

Abstract
Glioblastoma multiforme (GBM) is a devastating tumour with abysmal prognoses. We desperately need novel approaches to understand GBM biology and therapeutic vulnerabilities. Extracellular vesicles (EVs) are membrane-enclosed nanospheres released locally and systemically by all cells, including tumours, with tremendous potential for intercellular communication. Tumour EVs manipulate their local environments as well as distal targets; EVs may be a mechanism for tumourigenesis in the recurrent GBM setting. We hypothesized that GBM EVs drive molecular changes in normal human astrocytes (NHAs), yielding phenotypically tumour-promoting, or even tumourigenic, entities. We incubated NHAs with GBM EVs and examined the astrocytes for changes in cell migration, cytokine release and tumour cell growth promotion via the conditioned media. We measured alterations in intracellular signalling and transformation capacity (astrocyte growth in soft agar). GBM EV-treated NHAs displayed increased migratory capacity, along with enhanced cytokine production which promoted tumour cell growth. GBM EV-treated NHAs developed tumour-like signalling patterns and exhibited colony formation in soft agar, reminiscent of tumour cells themselves. GBM EVs modify the local environment to benefit the tumour itself, co-opting neighbouring astrocytes to promote tumour growth, and perhaps even driving astrocytes to a tumourigenic phenotype. Such biological activities could have profound impacts in the recurrent GBM setting.
This article is part of the discussion meeting issue ‘Extracellular vesicles and the tumour microenvironment’.
Keywords: exosomes, extracellular vesicles, glioblastoma multiforme, astrocytes, signalling, microenvironment
1. Introduction
Glioblastoma multiforme (GBM) is a devastating primary central nervous system (CNS) tumour with poor outcomes. The median survival is less than 15 months with a poor quality of life, despite aggressive surgical resection and combined chemo-radiation [1,2]. Current therapies display limited clinical success, especially in the setting of recurrent GBM [3–6]. Innovative strides in understanding GBM biology are necessary for improved therapy. Extracellular vesicles (EVs) are membrane-enclosed nanospheres released directly from the cell membrane (i.e. ‘microvesicles’) or from the endosomal system via fusion of multi-vesicular bodies with the cytoplasmic membrane (i.e. ‘exosomes’); this presumably occurs in all cell types [7,8]. The potential involvement of EVs in GBM pathobiology is becoming more and more recognized [9,10].
Tumour EVs have an extraordinary ability to modulate the tumour microenvironment [11–16], recruiting recipient cells to support tumour growth and progression. EV surface elements have been shown to mirror those of the tumour and remodel the extracellular matrix with matrix metalloproteinases, and transfer drug resistance with P-glycoprotein [17–19]. Given the wide variety of signalling molecules, bioactive lipids and functional RNAs within EVs, particularly tumour-derived EVs, they represent ideal candidates for transfer of information and activities that could generate delinquency in normal cells. However, no studies to date have characterized the effects of GBM EVs on peritumoural astrocytes. We lack almost any information on the impacts of GBM EVs on astrocytes in terms of phenotypic changes relating to proliferation, migration or signalling involving the recipient cells. Studies conducted on other cancer models, however, show that these effects on stromal/microenvironmental cells can be profound [20–24].
GBM EVs likely remain within the vicinity of the resection cavity following tumour excision, altering the behaviour of the surgically spared normal cells such as resident astrocytes. It is conceivable that ensuing therapeutic intervention (chemotherapy and targeted radiation) would leave degrees of damage to those normal cells that would enable the transformative impacts of tumour EVs in the vicinity. This may be due to the transfer of tumour-related RNAs, proteins, metabolites or by the tumour EVs promoting signalling changes in the astrocytes themselves. Thus, GBM EVs may play a role in co-opting neighbouring astrocytes to promote recurrent growth, or possibly, the astrocytes in the resection cavity may even be transformed. Understanding the effects of tumour-derived EVs on NHAs will likely become necessary in a full accounting of how to treat recurrent GBM. Here, we demonstrate that different concentrations of glioma-derived EVs have differential effects on the migratory capabilities, cytokine outputs, tumour cell growth promotion, intracellular signalling pathways and activation/transformation status of NHAs.
2. Material and methods
(a). Cell lines
E8-5 and F3-8 are GBM cell lines derived from surgically resected primary intracranial tumours from patients receiving treatment at the University of Colorado Hospital. Histologically, E8-5 originated in the patient's left parietal lobe, and is a World Health Organization (WHO) grade IV glioma, classic small cell variant, with loss of phosphatase and tensin homolog (PTEN) and gene amplification of epidermal growth factor receptor (EGFR). It is negative for 1p/19q co-deletion. F3-8 originated in the patient's left frontal lobe, and is an atypical small cell variant grade IV glioma without IDH1 mutation. Freshly resected tumours were placed in Neurobasal A (NBA) medium without phenol red (Thermo Fisher/Invitrogen Technologies, Waltham, MA, USA). Tumour cells were mechanically dissociated, filtered through a 100 µm cell strainer (Corning Life Sciences, Tewksbury, MA, USA) and were cultured under stem cell-like conditions in NBA supplemented with 2 mM l-glutamine, 1X B27 and N2, and 5 ng ml−1 basic fibroblast growth factor (FGF) and epidermal growth factor (EGF) (Thermo Fisher/Invitrogen). Normal human astrocytes (NHAs) were obtained from Lonza, and cultured in astrocyte basal growth media (ABM™) with astrocyte growth medium supplements (AGM; Lonza Inc., Anaheim, CA, USA). These cells were obtained from a human fetus, 18 weeks' gestation; the tissue is donated following permission for use in research applications by informed consent or legal authorization. Tissue origin is cerebral cortex (grey matter), and the cells are considered type I astrocytes. Cells are frozen after the second passage. The cells stain positive for glial fibrillary acidic protein, and show morphological characteristics of astrocytes. The cells are guaranteed for 10 additional doublings, and were expanded and used between passages 7 and 10. We purchased cells twice and received the same lot number (presumably the same astrocyte source) both times.
Normal human epithelial cells were obtained from healthy donor urine; cells were harvested from voided urine by centrifugation (400g, 10 min, RT). Cells were initially cultured in Dulbecco’s modified eagle medium (DMEM)/Ham's F12/10% fetal bovine serum (FBS)/Renal Cell Growth Medium supplements (Lonza). Cells were cultured on 0.1% gelatin-coated plates. Cells were switched to ‘proliferation medium’ (the aforementioned medium + DMEM/Glutamax + non-essential amino acids (Sigma-Aldrich, St Louis, MO, USA), + 5 ng ml−1 basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF) (Lonza)) before conversion to the aforementioned NBA with supplements. All cells were cultured at 37°C/5% CO2. This study was approved by the Colorado Combined Institutional Review Board (COMIRB, protocol no. 13-3007) and patients provided written consent, all consistent with NIH guidelines.
(b). Extracellular vesicle purification and characterization
EV extraction and purification from cell culture media were conducted as described [16], via filtration and differential centrifugation (diagrammed in figure 1). Note that the medium used was serum-free. Following resuspension of the pellet after high-speed centrifugation, we routinely extrude the materials through 20 and 25 G needles to break up EV clumps and disperse protein aggregates. Remaining aggregates are then removed by filtration through a 0.45 µm filter. EVs were verified using Exo-Check arrays (System Biosciences/SBI, Palo Alto, CA, USA) for eight purported exosome/EV markers. Nanoparticle tracking analysis (NTA) (NanoSight LM10 device, Malvern, Westborough, MA, USA) was used to verify that the particles were within the anticipated size range of 30–150 nm, although some are clearly larger (figure 1).
Figure 1.

Extraction and purification of EVs from GBM cell lines. (a) Two GBM cell lines viewed under light microscopy. E8-5 and F3-8, were dissociated and grown under stem cell-like conditions in supplemented NBA medium (Invitrogen Technologies). Scale bar, 100 µm. (b) EV purification from cell culture media was conducted as described [16]. (c) NTA (Nanosight) was used to determine sizes and concentrations of vesicles; EVs were verified using ExoCheck arrays for eight purported EV markers.
Vesicle concentrations used (50 and 500 µg ml−1) were based on previous studies [16,25] that were correlated with putative cellular microenvironment concentrations compared to blood concentrations. We also took into account other reports of blood and cerebral spinal fluid particle numbers derived from patients with brain tumours [26,27]. We then chose doses that would allow for a log increase that we could accommodate with EV purification yields.
(c). Normal human astrocyte migration/wound-healing assay
Migration capacity of NHAs exposed to 0 or 50 µg ml−1 GBM EVs was measured using standard procedures for a wound-healing assay, essentially as described [28]. Briefly, NHAs at 80–90% confluency in 6-well plates (Corning) were scratched with a 200 µl pipet tip to generate a gap between cells. GBM EVs were added (or not), and wound closure was measured at 24 h (compared to the distance at 0 h). Measurements of ‘wound healing’ were performed using NIH Image J.
(d). Cytokine release/‘secretome’ assays
Proteome Profiler Human XL Cytokine Array ARY022B and Human Cytokine Array Panel A ARY005 (R&D Systems, Minneapolis, MN, USA) were performed as per the manufacturer's protocols. Briefly, NHAs (2 × 106 per T75 flask in 10 ml medium) were washed with PBS and then treated with 0 or 500 µg ml−1 of GBM EVs or normal epithelial cell EVs (diluted into ABM medium) for 4 or 24 h. Culture media were collected (in addition, cell lysates were prepared) after the allotted incubation time. Arrays were developed with chemiluminescence and spot intensities quantified using a FluorChem Q developer with AlphaView software (ProteinSimple, San Jose, CA, USA). Background was subtracted from each membrane, and average luminosity from duplicate spots was compared between treatment groups.
(e). Glioblastoma multiforme cell proliferation in conditioned medium from glioblastoma multiforme extracellular vesicle-treated normal human astrocytes
F3-8 GBM cells were transferred from NBA to ABM medium, and were grown in 24-well plates at 5 × 104 cells well−1. Conditioned medium from NHAs that were treated with 0, 50 or 500 µg ml−1 GBM EVs (24 h) was used to replace the old medium. Conditioned astrocyte medium was centrifuged (2000g, 15 min) to remove any cells before adding the medium to the GBM cells. After 24 h, GBM cell proliferation was measured by MTS assay (Cell Titer 96 Aqueous One, Promega, Madison, WI, USA).
(f). Normal human astrocyte signalling profile following exposure to glioblastoma multiforme extracellular vesicles
NHAs (2 × 106 per T75 flask in 10 ml medium) were treated with 0, 50 or 500 µg ml−1 GBM EVs, and cells were harvested and lysed at 4, 8 or 24 h. Lysates were incubated on PathScan Intracellular Signaling Arrays (14471; Cell Signaling Technology, Danvers, MA, USA) following the manufacturer's instructions. Arrays were developed and quantified as described above.
(g). Normal human astrocyte growth in soft agar
NHAs were exposed to 0, 50 or 500 µg ml−1 GBM EVs for 24 h. Cells were collected and plated 1 × 106 ml−1 in soft agar (CytoSelect 96-Well Cell Transformation Assay, CBA 135; Cell Biolabs Inc., San Diego, CA, USA) as per the manufacturer's instructions. After 8 days, cell viability and proliferation were measured by MTT assay, per kit instructions.
(h). Ingenuity Pathway Analysis
Pathway and functional analysis predictions were based on ingenuity pathway analysis algorithms (IPA; http://www.ingenuity.com/; Ingenuity Systems/QIAGEN, Germantown, MD, USA). Core analyses and comparison analyses were used as described in the text or figure legends.
(i). Statistics
The fold change determinations are specified for the various experiments. Student's t-test was used to compare means. p<0.05 is considered statistically significant.
3. Results
(a). Glioblastoma multiforme cell lines and characterization of glioblastoma multiforme extracellular vesicles
GBM cell lines designated E8-5 and F3-8 were generated from freshly resected primary human gliomas acquired from patients undergoing therapeutic tumour resection surgery. Pathology, verified by a board-certified neuro-pathologist, showed the tumours as GBM small cell variant status; E8-5 showed EGFR gene expansion, while F3-8 was wild-type for IDH1. These cells were grown in serum-free, ‘stem cell’ media that promoted tumour sphere growth (figure 1a). EVs were harvested from conditioned media by differential centrifugation, filtration and concentration, and ultimately ultracentrifugation (figure 1b). NTA using NanoSight shows main peaks of particles with hydrodynamic radius of 100–120 nm (figure 1c), and antibody arrays for known EV marker proteins (ExoCheck) indicate materials with protein contents consistent with EVs (figure 1c).
Note, the ExoCheck kit refers to GM130/GOLGA2 as a marker of ‘cellular contamination’ in EV preps. This may derive from comments in the literature suggesting that proteins from non-endosomal compartments are considered ‘pollutants’ in EV preparations (e.g. [7,29]). However, GM130/GOLGA2 has been identified as an EV component in both Vesiclepedia (http://microvesicles.org/gene_summary?gene_id=2801) and Exocarta (http://www.exocarta.org/gene_summary?gene_id=2801), where both protein and mRNA were identified [13,30–32]. GM130/GOLGA2 is known to be on intracellular vesicles [33] along with Factor XIII-A/F13A1; the latter is also found in EVs (both Vesiclepedia (http://microvesicles.org/gene_summary?gene_id=2162) and Exocarta (http://www.exocarta.org/gene_summary?gene_id=2162)). Thus, we posit that GM130/GOLGA2 is likely to be a bona fide EV protein.
(b). Glioblastoma multiforme extracellular vesicles promote normal human astrocyte migration
Our previous work demonstrated that tumour EVs promote tumour cell migration [25], but impede lymphocyte migration [16]. With that background, we tested effects of GBM EVs on NHA migration using a wound-healing assay. NHAs were plated to 80–90% confluency, whereupon a scratch wound was introduced. Media containing 0 or 50 µg ml−1 E8-5 EVs replaced the conditioned media, and cells migrated into the gap for 24 h. Measurements of gap distance were taken at 0 and 24 h post-EV incubation; gap closure is listed as ‘%wound fill’ in figure 2a (results for three experiments are shown). GBM EV-treated cells covered 45–70% of the gap, compared to 15–25% for untreated cells in a 24 h period. Representative photos of the wound fill are shown in figure 2b.
Figure 2.

GBM EVs promote astrocyte migration. Astrocytes were obtained from a single individual. (a) Astrocytes were exposed to 50 µg ml−1 of tumour EVs and observed over a 24 h period in three separate wound fill assays (three biological replicates with three technical replicates per experiment). Compared to controls (0 µg ml−1 EVs), astrocytes displayed a significant increase in per cent wound closure in all three instances. Error bars, standard deviation. (b) Representative photos and ImageJ boundaries are shown.
(c). Glioblastoma multiforme extracellular vesicles alter normal human astrocyte cytokine production
One possible outcome of induced NHA migration driven by GBM EVs could be that NHAs may provide usable materials for the GBM. Thus, we assessed GBM EV-induced NHA cytokine release via cytokine antibody arrays measuring 105 different cytokines, chemokines and other secreted entities. We treated NHAs with 500 µg ml−1 of E8-5 EVs for 4 or 24 h, which resulted in upregulation of 40 different cytokines in the conditioned medium at over 5× control concentrations (untreated astrocyte secretions), as well as scores of others with 1.5–4× increases at 24 h. The probed arrays themselves are shown in figure 3 (left), and a heat map of the readouts is shown in right-hand side of figure 3. Note that the 4 h time point shows a very different profile when compared with control or 24 h/500 µg ml−1 GBM EV treatment. We based the time points used on our previous studies with signalling arrays and cytokine outputs [16]. We saw that there were both early and late effects that might take into account a time course of EV binding, internalization, and the potential de novo transcription, translation and release of cytokines. Additionally, we tested a different cytokine array that compared 500 µg ml−1 F3-8 GBM EV treatment to the same concentration of normal epithelial cell EVs for 24 h to determine if the effects were simply due to the quantity of EVs rather than the source. Of the 33 secretome proteins that overlapped with the previous array, 9–10 of them may be increased due to EV quantity, but the other 20+ are likely specific to GBM EV stimulation (electronic supplementary material, figure S1). Also, the tumour-derived EVs seemed to promote far greater changes in release of certain cytokines (e.g. interferon gamma (IFNG), interleukins 12, 1A, 8 and 1B), chemokine CXCL10 and factor C5.
Figure 3.

GBM EVs enhance astrocyte secretory outputs. Culture media from GBM EV-treated (500 µg ml−1) astrocytes were analysed on antibody microarrays (R&D Systems) for relative cytokine/chemokine/growth factor concentrations. (a) Representative arrays shown (no EV treatment (i), EV treatment (ii), with potentially important molecules noted; C, positive control spot). (b) Heat map of secretome of untreated, short-term-treated (4 h), and 24 h-treated astrocytes is shown. Results are from two biological replicates, with two technical replicates for each. Astrocytes were obtained from a single individual.
We assessed the changes in the GBM EV-stimulated NHA secretome by IPA. Table 1 presents the top canonical pathways, and diseases and biofunctions derived from the data. Notably, the themes include cell–cell communication, inflammatory responses, cell growth, movement and death. Embedded in those themes are terms related directly to cancer and tumour morphology, and more diverse inferences to immune signalling and responses. Of the top networks identified, the top three interactomes are shown in figure 4. Various cytokines and chemokines are heavily represented in those networks, but there are also notable nodes at major intracellular signalling proteins such as ERK1/2, PI3 K and AKT (figure 4a–c). Thus, analysis of the EV-stimulated NHA secretome validates the migratory capacity of the EV-treated NHAs, and suggests possible involvement of oncogenic signalling pathways.
Table 1.
Top canonical pathways, diseases and biofunctions, networks discerned from ingenuity pathway analysis for nha cytokine secretion following GBM EV treatment. EGF, epidermal growth factor; FLT3, receptor-type tyrosine-protein kinase FLT3/CD135; IL-22, interleukin 22; MAPK, mitogen activated protein kinase; UVA, ultraviolet radiation A.
| top canonical pathways | ||
|---|---|---|
| name | p-value | overlap |
| UVA-induced MAPK signaling | 2.92 × 10−17 | 8.8% 9/102 |
| FLT3 signaling in hematopoietic progenitor cells | 1.04 × 10−15 | 9.8% 8/82 |
| IL-22 signalling | 1.55 × 10−14 | 25.0% 6/24 |
| UVB-induced MAPK signaling | 4.71 × 10−14 | 10.6% 7/66 |
| EGF signaling | 8.89 × 10−14 | 9.7% 7/72 |
| top diseases and biofunctions | ||
| diseases and disorders | ||
| name | p-value | no. molecules |
| cancer | 7.06 × 10−4 to 3.09 × 10−12 | 17 |
| organismal injury and abnormalities | 7.06 × 10−4 to 3.09 × 10−12 | 17 |
| tumor morphology | 3.75 × 10−4 to 3.09 × 10−12 | 9 |
| renal and urological disease | 3.54 × 10−4 to 5.34 × 10−10 | 9 |
| infectious disease | 5.88 × 10−4 to 1.57 × 10−8 | 8 |
| molecular and cellular functions | ||
| name | p-value | no. molecules |
| cell death and survival | 6.49 × 10−4 to 7.96 × 10−14 | 17 |
| cellular development | 6.57 × 10−4 to 1.96× 10−11 | 17 |
| cellular growth and proliferation | 6.57 × 10−4 to 1.96× 10−11 | 9 |
| cell cycle | 5.93 × 10−4 to 3.92× 10−11 | 9 |
| cellular function and maintenance | 4.90 × 10−4 to 6.93× 10−11 | 8 |
| physiological system development and function | ||
| name | p-value | no. molecules |
| organismal functions | 1.92 × 10−6 to 1.88 × 10−9 | 4 |
| embryonic development | 4.19 × 10−4 to 2.16 × 10−9 | 10 |
| cardiovascular system development and function | 5.53 × 10−4 to 3.63 × 10−9 | 9 |
| organismal development | 4.19 × 10−4 to 3.63 × 10−9 | 10 |
| tissue development | 4.42 × 10−4 to 6.39 × 10−9 | 12 |
| top networks | ||
| ID associated network functions | score | |
| 1. cancer, cell death and survival, organismal injury and abnormalities | 13 | |
| 2. cardiac damage, cardiovascular disease, organismal injury and abnormalities | 8 | |
| 3. cancer, organismal functions, organismal injury and abnormalities | 8 | |
| 4. tissue morphology, dermatological diseases and conditions, hair and skin development and function | 6 | |
Figure 4.

Results of core analysis/comparison analysis in IPA. Values from figure 3 heat map were entered into a comparison analysis in IPA. The top three networks (interactomes) are shown (named in table 1). Proteins from the array are in larger bold font, with elevated values from the array depicted in shades of red. The ‘scores’ listed are −log (p-values) and are based on the probabilities of random associations of these proteins. The significance threshold by default = 1.25. ‘Focus molecules’ refer to nodes that initiate networks. Solid blue lines show direct connections between proteins found in the array, derived from the IPA knowledgebase. Broken lines represent indirect connections arising from reasoned speculation, or via known intermediaries. Light blue/turquoise lines connect identified proteins within the network with proteins not part of the array. Line lengths (edges) between the protein nodes correlate to the degree of supportive knowledge documenting interactions. Note, we have shortened some edges to fit the interactomes into the figure.
(d). Normal human astrocytes exposed to glioblastoma multiforme extracellular vesicles generate a growth-stimulating medium for tumour cells
As NHAs exposed to GBM EVs released an extraordinary cytokine/chemokine mixture into the extracellular milieu, we asked if that conditioned culture medium had effects on GBM cell growth. NHA-conditioned medium was prepared by incubating astrocytes in 0, 50 or 500 µg ml−1 F3-8 GBM EVs for 24 h. The medium was collected and used as growth medium for F3-8 GBM cells. F3-8 cells were adapted from NBA medium to astrocyte ABM medium for 24 h prior to replacement with the GBM EV-stimulated NHA conditioned medium. F3-8 cells were cultured for 24 h before MTS assay. Tumour cells displayed increased proliferation with NHA medium obtained from NHAs treated with increasing EV concentration (conditioned NHA medium) (figure 5). GBM cells grown in conditioned medium from the NHA cells treated with 500 µg ml−1 GBM EVs grew significantly more in that time frame compared with GBM cells grown in conditioned NHA medium from cells treated with 0 or 50 µg ml−1 GBM EVs (almost twofold higher than controls).
Figure 5.

GBM EV-treated astrocytes generate a growth-stimulating medium. Astrocytes are from a single individual. Astrocytes were exposed to 0, 50 or 500 µg ml−1 GBM EVs for 24 h. Conditioned astrocyte medium was collected, centrifuged to remove cells and debris, and then used as growth medium for GBM cells seeded at equal numbers. After 24 h, GBM cells were subjected to MTS assay (OD490 as readout); fold increase over control (set = 1) is presented. Results are from three biological replicates, with six technical replicates per experiment. Error bars, standard deviation.
(e). Glioblastoma multiforme extracellular vesicles modulate normal human astrocyte intracellular signalling
Given the observed changes in NHA migration and secretome release after exposure to GBM EVs, we assessed changes in NHAs via intracellular signalling (phospho-antibody) arrays. E8-5 GBM EVs (0, 50 or 500 µg ml−1) were incubated with recipient NHAs for 4, 8 or 24 h. NHAs were washed and lysed, and the lysates were exposed to antibody arrays for specific phosphorylation sites (or cleavage products) of 18 different proteins. The arrays were quantified, and are plotted as fold change over untreated control lysates (score set = 0, figure 6). Examples of developed arrays are shown in electronic supplementary material, figure S2.
Figure 6.

GBM EV-treated astrocytes induce tumour-like signalling patterns that are concentration- and time-dependent. Astrocytes are from a single individual. Astrocytes were incubated with different concentrations of GBM EVs (0, 50 or 500 µg ml−1) for 4, 8 and 24 h. Phospho-antibody arrays were incubated with lysates from EV-treated astrocytes; signal intensities were background-corrected and presented as fold change over control (untreated astrocytes at 4, 8 or 24 h = 0). Results are from two biological replicates with two technical replicates for each experiment.
Examples of increased phosphorylation state with increased EV concentration include ERK1/2, STATs 1 and 3, AKT1, and p53 at 24 h. Examples of low levels of EVs increasing protein phosphorylation until surpassing a threshold concentration where the effect is reversed and phosphorylation decreases include p53, JNK, GSK3B and also PARP cleavage. Similar data profiles were also observed over increased exposure time to GBM EVs. Protein BAD continually increased in phosphorylation as time progressed, while AMPKA activation only increased after 4 h before decreasing, as did HSP27 phosphorylation.
Again, using IPA algorithms, we note that the changes in phosphorylation profiles demonstrate strong canonical pathways involving MAPKs, and IL22 signalling (table 2), the latter being highly prevalent in the GBM EV-stimulated NHA secretome (figure 3). Other recurrent themes and terms include cancer, cell death and survival, and cell and tumour morphology. The top three interactomes derived from the top three networks are shown in figure 7, with the MAPKs, JNK, ERK, AKT and p53 found in prominent nodes.
Table 2.
Top canonical pathways, diseases and biofunctions, networks discerned from ingenuity pathway analysis for NHA intracellular signaling following GBM EV treatment.
| top canonical pathways | ||
|---|---|---|
| name | p-value | overlap |
| role of cytokines in mediating communication between immune cells | 5.23 × 10−44 | 48.1% 25/52 |
| hepatic fibrosis/hepatic stellate cell activation | 2.75 × 10−33 | 15.5% 28/181 |
| differential regulation of cytokine production in intestinal epithelial cells by IL-17A and IL-17F | 4.60 × 10−32 | 69.6% 16/23 |
| altered T cell and C cell signaling in rheumatoid arthritis | 4.63 × 10−32 | 27.2% 22/81 |
| communication between innate and adapative immune cells | 6.30 × 10−32 | 26.8% 22/82 |
| top diseases and biofunctions | ||
|---|---|---|
| diseases and disorders | ||
| name | p-value | no. molecules |
| inflammatory response | 5.56 × 10−20 to 2.50 × 10−88 | 84 |
| cancer | 4.40 × 10−21 to 6.81 × 10−57 | 88 |
| organismal injury and abnormalities | 4.40 × 10−21 to 6.81 × 10−57 | 89 |
| inflammatory disease | 9.74 × 10−21 to 3.60 × 10−54 | 78 |
| tumor morphology | 4.40 × 10−21 to 4.03 × 10−45 | 59 |
| molecular and cellular functions | ||
| name | p-value | no. molecules |
| cell-to-cell signaling and interaction | 5.84 × 10−20 to 5.36 × 10−84 | 95 |
| cellular movement | 7.79 × 10−20 to 1.13 × 10−77 | 91 |
| cellular growth and proliferation | 5.84 × 10−20 to 9.61 × 10−77 | 95 |
| cellular development | 5.16 × 10−20 to 1.46 × 10−56 | 88 |
| cell death and survival | 5.27 × 10−20 to 1.60 × 10−52 | 86 |
| physiological system development and function | ||
| name | p-value | no. molecules |
| immune cell trafficking | 5.56 × 10−20 to 1.13 × 10−77 | 80 |
| hematological system development and function | 5.84 × 10−20 to 1.38 × 10−76 | 89 |
| cardiovascular system development and function | 5.16 × 10−20 to 9.46 × 10−53 | 72 |
| organismal development | 5.16 × 10−20 to 9.46 × 10−53 | 68 |
| tissue morphology | 2.44 × 10−21 to 7.04 × 10−50 | 53 |
| top networks | ||
|---|---|---|
| ID associated network functions | score | |
| 1. cell-to-cell signaling and interaction, cellular movement, immune cell trafficking | 26 | |
| 2. cell-to-cell signaling and interaction, cellular growth and proliferation, hematological system development and function | 22 | |
| 3. cell-to-cell signaling and interaction, hematological system development and function, inflammatory response | 20 | |
| 4. cell-to-cell signaling and interaction, growth and proliferation, hematological system development and function | 20 | |
| 5. cellular movement, lipid metabolism, molecular transport | 18 | |
Figure 7.

Results of core analysis/comparison analysis in IPA. Values from figure 6 were entered into a comparison analysis in IPA. The top three networks (interactomes) are shown (named in table 2). Protein/gene names, edges, scores and focus molecules are presented as in figure 4, except that proteins with reduced values are shown in shades of green.
(f). Transformative effect of glioblastoma multiforme extracellular vesicles on normal human astrocytes
As GBM EVs induced signalling changes in recipient NHAs reminiscent of tumour-type pathway activation, we asked if GBM EV-treated NHAs might behave as if transformed. We exposed NHAs to 0, 50 or 500 µg ml−1 concentrations of F3-8 GBM EVs for 24 h. We then plated NHAs in a soft-agar assay for 8 days. We see that NHAs exposed to GBM EVs promoted colonization of the NHAs in soft agar; those exposed to the 500 µg ml−1 concentration of GBM EVs displayed a statistically significant increase in colony formation of 20% over the control (figure 8).
Figure 8.
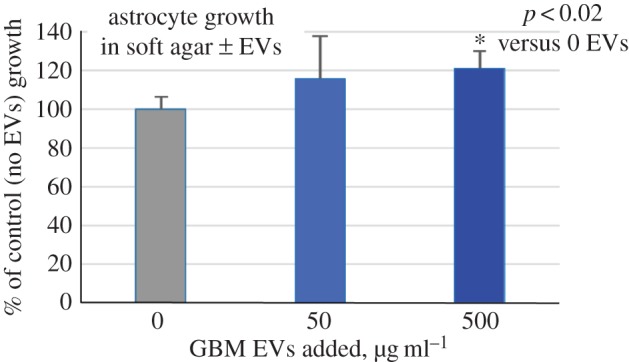
GBM EV treatment reduces sold-matrix growth dependence in astrocytes. Astrocytes are from a single individual. Astrocytes were incubated with 0, 50 or 500 µg ml−1 of GBM EVs for 24 h. Astrocytes were washed, detached and plated in a soft agar assay for eight days, after which colony viability was measured by MTT assay (OD570). Results are displayed as per cent growth over untreated astrocytes (control = 100%). Results are from two biological replicates with six technical replicates per experiment. Error bars, standard deviation.
4. Discussion
GBMs are known to profoundly alter their microenvironments, affecting infiltrating lymphocytes and monocytes [34–36], and endothelial cells [37,38]. Previous studies have implicated GBM EVs in the conditioning of tumour endothelial cells [39,40], and possibly of microglia [41], but we believe this is the first study characterizing the effects of GBM-derived EVs on astrocytes. Specifically, we show the potential for GBM EVs to drive tumour-like phenotypic changes in that normal cell population, possibly resulting in a transformed phenotype. The implications for tumourigenesis in the recurrent GBM setting would be of high importance.
Exposure to GBM EVs dramatically altered the intracellular signalling and cytokine profile (secretome) of once-normal astrocytes, resulting in internal and external effects. One such effect was the generation of a growth-stimulant medium by NHAs following exposure to GBM EVs. The resulting astrocyte conditioned medium was laden with growth factors (EGF, FGFs, CSFs, HGF, VEGF), chemokines (CXCL1, 9, 10, 11; CCL3, 5, 7, 20) and over a score of interleukins (figure 3). The conditioned medium accelerated growth of GBM cells (figure 5). As GBM EVs also promoted migration of NHAs (figure 2), one may envision a scenario where GBM cells ‘call out’ to astrocytes, prompting a close physical association in conjunction with a growth-stimulatory microenvironment. The proinflammatory nature of these secretions is also evident (table 1) [42,43]. Notably, MMP9 is also secreted by the EV-treated astrocytes, potentially providing a protease aid for GBM invasion [44–46]. Additionally, several cytokines secreted by the GBM EV-stimulated NHAs are considered drivers of T-helper 2 (Th2) phenotypes of T cells in GBM patients, such as CSF2 and 3, and ILs 4, 10 and 13 [47]. IL10 and CCL2 are also known glioma-derived inducers of regulatory T cells (Tregs) [48], so the cytokine/chemokine environment of the GBM EV-stimulated astrocytes could also promote immune suppression so prevalent in GBM patients [49]. Given the numbers of upregulated proteins in the EV-treated NHA secretome, and the multitudes of potential outcomes for GBMs and their microenvironments, further efforts are clearly needed to dissect this plethora of environmental cues.
Pathway analyses using IPA made provocative connections between the cytokine environment and well-known intracellular signalling molecules ERK, PI3 K and AKT (figure 4) [50,51]. Curiously, signalling triggered by CXCL12/SDF1 leads to activation of ERK and AKT pathways driving glioma versions of epithelial–mesenchymal transition [52,53], which are generally regarded as more invasive and tumourigenic phenotypes [54]. Also notable is that the intracellular signalling profile bears resemblance to GBM cells (U87MG) undergoing stress from the unfolded protein response [55], particularly ERK, STAT3, AKT, RPS6, mTOR, BAD, JNK and GSK3B phosphorylation.
While the directional extracellular-to-intracellular signalling is usually focused on the GBM cells, the IPA results here led us to profile intracellular signalling molecules in astrocytes following stimulation with GBM EVs. Those readouts (figure 6) also highlighted increased phosphorylation and presumed activation of ERK, STATs and AKT (and numerous other molecules) over time and EV dose. As key oncogenic entities in several cancers [56–59], activating these proteins in a juxtacrine/paracrine fashion (NHAs to GBM) would likely drive tumourigenicity. However, they may also promote a tumour-like phenotype in astrocytes, which was suggested by IPA Core Analysis (table 2) and resulting top network interactomes (figure 7), where ERK, mTOR, MAPKs, AKT and JNK are prominent signalling nodes.
Interestingly, proteins associated with apoptosis (cleaved PARP and CASP3) were also altered, leading to categories in IPA core analysis of molecular and cellular function such as ‘Cell Death and Survival’, with similar terms appearing in the top networks (table 2 and figure 7). The role of p53 (noted in figure 7c, with connections to PARP10 and CASP) may also involve apoptotic outcomes, but those pathways and networks are complicated [60]. Thus, there may be a fine line between highly activated states of the astrocytes versus cell death, but a number of the extracellular and intracellular proteins identified here are implicated in astrogliosis and glial scar formation [61]. Only one other publication reported on the effects of glial/glioma-derived EVs on astrocytes; Lo Cicero et al. [62] investigated the microvesicle-induced damage in astrocytes after exposure to G26-24 murine oligodendroglioma-derived EVs/microvesicles. In their hands, EVs from what is likely a low-grade tumour induced cell death in 40% of astrocytes. The major differences here are that we used EVs from primary human GBM cell lines grown in stem cell medium (whereas G26-24 was cloned from the parental murine G26 line, which was induced by methylcholanthrene [63]), our EVs show more exosome-like character (G26-24 EVs were not heavily characterized but appear to be of plasma membrane/‘shed-vesicle’ origin), and our EV-treated astrocytes did not suffer noticeable cell death in culture (including growth in soft agar, figure 8). Still, there may be some threshold in the cell-growth-versus-cell-death that astrocytes experience during prolonged exposure to GBM EVs. Precise profiling of the signalling and secretory patterns will require deeper analyses, but the NHA outputs suggest a co-opting of the NHAs by tumour that may go beyond simply aiding and abetting the tumour.
We and others have shown that tumour EVs effectively drive tumour cell migration [25,64], and tumour-derived EVs can remodel the extracellular matrix, inducing stromal cells to do the same, thus allowing and promoting tumour cell migration and invasion [65,66]; this is likely true for GBM EVs and peritumoural astrocytes [40]. Our findings here (figure 2) suggest that GBM EVs may attract surrounding astrocytes, facilitating cell recruitment and potential enhancement of the tumour microenvironment. Such tumour–stromal interactions are known in other systems where stromal cells as EV recipients may become cancer-associated fibroblasts [23,67,68]. These cells are no longer ‘normal’, and act to support tumourigenicity with loss of bystander capacity [69], much as we see with the GBM EV-treated astrocytes. The effects of GBM EVs on astrocytes may explain some of the infiltrative and reactive phenotypes seen in human GBM specimens [70].
Our findings from soft-agar assays where GBM EV-stimulated astrocytes produced viable colonies (figure 8) demonstrate the transforming capabilities of GBM EVs. Such treatment reduced astrocyte dependence on anchorage to a solid matrix—a hallmark of tumour-like growth. This has been demonstrated previously with retroviral transformation of astrocytes with additional tumour-promoting features (e.g. with SV40 large T antigen, and additional overexpression of EGFR [71]; SV40 T/t-Ag transduction with ectopic hTERT expression, and expression of oncogenic HRAS [72]). The similitudes between EVs and viruses in terms of respective release and uptake are noted [73,74], including viral hijacking of EVs/exosomes [75,76]. These features make vesicular transfer of oncogenic materials, including receptor tyrosine kinases of the EGFR family [77], worthy of increased focus (we note that GBM cell line E8-5 shows EGFR gene expansion). EV-driven conversion of nominally normal cells to outright tumorigenicity has been seen before [24], where breast cancer EVs promoted growth of MCA10F cells as solid tumours in a Dicer-dependent manner. The RNA components of GBM EVs and the recipient astrocytes await further work-up, but it is clear that RNAs such as microRNAs could be very important in the effects of tumour EVs on other cells [78].
The significance of GBM EVs driving phenotypic changes in normal astrocytes is profound; unsurprisingly, our current standard of care fails to address the influence of GBM EVs on peritumoural astrocytes. The Stupp protocol [79] typically combines temozolomide chemotherapy (an alkylating agent) with concurrent radiation (60 Gy fractionated over six weeks), with a four- to six-week break, and then adjuvant temozolomide (5 days out of a 28-day cycle) for as many cycles as achievable. Inevitably, tumours recur, and few viable treatment options exist, with much focus on novel experimental protocols in clinical trials [80,81]. Our findings here may suggest we look to a paradigm shift in current views of recurrent GBM development. Standard-of-care treatment aims for DNA damage; radiation is known to drive mutations in normal cells caught in the external beam radiation field [82]. Temozolomide treatment is known to either promote or lead to selection of mutations in DNA repair pathways [83]. While it is not clear that temozolomide induces mutations in treated astrocytes [84], the probability of mutational damage cannot be zero. Indeed, radiation-induced gliomas are well known in neuro-oncology [85], lending credence to the idea that normal cells incur damage. Gain-of-function mutations in p53 (TP53 gene) may play key roles in malignant transformation [86], and as noted (figure 7c), TP53/p53 is an important node in cancer-related pathways. The loss of p53 with constitutive activation of AKT and MYC also transforms astrocytes into tumourigenic entities [87], implying important roles for p53 with either gain- or loss-of-function. We suggest that GBM EV-exposed astrocytes in the context of therapeutic DNA damaging agents may be a p53 mutation away from becoming tumourigenic themselves. This concept suggests that prevention of such damage, or mitigation of signalling via AKT, p53 or other networks may be clinically relevant as means to protect astrocytes that have been subjected to GBM EV incubation in vivo. Feasible translational methods for preventing EV release in specific cell types (e.g. tumours) in vivo do not yet exist. Further studies on EV biology are clearly necessary to unravel the effects of tumour EVs and prevent the consequences of those EVs usurping normal cells to benefit tumour growth—or re-growth.
In summary, GBM-derived EVs impact peritumoural astrocytes, but the nature of those interactions and the molecular drivers remain unknown. Better characterization of GBM-derived EV contents and mechanisms of astrocyte phenotypic changes are necessary to understand the role of EVs in the recurrent GBM setting. Further exploration of induced signalling pathways in astrocytes may hold the key for novel interventions.
5. Conclusion
Our findings suggest that GBM EVs are capable of modifying their local environment by enhancing NHA cytokine production and promoting NHA migration. GBM EVs may drive molecular changes in NHAs that resemble known cancer signalling pathways, thereby enhancing NHA growth in a semi-solid matrix, an indicator of cellular transformation. These studies provide an impetus to find ways to mitigate effects of tumour EVs and may shift paradigms in our considerations of GBMs in the recurrent setting.
Supplementary Material
Supplementary Material
Data accessibility
While there are no specific datasets to share, any data inquiries may be directed to the corresponding author.
Authors' contributions
S.O. carried out a portion of the molecular laboratory work, participated in data analysis, participated in the design of the study and drafted the manuscript; J.E.H., M.W., T.A.H., D.G. and G.J.N. carried out the molecular laboratory work; J.E.H. and M.W. provided additional input for experiments and the manuscript draft; M.W.G. conceived of the study, designed the study, participated in data analysis, coordinated the study and helped draft the manuscript. T.J.A. provided scientific guidance and advice. All authors drafted the final manuscript and gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by the University of Colorado Neurosurgery Department, University of Colorado Cancer Center (J.E.H.) and by National Institutes of Health 4R01EB016378-04 (to T.J.A. and M.W.G.).
References
- 1.Stupp R, et al. 2009. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466. ( 10.1016/S1470-2045(09)70025-7) [DOI] [PubMed] [Google Scholar]
- 2.Sizoo EM, Dirven L, Reijneveld JC, Postma TJ, Heimans JJ, Deliens LJ.2014. Measuring health-related quality of life in high-grade glioma patients at the end of life using a proxy-reported retrospective questionnaire [Internet]. See http://www.ncbi.nlm.nih.gov/pubmed/24162875283–290 .
- 3.Lee EQ, Nayak L, Wen PY, Reardon DA. 2013. Treatment options in newly diagnosed glioblastoma. Curr. Treat. Options Neurol. 15, 281–288. ( 10.1007/s11940-013-0226-9) [DOI] [PubMed] [Google Scholar]
- 4.Cohen MH, Shen YL, Keegan P, Pazdur R. 2009. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 14, 1131–1138. ( 10.1634/theoncologist.2009-0121) [DOI] [PubMed] [Google Scholar]
- 5.Norden AD, Drappatz J, Wen PY. 2008. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol. 7, 1152–1160. ( 10.1016/S1474-4422(08)70260-6) [DOI] [PubMed] [Google Scholar]
- 6.Dixit S. 2014. Immunotherapy for high-grade glioma. Future Oncol. 10, 911–915. ( 10.2217/fon.14.20) [DOI] [PubMed] [Google Scholar]
- 7.Raposo G, Stoorvogel W. 2013. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383. ( 10.1083/jcb.201211138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yanez-Mo M, et al. 2015. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4, 27066 ( 10.3402/jev.v4.27066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Asti E, Chennakrishnaiah S, Lee TH, Rak J. 2016. Extracellular vesicles in brain tumor progression. Cell. Mol. Neurobiol. 36, 383–407. ( 10.1007/s10571-015-0296-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redzic JS, Ung TH, Graner MW. 2014. Glioblastoma extracellular vesicles: reservoirs of potential biomarkers. Pharmacogenomics Pers. Med. 7, 65–77. ( 10.2147/PGPM.S39768) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parolini I, et al. 2009. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 284, 34 211–34 222. ( 10.1074/jbc.M109.041152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. 2010. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 70, 9621–9630. ( 10.1158/0008-5472.CAN-10-1722) [DOI] [PubMed] [Google Scholar]
- 13.Skog J, et al. 2008. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476. ( 10.1038/ncb1800) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suetsugu A, Honma K, Saji S, Moriwaki H, Ochiya T, Hoffman RM. 2013. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 65, 383–390. ( 10.1016/j.addr.2012.08.007) [DOI] [PubMed] [Google Scholar]
- 15.Thery C, Ostrowski M, Segura E. 2009. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 9, 581–593. ( 10.1038/nri2567) [DOI] [PubMed] [Google Scholar]
- 16.Hellwinkel JE, Redzic JS, Harland TA, Gunaydin D, Anchordoquy TJ, Graner MW. 2016. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. 18, 497–506. ( 10.1093/neuonc/nov170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolo V, Ginestra A, Cassara D, Ghersi G, Nagase H, Vittorelli ML. 1999. Shed membrane vesicles and selective localization of gelatinases and MMP-9/TIMP-1 complexes. Ann. N Y Acad. Sci. 878, 497–499. ( 10.1111/j.1749-6632.1999.tb07707.x) [DOI] [PubMed] [Google Scholar]
- 18.Lv MM, et al. 2014. Exosomes mediate drug resistance transfer in MCF-7 breast cancer cells and a probable mechanism is delivery of P-glycoprotein. Tumour Biol. 35, 10 773–10 779. ( 10.1007/s13277-014-2377-z) [DOI] [PubMed] [Google Scholar]
- 19.Hakulinen J, Sankkila L, Sugiyama N, Lehti K, Keski-Oja J. 2008. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell Biochem. 105, 1211–1218. ( 10.1002/jcb.21923) [DOI] [PubMed] [Google Scholar]
- 20.Lugini L, Valtieri M, Federici C, Cecchetti S, Meschini S, Condello M, Signore M, Fais S. 2016. Exosomes from human colorectal cancer induce a tumor-like behavior in colonic mesenchymal stromal cells. Oncotarget 7, 50 086–50 098. ( 10.18632/oncotarget.10574) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corrado C, Saieva L, Raimondo S, Santoro A, De Leo G, Alessandro R. 2016. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell Mol. Med. 20, 1829–1839. ( 10.1111/jcmm.12873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maida Y, Takakura M, Nishiuchi T, Yoshimoto T, Kyo S. 2016. Exosomal transfer of functional small RNAs mediates cancer–stroma communication in human endometrium. Cancer Med. 5, 304–314. ( 10.1002/cam4.545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paggetti J, et al. 2015. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 126, 1106–1117. ( 10.1182/blood-2014-12-618025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melo SA, et al. 2014. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26, 707–721. ( 10.1016/j.ccell.2014.09.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Epple LM, Griffiths SG, Dechkovskaia AM, Dusto NL, White J, Ouellette RJ, Anchordoquy TJ, Bemis LT, Graner MW. 2012. Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS ONE 7, e42064 ( 10.1371/journal.pone.0042064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen WW, et al. 2013. BEAMing and droplet digital PCR analysis of mutant IDH1 mRNA in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol. Ther. Nucleic Acids 2, e109 ( 10.1038/mtna.2013.28) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akers JC, et al. 2013. MiR-21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): a platform for glioblastoma biomarker development. PLoS ONE 8, e78115 ( 10.1371/journal.pone.0078115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang CC, Park AY, Guan JL. 2007. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333. ( 10.1038/nprot.2007.30) [DOI] [PubMed] [Google Scholar]
- 29.Van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, Bracke M, De Wever O, Hendrix A. 2014. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 3, 24858 ( 10.3402/jev.v3.24858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong BS, et al. 2009. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 10, 556 ( 10.1186/1471-2164-10-556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demory Beckler M, et al. 2013. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell Proteomics 12, 343–355. ( 10.1074/mcp.M112.022806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skogberg G, et al. 2013. Characterization of human thymic exosomes. PLoS ONE 8, e67554 ( 10.1371/journal.pone.0067554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cordell PA, Kile BT, Standeven KF, Josefsson EC, Pease RJ, Grant PJ. 2010. Association of coagulation factor XIII-A with Golgi proteins within monocyte-macrophages: implications for subcellular trafficking and secretion. Blood 115, 2674–2681. ( 10.1182/blood-2009-08-231316) [DOI] [PubMed] [Google Scholar]
- 34.Crane CA, Ahn BJ, Han SJ, Parsa AT. 2012. Soluble factors secreted by glioblastoma cell lines facilitate recruitment, survival, and expansion of regulatory T cells: implications for immunotherapy. Neuro Oncol. 14, 584–595. ( 10.1093/neuonc/nos014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, ter Laan M, Wesseling P, Adema GJ. 2015. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J. Neuropathol. Exp. Neurol. 74, 390–400. ( 10.1097/NEN.0000000000000183) [DOI] [PubMed] [Google Scholar]
- 36.Antonios JP, et al. 2017. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol. 19, 796–807. ( 10.1093/neuonc/now287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dwyer J, Hebda JK, Le Guelte A, Galan-Moya EM, Smith SS, Azzi S, Bidere N, Gavard J. 2012. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLoS ONE 7, e45562 ( 10.1371/journal.pone.0045562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilbertson RJ, Rich JN. 2007. Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 7, 733–736. ( 10.1038/nrc2246) [DOI] [PubMed] [Google Scholar]
- 39.Giusti I, Delle Monache S, Di Francesco M, Sanita P, D'Ascenzo S, Gravina GL, Festuccia C, Dolo V. 2016. From glioblastoma to endothelial cells through extracellular vesicles: messages for angiogenesis. Tumour Biol. 37, 12 743–12 753. ( 10.1007/s13277-016-5165-0) [DOI] [PubMed] [Google Scholar]
- 40.Kucharzewska P, et al. 2013. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl Acad. Sci. USA 110, 7312–7317. ( 10.1073/pnas.1220998110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Vos KE, et al. 2016. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 18, 58–69. ( 10.1093/neuonc/nov244) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeung YT, McDonald KL, Grewal T, Munoz L. 2013. Interleukins in glioblastoma pathophysiology: implications for therapy. Br. J. Pharmacol. 168, 591–606. ( 10.1111/bph.12008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albulescu R, Codrici E, Popescu ID, Mihai S, Necula LG, Petrescu D, Teodoru M, Tanase CP. 2013. Cytokine patterns in brain tumour progression. Mediators Inflamm. 2013, 979748 ( 10.1155/2013/979748) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joseph JV, et al. 2015. Serum-induced differentiation of glioblastoma neurospheres leads to enhanced migration/invasion capacity that is associated with increased MMP9. PLoS ONE 10, e0145393 ( 10.1371/journal.pone.0145393) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, et al. 2015. Activation of toll-like receptor 2 promotes invasion by upregulating MMPs in glioma stem cells. Am. J. Transl. Res. 7, 607–615. [PMC free article] [PubMed] [Google Scholar]
- 46.Veeravalli KK, Rao JS. 2012. MMP-9 and uPAR regulated glioma cell migration. Cell Adh. Migr. 6, 509–512. ( 10.4161/cam.21673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harshyne LA, Nasca BJ, Kenyon LC, Andrews DW, Hooper DC. 2016. Serum exosomes and cytokines promote a T-helper cell type 2 environment in the peripheral blood of glioblastoma patients. Neuro Oncol. 18, 206–215. ( 10.1093/neuonc/nov107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sonabend AM, Rolle CE, Lesniak MS. 2008. The role of regulatory T cells in malignant glioma. Anticancer Res. 28, 1143–1150. [PubMed] [Google Scholar]
- 49.Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M. 2011. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. Clin. Dev. Immunol. 2011, 732413 ( 10.1155/2011/732413) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefranc F, Rynkowski M, DeWitte O, Kiss R. 2009. Present and potential future adjuvant issues in high-grade astrocytic glioma treatment. Adv. Tech. Stand. Neurosurg. 34, 3–35. ( 10.1007/978-3-211-78741-0_1) [DOI] [PubMed] [Google Scholar]
- 51.Lv S, Dai C, Liu Y, Sun B, Shi R, Han M, Bian R, Wang R. 2015. Cell surface protein C23 affects EGF-EGFR induced activation of ERK and PI3 K-AKT pathways. J. Mol. Neurosci. 55, 519–524. ( 10.1007/s12031-014-0375-7) [DOI] [PubMed] [Google Scholar]
- 52.Lv B, et al. 2015. CXCR4 signaling induced epithelial–mesenchymal transition by PI3 K/AKT and ERK pathways in glioblastoma. Mol. Neurobiol. 52, 1263–1268. ( 10.1007/s12035-014-8935-y) [DOI] [PubMed] [Google Scholar]
- 53.Liao A, et al. 2016. SDF-1/CXCR4 axis regulates cell cycle progression and epithelial–mesenchymal transition via up-regulation of survivin in glioblastoma. Mol. Neurobiol. 53, 210–215. ( 10.1007/s12035-014-9006-0) [DOI] [PubMed] [Google Scholar]
- 54.Iser IC, Pereira MB, Lenz G, Wink MR. 2016. The epithelial-to-mesenchymal transition-like process in glioblastoma: an updated systematic review and in silico investigation. Med. Res. Rev. 37, 271–313. ( 10.1002/med.21408) [DOI] [PubMed] [Google Scholar]
- 55.Redzic JS, Gomez JD, Hellwinkel JE, Anchordoquy TJ, Graner MW. 2016. Proteomic analyses of brain tumor cell lines amidst the unfolded protein response. Oncotarget 7, 47 831–47 847. ( 10.18632/oncotarget.10032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chetram MA, Hinton CV. 2012. PTEN regulation of ERK1/2 signaling in cancer. J. Recept. Signal Transduct. Res. 32, 190–195. ( 10.3109/10799893.2012.695798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steelman LS, et al. 2008. Contributions of the Raf/MEK/ERK, PI3 K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 22, 686–707. ( 10.1038/leu.2008.26) [DOI] [PubMed] [Google Scholar]
- 58.Dreesen O, Brivanlou AH. 2007. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 3, 7–17. ( 10.1007/s12015-007-0004-8) [DOI] [PubMed] [Google Scholar]
- 59.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. 2004. JAK/STAT, Raf/MEK/ERK, PI3 K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 18, 189–218. ( 10.1038/sj.leu.2403241) [DOI] [PubMed] [Google Scholar]
- 60.Meek DW. 2015. Regulation of the p53 response and its relationship to cancer. Biochem. J. 469, 325–346. ( 10.1042/BJ20150517) [DOI] [PubMed] [Google Scholar]
- 61.Sofroniew MV, Vinters HV. 2010. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. ( 10.1007/s00401-009-0619-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo Cicero A, Schiera G, Proia P, Saladino P, Savettieri G, Di Liegro CM, Di Liegro I. 2011. Oligodendroglioma cells shed microvesicles which contain TRAIL as well as molecular chaperones and induce cell death in astrocytes. Int. J. Oncol. 39, 1353–1357. ( 10.3892/ijo.2011.1160) [DOI] [PubMed] [Google Scholar]
- 63.Sundarraj N, Schachner M, Pfeiffer SE. 1975. Biochemically differentiated mouse glial lines carrying a nervous system specific cell surface antigen (NS-1). Proc. Natl Acad. Sci. USA 72, 1927–1931. ( 10.1073/pnas.72.5.1927) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vella LJ. 2014. The emerging role of exosomes in epithelial–mesenchymal-transition in cancer. Front. Oncol. 4, 361 ( 10.3389/fonc.2014.00361) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Atay S, Banskota S, Crow J, Sethi G, Rink L, Godwin AK. 2014. Oncogenic KIT-containing exosomes increase gastrointestinal stromal tumor cell invasion. Proc. Natl Acad. Sci. USA 111, 711–716. ( 10.1073/pnas.1310501111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mu W, Rana S, Zoller M. 2013. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 15, 875–887. ( 10.1593/neo.13786) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva TA, et al. 2016. AHNAK enables mammary carcinoma cells to produce extracellular vesicles that increase neighboring fibroblast cell motility. Oncotarget 7, 49 998–50 016. ( 10.18632/oncotarget.10307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baroni S, et al. 2016. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 7, e2312 ( 10.1038/cddis.2016.224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Franco OE, Shaw AK, Strand DW, Hayward SW. 2010. Cancer associated fibroblasts in cancer pathogenesis. Semin. Cell Dev. Biol. 21, 33–39. ( 10.1016/j.semcdb.2009.10.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen W, et al. 2015. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med. Oncol. 32, 43 ( 10.1007/s12032-015-0487-0) [DOI] [PubMed] [Google Scholar]
- 71.Frisa PS, Walter EI, Ling L, Kung HJ, Jacobberger JW. 1996. Stepwise transformation of astrocytes by simian virus 40 large T antigen and epidermal growth factor receptor overexpression. Cell Growth Differ. 7, 223–233. [PubMed] [Google Scholar]
- 72.Rich JN, Guo C, McLendon RE, Bigner DD, Wang XF, Counter CM. 2001. A genetically tractable model of human glioma formation. Cancer Res. 61, 3556–3560. [PubMed] [Google Scholar]
- 73.Medvedev R, Hildt E, Ploen D. 2016. Look who's talking–the crosstalk between oxidative stress and autophagy supports exosomal-dependent release of HCV particles. Cell Biol. Toxicol. 33, 211–231. ( 10.1007/s10565-016-9376-3) [DOI] [PubMed] [Google Scholar]
- 74.van Dongen HM, Masoumi N, Witwer KW, Pegtel DM. 2016. Extracellular vesicles exploit viral entry routes for cargo delivery. Microbiol. Mol. Biol. Rev. 80, 369–386. ( 10.1128/MMBR.00063-15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Anderson MR, Kashanchi F, Jacobson S. 2016. Exosomes in viral disease. Neurotherapeutics 13, 535–546. ( 10.1007/s13311-016-0450-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meckes DG., Jr 2015. Exosomal communication goes viral. J. Virol. 89, 5200–5203. ( 10.1128/JVI.02470-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. 2008. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 10, 619–624. ( 10.1038/ncb1725) [DOI] [PubMed] [Google Scholar]
- 78.Neviani P, Fabbri M. 2015. Exosomic microRNAs in the tumor microenvironment. Front. Med. 2, 47 ( 10.3389/fmed.2015.00047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stupp R, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl. J. Med. 352, 987–996. ( 10.1056/NEJMoa043330) [DOI] [PubMed] [Google Scholar]
- 80.Domingo-Musibay E, Galanis E. 2015. What next for newly diagnosed glioblastoma? Future Oncol. 11, 3273–3283. ( 10.2217/fon.15.258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tosoni A, Franceschi E, Poggi R, Brandes AA. 2016. Relapsed glioblastoma: treatment strategies for initial and subsequent recurrences. Curr. Treat. Options Oncol. 17, 49 ( 10.1007/s11864-016-0422-4) [DOI] [PubMed] [Google Scholar]
- 82.Braunstein S, Nakamura JL. 2013. Radiotherapy-induced malignancies: review of clinical features, pathobiology, and evolving approaches for mitigating risk. Front Oncol. 3, 73 ( 10.3389/fonc.2013.00073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, Louis DN. 2009. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 15, 4622–4629. ( 10.1158/1078-0432.CCR-08-3012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sato Y, Kurose A, Ogawa A, Ogasawara K, Traganos F, Darzynkiewicz Z, Sawai T. 2009. Diversity of DNA damage response of astrocytes and glioblastoma cell lines with various p53 status to treatment with etoposide and temozolomide. Cancer Biol. Ther. 8, 452–457. ( 10.4161/cbt.8.5.7740) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prasad G, Haas-Kogan DA. 2009. Radiation-induced gliomas. Expert Rev. Neurother 9, 1511–1517. ( 10.1002/pmic.200800802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu J, Zhang C, Feng Z. 2014. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin 46, 170–179. ( 10.1093/abbs/gmt144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Radke J, Bortolussi G, Pagenstecher A. 2013. Akt and c-Myc induce stem-cell markers in mature primary p53−/− astrocytes and render these cells gliomagenic in the brain of immunocompetent mice. PLoS ONE 8, e56691 ( 10.1371/journal.pone.0056691) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
While there are no specific datasets to share, any data inquiries may be directed to the corresponding author.