

Abstract
Blood vessels in the central nervous system (CNS) are controlled by neuronal activity; for example, widespread vessel constriction (vessel tone) is induced by brainstem neurons that release the monoamines serotonin and noradrenaline, and local vessel dilation is induced by glutamatergic neuron activity. Here, we examined how vessel tone adapts to the loss of neuron-derived monoamines after spinal cord injury (SCI) in rats. We find that, months after the imposition of SCI, the spinal cord below the site of injury is in a chronic state of hypoxia, due to paradoxical excess activity of monoamine receptors (5-HT1) on pericytes, despite the absence of monoamines. This monoamine receptor activity causes pericytes to locally constrict capillaries, reducing blood flow to ischemic levels. Receptor activation in the absence of monoamines results from the production of trace amines (such as tryptamine) by pericytes that ectopically express the enzyme aromatic-l-amino-acid-decarboxylase (AADC), which synthesizes trace amines directly from dietary amino acids (such as tryptophan). Inhibition of monoamine receptors or of AADC, or even increased inhaled oxygen, produces substantial relief from hypoxia and improves motoneuron and locomotor function after SCI.
Keywords: Trace amines, AADC, capillary, pericyte, motoneurons, spinal cord injury, neurovascular coupling, hypoxia, ischemia, 5-HT1B receptor, locomotion
Introduction
The central nervous system (CNS) is very susceptible to damage when its blood supply is compromised1-3. Accordingly, the CNS has numerous control systems to ensure adequate blood supply, including: 1) neurovascular coupling, whereby neuronal activity locally dilates blood vessels to increase blood flow via glutamate and NO signaling; 2) metabolic coupling, whereby energy systems (including O2) regulate neuronal activity and vessel diameter; and 3) basal vascular tone, whereby CNS-derived neuromodulators such as serotonin (5-HT), noradrenaline (NA) and dopamine produce widespread vasoconstriction (along with myogenic tone), which effectively conserves blood for locally active areas dilated by neurovascular coupling4-7. The latter phenomenon is analogous to the sympathetic control system in the periphery, where NA causes widespread vessel tone8,9; though in the CNS neurons in the brainstem provide most of the NA and 5-HT, except for sympathetic innervation of large pial arteries10-14.
In the setting of severe spinal cord injury (SCI), vessels and neurons caudal to the injury survive, but lose monoamine innervation that normally arises primarily from brainstem neurons15. Although vessels directly damaged by SCI undergo well-documented plasticity at the injury epicenter16-18, including angiogenesis19, little is known about the uninjured vessels caudal to the site of the SCI, especially long after injury2. Considering that uninjured spinal neurons undergo remarkable adaptations that compensate for loss of supraspinal monoamine innervation after SCI15,20,21, we investigated whether vessels undergo similar adaptations. Normally, 5-HT and other monoamines in the spinal cord are synthesized exclusively in brainstem-derived axons, via enzymatic pathways such as the conversion of tryptophan to 5-HTP and then to 5-HT by the action of tryptophan-hydroxylase (TPH) and aromatic-l-amino-acid-decarboxylase (AADC), respectively14. After SCI, with the loss of the axons in which these enzymes are expressed, the levels of these enzymes are dramatically reduced, with one notable exception: spinal capillaries, which normally only weakly express AADC and lack other relevant enzymes such as TPH, markedly upregulate AADC expression caudal to the injury22,23.
Although AADC activity alone is not sufficient for endogenous synthesis of 5-HT (and indeed vessels lack 5-HT after SCI14,23,24), AADC can directly synthesize trace amines (TAs; Supplementary-Fig-1), such as tryptamine and tyramine, from amino acids such as tryptophan and tyrosine. In general, the endogenous functions of TAs have remained largely elusive, especially in the spinal cord14,25-27. TAs are not packaged or released in vesicles, lack a high affinity transporter, and are accordingly rapidly metabolized by monoamine oxidase (MAO), leaving only trace (nanomolar) quantities23,25,26. However, when AADC is present at high levels, as within capillaries after SCI, TAs are likely to reach the high micromolar concentrations of their precursor amino acids circulating in the blood28,29, considering that amino acids are readily transported into all cells30. At these micromolar concentrations, TAs activate 5-HT1 receptors (tryptamine), α2-adrenergic receptors (tyramine, phenylethylamine)31,32, and specialized TA associated receptors (TAARs)33, and have known effects on heart and mesentery vasculature34,35. Thus, we hypothesized that TAs synthesized by AADC locally in capillaries might directly regulate vessel tone after SCI, replacing lost monoamine innervation.
Classically, CNS blood flow is thought to be controlled by smooth muscle cells (SMCs) of arterioles that constrict in response to vasoactive substances, including centrally-derived 5-HT and NA acting on 5-HT1 and α2-adrenergic receptors4,10-12,14,36-42, via similar mechanisms that occur in the periphery9,42. Arterioles branch to form small capillaries which lack SMCs, but which are associated with sparsely spaced pericytes that wrap around CNS capillaries41 (Fig-1a). Although pericytes have diverse functions, including contributing to scar formation after SCI43, many pericytes are contractile, containing smooth-muscle α-actin (α-SMA) and contracting in response to monoamines4,6,41,44. Indeed, pericytes have recently been shown to play a major role in both controlling normal CNS blood flow and the response to ischemic stroke6,7,14,37,44-46. Coincidentally, we noticed that when AADC is upregulated in capillaries after SCI, it is most intensely expressed in pericytes23. Thus, we explore here whether pericytes play a special role in regulating capillary tone and blood flow after SCI, by synthesis of TAs that replace lost monoamines.
Figure 1.
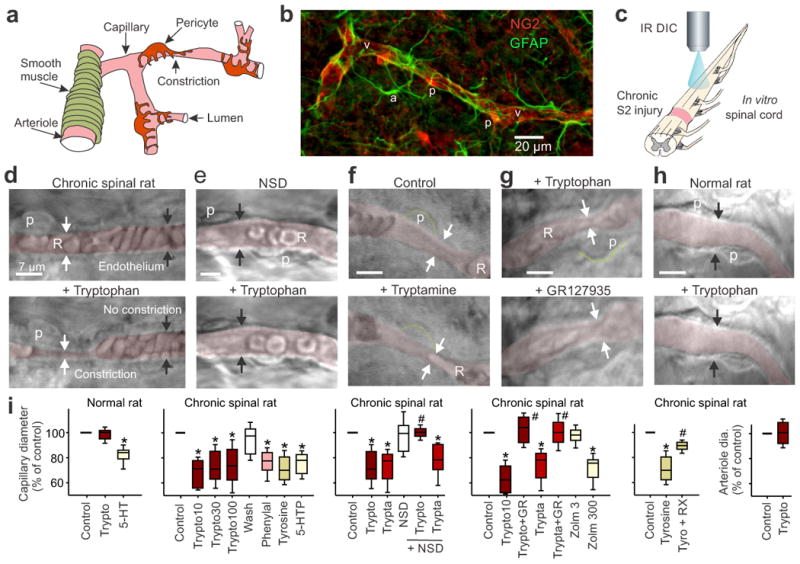
Trace amines constrict capillaries at pericytes after SCI. (a) Schematic of the spinal cord vasculature. (b) Immunolabeling for pericytes (NG2, p) on a spinal capillary (v) and nearby astrocytes (GFAP, a) caudal to the site of SCI. (c) Schematic of the DIC microscopy set-up used to image capillaries deep below the pial surface of cord after SCI. (d) Top, DIC image of capillary in the spinal cord caudal to a chronic sacral transection, with RBCs (R) and the lumen pseudo-colored red for clarity (arrows point to the endothelium). Bottom, application of tryptophan (30 μM) induced a tonic local vasoconstriction (white arrows; starting 1 min post application) adjacent to a pericyte (p), but not in regions lacking pericytes (black arrows). (e) Same as d, but with AADC inhibitor (NSD1015 [NSD]; 300 μM) applied prior to tryptophan, to prevent tryptamine production. (f) Tryptamine application mimicked tryptophan-induced vasoconstriction (d), adjacent to pericytes (outlined in green). (g) Tryptophan-induced constriction (top, at arrows) reversed by 5-HT1B receptor antagonist GR127935 (3 μM; bottom). (h) Capillary in normal rat (top), lacking tryptophan-induced constrictions (bottom). (i) Plots of group data for capillary diameters (normalized to pre-drug control) after bath application of amino acids (10, 30, 100 μM tryptophan [trypto10, trypto30, trypto100], 30 μM tyrosine [tyro], 50 μM phenylalanine [phenyl], or 0.1 μM 5-HTP), AADC products (10-100 μM tryptamine [trypta], or 0.3 μM 5-HT) or zolmitriptan (3 and 300 nM: zolm3 and zolm300), with and without inhibition of AADC (NSD1015) or antagonism of the 5-HT1B and α2 receptors (with GR127935 and 0.5 μM RX821002, respectively) are shown, and n values are detailed in the Methods. Right plot, arteriole diameter with tryptophan. * P < 0.05: significant change relative to pre-drug control (100%). # P < 0.05 relative change with antagonist or blocker. Box plots and horizontal bar within represent the interquartile range and median, respectively. Error bars extend to the most extreme data point that is within 1.5 times the interquartile range.
Results
Endogenously produced trace amines (TAs) constrict capillaries at pericytes after SCI
In the spinal cord of normal and injured rats, immunolabeling with the pericyte marker NG2 revealed pericytes spaced every ∼50 μm along capillaries (Fig-1b), as previously described elsewhere in the brain6. These pericytes have a characteristic hemispherical soma, unlike astrocytes, endothelial cells or SMCs (Fig-1a,b, Supplementary-Fig-2), allowing us to identify them morphologically, while imaging whole spinal cords maintained in vitro, using live tissue differential interference microscopy (DIC; Fig-1c,d)6. In spinal cords caudal to a chronic spinal transection, application of physiological concentrations of tryptophan, tyrosine or phenylalanine (10–100 μM)28,29 induced tonic local constrictions of capillaries adjacent to pericytes (Fig-1d,i; in 72%, 66% and 78% of pericytes, respectively), consistent with the notion that the increased expression of AADC in capillaries caudal to a SCI23 leads to production of TAs. Capillary regions without pericytes lacked constrictions (Fig-1d), consistent with the concept that pericytes are responsible for the vasoconstrictions, as previously demonstrated in the uninjured CNS6,45. The tryptophan-induced constrictions produced changes in vessel morphology and cord opacity, and widespread displacement of red blood cells (RBCs; Supplementary Fig 3 and Video). In summary, after SCI, application of tryptophan led to a decrease in capillary diameter adjacent to pericytes by ∼25%, halving the local cross-sectional area (Fig-1i), which should severely impair blood flow.
The AADC inhibitor NSD1015 eliminated tryptophan-induced vasoconstriction (Fig-1e,i), consistent with the conclusion that AADC conversion of tryptophan to tryptamine is responsible for inducing vasoconstriction. Indeed, exogenously applied tryptamine induced vasoconstrictions at 70% of pericytes, mimicking the effect of tryptophan, and tryptamine-induced vasoconstriction was resistant to NSD1015 (Fig-1f,i). NSD1015 alone had no effect on vessel diameter (Fig 1i), indicating that in vitro there is inadequate endogenous free tryptophan for AADC action (unlike in vivo, as described below). Tryptophan did not constrict arterioles after SCI (Fig-1i), ruling out the possibility that endogenously produced tryptamine can constrict these vessels. Tryptophan had no effect on capillaries or arterioles in spinal cords from normal uninjured rats (Fig-1h,i), consistent with the relative lack of vessel-associated AADC in uninjured spinal cords (as described below).
Trace amines activate 5-HT1B and α2-adrenergic receptors to constrict capillaries
We next tested whether TAs replace the function of monomines lost with injury by activating monoamine receptors. Indeed, tryptamine- and tryptophan-induced capillary vasoconstriction in chronic spinal rats were blocked by the selective 5-HT1B/D receptor antagonist GR127935 (Fig-1g,i), and were mimicked by the 5-HT1B/D receptor agonist zolmitriptan (Fig-1i), at doses consistent with zolmitriptan's moderate affinity for 5-HT1B receptors, and not its high affinity for 5-HT1D receptors20. Tyrosine-induced vasoconstriction (via AADC-mediated synthesis of tyramine) was blocked by the α2 receptor antagonist RX821002 (Fig-1i). 5-HTP, which is converted to 5-HT by AADC, had mixed effects on capillaries, constricting them at low doses, consistent with action at the 5-HT1B receptor, but dilating them at higher doses (Fig-1i, Supplementary-Fig-4). In spinal cords from uninjured rats, 5-HT generally induced capillary constrictions at pericytes (Fig-1i).
Mechanisms of pericyte action after SCI
In the spinal cord from uninjured rats (or in the spinal cord rostral to the site of injury after SCI), AADC immunolabeling was predominantly in axons (descending monoamine fibers), whereas it was absent (or only weakly expressed) in most vessels, as previously detailed23 (Supplemental Fig-5a). In contrast, caudal to a spinal cord transection, AADC immunolabeling was highly upregulated in vessels and not generally present in axon tracts23 (Supplementary Fig 5b). Double immunolabeling with the pericyte markers CD13 or NG2 revealed that this vessel AADC expression was entirely co-localized with pericytes on capillaries, with more intense expression near the pericyte membrane, as compared to the cytoplasm, especially adjacent to the vessel wall (forming a flat disc; Fig-2a,c,d; Supplementary Figs 5-7). In contrast, AADC was not co-expressed with an endothelial cell marker (Supplementary Fig 6), indicating a lack of endothelial AADC. AADC and its enzymatic products were present in capillary pericytes throughout the white and gray matter, including the dorsal horn and dorsal columns (Fig-2a, Supplementary Figs 5,8). AADC immunolabeling was not seen on arteries or arterioles (Fig 2a).
Figure 2.
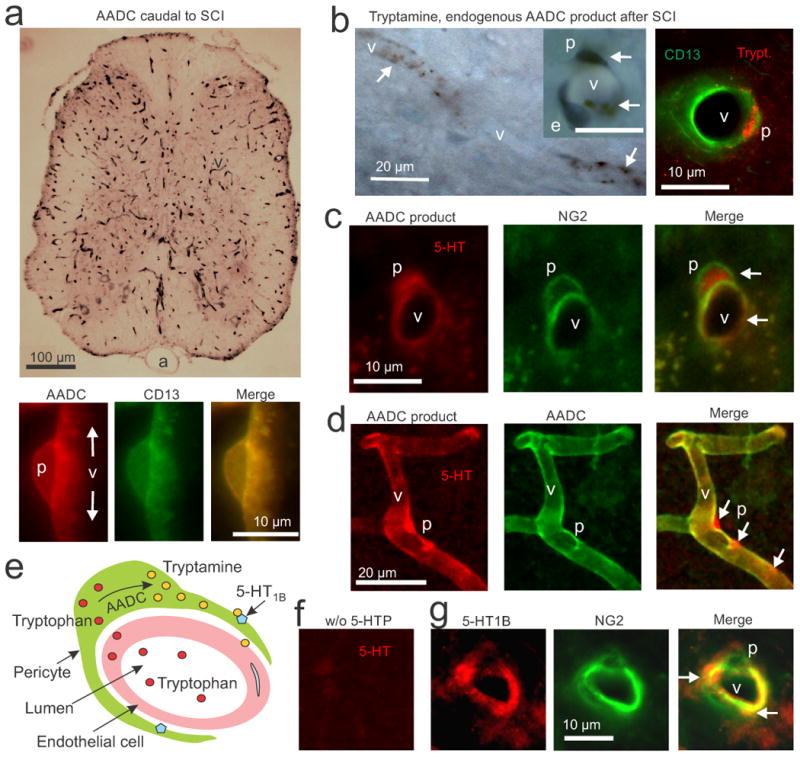
AADC, trace amines and 5-HT1B receptors are co-expressed in pericytes after SCI. (a) Top, immunolabeling with an AADC antibody (black, DAB, upper panel) in a transverse section of a spinal cord caudal to a chronic spinal transection, showing that AADC is widely expressed on capillaries (v), but not arteries (a). Bottom, immunofluorescence for AADC (red) and CD13 (green, pericyte marker) in a lengthwise section of capillary, showing exclusive colocalization of AADC and CD13 in pericytes (p; yellow). (b) Left, DAB immunolabeling of endogenous tryptamine (black) caudal the site of injury, showing dense staining for tryptamine in pericytes (arrows) of capillaries (v), especially in the soma. Inset shows a higher magnification view of a capillary cross-section (scale bar, 10 μm), showing that pericyte (p) cell bodies and processes stain for tryptamine (arrows), but an endothelial cell (e) does not (blue, cresyl-violet stain of the endothelial cell nucleus). Right, immunofluorescent staining for tryptamine (red) and CD13 (green) further showing tryptamine staining in a pericyte. (c) Immunolabeling for the AADC product 5-HT and for the pericyte marker NG2 caudal to a chronic transection injury after pre-treatment with 5-HTP (30 mg/kg, i.p., 25 min prior to fixation). Arrows, staining of 5-HT in NG2-labeled pericytes. (d) This AADC product 5-HT (red) is shown densely accumulated in pericyte cell bodies and processes labelled for AADC (green; at arrows). (e) Schematic of pericyte action on capillaries after SCI, showing diffusion of tryptophan (red) from blood into pericytes, synthesis of tryptamine (yellow) from AADC, and the action of tryptamine on nearby 5HT1B receptors (blue) to constrict the capillary. (f) Immunolabeling for 5-HT as in panels c and d, but without 5-HTP pre-treatment. (g) Immunolabeling for the 5-HT1B receptor and NG2 caudal to the site of injury. Arrows show localization of 5-HT1B receptor on NG2-labelled pericytes, with dense areas of receptor staining on pericyte processes. n = 5 rats tested per condition.
Immunolabeling for tryptamine revealed that this TA was endogenously expressed in capillaries caudal to the site of injury after an SCI, with dense punctate expression in the pericyte soma (Fig 2b; Supplementary Fig 7). Furthermore, this tryptamine staining was eliminated by pretreatment of the rats with NSD1015 (Supplementary Fig 7d). These results are consistent with the concept that AADC in pericytes produce tryptamine from endogenous tryptophan (Fig-2e). In contrast, tryptamine immunolabeling was largely absent from most capillaries rostral to the site of injury or in uninjured spinal cords (Supplementary-Fig 7). Immunolabeling for 5-HT1B receptors revealed that these receptors were densely expressed on pericytes (Fig-2g; Supplementary-Figure 10), where they could be readily activated by tryptamine synthesized locally by the pericyte itself (Fig-2e).
Immunolabeling for 5-HT confirmed previous findings that 5-HT is completely absent caudal to the spinal cord transection (Fig-2f, Supplementary-Fig 8b)23. However, pre-treatment of these chronic spinal rats by injection of the 5-HT precursor 5-HTP led to pronounced 5-HT immunolabeling in the microvasculature (Fig 2c,d; Supplementary Fig-8a). This 5-HT staining is eliminated by injection of an AADC blocker23, showing that the 5-HT is entirely produced by AADC and is a useful surrogate marker of AADC's ability to produce amines after SCI. This AADC product (5-HT staining) co-localized with pericyte AADC staining (Fig-2d, and CD13or NG2 pericyte labelling), and accumulated densely in the cytoplasm of the pericyte soma, adjacent to patches of dense AADC labeling (Fig-2d, Supplementary Fig-9), similar to tryptamine's localization. The mRNA expression of the pericyte marker CD13 was increased 45% caudal to the SCI, and expression of other pericyte signaling molecules was also altered, suggesting a proliferation of pericytes with SCI (Supplementary-Fig-11) 47.
Endogenous trace amines induce hypoxia after SCI
Considering that physiological concentrations of tryptophan constrict capillaries via endogenous TA production, we next examined blood flow in vivo. In the spinal cord below a chronic transection injury, the vasculature perfusion rate was half that in the cord above the injury or in normal spinal cords, as quantified by the time taken to fill all vessels with the vital dye methylene blue after intracardial injection (Fig-3a,b). Furthermore, when we injected a fluorescent dextran into the blood, two-photon-fluorescence microscopy revealed that the flow rates in spinal capillaries were abnormally low after SCI (Fig-3c), compared to in uninjured CNS48. Blocking TA production (with an AADC blocker; Fig-3c) or action (with a 5-HT1B receptor antagonist; Fig-3b) restored blood flow to near normal rates (doubling flow), consistent with the concept that endogenous tryptophan acts via AADC to restrict capillary flow under in vivo conditions. In a control experiment, application of an NO donor to fully dilate vessels showed that the unconstricted vasculature had equal perfusion capacity in normal uninjured and SCI rats (Fig-3b).
Figure 3.
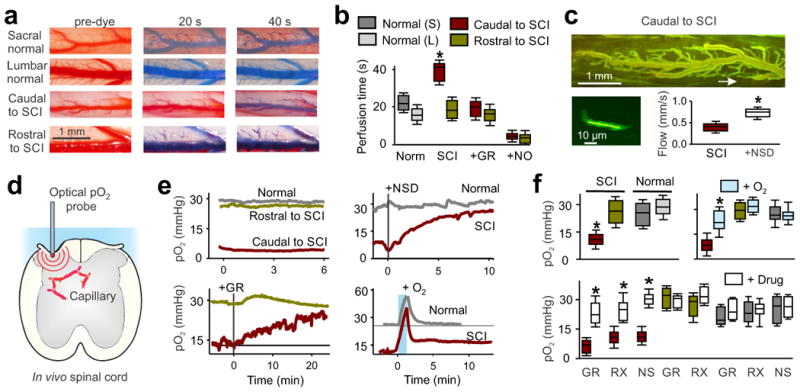
Poor blood flow and hypoxia after chronic SCI. (a) In vivo images of sacral and lumbar spinal cord dorsal vasculature in normal and chronic spinal rats, before and after (20 and 40 s) intracardial injection of methylene blue dye (2% in saline). (b) Perfusion times in sacral (S, caudal to injury) or lumbar (L, rostral) cords of injured and normal uninjured rats, and changes with the 5-HT1B antagonist GR127935 (GR, 30 μM topically applied to caudal cord) or sodium nitrate (NO donor; see Methods); n = 5 rats per group in box plots. (c) Top, two-photon microscopy image of the in vivo sacral spinal cord vasculature caudal to the site of injury, after FITC-dextran injection (i.v.). The arrow indicates location of a sub-pial spinal capillary imaged. Bottom left, higher magnification and brightened view of the indicated capillary where RBC flow computed. Bottom right, box plots of group capillary RBC flow rate in untreated and NSD1015 (NSD)-treated spinal cords (3 mM topically administered) in chronic spinal rats, n = 5 per group. (d) Schematic of in vivo oxygen measurement (pO2) in the spinal cord. (e) Low pO2 caudal to chronic SCI (hypoxia, red), compared to rostral to the SCI or in normal uninjured rats, and changes in pO2 after dilating vessels with NSD1015 or GR127935 (GR, topical 30 μM), or transient high O2 breathing (95% O2, with 5% CO2 for 1 min). (f) Box plots of pO2 in uninjured (normal) and injured rats before and after treatments with transient oxygen (95% for 1 min; measured at 10-20 min), GR127935, RX821002 (RX; 5 μM topical), or NSD1015 (NS); n = 5 - 20 per treatment, as detailed in the Methods. Drug effects peaked within minutes and sustained for the duration of the recording 10 - 90 min (peak reported); pO2 values are means from L4–L6 (rostral to injury or normal lumbar) and S2–S4, Ca1 (caudal to injury or normal sacral) spinal segments. *P < 0.05, significant difference relative to pre-treatment control (f) or normal tissue (b).
We next measured tissue oxygenation (pO2) in vivo. The spinal cord caudal to the site of injury was chronically hypoxic along its entire length (pO2 ≪ 20 mmHg49-51), with pO2 values caudal to the injury less than half of those rostral to the injury or of the uninjured spinal cord (Fig-3d,e). Treatments that dilated the capillaries—inhibition of AADC or blockade of 5-HT1B or α2-adrenergic receptors—restored normal pO2 caudal to the site of injury, while not affecting pO2 rostral to the site of injury (Fig-3e,f). Tryptophan application itself did not alter pO2, likely because endogenous tryptophan levels in the CNS and circulation (∼50 μM)28,29 are already well above the concentration that we found produces peak effects in the isolated spinal cord (10 μM; Fig-1i). A short period of hyperoxia (1 min) in which the concentration of inhaled O2 was raised to 100% or 95% (with 5% CO2 to maintain breathing rate) rapidly increased arterial pO2 from ∼100 to ∼400 mmHg and accordingly increased spinal cord pO2 to well above normal (Fig-3e, blue bar), demonstrating that caudal blood flow is inadequate to supply normal oxygenation without an artificial increase in arterial oxygen levels.
Unexpectedly, after this brief 1 min period of hyperoxic breathing, cord pO2 caudal to the site of injury remained elevated for 20 min, even though rostral to the injury the cord pO2 returned rapidly (within 1 min) to normal (Fig-3e), as did breathing rate, exhaled CO2 and arterial pO2 (Methods). This prolonged elevation in cord pO2 caudal to the site of injury is likely due to the effect of increased oxygenation on raising neural activity (see below), which in turn triggers neurovascular coupling that dilates vessels and further increases oxygen and neural activity in a positive feedback loop (especially in the hyperexcitable cord after SCI15). Indeed, we observed prominent neurovascular coupling after SCI, caudal to the site of injury (Supplementary-Fig-12). Transient breathing of 100% O2 (1 min) likewise led to a prolonged (20 min) elevation in pO2 caudal to the site of injury, as did air with increased CO2 levels (10% CO2 in air for 30 s; because CO2 dilates vessels1) or even mild hypoxia (10% O2 in air for 1 min) (Supplementary-Fig-13).
Motor function is enhanced by blocking trace amine action and preventing hypoxia
To evaluate the functional effects of the impaired blood flow and chronic hypoxia after SCI, we recorded EMG activity (electromyography) in the paralyzed tail muscles of the awake chronic spinal rat (sacral transection)15. Treatments that dilated spinal capillaries—the receptor antagonists GR127935 or RX821002, or inhaled 10% CO2—increased spontaneous EMG activity (Fig-4a-c,f). Likewise, treatments that indirectly increase spinal cord blood flow by systemically augmenting blood pressure (peripherally-acting dobutamine or vasopressin) also increased EMG activity (Supplementary-Fig-14). Transient hyperoxic breathing (95% O2 for 1 min) to increase spinal cord oxygenation by increasing arterial pO2 also increased spontaneous EMG activity, with effects lasting 20 min beyond the treatment period (Fig-4d,f), consistent with the cord pO2 measurements (Fig 3e). Each of these treatments also increased sensory-evoked EMG responses (long-lasting reflexes, LLRs in Fig-4b-f), and increased the incidence of rhythmic locomotor-like bursting (Fig-4d, blue thin line; Supplementary-Fig-14). Hyperoxia or hypercapnia produced similar increases in EMG when repeated at >60 min intervals.
Figure 4.

Treatments that dilate vessels and improve oxygenation after SCI lead to increased motor activity. (a) Schematic of awake chronic spinal rat in Plexiglas bottle for tail muscle EMG recording and electrical stimulation of the tip of the tail (50×T) to evoke reflexes. (b) Representative muscle activity in a chronic spinal rat, showing baseline EMG activity prior to stimulation (arrow) and long-lasting reflex (LLR) evoked by stimulation, before (top) and after GR127935 treatment (bottom) (intrathecal [i.t.], 30 μl, 10 mM). (c) Box plots of the change in the LLR measured in vitro (rectified-average) induced by blocking the TA-mediated vasoconstriction of capillary flow with either GR127935 (GR, i.p. 8 mg/kg, n = 3 or i.t. 10 mM in 30 μl; n = 2; combined) or RX821002 (RX, i.p. 1 mg/kg, n = 11; or i.t. 3 mM in 30 μl, n = 5), and compared to the change in LLR studied in the isolated in vitro spinal cord, with application of GR127935 (3 μM, n = 18), RX821002 (1 μM, n = 42) and NSD1015 (NS, 300 μM, n = 9) (see Supplementary-Fig-15). LLR normalized to LLR prior to drugs, 100%. (d) Representative EMG trace in a chronic spinal rat before and 15 min after transient breathing of 95% O2 (1 min, with 5% CO2). Smoothed rectified rhythmic activity is indicated by the blue line. (e) Time course of mean LLR before and after transiently increased O2 breathing in vivo (1 min), compared to LLR response to increased O2 in the isolated in vitro spinal cord (pO2 increased in nACSF). (f) Box plots of LLR in injured rats in vivo, either prior to treatment (Pre), during treatment with O2 or CO2, or 10–20 min post-treatment (post). Treatments were transient hyperoxia (O2, 95% for 1 min, n = 8) or hypercapnia (CO2, 10% in air for 30 sec, n = 10) breathing. Also shown are data for in vitro isolated spinal cords during or after treatment with O2 (n = 6). All conditions normalized as in c. * P < 0.05 significant difference relative to pre-drug condition.
To rule out direct actions of TAs on neurons, we also examined motor activity and sensory-evoked motor responses (LLRs) in the ventral roots of isolated spinal cords from chronic spinal rats. The spinal cords were maintained in vitro in oxygenated artificial cerebrospinal fluid (nACSF), where vessels have no influence (Supplementary-Fig-15a,b). Notably, in these isolated cords, motor activity (LLRs) was not altered by treatment with NSD1015, GR127935, RX821002 (Fig-4c) or tryptophan (Supplementary-Fig-15d,e). The lack of an effect of tryptophan indicates that TAs endogenously produced from tryptophan have no direct effect on neurons. Though exogenous application of high doses of TAs did increase LLRs, this effect was mediated by a different receptor subtype (blocked by 5-HT2C receptor antagonist) than the monoamine receptors that control capillaries (Supplementary-Fig-15f,i). Furthermore, transient alteration of the pO2 of the ACSF in the in vitro preparation, from normoxic to hyperoxic levels, transiently altered motoneuron activity (LLRs; Fig-4e,f): an increase in pO2 produced an increase in LLRs, consistent with a previous report52. However, this increase in LLRs did not outlast the O2 application, consistent with the concept that increased blood flow via neurovascular coupling mediates the sustained pO2 and LLRs observed in vivo after O2 breathing (Fig-4e,f).
Locomotor function after SCI is improved by treatments that improve capillary blood flow
To evaluate locomotion after SCI, we studied rats with a thoracic SCI, from either a severe contusion or a staggered hemisection injury (Fig. 5a); the latter type of injury transects most descending axons, including removing 5-HT15,53 (Supplementary-Fig-16). A month after these injuries, rats regained some voluntary hindlimb locomotor ability, though weight support was impaired, plantar foot placement was poor and step timing was slow, in part because of leg extensor spasms (Fig-5b). In the lumbar region caudal to the injury, pO2 was hypoxic, significantly lower than rostral to the site of injury or in uninjured spinal cord (Fig 5f-g). Dilation of capillaries with locally applied GR127935 or RX821002 restored near normal pO2 (Fig 5f-g), with effects lasting as long as the drug was present (durations of 30 - 90 min were tested). Transient O2 breathing (95%, 90 s) likewise restored normal pO2, with effects lasting 20 min, as for the chronic spinal rats in Fig 3 (Fig 5f-g). Intrathecal injection of GR127935 or NSD1015 to dilate capillaries, or transient O2 breathing, significantly improved walking ability (Fig-5c-e). These improvements in locomotion with GR127935 or oxygen breathing lasted 10 - 30 min, consistent with the pO2 measurements and with the known washout time of intrathecal drug injections15. Consistent with the action of NSD1015 as an irreversible AADC inhibitor, its actions lasted for at least 24 hours, demonstrating sustained improvements in locomotor function with vessel dilation (Fig 5e).
Figure 5.

Thoracic contusion or staggered hemisection injury induces chronic hypoxia that impairs locomotion. (a) Schematic of thoracic staggered-hemisection SCI, which transects all descending axons from the brain, including those containing monoaminergic neurons. (b) Video image sequence of a rat walking one month after receiving a staggered hemisection injury. Impaired hindlimb function while walking is evident from poor weight support (quantified as torso height above ground), leg extensor spasms (quantified as spasm time relative to step-cycle duration), slow steps (number of hindlimb plantar steps per step-cycle of front leg) and poor foot placement (caudal to behind hip). Hip (iliac crest), knee and ankle joints are shown with dots and lines. Arrow shows foot movement. (c) Effects of intrathecal application of GR127935 (10 mM in 30 μl) on locomotion, with locomotion phases annotated above. (d) Plots of mean locomotor parameters for each rat (circles) with staggered hemisection injury (3 - 5 weeks post injury), including body height, extensor spasm time, number of successful hindlimb steps and foot placement error, before and after application of GR127935 or transient breathing of 95% O2 (90 s; with 5% CO2; measured 10–15 min post treatment). n = 7 rats per treatment group. Error bars: s.e.m, bars: group means (e) Plots of mean locomotor parameters for each rats with contusion injury, in a similar format as d. Effects 1 and 24 h after treatment with NSD1015 (100 mM in 30 μl i.t.; NSD) were also measured. n = 9 rats for GR127935 and oxygen, and n = 6 for NSD. (f-g) Plots of pO2 in uninjured (normal) rats (lumbar cord) and in SCI rats (caudal [lumbar] and rostral [thoracic] to the site of injury), before and 10-15 min after treatments with transient oxygen (95%, 90 s), GR127935 (GR, topically applied 30 μM), or RX821002 (RX, topical 5 μM). *P < 0.05: difference relative to normal cord or change with treatment.
Discussion
Our results demonstrate several new concepts that fundamentally alter the understanding of spinal cord function and rehabilitation after SCI: (i) after SCI, pericytes play a major role in regulating capillary tone and blood flow in the spinal cord; (ii) SCI leads to a chronic state of excess capillary tone, poor blood flow and hypoxia; (iii) the monoamine receptors 5-HT1B and α2 regulate capillary tone in the absence of the monoamines 5-HT and NA in transected cords; (iv) AADC expressed in capillary pericytes synthesize the TAs tryptamine and tyramine from physiological concentrations of dietary amino acids, and these TAs are responsible for monamine receptor activation, replacing the action of brainstem-derived monoamines lost with SCI, and (v) antagonism of monoamine receptors, inhibition of AADC enzyme function or augmentation of inhaled O2 or CO2 after SCI produces lasting improvements in blood flow and cord oxygenation, which ultimately improves motor function, including locomotion. Notably, we found that chronic spinal cord hypoxia occurs with clinically-relevant partial spinal cord injury (contusion injury) and that long-term improvement in locomotor function can be achieved with interventions that dilate vessels (e.g., AADC inhibitors). These results demonstrate that alleviation of chronic hypoxia is a promising approach to rehabilitation after SCI.
Our results also show that TAs produced endogenously from amino acids (via AADC) do not directly affect neuronal activity (see also 27), but instead act only on vessels caudal to the site of injury. Whereas exogenously applied TAs can directly affect neuronal function, via the 5-HT2 and TAAR receptors27, TAs endogenously produced in vessels are likely metabolized by MAO before they can diffuse to neurons23. Furthermore, the treatments we used to dilate capillaries after SCI—a selective 5-HT1B antagonist (GR127935) and an AADC inhibitor—act primarily on pericytes rather than neurons, because they did not affect neuronal activity in isolated cords maintained in vitro. Additionally, GR127935 could not have directly affected locomotor or motoneuron function, because after severe SCI there is little to no 5-HT present to activate locomotor-related 5-HT receptors, and even if these receptors were somehow activated (by residual 5-HT in the contusion rats), the primary locomotor-related receptors include 5-HT2A/C and 5-HT1A receptors53, which are not antagonized by GR127935.
The concept that pericytes play a major role in controlling blood flow has only recently been described in the brain (cerebellum and retina)6,7,44,45 and has not been previously demonstrated in the spinal cord. Pericytes and SMCs can both regulate CNS blood flow, but controversy exists as to whether pericytes or SMC play the primary role54, particularly as pericytes are highly diverse, with many containing α-SMA, but others not43-46,54,55. Our results demonstrate that capillary pericytes can play a primary role in regulating blood flow, because treatment with tryptophan after SCI induces widespread constrictions of capillary pericytes, but not of arterioles, via the action of AADC that is expressed exclusively in pericytes, and is not present in arterioles, arteries or SMCs14,23 Furthermore, blocking the action of pericyte AADC (with NSD1015) or of associated TAs on the 5-HT1B receptor (with GR127935) markedly improves blood flow, oxygenation and motor function in vivo. Although arterioles (with SMCs) express monoamine receptors and respond to exogenously applied monoamines (5-HT and tryptamine)11,14,36, AADC products produced by pericytes likely do not diffuse sufficiently far to influence distant arterioles or neurons23.
The marked chronic hypoxia (pO2 ∼10 mmHg) that we observed in the cord far caudal to the site of injury was unexpected, especially in the case of partial injury (contusion injury), as previous studies have focused on the acute stage of injury and the injury epicenter2,16,19,43. In contrast to hypoxia caudal to the site of injury, the cord rostral to the injury is well oxygenated (28 mmHg), similar to that reported for the uninjured spinal cord and brain49-51. Based on these observations, any treatment that increases systemic blood flow or arterial pO2, including exercise53,56, should also increase cord oxygenation and indirectly modulate neuronal function52, consistent with our findings after manipulation of blood pressure and inhaled O2 concentrations.
Chronic spinal cord hypoxia likely contributes to long-term disabilities that slowly emerge after SCI, including metabolic plasticity19, chronic spinal cord inflammation18,47, long-term deterioration and loss of neurons57, and exhaustion during locomotor activity 58. Increasing cord perfusion (and pO2) may reduce these clinical problems, although the adverse effects of reactive oxidative species should be considered if oxygenation is abruptly increased1. Paradoxically, we found that transient hypoxic breathing produces a rebound increase in spinal pO2 that lasts > 20 min, which might help explain why intermittent hypoxic breathing improves rehabilitation after SCI59.
Our finding that TAs control capillary tone, arguably represents the first time that endogenous TAs have been shown to have a stand-alone spinal cord function, in the absence of classical monoamines. Previously, secondary functions have been ascribed to TAs in the CNS involving their modulation of monoamine function25,26, including TAs modulating the dopamine transporter, to compensate for reductions in dopamine33,60. The latter dopamine mechanism, involves TAs acting on the same cell in which they are produced (the dopamine neuron). Similarly, our results suggest that TAs produced by AADC in pericytes act locally to induce constriction of the same pericyte. AADC is expressed densely in the pericyte, especially adjacent to the vessel wall, allowing for ready access to blood-borne tryptophan30 to produce tryptamine, which in turn diffuses throughout the pericyte (filling the soma) and acts on the 5HT1B receptor on the pericyte itself to produce vasoconstriction (Fig 2e schematic). Likely, this requires tryptamine to leave the pericyte (via a transporter like OCT27) to act extracellularly on 5-HT1B receptors in the pericyte membrane, though we cannot rule out direct action on intracellular 5-HT1B receptors27. After SCI, a few neurons also upregulate AADC expression23,24, which may complement pericyte function in regulating vessel tone, since these neurons contact vessels23.
The remarkable adaptation of capillary pericytes to SCI is functionally similar to the adaptation of motoneurons to SCI: motoneurons become hyperactive due to paradoxical monoamine receptor activity, ultimately leading to muscle spasms15,21. However, the mechanisms underlying the effects of SCI on pericytes and motoneurons are very different, as motoneurons upregulate isoforms of 5-HT2C receptors that are constitutively active15, whereas pericytes upregulate AADC that produces TAs, leading to 5-HT1B receptor activation.
Online Methods
All rat experiments were approved by the Health Sciences University of Alberta Animal Care and Use Committee. Most of the studies, except the locomotor-related studies, were performed using the sacral transected injury model, where the S2 sacral spinal cord was transected in adult female Sprague-Dawley rats at ∼ 2 months of age, as described previously15,23. Briefly, under general anesthetic (sodium pentobarbital, 58.5 mg/kg, and isoflurane) and sterile conditions, a laminectomy was performed on the L2 vertebrae to expose the S2 spinal cord. The dura was slit transversely, and 0.1–0.3 ml Xylocaine (1%) was applied topically. Using a surgical microscope, the spinal cord was transected by holding the pia with forceps and removing tissue using suction (using a 1 cc syringe that had been melted and pulled to a fine tip). Caution was taken to avoid damaging the anterior artery or posterior/dorsal vein, since the sacrocaudal spinal cord dies without this midline vasculature. The dura was closed with two 8-0 silk sutures, the muscle layers and skin were tightly sutured over the cord, and the rat was allowed to recover. All experiments were performed 2 - 4 months post injury, when tail muscle spasticity was full developed and rats were considered to be in a chronic spinal state (chronic spinal rats)15. Chronic spinal rats were compared to age-matched normal uninjured rats (rats were randomly assigned to these two groups).
Contusion and staggered hemisection injury models were used for locomotion studies15. Under anesthesia with fentanyl (Hypnorm 120 μl/200g; Janssen, Canada) and midazolam (0.75 mg/200g; Sabex, Belgium), adult female Sprague-Dawley rats were injured at ∼ 6 weeks of age. Rats received either a severe contused injury at spinal T8 vertebrae (impact force 200 kDyn) or underwent a staggered hemisection. The latter rats were first hemisected on the right at the T10 spinal vertebrae (T10 -T11 cord level). Then two weeks later, rats received an over-hemisection on the left at spinal T7 - T8 vertebrae (T7-T8 cord). In this staggered hemisection model, all direct descending supraspinal inputs, including 5-HT fibers, are cut, whereas spared local propriospinal neurons relay descending signals around the lesion site (Fig-6a; Supplementary Fig 16)15. This procedure allows for some spontaneous recovery of voluntary locomotion (unlike in transected animals) in the absence of 5-HT. Bladders were expressed 3 times daily for at least 3 days, until bladder function recovered. Locomotion was evaluated 3 - 5 weeks after contusion or the second hemisection using parameters of plantar foot placement, step frequency, and iliac crest height (hip; see below).
In vitro preparation
Under urethane anesthesia (1.8 g/kg, with a maximum dose of 0.45 g), the whole spinal cord caudal to the S2 injury level was removed from chronic spinal rats and immersed in oxygenated modified artificial cerebrospinal fluid (mACSF). For ventral root recording and reflex testing, the sacral S4 and caudal Ca1 ventral roots and the Ca1 dorsal roots were used, and remaining roots removed. For imaging vessels in the cord, all roots were removed from the cord. After 0.5 - 1.5 h in mACSF (at 20°C), the cord was transferred to a recording chamber containing normal ACSF (nACSF) saturated with carbogen (95% O2-5% CO2) and maintained near 21°C, with a flow rate of 5 ml/min. In some cords, pO2 was measured using the same sensor as used in vivo (Optode, see below), and the pO2 was adjusted to a desired level (cord pO2 : 10 - 50 mmHg) by mixing small amounts of N2 with the carbogen.
In vitro ventral root reflex recording and averaging
Dorsal and ventral roots were mounted on silver-silver chloride wires above the nACSF of the recording chamber and covered with a 5:1 mixture of petroleum jelly and mineral oil for monopolar stimulation and recording. The dorsal root was stimulated with a single pulse (0.1 ms, 0.02 mA, 3×T [reflex Threshold]; repeated 5 times at 10 s intervals for one trial; trials repeated every 12 min), and the long-lasting reflex (LLR) response was recorded on the ventral roots, which corresponded to spasms in vivo, as previous described15. The ventral root recordings were amplified (2,000×), high-pass filtered at 100 Hz, low-pass filtered at 3 kHz, and recorded with a data acquisition system sampling at 6.7 kHz (Axonscope 8, Axon Instruments, Burlingame, CA). We quantified the LLR with custom-written software (MATLAB, MathWorks, Natick, MA) by averaging the rectified ventral root activity over a time-window of 500–4000 ms post stimulus, an interval corresponding to in vivo muscle spasms15. Because of slow diffusion in whole spinal cord preparations, drug treatment required 10-fold higher concentrations of the drug than are used in thin-slice preparations, and peak effects required 10–15 min. To assure selectivity of the drugs, they were titrated to a minimal dose that produced their peak effect, and results are reported at 10–45 min after drug application. Cumulative dose-response relations were computed by increasing drug doses at 12 min intervals (at doses of 0.003, 0.01, 0.03, 0.1 μM, down to 1 nM). The effects of drugs on the LLR were reversible on washout of the drug, but full recovery to baseline occurred only after several hours (data not shown), likely because of the large size of the whole cord preparation. Thus, washout of drugs was not feasible between doses of the drugs used in the construction of their dose-response curves.
IR-DIC microscopy of vessels, in vitro
The sacrocaudal spinal cord was removed from rats, put in a recording chamber and imaged using an infrared differential-interference-contrast (IR-DIC) microscope (Leica Axioskop2 FC Plus); the cord was maintained in nACSF, as described above in the section above. Spinal capillaries were imaged both in the dorsal horn of the whole sacrocaudal cord preparation (viewing through the dorsal surface, 200 - 300 μm below the pia), and throughout the cord in transverse sections or sagittal hemisected cords (0.5 mm transverse sections were cut on a vibratome; sagittal hemisection performed by pulling the cord gently in half from rostral to caudal). Similar results were obtained for all methods of imaging spinal capillaries, and so the data using all methods were combined. Spinal capillaries that were chosen for imaging were mostly in thin sacral S4 and coccygeal Co1 segments where light could readily traverse the tissue, with the following criteria for capillary identification: 1) they had a diameter of <11 μm; 2) they were located deep under the pia and white matter axons, assuring that they were not pial vessels; 3) they lacked continuous smooth muscle coverage as seen on arterioles; and 4) they were wrapped with sparsely-spaced, hemispherical-shaped pericytes. We analyzed pericytes on straight stretches of capillaries6, where drug-induced constrictions were maximal, rather than at vessel branch points, which had pericytes that did not always constrict. Drug application was the same as for in vitro root reflex recording methods. For normal rats we applied tryptophan (n = 24 vessel preparations) and 5-HT (n = 7). For chronic spinal rats we applied tryptophan (at 3 doses:10, 30 and 100 μM; n = 6, 34 and 44), phenylalanine (n=22), tyrosine (n = 20), 5-HTP (n = 23), tryptamine (n = 19), NSD1015 (with tryptamine or tryptophan; n = 16 each), GR127937 (with tryptamine or tryptophan, n = 10 and 21), zolmitriptan (at 2 doses, 3 and 300 nM; n = 8 and 10), RX821002 (with tyrosine, n = 5). Doses not provided here are quoted in the text or figure legends. Video was taken of vessels with an infrared camera and streamed to a computer via a high-definition video H.264 recorder (Blackmagic Design), and vessel diameter computed offline at common focal planes, before and after drug application effects reached steady state (5 - 10 min).
Spasms in awake chronic spinal rats
Tail muscle activity and spasms were evoked with brief electrical stimulation of the skin of the tail tip and recorded with tail muscle EMG. Percutaneous EMG wires (50 μm stainless steel, Cooner Wires) were inserted in segmental tail muscles at the midpoint of the tail, and recordings were made while the rat was in a Plexiglas tube, as detailed previously15. Muscle spasms (long-lasting reflexes; LLR) were evoked with electrical stimulation of the skin at the distal tip of the tail (percutaneous stimulation; 0.2 ms, 10 mA pulse; 50 reflex threshold [50 × T]; spasms evoked at 40 s intervals), and the tail was partly restrained from moving with a piece of masking tape connecting the midpoint of the tail to a rigid stand. EMG was sampled at 5 kHz, rectified and averaged over a 500 to 4,000 ms interval to quantify spasms (LLR) using an Axoscope instrument (Axon Instruments) and MATLAB software (MathWorks). EMG over the period of 300 ms prior to stimulation was also averaged (background EMG). Drugs were dissolved in sterile saline and applied in vivo in chronic spinal rats by both intraperitoneal (i.p.) and intrathecal (i.t.) injections, and EMG was recorded both before and after drug injection. In other chronic spinal rats, the inhaled air was transiently switched to 95% oxygen (with 5% CO2) for 60 s, or to 10% CO2 (in air for 30 s). Both gases were delivered into the plexiglass tube holding the rat.
Spinal vascular imaging and blood flow, in vivo
To evaluate blood flow rate in capillaries, in vivo two-photon laser scanning microscopy (TPLSM) was performed in chronic spinal rats48. Briefly, rats were anesthetized with urethane (1.8 g/kg, with a maximum dose of 0.45 g), and then a laminectomy was performed for T10-L4 vertebrae to expose the injured sacrocaudal spinal cord and the uninjured lumbar spinal cord. The exposed spinal segments were bathed in 0.9% saline and the rat was positioned under the microscope with a stereotactic frame stabilizing the vertebrae. Fluorescein isocyanate-dextran (FITC-dextran; 70,000 MW, Sigma-Aldrich) was injected via the lateral tail vein (0.3 mL, 5% [w/v] in saline), after warming the tail in water to dilate the vein. In vivo TPLSM was performed to track RBC velocity in capillaries (8 - 11 μm diameter), using a Leica SP5 MP TPLSM microscope and a Coherent Chameleon Vision II pulse laser tuned to 800 nm. Z-stacks and line scans through the first 200-400 μm of dorsal horn spinal tissue were acquired through the spinal preparation using a 20× 1.0 N.A. water-dipping objective. RBC velocity measurements were made from line scans using Leica MM AF software, with a high frame rate adequate to accurately estimate velocity (∼ 500 fps)48. Velocity was estimated before and after NSD1015 (dissolved in saline) was topically applied to the spinal cord. Velocity was measured in sections of the capillary between pericytes, chosen to be without obvious overall vessel diameter changes, so that the velocity corresponds to the overall capillary flow rate (volume).
We also measured global blood flow rates, using dye injection methods, in chronic spinal rats in the cord below the injury as compared to above the injury, and as compared to normal uninjured rats. Rats were anesthetized and laminectomy performed, as in the in vivo TPLSM experiments, to expose the lumbar and sacrocaudal spinal cord, which was filmed using a Panasonic G3 digital camera with a Leica macro-lens. Then rats were perfused intracardially with the vital dye methylene blue (5% Methylene blue solution, 1 g/ml, Sigma) in saline at room temperature using gravity-fed perfusion (at a height of 1.1 m to provide a common blood pressure close to normal; ∼90 mmHg). Global flow above and below the site of injury was estimated by the time taken for the methylene blue to turn major dorsal surface vessels blue (including the dorsal vein; the time at which the blue signal intensity quantitatively reached 90% of maximum steady state value), thus assuring that the dye fully traversed spinal capillary beds and filled the venous return. Video images were processed in ImageJ. GR127935 (30 μM in saline absorbed into a 4 mm3 piece of Gelfoam, Tocris) was applied locally onto the sacrocaudal spinal cord 5 min before perfusion (by putting the Gelfoam on the cord). For nitric oxide (NO) control experiments, rats were perfused for 2 min (via a pump) with ∼100 ml oxygenated nACSF containing the NO donor sodium nitrite (1 g/l; Fisher) and heparin (300 IU/l) to pre-dilate the vessels. Then the perfusion solution was switched to the 5% methylene blue solution (gravity-fed), and the time taken for cord perfusion was again measured. We have previously anecdotally noticed that, after SCI, the cord caudal to the injury was perfused poorly during preparation for histology, and thus the above methylene blue experiments were initially motivated by these observations. As we found that sodium nitrite in oxygenated nACSF reversed this poor flow problem (see Results), sodium nitrite in nACSF was injected into some rats prior to fixation to improve immunolabeling (see below).
Oxygen concentration measurements, in vivo
Oxygen concentrations were measured in normal uninjured and SCI rats in vivo. Anesthesia (urethane), laminectomy and stereotactic stabilizing were similar to the setup for spinal vascular imaging, described above. The partial pressure of oxygen (pO2) in the spinal cord was measured with an optical sensor49 (Optode, MicroOptode; Unisense) with a 50 μm tip coated with a fluorophore that when excited with 610 nm red light pulses emitted 780 nm infrared light that varied in intensity proportionally to the concentration of nearby oxygen quenching this light (where 1 μM O2 = 0.62 mmHg pO2; calibrated with a VetStat blood gas analyzer in serum, saline and nACSF). The sensor was mounted on a micromanipulator to advance the probe vertically to the dorsal cord surface. The probe either penetrated the pia and then penetrated deep into the grey matter of the dorsal and ventral horn, or dimpled the pia without penetrating it (by > 200 μm) in the dorsal horn; similar pO2 measurements were obtained with either method. The pia dimple method (as diagrammed in Fig-3d) caused less local damage and was thus preferred. Oxygen (pO2) was measured in the cord caudal to the SCI (n = 52; sacral cord), and compared to that rostral to the SCI (n = 39; lumbar cord) and in uninjured rats at equivalent sacral and lumbar regions (n = 34 and 12, respectively). Drugs were dissolved in saline and applied topically onto the cord, in normal rats (GR127935, n = 7; RX821001, n = 15; NSD1015, n = 6) and in chronic spinal rats (GR127935, n = 8; RX821001, n = 14; NSD1015, n = 16). Arterial blood gases (from the carotid artery: pO2, pCO2, pH and hemoglobin oxygen saturation [Sat] were measured; VetStat, IDEXX), end tidal CO2 (CWE, CapStar-100) and respiration rate were monitored to assure that they were in their normal ranges (95 - 105 mmHg, 35 - 45 mmHg, 7.3 - 7.4, 94 - 96%, 3.5 - 4.5% and 60 - 80 bps, respectively), and as necessary were maintained with artificial respiration (via a trachea tube and respirator, CWE SAR1000). Under standard conditions, rats inhaled room air. As a control to test the sensitivity of the spinal cord pO2 measures, rats briefly (∼1 min) inhaled differing mixtures of O2 and CO2, including: oxygen (100%, n = 7; hyperoxia; arterial blood gas pO2: 390 mmHg, pCO2: 35 mmHg; Sat:100%), carbogen (95% O2, 5% CO2; n = 20; blood gas pO2: ∼350 mmHg, pCO2: 50 mmHg; Sat: 100%), air with 10.5% O2 (10.5% O2, 89.5% N2; n = 7; hypoxic condition; blood gas pO2: 50 mmHg, pCO2: 28 mmHg; Sat: 81%), or air with 10% CO2 (n = 5; hypercapnia condition; blood gas pO2: 91 mmHg, PCO2: 57 mmHg; Sat: 93%). Blood gases rapidly returned to normal (within 1 - 2 min) after these manipulations. There were also rapid proportional changes in spinal cord pO2 as arterial pO2 changed, though the spinal pO2 showed sustained increases in pO2, outlasting the arterial changes (see Results). Also, when the sacral spinal cord blood flow was acutely terminated at the end of the experiment (by transection), the spinal cord pO2 dropped to zero within a few seconds. These inhaled gas and blood flow manipulations were routinely used to confirm the calibration and functioning of the pO2 measurement device.
Locomotion analysis and i.t. drug injections
Rats with high thoracic contusions or staggered-hemisections were filmed with a high-speed digital camera (Panasonic, 120 frames/s) while walking across a 1.5 m long Plexiglas runway with a mirror underneath, with markers on their hindlimbs to estimate hip (iliac crest), knee, ankle, and foot (fifth metatarsal) position. The following parameters were used to evaluate locomotion: 1) weight support (height; hip height minus torso width, equivalent to torso clearance above the ground), 2) leg extensor spasms (quantified as the time that the foot over-extends and drags immobile on its dorsal surface, relative to step-cycle duration), 3) plantar steps (the number of plantar steps counted relative to the number of front steps) and 4) foot placement in stance (measured as foot deviation behind the hip at onset of stance). Walking step-cycle cadence was defined by the front legs (front-leg steps/sec) and was chosen to be similar before and after treatments with oxygen or GR127935. To inhale oxygen, rats were put into a chamber filled with 95% O2 (carbogen, not pressurized) for 90 s. GR127935 was given intrathecally (i.t., with one exception given i.p. at 8 mg/kg). The i.t. injection was done under very brief isoflurane anesthesia as previously described21. That is, a standard 26 gauge injection needle (5/8 in) connected to a Hamilton-syringe was inserted by sliding the needle down the caudal aspect of the L5 vertical process to a small opening between the L5 and L6 vertebrae (at cauda equina), obtained by lifting the hips 5 - 10 cm with one hand (rostral) while injecting with the other (caudal). Rats received isoflurane and a saline i.t. injection (30 μl) as a control and 1 h later a drug i.t. injection (GR127935, 10 mM, 30 μl, dissolved in sterile saline) in one experimental session. We quantified walking in control and drug treated rats at ∼5 - 30 min after the i.t. injection, long enough for the rat to be fully alert (rats awoke and walked in < 2 min), but prior to washout of the i.t. injected drugs. A group of rats with contusions also received an i.t. injection of NSD1015 (100 mM in 30 μl), and its effects were determined at 1 and 24 h after injection.
The BBB score is a qualitative 1 - 20 scoring system useful for studying the progression of locomotor recovery as rats go from no walking ability acutely after injury to moderately improved walking in the chronic SCI state. However, in the chronic injured state of severely injured animals, like we studied, the BBB score is much less useful for detecting further improvements in locomotion, because the score saturates and is insensitive to improvements. Specifically, once rats can make occasional weight-supported plantar steps they have a score of 10 and to go beyond 11 they need to coordinate the forelimb and hindlimbs, which never happens after severe injury. Thus, in the chronic SCI state rats remain at about a score of 11, regardless of treatment. Thus, we could not use this score.
Drugs and solutions
Two kinds of ACSF were used in in vitro experiments: mACSF in the dissection dish before recording and nACSF in the recording chamber. mACSF was composed of (in mM) 118 NaCl, 24 NaHCO3, 1.5 CaCl2, 3 KCl, 5 MgCl2, 1.4 NaH2PO4, 1.3 MgSO4, 25 d-glucose, and 1 kynurenic acid. nACSF was composed of (in mM) 122 NaCl, 25 NaHCO3, 2.5 CaCl2, 3 KCl, 1 MgCl2, 0.5 NaH2PO4, and 12 d-glucose. Both types of ACSF were saturated with carbogen (95% O2-5% CO2) and maintained at pH 7.4. The drugs added to the nACSF were 5-HTP, clorgyline, pargyline, tryptamine, L-tryptophan, tyramine, L-tyrosine, 2-phenylethylamine, L-phenyalanine, NSD1015 (Sigma-Aldrich), α-methyl-5-HT, octopamine, SB206553, RX821002, GR127935, dobutamine, vasopressin (Tocris) and zolmitriptan (AstraZenica). All drugs were first dissolved as a 10 – 50 mM stock solution in water before final dilution in ACSF for in vitro ventral root reflexes recording and DIC microscopy or in saline for in vivo oxygen measurements and two photon microscopy, with the exception of zolmitriptan, which was dissolved in minimal amounts of DMSO (DMSO final concentration in nACSF was 0.04%). DMSO alone had no effect on in vitro LLR in vehicle controls, as compare to nACSF control state.
mRNA analysis
Gene expression for pericyte-related signaling proteins (including CD13, encoded by Anpep) was determined from data generated by our recent RNA-seq study of 8 chronic spinal rats (sacral spinal) and 8 age-matched control animals, which were sacrificed 8 weeks after SCI47. Briefly, mRNA was extracted from the whole sacral cord caudal to the SCI site (sacral S2, not including tissue at the site of injury) for each rat sample. This was converted to cDNA, amplified with PCR and sequenced, yielding a library of 14 million mRNA sequences per rat sample. Sequences were compared to the rat genome version rn4 obtained from UCSC, and the number of mRNA sequences corresponding to each gene was counted, with 15,000 different genes detected. To compensate for variability in the amount of total mRNA isolated in each sample, library-size-adjusted read counts were obtained using the ‘estimateSizeFactors’ function of the DESeq R package (Supplementary Figure 12). Differentially expressed (DE) genes after SCI were defined as those with fold change of > 1.2 or < 0.8 (for positively and negatively DE genes, respectively) and with a false discovery rate adjusted p value < 0.05.
Immunolabeling
Rats were euthanized with Euthanyl (BimedaMTC; 700 mg/kg) and perfused intracardially with 100 ml of saline containing sodium nitrite (1 g/l; Fisher) and heparin (300 IU/l, from 1,000 U/ml stock; Leo Pharma) for 3–4 min, followed by 400 ml of 4% paraformaldehyde (PFA; in phosphate buffer at room temperature), over 15 min. Spinal cords were postfixed in PFA overnight at 4°C, cryoprotected in 30% sucrose in phosphate buffer, frozen, and cut on a NX70 cryostat (Fisher Scientific) in horizontal or transverse 20 μm-thick sections. We mounted spinal cord sections on slides and rinsed with Tris-buffered saline (TBS, 50 mM) containing 0.3% Triton X-100 (TBS-TX, 3× 15 min rinses used for all TBS-TX rinses). Sections were incubated overnight at 4°C with the following primary antibodies in TBS-TX: rabbit anti-5-HT (1:5,000; Sigma S5545), mouse anti-glial fibrillary acidic protein (GFAP) (1:500; Millipore MAB360), mouse (1:200; Millipore MAB5384) and rabbit (1:200, Millipore; AB5320) anti-NG2 Chondroitin Sulfate Proteoglycan, rabbit anti-Tryptamine conjugated to glutaraldehyde (1:1000; Advanced Targeting Systems AB-T04), rabbit anti-5-HT1B (1:100; Abcam AB13896), goat anti-Aminopeptidese N/CD13 (CD13, 1:250; R&D systems AF233), rabbit Anti-von Willebrand Factor (VF, 1:100; Millipore AB7356), and sheep anti-AADC (1:200; Millipore AB119). The slides were rinsed again in TBS-TX. For staining of AADC, antigen retrieval was performed by incubating slides in 10 mM citrate buffer (pH 8.5) at 80°C for 30 min prior to primary antibody incubation23. For staining of 5-HT1B, antigen retrieval was performed by immersing slides in TBS with 0.05% tween 20 (pH 9) at 80°C for 40 min; then after 20 min at room temperature, slides were incubated 1 hr with 10% normal goat serum (NGS; Vector, S-1000; blocking procedure). For staining of tryptamine, rats were instead perfused with 0.5-1.0% glutaraldehyde (GA) and 4% formaldehyde (FA) and postfixed in the same GA/FA solution overnight, and cut in transverse sections as before. Then spinal sections (on slides) were incubated in 1% sodium borohydride for 20 minutes, and then underwent 1 h blocking in 10% NGS (Vector, S-1000; or normal donkey serum) in TBS-TX at room temperature. Finally, all sections (for all antibodies) were incubated overnight in TBS-TX with 1% NGS at room temperature. To visualize the labeling of 5-HT, VF, CD13, GFAP, 5-HT1B and NG2, fluorescent secondary antibodies were used, including goat anti-rabbit Texas red (1:200-1:500; Vector T-1000), goat anti-mouse Alexa Fluor 488 (1:200-1:500; Invitrogen A11029), donkey anti-rabbit Alexa Fluor 488 (1:200; Abcam AB1501529), and donkey anti-rabbit Alexa Fluor 555 (1:200, Abcam AB150174) in TBS-TX, applied on slides for 2 h at room temperature. To visualize AADC with fluorescent methods, tyramide amplification was sometimes performed (Invitrogen TSA Kit no. 12), which included tyramide, ABC amplification, and biotinylated Alexa Fluor 488 (Vector PK-6101) or Alexa Fluor 647 (1:200, AB150179; donkey anti-sheep). Alternatively, to view DAB labeling of AADC or tryptamine, biotinylated donkey anti-sheep secondary antibody (1:2,000; Millipore AP184B) or goat anti-rabbit secondary antibody (1:200; Vector ABC kit) was applied at room temperature for 2 h in TBS-TX, followed by DAB-ABC amplification according to the manufacturer's guidelines (ABC, Vector PK-6101; DAB, Vector SK-4100). At times, an 0.1% cresyl-violet counter-stain (1 min incubation) was applied with tryptamine DAB staining. After rinsing with TBS-TX, the slides were serially dehydrated with alcohol, cleared with xylene, and coverslipped in Permount (Sakura Finetek USA, Torrance, CA, USA). Image acquisition was performed by both conventional microscopy (for DAB) and confocal microscopy with a Leica TCS SP2 II Spectral Confocal System (for fluorescence). The latter used 1.3 μm optical sections that were collected into a z-stack of 5–20 μm and subsequently projected into a single image with maximum-intensity sorting (with ImageJ).
To quantify the function of AADC, we measured the AADC-mediated production of 5-HT in chronic spinal rats injected with 5-hydroxytryptophan (5-HTP), followed by fixation for immunolabeling. 5-HT synthesis is a useful surrogate marker of AADC and tryptamine production, because: 1) 5-HT is completely absent after spinal transection23 (and loss of brainstem innervation); 2) exogenous application of 5-HTP, 25 - 30 min prior to fixation, leads to 5-HT synthesis by AADC that is readily visualized with 5-HT immunolabeling23; 3) 5-HT is a tryptamine analog (5-hydroxytryptamine) equally well produced by AADC, compared to tryptamine, so the OH- group on 5-HT can be viewed as a marker that allows us to image tryptamine production; and 4) the 5-HT antibody is readily combined with other antibody labelling (NG2), because it does not rely on glutaraldehyde (GA) fixation, unlike the tryptamine antibody which binds only to glutaraldehyde-conjugated tryptamine and thus requires GA fixation, which is not compatible with many other antibodies (NG2).
In general, we found that antibody binding and labeling was reduced in over-fixed tissue, especially in vasculature that necessarily received the most direct fixative during perfusion fixation. Thus, rather than our standard procedure of perfusing vessels with fixative, vessel labeling with some antibodies (NG2, tryptamine, 5-HT, AADC) was confirmed and improved by an alternative method: after rinsing out the blood from cords by perfusion with ASCF with sodium nitrite (2 min; see above), the cords were directly immersed in fixative (e.g. 4 % PFA), or were perfused with a lower concentration of PFA (2%). This over-fixation problem likely explains the quantitative discrepancies in the literature seen for expression of AADC and its products in vessels23,24. Standard controls in which the primary antibody was omitted were used to confirm the selectivity of the antibody staining. Also, clear labeling of monoamine fibers in normal rats was used as a positive control for AADC and 5-HT staining, and loss of these fibers in chronic spinal rats was used as a negative control.
Data analysis
Data were analyzed in Clampfit 8.0 (Axon Instruments, USA) and Sigmaplot (Jandel Scientific, USA) software. Data are mostly presented as box plots, with a box and horizontal bar within representing the interquartile range and median, respectively. Whiskers (error bars) extend to the most extreme data point that is within 1.5 times the interquartile range from the box. When there are less than ten data points then individual data points are shown (Fig 5) or error bars represent one standard deviation from the mean (Fig 4). Elsewhere data is quoted as mean ± standard error mean (s.e.m.), as indicated. A Student's t-test was used to test for statistical differences before and after treatments, or between normal and injured rats, with a significance level of P < 0.05. Power of tests was computed with α = 0.05. A Kolmogorov-Smirnov test for normality was applied to each data set, with a P < 0.05 level set for significance. Most data sets were found to be normally distributed, as is required for a t-test. For those that were not normal a Wilcoxon Signed Rank Test was instead used with P < 0.05.
Supplementary Material
Editorial summary.
In rat models of spinal cord injury, the region of the spinal cord below the site of injury becomes hypoxic due to inadequate blood flow, due to increased production of neurotransmitters known as ‘trace amines’ that act on pericytes to constrict blood vessels. Alleviation of hypoxia by hyperoxic breathing or inhibition of trace amine synthesis or action improves locomotor function in the injured rats.
Acknowledgments
We thank Frank Geddes and Yonglie Ma for technical assistance. This research was supported by the Canadian Institutes of Health Research (MOP 14697) and the National Institutes of Health (NIH, R01NS47567).
Footnotes
Author Contributions: Y.L. performed all in vitro rat experiments and in vivo pO2 measurements, contributed to all other rat studies, and co-wrote the paper. R.V. and K.F. contributed to the in vivo rat locomotor experiments. I.R.W., K.F., R.V., L.S. and A.M.L.-O. contributed to immunolabelling experiments. I.W., L.S. and M.V.B. contributed to blood flow measurements. L.S. performed all sacral SCI surgeries. M.J.S., S.B. and K.K.F. contributed to analysis and editing. A.F.D. and S.D. performed mRNA-seq analysis. D.J.B. performed in vitro and in vivo rat experiments, directly supervised all the experiments, and co-wrote the paper. K.F. and D.J.B. shared senior authorship.
Competing Financial Interest: The authors have no competing interests as defined by Nature Publishing Group, or other interests that might be perceived to influence the results and/or discussion reported in this manuscript.
References
- 1.Acker T, Acker H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol. 2004;207:3171–3188. doi: 10.1242/jeb.01075. [DOI] [PubMed] [Google Scholar]
- 2.Martirosyan NL, et al. Blood supply and vascular reactivity of the spinal cord under normal and pathological conditions. J Neurosurg Spine. 2011;15:238–251. doi: 10.3171/2011.4.SPINE10543. [DOI] [PubMed] [Google Scholar]
- 3.Pena F, Ramirez JM. Hypoxia-induced changes in neuronal network properties. Mol Neurobiol. 2005;32:251–283. doi: 10.1385/MN:32:3:251. [DOI] [PubMed] [Google Scholar]
- 4.Hamilton NB, Attwell D, Hall CN. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics. 2010;2 doi: 10.3389/fnene.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh Y, Suzuki N. Control of brain capillary blood flow. J Cereb Blood Flow Metab. 2012;32:1167–1176. doi: 10.1038/jcbfm.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–704. doi: 10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reber F, Gersch U, Funk RW. Blockers of carbonic anhydrase can cause increase of retinal capillary diameter, decrease of extracellular and increase of intracellular pH in rat retinal organ culture. Graefes Arch Clin Exp Ophthalmol. 2003;241:140–148. doi: 10.1007/s00417-002-0560-1. [DOI] [PubMed] [Google Scholar]
- 8.Barcroft H, Bonnar WM, Edholm OG, Effron AS. On sympathetic vasoconstrictor tone in human skeletal muscle. J Physiol. 1943;102:21–31. doi: 10.1113/jphysiol.1943.sp004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westcott EB, Segal SS. Perivascular innervation: a multiplicity of roles in vasomotor control and myoendothelial signaling. Microcirculation. 2013;20:217–238. doi: 10.1111/micc.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonvento G, et al. Evidence for differing origins of the serotonergic innervation of major cerebral arteries and small pial vessels in the rat. J Neurochem. 1991;56:681–689. doi: 10.1111/j.1471-4159.1991.tb08203.x. [DOI] [PubMed] [Google Scholar]
- 11.Cohen Z, Bonvento G, Lacombe P, Hamel E. Serotonin in the regulation of brain microcirculation. Prog Neurobiol. 1996;50:335–362. doi: 10.1016/s0301-0082(96)00033-0. [DOI] [PubMed] [Google Scholar]
- 12.Cohen Z, Molinatti G, Hamel E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab. 1997;17:894–904. doi: 10.1097/00004647-199708000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Lincoln J. Innervation of cerebral arteries by nerves containing 5-hydroxytryptamine and noradrenaline. Pharmacol Ther. 1995;68:473–501. doi: 10.1016/0163-7258(95)02017-9. [DOI] [PubMed] [Google Scholar]
- 14.Hardebo JE, Owman C. Barrier mechanisms for neurotransmitter monoamines and their precursors at the blood-brain interface. Ann Neurol. 1980;8:1–31. doi: 10.1002/ana.410080102. [DOI] [PubMed] [Google Scholar]
- 15.Murray KC, et al. Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nat Med. 2010;16:694–700. doi: 10.1038/nm.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown A, Nabel A, Oh W, Etlinger JD, Zeman RJ. Perfusion imaging of spinal cord contusion: injury-induced blockade and partial reversal by beta2-agonist treatment in rats. J Neurosurg Spine. 2014;20:164–171. doi: 10.3171/2013.10.SPINE13113. [DOI] [PubMed] [Google Scholar]
- 17.Kang CE, Clarkson R, Tator CH, Yeung IW, Shoichet MS. Spinal cord blood flow and blood vessel permeability measured by dynamic computed tomography imaging in rats after localized delivery of fibroblast growth factor. J Neurotrauma. 2010;27:2041–2053. doi: 10.1089/neu.2010.1345. [DOI] [PubMed] [Google Scholar]
- 18.Sinescu C, et al. Molecular basis of vascular events following spinal cord injury. J Med Life. 2010;3:254–261. [PMC free article] [PubMed] [Google Scholar]
- 19.Kundi S, Bicknell R, Ahmed Z. The role of angiogenic and wound-healing factors after spinal cord injury in mammals. Neurosci Res. 2013;76:1–9. doi: 10.1016/j.neures.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 20.Murray KC, et al. Polysynaptic excitatory postsynaptic potentials that trigger spasms after spinal cord injury in rats are inhibited by 5-HT1B and 5-HT1F receptors. J Neurophysiol. 2011;106:925–943. doi: 10.1152/jn.01011.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rank MM, et al. Adrenergic receptors modulate motoneuron excitability, sensory synaptic transmission and muscle spasms after chronic spinal cord injury. J Neurophysiol. 2011;105:410–422. doi: 10.1152/jn.00775.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Commissiong JW. The synthesis and metabolism of catecholamines in the spinal cord of the rat after acute and chronic transections. Brain Res. 1985;347:104–111. doi: 10.1016/0006-8993(85)90893-5. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, et al. Synthesis, transport, and metabolism of serotonin formed from exogenously applied 5-HTP after spinal cord injury in rats. J Neurophysiol. 2014;111:145–163. doi: 10.1152/jn.00508.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wienecke J, et al. Spinal cord injury enables aromatic L-amino acid decarboxylase cells to synthesize monoamines. J Neurosci. 2014;34:11984–12000. doi: 10.1523/JNEUROSCI.3838-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry MD. Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J Neurochem. 2004;90:257–271. doi: 10.1111/j.1471-4159.2004.02501.x. [DOI] [PubMed] [Google Scholar]
- 26.Burchett SA, Hicks TP. The mysterious trace amines: protean neuromodulators of synaptic transmission in mammalian brain. Prog Neurobiol. 2006;79:223–246. doi: 10.1016/j.pneurobio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 27.Gozal EA, et al. Anatomical and functional evidence for trace amines as unique modulators of locomotor function in the mammalian spinal cord. Front Neural Circuits. 2014;8:134. doi: 10.3389/fncir.2014.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glaeser BS, Maher TJ, Wurtman RJ. Changes in brain levels of acidic, basic, and neutral amino acids after consumption of single meals containing various proportions of protein. J Neurochem. 1983;41:1016–1021. doi: 10.1111/j.1471-4159.1983.tb09045.x. [DOI] [PubMed] [Google Scholar]
- 29.Gessa GL, Biggio G, Fadda F, Corsini GU, Tagliamonte A. Effect of the oral administration of tryptophan-free amino acid mixtures on serum tryptophan, brain tryptophan and serotonin metabolism. J Neurochem. 1974;22:869–870. doi: 10.1111/j.1471-4159.1974.tb04308.x. [DOI] [PubMed] [Google Scholar]
- 30.Hawkins RA, O'Kane RL, Simpson IA, Vina JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136:218S–226S. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- 31.Boess FG, Martin IL. Molecular biology of 5-HT receptors. Neuropharmacology. 1994;33:275–317. doi: 10.1016/0028-3908(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 32.U'Prichard DC, Greenberg DA, Snyder SH. Binding characteristics of a radiolabeled agonist and antagonist at central nervous system alpha noradrenergic receptors. Mol Pharmacol. 1977;13:454–473. [PubMed] [Google Scholar]
- 33.Bunzow JR, et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- 34.Anwar MA, Ford WR, Broadley KJ, Herbert AA. Vasoconstrictor and vasodilator responses to tryptamine of rat-isolated perfused mesentery: comparison with tyramine and beta-phenylethylamine. Br J Pharmacol. 2012;165:2191–2202. doi: 10.1111/j.1476-5381.2011.01706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Broadley KJ, Fehler M, Ford WR, Kidd EJ. Functional evaluation of the receptors mediating vasoconstriction of rat aorta by trace amines and amphetamines. Eur J Pharmacol. 2013;715:370–380. doi: 10.1016/j.ejphar.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 36.Cohen Z, et al. Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab. 1999;19:908–917. doi: 10.1097/00004647-199908000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Rennels ML, Nelson E. Capillary innervation in the mammalian central nervous system: an electron microscopic demonstration. Am J Anat. 1975;144:233–241. doi: 10.1002/aja.1001440208. [DOI] [PubMed] [Google Scholar]
- 38.Busija DW, Leffler CW. Postjunctional alpha 2-adrenoceptors in pial arteries of anesthetized newborn pigs. Dev Pharmacol Ther. 1987;10:36–46. doi: 10.1159/000457726. [DOI] [PubMed] [Google Scholar]
- 39.Edvinsson L, Degueurce A, Duverger D, MacKenzie ET, Scatton B. Central serotonergic nerves project to the pial vessels of the brain. Nature. 1983;306:55–57. doi: 10.1038/306055a0. [DOI] [PubMed] [Google Scholar]
- 40.Attwell D, et al. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong Z, Sperelakis N. Regulation of L-type calcium channels of vascular smooth muscle cells. J Mol Cell Cardiol. 1995;27:75–91. doi: 10.1016/s0022-2828(08)80009-0. [DOI] [PubMed] [Google Scholar]
- 43.Goritz C, et al. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–242. doi: 10.1126/science.1203165. [DOI] [PubMed] [Google Scholar]
- 44.Dalkara T, Gursoy-Ozdemir Y, Yemisci M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011;122:1–9. doi: 10.1007/s00401-011-0847-6. [DOI] [PubMed] [Google Scholar]
- 45.Hall CN, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60. doi: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burdyga T, Borysova L. Calcium signalling in pericytes. J Vasc Res. 2014;51:190–199. doi: 10.1159/000362687. [DOI] [PubMed] [Google Scholar]
- 47.Narzo AF, et al. Decrease of mRNA Editing after Spinal Cord Injury is Caused by Down-regulation of ADAR2 that is Triggered by Inflammatory Response. Sci Rep. 2015;5:12615. doi: 10.1038/srep12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unekawa M, et al. RBC velocities in single capillaries of mouse and rat brains are the same, despite 10-fold difference in body size. Brain Res. 2010;1320:69–73. doi: 10.1016/j.brainres.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 49.Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15:1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marina N, et al. Brainstem hypoxia contributes to the development of hypertension in the spontaneously hypertensive rat. Hypertension. 2015;65:775–783. doi: 10.1161/HYPERTENSIONAHA.114.04683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schroeder JL, Highsmith JM, Young HF, Mathern BE. Reduction of hypoxia by perfluorocarbon emulsion in a traumatic spinal cord injury model. J Neurosurg Spine. 2008;9:213–220. doi: 10.3171/SPI/2008/9/8/213. [DOI] [PubMed] [Google Scholar]
- 52.Wilson RJ, Chersa T, Whelan PJ. Tissue PO2 and the effects of hypoxia on the generation of locomotor-like activity in the in vitro spinal cord of the neonatal mouse. Neuroscience. 2003;117:183–196. doi: 10.1016/s0306-4522(02)00831-x. [DOI] [PubMed] [Google Scholar]
- 53.van den Brand R, et al. Restoring voluntary control of locomotion after paralyzing spinal cord injury. Science. 2012;336:1182–1185. doi: 10.1126/science.1217416. [DOI] [PubMed] [Google Scholar]
- 54.Attwell D, Mishra A, Hall CN, O'Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab. 2016;36:451–455. doi: 10.1177/0271678X15610340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill RA, et al. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron. 2015;87:95–110. doi: 10.1016/j.neuron.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gorassini MA, Norton JA, Nevett-Duchcherer J, Roy FD, Yang JF. Changes in locomotor muscle activity after treadmill training in subjects with incomplete spinal cord injury. J Neurophysiol. 2009;101:969–979. doi: 10.1152/jn.91131.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kapitza S, et al. Tail spasms in rat spinal cord injury: changes in interneuronal connectivity. Exp Neurol. 2012;236:179–189. doi: 10.1016/j.expneurol.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 58.Beauparlant J, et al. Undirected compensatory plasticity contributes to neuronal dysfunction after severe spinal cord injury. Brain. 2013;136:3347–3361. doi: 10.1093/brain/awt204. [DOI] [PubMed] [Google Scholar]
- 59.Navarrete-Opazo A, Mitchell GS. Therapeutic potential of intermittent hypoxia: a matter of dose. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1181–1197. doi: 10.1152/ajpregu.00208.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller GM. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J Neurochem. 2011;116:164–176. doi: 10.1111/j.1471-4159.2010.07109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.