

Abstract
Maintenance of protein homeostasis is vitally important in post-mitotic cells, particularly neurons. Neurodegenerative diseases such as polyglutamine expansion disorders—like Huntington's disease or spinocerebellar ataxia (SCA), Alzheimer's disease, fronto-temporal dementia (FTD), amyotrophic lateral sclerosis (ALS) and Parkinson's disease—are often characterized by the presence of inclusions of aggregated protein. Neurons contain complex protein networks dedicated to protein quality control and maintaining protein homeostasis, or proteostasis. Molecular chaperones are a class of proteins with prominent roles in maintaining proteostasis, which act to bind and shield hydrophobic regions of nascent or misfolded proteins while allowing correct folding, conformational changes and enabling quality control. There are many different families of molecular chaperones with multiple functions in proteostasis. The DNAJ family of molecular chaperones is the largest chaperone family and is defined by the J-domain, which regulates the function of HSP70 chaperones. DNAJ proteins can also have multiple other protein domains such as ubiquitin-interacting motifs or clathrin-binding domains leading to diverse and specific roles in the cell, including targeting client proteins for degradation via the proteasome, chaperone-mediated autophagy and uncoating clathrin-coated vesicles. DNAJ proteins can also contain ER-signal peptides or mitochondrial leader sequences, targeting them to specific organelles in the cell. In this review, we discuss the multiple roles of DNAJ proteins and in particular focus on the role of DNAJ proteins in protecting against neurodegenerative diseases caused by misfolded proteins. We also discuss the role of DNAJ proteins as direct causes of inherited neurodegeneration via mutations in DNAJ family genes.
This article is part of the theme issue ‘Heat shock proteins as modulators and therapeutic targets of chronic disease: an integrated perspective’.
Keywords: DNAJ, molecular chaperones, neurodegeneration
1. Introduction
Intracellular or extracellular proteinaceous inclusions in specific brain regions are a pathological hallmark of many neurodegenerative diseases [1]. These inclusions are generally composed of misfolded and aggregated forms of specific disease-associated proteins. For example, Alzheimer's disease (AD) is characterized by the accumulation of extracellular amyloid-β plaques and intracellular tangles of phosphorylated tau; Parkinson's disease (PD) is associated with intracellular deposits of α-synuclein known as Lewy bodies; and in Huntington's disease (HD) intracellular aggregates of polyglutamine-expanded forms of the huntingtin protein are present. Protein aggregation in these neurodegenerative diseases can arise from genetic variations in the disease-related proteins (either as directly causative mutations or polymorphisms that shift the folding equilibrium of the disease-linked protein); genetic alterations that lead to elevated levels of the protein expression; or can be triggered by environmental stress and ageing [2].
It is not always clear whether protein aggregation into inclusions is a cause or consequence of neurodegeneration; however, in inherited forms of neurodegeneration many of the causative mutations disrupt the folding of the disease protein, leading to increased aggregation and inclusion formation. The pathological inclusions seen in all neurodegenerative disorders are thought to represent the endpoint of the protein aggregation process. Prior to the formation of large aggregates, mutated or misfolded proteins are believed to form small soluble oligomers, which some studies have demonstrated to be the more toxic species [3,4]. It has been suggested, therefore, that the proteinaceous inclusions seen pathologically are not the primary cause of neurotoxicity, and their formation is a protective defence mechanism employed by the cell to sequester the potentially more toxic soluble oligomers [2]. Nevertheless, it is likely that these inclusions can also contribute to toxicity in neurons by physically obstructing axonal transport, sequestering other essential proteins and disrupting overall protein homeostasis of the cell. Neurons are particularly vulnerable to this toxicity as they rely heavily on axonal transport between the cell body and synaptic terminals, and being a post-mitotic cell type, they do not have an ability to disperse protein aggregates via cell division, or be readily replaced [5].
Given their vulnerability to toxicity induced by aggregated oligomers and proteinaceous inclusions, neurons depend heavily on an intrinsic network of protein quality control mechanisms designed to maintain proteostasis, a state in which all proteins in the proteome are in the conformation, concentration and location that are required for correct functioning of the cell [6]. Cells have several mechanisms to regulate the biogenesis, folding, trafficking and degradation of proteins to ensure that proteostasis is maintained, and disruptions to these processes, or an imbalance in protein folding caused by mutations or stress, can lead to disease. Cells respond to stress through compartment-related signalling pathways. In the cytoplasm and nucleus, the heat shock response (HSR) mediates a transcriptional response to stress through heat shock factors (e.g. HSF1), whereas the endoplasmic reticulum (ER) has the unfolded protein response (UPR) to respond to stress [7,8]. Intrinsic degradation mechanisms employed to maintain proteostasis include clearance systems such as autophagy and the ubiquitin-proteasome system (UPS), which involve the compartmentalization, degradation and recycling of misfolded or unfolded proteins by lysosomes or proteasome, respectively [5,9–11]. The HSR and UPR act to restore protein homeostasis by reducing protein translation and activating signalling pathways that increase production of protective factors, such as molecular chaperones [8].
Molecular chaperones are heterogeneous and functionally diverse families of proteins that are involved in many critical cellular processes, including protein folding, trafficking, quality control and degradation. A common classification of molecular chaperones (also known as heat shock proteins; HSPs) is according to their molecular weight. The major families are HSP90, HSP70, HSP40 (DNAJ), HSP60 and the small HSPs. In this review, the focus will be on the DNAJ family members and their relation to neuronal proteostasis and neurodegeneration [12].
2. DNAJ proteins
DNAJ proteins (also known as J proteins or HSP40 proteins) are a family of chaperones that regulate HSP70 chaperones through stimulating ATP hydrolysis. The defining feature of DNAJ proteins is the J-domain, an approximately 70 amino acid highly conserved region containing 4 α-helices (figure 1). The linker region between helices 2 and 3 is especially well conserved and contains the histidine-proline-aspartic acid (HPD) motif that is absolutely required for stimulation of ATP hydrolysis in HSP70 [13]. There are approximately 50 different members of the DNAJ protein family in man, ranging in size from 10 to 520 kDa, suggesting that the HSP40 designation might not be an accurate description of this family of proteins [14]. The variety in size reflects the diversity in function of DNAJ proteins due to their varying domain structure [15].
Figure 1.

The J domain and DNAJ subfamilies. (a) The amino acid sequence of the DNAJB2 J domain with α-helixes (green), β-sheet (blue) and histidine–proline–aspartic acid (HPD) motif (boxed) highlighted. (b–d) Phylograms of DNAJ subfamilies and schematic illustrations of their conserved domains. Protein sequence alignments were performed using a Blosum scoring matrix in ClustalX. Bootstrap value is presented at right corner in (c). Numbers represent the degree of homology (0–1000). (e) The tertiary structure of J domain of DNAJB2 (PDB 2LGW) from N-terminus (dark blue) to C-terminus (yellow) is shown with the 4 α-helixes and HPD motif highlighted. (f) Illustration of how the J-domain (green) can facilitate substrate (dark blue) loading onto Hsp70 (grey). When the ATP is bound, the C-terminal substrate-binding domain (SBD) is docked onto the N-terminal nucleotide-binding domain (NBD). DNAJ proteins simulate Hsp70 ATPase hydrolysis, as well as recruiting substrates. When the ADP is bound, the lid closes and stabilizes the cleft-substrate binding. Nucleotide exchange factors (NEF) (brown) complete the cycle by stimulating the exchange of ADP for ATP and substrate release. (Online version in colour.)
DNAJ protein family members can be divided into three subtypes depending on their domain composition (class I, II or III, also called A, B or C; [16]) (figure 1). Class I (DNAJA) DNAJ proteins are the most similar to the eponymous E. coli DnaJ protein and contain the canonical domain structure of an N-terminal J-domain followed by a glycine/phenylalanine (G/F)-rich region, a zinc-finger motif and C-terminal client-binding domain (CBD). Class II (DNAJB) DNAJ proteins contain an N-terminal J-domain and G/F-rich region. Class III (DNAJC) DNAJ proteins only have the J-domain with no other canonical domains, and the J-domain may be located anywhere in the structure of the protein. DNAJC proteins are the largest subtype of DNAJ proteins and have the greatest diversity in their size, structure and domain architecture, reflecting highly specialized functions. Among the wide variety of protein domains found in DNAJ proteins are ubiquitin-interacting motifs (UIMs), cysteine-rich regions, GTP-binding domains, tetratricopepetide repeats (TPRs) and clathrin-binding domains [17].
3. Mutations in DNAJ proteins as a cause of disease
Mutations in DNAJ proteins can cause disease, as part of a larger collection of genetically inherited disorders caused by mutations in molecular chaperones known as chaperonopathies [18]. Furthermore, the majority of chaperonopathies result in neurodegenerative-like phenotypes, emphasizing the important role of molecular chaperones in neuronal proteostasis, in particular motor neurons [19]. Currently mutations are known to occur in fourteen DNAJ proteins (table 1, figure 2), leading to diseases such as cerebellar ataxia, distal hereditary motor neuropathy, Charcot Marie Tooth disease and Parkinson's disease [60]. However, mutations in some DNAJ proteins cause non-neurodegenerative disorders; for example, mutations in DNAJB13 cause primary ciliary dyskinesia [35], mutations in DNAJC12 cause hyperphenylalanemia [45] and mutations in DNAJC21 cause bone marrow failure syndrome [53]. In this section, we will focus on the role of DNAJ mutations in contributing to neurodegeneration and the consequences for neuronal proteostasis.
Table 1.
Mutations in DNAJ proteins cause a range of diseases.
| DNAJ gene (AKA) | mutation/result | disease | inheritance | references |
|---|---|---|---|---|
| DNAJB1 (HDJ1/HSP40) | 400 kb deletion on chromosome 19 resulting in N-terminal DNAJB1 chimeric in-frame fusion with PKA catalytic domain | fibrolamellar hepatocellular carcinoma | somatic | [20] |
| DNAJB2 (HSJ1) | c.352+1G>A resulting in intron 5 retention | distal hereditary motor neuropathy | recessive | [21–25] |
| c.229+1G>A resulting in intron 4 retention | ||||
| c.14A>G, p.Y5C (J-domain mutation) | Charcot Marie Tooth disease type 2 | |||
| c.619-1G>A resulting in splice site deletion | ||||
| c.309delC, p.F103fsX | ||||
| 3.8 kb deletion resulting in J-domain deletion | spinal muscular atrophy/juvenile Parkinsonism | |||
| DNAJB5 (HSC40) | c.43C>T, p.P15S (J-domain mutation) | hereditary myoclonus and progressive distal muscular atrophy | recessive | [25] |
| DNAJB6 (MRJ) | c.265T>A, p.F89I | limb-girdle muscular dystrophy | dominant | [26,27–34] |
| c.271T>A, p.F91I | ||||
| c.271T>C, p.F91L | ||||
| c.273C>G, p.F93I | ||||
| c.277T>A, c.277T>C, c.279C>A, c.279C>G, p.F93L | ||||
| c.287C>G, p.P96R | ||||
| c.287TC>T, p.P96L | ||||
| c.298T>G, p.F100V | ||||
| c.346+5G>A | ||||
| DNAJB13 | c.833T>G, p.M278R | primary ciliary dyskinesia type 34 | recessive | [35] |
| c.68+1G>C, p.Y24X | ||||
| DNAJC3 (p58) | c.508C>T, p.R194X | combined cerebellar and peripheral ataxia with hearing loss and diabetes mellitus | recessive | [36] |
| 72 kb deletion resulting in loss of exons 6–12 | ||||
| DNAJC5 (CSPα) | c.346_348delCTC, p.L116Δ | adult-onset neuronal ceroid lipofuscinosis | dominant | [37,38] |
| c.344T>G, p.L115R | ||||
| DNAJC6 (auxilin) | c.801-2A>G | autosomal recessive juvenile Parkinsonism | recessive | [39–42,43] |
| c.2371C>T, p.G791X | ||||
| c.397A>T, p.M133L | early-onset Parkinson's disease | |||
| c.626T>C, p.L209P | ||||
| c.1468+83del | ||||
| c.1855C>T, p.R619C | ||||
| c.2038+3A>G resulting in loss of splice donor site | ||||
| c.2200C>T, p.G734X | ||||
| c.2365C>T, p.G789X | ||||
| c.2223A>T, p.T741X | ||||
| c.2517del, p.F389LfsX22 | ||||
| c.2779A>G, p.R927G (J-domain mutation) | ||||
| 80 kb deletion of exons 5–19 | early-onset obesity, mental retardation and epilepsy | |||
| Dnajc11 | c.1524+56T>A (mice only) resulting in cryptic splicing, p.K508fsX43 | spasticity, MN pathology | recessive | [44] |
| DNAJC12 (JDP1) | c.298-968_503-2603del resulting in exon 4 deletion | hyperphenylalaninemia, mild, non-BH4 deficient | recessive | [45] |
| c.215G>C, p.R72P | ||||
| c.158-2A>T resulting in intron 3 splice site mutation | ||||
| DNAJC13 (RME-8) | c.2564A>G, p.N855R | autosomal dominant Parkinson's disease | dominant | [46] |
| DNAJC17 | c.681G>A (r.601_681del), p.Y201_A227del | retinitis pigmentosa and hypogammaglobulinemia | recessive | [47] |
| DNAJC19 (TIM14) | IVS3-1G>C resulting in skip exon 4 and frameshift truncation | dilated cardiomyopathy and ataxia | recessive | [48–52] |
| c.300delA, p.A100fsX11 | ||||
| c.63delC, p.Y21X | ||||
| c.280+1_280+5delGTAAG resulting in splice site deletion | ||||
| DNAJC21 | c.517C>T, p.R173X | bone marrow failure syndrome type 3 | recessive | [53] |
| c.983+1G>T, p.G299AfsX2 | ||||
| c.94C>G, p.P32A | ||||
| c.793G>T, p.Q265X | ||||
| DNAJC29 (sacsin) | c.7504C>T, p.R2502X | autosomal recessive spastic ataxia of Charlevoix-Saguenay | recessive | [54–59] |
| c.8844delT, p.P2948fsX3 | ||||
| c.12992G>A, p.R4331Q (J-domain mutation) | ||||
| c.12991C>T, p.R4331W (J-domain mutation) | ||||
| c.13027G>A, p.E4343K (J-domain mutation) | ||||
| more than 150 other mutations |
Figure 2.
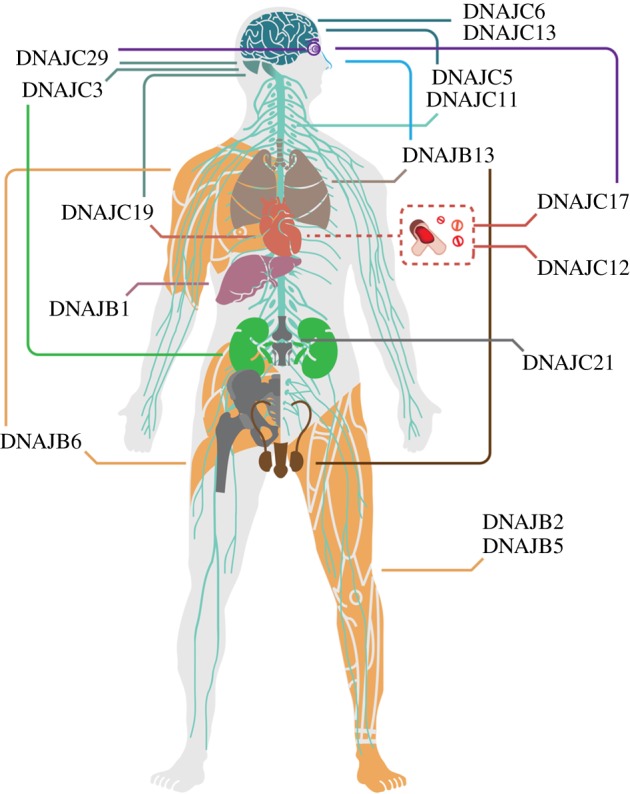
Pathogenic consequences of DNAJ family mutations. Schematic illustration of a human body demonstrating the tissues and organs affected by DNAJ mutations, including nerve system composed of brain (dark cyan), cerebellum (cyan), spinal cord and peripheral nervous system (light cyan), eyes (purple), nose (blue), lungs (beige), heart, blood vessels (red), liver (dark pink), kidneys (green), muscles (orange), reproductive system (brown) and bones (grey). (Online version in colour.)
(a). DNAJB2 (HSJ1)
DNAJB2 is an alternatively spliced neuronal protein forming two isoforms: a 36 kDa cytosolic/nuclear form (DNAJB2a; HSJ1a) and a larger 42 kDa isoprenylated membrane associated form (DNAJB2b; HSJ1b) [61]. As a type II DNAJ protein, it contains an N-terminal J-domain and G/F-rich region, but also a CBD (that is not conserved with DNAJ) and two UIMs, which can bind ubiquitylated client proteins and target them to the proteasome for degradation [62]. There are several biallelic mutations known in DNAJB2 that are associated with a range of neurodegenerative diseases. Charcot Marie Tooth (CMT) disease results in progressive degeneration of spinal cord motor neurons, leading to weakness and muscle atrophy in the lower limbs [63]. Patients also show distal sensory loss. A homozygous missense mutation c.14A>G in DNAJB2 resulting in a substitution of tyrosine for cysteine at residue five (Y5C) in the DNAJB2 J-domain causes CMT type 2 [64,21]. There are also reports of splicing mutations in DNAJB2 resulting in distal hereditary motor neuropathies (dHMN), which are a genetically and clinically heterogeneous group of disorders similar to CMT, but without the sensory abnormalities [65,66]. A homozygous splice site mutation (c.352+1G>A) was identified in DNAJB2 leading to either partial or total retention of intron 5, resulting in reduced DNAJB2 protein expression [22]. There have been additional reports of patients with this mutation recently [67]. This mutation has been suggested to be a potential founder mutation, because in another study of CMT/dHMN the five affected individuals with this mutation shared a common haplotype [23]. Similarly, Gess et al. reported a dHMN patient with a homozygous c.229+1G>A DNAJB2 splice site mutation, leading to the retention of intron 4 and subsequent loss of DNAJB2 protein expression [21]. A recent study identified a large-scale deletion incorporating the first four exons of DNAJB2 (including the entire J-domain) as causing spinal muscular atrophy (SMA) and atypical juvenile parkinsonism (AJP) [24]. A recent exome analysis of peripheral neuropathy patients identified two new mutations in DNAJB2; a frameshift truncation (F103fsX) and a splice site mutation (c.619-1G>A; [25]).
(b). DNAJB5 (HSC40)
DNAJB5 was originally identified as being similar to DNAJB1 [68] and has since been shown to interact with HSP70 [69]. A whole-exome sequencing analysis of CMT-like patients identified a mutation in the J-domain of DNAJB5 (P15S) as a novel cause of neuropathy [25]. Morpholino-mediated knockdown of DNAJB5 in zebrafish revealed abnormalities in peripheral nerve axon structure, but no effect on muscle architecture [25]
(c). DNAJB6 (MRJ)
DNAJB6 is a ubiquitous protein with high expression levels in the brain and detectable protein in muscle [70]. Alternative splicing of the DNAJB6 gene produces two isoforms: a 36 kDa nuclear isoform and a 26 kDa cell stress-responsive cytosolic form [71]. Mutations in DNAJB6 cause limb-girdle muscular dystrophy type 1 (LGMD1). LGMD1 is an autosomal dominant disease characterized by progressive distal and occasionally proximal muscle atrophy caused by myofibrillar myopathy. There is also a report of a DNAJB6 patient with frontotemporal dementia alongside LGMD1 [72]. There are currently twelve mutations known in DNAJB6 (table 1); interestingly, all of the mutations are found in exon 5, which codes for the G/F-rich region of the protein. Ruggieri and colleagues have suggested that there might be a genotype–phenotype correlation between both the severity of the disease and the location (proximal-distal) and the mutated residue involved, with C-terminal mutations leading to a distal phenotype [73]. Patients with DNAJB6 mutations have myofibrillar aggregates containing ubiquitin, TDP-43 and p62, suggesting defective protein clearance [26,74], which are also observed in Dnajb6 F93L transgenic mice [75]. Drosophila mutants recapitulating patient mutations result in loss of DNAJB6-dependent anti-aggregation activity [76].
(d). DNAJC3 (p58)
DNAJC3 is a 58 kDa DNAJ protein that is targeted to the cytoplasmic face of the ER [77,78]. DNAJC3 can also bind and inhibit the UPR sensor PERK in the ER, suggesting a role in regulating the UPR [79,80]. DNAJC3 can recruit cytosolic HSP70 to the face of the ER and work with Sec61 as part of the translocation machinery [81]. Knockdown of DNAJC3 results in accumulation of misfolded protein in the ER and activation of the UPR [82] and Dnajc3 knockout mice have decreased ability to cope with ER stress [81,83]. Mutations in DNAJC3 cause multisystemic neurodegeneration, including early-onset cerebellar ataxia and peripheral neuropathy, alongside diabetes mellitus [36]. Interestingly, Dnajc3 knockout mice also show a diabetic phenotype [83] and recent work has also shown that the ubiquitin ligase CHIP is involved in the turnover of the insulin receptor, suggesting a link between proteostasis network control and insulin regulation [84].
(e). DNAJC5 (CSPα)
DNAJC5 is a secretory vesicle protein found in both neuronal and non-neuronal tissues; however, the main α-isoform is only expressed in the brain [85]. DNAJC5 is characterized by a cysteine-rich region and is targeted to post-Golgi membranes via palmitoylation [86]. DNAJC5 has a role in binding and folding many proteins required at the synapse, such as SNAP-25, syntaxins and synaptotagmins [87–89], where it acts as a co-chaperone with the constitutive HSP70, HSC70 (HSPA8) [90]. DNAJC5, therefore, most likely plays a key role at the synapse as a chaperone [91,92]. Mutations in DNAJC5 cause autosomal dominant adult onset neuronal ceroid lipofusinosis (ANCL), an accumulation of autofluorescent lysosomal waste (known as lipofuscin) that causes a progressive neurodegenerative disorder characterized by ataxia, seizures and dementia [93]. ANCL is a rare disease and to date only two distinct mutations in DNAJC5 (deletion of leucine 116 and missense change L115R) have been identified in a handful of families [37,38,94,95]. The location of the mutations in the cysteine-rich region suggests a defect in the membrane trafficking of patient DNAJC5 and subsequent protein aggregation [96,97]. DNAJC5 interacts with another ANCL disease-causing protein, palmitoyl-protein thioesterase 1 (PPT1). PPT1 is accumulated in DNAJC5 patient brains and has decreased activity, suggesting a link between ANCL and palmitoylation of synaptic proteins [98]. Dnajc5 KO mice have deficient neuromuscular function and sensorimotor impairment. Indeed, these mice have specific degeneration of the neuromuscular junctions, implying that KO of Dnajc5 leads to synapse dysfunction [85]. Interestingly, DNAJC5 mutations in Caenorhabditis elegans lead to sensory neuron dysfunction that could be rescued by treatment with resveratrol [99].
(f). DNAJC6 (auxilin)
Another well-characterized vesicle-associated protein is DNAJC6, which has a role in uncoating clathrin-coated vesicles [100]. DNAJC6 binds clathrin via its C-terminal clathrin binding-domain [101,102]. The clathrin-coating and uncoating cycle is well characterized; clathrin triskelions form coated pits at the pre-synaptic membrane around the intended cargo. Before fusing with the endosome, the vesicles need to be uncoated by HSC70, following recruitment and activation by DNAJC6 [103,104]. In neurons, this process is vital for synaptic vesicle recycling. Mutations in DNAJC6 were first associated with autosomal recessive juvenile parkinsonism (ARJP) [39,40]. Symptoms of ARJP include typical PD features, but also include mental retardation and seizures. ARJP typically manifests in the first decade and rapidly leaves patients wheelchair-bound. There is also a report of a 80 kb large-scale deletion including DNAJC6 that results in ARJP [41]. A recent study also identified variants in DNAJC6 that are associated with early-onset PD, which has a later onset than ARJP [42]. The authors suggest that this may be due to residual DNAJC6 activity compared to the ARJP mutations, which likely cause complete loss of function, and therefore represents a genotype–phenotype correlation of DNAJC6 mutations. Interestingly, a mutation in a highly conserved residue (R927G) in the J-domain was found that potentially disrupts the HSC70 interaction.
(g). DNAJC11
DNAJC11 was originally described as a 63 kDa protein containing an N-terminal J-domain that is often deleted in neuroblastoma [105,106]. DNAJC11 was later identified as a mitochondrial protein [107], specifically as a member of the mitochondrial complex I, involved in the electron transport chain, although siRNA-mediated knockdown had no effect on the assembly of the complex [108]. Using random N-ethyl-N-nitrosurea (ENU) mutagenesis, Ioakeimidis et al. created a spastic mouse model with a deep intronic mutation in Dnajc11, resulting in the addition of a 109 bp cryptic exon and a frameshift truncation and reduction of Dnajc11 protein [44]. These mice had abnormal locomotion and progressive muscle wasting and spasticity resulting in death at five weeks of age. They also had highly vacuolated motor neurons in the lumbar spinal cord, generated from either abnormal mitochondrial cristae or ER, as mitochondria in these motor neurons were severely disrupted [44].
(h). DNAJC13 (RME-8)
DNAJC13 is an endocytic protein that has been shown to localize to early and recycling endosomes [109]. The J-domain of DNAJC13 is located in the middle of the protein, with a membrane-binding region at the N-terminus and four potential clathrin-binding motifs [109]. DNAJC13 interacts with the retromer complex [110] and thus may have a role in recruiting HSC70 to vesicle formation sites. An inherited variant (N855S) in DNAJC13 was originally thought to cause autosomal-dominant PD [46]; however, two affected family members did not have this variant and subsequent whole-exome sequencing identified two causative changes in another endosomal/synaptic protein TMEM230, questioning the importance of this variant for PD [111]. However, sequence analysis of exon 24 of DNAJC13 in a Caucasian population study has suggested that N855S could be a rare variant associated with PD [112]. Further analysis revealed that other DNAJC13 variants (E1740Q, R1615H, L2120W) might be associated with increased risk of PD [113,114].
(i). DNAJC19 (TIM14)
DNAJC19 is one of several mitochondrial DNAJ proteins, found at the inner mitochondrial membrane. It recruits and activates mitochondrial HSP70 (HSPA9) to function as part of the mitochondrial import machinery [115]. Mutations in DNAJC19 cause autosomal recessive dilated cardiomyopathy and cerebellar ataxia (DMCA). A splice site change that leads to the loss of exon 4 and subsequent truncation of the protein was the first mutation identified [48]. Recently, single nucleotide deletions and splice deletions have also been identified with associated disease features [49,50,51].
(j). DNAJC29 (sacsin)
The largest known DNAJ protein is DNAJC29 (520 kDa), which contains a C-terminal J-domain, an N-terminal ubiquitin-like (UbL) domain, three sacsin repeat regions (SRRs), which show homology to the ATP-binding domain of HSP90, and a C-terminal higher eukaryote and prokaryote (HEPN) domain [116,117]. DNAJC29 is a neuronal protein that is localized to the cytoplasmic face of the mitochondria; knockdown of DNAC29 results in disruption of the mitochondrial network [118]. Mutations in DNAJC29 cause autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), an early-onset disorder characterized by cerebellar ataxia and peripheral neuropathy, with prominent Purinkje cell death in the cerebellum [119]. There is a large founder effect in the patient population; the vast majority of patients are from the Quebec region in Canada and have the R2502X mutation [54], although more than 150 other patient mutations are now known worldwide, including large-scale deletions [120,121]. Mutations in DNAJC29 are the second most common cause of autosomal recessive ataxia after mutations in frataxin, which causes Freidrich's ataxia. The J-domain of DNAJC29 has been shown to functional via a bacterial complementation assay in an E. coli DnaJ and CbpA temperature-sensitive mutant, and interestingly there are two patient missense mutations located in the J-domain (R4331Q and E4343K) [55,56]. Dnajc29 knockout mice have ataxic symptoms with peripheral neuropathy and progressive Purkinje cell loss, recapitulating the human disorder [122]. Furthermore, Dnajc29 null mice motor neurons have elongated mitochondria and accumulations of neurofilaments [122]. DNAJC29 interacts with the mitochondrial fission protein DRP1 [118] and recent work using patient fibroblasts has shown that there is a reduction of DRP1 foci at the mitochondria and mitochondrial health and function in ARSACS are decreased, suggesting impairment in the ability of the mitochondrial network in affected neurons [123].
4. Manipulation of DNAJ proteins in models of neurodegeneration
The late onset of many neurodegenerative diseases has been suggested to correlate with a reduced efficiency of the protein quality control machinery as a result of ageing. Correspondingly, the manipulation of molecular chaperones is a promising therapeutic approach for many neurodegenerative diseases [12]. Recently, several studies have focused on increasing the expression of chaperones in different neurodegeneration models and the data support the potential of chaperone manipulation, and in particular DNAJ proteins, in the battle against neurodegenerative diseases. In this section, the focus will be on targeting different disease-related proteins in neurodegeneration with members of the DNAJ chaperone family.
(a). Polyglutamine (polyQ) expansion disorders
The polyglutamine (polyQ) disorders are a group of neurodegenerative diseases caused by a trinucleotide CAG repeat expansion that confers a toxic gain-of-function, with a direct relationship between the length of the polyQ expansion and the propensity to aggregate. PolyQ expansions have been identified in Huntington's disease (HD; huntingtin, htt), spinal and bulbar muscular dystrophy (SMBA; androgen receptor, AR) and spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA7, ataxin; SCA6, CACNA1A; SCA17, TATA box-binding protein (TBP)) (figure 3). Ubiquitylated inclusions of aggregated protein are characteristic of these diseases [124].
Figure 3.

DNAJ proteins modulate neurodegeneration in model systems. Polyglutamine (polyQ) expansion models of huntingtin (htt), ataxin-1 (Atx1), ataxin-3 (Atx3), androgen receptor (AR) and spinocerebellar ataxia type 6 (SCA6) shown in red. α-synuclein and parkin implicated in Parkinson's disease and amyloid-β (Aβ) and tau present in Alzheimer's disease in green and blue, respectively. TAR DNA-binding protein-43 kDa (TDP-43) and superoxide dismutase 1 (SOD1) present in amyotrophic lateral sclerosis in magenta. (Online version in colour.)
Manipulation of polyQ protein aggregates by molecular chaperones was first reported by Cummings et al. in 1998: overexpression of DNAJA1 in cells reduced aggregation of polyQ-expanded ataxin-1 (SCA1) [125]. In cells overexpressing polyQ-expanded ataxin-1, knockdown of DNAJC29 has been shown to enhance ataxin-1-mediated toxicity, indicating a protective role against polyQ-expanded ataxin-1 toxicity [116]. DNAJB2a has been shown to have a dual role on ataxin-3 (Atx3; SCA3) depending on the chaperone's domains. In cells, DNAJB2a can either reduce the protein levels of Atx3 by promoting proteosomal degradation through J-domain, or diminish this process by preserving ubiquitylated Atx3 via the UIM domain [126]. Interestingly, DNAJA1 has also been reported to increase polyQ aggregation depending on the cell line used, an effect attributed to the J-domain that is responsible for the recruitment of endogenous HSP70 [127]. Since DNAJ chaperones are co-chaperones of HSP70, differences in levels of endogenous expression of HSP70 and DNAJ proteins between cell lines could explain this effect, as it could depend on the cellular chaperone balance. Similarly, overexpression of DNAJB1 in Neuro2a cells suppressed htt inclusion formation, while simultaneous overexpression of HSP70 improved folding efficiency and cellular proliferation and reduced cytotoxicity [128,129]. DNAJB1 has been also shown to increase solubility of polyQ-expanded AR and also enhance proteasome-mediated degradation in cells, an effect again amplified in the presence of HSP70 [130,131].
A screen of several different DNAJA and DNAJB proteins revealed that a subfamily of DNAJB proteins were the most efficient at reducing polyQ aggregation [69,132]. This subfamily includes DNAJB2a, DNAJB6b and DNAJB8, which are closely related (figure 1c), but the effect is also dependent on their sub-cellular localization. In vitro studies with purified proteins have shown that DNAJB6b can supress the formation of amyloid-like fibrils of polyQ peptides [133]. Moreover, DNAJB6b and DNAJB8 were shown to suppress polyQ aggregation and related toxicity in cells and transgenic Xenopus laevis models [132]. Genome-wide RNA interference screen on transgenic C. elegans expressing polyQ proteins identified DNAJ as a suppressor of polyQ aggregation [134]. Interestingly, DNAJB6b and DNAJB8 are effective in suppressing the aggregation not only of polyQ-expanded htt, but also of other disease-related polyQ-expanded proteins, such as Atx3 and the androgen receptor (SBMA) [132]. DNAJB6 and DNAJB8 were suggested to act on earlier stages of aggregation in cells despite their irreversible recruitment on larger aggregates in an unsuccessful attempt to prevent aggregation [135]. Furthermore, the cytoplasmic/nuclear DNAJB2 isoform, DNAJB2a, is recruited to polyQ inclusions and can reduce the polyQ aggregation and inclusion incidence in a cellular overexpression model in a J-domain and UIM independent manner by promoting degradation via the proteasome [62,136]. DNAJB2b has been also shown to inhibit neuronal death caused from mutant htt in vitro and also improve neuronal dysfunction in a C. elegans model of HD independent of any effect on polyQ aggregation [136]. In vitro studies have suggested that HSP70 and DNAJB1 can act on early stages of polyQ aggregation by halting or suppressing the formation of detergent-insoluble amyloid-like fibrils of polyQ [137].
In vivo investigation in Drosophila models for HD identified dHDJ1, the homologue of DNAJB1, as a suppressor of polyQ-driven toxicity [138]. A separate study showed that the DNAJB1-induced reduction of eye degeneration in transgenic polyQ Drosophila was enhanced by Drosophila HSC70cb and its human homologue APG-1, while DNAJB1 also had an effect in the absence of HSP70 [139]. Moreover, dMRJ, the Drosophila orthologue of the human DNAJB6, was recruited in the polyQ inclusions and was shown to suppress polyQ-mediated toxicity in flies [140]. In the same model, early expression of dHDJ1 dramatically promoted cytoplasmic aggregation of polyQ, while both DNAJ chaperones increased the level of detergent-soluble polyQ, illustrating the similarities and diversity of DNAJ chaperones [140]. Expression of dHDJ1 on mutant Atx3-expressing flies restored eye structure, an effect attributed to both J- and C-terminal domains. Interestingly, the effect of dHDJ1 on toxicity is enhanced in the presence of HSP70 and abolished in the presence of mutant HSP70. Both dHDJ1 and HSP70 overexpression altered the solubility of polyQ; however, expression of dHDJ2, which has the same J-domain but different C-terminal domains, resulted in weak suppression of eye degeneration in the flies, suggesting a role of the C-terminal domain [141]. In Drosophila models of SCA6 that express a CAG expansion in exon 47 of CACNA1A (a1ACT), DNAJ-1 was shown to suppress a1ACT-induced toxicity in the eye, while DNAJ-1 knockdown dramatically accelerated eye degeneration [142]. Interestingly, normal Atx3 has been shown to alleviate toxicity of several polyQ-expanded disease proteins including itself and mutated htt. Atx3 interacts with Rab23, which leads to increased DNAJ levels, which in turn leads to reduced eye degeneration in flies [143].
Despite these promising effects in other models, few direct chaperone overexpression experiments have successfully translated to the mammalian nervous system. DNAJB2a was effective in reducing polyQ inclusion formation in a rat brain model of SBMA using viral delivery, by increasing ubiquitylation and targeting to the UPS [144]. Moreover, two members of the DNAJ family have been shown to be effective on polyQ aggregation in the R6/2 transgenic mouse model of HD [145,146]. Transgenic overexpression of human DNAJB2a led to a reduction in polyQ aggregation and inclusion size in the cortex and striatum of R6/2 mice at 15 weeks of age and led an increase in htt solubility; however, the improvement in the neurological performance was relatively modest and there was no increase in lifespan [145]. Immunopurification of htt from mouse brain and combinations of purified polyQ protein with cell or mouse brain extracts suggested that the maximal DNAJB2 effect required functional J and UIM domains, and that the effect was mainly being mediated on preformed aggregates, preventing further seeding of aggregation [145]. A recent study on transgenic R6/2 mice overexpressing human DNAJB6 also showed a reduction in inclusion formation in the brain accompanied by improved neurological performance and increased lifespan [146]. In vitro studies suggest this was through an effect on primary nucleation of polyQ aggregation [146]. The differences in the magnitude of the neurological effect between the DNAJB2a and DNAJB6 R6/2 mice could be attributed either to differences in the mechanism of action of the two chaperones, differences in the level of the transgene expression (as different promoters were used), or differences in chaperone regulation. For example, recently DNAJB2a has been shown to be a target of the ubiquitously expressed kinase CK2. CK2 phosphorylated DNAJB2 in the second UIM and reduced its ability to bind ubiquitylated clients [147]. Therefore, it is possible that the maximal activity of DNAJB2a was repressed by CK2 and that inhibition of CK2 could amplify the effect of DNAJB2.
(b). α-synuclein and Parkin in Parkinson's disease
Parkinson's disease (PD) is the second most common neurodegenerative disorder. Although most cases of Parkinson's disease are sporadic, α-synuclein is the main component of Lewy bodies, which are ubiquitin-positive cytoplasmic inclusions formed in patients with PD, Lewy body dementia and other disorders [148]. Furthermore, mutations in SNCA, which encodes α-synuclein, have been associated with PD and mutations in PARK2, which encodes Parkin, lead to the autosomal recessive juvenile form of the disease ARJPD [149].
DNAJB1 has been shown to slow down the assembly of α-synuclein fibrils and increase the binding of HSC70 to fibrillar α-synuclein in vitro [150]. In addition, DNAJA1 was reported to bind α-synuclein fibrils and increase binding of HSC70 to preformed fibrils in vitro; however, DNAJA1 alone had no effect on the assembly of α-synuclein fibrils [150]. Both DNAJA1 and DNAJB1 have been shown to co-localize with α-synuclein inclusions in cells [151]. Post-mortem PD brain tissues showed immunoreactivity for both DNAJB1 and DNAJB6 in Lewy bodies, while DNAJB1 was also present in Lewy neurites and DNAJB6 was upregulated in astrocytes, indicating a potential role in the disease [152,153]. Moreover, co-expression of α-synuclein and either DNAJA1 or DNAJB1 dramatically decreased α-synuclein aggregates in cells [151]. Finally, DNAJB1 combined with HSP70 and HSP110 can recover amorphous α-synuclein aggregates, while they also enhance the effect of non-mammalian Hsp104 to remodel α-synuclein amyloids in vitro [154]. In vivo overexpression of the human homologue DNAJC10 in C. elegans decreased α-synuclein aggregates and toxicity [155].
Mutations in PARK2 cause ARJPD. PARK2 encodes Parkin, a ubiquitin E3 protein ligase containing a N-terminal ubiquitin-like domain and two C-terminal RING finger domains that play an important role in mitochondria dynamics and function [156]. DNAJB2a expression was effective in reducing misfolding and aggregation of RING1 domain mutant Parkin in cells. Furthermore, in the presence of DNAJB2a, mutant Parkin was relocalised to mitochondria and its ability to promote mitophagy of damaged mitochondria was significantly restored [157]. In contrast to polyQ, most cytosolic DNAJ proteins tested could reduce Parkin RING1 domain mutant (C289G) aggregation, and for DNAJB6 and DNAJB8 this was less reliant on their S/T region and more dependent on HSP70 [158]. This illustrates that chaperone manipulation can be versatile and unique to individual protein clients.
(c). Tau and amyloid-β
Extracellular amyloid plaques composed of amyloid-β (Aβ) peptides and intraneuronal tau neurofibrillary tangles form the characteristic pathophysiological profile of Alzheimer's disease (AD) [159]. Although accumulation of Aβ fibrils occurs extracellularly on senile plaques, intraneuronal generation of Aβ has been correlated to synapse damage and enhanced intracellular accumulation in AD-transgenic mice [160].
Interestingly, DNAJB1 has been shown to enhance the effect of HSP70 in vitro in reducing Aβ aggregation through targeting smaller species such as oligomers [161]. Moreover, DNAJB6, and specifically the DNAJB6b isoform, which is localized in both the nucleus and the cytosol, has been shown to be a potent suppressor of Aβ42 aggregation in vitro preventing the formation of amyloid fibrils by interacting with the early formed aggregates during nucleation [162]. In a cellular model of AD overexpressing GFP-tagged Aβ42, DNAJB6 was shown to reduce intracellular Aβ aggregation and required interaction with HSP70. In C. elegans models of Aβ, overexpression of DNAJ27 (orthologue of mammalian DNAJC10) had a protective role against Aβ-induced toxicity; however, overexpression of human DNAJC10 in Aβ worms had no effect [155].
DNAJA1 has been shown to act as a regulator of tau fate depending on HSP70 levels. More specifically, in the absence of HSP70, DNAJA1 enhanced ubiquitin-mediated proteolysis of mutant tau, while in the presence of HSP70, DNAJA1 stabilized tau and halted degradation [163]. Considering that it is still not clear whether aggregation of misfolded proteins is a protective or pathogenic mechanism for neurons, this dual potential of DNAJA1 could be of value in targeting AD pathogenesis. DNAJB1 had a dose-dependent effect on tau aggregation in vitro [164]. Finally, Brehme et al. have shown that knockdown of DNAJA1 and DNAJA4 or the C. elegans homologues can increase the aggregation and toxicity of Aβ42 [165].
(d). SOD1 and TDP-43
The misfolding and aggregation of TAR DNA-binding protein-43 kDa (TDP-43) and superoxide dismutase 1 (SOD1) are associated with amyotrophic lateral sclerosis (ALS), which presents with degeneration of the upper and lower motor neurons. In healthy individuals, TDP-43 appears predominantly in the nucleus, while in disease TDP-43 forms ubiquitin-positive nuclear and cytoplasmic inclusions with abnormal phosphorylation. In familial ALS, SOD1 mutations lead to the formation of ubiquitin-positive SOD1 inclusions in ALS patient spinal cord and in mouse models [166].
Both DNAJB2 isoforms have been shown to significantly reduce mutant SOD aggregation in an overexpression cell model [22,167]. In vivo investigation of DNAJB2a overexpression in double transgenic SOD1G93A mice has shown that DNAJB2a can improve muscle function in late stages of the disease by improving the survival of motor neurons and muscle weight [167].
DNAJB1 co-immunopurified with mutant SOD1, but not with wild type or endogenous SOD1 from cell extracts [168]. In the presence of Hsp70, DNAJB1 can reduce the formation of cytoplasmic aggregates of SOD1 and improve neurite outgrowth in a neuronal cell model (Neuro2a) [169]. Finally, Chen et al. [170] showed that HSF-1 overexpression could reduce TDP-43 aggregation in HEK293 cells. A screen of several DNAJ chaperones revealed that overexpression of DNAJB2a was the most efficient at suppressing TDP-43 aggregation at similar levels to HSF-1 activation. It was suggested that DNAJB2a binds TDP-43 aggregates and delivers them to HSP70 for refolding via its J-domain and not for degradation [170].
5. Conclusion
The essential role of molecular chaperones in maintaining neuronal proteostasis is highlighted by the disease-causing mutations in members of the DNAJ family. Moreover, several DNAJ proteins have been shown to be beneficial for restoring neuronal proteostasis and reducing neurotoxicity associated with a wide range of neurodegeneration proteins both in vitro and in vivo. The great diversity among DNAJ proteins might enable individual DNAJ proteins to be tailored to distinct aggregation-prone proteins. Conversely, some members of the DNAJ family, such as DNAJB2 and DNAJB6, appear to have the ability to affect a wide range of neurodegeneration-related protein clients for potential therapeutic benefit. Enhanced understanding of the DNAJ family function and regulation in neurons is likely to lead to better application of these potentially critical architects of neuronal proteostasis.
Data accessibility
This article has no additional data.
Competing interests
We declare we have no competing interests.
Funding
Research on proteostasis in the Cheetham lab is supported by the MRC, Wellcome Trust and MNDA. C.Z. is an MNDA funded PhD student, L.M.G. is a Leonard Wolfson Experimental Neurology PhD student and W.L. was a CSC PhD student.
References
- 1.Soto C. 2003. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. ( 10.1038/nrn1007) [DOI] [PubMed] [Google Scholar]
- 2.Ross CA, Poirier MA. 2005. Opinion: what is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 6, 891–898. ( 10.1038/nrm1742) [DOI] [PubMed] [Google Scholar]
- 3.Winner B. et al. 2011. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl Acad. Sci. USA 108, 4194–4199. ( 10.1073/pnas.1100976108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. 2002. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. ( 10.1038/416535a) [DOI] [PubMed] [Google Scholar]
- 5.Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, Cashman NR, Wilson MR, Ecroyd H. 2016. Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem. 137, 489–505. ( 10.1111/jnc.13575) [DOI] [PubMed] [Google Scholar]
- 6.Balch WE, Morimoto RI, Dillin A, Kelly JW. 2008. Adapting proteostasis for disease intervention. Science 319, 916–919. ( 10.1126/science.1141448) [DOI] [PubMed] [Google Scholar]
- 7.Muchowski PJ, Wacker JL. 2005. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 6, 11–22. ( 10.1038/nrn1587) [DOI] [PubMed] [Google Scholar]
- 8.Hetz C, Chevet E, Oakes SA. 2015. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. ( 10.1038/ncb3184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravikumar B, et al. 2010. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435. ( 10.1152/physrev.00030.2009) [DOI] [PubMed] [Google Scholar]
- 10.Diaz-Villanueva JF, Diaz-Molina R, Garcia-Gonzalez V. 2015. Protein folding and mechanisms of proteostasis. Int. J. Mol. Sci. 16, 17 193–17 230. ( 10.3390/ijms160817193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka K, Matsuda N. 2014. Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 1843, 197–204. ( 10.1016/j.bbamcr.2013.03.012) [DOI] [PubMed] [Google Scholar]
- 12.Hartl FU, Bracher A, Hayer-Hartl M. 2011. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. ( 10.1038/nature10317) [DOI] [PubMed] [Google Scholar]
- 13.Tsai J, Douglas MG. 1996. A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J. Biol. Chem. 271, 9347–9354. ( 10.1074/jbc.271.16.9347) [DOI] [PubMed] [Google Scholar]
- 14.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. 2009. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111. ( 10.1007/s12192-008-0068-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craig EA, Marszalek J. 2017. How do J-Proteins get Hsp70 to do so many different things? Trends Biochem. Sci. 42, 355–368. ( 10.1016/j.tibs.2017.02.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheetham ME, Caplan AJ. 1998. Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones 3, 28–36. ( 10.1379/1466-1268(1998)003%3C0028:SFAEOD%3E2.3.CO;2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592. ( 10.1038/nrm2941) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macario AJ, Conway de Macario E. 2007. Chaperonopathies and chaperonotherapy. FEBS Lett. 581, 3681–3688. ( 10.1016/j.febslet.2007.04.030) [DOI] [PubMed] [Google Scholar]
- 19.Smith HL, Li W, Cheetham ME. 2015. Molecular chaperones and neuronal proteostasis. Semin. Cell Dev. Biol. 40, 142–152. ( 10.1016/j.semcdb.2015.03.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honeyman JN, et al. 2014. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343, 1010–1014. ( 10.1126/science.1249484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gess B, et al. 2014. HSJ1-related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology 83, 1726–1732. ( 10.1212/WNL.0000000000000966) [DOI] [PubMed] [Google Scholar]
- 22.Blumen SC, et al. 2012. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann. Neurol. 71, 509–519. ( 10.1002/ana.22684) [DOI] [PubMed] [Google Scholar]
- 23.Lupo V, et al. 2016. Assessment of targeted next-generation sequencing as a tool for the diagnosis of charcot-marie-tooth disease and hereditary motor neuropathy. J. Mol. Diagn. 18, 225–234. ( 10.1016/j.jmoldx.2015.10.005) [DOI] [PubMed] [Google Scholar]
- 24.Sanchez E, Darvish H, Mesias R, Taghavi S, Firouzabadi SG, Walker RH, Tafakhori A, Paisan-Ruiz C. 2016. Identification of a large DNAJB2 deletion in a family with spinal muscular atrophy and parkinsonism. Hum. Mutat. 37, 1180–1189. ( 10.1002/humu.23055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzaga-Jauregui C, et al. 2015. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep. 12, 1169–1183. ( 10.1016/j.celrep.2015.07.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato T, et al. 2013. DNAJB6 myopathy in an Asian cohort and cytoplasmic/nuclear inclusions. Neuromuscul. Disord. 23, 269–276. ( 10.1016/j.nmd.2012.12.010) [DOI] [PubMed] [Google Scholar]
- 27.Harms MB, Sommerville RB, Allred P, Bell S, Ma D, Cooper P, Lopate G, Pestronk A, Weihl CC, Baloh RH. 2012. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann. Neurol. 71, 407–416. ( 10.1002/ana.22683) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarparanta J, et al. 2012. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450–455, S451-452 ( 10.1038/ng.1103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suarez-Cedeno G, Winder T, Milone M. 2014. DNAJB6 myopathy: a vacuolar myopathy with childhood onset. Muscle Nerve 49, 607–610. ( 10.1002/mus.24106) [DOI] [PubMed] [Google Scholar]
- 30.Palmio J, et al. 2015. Novel mutations in DNAJB6 gene cause a very severe early-onset limb-girdle muscular dystrophy 1D disease. Neuromuscul. Disord. 25, 835–842. ( 10.1016/j.nmd.2015.07.014) [DOI] [PubMed] [Google Scholar]
- 31.Nam TS, et al. 2015. A novel mutation in DNAJB6, p.(Phe91Leu), in childhood-onset LGMD1D with a severe phenotype. Neuromuscul. Disord. 25, 843–851. ( 10.1016/j.nmd.2015.08.002) [DOI] [PubMed] [Google Scholar]
- 32.Ruggieri A, et al. 2015. Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal-onset DNAJB6 myopathy. Acta Neuropathol. Commun. 3, 44 ( 10.1186/s40478-015-0224-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsai PC, Tsai YS, Soong BW, Huang YH, Wu HT, Chen YH, Lin KP, Liao YC, Lee YC. 2017. A novel DNAJB6 mutation causes dominantly-inherited distal-onset myopathy and compromises DNAJB6 function. Clin. Genet. ( 10.1111/cge.13001) [DOI] [PubMed] [Google Scholar]
- 34.Monies D, et al. 2016. A first-line diagnostic assay for limb-girdle muscular dystrophy and other myopathies. Hum. Genomics 10, 32 ( 10.1186/s40246-016-0089-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El Khouri E, et al. 2016. Mutations in DNAJB13, encoding an HSP40 family member, cause primary ciliary dyskinesia and male infertility. Am. J. Hum. Genet. 99, 489–500. ( 10.1016/j.ajhg.2016.06.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Synofzik M, et al. 2014. Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Genet. 95, 689–697. ( 10.1016/j.ajhg.2014.10.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benitez BA, et al. 2011. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE 6, e26741 ( 10.1371/journal.pone.0026741) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noskova L, et al. 2011. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 89, 241–252. ( 10.1016/j.ajhg.2011.07.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edvardson S, et al. 2012. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 7, e36458 ( 10.1371/journal.pone.0036458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koroglu C, Baysal L, Cetinkaya M, Karasoy H, Tolun A. 2013. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat. Disord. 19, 320–324. ( 10.1016/j.parkreldis.2012.11.006) [DOI] [PubMed] [Google Scholar]
- 41.Vauthier V, Jaillard S, Journel H, Dubourg C, Jockers R, Dam J. 2012. Homozygous deletion of an 80 kb region comprising part of DNAJC6 and LEPR genes on chromosome 1P31.3 is associated with early onset obesity, mental retardation and epilepsy. Mol. Genet. Metab. 106, 345–350. ( 10.1016/j.ymgme.2012.04.026) [DOI] [PubMed] [Google Scholar]
- 42.Olgiati S, et al. 2016. DNAJC6 mutations associated with early-onset Parkinson's disease. Ann. Neurol. 79, 244–256. ( 10.1002/ana.24553) [DOI] [PubMed] [Google Scholar]
- 43.Elsayed LE, et al. 2016. A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal–recessive juvenile-onset Parkinson's disease. Ann. Neurol. 79, 335–337. ( 10.1002/ana.24591) [DOI] [PubMed] [Google Scholar]
- 44.Ioakeimidis F, et al. 2014. A splicing mutation in the novel mitochondrial protein DNAJC11 causes motor neuron pathology associated with cristae disorganization, and lymphoid abnormalities in mice. PLoS ONE 9, e104237 ( 10.1371/journal.pone.0104237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anikster Y, et al. 2017. Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am. J. Hum. Genet. 100, 257–266. ( 10.1016/j.ajhg.2017.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vilarino-Guell C, et al. 2014. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 23, 1794–1801. ( 10.1093/hmg/ddt570) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel N, et al. 2016. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 18, 554–562. ( 10.1038/gim.2015.127) [DOI] [PubMed] [Google Scholar]
- 48.Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP. 2006. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 43, 385–393. ( 10.1136/jmg.2005.036657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ojala T, Polinati P, Manninen T, Hiippala A, Rajantie J, Karikoski R, Suomalainen A, Tyni T. 2012. New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr. Res. 72, 432–437. ( 10.1038/pr.2012.92) [DOI] [PubMed] [Google Scholar]
- 50.Al Teneiji A, Siriwardena K, George K, Mital S, Mercimek-Mahmutoglu S. 2016. Progressive cerebellar atrophy and a novel homozygous pathogenic DNAJC19 variant as a cause of dilated cardiomyopathy ataxia syndrome. Pediatr. Neurol. 62, 58–61. ( 10.1016/j.pediatrneurol.2016.03.020) [DOI] [PubMed] [Google Scholar]
- 51.Ucar SK, Mayr JA, Feichtinger RG, Canda E, Coker M, Wortmann SB. 2016. Previously unreported biallelic mutation in DNAJC19: are sensorineural hearing loss and basal ganglia lesions additional features of dilated cardiomyopathy and ataxia (DCMA) syndrome? JIMD Rep. 35, 39–45. ( 10.1007/8904_2016_23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sparkes R, Patton D, Bernier F. 2007. Cardiac features of a novel autosomal recessive dilated cardiomyopathic syndrome due to defective importation of mitochondrial protein. Cardiol. Young 17, 215–217. ( 10.1017/S1047951107000042) [DOI] [PubMed] [Google Scholar]
- 53.Tummala H, et al. 2016. DNAJC21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60S ribosome subunit maturation. Am. J. Hum. Genet. 99, 115–124. ( 10.1016/j.ajhg.2016.05.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Engert JC, et al. 2000. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 24, 120–125. ( 10.1038/72769) [DOI] [PubMed] [Google Scholar]
- 55.Vermeer S, et al. 2008. ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics 9, 207–214. ( 10.1007/s10048-008-0131-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baets J, et al. 2010. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 75, 1181–1188. ( 10.1212/WNL.0b013e3181f4d86c) [DOI] [PubMed] [Google Scholar]
- 57.Prodi E, et al. 2013. Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur. J. Neurol. 20, 138–146. ( 10.1111/j.1468-1331.2012.03815.x) [DOI] [PubMed] [Google Scholar]
- 58.Pilliod J, et al. 2015. New practical definitions for the diagnosis of autosomal recessive spastic ataxia of Charlevoix-Saguenay. Ann. Neurol. 78, 871–886. ( 10.1002/ana.24509) [DOI] [PubMed] [Google Scholar]
- 59.Li X, Menade M, Kozlov G, Hu Z, Dai Z, McPherson PS, Brais B, Gehring K. 2015. High-throughput screening for ligands of the HEPN domain of Sacsin. PLoS One 10, e0137298 ( 10.1371/journal.pone.0137298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koutras C, Braun JE. 2014. J protein mutations and resulting proteostasis collapse. Front. Cell. Neurosci. 8, 191 ( 10.3389/fncel.2014.00191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chapple JP, Cheetham ME. 2003. The chaperone environment at the cytoplasmic face of the endoplasmic reticulum can modulate rhodopsin processing and inclusion formation. J. Biol. Chem. 278, 19 087–19 094. ( 10.1074/jbc.M212349200) [DOI] [PubMed] [Google Scholar]
- 62.Westhoff B, Chapple JP, van der Spuy J, Hohfeld J, Cheetham ME. 2005. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 15, 1058–1064. ( 10.1016/j.cub.2005.04.058) [DOI] [PubMed] [Google Scholar]
- 63.Tazir M, Hamadouche T, Nouioua S, Mathis S, Vallat JM. 2014. Hereditary motor and sensory neuropathies or charcot-marie-tooth diseases: an update. J. Neurol. Sci. 347, 14–22. ( 10.1016/j.jns.2014.10.013) [DOI] [PubMed] [Google Scholar]
- 64.Schabhuttl M, et al. 2014. Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J. Neurol. 261, 970–982. ( 10.1007/s00415-014-7289-8) [DOI] [PubMed] [Google Scholar]
- 65.Rossor AM, Kalmar B, Greensmith L, Reilly MM. 2012. The distal hereditary motor neuropathies. J. Neurol. Neurosurg. Psychiatry 83, 6–14. ( 10.1136/jnnp-2011-300952) [DOI] [PubMed] [Google Scholar]
- 66.Bansagi B, et al. 2017. Genetic heterogeneity of motor neuropathies. Neurology 88, 1226–1234. ( 10.1212/WNL.0000000000003772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frasquet M, Chumillas MJ, Vilchez JJ, Marquez-Infante C, Palau F, Vazquez-Costa JF, Lupo V, Espinos C, Sevilla T. 2016. Phenotype and natural history of inherited neuropathies caused by HSJ1 c.352+1G>A mutation. J. Neurol. Neurosurg. Psychiatry 87, 1265–1268. ( 10.1136/jnnp-2015-312890) [DOI] [PubMed] [Google Scholar]
- 68.Chen MS, Roti JR, Laszlo A. 1999. Hsc40, a new member of the hsp40 family, exhibits similar expression profile to that of hsc70 in mammalian cells. Gene 238, 333–341. ( 10.1016/S0378-1119(99)00333-9) [DOI] [PubMed] [Google Scholar]
- 69.Hageman J, van Waarde MA, Zylicz A, Walerych D, Kampinga HH. 2011. The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem. J. 435, 127–142. ( 10.1042/BJ20101247) [DOI] [PubMed] [Google Scholar]
- 70.Chuang JZ, Zhou H, Zhu M, Li SH, Li XJ, Sung CH. 2002. Characterization of a brain-enriched chaperone, MRJ, that inhibits Huntingtin aggregation and toxicity independently. J. Biol. Chem. 277, 19 831–19 838. ( 10.1074/jbc.M109613200) [DOI] [PubMed] [Google Scholar]
- 71.Mitra A, et al. 2008. Large isoform of MRJ (DNAJB6) reduces malignant activity of breast cancer. Breast Cancer Res. 10, R22 ( 10.1186/bcr1874) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yabe I, et al. 2014. Pathology of frontotemporal dementia with limb girdle muscular dystrophy caused by a DNAJB6 mutation. Clin. Neurol. Neurosurg. 127, 10–12. ( 10.1016/j.clineuro.2014.09.013) [DOI] [PubMed] [Google Scholar]
- 73.Ruggieri A, Saredi S, Zanotti S, Pasanisi MB, Maggi L, Mora M. 2016. DNAJB6 myopathies: focused review on an emerging and expanding group of myopathies. Front. Mol. Biosci. 3, 63 ( 10.3389/fmolb.2016.00063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sandell S, Huovinen S, Palmio J, Raheem O, Lindfors M, Zhao F, Haapasalo H, Udd B. 2016. Diagnostically important muscle pathology in DNAJB6 mutated LGMD1D. Acta Neuropathol. Commun. 4, 9 ( 10.1186/s40478-016-0276-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bengoechea R, Pittman SK, Tuck EP, True HL, Weihl CC. 2015. Myofibrillar disruption and RNA-binding protein aggregation in a mouse model of limb-girdle muscular dystrophy 1D. Hum. Mol. Genet. 24, 6588–6602. ( 10.1093/hmg/ddv363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li S, et al. 2016. Genetic interaction of hnRNPA2B1 and DNAJB6 in a Drosophila model of multisystem proteinopathy. Hum. Mol. Genet. 25, 936–950. ( 10.1093/hmg/ddv627) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Melville MW, Tan SL, Wambach M, Song J, Morimoto RI, Katze MG. 1999. The cellular inhibitor of the PKR protein kinase, P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. J. Biol. Chem. 274, 3797–3803. ( 10.1074/jbc.274.6.3797) [DOI] [PubMed] [Google Scholar]
- 78.Tao J, Wu Y, Ron D, Sha B. 2008. Preliminary X-ray crystallographic studies of mouse UPR responsive protein P58(IPK) TPR fragment. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 108–110. ( 10.1107/S1744309108000833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, Katze MG. 2002. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl Acad. Sci. USA 99, 15 920–15 925. ( 10.1073/pnas.252341799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, Kaufman RJ, Hegde RS. 2007. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 18, 3681–3691. ( 10.1091/mbc.E07-03-0272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oyadomari S, et al. 2006. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 126, 727–739. ( 10.1016/j.cell.2006.06.051) [DOI] [PubMed] [Google Scholar]
- 82.Petrova K, Oyadomari S, Hendershot LM, Ron D. 2008. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 27, 2862–2872. ( 10.1038/emboj.2008.199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ladiges WC, et al. 2005. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54, 1074–1081. ( 10.2337/diabetes.54.4.1074) [DOI] [PubMed] [Google Scholar]
- 84.Tawo R, Pokrzywa W, Kevei E, Akyuz ME, Balaji V, Adrian S, Hohfeld J, Hoppe T. 2017. The ubiquitin ligase CHIP integrates proteostasis and aging by regulation of insulin receptor turnover. Cell 169, 470–482.e413. ( 10.1016/j.cell.2017.04.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fernandez-Chacon R, et al. 2004. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 42, 237–251. ( 10.1016/S0896-6273(04)00190-4) [DOI] [PubMed] [Google Scholar]
- 86.Greaves J, Chamberlain LH. 2006. Dual role of the cysteine-string domain in membrane binding and palmitoylation-dependent sorting of the molecular chaperone cysteine-string protein. Mol. Biol. Cell 17, 4748–4759. ( 10.1091/mbc.E06-03-0183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang YQ, Henderson MX, Colangelo CM, Ginsberg SD, Bruce C, Wu T, Chandra SS. 2012. Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150. ( 10.1016/j.neuron.2012.01.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chamberlain LH, Graham ME, Kane S, Jackson JL, Maier VH, Burgoyne RD, Gould GW. 2001. The synaptic vesicle protein, cysteine-string protein, is associated with the plasma membrane in 3T3-L1 adipocytes and interacts with syntaxin 4. J. Cell Sci. 114, 445–455. [DOI] [PubMed] [Google Scholar]
- 89.Evans GJ, Morgan A. 2002. Phosphorylation-dependent interaction of the synaptic vesicle proteins cysteine string protein and synaptotagmin I. Biochem. J. 364, 343–347. ( 10.1042/bj20020123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tobaben S, Thakur P, Fernandez-Chacon R, Sudhof TC, Rettig J, Stahl B. 2001. A trimeric protein complex functions as a synaptic chaperone machine. Neuron 31, 987–999. ( 10.1016/S0896-6273(01)00427-5) [DOI] [PubMed] [Google Scholar]
- 91.Rozas JL, Gomez-Sanchez L, Mircheski J, Linares-Clemente P, Nieto-Gonzalez JL, Vazquez ME, Lujan R, Fernandez-Chacon R. 2012. Motorneurons require cysteine string protein-alpha to maintain the readily releasable vesicular pool and synaptic vesicle recycling. Neuron 74, 151–165. ( 10.1016/j.neuron.2012.02.019) [DOI] [PubMed] [Google Scholar]
- 92.Burgoyne RD, Morgan A. 2015. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 40, 153–159. ( 10.1016/j.semcdb.2015.03.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benitez BA, Cairns NJ, Schmidt RE, Morris JC, Norton JB, Cruchaga C, Sands MS. 2015. Clinically early-stage CSPalpha mutation carrier exhibits remarkable terminal stage neuronal pathology with minimal evidence of synaptic loss. Acta Neuropathol. Commun. 3, 73 ( 10.1186/s40478-015-0256-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cadieux-Dion M, et al. 2013. Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin. Genet. 83, 571–575. ( 10.1111/cge.12020) [DOI] [PubMed] [Google Scholar]
- 95.Velinov M. et al 2012. Mutations in the gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS ONE 7, e29729 ( 10.1371/journal.pone.0029729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Diez-Ardanuy C, Greaves J, Munro KR, Tomkinson NC, Chamberlain LH. 2017. A cluster of palmitoylated cysteines are essential for aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. Sci. Rep. 7, 10 ( 10.1038/s41598-017-00036-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Greaves J, Lemonidis K, Gorleku OA, Cruchaga C, Grefen C, Chamberlain LH. 2012. Palmitoylation-induced aggregation of cysteine-string protein mutants that cause neuronal ceroid lipofuscinosis. J. Biol. Chem. 287, 37 330–37 339. ( 10.1074/jbc.M112.389098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Henderson MX, et al. 2016. Neuronal ceroid lipofuscinosis with DNAJC5/CSPalpha mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol. 131, 621–637. ( 10.1007/s00401-015-1512-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kashyap SS, et al. 2014. Caenorhabditis elegans dnj-14, the orthologue of the DNAJC5 gene mutated in adult onset neuronal ceroid lipofuscinosis, provides a new platform for neuroprotective drug screening and identifies a SIR-2.1-independent action of resveratrol. Hum. Mol. Genet. 23, 5916–5927. ( 10.1093/hmg/ddu316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ahle S, Ungewickell E. 1990. Auxilin, a newly identified clathrin-associated protein in coated vesicles from bovine brain. J. Cell Biol. 111, 19–29. ( 10.1083/jcb.111.1.19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fotin A, Cheng Y, Grigorieff N, Walz T, Harrison SC, Kirchhausen T. 2004. Structure of an auxilin-bound clathrin coat and its implications for the mechanism of uncoating. Nature 432, 649–653. ( 10.1038/nature03078) [DOI] [PubMed] [Google Scholar]
- 102.Bocking T, Aguet F, Harrison SC, Kirchhausen T. 2011. Single-molecule analysis of a molecular disassemblase reveals the mechanism of Hsc70-driven clathrin uncoating. Nat. Struct. Mol. Biol. 18, 295–301. ( 10.1038/nsmb.1985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Holstein SE, Ungewickell H, Ungewickell E. 1996. Mechanism of clathrin basket dissociation: separate functions of protein domains of the DnaJ homologue auxilin. J. Cell Biol. 135, 925–937. ( 10.1083/jcb.135.4.925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Massol RH, Boll W, Griffin AM, Kirchhausen T. 2006. A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc. Natl Acad. Sci. USA 103, 10 265–10 270. ( 10.1073/pnas.0603369103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Katoh M, Katoh M. 2003. Identification and characterization of FLJ10737 and CAMTA1 genes on the commonly deleted region of neuroblastoma at human chromosome 1p36.31-p36.23. Int. J. Oncol. 23, 1219–1224. [PubMed] [Google Scholar]
- 106.Henrich KO, Claas A, Praml C, Benner A, Mollenhauer J, Poustka A, Schwab M, Westermann F. 2007. Allelic variants of CAMTA1 and FLJ10737 within a commonly deleted region at 1p36 in neuroblastoma. Eur. J. Cancer 43, 607–616. ( 10.1016/j.ejca.2006.09.023) [DOI] [PubMed] [Google Scholar]
- 107.Xie J, Marusich MF, Souda P, Whitelegge J, Capaldi RA. 2007. The mitochondrial inner membrane protein mitofilin exists as a complex with SAM50, metaxins 1 and 2, coiled-coil-helix coiled-coil-helix domain-containing protein 3 and 6 and DnaJC11. FEBS Lett. 581, 3545–3549. ( 10.1016/j.febslet.2007.06.052) [DOI] [PubMed] [Google Scholar]
- 108.Andrews B, Carroll J, Ding S, Fearnley IM, Walker JE. 2013. Assembly factors for the membrane arm of human complex I. Proc. Natl Acad. Sci. USA 110, 18 934–18 939. ( 10.1073/pnas.1319247110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Girard M, Poupon V, Blondeau F, McPherson PS. 2005. The DnaJ-domain protein RME-8 functions in endosomal trafficking. J. Biol. Chem. 280, 40 135–40 143. ( 10.1074/jbc.M505036200) [DOI] [PubMed] [Google Scholar]
- 110.Popoff V, et al. 2009. Analysis of articulation between clathrin and retromer in retrograde sorting on early endosomes. Traffic 10, 1868–1880. ( 10.1111/j.1600-0854.2009.00993.x) [DOI] [PubMed] [Google Scholar]
- 111.Deng HX, et al. 2016. Identification of TMEM230 mutations in familial Parkinson's disease. Nat. Genet. 48, 733–739. ( 10.1038/ng.3589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lorenzo-Betancor O, et al. 2015. DNAJC13 p.Asn855Ser mutation screening in Parkinson's disease and pathologically confirmed Lewy body disease patients. Eur. J. Neurol. 22, 1323–1325. ( 10.1111/ene.12770) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gustavsson EK, et al. 2015. DNAJC13 genetic variants in parkinsonism. Mov. Disord. 30, 273–278. ( 10.1002/mds.26064) [DOI] [PubMed] [Google Scholar]
- 114.Ross JP, et al. 2016. Analysis of DNAJC13 mutations in French-Canadian/French cohort of Parkinson's disease. Neurobiol. Aging 45, 212.e13–212.e17. ( 10.1016/j.neurobiolaging.2016.04.023) [DOI] [PubMed] [Google Scholar]
- 115.Mokranjac D, Sichting M, Neupert W, Hell K. 2003. Tim14, a novel key component of the import motor of the TIM23 protein translocase of mitochondria. EMBO J. 22, 4945–4956. ( 10.1093/emboj/cdg485) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parfitt DA, et al. 2009. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 18, 1556–1565. ( 10.1093/hmg/ddp067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Anderson JF, Siller E, Barral JM. 2010. The sacsin repeating region (SRR): a novel Hsp90-related supra-domain associated with neurodegeneration. J. Mol. Biol. 400, 665–674. ( 10.1016/j.jmb.2010.05.023) [DOI] [PubMed] [Google Scholar]
- 118.Girard M, et al. 2012. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl Acad. Sci. USA 109, 1661–1666. ( 10.1073/pnas.1113166109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. 1978. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. 5, 61–69. [PubMed] [Google Scholar]
- 120.Thiffault I, et al. 2013. Diversity of ARSACS mutations in french-canadians. Can. J. Neurol. Sci. 40, 61–66. ( 10.1017/S0317167100012968) [DOI] [PubMed] [Google Scholar]
- 121.Breckpot J, Takiyama Y, Thienpont B, Van Vooren S, Vermeesch JR, Ortibus E, Devriendt K. 2008. A novel genomic disorder: a deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur. J. Hum. Genet. 16, 1050–1054. ( 10.1038/ejhg.2008.58) [DOI] [PubMed] [Google Scholar]
- 122.Lariviere R, et al. 2015. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 24, 727–739. ( 10.1093/hmg/ddu491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bradshaw TY, Romano LE, Duncan EJ, Nethisinghe S, Abeti R, Michael GJ, Giunti P, Vermeer S, Chapple JP. 2016. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 25, 3232–3244. ( 10.1093/hmg/ddw173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shao J, Diamond MI. 2007. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 16, R115–R123. ( 10.1093/hmg/ddm213) [DOI] [PubMed] [Google Scholar]
- 125.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. 1998. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 19, 148–154. ( 10.1038/502) [DOI] [PubMed] [Google Scholar]
- 126.Gao X-C, Zhou C-J, Zhou Z-R, Zhang Y-H, Zheng X-M, Song A-X, Hu H-Y. 2011. Co-Chaperone HSJ1a dually regulates the proteasomal degradation of Ataxin-3. PLOS ONE 6, e19763 ( 10.1371/journal.pone.0019763) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wyttenbach A, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC. 2000. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc, Natl Acad. Sci. USA 97, 2898–2903. ( 10.1073/pnas.97.6.2898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ormsby AR, Ramdzan YM, Mok YF, Jovanoski KD, Hatters DM. 2013. A platform to view huntingtin exon 1 aggregation flux in the cell reveals divergent influences from chaperones hsp40 and hsp70. J. Biol. Chem. 288, 37 192–37 203. ( 10.1074/jbc.M113.486944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jana NR, Tanaka M, Wang G, Nukina N. 2000. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9, 2009–2018. ( 10.1093/hmg/9.13.2009) [DOI] [PubMed] [Google Scholar]
- 130.Bailey CK, Andriola IF, Kampinga HH, Merry DE. 2002. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 11, 515–523. ( 10.1093/hmg/11.5.515) [DOI] [PubMed] [Google Scholar]
- 131.Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. 2000. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J. Biol. Chem. 275, 8772–8778. ( 10.1074/jbc.275.12.8772) [DOI] [PubMed] [Google Scholar]
- 132.Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. 2010. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369. ( 10.1016/j.molcel.2010.01.001) [DOI] [PubMed] [Google Scholar]
- 133.Mansson C, Kakkar V, Monsellier E, Sourigues Y, Harmark J, Kampinga HH, Melki R, Emanuelsson C. 2014. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones 19, 227–239. ( 10.1007/s12192-013-0448-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nollen EAA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, Plasterk RHA. 2004. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl Acad. Sci. USA 101, 6403–6408. ( 10.1073/pnas.0307697101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gillis J, et al. 2013. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J. Biol. Chem. 288, 17 225–17 237. ( 10.1074/jbc.M112.421685) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Borrell-Pages M, et al. 2006. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Invest. 116, 1410–1424. ( 10.1172/jci27607) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. 2000. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl Acad. Sci. USA 97, 7841–7846. ( 10.1073/pnas.140202897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kazemi-Esfarjani P, Benzer S. 2000. Genetic suppression of polyglutamine toxicity in Drosophila. Science 287, 1837–1840. ( 10.1126/science.287.5459.1837) [DOI] [PubMed] [Google Scholar]
- 139.Kuo Y, Ren S, Lao U, Edgar BA, Wang T. 2013. Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis. 4, e833 ( 10.1038/cddis.2013.351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fayazi Z, Ghosh S, Marion S, Bao X, Shero M, Kazemi-Esfarjani P. 2006. A Drosophila ortholog of the human MRJ modulates polyglutamine toxicity and aggregation. Neurobiol. Dis. 24, 226–244. ( 10.1016/j.nbd.2006.06.015) [DOI] [PubMed] [Google Scholar]
- 141.Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM. 2000. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet. 9, 2811–2820. ( 10.1093/hmg/9.19.2811) [DOI] [PubMed] [Google Scholar]
- 142.Tsou W.-L., Hosking RR, Burr AA, Sutton JR, Ouyang M, Du X, Gomez CM, Todi SV. 2015. DnaJ-1 and karyopherin α3 suppress degeneration in a new Drosophila model of Spinocerebellar Ataxia Type 6. Hum. Mol. Genet. 24, 4385–4396. ( 10.1093/hmg/ddv174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tsou W.-L., Ouyang M, Hosking RR, Sutton JR, Blount JR, Burr AA, Todi SV. 2015. The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 82, 12–21. ( 10.1016/j.nbd.2015.05.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Howarth JL, et al. 2007. Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol. Ther. 15, 1100–1105. ( 10.1038/sj.mt.6300163) [DOI] [PubMed] [Google Scholar]
- 145.Labbadia J, Novoselov SS, Bett JS, Weiss A, Paganetti P, Bates GP, Cheetham ME. 2012. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain 135, 1180–1196. ( 10.1093/brain/aws022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kakkar V. et al 2016. The S/T-Rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol. Cell 62, 272–283. ( 10.1016/j.molcel.2016.03.017) [DOI] [PubMed] [Google Scholar]
- 147.Ottaviani D, et al. 2016. Protein kinase CK2 modulates HSJ1 function through phosphorylation of the UIM2 domain. Hum. Mol. Genet. ( 10.1093/hmg/ddw420) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang B, Abraham N, Gao G, Yang Q. 2016. Dysregulation of autophagy and mitochondrial function in Parkinson's disease. Transl. Neurodegener. 5, 19 ( 10.1186/s40035-016-0065-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. ( 10.1038/33416) [DOI] [PubMed] [Google Scholar]
- 150.Pemberton S, Madiona K, Pieri L, Kabani M, Bousset L, Melki R. 2011. Hsc70 protein interaction with soluble and fibrillar alpha-synuclein. J. Biol. Chem. 286, 34 690–34 699. ( 10.1074/jbc.M111.261321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT. 2002. TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J. Neurochem. 83, 846–854. ( 10.1046/j.1471-4159.2002.01190.x) [DOI] [PubMed] [Google Scholar]
- 152.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. 2002. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295, 865–868. ( 10.1126/science.1067389) [DOI] [PubMed] [Google Scholar]
- 153.Durrenberger PF, Filiou MD, Moran LB, Michael GJ, Novoselov S, Cheetham ME, Clark P, Pearce RK, Graeber MB. 2009. DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. J. Neurosci. Res. 87, 238–245. ( 10.1002/jnr.21819) [DOI] [PubMed] [Google Scholar]
- 154.Shorter J. 2011. The Mammalian disaggregase machinery: Hsp110 Synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 6, e26319 ( 10.1371/journal.pone.0026319) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Munoz-Lobato F, et al. 2014. Protective role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases. Antioxid Redox Signal. 20, 217–235. ( 10.1089/ars.2012.5051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dawson TM, Dawson VL. 2010. The role of parkin in familial and sporadic Parkinson's disease. Mov. Disord. 25(Suppl 1), S32–S39. ( 10.1002/mds.22798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rose JM, Novoselov SS, Robinson PA, Cheetham ME. 2011. Molecular chaperone-mediated rescue of mitophagy by a Parkin RING1 domain mutant. Hum. Mol. Genet. 20, 16–27. ( 10.1093/hmg/ddq428) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kakkar V, Kuiper EF, Pandey A, Braakman I, Kampinga HH. 2016. Versatile members of the DNAJ family show Hsp70 dependent anti-aggregation activity on RING1 mutant parkin C289G. Sci. Rep. 6, 34830 ( 10.1038/srep34830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bloom GS. 2014. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508. ( 10.1001/jamaneurol.2013.5847) [DOI] [PubMed] [Google Scholar]
- 160.Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK. 2010. Effects of synaptic modulation on β-amyloid, synaptophysin and memory performance in Alzheimer's disease transgenic mice. J. Neurosci. 30, 14 299–14 304. ( 10.1523/JNEUROSCI.3383-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Evans CG, Wisen S, Gestwicki JE. 2006. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. J. Biol. Chem. 281, 33 182–33 191. ( 10.1074/jbc.M606192200) [DOI] [PubMed] [Google Scholar]
- 162.Mansson C, et al. 2014. Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Abeta42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31 066–31 076. ( 10.1074/jbc.M114.595124) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Abisambra JF, Jinwal UK, Suntharalingam A, Arulselvam K, Brady S, Cockman M, Jin Y, Zhang B, Dickey CA. 2012. DnaJA1 antagonizes constitutive Hsp70-mediated stabilization of tau. J. Mol. Biol. 421, 653–661. ( 10.1016/j.jmb.2012.02.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sahara N, et al. 2007. Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J. Neurosci. Res. 85, 3098–3108. ( 10.1002/jnr.21417) [DOI] [PubMed] [Google Scholar]
- 165.Brehme M, et al. 2014. A chaperome sub-network safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. ( 10.1016/j.celrep.2014.09.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ling SC, Polymenidou M, Cleveland DW. 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. ( 10.1016/j.neuron.2013.07.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Novoselov SS, Mustill WJ, Gray AL, Dick JR, Kanuga N, Kalmar B, Greensmith L, Cheetham ME. 2013. Molecular chaperone mediated late-stage neuroprotection in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. PLoS ONE 8, e73944 ( 10.1371/journal.pone.0073944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shinder GA, Lacourse M.-C., Minotti S, Durham HD. 2001. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J. Biol. Chem. 276, 12 791–12 796. ( 10.1074/jbc.M010759200) [DOI] [PubMed] [Google Scholar]
- 169.Takeuchi H, Kobayashi Y, Yoshihara T, Niwa J, Doyu M, Ohtsuka K, Sobue G. 2002. Hsp70 and Hsp40 improve neurite outgrowth and suppress intracytoplasmic aggregate formation in cultured neuronal cells expressing mutant SOD1. Brain Res. 949, 11–22. ( 10.1016/S0006-8993(02)02568-4) [DOI] [PubMed] [Google Scholar]
- 170.Chen HJ, Mitchell JC, Novoselov S, Miller J, Nishimura AL, Scotter EL, Vance CA, Cheetham ME, Shaw CE. 2016. The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain 139, 1417–1432. ( 10.1093/brain/aww028) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.