

ABSTRACT
Campylobacter jejuni is a leading cause of foodborne illnesses worldwide. As a microaerophilic organism, C. jejuni must be able to defend against oxidative stress encountered both in the host and in the environment. How Campylobacter utilizes a mutation-based mechanism for adaptation to oxidative stress is still unknown. Here we present a previously undescribed phenotypic and genetic mechanism that promotes the emergence of oxidative stress-resistant mutants. Specifically, we showed that a naturally occurring mutator phenotype, resulting from a loss of function mutation in the DNA repair enzyme MutY, increased oxidative stress resistance (OXR) in C. jejuni. We further demonstrated that MutY malfunction did not directly contribute to the OXR phenotype but increased the spontaneous mutation rate in the peroxide regulator gene perR, which functions as a repressor for multiple genes involved in oxidative stress resistance. Mutations in PerR resulted in loss of its DNA binding function and derepression of PerR-controlled oxidative stress defense genes, thereby conferring an OXR phenotype and facilitating Campylobacter survival under oxidative stress. These findings reveal a new mechanism that promotes the emergence of spontaneous OXR mutants in bacterial organisms.
IMPORTANCE Although a mutator phenotype has been shown to promote antibiotic resistance in many bacterial species, little is known about its contribution to the emergence of OXR mutants. This work describes the link between a mutator phenotype and the enhanced emergence of OXR mutants as well as its underlying mechanism involving DNA repair and mutations in PerR. Since DNA repair systems and PerR are well conserved in many bacterial species, especially in Gram positives, the same mechanism may operate in multiple bacterial species. Additionally, we developed a novel method that allows for rapid quantification of spontaneous OXR mutants in a bacterial population. This method represents a technical innovation and may also be applied to other bacterial species. These findings significantly advance our understanding of bacterial mechanisms for survival under oxidative stress.
KEYWORDS: Campylobacter, DNA repair, oxidative stress
INTRODUCTION
Campylobacter jejuni is a major enteric pathogen, and it is considered to be the most common bacterial cause of human gastroenteritis in the world (1). The Centers for Disease Control and Prevention (CDC) of the United States estimated that it causes over 1.3 million cases of foodborne illnesses in the United States annually (2). As a microaerophilic organism and a pathogen transmitted mainly via the food chain, C. jejuni is exposed to highly variable oxygen concentrations. In order to survive, C. jejuni must be able to cope with high environmental oxygen tensions and resist the oxidative stresses encountered both in the host and in the environment (3). Reactive oxygen species (ROS), including superoxide radicals, hydrogen peroxide, and hydroxyl radicals, are generated during aerobic metabolism by the stepwise one-electron reduction of molecular oxygen (4). Additionally, C. jejuni cells are also exposed to ROS produced by the host immune system (5, 6). ROS damages DNA and proteins and causes peroxidation of lipids (4, 7). To survive the stress from ROS, microorganisms, including C. jejuni, have developed various mechanisms to detoxify ROS (3, 8).
In many enteric Gram-negative bacteria such as Escherichia coli and Salmonella enterica serovar Typhimurium, cells defend against oxidative stress by inducing two distinct stress responses, the peroxide stimulon and superoxide stimulon, which are regulated by the superoxide- and peroxide-sensing regulators, SoxRS and OxyR, respectively (7, 8). However, these regulators are not found in the sequenced C. jejuni genome (9), which suggests that oxidative stress defense in Campylobacter is regulated by different systems. van Vliet et al. identified a C. jejuni gene encoding a peroxide regulator (PerR) homolog, which was originally characterized in a Gram-positive bacterium, Bacillus subtilis (10). PerR was then found to regulate a number of oxidative stress-related genes in C. jejuni (10, 11). For example, catalase (KatA) and alkyl hydroperoxide reductase (AhpC), two of the most important factors in defending against oxidative stress, are negatively regulated by PerR in C. jejuni in an iron-dependent manner (10). Several other oxidative-stress-related genes are also regulated by PerR, including superoxide dismutase (SodB) and even perR itself (11, 12). It was later discovered that Fur (ferric uptake regulator), a PerR homolog, coregulates several oxidative stress defense genes (12, 13). Additionally, an OmpR-type response regulator, CosR, was also found to play an important role in oxidative stress defense in C. jejuni (14, 15). Thus, various regulons and enzymes function together in modulating oxidative stress defense in C. jejuni.
DNA base excision repair (BER) is also involved in defending against oxidative stress in living organisms. Specifically, the DNA glycosylase MutYH in humans locates and repairs 8-oxoguanine (8-oxoG) lesions, a common product of oxidative damage to DNA (16). Defects in the DNA glycosylase MutYH are shown to be directly associated with colorectal cancer, which underlines the importance of preventing mutations associated with 8-oxoG (17). The adenine glycosylase MutY in different bacterial species was also reported to be involved in the protection against oxidative stress (8, 18 – 20). Epsilonproteobacteria, including Campylobacter and Helicobacter, have a homolog of the MutY enzyme. A Helicobacter pylori mutY mutant exhibited a greater spontaneous mutation rate than its parent strain when incubated at 5% O2. Interestingly, the mutation rate is further increased by exposing the mutY mutant to atmospheric levels of oxygen, which was not observed in an E. coli mutY mutant (19). Therefore, it is suggested that the H. pylori DNA repair system plays a significant role in defending against oxidative DNA damage.
Recently, we identified a C. jejuni mutator phenotype in the isolate named CMT, which carried a naturally occurring loss-of-function mutation (corresponding to an amino acid change) in the DNA repair gene mutY and showed enhanced emergence of spontaneous antibiotic-resistant mutants (21). Interestingly, we also observed that CMT demonstrated an increased oxidative stress resistance (OXR) phenotype compared to the wild-type (WT) strain with a functional MutY system. This phenotype appeared to be contradictory to that in other bacteria such as H. pylori, since it was reported that H. pylori mutants lacking a functional DNA repair protein are more sensitive than WT cells to oxidative stress induced by agents such as H2O2 (22). Therefore, the unexpected OXR phenotype in the C. jejuni CMT isolate prompted us to study the relationship between the DNA repair system and oxidative stress defense and understand how MutY affects Campylobacter survival under oxidative stress. By utilizing various genetic and biochemical methods, we found that MutY malfunction did not directly contribute to OXR in C. jejuni; instead, it enhanced the spontaneous mutation rate in the perR gene, which consequently results in the loss of function of PerR and derepression of PerR-controlled oxidative stress defense genes, thereby conferring an OXR phenotype in Campylobacter.
RESULTS
Loss-of-function mutation in MutY leads to an oxidative stress resistance phenotype.
All bacterial strains used in this study are listed in Table 1. C. jejuni 11168 (9) and its MutY mutant CMT (21) were tested for the susceptibility to a mixture of paraquat (0.2 mM) and H2O2 (2 mM). Interestingly, the CMT isolate was more resistant to the killing by the mixture of oxidants than C. jejuni 11168 under the same experimental conditions (Fig. 1). This finding suggested that the loss-of-function mutation (an amino acid change) in MutY of CMT might have contributed to the enhanced resistance to oxidative stress. To further investigate the role of MutY in the OXR phenotype, we constructed insertional mutY mutants in C. jejuni 11168 and 81-176 as well as their complements. Using the same oxidative stress susceptibility assay, it was shown that the insertional mutY mutants of C. jejuni 11168 and 81-176 were indeed more resistant to the killing by paraquat and H2O2 than their parent (WT) strains (Fig. 1). Complementation of the mutY mutants fully restored their peroxide susceptibility to the WT level (Fig. 1).
TABLE 1.
C. jejuni strains used in this study
| Strain | Relevant genotype or phenotypea | Source |
|---|---|---|
| 11168 | C. jejuni WT isolate | 9 |
| CMT | A naturally occurring mutant of C. jejuni 11168 with the G199→W change in MutY | 21 |
| 81-176 | C. jejuni WT isolate | 52 |
| 11168ΔmutY | 11168 derivative; ΔmutY::cat insertional mutation | This study |
| 11168ΔmutYC | 11168ΔmutY complement; 11168 ΔmutY::cat 16S::mutY | This study |
| 81-176ΔmutY | 81-176 derivative; ΔmutY::cat insertional mutation | This study |
| 81-176ΔmutYC | 81-176ΔmutY complement; 81-176 ΔmutY::cat 16S::mutY | This study |
| CMT perRC143A | CMT spontaneous OXR mutant; C143→A substitution in perR | This study |
| CMT perRC250A | CMT spontaneous OXR mutant; C250→A substitution in perR | This study |
| CMT perRG319T | CMT spontaneous OXR mutant; G319→T substitution in perR | This study |
| 11168 perRA338Del | 11168 spontaneous OXR mutant; A338 deletion in perR | This study |
| 11168ΔperR | 11168 derivative; ΔperR::cat insertional mutation | This study |
| 11168ΔperRCperR11168 | 11168ΔperR complement; 11168 ΔperR::cat 16S::perR11168 | This study |
| 11168ΔperRCperRC143A | 11168ΔperR complement; 11168 ΔperR::cat 16S::perRC143A | This study |
| 11168ΔperRCperRC250A | 11168ΔperR complement; 11168 ΔperR::cat 16S::perRC250A | This study |
| 11168ΔperRCperRG319T | 11168ΔperR complement; 11168 ΔperR::cat 16S::perRG319T | This study |
| 11168ΔperRCperRA338Del | 11168ΔperR complement; 11168 ΔperR::cat 16S::perRA338Del | This study |
| CMT perRC143ARV | CMT perRC143A derivative, perRC143A reverted to perR11168 | This study |
| CMT perRC250ARV | CMT perRC250A derivative, perRC250A reverted to perR11168 | This study |
| CMT perRG319TRV | CMT perRG319T derivative, perRG319T reverted to perR11168 | This study |
| 11168 perRA338DelRV | 11168 perRA338Del derivative, perRA338Del reverted to perR11168 | This study |
| CMT perRC250ACT | CMT perRC250A derivative, control for perR mutant reversion | This study |
| 11168 PkatA-cat | 11168 fusion construct; 11168 16S::PkatA-cat | This study |
| CMT PkatA-cat | CMT fusion construct; CMT 16S::PkatA-cat | This study |
16S, 16S rRNA gene.
FIG 1.
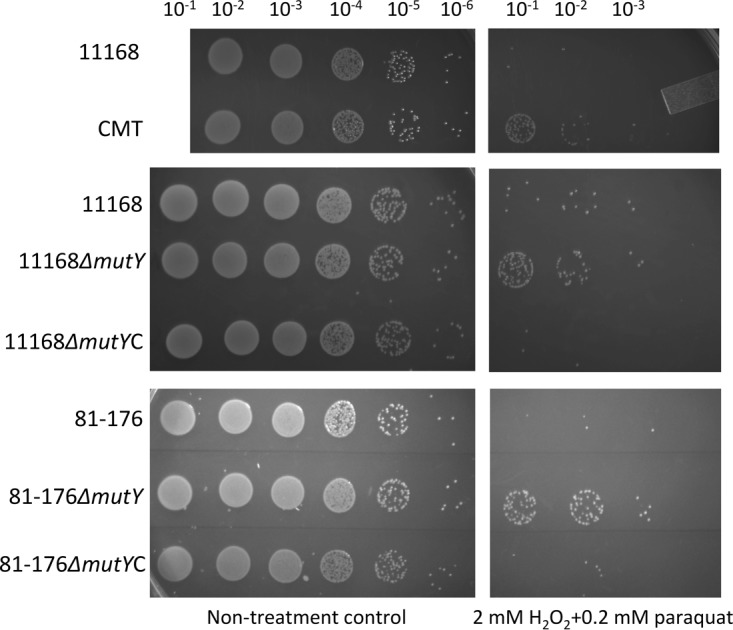
Enhanced resistance of the MutY mutants to oxidative stress as determined by a plate assay. The strains used in the test are labeled on the left. Their 10-fold serial dilutions are indicated at the top. The treatment was done with a mixture of 2 mM H2O2 and 0.2 mM paraquat for 30 min. Each set of assays was carried out in triplicate, and representative results are shown.
Detection of spontaneous OXR mutants after peroxide treatment.
Although the oxidative stress susceptibility assay (Fig. 1) clearly showed that strain CMT was more resistant to the peroxide treatment than strain 11168, the disk inhibition assay did not show a significant difference between the diameter of zone inhibition for the tested oxidants of C. jejuni 11168 and that of the CMT isolate (Table 2). This discrepancy prompted us to hypothesize that the mutator isolate CMT might generate more spontaneous mutants that somehow survive the oxidant treatment better than do WT isolates. To evaluate this possibility, single colonies were randomly picked from the two strains grown on Mueller-Hinton (MH) plates with or without peroxide treatment (see “Oxidative stress sensitivity assay and selection of spontaneous OXR mutants” in Materials and Methods). All picked colonies from MH plates without peroxide treatment (control group) showed similar susceptibility to their parent C. jejuni strains 11168 and CMT (data not shown). However, all three picked colonies from the CMT isolate grown on MH plates after the peroxide treatment were highly resistant to the tested oxidants in the disk inhibition assay, especially to H2O2 (Table 2). For strain 11168, few colonies grew on MH plates after the peroxide treatment, and it was possible to pick only three colonies in total from multiple plates. One of the three colonies was confirmed to have increased peroxide resistance by disk diffusion assay, while the other two remained susceptible to peroxide (Table 2). These results revealed the existence of spontaneous OXR mutants in C. jejuni, which were observed after treating C. jejuni cultures with oxidants. This prompted us to further characterize the OXR phenotype in Campylobacter.
TABLE 2.
Oxidative stress sensitivity of various C. jejuni strains and constructs as measured by disk diffusion assaya
| Strain | Mean diam (mm) of zone of inhibition |
||
|---|---|---|---|
| H2O2 | Cumene hydroperoxide | Paraquat dichloride | |
| 11168 | 22 | 32 | 11 |
| CMT | 23.5 | 32 | 10.5 |
| 11168 perRA338Del | 6 | 25.5 | 6 |
| CMT perRC143A | 6 | 25.5 | 6 |
| CMT perRC250A | 6 | 26 | 6 |
| CMT perRG319T | 6 | 25 | 6 |
| 11168ΔperR | 6 | NDb | ND |
| 11168ΔperRCperR11168 | 19.5 | ND | ND |
| 11168ΔperRCperRC143A | 6 | ND | ND |
| 11168ΔperRCperRC250A | 6 | ND | ND |
| 11168ΔperRCperRG319T | 6 | ND | ND |
| 11168ΔperRCperRA338Del | 6 | ND | ND |
| CMT perRC143ARV | 24 | ND | ND |
| CMT perRC250ARV | 24 | ND | ND |
| CMT perRG319TRV | 26 | ND | ND |
| 11168 perRA338DelRV | 23.5 | ND | ND |
| CMT perRC250ACT | 6 | ND | ND |
Data are means from triplicate plates. The diameter of the disk itself is 6 mm; therefore, 6 mm indicates no obvious inhibition.
ND, not determined.
Increased catalase activity in the spontaneous OXR mutants.
To understand how the OXR mutants were resistant to peroxides, we analyzed cellular catalase activities in the mutants and their parent strains. Whole-protein extracts were prepared from C. jejuni 11168, CMT, and their corresponding spontaneous OXR mutants and then tested for catalase activity. As depicted in Fig. 2, extracts from the spontaneous OXR mutants exhibited significantly more rapid depletion of H2O2 than their parent strains (C. jejuni 11168 and CMT). Additionally, no obvious difference between C. jejuni 11168 and CMT was observed, indicating the MutY mutation itself did not contribute to the increased catalase activity in Campylobacter. Together, these results showed that the spontaneous OXR mutants exhibited significantly elevated catalase activity compared with their parent isolates.
FIG 2.
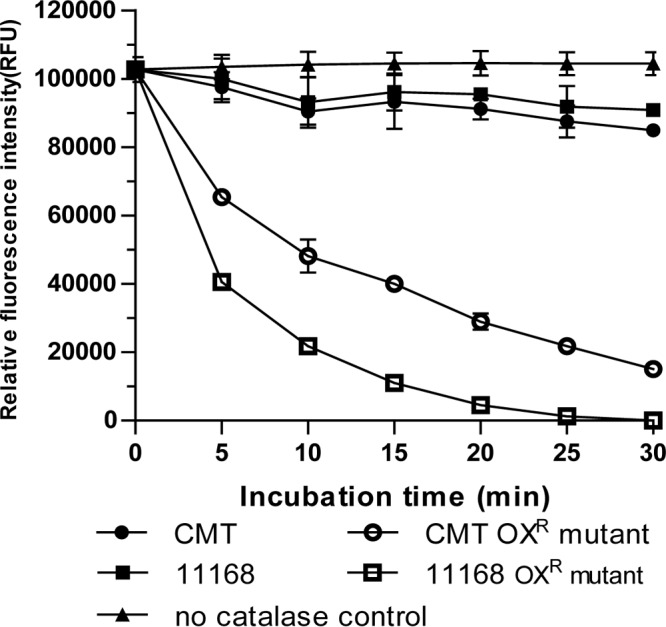
Catalase activities measured by the Amplex Red-based assay. PBS control (filled triangle), 11168 (filled square), CMT (filled circle), 11168 OXR mutant 11168 perRA338Del (open square), and CMT OXR mutant CMT perRC143A (open circle) were used in the assay. Amplex Red reagent was added to the reaction mixtures at 5-min intervals, and fluorescence signals from the oxidation indicator product resorufin were measured in triplicate wells. The experiments were carried out at least three times, and a representative result is shown.
Elevated transcription of oxidative stress resistance genes in the spontaneous OXR mutants.
It was previously reported that catalase (KatA) and alkyl hydroperoxide reductase (AhpC) are two of the most important enzymes in the defense against oxidative stress in Campylobacter (10, 12, 23). In C. jejuni, PerR was shown to mediate iron-dependent regulation of katA and ahpC (10). In this study, the spontaneous OXR mutants showed markedly elevated catalase activity compared with their parent isolates (Fig. 2), suggesting an increased expression of the catalase-producing genes. Therefore, transcription levels of the PerR regulon, including the perR, katA, and ahpC genes in the spontaneous OXR mutants and their parent strains, were compared by utilizing real-time quantitative reverse transcription (RT)-PCR. Results revealed that the transcription of katA and ahpC were significantly upregulated in the OXR mutants compared to 11168 or CMT (Table 3). Particularly, a >500-fold increase in the transcription of katA was detected in the OXR mutants, consistent with their increased catalase production (Fig. 2). Transcription of the perR gene was also upregulated in the spontaneous OXR mutants (Table 3), consistent with the previous finding that PerR is transcriptionally autoregulated in C. jejuni (11). There was no difference in the transcription of the three genes in C. jejuni 11168 and in the CMT isolate (Table 3), indicating that the MutY mutation itself did not affect gene expression of the PerR regulon. Taken together, the results strongly suggest that the hyperresistance to peroxide in the OXR mutants is due to the upregulation of the PerR regulon in Campylobacter.
TABLE 3.
qRT-PCR analysis of katA, perR, and ahpC transcription
| Gene | Fold change ± SD (2−ΔΔCT)a |
||
|---|---|---|---|
| 11168/CMT | CMT perRC143A/CMT | 11168 perRA338Del/11168 | |
| katA | 0.91 ± 0.14 | 564.93 ± 28.99 | 833.24 ± 151.17 |
| perR | 0.89 ± 0.09 | 2.32 ± 0.55 | 4.12 ± 0.53 |
| ahpC | 0.96 ± 0.21 | 12.16 ± 1.49 | 15.30 ± 3.32 |
Fold change of gene transcription for the strain on the left of the slash relative to that on the right of the slash (e.g., the values for 11168/CMT represent fold change for 11168 gene transcription relative to CMT gene transcription).
Occurrence of point mutations in perR of the spontaneous OXR mutants.
The spontaneous OXR mutants from C. jejuni 11168 or CMT were sequenced for mutations in oxidative stress defense-related genes, including katA, ahpC, sodB, perR, and fur. The three OXR mutants originated from CMT were found to carry a single substitution in the perR gene, namely, a C143→A, C250→A, or G319→T transversion, which led to a 48Ser→Tyr, 86Thr→Lys, or 107Glu→Stop codon change, respectively, in the PerR protein (Table 1). Except for the point mutations in perR, no additional mutations were detected in other sequenced genes, including their promoter regions in the OXR mutants of the CMT isolate. The only OXR mutant from 11168 was found to carry a 1-bp deletion (338A) in the perR encoding sequence, resulting in a frameshift after 112 amino acids and truncation of PerR. No mutations were detected in the other analyzed genes. Therefore, all of the analyzed spontaneous OXR mutants carried point mutations in perR, suggesting that these point mutations might have affected the function of PerR, which consequently led to the peroxide resistance phenotype in Campylobacter.
The point mutations in perR of the spontaneous OXR mutants abolished PerR function.
In order to investigate whether PerR from the spontaneous OXR mutants is still functional in Campylobacter, the perR gene sequences from the spontaneous OXR mutants were cloned into a C. jejuni perR knockout strain (ΔperR::cat). These constructs were assayed for H2O2 susceptibility using the disk inhibition assay. The ΔperR::cat mutant complemented with the wild-type 11168 perR sequence served as a control. All the constructs were confirmed with the desired perR insertions in the chromosomal location by DNA sequencing. As shown in Table 2, 11168ΔperR exhibited significantly reduced susceptibility to exposure to H2O2 compared with the wild-type strain 11168. Complementation of the mutant with perR from 11168 (11168ΔperRCperR11168) restored its susceptibility to H2O2. In contrast, the complementation constructs 11168ΔperRCperRC143A, 11168ΔperRCperRC250A, 11168ΔperRCperRG319T, and 11168ΔperRCperRA338Del (Table 1) with a perR containing either a perRC143A, perRC250A, perRG319T, or perRA338Del mutation failed to restore the susceptibility to H2O2 (Table 2). These results indicated that the mutated perR sequences failed to restore the H2O2 susceptibility, suggesting that the point mutations in PerR compromised its function.
Reversion of the point mutations in perR of the OXR mutants restored their susceptibility to H2O2.
To further define the roles of the perR point mutations in oxidative stress resistance in C. jejuni, the point mutations were reverted to the wild-type perR sequence by homologous recombination, creating constructs CMT perRC143ARV, CMT perRC250ARV, CMT perRG319TRV, and 11168ΔperRA338DelRV (Table 1), in which the reversion was accompanied by an insertion of a kanamycin resistance cassette in the adjacent gene cj0323. To ensure that insertion of a kanamycin resistance cassette in cj0323 itself did not affect the susceptibility to oxidative stress, a control construct CMT perRC250ACT (Table 1) was also made, in which the kanamycin resistance cassette was inserted into cj0323 without reversion of the mutation in perR. As shown in Table 2, the control construct did not show any change in H2O2 susceptibility compared with that of the OXR mutants, while all the revertants showed a drastic increase in H2O2 susceptibility, to a level comparable to that of wild-type C. jejuni 11168. Together, these results clearly indicated that point mutations in perR were responsible for the oxidative stress resistance phenotype.
Mutated PerR failed to bind to promoter DNA of katA.
To determine if the point mutations in perR affect its direct interactions with target promoter DNA, a gel mobility shift assay was performed using purified rPerR and the promoter region of katA. Purification was successful with only three of the five rPerRs made in E. coli, including rPerR from 11168, CMT perRC250A, and 11168 perRA338Del (Fig. 3a). Multiple trials for purifying rPerR from CMT perRC143A and CMT perRG319T under the same conditions failed to yield any purified rPerR (see Discussion). Therefore, only three rPerRs were further evaluated by electrophoretic mobility shift assays (EMSAs). As shown in Fig. 3b, the wild-type 11168 rPerR bound to the katA promoter, shown by shift of bands, but not to the control dnaE promoter. However, rPerR from both CMT perRC250A and 11168 perRA338Del did not bind to either katA or dnaE promoter DNA. Together, these findings indicate that the point mutations in rPerR proteins of CMT perRC250A and 11168 perRA338Del resulted in loss of binding of PerR to the katA promoter, explaining the derepression of the PerR regulon.
FIG 3.

Production of rPerR proteins in E. coli and analysis of their binding to the promoter DNA of katA. (a) SDS-PAGE analysis of rPerR produced in E. coli. Lane M, prestained molecular mass markers (Bio-Rad); lanes 1, 3, and 5, whole-cell lysates of E. coli constructs (induced with 1 mM IPTG) expressing PerR of 11168, CMT perRC250A, and 11168 perRA338Del, respectively; lanes 2, 4, and 6, purified rPerR of 11168, CMT perRC250A, and 11168 perRA338Del, respectively, purified by Ni-nitrilotriacetic acid affinity chromatography. (b) EMSAs using purified rPerR and promoter DNA of katA. The various rPerR proteins and their concentrations are indicated above the panel. The dnaE promoter DNA is used as a negative control.
Elevated spontaneous OXR mutation frequency in the CMT isolate.
It was not possible to determine the spontaneous OXR mutation frequency directly by using H2O2 as a selection agent, since it is not stable and tends to be degraded in the culture medium. To quantify the differences in the frequencies of emergence of spontaneous OXR mutants between C. jejuni 11168 and the CMT isolates, we developed a reporter system by fusing the promoter of katA with the chloramphenicol resistance gene cat, yielding constructs 11168 PkatA-cat and CMT PkatA-cat. The Cmr mutation frequency for the CMT PkatA-cat isolate was 5.90 × 10−7, >100-fold higher than that of the C. jejuni 11168 PkatA-cat isolate (5.14 × 10−9). This difference is statistically significant (P < 0.0001; Student's t test) and indicates that the CMT isolate is much more mutable to oxidative stress.
For each of 11168 PkatA-cat and CMT PkatA-cat, 15 or 16 Cmr colonies grown on the selective plates were randomly picked and subsequently sequenced for the perR gene sequence and tested for H2O2 susceptibility. As expected, all sequenced Cmr colonies carried point mutations in the perR gene (see Table S1 in the supplemental material). For strain 11168 PkatA-cat, most (except one) of its Cmr mutants had a 1- to 2-bp insertion or deletion in perR, which resulted in frameshift and truncation of the PerR protein. However, all mutant colonies sequenced for strain CMT PkatA-cat carried a G→T or C→A transversion in the perR encoding region. Interestingly, 13 of the 16 G→T or C→A transversions led to codon changes from an amino acid to termination codons (TAA or TAG), resulting in early translational termination of the PerR protein. The fact that all OXR mutants from the C. jejuni CMT isolate carried a C→A or G→T transversion in perR (Table S1) is consistent with the previous finding that the MutY mutation in CMT promoted G→T and C→A transversions (21). These results revealed that the Cmr mutants generated from strain CMT had distinct mutation patterns compared with those of 11168, but the end outcomes of these mutations in the two strains were the same, i.e., loss of function for PerR. The disk diffusion assay showed that all the Cmr mutants carrying perR mutations from either 11168 PkatA-cat or CMT PkatA-cat were also highly resistant to H2O2 (data not shown), which indicated that the promoter fusions (Cmr) allowed for accurate selection of OXR mutants. Together, these results revealed that spontaneous OXR mutants occurred much more frequently in the CMT isolate than in 11168 and the OXR mutants carried loss-of-function mutations in PerR.
DISCUSSION
Unlike the spontaneous mutants resistant to antibiotics, spontaneous OXR mutants have been rarely reported in bacteria. It was previously reported that a point mutation in the oxyR gene, which encodes the peroxide sensor OxyR, led to the activation of the oxidative stress defense genes and peroxide resistance in a plant pathogen, Xanthomonas campestris (24). With Gram-positive Bacillus subtilis, Chen et al. discovered that spontaneous PerR mutants overproduced KatA and AhpC and displayed an H2O2 resistance phenotype (25). Interestingly, C. jejuni, a Gram-negative bacterium, lacks OxyR and instead possesses the metalloregulator PerR, which is normally found in Gram-positive bacteria such as B. subtilis (26), Staphylococcus aureus (27), Enterococcus faecalis (28), and Streptococcus pyogenes (29). Although the insertional inactivation of PerR has been linked to increased aerotolerance and hyperresistance to H2O2 in C. jejuni (10, 12, 30), spontaneous mutations conferring peroxide resistance in Campylobacter have not been described. In this study, the MutY mutation in the mutator isolate CMT was found to increase the spontaneous mutation frequency in perR, leading to malfunction of PerR, derepression of the PerR regulon, and consequently the increased emergence of OXR mutants in Campylobacter. These findings reveal a new role for a mutator phenotype and the underlying mechanism in promoting the emergence of OXR mutants.
Previously, we found that the MutY mutation in C. jejuni increased the frequencies of G-T or C-A mutation, which resulted in an increase of the spontaneous mutation rate in the gyrA gene and consequently the enhanced occurrence of fluoroquinolone-resistant mutants in C. jejuni (21). The MutY mutation also elevated the mutation rate for β-lactam resistance in C. jejuni (21). In this study, the same MutY mutant isolate was found to increase the emergence of spontaneous OXR mutants, and this was due to the increased G-T or C-A mutation rate in the perR gene. Since the antibiotic-resistant mutants and OXR mutants were developed from spontaneous mutations in different target genes, the two phenotypes did not overlap, and the OXR mutants analyzed in this study did not show enhanced resistance to several tested antibiotics, including fluoroquinolones and β-lactams (data not shown). However, this does not necessarily exclude the possibility that a mutant isolate may harbor mutations in both perR and other genes (e.g., gyrA) targeted by antibiotics, which is expected to occur less frequently and requires the use of both antibiotics and oxidants for selection. Additional studies are needed to assess this possibility.
PerR plays an important role in defense against oxidative stress in C. jejuni (3, 12, 30). PerR represses the expression of katA and ahpC, and insertional mutation of perR results in overexpression of the KatA and AhpC proteins, which makes C. jejuni hyperresistant to peroxide stresses such as cumene hydroperoxide and hydrogen peroxide (10). The oxidative-stress-sensing mechanism by PerR has not yet been fully investigated in Campylobacter, but it was suggested that PerR senses peroxide stress by oxidation of the metal cofactors, including iron and manganese and subsequent oxidation of histidine residues in PerR protein (31). Interestingly, the expression and activity of KatA are still partially regulated by iron in the perR mutant but not in the fur and perR double mutant background (10). Additionally, inactivation of either perR or fur led to a partial reduction in C. jejuni colonization of chicken, but colonization was fully compromised in the PerR and Fur double mutant (12), suggesting that Fur and PerR have overlapping functions in modulating Campylobacter iron regulation and colonization in animals (31). Notably, all C. jejuni spontaneous OXR mutants analyzed in this study carried mutations in perR, not in fur, suggesting a more prominent role of PerR in modulating gene expression associated with oxidative stress defense in C. jejuni.
Analysis of the perR gene sequences in spontaneous C. jejuni OXR mutants revealed amino acid changes and frameshift mutations in PerR (Table 1; see also Table S1 in the supplemental material). The crystal structure of PerR from B. subtilis revealed the structural basis for peroxide sensing by PerR (32). Interestingly, alignment of C. jejuni PerR with the B. subtilis homolog indicated that 48Ser and 84His are conserved in both PerR proteins, with the 48Ser located in the DNA binding helix and 84His as an identified important metal binding site in PerR (see Fig. S1 in the supplemental material), suggesting that mutation of the two amino acids may affect the function of the PerR regulator. Other mutations produced frameshifts and truncation of the PerR protein (136 amino acids [aa] in total). Results of EMSAs indicated that the rPerR protein with either the 84His→Asp mutation or the truncation after the N-terminal 112 aa failed to bind to the katA promoter, indicating that these two mutated forms of PerR lacked DNA binding activity, which implies the derepression of the PerR regulon in C. jejuni. Indeed, real-time PCR data (Table 3) revealed a drastic overexpression of the katA gene in the OXR mutants, which is further correlated with the increased production of catalase activities in OXR mutants (Fig. 2), explaining the enhanced resistance to peroxides. The specific role of the PerR mutations in oxidative stress resistance was further demonstrated by reverting the changes back to the wild-type sequences (Table 1). Together, these results convincingly established the molecular mechanisms involved in the OXR phenotype.
Several attempts to purify the rPerR proteins with the 48Ser→Tyr mutation or the truncated protein with the N-terminal 107 aa from the E. coli host were not successful. IPTG (isopropyl-β-d-thiogalactopyranoside) induction indicated that the two mutant PerR proteins were expressed as determined by SDS-PAGE (see Fig. S2 in the supplemental material). It is unknown whether the two mutations affected the folding or solubility of rPerR in E. coli. Although they could not be purified for the EMSA, it is very likely that they lost the ability to bind to DNA, as the OXR mutants carrying the PerR mutations showed drastically increased expression of katA and ahpC.
The MutY protein specifically repairs the G→T or C→A transversion (33). The C. jejuni CMT isolate has lost the repair function due to a mutation in MutY and consequently shows an elevated mutation rate with the G→T or C→A transversion (21). This was also seen in this study, as all OXR mutants from strain 11168 carried a 1- to 2-bp deletion or insertion in perR, but all the OXR mutants from the CMT isolate carried either a G→T or a C→A transversion (Tables 1 and S1). Reversion of the point mutations in perR to the WT 11168 sequence fully restored their susceptibility to H2O2 (Table 2), indicating that these mutations are responsible for the peroxide resistance of C. jejuni OXR mutants. Thus, it can be concluded that the MutY mutation in CMT promotes oxidative stress resistance via enhancing the spontaneous loss of function mutation rates in perR with a G→T or C→A transversion, leading to overexpression of oxidative defense genes and consequently the increased emergence of OXR mutants in Campylobacter.
In summary, the results in this study revealed how a mutator phenotype elevates the spontaneous mutation rate in perR and consequently promotes the emergence of OXR mutants in C. jejuni. Given the importance of PerR in Campylobacter physiology and colonization of an animal host, permanent loss of PerR function is likely to be detrimental for the long-term adaptation of Campylobacter to various environments. However, elevated mutations in perR may facilitate Campylobacter survival under certain conditions, such as the food production environment, where Campylobacter is exposed to high-level oxidative stress. Multiple studies showed that C. jejuni perR mutants displayed significantly increased peroxide resistance and aerotolerance (10, 12, 30). For example, there were 2 to 3 log more surviving C. jejuni perR insertional mutants than there were wild-type cells after 9 h of incubation under aerobic conditions (30). Recently, aerotolerant C. jejuni and Campylobacter coli have been increasingly isolated from various sources, including chicken or retail meat samples (34 – 36). However, the mechanisms underlying the aerotolerant phenotype remain to be deciphered. Thus, it would be interesting to investigate if perR mutations are involved in aerotolerance under natural conditions. Since PerR is well conserved (26 – 29, 37) and a mutator phenotype due to defects in the DNA repair system has been widely reported in many bacterial species (38 – 42), it would also be intriguing to determine whether mutator isolates also promote the emergence of spontaneous OXR mutants in other bacterial species. Further in-depth examination of the relation between mutators and spontaneous OXR mutants should provide novel insights into the adaptive mechanisms against oxidative stress in bacterial organisms.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. jejuni was cultured using Mueller-Hinton (MH) broth or agar (Difco) at 42°C under microaerobic conditions (5% O2, 10% CO2, and 85% N2). E. coli was grown on Luria-Bertani (LB) agar or in LB broth at 37°C for 24 h under aerobic conditions.
Oxidative stress sensitivity assay and selection of spontaneous OXR mutants.
The oxidative stress sensitivity assay was carried out as previously described with some modification (22). Fresh overnight cultures of C. jejuni strains under microaerobic conditions were adjusted to a final optical density at 600 nm (OD600) of 0.5 (109 CFU/ml) in MH broth. Cultures for each isolate were divided into two groups, with one group containing the addition of a final concentration of a mixture of 0.2 mM paraquat dichloride and freshly prepared 2 mM H2O2 and the other group with no treatment, as control. All cultures were incubated at room temperature for 30 min and then serially diluted and plated on MH agar plates, which were incubated for 2 to 4 days under microaerobic conditions. Colonies on the plates were counted for CFU calculation, and single colonies were randomly picked for detection of spontaneous OXR mutants. The picked colonies were subsequently subjected to disk inhibition assay with oxidants as described below. Those colonies that were highly resistant to the tested oxidants in the disk inhibition assay were stocked for further analysis and characterization.
Disk inhibition assay.
In order to quantitatively compare the oxidative stress sensitivities of different C. jejuni isolates, a disk inhibition assay was performed as described previously (12). Each C. jejuni isolate was tested for susceptibility to 3% H2O2 and 3% cumene hydroperoxide in dimethyl sulfoxide (DMSO) and 100 mM paraquat dichloride in H2O, respectively. DMSO, which was used for preparing the cumene hydroperoxide solution, was used as a control.
Determination of catalase activities in C. jejuni WT and spontaneous OXR mutants.
Overnight cultures of C. jejuni WT strains and their corresponding spontaneous OXR mutants grown on MH plates were washed twice with phosphate-buffered saline (PBS) buffer (pH 7.4) and then resuspended and normalized to an OD600 of 5 in PBS. One milliliter of the suspension was centrifuged at 6,000 × g for 5 min, and the supernatant was discarded. To the cell pellet was added 500 μl of B-PER II bacterial protein extraction reagent (Thermo Fisher Scientific). Whole-cell proteins were extracted using the protocol recommended by the manufacturer. Soluble lysates were then obtained by centrifugation at 10,000 × g for 20 min at 4°C. The catalase activity in the whole-cell extract was determined with the Amplex Red catalase assay kit from Molecular Probes using the methods indicated in the kit with some modifications. For the assay (Molecular Probes), catalase in a sample first reacts with H2O2 to produce water and oxygen (O2). Then, the Amplex Red reagent is added to the reaction mixture and detects any unreacted H2O2 in the presence of horseradish peroxidase (HRP) to produce the fluorescent oxidation product resorufin. Therefore, as the concentration of H2O2 decreases in the reaction mixture due to catalase activity, the fluorescent signal from the oxidation indicator product, resorufin, decreases correspondingly. In this study, the experiment was carried out in a 96-well plate. First, 100 μl of 20 μM H2O2 in PBS buffer (pH 7.4) was aliquoted into each well. Subsequently, 10 μl of 1:4,000-diluted whole-cell extracts of Campylobacter or PBS buffer was quickly added into the wells containing H2O2. For each cell extract, three wells were used for each time point measurement. At 5-min intervals for 30 min, the Amplex Red reagent was added into the designated wells. The fluorescence of resorufin was measured using FLUOstar Omega (BMG Labtech, Offenburg, Germany) with excitation and emission wavelengths of 545 nm and 590 nm, respectively.
qRT-PCR analysis of transcription of oxidative-stress-related genes.
Specific primers for the 16S rRNA gene, katA, ahpC, and perR in Campylobacter were designed using the Primer3 online interface (http://bioinfo.ut.ee/primer3/) and are listed in Table S2 in the supplemental material. C. jejuni 11168, MutY-deficient CMT, and their spontaneous OXR mutants were grown in MH broth for 16 h under microaerobic conditions. Total RNA purification from the cultures and subsequent real-time quantitative RT-PCRs (qRT-PCRs) were carried out as previously described (43). The relative changes (n-fold) in gene transcriptions between the parent strains and their spontaneous OXR mutants were calculated using the 2−ΔΔCT method (where CT is threshold cycle) (44).
DNA sequence analysis of oxidative-stress-related genes.
The total DNA from each C. jejuni strain was prepared by boiling the cells for 15 min. The supernatant was directly used as the template for PCRs. The encoding sequences and the promoter regions of katA, ahpC, sodB, perR, and fur were amplified using a DNA template prepared from the parent strains and their spontaneous OXR mutants. The PCR products were sequenced in both directions with PCR primers used for the amplification (Table S2).
Construction of insertional perR and mutY mutants in C. jejuni isolates.
Insertional mutants of perR and mutY were produced using natural transformation and homologous recombination. Primer pair perR-5F/perR-5R was used to amplify the 5′ part of perR and its upstream region (perR-5′ fragment), while primer pair perR-3F/perR-3R was used to amplify the 3′ part of perR and its downstream region (perR-3′ fragment). The primer pair cat-F/cat-R was used to amplify the cat gene from the pRY112 plasmid, encoding chloramphenicol resistance (45). All PCRs were performed using the Phusion High-Fidelity DNA Polymerase (New England BioLabs, USA). The PCR-amplified perR-5′ fragment and the perR-3′ fragment were linked with the cat gene between them by overlap PCR using the primers listed in Table S2, resulting in the generation of the perR-5′-cat-perR-3′ construct, which was then purified and used to naturally transform C. jejuni 11168. Transformants were screened on MH agar plates containing 10 mg/liter chloramphenicol. The construct 11168ΔperR with the insertion of the cat cassette in the perR gene was confirmed by chromosomal DNA amplification using primers perR-5F/perR-3R. The same strategies were utilized to construct the insertional MutY mutants 11168ΔmutY and 81-176ΔmutY in C. jejuni 11168 and 81-176, respectively.
Complementation of the perR and mutY mutants.
The 11168ΔperR mutant was complemented by inserting a perR gene from C. jejuni OXR mutants in a chromosomal location between the 16S and 23S rRNAs as described by Muraoka and Zhang (46), with some modifications. Briefly, the pRRK plasmid, which contains an aphA3 cassette in the orientation opposite to that of the ribosomal genes, was linearized by PCR using primers pRRK-lF and pRRK-lR. Primers perR-cF and perR-cR were used to amplify the intact perR, including its promoter region and ribosome binding site from C. jejuni 11168 and spontaneous OXR mutants, i.e., 11168 perRA338Del, CMT perRC143A, CMT perRC250A, and CMT perRG319T (Table 1). The perR amplicons and linearized pRRK plasmid were ligated utilizing the SLiCE cloning method (47), to obtain plasmid construct pRRK-perR with perR in the transcriptional direction opposite to that of the ribosomal genes. The nucleotide sequences of the inserted perR gene on pRRK plasmid were confirmed by sequencing using primers pRRK-seqF and pRRK-seqR. The pRRK-perR constructs were then used as suicide vectors to insert perR into the chromosome of the 11168ΔperR isolate. The complemented constructs 11168ΔperRCperR11168, 11168ΔperRCperRC143A, 11168ΔperRCperRC250A, 11168ΔperRCperRG319T, and 11168ΔperRCperRA338Del (Table 1) were selected on MH agar containing 30 μg/ml of kanamycin and 10 μg/ml of chloramphenicol. The same strategies were utilized to insert an intact mutY gene into the chromosome of 11168ΔmutY and 81-176ΔmutY, resulting in constructs 11168ΔmutYC and 81-176ΔmutYC (Table 1).
Site-specific reversion in PerR of C. jejuni OXR mutant isolates.
In order to investigate the role of the single amino acid change in PerR in mediating oxidative stress resistance, the perR mutations in four of the C. jejuni OXR mutants were reverted to the WT perR sequence by using a previously reported method with some modifications (21, 48). perR (cj0322) and cj0323 are tandemly positioned on the chromosome of C. jejuni and transcribed in the same direction. cj0323 encodes a hypothetical protein with an unknown function. A cat cassette was inserted in the cj0323 gene downstream of perR to facilitate the reversion of the specific mutation in perR by homologous recombination. Briefly, a 949-bp fragment containing the entire perR encoding sequence and part of its upstream gene cj0321, and another 802-bp fragment containing most of the cj0323 encoding sequence, were amplified by primer pairs perR-rF/perR-rR and 0323-rF/0323-rR, respectively (Table S2), using C. jejuni 11168 DNA as the template. These two PCR fragments were then linked with the cat cassette by overlap PCR using the listed primers. The overlap PCR product was purified and used to naturally transform different C. jejuni OXR mutants. Transformants were screened on MH agar plates containing 10 mg/liter chloramphenicol and confirmed by PCR amplification of the gene flanking the insertion site. This resulted in C. jejuni constructs 11168perRA338DelRV, CMT perRC143ARV, CMT perRC250ARV, and CMT perRG319TRV (Table 1), in which perR was reverted to the WT sequence at the original site in the genome of C. jejuni OXR mutants through homologous recombination. The reversions were confirmed by DNA sequencing. As a control, CMT perRC250A was inserted with the cat cassette in cj0323 without the perR reversion, and this construct, CMT perRC250ACT, served to demonstrate that neither the presence nor the location of the cat cassette in cj0323 had an effect on the peroxide susceptibility in C. jejuni.
Expression and purification of rPerR from WT and C. jejuni OXR mutants.
Full-length histidine-tagged recombinant PerR (rPerR) from the WT and its C. jejuni OXR mutants was produced in the E. coli JM109 strain by using the pQE-30 vector (Qiagen). The complete coding sequences of perR in the isolates were amplified with primers perR-HisF and perR-HisR (Table S2). The amplified PCR products were ligated into the pQE-30 vector, which had previously been digested with BamHI and HindIII, utilizing the SLiCE cloning method as mentioned above. The plasmids in the E. coli clones producing different PerR proteins were sequenced, confirming the cloned sequences of perR. E. coli harboring pQE-30-perR was grown in LB broth at 37°C, with shaking at 180 rpm to an OD600 of 1.0. The expression of recombinant PerR was induced by addition of 1.0 mM IPTG for 5 h at 28°C. Purification of recombinant PerR proteins was performed by following procedures described previously (49, 50).
Electrophoretic mobility shift assays.
In order to investigate the role of the single-nucleotide changes in affecting the binding of PerR to the promoter regions regulated by PerR, EMSAs were performed by a procedure described previously (30, 51), with some modifications. Briefly, primers KatAPromF/KatAPromR and DnaEPromF/DnaEPromR (30) were used to amplify the promoter regions of katA and dnaE, respectively. The purified PCR products were then labeled at the 3′ end with digoxigenin-11-ddUTP (DIG-11-ddUTP) by using the DIG oligonucleotide 3′-End Labeling kit (Roche Molecular Biochemicals). DIG-labeled PCR products were incubated in 0.5 nM aliquots with 5 to 20 nM purified recombinant PerR (rPerR) from either C. jejuni WT or OXR mutant strains in 10 μl of binding buffer containing 50 μM MnCl2, 20 mM Tris-Cl (pH 7.4), 50 mM KCl, 3 mM MgCl2, 5% (vol/vol) glycerol, and 0.1% Triton X-100 (vol/vol). The reaction mixtures were incubated for 1 h at room temperature. The reaction mixtures were then subjected to electrophoresis on a nondenaturing 6% (wt/vol) polyacrylamide gel in 0.5× Tris-borate-EDTA (TBE; 44 mM Tris, 44 mM boric acid, 1 mM EDTA [pH 8.0]) at 200 V for 45 min. The DNA in the gel was transferred to a nylon membrane with a vacuum blotter. DIG-labeled DNA was detected and visualized by using alkaline phosphatase-conjugated anti-DIG antibody and the chemiluminescent substrate CDP-Star (Roche Molecular Biochemicals).
Determination of spontaneous OXR mutation frequencies in C. jejuni isolates.
The traditional method for measuring spontaneous mutation frequencies in Campylobacter (21) failed to determine spontaneous H2O2 resistance mutation frequencies, simply due to the rapid decomposition of H2O2 during medium preparation and culture incubation. Therefore, to quantify spontaneous OXR mutation frequencies in Campylobacter, promoter-reporter fusion PkatA-cat, with the promoter region of the katA gene fused with promoterless chloramphenicol resistance gene cat, was constructed and cloned into the pRRK plasmid by SLiCE cloning using the listed primer pairs catORF-F/catORF-R and PkatA-F/PkatA-R (Table S2). The fusion gene PkatA-cat was then inserted into the genome of C. jejuni 11168 and CMT isolates, generating 11168 PkatA-cat and CMT PkatA-cat constructs. Both constructs showed chloramphenicol susceptibility (MIC = 1 to 2 μg/ml) similar to that of C. jejuni 11168 and CMT isolates, since transcription of the PkatA-cat gene is inhibited by the functional PerR protein in strains 11168 PkatA-cat and CMT PkatA-cat. However, those spontaneous perR mutations, which compromise PerR function, were expected to result in the derepression of the katA promoter of PkatA-cat and significantly increased expression of cat, reducing the susceptibility to chloramphenicol (MIC ≥ 16 μg/ml). Therefore, 4 μg/ml of chloramphenicol was used in this study to detect spontaneous perR loss-of-function mutations in C. jejuni 11168 PkatA-cat and CMT PkatA-cat constructs. The spontaneous chloramphenicol resistance (Cmr) mutation frequencies were determined as previously described for spontaneous ciprofloxacin and ampicillin resistance mutations in C. jejuni (21). Several colonies growing on selective MH plates from the spontaneous Cmr mutation frequency test were randomly picked to sequence the perR gene. Meanwhile, these colonies were subcultured and tested for their susceptibility to H2O2 using the disk diffusion assay, as described above.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant no. R01AI118283 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01685-17.
REFERENCES
- 1.WHO. 2016. Campylobacter fact sheet, 2016. http://www.who.int/mediacentre/factsheets/fs255/en/ WHO, Geneva, Switzerland. [Google Scholar]
- 2.CDC. 2014. Antibiotic resistance threats in the United States. http://www.cdc.gov/drugresistance/threat-report-2013/. [PubMed]
- 3.Atack JM, Kelly DJ. 2009. Oxidative stress in Campylobacter jejuni: responses, resistance and regulation. Future Microbiol 4:677–690. doi: 10.2217/fmb.09.44. [DOI] [PubMed] [Google Scholar]
- 4.Imlay JA. 2003. Pathways of oxidative damage. Annu Rev Microbiol 57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 5.Segal AW. 2005. How neutrophils kill microbes. Annu Rev Immunol 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosen H. 2004. Bacterial responses to neutrophil phagocytosis. Curr Opin Hematol 11:1–6. [DOI] [PubMed] [Google Scholar]
- 7.Imlay JA. 2008. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farr SB, Kogoma T. 1991. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol Rev 55:561–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. 2000. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 10.van Vliet AH, Baillon ML, Penn CW, Ketley JM. 1999. Campylobacter jejuni contains two fur homologs: characterization of iron-responsive regulation of peroxide stress defense genes by the PerR repressor. J Bacteriol 181:6371–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim M, Hwang S, Ryu S, Jeon B. 2011. Regulation of perR expression by iron and PerR in Campylobacter jejuni. J Bacteriol 193:6171–6178. doi: 10.1128/JB.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palyada K, Sun YQ, Flint A, Butcher J, Naikare H, Stintzi A. 2009. Characterization of the oxidative stress stimulon and PerR regulon of Campylobacter jejuni. BMC Genomics 10:481. doi: 10.1186/1471-2164-10-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palyada K, Threadgill D, Stintzi A. 2004. Iron acquisition and regulation in Campylobacter jejuni. J Bacteriol 186:4714–4729. doi: 10.1128/JB.186.14.4714-4729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang S, Kim M, Ryu S, Jeon B. 2011. Regulation of oxidative stress response by CosR, an essential response regulator in Campylobacter jejuni. PLoS One 6:e22300. doi: 10.1371/journal.pone.0022300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang S, Zhang Q, Ryu S, Jeon B. 2012. Transcriptional regulation of the CmeABC multidrug efflux pump and the KatA catalase by CosR in Campylobacter jejuni. J Bacteriol 194:6883–6891. doi: 10.1128/JB.01636-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.David SS, O'Shea VL, Kundu S. 2007. Base-excision repair of oxidative DNA damage. Nature 447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. 2002. Inherited variants of MYH associated with somatic G:C–>T:A mutations in colorectal tumors. Nat Genet 30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 18.Robles AG, Reid K, Roy F, Fletcher HM. 2011. Porphyromonas gingivalis mutY is involved in the repair of oxidative stress-induced DNA mispairing. Mol Oral Microbiol 26:175–186. doi: 10.1111/j.2041-1014.2011.00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eutsey R, Wang G, Maier RJ. 2007. Role of a MutY DNA glycosylase in combating oxidative DNA damage in Helicobacter pylori. DNA Repair (Amst) 6:19–26. doi: 10.1016/j.dnarep.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davidsen T, Bjoras M, Seeberg EC, Tonjum T. 2005. Antimutator role of DNA glycosylase MutY in pathogenic Neisseria species. J Bacteriol 187:2801–2809. doi: 10.1128/JB.187.8.2801-2809.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai L, Muraoka WT, Wu Z, Sahin O, Zhang Q. 2015. A single nucleotide change in mutY increases the emergence of antibiotic-resistant Campylobacter jejuni mutants. J Antimicrob Chemother 70:2739–2748. doi: 10.1093/jac/dkv190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Rourke EJ, Chevalier C, Pinto AV, Thiberge JM, Ielpi L, Labigne A, Radicella JP. 2003. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc Natl Acad Sci U S A 100:2789–2794. doi: 10.1073/pnas.0337641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes K, Mulholland F, Pearson BM, Pin C, McNicholl-Kennedy J, Ketley JM, Wells JM. 2005. Campylobacter jejuni gene expression in response to iron limitation and the role of Fur. Microbiology 151:243–257. doi: 10.1099/mic.0.27412-0. [DOI] [PubMed] [Google Scholar]
- 24.Mongkolsuk S, Whangsuk W, Fuangthong M, Loprasert S. 2000. Mutations in oxyR resulting in peroxide resistance in Xanthomonas campestris. J Bacteriol 182:3846–3849. doi: 10.1128/JB.182.13.3846-3849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Keramati L, Helmann JD. 1995. Coordinate regulation of Bacillus subtilis peroxide stress genes by hydrogen peroxide and metal ions. Proc Natl Acad Sci U S A 92:8190–8194. doi: 10.1073/pnas.92.18.8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bsat N, Herbig A, Casillas-Martinez L, Setlow P, Helmann JD. 1998. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol Microbiol 29:189–198. doi: 10.1046/j.1365-2958.1998.00921.x. [DOI] [PubMed] [Google Scholar]
- 27.Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun 69:3744–3754. doi: 10.1128/IAI.69.6.3744-3754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verneuil N, Rince A, Sanguinetti M, Posteraro B, Fadda G, Auffray Y, Hartke A, Giard JC. 2005. Contribution of a PerR-like regulator to the oxidative-stress response and virulence of Enterococcus faecalis. Microbiology 151:3997–4004. doi: 10.1099/mic.0.28325-0. [DOI] [PubMed] [Google Scholar]
- 29.King KY, Horenstein JA, Caparon MG. 2000. Aerotolerance and peroxide resistance in peroxidase and PerR mutants of Streptococcus pyogenes. J Bacteriol 182:5290–5299. doi: 10.1128/JB.182.19.5290-5299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handley RA, Mulholland F, Reuter M, Ramachandran VK, Musk H, Clissold L, Le Brun NE, van Vliet AH. 2015. PerR controls oxidative stress defence and aerotolerance, but not motility-associated phenotypes of Campylobacter jejuni. Microbiology 161:1524–1536. doi: 10.1099/mic.0.000109. [DOI] [PubMed] [Google Scholar]
- 31.Stintzi A, Vliet AHMV, Ketley JM. 2008. Iron metabolism, transport, and regulation, p 591–610. In Nachamkin I, Szymanski CM, Blaser MJ (ed), Campylobacter, 3rd ed ASM Press, Washington, DC. [Google Scholar]
- 32.Lee JW, Helmann JD. 2006. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature 440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 33.Michaels ML, Miller JH. 1992. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J Bacteriol 174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodrigues RC, Pocheron AL, Hernould M, Haddad N, Tresse O, Cappelier JM. 2015. Description of Campylobacter jejuni Bf, an atypical aero-tolerant strain. Gut Pathog 7:30. doi: 10.1186/s13099-015-0077-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh E, McMullen L, Jeon B. 2015. High prevalence of hyper-aerotolerant Campylobacter jejuni in retail poultry with potential implication in human infection. Front Microbiol 6:1263. doi: 10.3389/fmicb.2015.01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Kane PM, Connerton IF. 2017. Characterisation of aerotolerant forms of a robust chicken colonizing Campylobacter coli. Front Microbiol 8:513. doi: 10.3389/fmicb.2017.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rea RB, Gahan CG, Hill C. 2004. Disruption of putative regulatory loci in Listeria monocytogenes demonstrates a significant role for Fur and PerR in virulence. Infect Immun 72:717–727. doi: 10.1128/IAI.72.2.717-727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bjorkholm B, Sjolund M, Falk PG, Berg OG, Engstrand L, Andersson DI. 2001. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc Natl Acad Sci U S A 98:14607–14612. doi: 10.1073/pnas.241517298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morosini MI, Baquero MR, Sanchez-Romero JM, Negri MC, Galan JC, del Campo R, Perez-Diaz JC, Baquero F. 2003. Frequency of mutation to rifampin resistance in Streptococcus pneumoniae clinical strains: hexA and hexB polymorphisms do not account for hypermutation. Antimicrob Agents Chemother 47:1464–1467. doi: 10.1128/AAC.47.4.1464-1467.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denamur E, Bonacorsi S, Giraud A, Duriez P, Hilali F, Amorin C, Bingen E, Andremont A, Picard B, Taddei F, Matic I. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J Bacteriol 184:605–609. doi: 10.1128/JB.184.2.605-609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oliver A, Canton R, Campo P, Baquero F, Blazquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 42.Richardson AR, Yu Z, Popovic T, Stojiljkovic I. 2002. Mutator clones of Neisseria meningitidis in epidemic serogroup A disease. Proc Natl Acad Sci U S A 99:6103–6107. doi: 10.1073/pnas.092568699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin J, Cagliero C, Guo B, Barton YW, Maurel MC, Payot S, Zhang Q. 2005. Bile salts modulate expression of the CmeABC multidrug efflux pump in Campylobacter jejuni. J Bacteriol 187:7417–7424. doi: 10.1128/JB.187.21.7417-7424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Yao R, Alm RA, Trust TJ, Guerry P. 1993. Construction of new Campylobacter cloning vectors and a new mutational cat cassette. Gene 130:127–130. doi: 10.1016/0378-1119(93)90355-7. [DOI] [PubMed] [Google Scholar]
- 46.Muraoka WT, Zhang Q. 2011. Phenotypic and genotypic evidence for L-fucose utilization by Campylobacter jejuni. J Bacteriol 193:1065–1075. doi: 10.1128/JB.01252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Werling U, Edelmann W. 2012. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res 40:e55. doi: 10.1093/nar/gkr1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge B, McDermott PF, White DG, Meng J. 2005. Role of efflux pumps and topoisomerase mutations in fluoroquinolone resistance in Campylobacter jejuni and Campylobacter coli. Antimicrob Agents Chemother 49:3347–3354. doi: 10.1128/AAC.49.8.3347-3354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin J, Michel LO, Zhang Q. 2002. CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob Agents Chemother 46:2124–2131. doi: 10.1128/AAC.46.7.2124-2131.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Q, Meitzler JC, Huang S, Morishita T. 2000. Sequence polymorphism, predicted secondary structures, and surface-exposed conformational epitopes of Campylobacter major outer membrane protein. Infect Immun 68:5679–5689. doi: 10.1128/IAI.68.10.5679-5689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin J, Akiba M, Sahin O, Zhang Q. 2005. CmeR functions as a transcriptional repressor for the multidrug efflux pump CmeABC in Campylobacter jejuni. Antimicrob Agents Chemother 49:1067–1075. doi: 10.1128/AAC.49.3.1067-1075.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hofreuter D, Tsai J, Watson RO, Novik V, Altman B, Benitez M, Clark C, Perbost C, Jarvie T, Du L, Galan JE. 2006. Unique features of a highly pathogenic Campylobacter jejuni strain. Infect Immun 74:4694–4707. doi: 10.1128/IAI.00210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.