

Abstract
The conserved cell division protein SepF aligns polymers of FtsZ, the key cell division protein in bacteria, during synthesis of the (Fts)Z-ring at midcell, the first stage in cytokinesis. In addition, SepF acts as a membrane anchor for the Z-ring. Recently, it was shown that SepF overexpression in Mycobacterium smegmatis blocks cell division. Why this is the case is not known. Surprisingly, we found in Bacillus subtilis that SepF overproduction does not interfere with Z-ring assembly, but instead blocks assembly of late division proteins responsible for septum synthesis. Transposon mutagenesis suggested that SepF overproduction suppresses the essential WalRK two-component system, which stimulates expression of ftsZ. Indeed, it emerged that SepF overproduction impairs normal WalK localization. However, transcriptome analysis showed that the WalRK activity was in fact not reduced in SepF overexpressing cells. Further experiments indicated that SepF competes with EzrA and FtsA for binding to FtsZ, and that binding of extra SepF by FtsZ alleviates the cell division defect. This may explain why activation of WalRK in the transposon mutant, which increases ftsZ expression, counteracts the division defect. In conclusion, our data shows that an imbalance in early cell division proteins can interfere with recruitment of late cell division proteins.
Introduction
Bacterial cell division is initiated by polymerization of the tubulin homologue FtsZ into a ring-like structure at midcell. This so-called Z-ring or protoring provides the scaffold for the late cell division proteins that synthesize the division septum. In many bacteria FtsZ polymers are linked to each other by the protein ZapA1,2, and attached to the cell membrane by the peripheral membrane protein FtsA3,4. In Gram-positive and cyanobacteria this function is supported by the peripheral membrane protein SepF5. Most bacteria contain a transmembrane protein that interacts with FtsZ, e.g. ZipA in many proteobacteria, and EzrA in most firmicutes. However, these proteins do not seem to function as membrane anchors, but fulfill a regulatory role in the assembly of the Z-ring5–7. Once the Z-ring is assembled, the so called late cell division proteins are recruited responsible for synthesize of the division septum, such as the peptidoglycan glycosyltransferase FtsW, and the peptidoglycan transpeptidases Pbp2B in Bacillus subtilis and FtsI in Escherichia coli 8–12. Assembly of the late proteins requires the presence of the conserved transmembrane proteins FtsL, DivIC and DivIB in B. subtilis and the homologous proteins FtsL, FtsB and FtsQ in E. coli 13–15. These proteins do not have clear catalytic domains and it is assumed that they play a structural role and somehow regulate the recruitment of late cell division proteins to the Z-ring. In B. subtilis, this recruitment is a strongly cooperative process and the absence of one of the late proteins (except for the non-essential protein DivIB) inhibits assembly of all late cell division proteins16,17. For a recent review on bacterial cell division see e.g.18.
Recently, it was shown that overproduction of SepF in Mycobacterium smegmatis interferes with cell division, resulting in filamentous cells19. Actinomycetes, including the mycobacteria, lack FtsA, and SepF is presumably the only membrane anchor for the Z-ring in these bacteria20. It is therefore probable that a too high concentration of SepF interferes with the formation of Z-rings. We set out to investigate this using the genetically more tractable bacterium Bacillus subtilis.
SepF is highly conserved in Gram-positive bacteria and cyanobacteria, and was first discovered to play a role in cell division in Streptococcus pneumoniae and Synechococcus elongates 21,22. Inactivation of sepF in these organisms leads to severe cell division defects. Deletion of sepF in B. subtilis has a more subtle effect, and only with transmission electron microscopy it becomes apparent that the division septum is strongly deformed when SepF is absent23. Purified SepF assembles into large and regular protein rings with an inner diameter of about 40 nm, which is close to the average thickness of septa. Electron microscopic analyses suggested that the membrane binding domain, an N-terminally located amphipathic helix, is located inside the SepF ring. When SepF rings are mixed with polymerizing FtsZ, FtsZ polymers become attached to the rings and long tubular structures are formed24. Based on these observations, it was postulated that SepF forms arcs on top of the leading edge of nascent septa, thereby attaching FtsZ polymers to the cell membrane and helping them to align parallel to the plane of division5.
Because of these characteristics, it is not surprising that overproduction of SepF in M. smegmatis causes a cell division defect. We found that SepF overproduction has the same effect in B. subtilis. Surprisingly, however, it appeared that the formation of Z-rings was not disturbed when SepF was overproduced. Extensive genetic analyses, including transcriptomics and transposon mutagenesis, show that SepF overproduction blocks the assembly of late cell division proteins and occurs when excess SepF is not interacting with FtsZ. These data indicate that an imbalance in early cell division proteins can interfere with the assembly of late cell division proteins, and may suggest that SepF also plays a role in the recruitment of these proteins.
Results
SepF overexpression is lethal in B. subtilis
Overexpression of SepF in M. smegmatis is lethal and blocks cell division19. Why this is the case is not clear. To investigate this in more detail, we tested whether the same phenotype occurred in the more tractable model organism B. subtilis. A B. subtilis strain was constructed harboring an extra copy of sepF under control of the strong xylose-inducible promoter Pxyl (strain GYQ215). The presence of 1% xylose resulted in a strong overproduction of SepF (Fig. S1). As shown in Fig. 1, overexpression of SepF in B. subtilis resulted in filamentous cells. After approximately 3 h induction, these filamentous cells were unable to grow any further (Fig. 1). This phenotype is comparable to what has been found for M. smegmatis 19.
Figure 1.
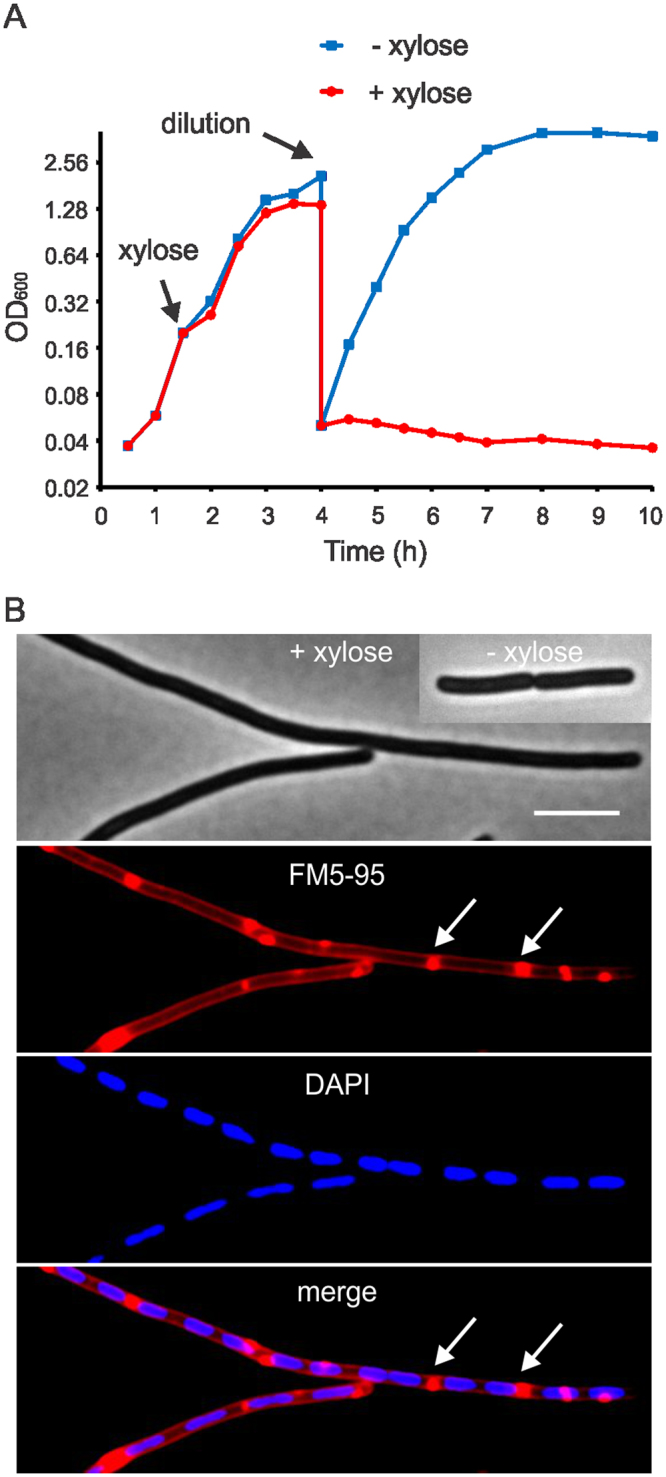
SepF overexpression in B. subtilis. (A) Growth curve of B. subtilis strain GYQ215 containing an extra xylose-inducible sepF gene (amyE::Pxyl-sepF) grown with or without 1% xylose. After 3 h induction, cultures were diluted into fresh medium. (B) Fluorescence light microscopy images of cells after 3 h induction. Membrane and DNA were stained with FM5-95 and DAPI, respectively. Membrane invaginations are indicated by arrows. Scale bar is 5 µm. More images of membrane stained SepF-overexpressing cells can be found in Fig. S2.
Membrane invaginations
Despite the filamentous phenotype, cytosolic separation still seems to occur according to the fluorescent membrane stain (Fig. 1B, white arrows). To gain a better view of these apparent cell division sites, we resorted to Structured Illumination Microscopy (SIM) to increase the resolution (Fig. 2A). At higher resolution, these cell division sites appeared to constitute large membrane invaginations, but no clear cell dividing membranes. The absence of cytoplasmic GFP from these areas indicates that these are not SIM artefacts that can originate from the strong fluorescence of membrane probes25. Finally, transmission electron microscopy also revealed large membrane invagination in all (>20) cells analyzed (Figs 2B, S3). These large membrane structures are reminiscent of the extra membrane material that is formed when the carboxyltransferase AccDA is overproduced (Fig. S4A). AccDA is involved in the first step of fatty acid synthesis26, and its upregulation results in enhanced fatty acid levels and extra membrane synthesis27. To test whether the expression of accDA was increased due to SepF overexpression, we fused the promoter of accDA to the lacZ expression reporter. However, β-galactosidase activity measurements did not reveal any significant lacZ expression differences when SepF was overexpressed (Fig. S4B).
Figure 2.
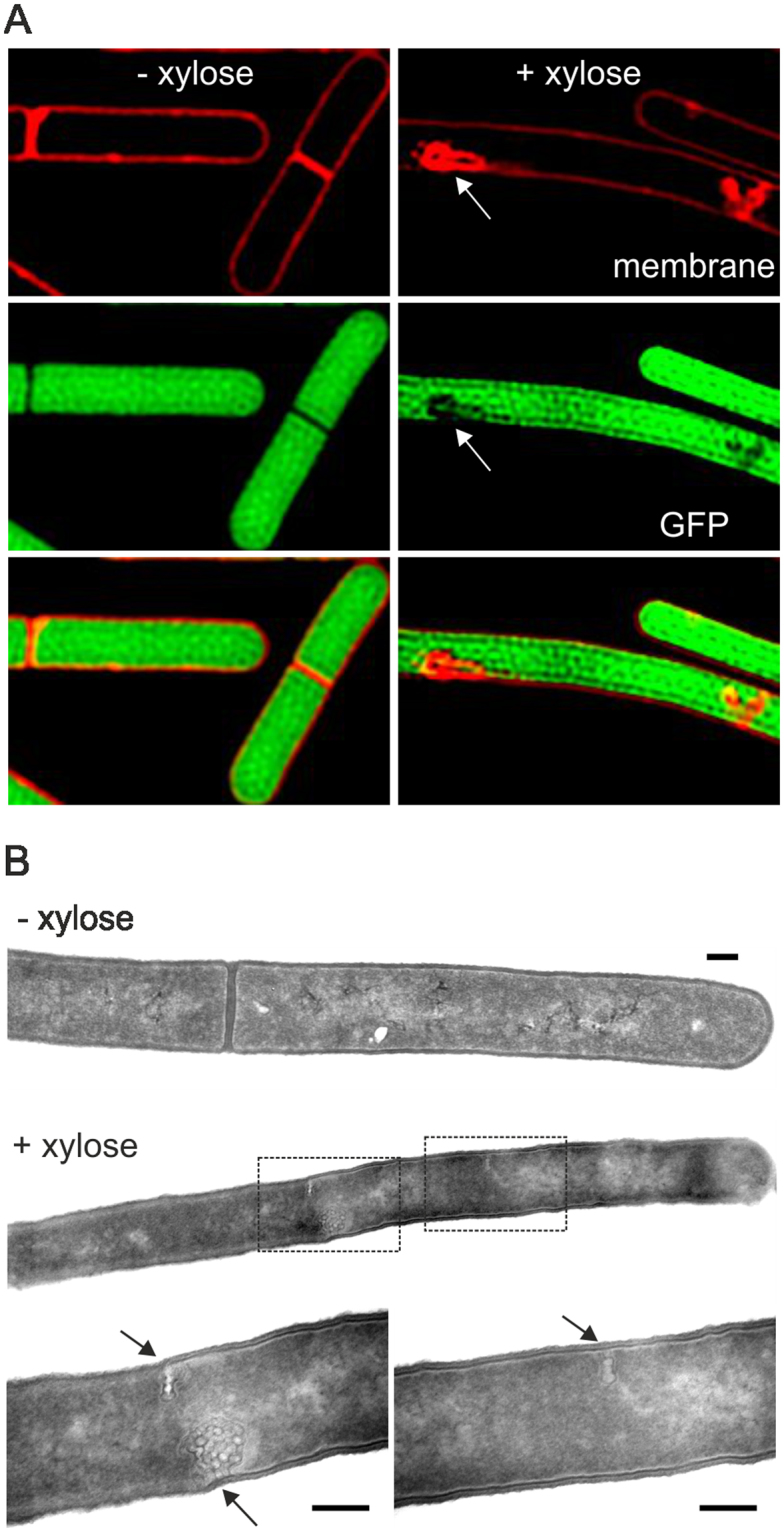
SepF overexpression results in membrane invagination. (A) N-SIM microscopy images of strain GYQ257 (amyE::Pxyl-sepF aprE::PrpsD-gfp) grown without or with 1% xylose for 3 h to induce SepF expression. Membrane was stained with Nile Red. Membrane invaginations indicated by arrows. (B) Transmission electron microscopy images of strain GYQ215 (amyE::Pxyl-sepF) grown without or with 1% xylose. Only longitudinally cut cells revealing whole cells were evaluated. Enlarged images are shown in the lower panels. Arrows indicate membrane invaginations. Scale bar is 200 nm. More examples of these membrane invaginations are shown in Fig. S3.
Localization of cell division proteins
To examine whether Z-ring formation is affected by these membrane invaginations, a fluorescent FtsZ-GFP reporter fusion was introduced into the strain (strain GYQ298). Surprisingly, Z-rings are still being formed, and most of them (~76%) do not co-localize with the large membrane invaginations (Figs 3A, S5).
Figure 3.

Effect of SepF overexpression on FtsZ, EzrA, ZapA and FtsA localization. Fluorescence light microscopy images of (A) strain GYQ298 (amyE::Pxyl-sepF ftsZ::ftsZ-gfp) expressing Ftsz-GFP, (B) strain GYQ30 (amyE::Pxyl-sepF ezrA::ezrA-gfp) expressing EzrA-GFP, and (C) strain GYQ212 (amyE::Pxyl-sepF aprE::Pspac-yfp-ftsA zapA:Pxyl-mcherry-zapA) expressing YFP-FtsA and mCherry-ZapA, grown without or with 1% xylose to induce SepF expression. Membranes were stained with FM5-95. Scale bar is 5 µm.
Although overproduction of SepF does not affect Z-ring assembly, there seems to be a problem with Z-ring closure since small Z-rings, indicative of a closing septum, were never observed (~50 cells observed), also not when localization of the early cell division proteins EzrA, FtsA and ZapA were monitored (Fig. 3B,C). This raised the question whether there was a problem with the assembly of the late cell division proteins. When the localization of the transpeptidase Pbp2B was followed using a GFP-Pbp2B reporter fusion, it appeared that overexpression of SepF completely delocalized this protein (Fig. 4A). Strongly fluorescent Pbp2B foci are observed that colocalizes with membrane invaginations (arrow, Fig. 4A). The correlation between the GFP intensity and the membrane dye (FM5-95) intensity indicated that the strongly fluorescent Ppb2B foci are caused by extra cell membrane material (Fig. S6). Importantly, no fluorescent ring-like structures can be seen. The same results were found when the localization of the late division proteins FtsW and FtsL were tracked using fluorescently labelled reporter fusions (Fig. 4B,C). Thus, overproduction of SepF does not impair Z-ring assembly but interferes with the recruitment of late cell division proteins.
Figure 4.
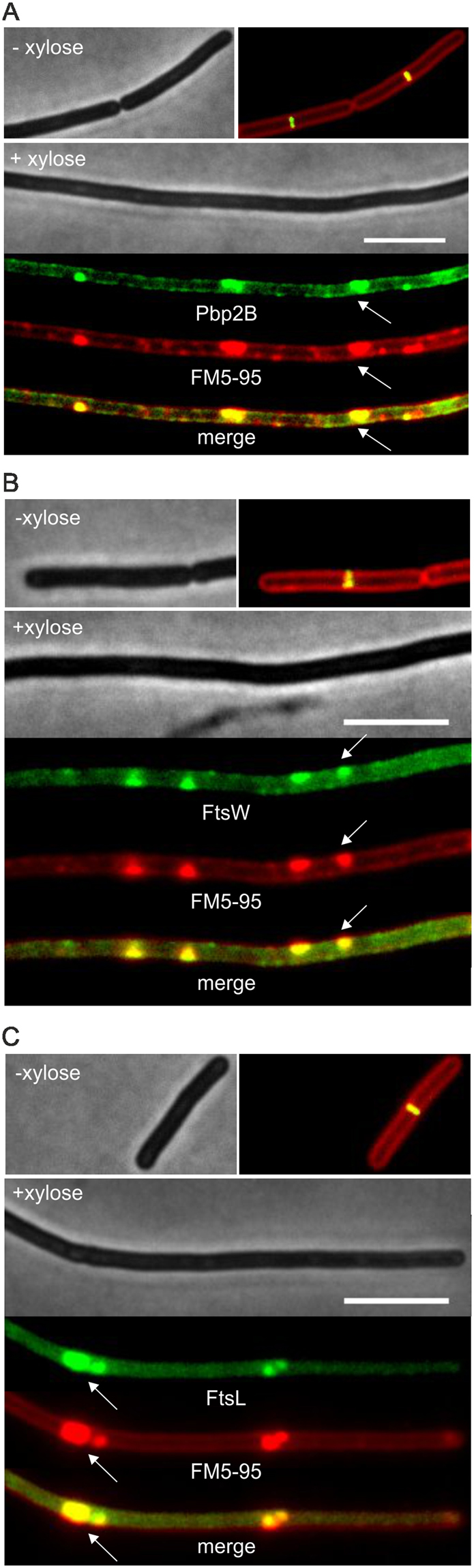
Effect of SepF overexpression on Pbp2B, FtsW and FtsL localization. Microscopic images of (A) strain GYQ72 (amyE::Pxyl-sepF aprE::Pspac-gfp-pbpB) expressing GFP-Pbp2B, (B) strain GYQ74 (amyE::Pxyl-sepF aprE::Pspac-gfp-ftsW) expressing GFP-FtsW, and (C) Strain GYQ204 (amyE::Pxyl-sepF aprE::Pspac-gfp-ftsL) expressing GFP-FtsL, grown in the absence (−) or presence (+) of 1% xylose to induce SepF expression. Membranes were fluorescently stained with FM5-95. Scale bar is 5 µm.
Suppressor mutagenesis
A possible explanation for the failure to form a complete cell division complex could be that high levels of SepF activate a negative regulator that controls assembly of the late cell division proteins. It is important to emphasize that the assembly process of late cell division proteins is not well understood. In fact, after completion of the Z-ring, it takes quite some time, up to 20% of the cell cycle, before the late cell division proteins assemble28,29. It is unknown what is responsible for this delay. To identify a possible assembly regulator, we employed transposon suppressor mutagenesis screening. To this end, plasmid pMarB, carrying the mariner transposon TmYLB-1, was transformed into the SepF overproducing strain GYQ215, and approximately 70,000 transposon mutants were screened on xylose containing plates. After backcrossing of several potential suppressor clones, two stable suppressor mutants were isolated that rescued growth when SepF was overproduced. Sequencing of the insertion sites showed that one mutant harbored a transposon insertion into Pxyl-sepF, and the other transposon mutant contained an insertion into the gene yycH (Fig. 5A).
Figure 5.

Deletion of yycH or yycI rescues growth and cell division. (A) Map of the WalRK locus. (B) Growth curve of strains 168 (wild type), GYQ215 (amyE::Pxyl-sepF), GYQ17 (amyE::Pxyl-sepF ∆yycH), GYQ67 (amyE::Pxyl-sepF ∆yycI) and GYQ471 (amyE::Pxyl-sepF ∆yycJ) grown without or with 1% xylose to induce SepF expression. (C) Phase contrast images of GYQ17 (amyE::Pxyl-sepF ∆yycH) and GYQ67 (amyE::Pxyl-sepF ∆yycI) cells sampled from the cultures in (A) at t = 3 h. Cells were stained with membrane dye FM5-95. Scale bar is 5 µm.
YycH is a transmembrane protein that negatively regulates phosphorylation of the essential WalRK two component system required for cell wall homeostasis and cell division30,31. The response regulator WalR activates the expression of several essential enzymes involved in cell wall remodeling, cell division and cell separation31,32. Activated WalR also induces transcription of ftsZ 33. WalR is phosphorylated by the transmembrane sensor histidine kinase WalK, which is recruited to cell division sites34. To confirm that inhibition of yycH confers resistance to high SepF levels, a SepF-overproducing strain was constructed in which the complete yycH gene was replaced by an erythromycin resistance marker. Indeed, the resulting strain (strain GYQ17) grew normally when 1% xylose was present in the medium and large membrane invaginations were no longer observed (Fig. 5B,C, ~100 cells observed).
YycH is the third gene in the walRK operon (Fig. 5A). The downstream located gene, yycI, codes for another transmembrane protein that interacts with and suppresses WalK35. Interestingly, replacement of the yycI ORF by an erythromycin resistance marker (strain GYQ67) also restored growth (Fig. 5B), while insertion of an erythromycin resistance marker into the downstream located yycJ ORF failed to rescue the growth defect caused by SepF overproduction (not shown). These data might suggest that SepF overproduction reduces the activity of WalR. In line with this, when we introduced a constitutively active WalR variant containing the R204C mutation36 into the SepF overexpression strain (GYQ159), growth was restored and cells divided again, although not with the same frequency compared to wild type cells as the increase in cell lengths indicated (Fig. 6C).
Figure 6.
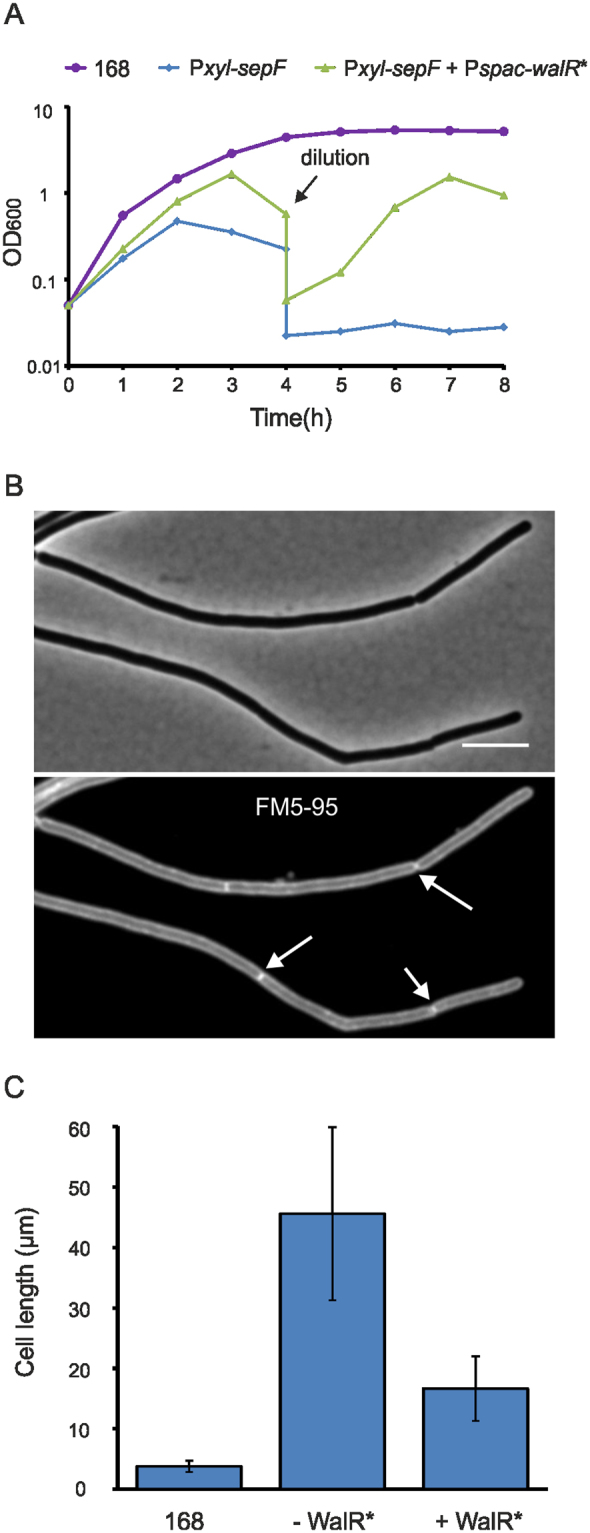
Expression of constitutively activate WalR-R204C. (A) Growth of strain 168, GYQ215 (amyE::Pxyl-sepF) and strain GYQ159 (amyE::Pxyl-sepF aprE::Pspac-walR-R204C) in medium with 1% xylose to induce SepF. In case of strain GYQ159, 1 mM IPTG was added to induce the constitutively active WalR variant R204C (walR*). After 4 h, the culture was diluted. (B) Microscopy images of GYQ159 at t = 6 h. Septa are indicated by arrows. Cells were stained with the membrane dye FM5-95. Scale bar is 5 µm. (C) Cell length measurement of 168, GYQ215 (−WalR*) and GYQ159 (+WalR*). Samples were taken at t = 3 h.
WalK delocalization
WalK is recruited to the Z-ring37. Possibly, overproduction of SepF interferes with the localization of WalK. To examine this, we introduced a Mcherry-WalK reporter fusion into a SepF overproducing strain that also contained a FtsZ-GFP reporter fusion. Interestingly, the Mcherry-WalK fusion completely delocalized from Z-rings when SepF was overexpressed (Fig. 7A, >50 cells observed).
Figure 7.

SepF overexpression and pbpB depletion delocalize WalK. (A) Microscopic images of strain GYQ571 (amyE::Pxyl-sepF ftsZ-gfp aprE::Pspac-mcherry-walK) expressing both FtsZ-GFP and mCherry-WalK, grown without or with 1% xylose to induce SepF overexpression. Additional images shown in Fig. S7. (B) Septum-localization of WalK is abolished in cells depted for pbpB (strain GYQ174, pbpB::Pspac-pbpB amyE::Pxyl-gfp-walK). Membranes were stained with FM5-95. Scale bar is 5 µm.
In a previous study it was shown that the septal localization of WalK requires FtsZ but does not depend on the presence of late cell division proteins37. This finding seems to contradict our results since Z-rings are still being formed when SepF is overproduced, yet WalK is delocalized. To confirm that localization of WalK depends on the presence of late cell division proteins, strain GYQ174 was constructed containing the GFP-WalK reporter fusion and an IPTG-inducible pbpB gene, coding for the late cell division protein Pbp2B. In B. subtilis, assembly of the late division proteins is highly cooperative, and depletion of Pbp2B prevents recruitment of other late cell division proteins to the Z-ring9. As shown in Fig. 7B, depletion of Pbp2B also results in complete delocalization of WalK. Since depletion of Pbp2B does not affect Z-ring formation9, our data suggest that WalK requires late cell division proteins for septal localization. Therefore, delocalization of WalK due to overexpression of SepF might be a consequence of the failed assembly of late cell division proteins.
Transcriptome analysis of SepF overexpression
To confirm that SepF overproduction leads to WalR repression, we determined the genome-wide expression profile of SepF overexpressing cells. To this end, wild type strain 168 and a SepF overproduction strain (YK240) where grown in LB to an OD600 of ~0.5 (log-phase) in the presence of 1.5% xylose, after which cells were harvested and mRNA isolated for transcriptome analysis. Table 1 lists 64 genes that showed a more than 4-fold expression difference between both strains, with an adjusted p-value of less than 0.05 (reliability measure). To verify the transcriptome data, lacZ reporter fusions were constructed with the promoter of the upregulated ydcFG and srfAA operons, and the down-regulated mtnKA operon. The expression response of these fusions were in line with the transcriptome data (Fig. S8). Surprisingly, none of the known WalR-activated genes (e.g. ftsA, ftsZ, tagA-F, Table S1) appeared to be affected by SepF overproduction, and even more puzzling, all the known WalR-repressed genes (iseA, pdaC, wapA, wapI) were down-regulated (Table 1, Table S1). Apparently, the cell division defect caused by SepF overproduction does not seem to originate from the repression of WalR.
Table 1.
Transcriptome analysis of SepF overexpression.
| gene | YK240/wt | p.val | function |
|---|---|---|---|
| ydcF | 65 | 0.000 | hypothetical protein |
| ydcG | 37 | 0.000 | hypothetical protein |
| ydcH | 36 | 0.000 | hypothetical protein |
| sepF | 11 | 0.000 | cell division protein SepF |
| bmrC | 7 | 0.000 | multidrug ABC transporter |
| bmrD | 6 | 0.000 | multidrug ABC transporter |
| catD | 5 | 0.016 | viability in the presence of catechol |
| catE | 5 | 0.016 | essential for the viability in the presence of catechol |
| srfAC | 4 | 0.016 | surfactin synthesis |
| srfAD | 4 | 0.012 | surfactin synthesis |
| ycxA | 4 | 0.001 | hypothetical protein |
| mtnK | −90 | 0.000 | methionine salvage |
| mtnA | −70 | 0.000 | methionine salvage |
| gsiB* | −16 | 0.023 | general stress protein, response to water deficits |
| gspA* | −12 | 0.031 | general stress protein |
| yflT* | −10 | 0.023 | general stress protein, ethanol stress |
| ydaC* | −10 | 0.007 | hypothetical protein |
| ydaD* | −10 | 0.029 | general stress protein, similar to alcohol dehydrogenase |
| wapA | −10 | 0.000 | contact-dependent growth inhibition protein |
| wapI | −10 | 0.000 | immunity protein against toxic activity of WapA |
| yxzC | −10 | 0.000 | hypothetical protein |
| yxiF | −9 | 0.000 | hypothetical protein |
| rapB* | −9 | 0.035 | response regulator, control of sporulation initiation |
| csbC* | −9 | 0.037 | protection against paraquat stress |
| csbD* | −9 | 0.023 | general stress protein, salt and low temperature stress |
| mtnU | −9 | 0.016 | methionine salvage |
| ywzA* | −9 | 0.040 | general stress protein |
| ydaE* | −9 | 0.023 | general stress protein, ethanol and low temperature stress |
| yhxD* | −9 | 0.022 | general stress protein, salt and ethanol stress |
| yxbG* | −8 | 0.016 | general stress protein, similar to glucose 1-dehydrogenase |
| yxiG | −8 | 0.000 | hypothetical protein |
| yxiH | −8 | 0.000 | hypothetical protein |
| yxiJ | −8 | 0.000 | hypothetical protein |
| ysnF* | −8 | 0.007 | general stress protein, ethanol stress |
| ygxB* | −8 | 0.021 | general stress protein |
| ydaS* | −8 | 0.020 | hypothetical protein |
| ytaB* | −8 | 0.023 | general stress protein, salt and ethanol stress |
| katE* | −8 | 0.043 | general stress protein, catalase |
| yxzG | −7 | 0.000 | hypothetical protein |
| yxzI | −7 | 0.000 | hypothetical protein |
| yxiI | −7 | 0.000 | hypothetical protein |
| yxiK | −7 | 0.000 | hypothetical protein |
| yxiM | −7 | 0.000 | similar to rhamnogalacturonan acetylesterase |
| yjgD* | −7 | 0.046 | general stress protein, ethanol and paraquat stress |
| cypC* | −7 | 0.039 | protection against paraquat stress |
| ywtG* | −7 | 0.015 | general stress protein, similar to metabolite transport protein |
| yybO* | −7 | 0.023 | similar to permease |
| chaA* | −6 | 0.034 | calcium export via proton antiporter |
| ywiE* | −6 | 0.036 | cardiolipin synthesis, protection against paraquat stress |
| ycbP* | −6 | 0.023 | general stress protein |
| yqhB* | −6 | 0.016 | general stress protein, protection against oxidative stress |
| ydaT* | −6 | 0.023 | general stress protein, ethanol and low temperature stress |
| bmrU* | −6 | 0.023 | general stress protein, multidrug resistance protein |
| ohrB* | −6 | 0.036 | general stress protein, organic peroxide resistance |
| katX* | −6 | 0.032 | general stress protein, catalase |
| yerD* | −6 | 0.024 | general stress protein, protection against paraquat stress |
| corA* | −6 | 0.021 | general stress protein, similar to magnesium transporter |
| yfkM* | −5 | 0.038 | general stress protein, detoxification of methylglyoxal |
| ykgA* | −5 | 0.006 | general stress protein, salt and ethanol stress |
| yvbG* | −5 | 0.020 | hypothetical protein |
| ydaG* | −4 | 0.021 | general stress protein, protection against paraquat stress |
| ydaP* | −4 | 0.029 | general stress protein, ethanol stress |
| yfkD* | −4 | 0.018 | hypothetical protein |
| yxzF* | −4 | 0.013 | general stress protein |
| ycdF* | −4 | 0.047 | general stress protein, ethanol and low temperature stress |
Genes are listed based on expression fold difference and selected for >4-fold expression difference between wild type (wt) and strain YK240 (Pxyl-sepF) grown in the presence of 1.5% xylose to induce SepF. Genes with adjusted P-value (p.val) larger than 0.05 were discarded. Genes marked with ‘*’ belong to the SigB regulon.
Almost ¾ of the 54 down-regulated genes belong to the SigB regulon. Normally expression of the general stress sigma factor SigB is triggered by a wide variety of intra- and extracellular stresses among which membrane integrity affecting substances such as ethanol38. Why the production of large membrane invaginations by SepF would reduce rather than induce the SigB stress response is unclear.
FtsZ and membrane interaction
The transcriptome data did not show a clear down regulation of cell division genes and Z-rings were still formed when SepF was overexpressed. This raised the question whether binding of SepF to FtsZ is actually important for the observed cell division defects. To test this, phenylalanine 126 of SepF was mutated to a serine. It has been shown that this mutation impairs interaction with FtsZ but neither interferes with SepF polymerization (ring formation) nor with membrane binding5. Surprisingly, when SepF-F126S was overproduced in a background strain lacking wild type sepF, the inhibiting effective on growth was even more severe (Fig. 8A), and the filamentous cells still showed clear membrane invaginations (Fig. 8B). Apparently, SepF does not cause cell filamentation while interacting with FtsZ, which explains why Z-rings are not affected when SepF is overproduced.
Figure 8.
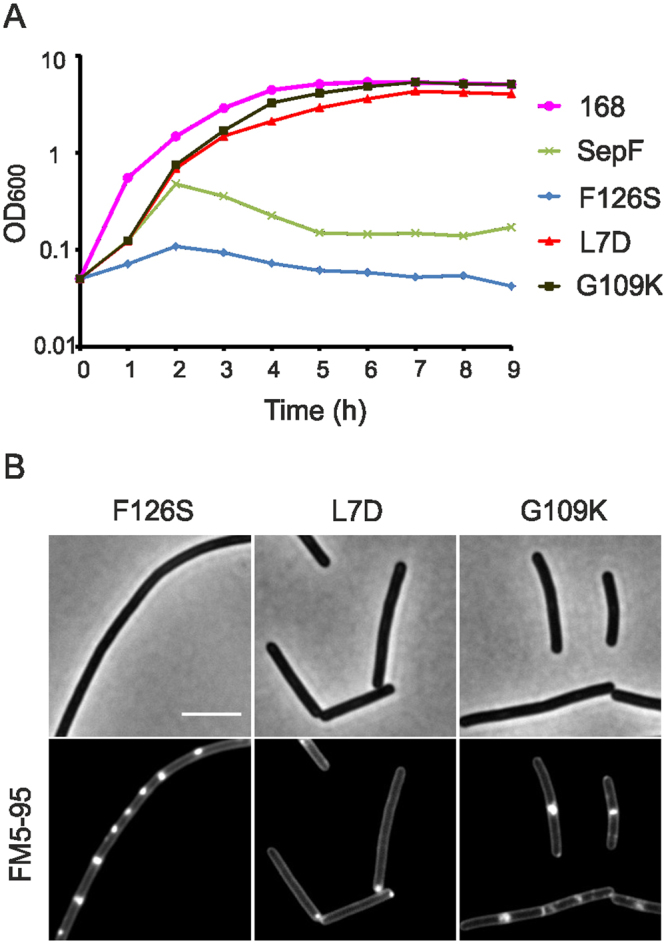
Importance of FtsZ interaction, membrane binding and multimerization domains of SepF. (A) Growth of ∆sepF strains containing ectopic copies of SepF mutants F126S, L7D or G109K, which either impair FtsZ binding, membrane binding or multimerization, respectively (strain GYQ215 amyE::Pxyl-sepF, strain GYQ207 amyE::Pxyl-sepF-F126S ∆sepF, strain GYQ205 amyE::Pxyl-sepF-L7D ∆sepF, strain GYQ206 amyE::Pxyl-sepF-G109K ∆sepF). Cells were grown in medium supplemented with 1% xylose to induce the SepF mutants. (B) Microscopic images of strains GYQ205, GYQ206 and GYQ207 after 3 h growth in the presence of xylose. Membrane invaginations are most apparent when SepF-F126S is expressed. Cells were stained with membrane dye FM5-95. Scale bar is 5 µm.
This finding raised the question whether the lethal effect of too much SepF depends on its interaction with the cell membrane. To test this, a L7 to D7 substitution was made in the N-terminal amphipathic helix of SepF. This mutation has been shown to impair membrane interaction5. When SepF-L7D was overexpressed neither growth nor division was affected (Fig. 8). Induction of SepF containing a G109K mutation, which has been shown to impair polymerization5, also had no effect on growth and division, indicating that polymerization of SepF is required for the cell division defect, as well.
Competition for FtsZ
Overproduction of SepF mutant F126S appeared to be more toxic than overproduction of wild type SepF (Fig. 8A). Possibly, SepF can only exert its inhibiting effect when the protein is not engaged in Z-ring formation. To test this, an extra IPTG-inducible copy of ftsZ (Pspac-ftsZ) was introduced into the SepF overproduction strain (strain GYQ77). As shown in Fig. 9A, increasing FtsZ levels completely mitigated the growth defect. In line with this finding, when FtsZ levels were increased in cells overexpressing the FtsZ-binding mutant SepF-F126S, growth and cell division were still impaired (Fig. 9B,C).
Figure 9.
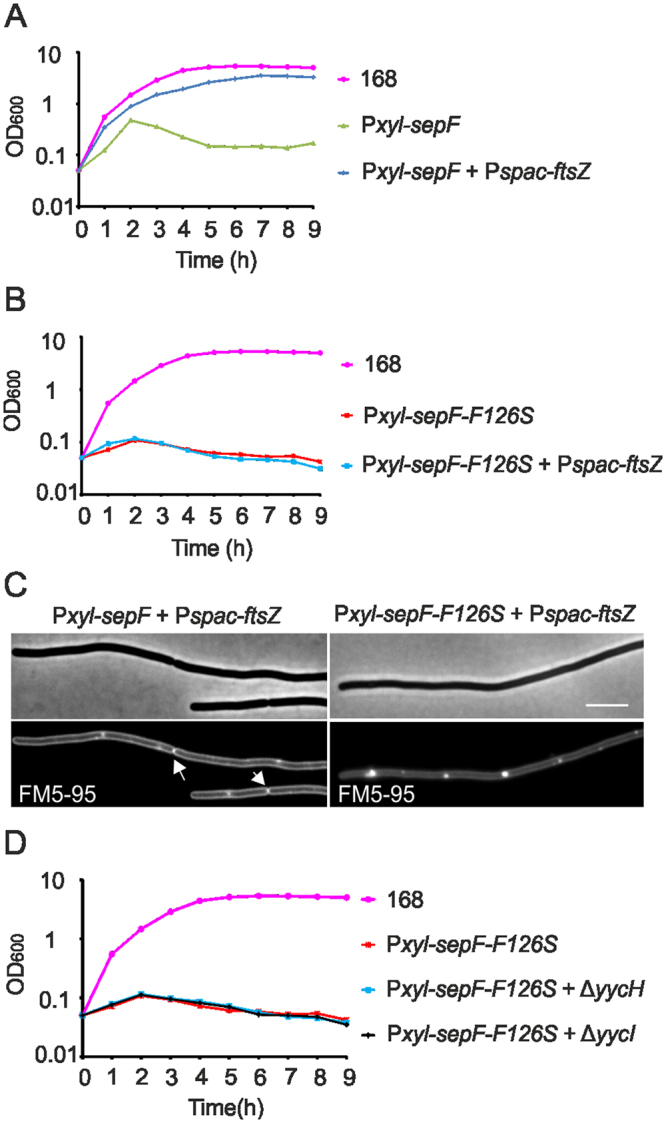
FtsZ induction suppresses division defect. (A) Growth curves of strain 168 (wild type), strain GYQ215 (amyE::Pxyl-sepF), and strain GYQ77 (amyE::Pxyl-sepF aprE::Pspac-ftsZ) in medium containing 1% xylose to induce SepF and 5 mM IPTG to induce FtsZ expression. (B) Growth curves of strain 168 (wild type), strain GYQ207 (amyE::Pxyl-sepF-F126S ∆sepF) and strain GYQ210 (amyE::Pxyl-sepF-F126S ∆sepF aprE::Pspac-ftsZ) in medium containing 1% xylose to induce SepF-F126S and 5 mM IPTG to induce FtsZ expression. (C) Phase contrast and membrane stain of cells from culture GYQ77 (Fig. 9A), and culture GYQ210 (Fig. 9B). Cells were stained with the membrane dye FM5-95. Scale bar is 5 µm. Normal septa are indicated by arrows. (D) Growth curves of strain 168 (wild type), strain GYQ185 (amyE::Pxyl-sepF-F126S ∆sepF), strain GYQ223 (amyE::Pxyl-sepF-F126S ∆sepF ∆yycH) and strain GYQ224 (amyE::Pxyl-sepF-F126S ∆sepF ∆yycI) in medium containing 1% xylose to induce SepF and SepF-F126S.
It appeared that SepF can only perform its cell division inhibiting activity when it is not interacting with FtsZ. Interestingly, it has been shown that overproduction of SepF in a ftsA mutant is not lethal (Fig. S9)39. Both FtsA and SepF interact with the C-terminal tail of FtsZ4,40, suggesting that in a ftsA mutant strain more FtsZ is available for binding to SepF. EzrA too binds to the flexible C-terminal domain of FtsZ, and introduction of an ezrA deletion into the SepF overexpression strain restored growth and cell division as well (Fig. S9). Apparently, SepF competes with both EzrA and FtsA for binding to the C-terminal domain of FtsZ.
The finding that binding to FtsZ blocks the lethal effect of excess SepF provided an explanation as to why deletion of either yycH or yycI restored growth when SepF was overexpressed. The absence of these WalK repressors increases WalR phosphorylation, resulting in increased expression of ftsZ 33. If this is true then a ∆yycH or ∆yycI mutation should not be able to restore the lethal effect of the SepF-F126S mutant (impaired in FtsZ binding). Indeed, overexpression of SepF-F126S was still lethal when either yycH or yycI were absent (Fig. 9D).
Discussion
Overproduction of SepF in the actinomycete M. smegmatis and the firmicute B. subtilis blocks cell division and results in filamentous cells. Here we provide evidence that this is caused by membrane-associated SepF polymers that are not interacting with FtsZ molecules. These free SepF polymers block recruitment of the late cell division proteins to the Z-ring.
Not much is known about the recruitment of these proteins to the Z-ring, but it is assumed that there is no direct contact between FtsZ and late cell division proteins. In E. coli the protein FtsK links early and late division proteins41,42, and FtsA interacts with the late cell division proteins FtsN and FtsI43,44. B. subtilis has two FtsK homologues, SpoIIIE and SftA, but none of these proteins is required for cell division45,46. Gram-positive bacteria do not contain an FtsN homologue, and so far there is no clear evidence for a physical interaction between FtsA and late cell division proteins in Gram-positive bacteria. In fact, actinomycetes do not contain a FtsA homologue. A possible explanation for the observed cell division defect is that SepF interacts with one of the late cell division proteins and that overexpression of SepF sterically hinders the formation of a functional link between FtsZ and the late cell division protein.
Alternatively, the large membrane invaginations formed when SepF is overexpressed might hamper the cooperative assembly of late cell division proteins. These membrane invaginations are reminiscent of the excess membrane structures synthesized when the fatty acid synthase subunits AccDA are overexpressed27. However, the accDA promoter is not activated by SepF overproduction. Also the transcriptome data did not reveal a clear upregulation of genes involved in fatty acid synthesis (Table S2). Presumably, the generation of membrane invagination is related to the mechanism by which SepF binds to the cell membrane. Purified SepF has been shown to strongly deform liposomes and even to cause membrane fusion in vitro 5. The N-terminus of SepF contains a 10 amino acid long amphipathic helix that inserts into the inner phospholipid layer of the cell membrane. Insertion of such bulky peptide moiety will push tightly packed lipid molecules apart, resulting in substantial bending of the membrane47. This effect is enhanced when SepF forms polymers (rings). Therefore, insertion of SepF will curve the membrane towards the cytoplasm and at high concentrations this will lead to invagination of the cell membrane. Possibly, these membrane invaginations, which do not colocalizes with Z-rings (Figs 3, S5), lower the critical concentration of late cell division proteins necessary for the cooperative assembly at Z-rings.
Our transposon experiment suggested that SepF overproduction impairs phosphorylation of WalK, however, this was refuted by the transcriptome data. It turned out that the transposon mutant in yycH very likely suppresses the growth defect by increasing the WalKR activity and ftsZ expression, thus lowering the concentration of free SepF. Nevertheless, we also found that SepF overproduction prevents binding of WalK to the Z-ring. The likely reason for this is that WalK recruitment relies on the assembly of late cell division proteins. This finding contradicts an earlier study in which it was shown that accumulation of WalK at division sites does not depend on the presence of late proteins37. However, in that same study a bacterial two-hybrid assay was used to show that WalK interacts with the late cell division protiens FtsL, DivIB, Pbp2B and FtsW, and in this assay no interaction between WalK and FtsZ could be observed37. These bacterial two-hybrid data seems to be more in line with our finding that WalK localization depends on the presence of the late division proteins.
FtsZ binds several proteins during Z-ring formation, four in the case of B. subtilis: ZapA, EzrA, FtsA and SepF. The latter three proteins all bind to the flexible C-terminus of FtsZ4,40,48. Thus far, it is unknown whether these proteins compete for binding, or whether there is enough space in this ~20 amino acid long domain to simultaneously bind all three proteins. The fact that removing either FtsA or EzrA suppresses the cell division defect of SepF overexpression, as does the induction of FtsZ, suggests that SepF competes with these two proteins for FtsZ binding. Our knowledge of the cell division protein complex, also referred to as the divisome, is still rather limited and it is unclear whether such competition could play a role in the recruitment of late cell division proteins. It has been shown that overexpression of the early cell division proteins ZipA and FtsA in E. coli blocks cell division7,49,50. However, in these studies it was not further investigated which stage in the division process was impaired. Other studies showed that overexpression of EzrA in B. subtilis prevents the formation of Z-rings6, and that overexpression of FtsA in Streptococcus pneumoniae stimulates Z-ring formation51. Our study is the first to show that an imbalance in early cell division proteins affects specifically the recruitment of late cell division proteins.
Methods
Bacterial strains and general methods
Strains used in this study are listed in Table S3. B. subtilis and its derivatives were grown in LB medium, supplemented with the appropriate antibiotic at the following concentrations: kanamycin 5 µg/ml, chloramphenicol 5 µg/ml, erythromycin 2 µg/ml, spectinomycin 50 µg/ml, and phleomycin 2 µg/ml. E. coli Top10 was used for plasmid construction and propagated in LB containing 100 µg/ml ampicillin. All the above strains were cultivated at 37 °C except for the ones harboring FtsZ-GFP, whose detectable fluorescence is stronger at 30 °C. PCR and E. coli transformation were carried out using standard methods, and purification of B. subtilis chromosomal DNA has been described by Venema et al.52. Transformation of competent B. subtilis was accomplished based on the method of the optimized two-step starvation procedure53,54. Gibson assembly cloning was applied for plasmid construction55.
Plasmids construction
PCR primers are listed in Table S4, and all constructs were sequenced to omit possible mutations originating from the PCR reaction. To obtain plasmid pYQ10 harboring the cassette Pxyl-gfp-walK for integration into the amyE locus, walK was amplified using primers YQ99 and YQ101, and genomic DNA of strain 168 as template. The amyE-integration vector part was derived by PCR from pHJS105 using primers EKP22 and YQ100. The two fragments, with 20 bp overlapping sequences, were assembled using Gibson assembly (see New England Biolabs protocol for details). pYQ10 was used as vector template using PCR and primers YQ451 and YQ452, and the mCherry region was derived from genomic DNA of EKB36 using primers YQ450 and YQ453. The two fragments were assembled, resulting in the mCherry-WalK fusion (pYQ141).
GFP reporter fusions integrated into the aprE locus were constructed as follows. Firstly, plasmids carrying Pxyl-gfp-pbpB, Pxyl-gfp-ftsW and Pxyl-gfp-ftsL for integration into the amyE locus were constructed, whereby, pbpB, ftsW, and ftsL were amplified using genomic DNA and primer pairs EKP31 & EKP32, EKP38 & EKP39, EKP33 & EKP34, respectively. The amyE-integration vector part was derived from pHJS105 using primers EKP22 and EKP30. The fragments were assembled using Gibson assembly, resulting in pEKC12, pEKC14 and pEKC13, respectively. Plasmid pTNV9 containing Erm-lacI-Pspac-gfp for aprE locus integration was assembled using two fragments. The plasmid fragment carrying erm-lacI-Pspac and aprE flanking sequences was derived from pAPNC213 erm using primers TerS135 and TerS136, and the GFP fragment was amplified with primers TerS139 and TerS140, and pHJS105 as template. The aprE integration plasmids carrying the GFP reporter fusions aprE::Pspac-gfp-ftsW, aprE::Pspac-gfp-ftsL, aprE::Pspac-gfp-pbpB and aprE::Pspac-gfp-walK, were constructed in the following way. Firstly, the gfp-ftsW, gfp-ftsL, gfp-pbpB and gfp-walK gene fusions were isolated by PCR using primer pairs YQ41 and YQ42 and template DNA pEKC14, pEKC13, pEKC12 and pYQ10, respectively. The vector part containing the Pspac promoter and aprE flanking sequences was amplified with primers YQ43 and YQ44 and plasmid pTNV9 as template. Subsequently, the GFP reporter fusions and the vector part were ligated using Gibson assembly, resulting in pYQ01, pYQ02, pYQ03 and pYQ11, respectively.
To construct plasmid pYQ14 harboring an IPTG-inducible WalR-R204C mutant, full length walR with its own ribosome-binding site was isolated using primers YQ142 and YQ75, and the vector fragment was amplified using primers YQ76 and YQ143 and the aprE-integration plasmid pAPNC213 Kan as template. The two PCR products were fused using Gibson assembly resulting in plasmid pYQ13. Plasmid pYQ14 was constructed with quick-change mutagenesis of plasmid pYQ13, using primers YQ78 and YQ79.
The accDA and ydcF promoter-lacZ reporter fusions were constructed by amplifying the accDA or ydcF promoter regions from the chromosome using primer pairs YQ102 & YQ103, and YQ94 & YQ95, respectively. PCR products were digested with BamHI and HindIII, and ligated into digested pMutin4, resulting in plasmids pYQ40 and pYQ05, respectively. pYQ87 the promoter-lacZ reporter plasmid for aprE locus integration, was assembled from 4 PCR fragments. The plasmid fragment carrying the aprE flanking sequences was derived from pTNV9 using primers YQ73 and YQ218. The spectinomycin resistance cassette was amplified using primers YQ72 and YQ219 and pHJS105 as template. The terminator fragment containing three terminators (t1, t2, t0 from rrnB operon of E. coli) was amplified from pMutin4 with primers YQ220 and YQ216. The lacZ reporter fragment was amplified with primers YQ214 and YQ217 using pMutin4 as template. These 4 PCR fragments were ligated with Gibson assembly resulting in pYQ87. The three terminators are positioned upstream of the promoter-lacZ reporter and downstream of the spectinomycin cassette. PsrfAA and PmtnK promoter regions were amplified from genomic DNA with primer pairs YQ236 & YQ237, and YQ246 & YQ247, respectively, and assembled with a vector fragment derived from pYQ87 using primers YQ214 and YQ215, resulting in pYQ47 and pYQ56, respectively.
The PrpsD-gfp reporter fusion was amplified with primers YQ72 and YQ42, using chromosomal DNA of strain BSS421 as template. The vector containing aprE flanking sequences was amplified with YQ43 and YQ73, and pTNV9 as template. The two fragments were assembled resulting in pYQ73. To construct plasmid pTNV60, for replacement of the chloramphenicol resistance marker (Cm) with the kanamycin resistance marker (Kan) in the B. subtilis genome, first plasmid pTNV42 was constructed using pUC19 amplified by primer pair TerS117 and TerS118, in which the Cm region was inserted. This Cm cassette was amplified with TerS125 and TerS126, and plasmid pAPNC213 Cm as template. Subsequently, pTNV42 was used as vector template in a PCR amplification reaction using primers TerS257 and TerS258. The Kan region was derived from pMarB using primers TerS259 and TerS260. Both fragments were assembled, resulting in pTNV60.
Strain construction
The relevant B. subtilis strains are listed in Table S3. GYQ215 was constructed by transforming chromosomal DNA of YK240 to 168. To construct the mutant SepF-L7D controlled by Pxyl at the amyE locus, the upstream region was amplified by PCR using primers YQ53 and YQ150, and the downstream region was amplified with primers YQ149 and YQ52. Chromosomal DNA of strain GYQ215 was used as template for the PCR reactions, and the two PCR fragments were assembled by Gibson assembly and directly transformed to competent 168 cells, resulting in strain GYQ178. The same procedure was followed for the construction of Pxyl-sepF-G109K and Pxyl-sepF-F126S. Upstream regions of Pxyl-sepF-G109K and Pxyl-sepF-F126S were amplified with primer pairs YQ53 & YQ152, and YQ53 & YQ154, and the downstream regions were amplified using primer pairs YQ52 & YQ151, and YQ52 & YQ153, respectively. The upstream and downstream fragments were ligated with Gibson assembly and transformed to competent 168 cells, generating strain GYQ179 and GYQ180, respectively.
Strain GYQ124 carrying Pxyl-gfp-walK at the amyE locus was constructed by transforming pYQ10 to competent 168 cells. All the integrations at the amyE locus were verified using colony PCR with primers TerS350 and TerS351 and by sequencing if necessary. Plasmids pYQ01, pYQ02, pYQ03, pYQ11 and pYQ141 were transformed to competent 168 cells, resulting in strains GYQ81, GYQ203, GYQ73, GYQ144 and GYQ570, respectively. Strains GYQ195, GYQ217 and GYQ254 were constructed by transforming pYQ47, pYQ56 and pYQ73, respectively to competent 168 cells. To construct strain GYQ152 carrying the Pspac-walR-R204C reporter at the aprE locus, pYQ14 was transformed to competent 168 cells. All aprE locus integrations were verified by colony PCR with primers TerS352 and TerS353.
Strain GYQ135 harbouring the PaccDA-lacZ reporter and GYQ132 harbouring the PydcF-lacZ reporter at their native locus were constructed by transforming plasmid pYQ40 and pYQ05, respectively to competent 168 cells followed by single crossover. Integration of reporter PydcF-lacZ was checked by colony PCR using primers YQ105 and YQ98, and integration of reporter PaccDA-lacZ was verified by colony PCR with primers YQ104 and YQ98. To replace the genomic Cm of EKB36 with Kan, the plasmid pTNV60 was transformed into competence EKB36 carrying Pxyl-mcherry-zapA, resulting in GYQ29. To replace the genomic Cm of 4057 to Kan, the plasmid pTNV60 was transformed into competence 4057 carrying ezrA-gfp, resulting in GYQ28.
Fluorescence light microscopy
Overnight cultures in LB medium were inoculated into fresh LB medium supplemented with the relevant antibiotics and inducers. For the strains harbouring the pbpB depletion construct (e.g. strain GYQ174), the overnight culture was washed by centrifugation to remove IPTG, and inoculated into fresh LB without IPTG. For fluorescence microscopy, samples were taken at exponentially growth and mounted onto microscope slides coated with a thin layer of 1.2% agarose. Images were acquired with Nikon CoolSnap camera with a Zeiss Axiovert 200 M epifluorescence microscope running MetaMorph software, or a Nikon N-SIM microscope equipped with a Nikon APO TIRF × 100/1.49 lens in both EPI and 2D-SIM modes with 488 nm solid-state lasers. When required, cells were incubated with membrane dye FM5-95 (90 µg/ml) or MitoTracker green (500 nM) for 5 min prior to immobilization on microscope slides. When membranes were visualized with Nile Red under the SIM microscope, the coverslip was coated with a poly-dopamine film freshly prepared with 2 g/l dopamine solution56. For DNA staining, 2 µg/ml DAPI was added to the medium after which cells were prepared for microscopy. For cell length measurement, cells were mixed with membrane dye FM5-95 (90 µg/ml), prior to microscopic examinations, and the ChainTracer, based on ImageJ Plugin ObjesctJ, was used for the measurement of cell length57.
Transposon mutagenesis screen
Random transposon mutagenesis of the strain GYQ215 (amyE::Pxyl-sepF) was performed using the plasmid pMarB carrying mariner transposable element TnYLB-1 as described58. The competence GYQ215 cells were transformed with plasmid pMarB and incubated at 30 °C. Several individual colonies carrying pMarB were picked and grown at 30 °C overnight, and then inoculated into fresh LB medium at 30 °C until OD600 reached 0.2–0.3, after which growth was continued at 48 °C for 2 h. Aliquots were frozen and stored at −80 °C. Serial dilutions of each culture were spread on LB plates with kanamycin or erythromycin, and incubated at 50 °C overnight to lose the plasmid pMarB. The aliquot with the highest ratio of KanR/ermR colonies was chosen for further experiment. This aliquot was plated on LB plates supplemented with kanamycin and 1% xylose and incubated at 50 °C overnight. Individual colonies that grew with kanamycin and 1% xylose but failed to survive on plate containing erythromycin and 1% xylose, were picked and checked under the microscope for loss of filamentous phenotype. Two rounds of backcrosses were performed as follows. Chromosome DNA of selected colonies was transformed into wild type 168, and chromosome DNA from the resulting transformant was transformed into GYQ215 to confirm the linkage between transposon insertion and loss of filamentous phenotype. Finally, inverse PCR amplification and subsequent sequencing was performed to determine the transposon insertion site.
Transcriptome analysis
For transcriptome analyses a 8 × 15 k Custom Agilent microarray was used. The NCBI annotation BSU41030 B. subtilis 168 genome 2006-05-02 GenBank, containing information for 4105 transcripts, was used to design three probes per transcript. Overnight cultures of wild type strain 168 and strain strain YK240 were diluted (1:100) into fresh LB medium supplemented with 1.5% xylose and grown at 37 °C until OD600 ~0.5, after which the cells were collected by centrifugation, flash frozen in liquid nitrogen and stored at −80 °C. Frozen pellets were grounded and subjected to RNA extraction as described previously59, yielding RIN values of ≥9.6. Labelling was performed by reverse transcription using random octamers, incorporating Cy3 for the test samples and Cy5 for the common reference, as described60. The common reference was a pool of equal amounts of total RNA taken from all test samples. Hybridization, washing, and scanning was performed as described in the Two-Color Microarray-Based Gene Expression Analysis manual (Version 6.6, Agilent Technologies). Briefly, hybridization mixtures were made by combining 300 ng test (Cy3) and 300 ng common reference (Cy5) material and were subsequently hybridized to the Agilent SurePrint Custom 8 × 15 k microarrays G2509F (Agilent Technologies). Three biological replicates were used for each strain. Raw and normalized data from all arrays were subjected to various quality control checks59. Normalized expression values were calculated using the robust multi-array average (RMA) algorithm61, and collecting and summarizing the intensity values of probes associated with a specific BSU locus tag. Differences in gene expression were statistically analysed using the Limma package in R 2.14.1 (http://cran.r-project.org/). Empirical Bayes test statistics were used for calculating P-values62, and for calculating false discovery rate corrected P-values (adjusted P-values)63. Gene expression data and array design have been deposited at the public repository Gene Expression Omnibus, accession number. Functionality of selected genes (Table 1) were assigned using Subtiwiki64.
Expression of several genes was verified by lacZ promoter fusions and β-galactosidase assays. These were performed using exponentially growing cultures according to the method described by Daniel et al.65.
Western blotting
Samples of exponentially growing cultures were collected, spun down, and washed twice with TE buffer (100 mM Tris-HCl pH 7.5, 1 mM EDTA) and stored on ice. Cell pellets were resuspended in 500 µl buffer (10 mM Tris-HCl PH 7.5) and disrupted by sonication. Cell debris was removed by centrifugation. Protein concentrations were determined with Bio-Rad protein assay and equal amounts of protein were loaded onto a SDS-PAGE gel. Proteins were transferred onto a Hybond-P PVDE membrane (GE Healthcare) using a wet procedure according to standard Western Blotting protocols. A 1:3000 dilution of rabbit polyclonal anti-SepF was used, and anti-rabbit horseradish peroxidase-linked antiserum as secondary antibody with a dilution of 1:10000. Protein bands were imaged using the ImageQuant LAS 4000 mini digital imaging system (GE Healthcare).
Transmission electron microscopy
Cells were placed on an agarose patch, allowed to dry for 2 min, and subsequently fixed with 5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 20 min. Agarose-embedded samples were washed three times with 0.1 M cacodylate buffer (pH 7.4) and then stained with a 1:1 mixture of osmium tetroxide (1%) and K3[Ru(III)(CN)6] (1%) for 30 min, followed by three times washing with water. Samples were then dehydrated in an incubation series with rising concentrations of ethanol as follows: 5 min 30% ethanol, 5 min 50% ethanol, 15 min 70% ethanol (twice), 60 min 80% ethanol, 15 min 90% ethanol, 15 min 96% ethanol, 15 min 100% ethanol, 30 min 100% ethanol (water-free), 5 min propylene oxide, 30 min 1:1 EPON/propylene oxide, 30 min 2:1 EPON/propylene oxide. Samples were then covered with fresh EPON, incubated over night at room temperature, and subsequently allowed to polymerize at 65 °C for 36 h prior to ultrathin sectioning. Pictures were taken with a Jeol 1010 TEM at an electron voltage of 80 kV.
Electronic supplementary material
Acknowledgements
We would like to thank Tjalling Siersma for help with the transcriptome analysis, Edward de Koning for construction of pEKC12, pEKC13 and pEKC14, Terrens N. V. Saaki for construction of pTNV9 and pTNV60, and other members of the Bacterial Cell Biology groups for helpful discussions. We would like to thank Marien Dekker and Jan van Weering (VU University) for support with the electron microscopy, which was performed at the VU/VUmc EM facility, supported by the Netherlands Organization for Scientific Research (NWO, middelgroot 91111009). We would like to thank Yoshi Kawai for providing us with strain YK420, and the Bacillus Genetic Stock Center for providing us with strains BKE40380 and BKE40370. This work was funded by a China Scholarship Council fellowship provided to Y.G. and a Netherlands Organization for Scientific Research (NWO) STW-Vici vici grant (#12128) of L.H.
Author Contributions
Y.G. designed, performed, analysed experiments and wrote the manuscript, M.W. performed and analysed SIM and TEM experiments, M.J. analysed the transcriptome data, and L.H. conceived the project, designed and analysed experiments and edited the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17155-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gueiros-Filho FJ, Losick R. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes & development. 2002;16:2544–2556. doi: 10.1101/gad.1014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Small E, et al. FtsZ polymer-bundling by the Escherichia coli ZapA orthologue, YgfE, involves a conformational change in bound GTP. Journal of molecular biology. 2007;369:210–221. doi: 10.1016/j.jmb.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Pichoff S, Lutkenhaus J. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Molecular microbiology. 2005;55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 4.Jensen SO, Thompson LS, Harry EJ. Cell division in Bacillus subtilis: FtsZ and FtsA association is Z-ring independent, and FtsA is required for efficient midcell Z-Ring assembly. J Bacteriol. 2005;187:6536–6544. doi: 10.1128/JB.187.18.6536-6544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duman R, et al. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4601–4610. doi: 10.1073/pnas.1313978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haeusser DP, Schwartz RL, Smith AM, Oates ME, Levin PA. EzrA prevents aberrant cell division by modulating assembly of the cytoskeletal protein FtsZ. Mol Microbiol. 2004;52:801–814. doi: 10.1111/j.1365-2958.2004.04016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hale CA, de Boer PA. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell. 1997;88:175–185. doi: 10.1016/S0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- 8.Mohammadi T, et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. The EMBO journal. 2011;30:1425–1432. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daniel RA, Harry EJ, Errington J. Role of penicillin‐binding protein PBP2B in assembly and functioning of the division machinery of Bacillus subtilis. Molecular microbiology. 2000;35:299–311. doi: 10.1046/j.1365-2958.2000.01724.x. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Khattar MK, Donachie W, Lutkenhaus J. FtsI and FtsW Are Localized to the Septum in Escherichia coli. Journal of bacteriology. 1998;180:2810–2816. doi: 10.1128/jb.180.11.2810-2816.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercer KL, Weiss DS. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. Journal of bacteriology. 2002;184:904–912. doi: 10.1128/jb.184.4.904-912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meeske AJ, et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature. 2016;537:634–638. doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniel RA, Errington J. Intrinsic instability of the essential cell division protein FtsL of Bacillus subtilis and a role for DivIB protein in FtsL turnover. Molecular microbiology. 2000;36:278–289. doi: 10.1046/j.1365-2958.2000.01857.x. [DOI] [PubMed] [Google Scholar]
- 14.Katis V, Wake R, Harry E. Septal Localization of the Membrane-Bound Division Proteins of Bacillus subtilis DivIB and DivIC Is Codependent Only at High Temperatures and Requires FtsZ. Journal of bacteriology. 2000;182:3607–3611. doi: 10.1128/JB.182.12.3607-3611.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buddelmeijer N, Beckwith J. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Molecular microbiology. 2004;52:1315–1327. doi: 10.1111/j.1365-2958.2004.04044.x. [DOI] [PubMed] [Google Scholar]
- 16.Errington J, Daniel RA, Scheffers D-J. Cytokinesis in bacteria. Microbiology and Molecular Biology Reviews. 2003;67:52–65. doi: 10.1128/MMBR.67.1.52-65.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamba P, Hamoen LW, Daniel RA. Cooperative Recruitment of FtsW to the Division Site of Bacillus subtilis. Frontiers in microbiology. 2016;7:1808. doi: 10.3389/fmicb.2016.01808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haeusser DP, Margolin W. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nature reviews. Microbiology. 2016;14:305–319. doi: 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gola S, Munder T, Casonato S, Manganelli R, Vicente M. The essential role of SepF in mycobacterial division. Mol Microbiol. 2015;97:560–576. doi: 10.1111/mmi.13050. [DOI] [PubMed] [Google Scholar]
- 20.Gupta S, et al. Essential protein SepF of mycobacteria interacts with FtsZ and MurG to regulate cell growth and division. Microbiology. 2015;161:1627–1638. doi: 10.1099/mic.0.000108. [DOI] [PubMed] [Google Scholar]
- 21.Fadda D, et al. Characterization of divIVA and other genes located in the chromosomal region downstream of the dcw cluster in Streptococcus pneumoniae. Journal of bacteriology. 2003;185:6209–6214. doi: 10.1128/JB.185.20.6209-6214.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyagishima Sy, Wolk CP, Osteryoung KW. Identification of cyanobacterial cell division genes by comparative and mutational analyses. Molecular microbiology. 2005;56:126–143. doi: 10.1111/j.1365-2958.2005.04548.x. [DOI] [PubMed] [Google Scholar]
- 23.Hamoen LW, Meile JC, de Jong W, Noirot P, Errington J. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol Microbiol. 2006;59:989–999. doi: 10.1111/j.1365-2958.2005.04987.x. [DOI] [PubMed] [Google Scholar]
- 24.Gündoğdu ME, et al. Large ring polymers align FtsZ polymers for normal septum formation. The EMBO journal. 2011;30:617–626. doi: 10.1038/emboj.2010.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jahn N, Brantl S, Strahl H. Against the mainstream: the membrane-associated type I toxin BsrG from Bacillus subtilis interferes with cell envelope biosynthesis without increasing membrane permeability. Molecular microbiology. 2015;98:651–666. doi: 10.1111/mmi.13146. [DOI] [PubMed] [Google Scholar]
- 26.Cronan JE, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Progress in lipid research. 2002;41:407–435. doi: 10.1016/S0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 27.Mercier R, Kawai Y, Errington J. Excess membrane synthesis drives a primitive mode of cell proliferation. Cell. 2013;152:997–1007. doi: 10.1016/j.cell.2013.01.043. [DOI] [PubMed] [Google Scholar]
- 28.Aarsman ME, et al. Maturation of the Escherichia coli divisome occurs in two steps. Mol Microbiol. 2005;55:1631–1645. doi: 10.1111/j.1365-2958.2005.04502.x. [DOI] [PubMed] [Google Scholar]
- 29.Gamba P, Veening JW, Saunders NJ, Hamoen LW, Daniel RA. Two-step assembly dynamics of the Bacillus subtilis divisome. J Bacteriol. 2009;191:4186–4194. doi: 10.1128/JB.01758-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szurmant H, Nelson K, Kim EJ, Perego M, Hoch JA. YycH regulates the activity of the essential YycFG two-component system in Bacillus subtilis. Journal of bacteriology. 2005;187:5419–5426. doi: 10.1128/JB.187.15.5419-5426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bisicchia P, et al. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Molecular microbiology. 2007;65:180–200. doi: 10.1111/j.1365-2958.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- 32.Salzberg LI, et al. TheWalRK (YycFG) and σI RsgI regulators cooperate to control CwlO and LytE expression in exponentially growing and stressed Bacillus subtilis cells. Molecular microbiology. 2013;87:180–195. doi: 10.1111/mmi.12092. [DOI] [PubMed] [Google Scholar]
- 33.Fukuchi K, et al. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiology. 2000;146:1573–1583. doi: 10.1099/00221287-146-7-1573. [DOI] [PubMed] [Google Scholar]
- 34.Fukushima T, Szurmant H, Kim EJ, Perego M, Hoch JA. A sensor histidine kinase co-ordinates cell wall architecture with cell division in Bacillus subtilis. Molecular microbiology. 2008;69:621–632. doi: 10.1111/j.1365-2958.2008.06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szurmant H, Mohan MA, Imus PM, Hoch JA. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. Journal of bacteriology. 2007;189:3280–3289. doi: 10.1128/JB.01936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domínguez‐Cuevas P, Mercier R, Leaver M, Kawai Y, Errington J. The rod to L‐form transition of Bacillus subtilis is limited by a requirement for the protoplast to escape from the cell wall sacculus. Molecular microbiology. 2012;83:52–66. doi: 10.1111/j.1365-2958.2011.07920.x. [DOI] [PubMed] [Google Scholar]
- 37.Fukushima T, et al. A role for the essential YycG sensor histidine kinase in sensing cell division. Molecular microbiology. 2011;79:503–522. doi: 10.1111/j.1365-2958.2010.07464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hecker M, Pane-Farre J, Volker U. SigB-dependent general stress response in Bacillus subtilis and related gram-positive bacteria. Annu Rev Microbiol. 2007;61:215–236. doi: 10.1146/annurev.micro.61.080706.093445. [DOI] [PubMed] [Google Scholar]
- 39.Ishikawa S, Kawai Y, Hiramatsu K, Kuwano M, Ogasawara N. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol Microbiol. 2006;60:1364–1380. doi: 10.1111/j.1365-2958.2006.05184.x. [DOI] [PubMed] [Google Scholar]
- 40.Król E, et al. Bacillus subtilis SepF binds to the C-terminus of FtsZ. PloS one. 2012;7:e43293. doi: 10.1371/journal.pone.0043293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goehring NW, Gonzalez MD, Beckwith J. Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Molecular microbiology. 2006;61:33–45. doi: 10.1111/j.1365-2958.2006.05206.x. [DOI] [PubMed] [Google Scholar]
- 42.Geissler B, Margolin W. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Molecular microbiology. 2005;58:596–612. doi: 10.1111/j.1365-2958.2005.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen JC, Beckwith J. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Molecular microbiology. 2001;42:395–413. doi: 10.1046/j.1365-2958.2001.02640.x. [DOI] [PubMed] [Google Scholar]
- 44.Corbin BD, Geissler B, Sadasivam M, Margolin W. Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. Journal of bacteriology. 2004;186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biller SJ, Burkholder WF. The Bacillus subtilis SftA (YtpS) and SpoIIIE DNA translocases play distinct roles in growing cells to ensure faithful chromosome partitioning. Mol Microbiol. 2009;74:790–809. doi: 10.1111/j.1365-2958.2009.06893.x. [DOI] [PubMed] [Google Scholar]
- 46.Kaimer C, Gonzalez-Pastor JE, Graumann PL. SpoIIIE and a novel type of DNA translocase, SftA, couple chromosome segregation with cell division in Bacillus subtilis. Mol Microbiol. 2009;74:810–825. doi: 10.1111/j.1365-2958.2009.06894.x. [DOI] [PubMed] [Google Scholar]
- 47.Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nature reviews Molecular cell biology. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 48.Cleverley RM, et al. Structure and function of a spectrin-like regulator of bacterial cytokinesis. Nat Commun. 2014;5:5421. doi: 10.1038/ncomms6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H, Gayda RC. High-level expression of the FtsA protein inhibits cell septation in Escherichia coli K-12. Journal of bacteriology. 1990;172:4736–4740. doi: 10.1128/jb.172.8.4736-4740.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai K, Lutkenhaus J. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. Journal of bacteriology. 1992;174:6145–6151. doi: 10.1128/jb.174.19.6145-6151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mura, A. et al. Roles of the essential protein FtsA in cell growth and division in Streptococcus pneumoniae. Journal of bacteriology (2016). [DOI] [PMC free article] [PubMed]
- 52.Venema G, Pritchard R, Venema-Schröder T. Fate of transforming deoxyribonucleic acid in Bacillus subtilis. Journal of bacteriology. 1965;89:1250–1255. doi: 10.1128/jb.89.5.1250-1255.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. Journal of bacteriology. 1961;81:741. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamoen LW, Smits WK, de Jong A, Holsappel S, Kuipers OP. Improving the predictive value of the competence transcription factor (ComK) binding site in Bacillus subtilis using a genomic approach. Nucleic acids research. 2002;30:5517–5528. doi: 10.1093/nar/gkf698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, et al. Surface and tribological behaviors of the bioinspired polydopamine thin films under dry and wet conditions. Biomacromolecules. 2013;14:394–405. doi: 10.1021/bm3015768. [DOI] [PubMed] [Google Scholar]
- 57.Syvertsson S, Vischer NO, Gao Y, Hamoen LW. When Phase Contrast Fails: ChainTracer and NucTracer, Two ImageJ Methods for Semi-Automated Single Cell Analysis Using Membrane or DNA Staining. PLoS ONE. 2016;11:e0151267. doi: 10.1371/journal.pone.0151267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le Breton Y, Mohapatra NP, Haldenwang WG. In vivo random mutagenesis of Bacillus subtilis by use of TnYLB-1, a mariner-based transposon. Appl Environ Microbiol. 2006;72:327–333. doi: 10.1128/AEM.72.1.327-333.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Surdova K, et al. The conserved DNA-binding protein WhiA is involved in cell division in Bacillus subtilis. Journal of bacteriology. 2013;195:5450–5460. doi: 10.1128/JB.00507-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Knegt GJ, et al. Rifampicin-induced transcriptome response in rifampicin-resistant Mycobacterium tuberculosis. Tuberculosis. 2013;93:96–101. doi: 10.1016/j.tube.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 61.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 62.Berkeley, C. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. E-book available athttp://www.bepress.com/sagmb/vol3/iss1/art3.[PubMed] (2004). [DOI] [PubMed]
- 63.Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological), 289–300 (1995).
- 64.Michna RH, Zhu B, Mader U, Stulke J. SubtiWiki 2.0–an integrated database for the model organism Bacillus subtilis. Nucleic Acids Res. 2016;44:D654–662. doi: 10.1093/nar/gkv1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daniel RA, Williams AM, Errington J. A complex four-gene operon containing essential cell division gene pbpB in Bacillus subtilis. Journal of bacteriology. 1996;178:2343–2350. doi: 10.1128/jb.178.8.2343-2350.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.