

Abstract
Background
Diagnosis of monogenic as well as atypical forms of diabetes mellitus has important clinical implications for their specific diagnosis, prognosis, and targeted treatment. Single gene mutations that affect beta-cell function represent 1–2% of all cases of diabetes. However, phenotypic heterogeneity and lack of family history of diabetes can limit the diagnosis of monogenic forms of diabetes. Next-generation sequencing technologies provide an excellent opportunity to screen large numbers of individuals with a diagnosis of diabetes for mutations in disease-associated genes.
Methods
We utilized a targeted sequencing approach using the Illumina HiSeq to perform a case-control sequencing study of 22 monogenic diabetes genes in 4016 individuals with type 2 diabetes (including 1346 individuals diagnosed before the age of 40 years) and 2872 controls. We analyzed protein-coding variants identified from the sequence data and compared the frequencies of pathogenic variants (protein-truncating variants and missense variants) between the cases and controls.
Results
A total of 40 individuals with diabetes (1.8% of early onset sub-group and 0.6% of adult onset sub-group) were carriers of known pathogenic missense variants in the GCK, HNF1A, HNF4A, ABCC8, and INS genes. In addition, heterozygous protein truncating mutations were detected in the GCK, HNF1A, and HNF1B genes in seven individuals with diabetes. Rare missense mutations in the GCK gene were significantly over-represented in individuals with diabetes (0.5% carrier frequency) compared to controls (0.035%). One individual with early onset diabetes was homozygous for a rare pathogenic missense variant in the WFS1 gene but did not have the additional phenotypes associated with Wolfram syndrome.
Conclusion
Targeted sequencing of genes linked with monogenic diabetes can identify disease-relevant mutations in individuals diagnosed with type 2 diabetes not suspected of having monogenic forms of the disease. Our data suggests that GCK-MODY frequently masquerades as classical type 2 diabetes. The results confirm that MODY is under-diagnosed, particularly in individuals presenting with early onset diabetes and clinically labeled as type 2 diabetes; thus, sequencing of all monogenic diabetes genes should be routinely considered in such individuals. Genetic information can provide a specific diagnosis, inform disease prognosis and may help to better stratify treatment plans.
Electronic supplementary material
The online version of this article (doi:10.1186/s12916-017-0977-3) contains supplementary material, which is available to authorized users.
Keywords: High-throughput sequencing, Monogenic diabetes, Pathogenic variants, Type 2 diabetes, MODY, DNA pooling, Targeted sequencing
Background
Diabetes mellitus is a heterogeneous disorder characterized by high fasting blood glucose levels or hyperglycemia that results from a combination of both genetic and environmental risk factors. Most individuals with diabetes are classified into type 1 (T1D) and type 2 diabetes (T2D). Compared to T1D, which presents early in life and is primarily an auto-immune disorder, T2D represents approximately 90% of all diabetes and typically manifests later in life. T2D is a complex polygenic disease caused by interactions between multiple genetic and environmental factors. Significant progress has been made in understanding the genetic architecture of T2D over the past 10 years [1]. A number of genome-wide association studies in diverse human populations have identified more than 60 common variants and loci associated with risk for T2D [2]. These studies have also revealed a significant overlap between traits and phenotypes of monogenic diabetes with related “common” T2D as a prototypic complex disease [3–6].
In contrast to T1D and T2D, monogenic diabetes represents a form of non-autoimmune, early onset diabetes that is primarily genetic. Maturity onset diabetes of the young (MODY), first reported in 1974 [7], is an autosomal dominant form of non-insulin dependent diabetes that is typically diagnosed before the age of 25. Using linkage analysis in families with a high prevalence of diabetes, mutations in more than 10 different genes have been shown to cause multiple types of monogenic diabetes, each with different clinical presentation [8, 9]. MODY is estimated to represent 1–2% of diabetes [10]. In addition, mutations in several genes are known to cause neonatal diabetes and rare syndromes such as Wolfram syndrome (WS) [11], which includes diabetes among other phenotypes. Common variants in the monogenic diabetes genes HNF4A [12] and WFS1 [13], and a low-frequency variant in the HNF1A gene [14] have also been associated with risk for T2D, highlighting the genetic overlap between monogenic diabetes and T2D.
Subjects suspected of having monogenic diabetes based on age of onset, family history, and additional phenotypes are referred for genetic screening. However, not all individuals with monogenic diabetes fulfill the classical criteria of MODY [7, 9, 10]. In addition, individuals without any family history of diabetes are likely to be misdiagnosed as having T1D or T2D [15, 16]. Further, many rare forms of diabetes share clinical features with T2D and are sometimes misdiagnosed as T2D due to a lack of genetic information and atypical clinical presentation. An accurate molecular diagnosis of monogenic forms of diabetes is important for determining the right treatment as well as genetic counseling for their families [17, 18]. For MODY, genetic diagnosis has important therapeutic implications [9]. Diabetic individuals with mutations in the glucokinase gene (MODY2) often require no treatment and have low prevalence of complications such as retinopathy and neuropathy despite lifelong hypergylcemia [19, 20]. On the other hand, individuals with a mutation in the ABCC8 or KCNJ11 genes can be successfully treated with sulfonylureas rather than with insulin therapy [9].
Screening of monogenic diabetes genes in a large cohort with a clinical diagnosis of T2D has the potential to identify subjects with misdiagnosed monogenic diabetes, in particular in subjects with early onset of the disease. In recent years, advances in high-throughput sequencing technologies have made it possible to sequence selected regions of the human genome in large numbers of individuals. These targeted sequencing approaches have been utilized to sequence genes associated with diabetes and obesity [21–23]. In this study, we utilized the Illumina high-throughput sequencing technology to sequence 225 diabetes associated genes, including genes implicated in monogenic forms of diabetes and genes near variants identified in genome-wide association studies for T2D. A total of 4016 individuals diagnosed with T2D, including 1346 individuals with diabetes diagnosed before the age of 40 years, and 2872 controls were sequenced in our study. In this paper, we focus on the analysis of variants in 22 genes (Additional file 1: Table S1) that have been associated with monogenic forms of diabetes. These include the 13 MODY genes (GCK, HNF1A, HNF4A, HNF1B, INS, NEUROD1, PDX1, PAX4, ABCC8, KCNJ11, KLF11, CEL, and BLK), 6 genes associated with recessive diseases that include diabetes as a phenotype (WFS1, NEUROG3, EIF2AK3, GLIS3, RFX6, and SLC19A2), and 3 genes in which heterozygous mutations have been shown to cause diabetes mellitus (PAX6, GATA6, and PPARG). Our primary objectives were to (1) identify subjects with potentially undiagnosed monogenic diabetes, (2) compare and contrast the frequency of deleterious mutations in monogenic diabetes genes between individuals with early-onset diabetes or adult-onset diabetes and population controls, and (3) assess the relationship between deleterious mutations in less frequently mutated monogenic diabetes genes and risk for early onset diabetes.
Methods
Cohorts
All samples were obtained through the Centre of Excellence for Metabolic Disorders, Division of Endocrinology and Diabetes, Ulm University Medical Centre. Diabetes was defined as fasting plasma glucose > 125 mg/dL or 2 hour glucose > 200 mg/dL after an oral glucose tolerance test. Furthermore, individuals with a history of diabetes or undergoing treatment with oral anti-diabetic drugs (primarily metformin and sulfonylureas) or insulin were considered as cases. All subjects studied were of Northern European ancestry. In addition, all diabetes subjects and the controls were tested for the presence of serum autoantibodies, including islet cell autoantibodies, glutamic acid decarboxylase, and islet antigen 2 antibodies, as previously described [24]. Positivity for islet-cell autoantibodies, insulin requirement, and evidence of ketosis at the time of diagnosis were criteria for exclusion. Exclusion criteria were also pregnancy and the presence of any other severe disease. Each study subject was interviewed regarding their family history; history taken included basic clinical information, namely age at diagnosis, sex, treatment (including time to insulin treatment), body mass index, current glycated hemoglobin (HbA1c), current age, and the presence or absence of a parent with diabetes [25]. No interview or biochemical test was performed in any of the relatives. All subjects diagnosed before 25 years of age did not have the MODY phenotype based on classical diagnostic criteria, namely no treatment with insulin for at least 2 years after diagnosis and multi-generational inheritance of diabetes [7]. All individuals with early onset diabetes were also screened for apparent WS based on phenotypes such as optic atrophy, diabetes insipidus, and deafness. Controls had normal fasting glucose (confirmed by HbA1c < 6%) and had no evidence of islet autoimmunity.
The first set of sequenced samples included 1880 individuals with T2D and 1840 controls. The mean age at diagnosis of diabetes in cases was 43.4 years, with 734 individuals classified as having early onset diabetes (age at diagnosis < 40 years). The second group of sequenced samples included 2136 individuals with T2D (612 individuals with age at diagnosis < 40 years) and 1032 population controls (age > 65 years) from the southern part of Germany. For a subset of individuals, additional phenotype information about the presence of diabetic complications (nephropathy, neuropathy, and retinopathy) was also available. Clinical characteristics and phenotype data (age at diagnosis, body mass index, and HbA1c or fasting blood glucose) from the case and control populations are reported in Additional file 1: Table S4.
Selection of genes for sequencing
Although high throughput sequencing technologies make it possible to sequence human genomes, it is still costly to sequence the entire human genomes of thousands of individuals. However, targeted sequencing of specific regions (e.g., exons of genes of interest) is feasible in thousands of individuals using the same sequencing throughput. We performed targeted sequencing of the exons and the 5’ and 3’ un-translated regions of genes that (1) are associated with monogenic or rare forms of diabetes, (2) are located near common variants associated with risk for T2D [6], (3) have been linked to diabetes in model organisms, or (4) have relevance for drugs used to treat diabetes. In total, a total of 225 genes were selected for sequencing based on these criteria (Additional file 1: Table S2).
Target enrichment and pooled sequencing
For enrichment of the targeted regions, we utilized the Agilent SureSelect solution hybridization method [26]. For each gene, exon coordinates were obtained from the RefSeq database to identify the coding and untranslated regions. Subsequently, baits were designed (120 bp length, 2× tiling) targeting the DNA sequence of the selected regions. Although targeted sequencing dramatically increases the cost-efficiency of sequencing, there is a significant cost associated with preparing DNA sequencing libraries for each individual sample. Therefore, to reduce the cost of sequencing per sample, DNA from multiple individuals was pooled prior to library preparation and hybridization. We have previously demonstrated that both rare (even singleton mutations present in moderate sized pools) and common mutations can be detected with high sensitivity and specificity from pooled sequence data [27, 28]. A number of studies have utilized pooled sequencing to search for disease risk variants in selected regions of the human genome for a number of diseases, including T1D [29], inflammatory bowel disease [30, 31], Crohn’s disease [32], anorexia nervosa [33], and breast cancer [34]. Similar to previous studies, the number of individuals in a pool was chosen to be small (20–24) since this significantly reduces the cost of library preparation per individual (8–10 times more individuals can be sequenced for the same cost [27, 28]) but still allows for the accurate detection of variants.
Study design
Sequencing of the DNA samples was performed in three stages (Fig. 1). In the first stage, selected regions of 136 genes were sequenced in 1880 individuals with T2D and 1840 controls using a pooled sequencing design (Additional file 1: Figure S2). All pools contained DNA from 20 individuals each and were designed to be homogeneous with respect to the presence or absence of T2D as well as additional phenotypes such as the age of onset (for cases) or current age (for controls) and diabetic complications. Subsequently, in Stage 2, an independent set of 2136 cases and 1032 controls was sequenced using pools of size 24. The pool size was increased to enable the sequencing of additional samples. Finally, to validate deleterious variants identified in Stages 1 and 2 and to identify the carriers of rare coding variants, we performed pooled sequencing of DNA from 2014 individuals with diabetes (1268 early-onset and 746 late-onset) that were also sequenced in Stage 1 and 2. Pools from Stage 1 and 2 with deleterious variants (e.g., missense mutations in GCK) were prioritized for sequencing in Stage 3. The pools in Stage 3 were designed to be orthogonal to pools in Stages 1 and 2 such that a pool from the first two stages of sequencing and a pool from the third stage shared at most 1–2 individuals (Additional file 1: Figure S2).
Fig. 1.
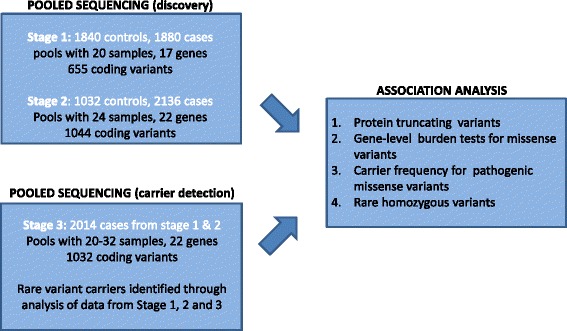
Overview of the sequencing study. A total of 2872 controls and 4016 cases (1346 individuals with age of onset < 40 years) for type 2 diabetes were sequenced using pools of 20 (Stage 1) and 24 (Stage 2) individuals. To validate rare functional variants and to identify the carriers of rare coding variants, 2014 cases selected from Stage 1 and 2 were sequenced again in Stage 3. The resulting variant data was analyzed to perform gene-level burden tests and compare the frequency of protein truncating variants and known pathogenic missense variants in monogenic diabetes genes between the case and control groups
Library preparation and sequencing
For each individual, DNA was quantified in duplicate (or triplicate if necessary) using PicoGreen. Subsequently, samples were pooled in equimolar concentrations to form pools with DNA from the selected number of individuals. The pools were then carried through the standard Illumina library preparation process using Adaptive Focused Acoustics for shearing (Covaris), end-repair, A-tailing, and ligation. Agilent SureSelect in-solution hybridization was performed on the pooled samples using the recommended protocol for a single genomic DNA sample as previously described [28]. Captured DNA was then sequenced using a 100 bp paired-end multiplexed read protocol on an Illumina HiSeq instrument.
Read alignment and variant calling
The paired-end reads for each pool were aligned to the human genome reference sequence (hg19) using the Novoalign alignment program [35] (with soft-clipping (v3.0) to generate a BAM file. The BAM file was sorted and PCR duplicates were removed using the Picard [36] MarkDuplicates command. Subsequently, the bam files for the pools were processed using the CRISP variant calling program [27] to identify variants (details in Additional file 2: Supplementary Methods). Variants were identified for pooled sequence data for each stage separately but jointly across all pools. Variant calls were restricted to the targeted regions and the 100 base pairs flanking the targeted regions.
Variant annotation
All identified variants were annotated using the Annovar annotation program using the RefSeq transcript database [37]. We focused primarily on coding variants that are predicted to impact the protein sequence, namely (1) missense variants (including non-frameshift insertions or deletions (indels)) and (2) protein truncating variants (nonsense, splice-site and frameshift indels). Missense variants were further annotated using the in silico prediction tools PolyPhen2 [38], SIFT [39], MutationTaster [40], and CADD [41]. Alignments for protein truncating variants were inspected visually and variants with weak read support were removed. We utilized variant calls and allele frequency data from the National Heart, Lung, and Blood Institute Exome Sequencing Project [42] and the Exome Aggregation Consortium (ExAC) database [43] to estimate the allele frequencies of the variants. Information about missense mutations that have been reported to be associated with early onset diabetes and MODY was obtained from published papers and the Human Gene Mutation Database [44]. Variants that have been shown to not impact gene function or with a high allele frequency in controls were not considered as pathogenic. Rare variants were further classified using a five-tier classification system as per the American College of Medical Genetics (ACMG) guidelines [45]. Each variant was classified as ‘Benign’ (class 1), ‘Likely benign’ (class 2), ‘Unknown significance’ (class 3), ‘Likely pathogenic’ (class 4), and ‘Pathogenic’ (class 5) using the bioinformatics tool InterVar [46], ClinVar [47] and clinical, functional and genotype-phenotype data from the literature.
Results
Description of variants identified and data quality
In the first stage, targeted sequencing was performed on 1880 individuals with diabetes and 1840 controls using 186 pools. Analysis of the read depth across the coding sequence of the sequenced genes showed that the median coverage per pool varied from 600× to 970× per base. The fraction of the targeted bases with a read depth of 200× or greater (10× per individual in a pool with 20 individuals) varied between 0.79 and 0.87 across the pools and was slightly higher in the control pools (0.84 ± 0.02) compared to cases (0.834 ± 0.02) (see Additional file 1: Figure S3 for a distribution of coverage across pools). A small number of targeted exons had a low read depth across all sequenced pools in Stage 1 as well in Stage 2 (Additional file 1: Table S8); 5/7 of these exons also had low sequence coverage (<10× median coverage) in large-scale exome sequence datasets and 3 of these exons correspond to GC-rich regions (GC% ≥ 70%, Additional file 1: Table S8). Excluding these 7 exons with low read depth, 88.7% of the targeted bases were well covered at a threshold of 200×. Further, using a stringent coverage criteria (≥ 90% pools with ≥ 200× coverage at each base), 79% of the targeted bases in the 17 monogenic diabetes genes (~25 kilobases of DNA sequence) were well covered. For two genes, PDX1 and INS, less than 40% of the bases were well covered. Both of these genes also had low sequence coverage in Stage 2 pools (Additional file 1, Table S1) and were difficult to sequence using target capture-based methods [22].
Analysis of the sequence data for the 186 pools using a pooled variant calling method, CRISP [27], identified 655 coding variants in 17 monogenic diabetes genes that included 253 (38.6%) synonymous single nucleotide variants (SNVs), 379 missense SNVs, 3 stop-gain mutations, and 18 indel variants (Additional file 1: Table S3). Most of the detected variants were very rare, with 54% of the variants having an estimated allele count of 1 (also known as singletons) and 81% of the variants estimated to have an allele frequency of 0.001 or lower (Additional file 1: Figure S1). To assess the sensitivity and specificity of variant detection from pooled sequencing, we sequenced 20 samples from one pool individually using the same target capture and library preparation protocols. Overall, 47 variants were identified from the analysis of individual-level sequence data of the 20 samples, while 45 variants were detected from the pooled data, 44 variants overlapped, and 2/3 variants unique to the individual sequence data had low coverage in the pooled data (2–3× per individual). From this data, we estimated a low rate of false positive variants per pool (< 3%) and a low false negative rate (< 7%) primarily due to low sequence coverage.
To assess the accuracy of the variants identified from the pooled sequence data, we compared the variants and their allele frequencies with exome sequence data from the National Heart, Lung, and Blood Institute Exome Sequencing Project [42]. This comparison demonstrated a high sensitivity for the detection of even low frequency variants (minor allele frequency ≥ 0.001) and high concordance of variant allele frequencies (r 2 = 0.998 for all SNVs, Additional file 2: Supplementary Methods). Furthermore, using Sequenom genotyping of 23 SNVs in 240 individuals, the allele counts at individual variant sites estimated from the pooled sequence data were observed to be highly accurate (r 2 = 0.998, see Additional file 2 for details).
In the second stage of the study, targeted sequencing was performed on DNA from 3168 individuals using 132 pools (43 control pools and 89 case pools with 24 individuals per pool). Of the 1044 variants detected, 602 (56.7%) were missense variants and 18 were insertion/deletion variants. The fraction of missense variants was very similar to the fraction of missense variants (0.6) in the first stage of the sequencing. The number of variants detected was greater than in Stage 1 since five additional monogenic diabetes genes (CEL, EIF2AK3, ABCC8, BLK, and KLF11) were sequenced (Additional file 1: Table S1). Overall, 88.9 ± 1.3% of the targeted bases across the 22 genes (38 kilobases of DNA sequence) had a median read depth of ≥ 240× (10× per individual in pools of size 24). The coverage was slightly higher in the control pools compared to the case pools (Additional file 1: Figure S3). The allele frequency distribution of rare variants (Additional file 1: Figure S1) and the proportion of singleton variants was similar to that observed in the first stage of sequencing.
In Stage 3, 1011 coding variants were identified from the sequencing of 2014 individuals with diabetes. These variants included 585 missense SNVs and 21 indel variants (Additional file 1: Table S3). Through joint analysis of pools sequenced in Stage 3 and the corresponding pools in Stages 1 and 2 as well as information about the overlap between pools, we identified the carrier(s) of each rare variant using a parsimonious approach (Additional file 2: Supplementary Methods). There was strong agreement between the observed number of carriers of the variant allele in Stage 3 data and the expected number of carriers from Stage 1 and 2 data. In the GCK, HNF1A, and HNF4A genes, 51 rare missense and protein truncating variants (allele counts < 5) were observed in pools from Stage 3. For 48 of the 51 variants, the expected and observed allele counts were perfectly consistent with data from Stages 1 and 2 and carriers could be identified with little or no ambiguity. Low sequence coverage in Stage 1 and 2 data explained the discrepancy for the three variants. Overall, less than 7% of rare coding variants in the 17 monogenic diabetes genes that were sequenced in all three stages had discrepancy between variant-positive pools, and these were primarily due to sites with low sequence coverage in Stage 1 data compared to Stage 3. The orthogonal pooled sequencing provided independent validation of the sequence variants detected in Stage 1 and 2 data since each individual with the variant allele was sequenced twice in two different pools and library preparation was also performed independently. For variants with multiple variant-positive pools in Stage 3 as well as in Stages 1 or 2, there was some ambiguity in identifying variant carriers. Nevertheless, even in such cases, the orthogonal pooled sequencing enabled partial carrier identification and provided useful information about the age of diagnosis of the individual(s) with the variant. Information about variants identified in Stage 3 pools was not used for comparison of variants between cases and controls but only to validate rare variants and identify the carriers (and age of onset) of rare deleterious variants.
Analysis of protein-truncating variants
Genetic variants that result in a premature stop codon in the transcript are commonly referred to as protein truncating or loss-of-function variants and typically result in a severe impact on gene function. Seven such mutations were observed in MODY genes in which heterozygous loss-of-function variants are known to be pathogenic for diabetes – three each in the GCK and HNF1A genes and one in the HNF1B gene (Table 1). All seven variants were singletons and each of the seven individuals with these mutations had diabetes (mean age at diagnosis = 27.5 years). The individual with the protein truncating variant (PTV) in the HNF1B gene was diagnosed at 14 years of age and likely has MODY5. Although the cases were screened for classical MODY phenotypes, some subjects with MODY can only be identified via genetic analysis. A recent study showed that the majority of individuals with early onset diabetes that were found to carry MODY mutations using genetic testing were clinically misdiagnosed [48].
Table 1.
List of protein truncating variants identified in monogenic diabetes genes in which heterozygous protein truncating variants are known to be pathogenic for diabetes. None of the variants were present in the ExAC database
| Counts | |||||||
|---|---|---|---|---|---|---|---|
| Gene | DNA change | AA change | Cases | Early onset | Controls | dbSNP 144 | ACMG classa |
| GCK | c.871A > T | p.K291* | 1 | 1 | 0 | rs193922335 | 5 |
| GCK | c.1340_1368del | p.R447fs | 1 | 1 | 0 | ─ | 4 |
| GCK | c.863 + 1G > T | p.? | 1 | 0 | 0 | ─ | 4 |
| HNF1A | c.994delG | p.E332fs | 1 | 1 | 0 | ─ | 4 |
| HNF1A | c.955 + 1G > T | p.? | 1 | 1 | 0 | ─ | 4 |
| HNF1A | c.1730_1733dupACCT | p.Q579fs | 1 | 0 | 0 | ─ | 4 |
| HNF1B | c.1005dupC | p.H336fs | 1 | 1 | 0 | ─ | 4 |
| PPARG | c.465delC | p.H155fs | 1 | 0 | 0 | ─ | 4 |
Compared to GCK, HNF1A, HNF4A, and HNF1B, protein truncating mutations in the other MODY genes are less frequent causes of MODY [49]. For some of these genes, only a few mutations linked to early onset diabetes have been reported. In the PDX1 gene, a frameshift insertion was detected in a diabetic individual diagnosed at the age of 48 years. Recently, a study from Iceland [50] identified a rare frameshift variant in PDX1 to be associated with an increased risk of T2D (odds ratio (OR), 2.47). Similarly, the individual with the PAX4 PTV had adult onset diabetes (age at diagnosis 56 years). In the KLF11 gene, two PTVs (one in an early onset diabetic patient and the second in a control individual) were detected (Additional file 1: Table S5). No such variants were detected in the INS, BLK, NEUROD1, and KCNJ11 genes.
The CEL gene was sequenced in a subset of the samples and four frame-shift mutations were observed (four carriers in 2136 cases and one carrier in 1032 controls; OR, 1.94; Additional file 1: Table S5). Only one of the four mutation carriers was from the sub-group with early onset diabetes, indicating that, unlike classical MODY genes, heterozygous protein truncating mutations that affect the CEL gene are unlikely to be a strong risk factor for early onset diabetes. This is not very surprising since both of the two single base deletions that have been reported to cause CEL-MODY impact a VNTR sequence located at the C-terminal of the gene [51] and result in a protein sequence that is unlikely to be affected by non-sense mediated decay [52].
In non-MODY genes with an autosomal dominant disease inheritance, we identified one protein truncating mutation in PPARG in an individual diagnosed with diabetes at 41 years of age. The frameshift mutation (deletion of a C) is located in exon 5 (transcript NM_138711) and is predicted to introduce 48 novel amino acids before ending in a premature stop codon. Protein-truncating mutations in PPARG are rare but have been described previously in individuals with T2D and partial lipodystrophy [53–55]. In addition, 19 rare PTVs were identified in five recessive monogenic diabetes genes (all carriers were heterozygous) but were not more frequent in individuals with diabetes (0.35% of cases and 0.31% of controls; Additional file 1: Table S5).
Gene-level association analysis for rare coding variants
To identify associations for rare coding variants with diabetes, we performed gene-level association tests using coding variants (missense and in-frame indel variants with minor allele frequency < 0.2%) detected in each gene (Additional file 2: Supplementary Methods). Association analysis was performed on sequence data from Stages 1 and 2 independently. The GCK gene showed a nominal association between rare coding variants and early onset diabetes (P = 0.0174 for early onset cases versus controls in Stage 1 and P = 0.0013 in Stage 2). Jointly across Stage 1 and 2 data, missense variants in GCK (including one in-frame deletion) were detected in 0.5% of cases and 0.035% of controls. Although seven individuals with a GCK mutation had early onset diabetes (Table 2), only four of these were diagnosed before 25 years of age. A large number of heterozygous missense mutations that cause GCK-MODY have been identified over the years and are distributed across the gene (> 600 mutations were tabulated by Osbak et al. [56]). Comparison to these known mutations revealed that 14 of the 20 missense variants in our cohort have previously been reported in at least one individual with MODY (Table 2). GCK has two protein isoforms that differ in the first 15 amino acids and two of the missense mutations were located at positions 10 and 12 in this region. The one missense variant (p.K12R) detected in an individual without diabetes was predicted to be a benign variant in the pancreas-specific splice isoform (Table 2).
Table 2.
List of missense (and in-frame indels) mutations detected in the GCK gene. All mutations (except p.A11T) were observed in a single individual in our dataset
| DNA change | AA change | Poly-Phen2a | SIFTb | MutationTasterc | CADDd | Age at diagnosis, years | Previously observed in MODY | ExAC AFe | dbSNP144 | ACMG classf |
|---|---|---|---|---|---|---|---|---|---|---|
| c.484G > A | p.G162S | Pr.D | tol | del | 26.5 | 13 | 1 family | ─ | ─ | 3 |
| c.952G > A | p.G318R | Pos.D | del | del | 27.2 | 14 | 4 families | ─ | ─ | 4 |
| c.617C > T | p.T206M | Pr.D | del | del | 33 | 19 | 13 families | ─ | ─ | 4 |
| c.238G > A | p.G80S | Pr.D | del | del | 32 | 24 | 2 families | ─ | rs193922317 | 4 |
| c.1349C > T | p.A450V | Pr.D | del | del | 29.7 | 27 | ─ | ─ | 3 | |
| c.911T > C | p.L304P | Pr.D | tol | del | 24.6 | 28 | 3 families | ─ | ─ | 4 |
| c.559G > T | p.D187Y | Pr.D | del | del | 33 | 28 | 3 families | ─ | ─ | 4 |
| c.214G > A | p.G72R | Pr.D | del | del | 34 | 29 | 18 families | ─ | rs193922289 | 5 |
| c.118G > A | p.E40K | Pr.D | del | del | 33 | 30 | 5 families | ─ | ─ | 4 |
| c.562G > A | p.A188T | Pr.D | del | del | 35 | 30 | 22 families | 0.0001 | rs751279776 | 4 |
| c.640T > G | p.Y214D | Pr.D | del | del | 27.2 | 33 | ─ | ─ | 3 | |
| c.131G > A | p.G44D | Pr.D | del | del | 29 | 34 | 4 families | ─ | rs193922279 | 4 |
| c.572G > A | p.R191Q | Pr.D | del | del | 35 | 37 | 9 families | ─ | ─ | 4 |
| c.787_801del | p.263_267del | ─ | ─ | ─ | ─ | 39 | ─ | ─ | 4 | |
| c.544G > A | p.V182M | Pr.D | del | del | 34 | 41 | 12 families | ─ | rs587780345 | 5 |
| c.706G > A | p.E236K | Pos.D | del | del | 33 | 42 | 2 families | ─ | rs587780347 | 4 |
| c.394G > A | p.D132N | benign | tol | del | 23 | 56 | 1 family | 0.000015 | ─ | 3 |
| c.757G > A | p.V253I | benign | tol | del | 18.4 | 61 | 0.00006 | rs748964205 | 3 | |
| c.31G > A | p.A11T | benign | tol | poly | 12.8 | 32, 45 | 0.024 | rs116093166 | 2 | |
| c.35A > G | p.K12R | benign | tol | poly | 16.8 | NA | 0.000015 | rs777958777 | 3 |
Reference sequence for GCK: NM_000162
aPolyPhen2 predictions are probably damaging (Pr.D), possibly damaging (Pos.D) and benign
bSIFT predictions are deleterious (del) and tolerated (tol)
cMutationTaster predictions are disease causing (del) and polymorphism (poly)
dCADD scaled C-scores range from 0 to 30. Higher CADD scores correspond to more deleterious variants; a CADD score of 20 (30) corresponds to the top 1% (0.1%) of deleterious substitutions in the human genome
eExAC allele frequency is the maximum allele frequency of the variant allele among the different populations
fACMG classification: 5 = pathogenic, 4 = likely pathogenic and 3 = uncertain significance (see Methods)
AA amino acid, ACMG American College of Medical Genetics, AF allele frequency, dbSNP Single Nucleotide Polymorphism Database, ExAC Exome Aggregation Consortium, NA not available
Frequency of missense variants in MODY genes
We did not detect a significant association between missense mutations in genes such as HNF1A and HNF4A, which are frequently mutated in early onset diabetes, likely due to the fact that not all missense mutations in these genes are pathogenic. Previously reported missense mutations in individuals with MODY or early onset diabetes have a strong prior likelihood of being pathogenic. To analyze the frequencies of the carriers of such mutations in our cohort, we analyzed genes (HNF1A, HNF4A, HNF1B, INS, ABCC8, and KCNJ11) in which a significant number of missense mutations have previously been reported in MODY, neonatal diabetes mellitus, or early onset diabetes [57]. We excluded the remaining MODY genes (CEL, PDX1, PAX4, BLK, KLF11, NEUROD1) from this analysis since either very few missense mutations in these genes have been associated with early onset diabetes or the genetic evidence for association is limited. Previously reported mutations that have been shown to be benign using functional assays or have high frequency in controls were also excluded (see Methods).
We identified 23 missense mutations in these six genes that have previously been reported in MODY or early onset diabetes and are likely pathogenic – 14 in HNF1A, 3 in HNF4A, 5 in ABCC8, and 1 in the INS gene (Table 3). Overall, 26 of the 29 individuals with these missense mutations had diabetes (OR, 6.24 for cases versus controls; 95% confidence interval 1.9–20.6; Fisher’s exact test P = 0.0004), demonstrating that previously reported pathogenic missense mutations in these genes are significantly over-represented in individuals diagnosed with diabetes compared to controls, particularly in the sub-group of individuals with early onset diabetes (OR, 1.99 for early onset versus late onset sub-group). Nevertheless, 50% of mutation carriers with diabetes were diagnosed at 40 years or later, indicating that not all mutations previously reported in individuals with a diagnosis of MODY or early-onset diabetes are fully penetrant. One such mutation, the p.R136W variant (also reported as p.R114W in literature, Table 3), is the most frequently reported HNF4A mutation and was detected in two pools consisting of individuals with late onset diabetes (age at diagnosis > 46 years). Recent analysis of this specific mutation has shown that this mutation causes MODY-like diabetes but has lower penetrance in comparison to classical MODY mutations [58]. Mutations that are pathogenic for early onset diabetes are expected to be very rare in the population. Indeed, analysis of the population allele frequencies showed that all variants were very rare and the minor allele frequency for 22 of the 23 variants was less than 0.0005 (Table 3).
Table 3.
List of missense mutations in the HNF1A, HNF4A, HNF1B, INS, and ABCC8 genes that have previously been reported in individuals or families with MODY or early onset diabetes. The ABCC8 gene was sequenced in a subset of individuals (2132 cases and 1024 controls)
| Counts | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | cDNA change | AA change | Cases | Early onset | Controls | PolyPhen2a | SIFTb | MutationTasterc | CADDd | Previously observed in MODY/diabetese | ExAC AFf | dbSNP 144 | ACMG classg |
| HNF1A | c.391C > T | p.R131W | 1 | 1 | 0 | Pr.D | del | del | 31 | 29 families | ─ | rs137853244 | 5 |
| HNF1A | c.608G > A | p.R203H | 2 | 1 | 0 | Pos.D | del | del | 29 | 19 individuals | ─ | rs587780357 | 4 |
| HNF1A | c.812G > A | p.R271Q | 1 | 1 | 0 | Pr.D | del | del | 34 | 13 individuals | 0.00007 | rs779184183 | 4 |
| HNF1A | c.779C > T | p.T260M | 1 | 1 | 0 | Pr.D | del | del | 33 | 13 families | ─ | ─ | 4 |
| HNF1A | c.1340C > T | p.P447L | 1 | 1 | 0 | Pr.D | del | del | 34 | 11 studies | ─ | rs137853236 | 5 |
| HNF1A | c.1135C > G | p.P379A | 1 | 1 | 0 | Pr.D | del | del | 25 | 10 studies | 0.0006 | rs754729248 | 4 |
| HNF1A | c.815G > A | p.R272H | 1 | 0 | 0 | Pr.D | del | del | 34 | 20 families | ─ | rs137853238 | 5 |
| HNF1A | c.1061C > T | p.T354M | 2 | 1 | 0 | benign | tol | poly | 23 | 3 individuals | 0.00006 | rs757068809 | 3 |
| HNF1A | c.1513C > A | p.H505N | 1 | 0 | 0 | Pos.D | tol | del | 26.1 | 3 individuals from one study | 0.00017 | rs577078110 | 4 |
| HNF1A | c.1400C > T | p.P467L | 1 | 0 | 0 | benign | del | del | 20.8 | 3 individuals | 0.000015 | ─ | 3 |
| HNF1A | c.481G > A | p.A161T | 0 | 0 | 1 | Pos.D | del | del | 31 | 1 individual | 0.00024 | rs201095611 | 3 |
| HNF1A | c.503G > A | p.R168H | 0 | 0 | 2 | Pos.D | del | del | 32 | 1 individual | 0.00006 | rs377110124 | 3 |
| HNF1A | c.403G > A | p.D135N | 1 | 1 | 0 | Pos.D | del | del | 32 | 1 individual | ─ | ─ | 3 |
| HNF1A | c.1699G > A | p.V567I | 1 | 0 | 0 | benign | tol | poly | 18.8 | 1 individual | 0.0001 | ─ | 3 |
| HNF4A | c.400C > T | p.R134W | 1 | 1 | 0 | Pos.D | del | del | 35 | 5 families | ─ | rs370239205 | 4 |
| HNF4A | c.406C > T | p.R136W | 2 | 0 | 0 | Pos.D | del | del | 34 | 36 families | 0.0001 | rs137853336 | 5 |
| HNF4A | c.929G > A | p.R310Q | 2 | 0 | 0 | Pr.D | tol | del | 24.7 | 1 family/co-segregation with diabetes [80] | 0.00003 | rs371124358 | 4 |
| ABCC8 | c.886G > A | p.G296R | 1 | 1 | 0 | benign | del | del | 27.1 | Individual with diabetes at 7 months [82] | 0.00006 | rs148529020 | 3 |
| ABCC8 | c.1067A > G | p.Y356C | 1 | 0 | 0 | Pr.D | del | del | 26.1 | Early onset diabetes family [78] | 0.00005 | rs59852838 | 4 |
| ABCC8 | c.2473C > T | p.R825W | 2 | 1 | 0 | Pr.D | del | del | 35 | Multiple individuals with NDM [83] | 0.00001 | rs779736828 | 4 |
| ABCC8 | c.4136G > A | p.R1379H | 1 | 1 | 0 | Pr.D | del | del | 34 | One individual with transient NDM [81] | ─ | ─ | 3 |
| ABCC8 | c.4516G > A | p.E1506K | 1 | 1 | 0 | Pr.D | del | del | 35 | Finnish family [77] | ─ | rs137852671 | 5 |
| INS | c.16C > T | p.R6C | 1 | 0 | 0 | ─ | del | del | 22.7 | Three-generation MODY family [76] | 0.00006 | rs121908278 | 5 |
Reference sequences: HNF1A, NM_000545; HNF4A, NM_000457; ABCC8, NM_000352; INS, NM_001185098
aPolyPhen predictions are probably damaging (Pr.D), possibly damaging (Pos.D) and benign
bSIFT predictions are deleterious (del) and tolerated (tol)
cMutationTaster predictions are disease causing (del) and polymorphism (poly)
dCADD scaled C-scores range from 0-30. Higher CADD scores correspond to more deleterious variants; a CADD score of 20 (30) corresponds to the top 1% (0.1%) of deleterious substitutions in the human genome
eInformation about previously observed MODY mutations in the HNF1A and HNF4A genes was obtained from Colclough et al. [79]
fExAC allele frequency is the maximum allele frequency of the variant allele among the different populations reported in the database
gACMG classification: 5 = pathogenic, 4 = likely pathogenic, and 3 = uncertain significance
AA amino acid, ACMG American College of Medical Genetics, AF allele frequency, dbSNP Single Nucleotide Polymorphism Database, ExAC Exome Aggregation Consortium, NA not available, NDM neonatal diabetes mellitus
Combined with the 14 missense mutations in the GCK gene, the overall frequency of previously reported pathogenic missense mutations was 1.8% in early onset diabetes (24/1346), 0.6% in late onset cases (16/2670), and 0.1% in controls (Additional file 1: Table S7). Overall, analysis of rare missense mutations in these genes indicated that previously reported pathogenic missense mutations were significantly over-represented in individuals with diabetes compared to controls (OR, 9.3; P = 5 × 10–7). Analysis of rare missense variants classified as likely pathogenic or pathogenic (class 4 or 5) using the ACMG guidelines [45] showed a similar trend, wherein 1.5% of individuals in the early onset diabetes sub-group, 0.4% of individuals in the late onset sub-group, and none of the controls carried such mutations in the GCK, HNF1A, HNF4A, ABCC8, and INS genes (Tables 2 and 3).
The detection of a significant number of individuals with previously reported pathogenic missense variants indicated that additional, previously unreported pathogenic mutations could also be present in the data. Therefore, we analyzed missense mutations that are predicted to be deleterious by the two leading in silico annotation tools (Polyphen2 and SIFT) and have low population allele frequency (minor allele frequency < 0.0005). All of these missense variants were also classified as deleterious by MutationTaster and CADD (C-scores > 20); 18 such missense mutations were observed in the sequence data with 18 carriers in cases and 6 in controls (OR, 2.15; Additional file 1: Table S6), indicating that additional pathogenic mutations likely exist in the sequenced data but are difficult to pinpoint without functional or genetic data.
Variants in recessive monogenic diabetes genes
Next, using information about the carriers of rare variants identified from Stage 3 sequence data, we searched for individuals who were homozygous for rare coding mutations in six recessive monogenic diabetes genes. We identified an individual who is likely homozygous for a rare missense variant (NM_001145853; exon 8; c.1672C > T; p.R558C) in the WFS1 gene. Homozygous or compound heterozygous mutations in WFS1 cause WS, which is characterized by a lack of insulin secretion leading to diabetes mellitus, optic atrophy, and several other phenotypes [11]. This individual was diagnosed with diabetes at the age of 14 years but does not have additional symptoms typically associated with WS such as diabetes insipidus, deafness, optic atrophy, or renal and neurological problems. This same variant has previously been reported in two individuals with WS, namely in an individual with an atypical presentation of the disease who was identified to be a homozygous carrier for this variant [59] and in another individual who carried this variant in combination with a coding deletion variant [60]. The second individual had a mild phenotype with diabetes and optic atrophy without other phenotypes of WS. The p.R558C variant is a rare variant with an allele frequency of 0.0008 in individuals of European ancestry from the ExAC database [43] and even lower in other populations. In our dataset, the frequency of this variant was 0.0007, similar to that observed in the ExAC database.
Discussion
In this study, we sequenced and analyzed mutations in monogenic diabetes genes in a large cohort of individuals with diabetes (n = 4016) and controls (n = 2872) from the southern part of Germany. Among individuals with young onset and adult onset diabetes, 40 individuals (1.8% of subjects with early onset diabetes and 0.6% with late onset) were carriers of known pathogenic missense mutations in the GCK, HNF1A, HNF4A, HNF1B, ABCC8, and INS genes. Additionally, protein truncating mutations in these genes were identified in seven individuals with diabetes. The diabetes phenotype of these individuals is likely “dominated by perturbation in a small number of processes” related to islet-cell function and hence their diagnosis and treatment can benefit from this knowledge [61]. Although pathogenic missense and PTVs in these genes were strongly enriched in individuals with early onset diabetes, none of these participants fulfilled classical Tattersall criteria of monogenic diabetes mellitus. To enable clinicians to discriminate between T2D and MODY, guidelines for selecting individuals for genetic testing based on clinical criteria have been established [25]. Our population study was not designed to include family members and to genotype or phenotype family members, which may have potentially limited the ability to identify individuals with MODY. Nevertheless, several studies have shown that clinical criteria alone are not sufficient to diagnose MODY and genetic testing is needed for a definitive diagnosis [49].
The most commonly mutated genes in MODY are HNF1A and GCK, followed by HNF4A and HNF1B [49]. In our data, the maximum number of pathogenic mutations was observed in the GCK gene (17 carriers with 14 in the early onset sub-group) followed by HNF1A. GCK-MODY is characterized by mild hyperglycemia typically without diabetes associated microvascular and macrovascular complications [20]. Therefore, GCK-MODY is perhaps the most likely form of MODY to be misdiagnosed as T2D [20]. Detection of a GCK mutation in an individual with T2D is important from a clinical perspective since no medications are necessary for such individuals except for females during pregnancy.
In addition, the frequency of missense pathogenic mutations in commonly mutated MODY genes observed in our cohort was much higher than the frequency of protein truncating mutations, likely due to the fact that individuals with early onset diabetes were screened for MODY using phenotypic criteria and, as a result, the cohort is depleted of individuals with protein truncating mutations in MODY genes likely to be fully penetrant. Approximately 0.6% of individuals with late onset diabetes were observed to be carriers of likely pathogenic missense mutations that have previously been associated in MODY or early onset diabetes, suggesting that individuals with late onset T2D can also harbor deleterious variants in monogenic diabetes genes with moderate penetrance. A recent study by Flannick et al. [22] sequenced seven MODY genes in two large population cohorts and found 0.5–1.5% of individuals to be carriers of rare missense mutations predicted to be deleterious by bioinformatics tools or previously reported in MODY. However, the majority of these individuals were found not to have diabetes. In contrast, our study was a case–control study and included a large number of individuals (1346) with early onset diabetes. Rare missense mutations that have been previously associated with MODY or early onset diabetes were strongly enriched in the sub-group with early onset diabetes. Another recent large-scale exome sequencing study found a modest but statistically significant enrichment of rare deleterious variants in monogenic diabetes genes in individuals with T2D compared to controls [62].
We detected multiple early onset diabetes subjects with pathogenic missense mutations in the ABCC8 gene. Such individuals can be treated effectively with sulfonylureas rather than insulin or other medications. The ABCC8 gene is considered for genetic testing in neonatal diabetes but several studies have identified ABCC8 missense mutations in individuals with early and late onset diabetes [63–65]. In addition, one individual with early onset diabetes was homozygous for a rare and pathogenic missense variant in the WFS1 gene, suggesting that genetic testing can identify individuals with an atypical presentation of WS.
Although sequencing can identify pathogenic mutations in genes strongly linked with disease, such as MODY genes, it is challenging to distinguish such mutations from the vast number of neutral mutations observed in large-scale sequencing studies [66]. In the GCK gene, our data suggests that the vast majority of missense mutations increase the risk for diabetes to a varying degree. However, in MODY genes such as HNF1A and HNF4A, not all missense mutations increase the risk for diabetes and, therefore, it is challenging to ascribe pathogenicity to a novel missense mutation based on predictions made by bioinformatics tools. In our data, we did not observe a significant association between rare missense mutations in the genes predicted to be deleterious by multiple bioinformatics tools and risk of diabetes. Recently, Najmi et al. [67] used functional assays to evaluate the missense mutations in the HNF1A gene identified by Flannick et al. [22], and showed that 11 of these mutations that reduced transcriptional activity were strongly associated with an increased risk of diabetes (OR, 5.04). Functional assays have been used to identify pathogenic variants in other genes linked with diabetes [68, 69]. Therefore, classification of novel missense variants identified in our study using functional assays has the potential to identify additional individuals with mutations that increase the risk of MODY or T2D.
Our study leveraged the massive throughput of high-throughput sequencing instruments and the ability to sequence selected regions of the human genome in large numbers of individuals. We utilized a pooled DNA sequencing approach to reduce the cost of DNA library preparation. Although pooled sequencing was highly cost-effective and allowed us to sequence nearly 6900 individuals with high sensitivity and specificity for the detection of rare variants, it is less informative than individual sequencing about individual genotypes and does not allow for the detection of copy number variants such as large deletions. In addition, some of the genes targeted for sequencing in our study had low sequence coverage (e.g., the INS gene) and we estimated a false negative rate of ~7% for the discovery of rare variants. As a result, a small number of pathogenic mutations were likely not detected. It is possible that additional pathogenic variants (e.g., in non-coding regions) in known monogenic diabetes genes as well as novel genes for early onset diabetes remain to be identified, thereby defining new variants with a large effect on the disease phenotype. Another limitation of our study is the lack of family data or access to DNA samples from first degree relatives of individuals with diabetes for further genotype and phenotype studies.
Our cohort represents a relatively homogeneous cohort of European ancestry from the southern region of Germany with well-defined criteria for classifying individuals as cases and controls. All subjects had been screened for the presence of islet cell autoimmunity to exclude the presence of classical autoimmune diabetes (T1D) and late onset/latent autoimmune diabetes in adult [70]. Many large scale studies of the genetics of T2D do not measure islet cell antibodies and, therefore, exclude subjects with an early age of onset to avoid including T1D cases. In addition, in view of the high prevalence of subjects with latent autoimmune diabetes in adult onset diabetes subjects a major confounding factor can be present in genetic studies of the so-called T2D [71]. There is growing evidence from genetic studies for the heterogeneity of the adult onset diabetes phenotype and overlap with monogenic diabetes [67] and T1D [70]. Multiple studies have shown that 5–15% of individuals with so called T2D are positive for islet cell antibodies [72, 73]. Recent work has addressed the question of a fine-grained categorization of adult onset diabetes using clinical data in large cohorts [74].
Conclusion
In our sequencing study involving 6888 individuals, 2.2% of individuals with early onset diabetes and 0.7% of individuals with late onset diabetes harbored a likely pathogenic mutation in monogenic diabetes genes. Our results confirm previous reports that MODY is under-diagnosed [19, 75], particularly in individuals presenting with early onset diabetes and clinically labeled as T2D and, in such cases, genetic testing can provide an etiological diagnosis. With the continuing reduction in costs of DNA sequencing, genetic screening of all known monogenic diabetes genes in individuals with early onset diabetes should be routinely considered since it can identify individuals with undiagnosed MODY as well as atypical forms of monogenic diabetes. Knowledge of mutations in monogenic diabetes genes has the potential to influence diagnosis and therapy for individuals with diabetes as well as to enable the genetic testing of relatives.
Additional files
List of 22 genes associated with monogenic forms of diabetes that were analyzed in this paper. Table S2. Criteria used to select genes for targeted sequencing. Table S3. Summary of samples sequenced in Stages 1, 2, and 3, and the coding variants identified in each stage. Table S4. Clinical data of the cases and controls for type 2 diabetes sequenced in this study. Table S5. List of all protein truncating mutations identified in the 22 monogenic diabetes genes. Table S6. Rare missense mutations in the HNF1A, HNF4A, HNF1B, ABCC8, and KCNJ11 genes predicted to be deleterious by PolyPhen2, SIFT, and MutationTaster. Table S7. Number of individuals with protein truncating variants and previously reported pathogenic missense variants in MODY genes. Table S8. List of exons with low sequence coverage in data from Stage 1 and 2 pools. Figure S1. Minor allele frequency distribution of variants identified from sequencing of pools in Stages 1 and 2. Figure S2. Pooled sequencing design of the study. Figure S3. Comparison of sequence coverage between cases and controls. (PDF 700 kb)
Supplementary Methods: Description of methods for pooled variant calling, gene-level tests for rare coding variants, statistical analyses, comparison of pooled sequence data with population exome data, comparison of pooled allele counts with individual genotypes, and identification of the carriers of rare variants. (PDF 671 kb)
Acknowledgements
VB is supported by start-up funds from the Department of Pediatrics, University of California San Diego. BOB is supported by Lee Kong Chian School of Medicine, Nanyang Technological University Start Up Grant, MOE AcRF Tier 1 (2015-T1-001-258) and NTU-NHG Metabolic Diseases Collaboration Grant (MDCG/15006); Deutsche Forschungsgemeinschaft (DFG, GrK 1041) and State Baden-Wuerttemberg, Germany. We thank Roza Blagieva for recruitment and phenotyping of diabetes patients and controls. We thank Silke Rosinger, Angelika Kurkhaus, Rosina Sing, and Silvia Hoepfer for sample collection, DNA preparation and aliquoting as well as for performing islet-cell antibody measurements. We thank Ryan Tewhey for help with DNA pooling.
Funding
This study was supported in part by Sanofi-Aventis. The funders had no role in data analysis and interpretation and in the writing of the manuscript.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACMG
American College of Medical Genetics
- ExAC
Exome Aggregation Consortium
- HbA1c
glycated hemoglobin
- Indel
insertion/deletion
- MODY
Maturity onset diabetes of the young
- PTV
protein truncating variant
- SNV
Single nucleotide variant
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- WS
Wolfram syndrome
Authors’ contributions
Study concept: VB, JG and BOB. Design and analysis of sequencing experiments: VB. Generation of sequence data: GO, TP, NV, RH, and EJT. Recruitment, phenotyping and follow-up of diabetes patients and controls: BOB and TS. Interpretation of data: VB, JG and BOB. Resources: BOB and EJT. Preparation of manuscript: VB. Revision of manuscript: VB, JG, TS and BOB. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All individuals gave informed consent for use of their DNA samples for genetic studies. The study was approved by the Institutional Review Board of Ulm University, Ulm, Germany (registration numbers 42/2004 and 189/2007) and the Chamber of Physicians, State Baden-Wuerttemberg, Germany (registration number 133-2002), and is in accordance with the ethical principles of the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
JG is an employee of Sanofi-Aventis.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12916-017-0977-3) contains supplementary material, which is available to authorized users.
Contributor Information
Vikas Bansal, Email: vibansal@ucsd.edu.
Bernhard O. Boehm, Email: bernhard.boehm@ntu.edu.sg
References
- 1.McCarthy MI. Genetics of T2DM in 2016: Biological and translational insights from T2DM genetics. Nat Rev Endocrinol. 2017;13(2):71–2. doi: 10.1038/nrendo.2016.212. [DOI] [PubMed] [Google Scholar]
- 2.Bonnefond A, Froguel P. Rare and common genetic events in type 2 diabetes: what should biologists know? Cell Metab. 2015;21(3):357–68. doi: 10.1016/j.cmet.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 3.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–5. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–89. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med. 2010;16:407–16. doi: 10.1016/j.molmed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Dorajoo R, Liu J, Boehm BO. Genetics of type 2 diabetes and clinical utility. Genes (Basel) 2015;6:372–84. doi: 10.3390/genes6020372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24:44–53. doi: 10.2337/diab.24.1.44. [DOI] [PubMed] [Google Scholar]
- 8.Vaxillaire M, Bonnefond A, Froguel P. The lessons of early-onset monogenic diabetes for the understanding of diabetes pathogenesis. Best Pract Res Clin Endocrinol Metab. 2012;26:171–87. doi: 10.1016/j.beem.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia. 2017;60(5):769–77. doi: 10.1007/s00125-017-4226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem. 2013;50:403–15. doi: 10.1177/0004563213483458. [DOI] [PubMed] [Google Scholar]
- 11.Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat Genet. 1998;20:143–8. doi: 10.1038/2441. [DOI] [PubMed] [Google Scholar]
- 12.Hegele RA, Cao H, Harris SB, Hanley AJ, Zinman B. The hepatic nuclear factor-1alpha G319S variant is associated with early-onset type 2 diabetes in Canadian Oji-Cree. J Clin Endocrinol Metab. 1999;84(3):1077–82. doi: 10.1210/jcem.84.3.5528. [DOI] [PubMed] [Google Scholar]
- 13.Sandhu MS, Weedon MN, Fawcett KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39(8):951–3. doi: 10.1038/ng2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Estrada K, Aukrust I, Bjorkhaug L, et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. 2014;311:2305–14. doi: 10.1001/jama.2014.6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearson ER, Flechtner I, Njølstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355:467–77. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 16.Sperling MA. ATP-sensitive potassium channels--neonatal diabetes mellitus and beyond. N Engl J Med. 2006;355:507–10. doi: 10.1056/NEJMe068142. [DOI] [PubMed] [Google Scholar]
- 17.Boycott KM, Innes AM. When one diagnosis is not enough. N Engl J Med. 2017;376:83–5. doi: 10.1056/NEJMe1614384. [DOI] [PubMed] [Google Scholar]
- 18.Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376(1):21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakera AJ, Steele AM, Gloyn AL, et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care. 2015;38:1383–92. doi: 10.2337/dc14-2769. [DOI] [PubMed] [Google Scholar]
- 20.Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311:279–86. doi: 10.1001/jama.2013.283980. [DOI] [PubMed] [Google Scholar]
- 21.Bonnefond A, Philippe J, Durand E, et al. Highly sensitive diagnosis of 43 monogenic forms of diabetes or obesity through one-step PCR-based enrichment in combination with next-generation sequencing. Diabetes Care. 2014;37(2):460–7. doi: 10.2337/dc13-0698. [DOI] [PubMed] [Google Scholar]
- 22.Flannick J, Beer NL, Bick AG, et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat Genet. 2013;45:1380–5. doi: 10.1038/ng.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellard S, Lango Allen H, De Franco E, et al. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia. 2013;56(9):1958–63. doi: 10.1007/s00125-013-2962-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howson JMM, Rosinger S, Smyth DJ, Boehm BO, ADBW-END Study Group. Todd JA. Genetic analysis of adult-onset autoimmune diabetes. Diabetes. 2011;60:2645–53. doi: 10.2337/db11-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia. 2012;55(5):1265–72. doi: 10.1007/s00125-011-2418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–9. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bansal V. A statistical method for the detection of variants from next-generation resequencing of DNA pools. Bioinformatics. 2010;26:i318–24. doi: 10.1093/bioinformatics/btq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bansal V, Tewhey R, Leproust EM, Schork NJ. Efficient and cost effective population resequencing by pooling and in-solution hybridization. PLoS One. 2011;6:e18353. doi: 10.1371/journal.pone.0018353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–73. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prescott NJ, Lehne B, Stone K, et al. Pooled sequencing of 531 genes in inflammatory bowel disease identifies an associated rare variant in BTNL2 and implicates other immune related genes. PLoS Genet. 2015;11:e1004955. doi: 10.1371/journal.pgen.1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong SN, Park C, Park SJ, et al. Deep resequencing of 131 Crohn's disease associated genes in pooled DNA confirmed three reported variants and identified eight novel variants. Gut. 2016;65(5):788–96. doi: 10.1136/gutjnl-2014-308617. [DOI] [PubMed] [Google Scholar]
- 33.Scott-Van Zeeland AA, Bloss CS, Tewhey R, et al. Evidence for the role of EPHX2 gene variants in anorexia nervosa. Mol Psychiatry. 2014;19:724–32. doi: 10.1038/mp.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruark E, Snape K, Humburg P, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–10. doi: 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novocraft.com. Novoalign short read mapper. http://www.novocraft.com/. Accessed 1 Sept 2013.
- 36.Picard. A Set of Tools for Working with High-Throughput Sequencing Data. https://sourceforge.net/projects/picard/. Accessed 1 Sept 2013.
- 37.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(Web Server issue):W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep sequencing age. Nat Methods. 2014;11(4):361–2. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 41.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Exome Variant Server. NHLBI Exome Sequencing Project (ESP), Seattle WA. http://evs.gs.washington.edu/EVS/. Accessed 1 Sept 2014.
- 43.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stenson P, Ball E, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–81. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 45.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. 2017;100(2):267–80. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landrum M, Lee J, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chambers C, Fouts A, Dong F, et al. Characteristics of maturity onset diabetes of the young in a large diabetes center. Pediatr Diabetes. 2016;17(5):360–7. doi: 10.1111/pedi.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–8. doi: 10.1007/s00125-010-1799-4. [DOI] [PubMed] [Google Scholar]
- 50.Steinthorsdottir V, Thorleifsson G, Sulem P, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46:294–8. doi: 10.1038/ng.2882. [DOI] [PubMed] [Google Scholar]
- 51.Raeder H, Johansson S, Holm PI, et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38:54–62. doi: 10.1038/ng1708. [DOI] [PubMed] [Google Scholar]
- 52.Torsvik J, Johansson BB, Dalva M, et al. Endocytosis of secreted carboxyl ester lipase in a syndrome of diabetes and pancreatic exocrine dysfunction. J Biol Chem. 2014;289:29097–111. doi: 10.1074/jbc.M114.574244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hegele RA, Ur E, Ransom TP, Cao H. A frameshift mutation in peroxisome-proliferator-activated receptor-gamma in familial partial lipodystrophy subtype 3 (FPLD3; MIM 604367) Clin Genet. 2006;70(4):360–2. doi: 10.1111/j.1399-0004.2006.00674.x. [DOI] [PubMed] [Google Scholar]
- 54.Francis GA, Li G, Casey R, et al. Peroxisomal proliferator activated receptor-gamma deficiency in a Canadian kindred with familial partial lipodystrophy type 3 (FPLD3) BMC Med Genet. 2006;7:3. doi: 10.1186/1471-2350-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Agostini M, Schoenmakers E, Mitchell C, et al. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab. 2006;4(4):303–11. doi: 10.1016/j.cmet.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osbak KK, Colclough K, Saint-Martin C, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–26. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- 57.Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4:200–13. doi: 10.1038/ncpendmet0778. [DOI] [PubMed] [Google Scholar]
- 58.Laver TW, Colclough K, Shepherd M, et al. The common p.R114W HNF4A mutation causes a distinct clinical subtype of monogenic diabetes. Diabetes. 2016;65(10):3212–7. doi: 10.2337/db16-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lieber DS, Vafai SB, Horton LC, et al. Atypical case of Wolfram syndrome revealed through targeted exome sequencing in a patient with suspected mitochondrial disease. BMC Med Genet. 2012;13:3. doi: 10.1186/1471-2350-13-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cano A, Rouzier C, Monnot S, et al. Identification of novel mutations in WFS1 and genotype-phenotype correlation in Wolfram syndrome. Am J Med Genet A. 2007;143A(14):1605–12. doi: 10.1002/ajmg.a.31809. [DOI] [PubMed] [Google Scholar]
- 61.McCarthy MI. Painting a new picture of personalised medicine for diabetes. Diabetologia. 2017;60(5):793–9. doi: 10.1007/s00125-017-4210-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536(7614):41–7. doi: 10.1038/nature18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patch AM, Flanagan SE, Boustred C, Hattersley AT, Ellard S. Mutations in the ABCC8 gene encoding the SUR1 subunit of the KATP channel cause transient neonatal diabetes, permanent neonatal diabetes or permanent diabetes diagnosed outside the neonatal period. Diabetes Obes Metab. 2007;9(Suppl 2):28–39. doi: 10.1111/j.1463-1326.2007.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bowman P, Flanagan SE, Edghill EL, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. 2012;55(1):123–7. doi: 10.1007/s00125-011-2319-x. [DOI] [PubMed] [Google Scholar]
- 65.Riveline JP, Rousseau E, Reznik Y, et al. Clinical and metabolic features of adult-onset diabetes caused by ABCC8 mutations. Diabetes Care. 2012;35:248–51. doi: 10.2337/dc11-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–76. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Najmi LA, Aukrust I, Flannick J, et al. Functional investigations of HNF1A identify rare variants as risk factors for type 2 diabetes in the general population. Diabetes. 2017;66(2):335–46. doi: 10.2337/db16-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bonnefond A, Clement N, Fawcett K, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. 2012;44:297–301. doi: 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Majithia AR, Flannick J, Shahinian P, et al. Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc Natl Acad Sci U S A. 2014;111(36):13127–32. doi: 10.1073/pnas.1410428111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mishra R, Chesi A, Cousminer DL, et al. Relative contribution of type 1 and type 2 diabetes loci to the genetic etiology of adult-onset, non-insulin-requiring autoimmune diabetes. BMC Med. 2017;15(1):88. doi: 10.1186/s12916-017-0846-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Merger SR, Leslie RD, Boehm BO. The broad clinical phenotype of type 1 diabetes at presentation. Diabet Med. 2013;30(2):170–8. doi: 10.1111/dme.12048. [DOI] [PubMed] [Google Scholar]
- 72.Turner R, Stratton I, Horton V, et al. UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet (London, England) 1997;350(9087):1288–93. doi: 10.1016/S0140-6736(97)03062-6. [DOI] [PubMed] [Google Scholar]
- 73.Ong YH, Koh WCA, Ng ML, et al. Glutamic acid decarboxylase and islet antigen 2 antibody profiles in people with adult-onset diabetes mellitus: a comparison between mixed ethnic populations in Singapore and Germany. Diabet Med. 2017;34(8):1145–53. doi: 10.1111/dme.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Storm P, Rosengren A, Groop L. A Novel Fine-Tuned Classification of Diabetes with Prognostic Value: Steps towards Precision Medicine [abstract]. Diabetes. 2016;65(suppl 1):A359.
- 75.Thanabalasingham G, Pal A, Selwood M, et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care. 2012;35(6):1206–12. doi: 10.2337/dc11-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–42. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M. A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet. 2003;361(9354):301–7. doi: 10.1016/S0140-6736(03)12325-2. [DOI] [PubMed] [Google Scholar]
- 78.Tarasov AI, Nicolson TJ, Riveline JP, et al. A rare mutation in ABCC8/SUR1 leading to altered ATP-sensitive K+ channel activity and beta-cell glucose sensing is associated with type 2 diabetes in adults. Diabetes. 2008;57(6):1595–604. doi: 10.2337/db07-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013;34(5):669–85. doi: 10.1002/humu.22279. [DOI] [PubMed] [Google Scholar]
- 80.Johansen A, Ek J, Mortensen HB, Pedersen O, Hansen T. Half of clinically defined maturity-onset diabetes of the young patients in Denmark do not have mutations in HNF4A, GCK, and TCF1. J Clin Endocrinol Metab. 2005;90:4607–14. doi: 10.1210/jc.2005-0196. [DOI] [PubMed] [Google Scholar]
- 81.Vaxillaire M, Dechaume A, Busiah K, et al. New ABCC8 mutations in relapsing neonatal diabetes and clinical features. Diabetes. 2007;56(6):1737–41. doi: 10.2337/db06-1540. [DOI] [PubMed] [Google Scholar]
- 82.Lin YW, Akrouh A, Hsu Y, Hughes N, Nichols CG, León DDD. Compound heterozygous mutations in the SUR1 (ABCC 8) subunit of pancreatic K(ATP) channels cause neonatal diabetes by perturbing the coupling between Kir6.2 and SUR1 subunits. Channels (Austin) 2012;6(2):133–8. doi: 10.4161/chan.19980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Wet H, Proks P, Lafond M, et al. A mutation (R826W) in nucleotide-binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetes. EMBO Rep. 2008;9(7):648–54. doi: 10.1038/embor.2008.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of 22 genes associated with monogenic forms of diabetes that were analyzed in this paper. Table S2. Criteria used to select genes for targeted sequencing. Table S3. Summary of samples sequenced in Stages 1, 2, and 3, and the coding variants identified in each stage. Table S4. Clinical data of the cases and controls for type 2 diabetes sequenced in this study. Table S5. List of all protein truncating mutations identified in the 22 monogenic diabetes genes. Table S6. Rare missense mutations in the HNF1A, HNF4A, HNF1B, ABCC8, and KCNJ11 genes predicted to be deleterious by PolyPhen2, SIFT, and MutationTaster. Table S7. Number of individuals with protein truncating variants and previously reported pathogenic missense variants in MODY genes. Table S8. List of exons with low sequence coverage in data from Stage 1 and 2 pools. Figure S1. Minor allele frequency distribution of variants identified from sequencing of pools in Stages 1 and 2. Figure S2. Pooled sequencing design of the study. Figure S3. Comparison of sequence coverage between cases and controls. (PDF 700 kb)
Supplementary Methods: Description of methods for pooled variant calling, gene-level tests for rare coding variants, statistical analyses, comparison of pooled sequence data with population exome data, comparison of pooled allele counts with individual genotypes, and identification of the carriers of rare variants. (PDF 671 kb)
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author on reasonable request.