M., Lopaschuk G., Opie L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet. 2014;383:1933–1943. doi: 10.1016/S0140-6736(14)60107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomez D., Owens G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha J.M., Yun S.J., Kim Y.W., Jin S.Y., Lee H.S., Song S.H., Shin H.K., Bae S.S. Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem. Biophys. Res. Commun. 2015;464:57–62. doi: 10.1016/j.bbrc.2015.05.097. [DOI] [PubMed] [Google Scholar]
- 10.Shawky N.M., Segar L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol. Res. 2017;119:251–264. doi: 10.1016/j.phrs.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai K.L., Kao C.L., Hung C.H., Cheng Y.H., Lin H.C., Chu P.M. Chicoric acid is a potent anti-atherosclerotic ingredient by anti-oxidant action and anti-inflammation capacity. Oncotarget. 2017;8:29600–29612. doi: 10.18632/oncotarget.16768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlernitzauer A., Oiry C., Hamad R., Galas S., Cortade F., Chabi B., Casas F., Pessemesse L., Fouret G., Feillet-Coudray C., Cros G., Cabello G., Magous R., Wrutniak-Cabello C. Chicoric acid is an antioxidant molecule that stimulates AMP kinase pathway in L6 myotubes and extends lifespan in Caenorhabditis elegans. PLoS One. 2013;8:e78788. doi: 10.1371/journal.pone.0078788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tousch D., Lajoix A.D., Hosy E., Azay-Milhau J., Ferrare K., Jahannault C., Cros G., Petit P. Chicoric acid, a new compound able to enhance insulin release and glucose uptake. Biochem. Biophys. Res. Commun. 2008;377:131–135. doi: 10.1016/j.bbrc.2008.09.088. [DOI] [PubMed] [Google Scholar]
- 14.Xiao H., Wang J., Yuan L., Xiao C., Wang Y., Liu X. Chicoric acid induces apoptosis in 3T3-L1 preadipocytes through ROS-mediated PI3K/Akt and MAPK signaling pathways. J. Agric. Food Chem. 2013;61:1509–1520. doi: 10.1021/jf3050268. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava S., Cahill D.M., Conlan X.A., Adholeya A. A novel in vitro whole plant system for analysis of polyphenolics and their antioxidant potential in cultivars of Ocimum basilicum. J. Agric. Food Chem. 2014;62:10064–10075. doi: 10.1021/jf502709e. [DOI] [PubMed] [Google Scholar]
- 16.Liu Q., Hu Y., Cao Y., Song G., Liu Z., Liu X. Chicoric acid ameliorates lipopolysaccharide-induced oxidative stress via promoting the Keap1/Nrf2 transcriptional signaling pathway in BV-2 microglial cells and mouse brain. J. Agric. Food Chem. 2017;65:338–347. doi: 10.1021/acs.jafc.6b04873. [DOI] [PubMed] [Google Scholar]
- 17.Sakellariou G.K., Pearson T., Lightfoot A.P., Nye G.A., Wells N., Giakoumaki I.I., Griffiths R.D., McArdle A., Jackson M.J. Long-term administration of the mitochondria-targeted antioxidant mitoquinone mesylate fails to attenuate age-related oxidative damage or rescue the loss of muscle mass and function associated with aging of skeletal muscle. FASEB J. 2016;30:3771–3785. doi: 10.1096/fj.201600450R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q., Chen Y., Shen C., Xiao Y., Wang Y., Liu Z., Liu X. Chicoric acid supplementation prevents systemic inflammation-induced memory impairment and amyloidogenesis via inhibition of NF-kappaB. FASEB J. 2017;31:1494–1507. doi: 10.1096/fj.201601071R. [DOI] [PubMed] [Google Scholar]
- 19.Zhu D., Wang Y., Du Q., Liu Z., Liu X. Cichoric acid reverses insulin resistance and suppresses inflammatory responses in the glucosamine-induced HepG2 cells. J. Agric. Food Chem. 2015;63:10903–10913. doi: 10.1021/acs.jafc.5b04533. [DOI] [PubMed] [Google Scholar]
- 20.Sun H.J., Zhao M.X., Ren X.S., Liu T.Y., Chen Q., Li Y.H., Kang Y.M., Wang J.J., Zhu G.Q. Salusin-beta promotes vascular smooth muscle cell migration and intimal hyperplasia after vascular injury via ROS/NFkappaB/MMP-9 pathway. Antioxid. Redox Signal. 2016;24:1045–1057. doi: 10.1089/ars.2015.6475. [DOI] [PubMed] [Google Scholar]
- 21.Pan S., Lin H., Luo H., Gao F., Meng L., Zhou C., Jiang C., Guo Y., Ji Z., Chi J., Guo H. Folic acid inhibits dedifferentiation of PDGF-BB-induced vascular smooth muscle cells by suppressing mTOR/P70S6K signaling. Am. J. Transl. Res. 2017;9:1307–1316. [PMC free article] [PubMed] [Google Scholar]
- 22.Blazevic T., Schwaiberger A.V., Schreiner C.E., Schachner D., Schaible A.M., Grojer C.S., Atanasov A.G., Werz O., Dirsch V.M., Heiss E.H. 12/15-lipoxygenase contributes to platelet-derived growth factor-induced activation of signal transducer and activator of transcription 3. J. Biol. Chem. 2013;288:35592–35603. doi: 10.1074/jbc.M113.489013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhi L., Ustyugova I.V., Chen X., Zhang Q., Wu M.X. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J. Immunol. 2012;189:1639–1647. doi: 10.4049/jimmunol.1200528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu F.F., Esworthy R.S., Doroshow J.H., Grasberger H., Donko A., Leto T.L., Gao Q., Shen B. Deficiency in Duox2 activity alleviates ileitis in GPx1- and GPx2-knockout mice without affecting apoptosis incidence in the crypt epithelium. Redox Biol. 2017;11:144–156. doi: 10.1016/j.redox.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu X., Zhou Y., Cai W., Sun H., Qiu L. Salusin-beta mediates high glucose-induced endothelial injury via disruption of AMPK signaling pathway. Biochem. Biophys. Res. Commun. 2017;491:515–521. doi: 10.1016/j.bbrc.2017.06.126. [DOI] [PubMed] [Google Scholar]
- 26.Sun H.J., Xu D.Y., Sun Y.X., Xue T., Zhang C.X., Zhang Z.X., Lin W., Li K.X. CO-releasing molecules-2 attenuates ox-LDL-induced injury in HUVECs by ameliorating mitochondrial function and inhibiting Wnt/beta-catenin pathway. Biochem. Biophys. Res. Commun. 2017;490:629–635. doi: 10.1016/j.bbrc.2017.06.089. [DOI] [PubMed] [Google Scholar]
- 27.Sun H., Zhu X., Zhou Y., Cai W., Qiu L. C1q/TNF-related protein-9 ameliorates Ox-LDL-induced endothelial dysfunction via PGC-1alpha/AMPK-mediated antioxidant enzyme induction. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun H.J., Liu T.Y., Zhang F., Xiong X.Q., Wang J.J., Chen Q., Li Y.H., Kang Y.M., Zhou Y.B., Han Y., Gao X.Y., Zhu G.Q. Salusin-beta contributes to vascular remodeling associated with hypertension via promoting vascular smooth muscle cell proliferation and vascular fibrosis. Biochim. Biophys. Acta. 2015;1852:1709–1718. doi: 10.1016/j.bbadis.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 29.Zhao M.X., Zhou B., Ling L., Xiong X.Q., Zhang F., Chen Q., Li Y.H., Kang Y.M., Zhu G.Q. Salusin-beta contributes to oxidative stress and inflammation in diabetic cardiomyopathy. Cell Death Dis. 2017;8:e2690. doi: 10.1038/cddis.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng J., Lv Z., Qiao X., Li X., Li Y., Zhang Y., Chen C. The decay of redox-stress response capacity is a substantive characteristic of aging: revising the redox theory of aging. Redox Biol. 2017;11:365–374. doi: 10.1016/j.redox.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Z., Zhang X., Chen C., Wen Z., Hoopes S.L., Zeldin D.C., Wang D.W. Cardiomyocyte-specific expression of CYP2J2 prevents development of cardiac remodelling induced by angiotensin II. Cardiovasc. Res. 2015;105:304–317. doi: 10.1093/cvr/cvv018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han Y., Su J., Liu X., Zhao Y., Wang C., Li X. Naringin alleviates early brain injury after experimental subarachnoid hemorrhage by reducing oxidative stress and inhibiting apoptosis. Brain Res. Bull. 2017;133:42–50. doi: 10.1016/j.brainresbull.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z., Li S., Cai Y., Wang A., He Q., Zheng C., Zhao T., Ding X., Zhou X. Manganese superoxide dismutase induces migration and invasion of tongue squamous cell carcinoma via H2O2-dependent Snail signaling. Free Radic. Biol. Med. 2012;53:44–50. doi: 10.1016/j.freeradbiomed.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menendez-Castro C., Cordasic N., Schmid M., Fahlbusch F., Rascher W., Amann K., Hilgers K.F., Hartner A. Intrauterine growth restriction promotes vascular remodelling following carotid artery ligation in rats. Clin. Sci. 2012;123:437–444. doi: 10.1042/CS20110637. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y., Xie G., Liu Q., Duan X., Liu Z., Liu X. Pharmacokinetics, tissue distribution, and plasma protein binding study of chicoric acid by HPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016;1031:139–145. doi: 10.1016/j.jchromb.2016.07.045. [DOI] [PubMed] [Google Scholar]
- 36.Jiang L., Li W., Wang Y., Zhang X., Yu D., Yin Y., Xie Z., Yuan Y. Effects of cichoric acid extract from Echinacea purpurea on collagen-induced arthritis in rats. Am. J. Chin. Med. 2014;42:679–692. doi: 10.1142/S0192415X1450044X. [DOI] [PubMed] [Google Scholar]
- 37.El-Tantawy W.H., Temraz A., Hozaien H.E., El-Gindi O.D., Taha K.F. Anti-hyperlipidemic activity of an extract from roots and rhizomes of Panicum repens L. on high cholesterol diet-induced hyperlipidemia in rats. Z. Nat. C. 2015;70:139–144. doi: 10.1515/znc-2014-4147. [DOI] [PubMed] [Google Scholar]
- 38.Zhu D., Zhang X., Niu Y., Diao Z., Ren B., Li X., Liu Z., Liu X. Cichoric acid improved hyperglycaemia and restored muscle injury via activating antioxidant response in MLD-STZ-induced diabetic mice. Food Chem. Toxicol. 2017;107:138–149. doi: 10.1016/j.fct.2017.06.041. [DOI] [PubMed] [Google Scholar]
- 39.Wang B., Zhang M., Takayama T., Shi X., Roenneburg D.A., Kent K.C., Guo L.W. BET bromodomain blockade mitigates intimal hyperplasia in rat carotid arteries. EBioMedicine. 2015;2:1650–1661. doi: 10.1016/j.ebiom.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y., McRobb L.S., Khachigian L.M. Inhibition of intimal thickening after vascular injury with a cocktail of vascular endothelial growth factor and cyclic Arg-Gly-Asp peptide. Int. J. Cardiol. 2016;220:185–191. doi: 10.1016/j.ijcard.2016.06.300. [DOI] [PubMed] [Google Scholar]
- 41.Han J.H., Lee S.G., Jung S.H., Lee J.J., Park H.S., Kim Y.H., Myung C.S. Sesamin inhibits PDGF-mediated proliferation of vascular smooth muscle cells by upregulating p21 and p27. J. Agric. Food Chem. 2015;63:7317–7325. doi: 10.1021/acs.jafc.5b03374. [DOI] [PubMed] [Google Scholar]
- 42.Liu J., Xiu J., Cao J., Gao Q., Ma D., Fu L. Berberine cooperates with adrenal androgen dehydroepiandrosterone sulfate to attenuate PDGF-induced proliferation of vascular smooth muscle cell A7r5 through Skp2 signaling pathway. Mol. Cell. Biochem. 2011;355:127–134. doi: 10.1007/s11010-011-0846-x. [DOI] [PubMed] [Google Scholar]
- 43.Xi G., Shen X., Wai C., Vilas C.K., Clemmons D.R. Hyperglycemia stimulates p62/PKCzeta interaction, which mediates NF-kappaB activation, increased Nox4 expression, and inflammatory cytokine activation in vascular smooth muscle. FASEB J. 2015;29:4772–4782. doi: 10.1096/fj.15-275453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cai H., Griendling K.K., Harrison D.G. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 45.Al Ghouleh I., Khoo N.K., Knaus U.G., Griendling K.K., Touyz R.M., Thannickal V.J., Barchowsky A., Nauseef W.M., Kelley E.E., Bauer P.M., Darley-Usmar V., Shiva S., Cifuentes-Pagano E., Freeman B.A., Gladwin M.T., Pagano P.J. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011;51:1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Escribano-Lopez I., Diaz-Morales N., Rovira-Llopis S., de Maranon A.M., Orden S., Alvarez A., Banuls C., Rocha M., Murphy M.P., Hernandez-Mijares A., Victor V.M. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016;10:200–205. doi: 10.1016/j.redox.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi N., Chen S.Y. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J. Biomed. Res. 2014;28:40–46. doi: 10.7555/JBR.28.20130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao X., Debenedittis P., Sun Y., Chen J., Yuan K., Jiao K., Chen Y. Vascular smooth muscle cell Smad4 gene is important for mouse vascular development. Arterioscler. Thromb. Vasc. Biol. 2012;32:2171–2177. doi: 10.1161/ATVBAHA.112.253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rzucidlo E.M., Martin K.A., Powell R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007;45(Suppl. A) doi: 10.1016/j.jvs.2007.03.001. (A25-32) [DOI] [PubMed] [Google Scholar]
- 50.Xie C., Guo Y., Zhu T., Zhang J., Ma P.X., Chen Y.E. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J. Biol. Chem. 2012;287:14598–14605. doi: 10.1074/jbc.M111.329268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding M., Xie Y., Wagner R.J., Jin Y., Carrao A.C., Liu L.S., Guzman A.K., Powell R.J., Hwa J., Rzucidlo E.M., Martin K.A. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler. Thromb. Vasc. Biol. 2011;31:1403–1410. doi: 10.1161/ATVBAHA.110.216804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jia G., Aroor A.R., Martinez-Lemus L.A., Sowers J.R. Overnutrition, mTOR signaling, and cardiovascular diseases. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014;307:R1198–1206. doi: 10.1152/ajpregu.00262.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun L., Zhao M., Liu A., Lv M., Zhang J., Li Y., Yang X., Wu Z. Shear stress induces phenotypic modulation of vascular smooth muscle cells via AMPK/mTOR/ULK1-mediated autophagy. Cell Mol. Neurobiol. 2017 doi: 10.1007/s10571-017-0505-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho J.R., Lee C.Y., Lee J., Seo H.H., Choi E., Chung N., Kim S.M., Hwang K.C., Lee S. MicroRNA-761 inhibits Angiotensin II-induced vascular smooth muscle cell proliferation and migration by targeting mammalian target of rapamycin. Clin. Hemorheol. Microcirc. 2015;63:45–56. doi: 10.3233/CH-151981. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J., Wang X., Vikash V., Ye Q., Wu D., Liu Y., Dong W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016;2016:4350965. doi: 10.1155/2016/4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J., Chen L., Ding J., Fan Z., Li S., Wu H., Zhang J., Yang C., Wang H., Zeng P., Yang J. MicroRNA-24 inhibits high glucose-induced vascular smooth muscle cell proliferation and migration by targeting HMGB1. Gene. 2016;586:268–273. doi: 10.1016/j.gene.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 57.Jeong I.K., Oh D.H., Park S.J., Kang J.H., Kim S., Lee M.S., Kim M.J., Hwang Y.C., Ahn K.J., Chung H.Y., Chae M.K., Yoo H.J. Inhibition of NF-kappaB prevents high glucose-induced proliferation and plasminogen activator inhibitor-1 expression in vascular smooth muscle cells. Exp. Mol. Med. 2011;43:684–692. doi: 10.3858/emm.2011.43.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan S., Zhang X., Zheng H., Hu D., Zhang Y., Guan Q., Liu L., Ding Q., Li Y. Clematichinenoside inhibits VCAM-1 and ICAM-1 expression in TNF-alpha-treated endothelial cells via NADPH oxidase-dependent IkappaB kinase/NF-kappaB pathway. Free Radic. Biol. Med. 2015;78:190–201. doi: 10.1016/j.freeradbiomed.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Liang C.J., Wang S.H., Chen Y.H., Chang S.S., Hwang T.L., Leu Y.L., Tseng Y.C., Li C.Y., Chen Y.L. Viscolin reduces VCAM-1 expression in TNF-alpha-treated endothelial cells via the JNK/NF-kappaB and ROS pathway. Free Radic. Biol. Med. 2011;51:1337–1346. doi: 10.1016/j.freeradbiomed.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 60.Park C.M., Jin K.S., Lee Y.W., Song Y.S. Luteolin and chicoric acid synergistically inhibited inflammatory responses via inactivation of PI3K-Akt pathway and impairment of NF-kappaB translocation in LPS stimulated RAW 264.7 cells. Eur. J. Pharmacol. 2011;660:454–459. doi: 10.1016/j.ejphar.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 61.Wang Y., Ji L., Jiang R., Zheng L., Liu D. Oxidized high-density lipoprotein induces the proliferation and migration of vascular smooth muscle cells by promoting the production of ROS. J. Atheroscler. Thromb. 2014;21:204–216. doi: 10.5551/jat.19448. [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez A.I., Csanyi G., Ranayhossaini D.J., Feck D.M., Blose K.J., Assatourian L., Vorp D.A., Pagano P.J. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2015;35:430–438. doi: 10.1161/ATVBAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chatterjee S., Fisher A.B. Mechanotransduction: forces, sensors, and redox signaling. Antioxid. Redox Signal. 2014;20:868–871. doi: 10.1089/ars.2013.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montezano A.C., Touyz R.M. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid. Redox Signal. 2014;20:164–182. doi: 10.1089/ars.2013.5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clempus R.E., Griendling K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lambert A.J., Buckingham J.A., Boysen H.M., Brand M.D. Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta. 2008;1777:397–403. doi: 10.1016/j.bbabio.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 67.Bulua A.C., Simon A., Maddipati R., Pelletier M., Park H., Kim K.Y., Sack M.N., Kastner D.L., Siegel R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J. Exp. Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuan X., Zhou Y., Wang W., Li J., Xie G., Zhao Y., Xu D., Shen L. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013;4:e794. doi: 10.1038/cddis.2013.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material