

Abstract
Background
Chlamydia (C.) gallinacea is a recently identified bacterium that mainly infects domestic chickens. Demonstration of C. gallinacea in human atypical pneumonia suggests its zoonotic potential. Its prevalence in chickens exceeds that of C. psittaci, but genetic and genomic research on C. gallinacea is still at the beginning. In this study, we conducted whole-genome sequencing of C. gallinacea strain JX-1 isolated from an asymptomatic chicken, and comparative genomic analysis between C. gallinacea strains and related chlamydial species.
Results
The genome of C. gallinacea JX-1 was sequenced by single-molecule, real-time technology and is comprised of a 1,059,522-bp circular chromosome with an overall G + C content of 37.93% and sequence similarity of 99.4% to type strain 08-1274/3. In addition, a plasmid designated pJX-1, almost identical to p1274 of the type strain, except for two point mutations, was only found in field strains from chicken, but not in other hosts. In contrast to chlamydial species with notably variable polymorphic membrane protein (pmp) genes and plasticity zone (PZ), these regions were conserved in both C. gallinacea strains. There were 15 predicted pmp genes, but only B, A, E1, H, G1 and G2 were apparently intact in both strains. In comparison to chlamydial species where the PZ may be up to 50 kbp, C. gallinacea strains displayed gene content reduction in the PZ (14 kbp), with strain JX-1 having a premature STOP codon in the cytotoxin (tox) gene, while tox gene is intact in the type strain. In multilocus sequence typing (MLST), 15 C. gallinacea STs were identified among 25 strains based on cognate MLST allelic profiles of the concatenated sequences. The type strain and all Chinese strains belong to two distinct phylogenetic clades. Clade of the Chinese strains separated into 14 genetically distinct lineages, thus revealing considerable genetic diversity of C. gallinacea strains in China.
Conclusions
In this first detailed comparative genomic analysis of C. gallinacea, we have provided evidence for substantial genetic diversity among C. gallinacea strains. How these genetic polymorphisms affect C. gallinacea biology and pathogenicity should be addressed in future studies that focus on phylogenetics and host adaption of this enigmatic bacterial agent.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4343-9) contains supplementary material, which is available to authorized users.
Keywords: Chlamydia gallinacea, Whole-genome sequence, Comparative genomics analysis, MLST, Phylogenetic analysis
Background
The obligate intracellular bacteria in the genus Chlamydia are globally widespread and represent successful pathogens that infect a wide range of animals as well as humans. However, some of them are frequently overlooked as these infections typically remain latent and only rarely lead to overt clinical signs. For a long time, Chlamydia (C.) psittaci, an avian pathogen with well-documented zoonotic potential, was considered the only chlamydial species infecting domestic and wild birds. However, recent reports showed that C. gallinacea and C. avium are two emerging chlamydial agents that can also be involved in avian chlamydiosis [1]. To date, C. avium has been detected in pigeons and psittacine birds, while C. gallinacea has been mainly detected in chickens, ducks, guinea fowl, turkey, backyard poultry and cattle [2, 3]. Interestingly, the high prevalence of C. gallinacea in poultry flocks across Europe and China determined by PCRs surpassed that of C. psittaci [4, 5]. This organism is known to occasionally be in transmission with C. psittaci in the same flock and also can co-infect individuals [6, 7]. Beyond the potential role of this emerging pathogen in animal health, an earlier study of an outbreak of atypical pneumonia in a slaughterhouse, where workers were exposed to C. gallinacea-infected chickens, raised questions over its zoonotic potential as well [8].
Whole-genome sequencing and subsequent comparative genomic analysis has become standard in analysis of the biology, virulence factors, evolution and phylogenetic relationships of chlamydial organisms [9, 10]. While there is plentiful genomic data on the related chlamydial species, the only completely assembled genomic sequence of C. gallinacea currently available is that of the type strain 08-1274/3, which was isolated from a chicken in France [11]. So far, this limited genomic information for C. gallinacea has allowed only little insight into its developmental cycle and potential virulence factors. Likewise, intra-species genetic diversity and phylogenetic relationships have yet to be investigated. Little information available from partial multi-locus sequence analysis (MLSA) of five C. gallinacea strains revealed limited genetic diversity within the species [12]. However, this contrasts with the findings of our own genotyping studies targeting the ompA gene, which encodes the chlamydial major outer membrane protein (MOMP), where we found 13 diverse ompA genotypes of C. gallinacea in Chinese poultry [4].
In the present study, we describe the second whole-genome sequence (WGS) of C. gallinacea, which originates from the Chinese chicken isolate JX-1, and report findings from comparative genomic analysis between C. gallinacea strains and closely related species in the genus Chlamydia. To understand the epidemiology and genetic diversity of C. gallinacea infections in chickens, we conducted previously described Chlamydiales multi-locus sequence typing (MLST) [13] on 23 C. gallinacea-positive samples from nine farms located in nine provinces across China. This enabled us to provide a detailed description of genomic features and assess naturally occurring genetic diversity of this pathogen.
Methods
Description of C. gallinacea isolate JX-1 and clinical samples used in this study
C. gallinacea JX-1 strain, used for genome sequencing and plasmid characterization, was isolated from a cloacal swab of an asymptomatic chicken in the Jiangxi province of China [4]. In the present study, we also used DNA from 45 previously tested C. gallinacea-positive clinical swabs taken from oral and cloacal anatomical sites of chickens, pigeons, ducks and geese from various farms across China [4] (Additional file 1: Table S1). Ethics approval was not needed as the DNA used in this study was extracted from the chickens in a previous study [4].
Whole-genome sequencing and assembly
C. gallinacea strain JX-1 was propagated via yolk sac inoculation on a 7-day-old chicken embryo followed by yolk membrane harvesting, in order to perform genomic DNA extraction using the QIAgen® DNA Mini Kit (Qiagen, Valencia, CA, USA). The obtained total DNA was subjected to quality control, by running 1 μl of DNA on an agarose gel and quantification by Qubit. The genome of C. gallinacea JX-1 was sequenced by Single-Molecule, Real-Time (SMRT) technology at the Beijing Novogene Bioinformatics Technology Co., Ltd. (China). SMRT Analysis 2.3.0 was used to filter low-quality reads, following assembly into a single gap-free contig using filtered reads. Low-quality reads were filtered by the SMRT Analysis v2.3.0 software, and then the genome was subjected to de novo assembly by the SMRT portal software according to the valid sequencing data. The draft C. gallinacea JX-1 genome was automatically annotated using the NCBI Prokaryotic Genomes Annotation Pipeline (NCBI_PGAP), and the genome sequence was deposited in the NCBI database under GenBank accession number CP019792.
C. gallinacea plasmid screening
In order to assess whether the C. gallinacea JX-1 carries a plasmid, 16 paired primers were designed based on the plasmid sequence of type strain 08-1274/3 (Additional file 2: Table S2) to amplify the complete plasmid sequence. PCR conditions and reaction mixes are described in the section below. Each amplified fragment was purified using the QIAquick PCR Purification Kit (Qiagen), and sent for Sanger sequencing to GenScript, Jiangsu, Nanjing, China. The chromatograms of the sequenced plasmid fragments were mapped against the p1274 sequence, and the complete C. gallinacea JX-1 plasmid (pJX-1) was extracted and annotated using RAST [14] and deposited in GenBank under accession number CP019793. We have also screened the C. gallinacea-positive clinical samples (Additional file 1: Table S1) for plasmid presence by amplifying a 661 bp fragment of the plasmid’s CDS1 (integrase) using primer pair plaF1 and plaR1 of the plasmid (Additional file 2: Table S2).
Macroscopic comparative genomic and phylogenetic analyses
The genome of C. gallinacea JX-1was compared in-depth to the reference genome of type strain 08-1274/3, as well as to publicly available genomes of other related chlamydial species, i.e. C. avium 10 DC88 (NZ_CP006571.1), C. pecorum E58 (CP002608), C. psittaci 6 BC (CP002586.1) and C. abortus S26/3 (CR848038.1). Pairwise genomic comparison was performed using the Artemis Comparison Tool (ACT) [15], and Geneious 9 [16] using alignments produced with progressive Mauve [17] and MAFFT [18]. The genomic regions of interest and/or loci were extracted from the analyzed genomes and aligned, in order to be used for further nucleotide and/or translated protein sequence analyses performed using DNASp 5.0 [19], as well as BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). In addition, we have also used open source TMHMM Server v. 2.0 (available from http://www.cbs.dtu.dk/services/TMHMM/) which predicts transmembrane helices in proteins, to predict chlamydial inclusion membrane proteins with a presence of bilobed hydrophobic domains using translated C. gallinacea hypothetical gene sequences. A mid-point rooted phylogenetic tree constructed from the alignment of all identified C. gallinacea pmp genes from both strains used in this study was generated with PhyML with 1000 bootstrap repetitions [20], as implemented in Geneious 9. Figures of the whole-genome comparison and specific genomic regions using blastn and tblastx algorithms were generated with Brig [21] and EasyFig [22], while the graphical representation of the C. gallinacea JX-1 genome and its elements was generated with the DnaPlotter [23].
The phylogenetic relationship of the two C. gallinacea strains was examined through comparison to each other and related chlamydial species using an 11.2 kbp alignment of concatenated sequences. The concatenated sequence consisted of 12 partial and full-length conserved chlamydial phylogenetic markers that were concatenated in the following order: six MLST house-keeping gene fragments (gatA, hflX, gidA, enoA, hemN, fumC), and full-length major outer membrane protein gene ompA, DNA-directed RNA polymerase subunit beta gene rpoB, 50S ribosomal protein L3 gene rplC, 50S ribosomal protein L4 gene rplD, DNA recombination/repair protein gene recA, and tyrosine—tRNA ligase gene tyrS. In addition to the two C. gallinacea genomes used in this study, each of these sequences were extracted from the genomes of the following related species: C. avium 10 DC88 (NZ_CP006571.1), C. caviae GPIC (NC_003361.3), C. felis F/C-56 (NC_007899.1), Candidatus C. ibidis 10-1398/6 (NZ_APJW00000000.1), C. pneumoniae LpColN (NC_017285.1), C. pecorum E58 (CP002608), C. psittaci 6 BC (CP002586.1), C. trachomatis AHAR-13 (CP000051.1), C. muridarum Nigg (NC_002620.2), C. suis MD56 (NZ_KI538658.1) and C. abortus S26/3 (CR848038.1). A mid-point rooted maximum-likelihood phylogenetic tree was constructed using PhyML with 1000 bootstrap repetitions, as integrated in Geneious 9.
MLST of C. gallinacea
In this study, we performed a complete MLST, based on a previously published scheme for chlamydiae [13]. The primers used to amplify seven C. gallinacea-specific house-keeping (HK) genes were designed in this study based on the sequence of C. gallinacea type strain 08-1274/3 and other related chlamydial species (Additional file 3: Table S3).
PCR amplification of the seven HK genes (as well as plasmid fragments) was performed in a LightCycler 480-II real-time PCR platform using a high-stringency 18-cycle step-down temperature protocol (Additional file 4: Table S4) as described [3, 4]. C. gallinacea JX-1 DNA was used as a positive control, while ultrapure H2O was used as a negative control in each assay. The PCR products were electrophoresed through 2% agarose gel and purified using the QIAquick PCR Purification Kit (Qiagen) for automated DNA sequencing (GenScript, Jiangsu, Nanjing, China).
After optimization and development, the C. gallinacea MLST was applied to sequences of a total of 45 C. gallinacea-positive oral and cloacal swabs from chickens (n = 20), hens (n = 4), ducks (n = 12), pigeons (n = 6), and geese (n = 3), collected in different provinces of China (Additional file 1: Table S1).
C. gallinacea MLST and phylogenetic analysis were performed using DNASp 5.0 and Geneious 9. Briefly, forward and reverse chromatograms for each sequenced HK gene fragment were aligned and trimmed, and the fragment sequence for that allele was obtained. Allele and sequence type (ST) assignment for 25 C. gallinacea strains were determined and deposited at http://pubmlst.org/chlamydiales/ [24] (Additional file 5: Table S5).
Sequences of individual genes and concatenated gene sets were aligned using ClustalX. DnaSP 5.0 was used to analyze sequence polymorphisms by determining the number of synonymous (ds) and non-synonymous (dn) substitutions per site, Jukes-Cantor corrected, the number of polymorphic sites and haplotypes (Additional file 5: Table S5).
Best-fit models of nucleotide substitution for our data set were estimated by considering eleven substitution (nst = 11) models using jModelTest v.2.2. [25]. A Bayesian phylogenetic tree using concatenated MLST sequences of 25 C. gallinacea strains was constructed with MrBayes [26] with the HKY + I model, as implemented in Geneious 9. Run parameters included four Markov Chain Monte Carlo (MCMC) chains with a million generations, sampled every 1000 generations and with the first 10,000 trees were discarded as burn-in. The C. avium MLST sequence was used as an outgroup.
Results
Description of the C. gallinacea JX-1 genome
Using SMRT sequencing technology, we have completely sequenced and assembled the genome of C. gallinacea strain JX-1. The whole genome is sized 1,059,522 bp with an overall GC content of 37.93%, encompassing 957 predicted CDSs that account for 91.21% of the genome and a 7.49 kbp plasmid (Table 1, Fig. 1). Alignment to the reference genome of C. gallinacea type strain 08-1274/3 confirmed 100% chromosome coverage for our newly described JX-1 genome.
Table 1.
Description of Chlamydia gallinacea JX-1 genome
| Strain | Chlamydia gallinacea JX-1 |
|---|---|
| Clinical manifestation and anatomical site sample type | Asymptomatic/Cloacal swab |
| Host and country of origin | Chicken, China |
| Total No. of filtered reads | 66,564 |
| Average read length | 11,688 bp |
| Average read depth | 100 × |
| Genome size (bp) and % GC | 1,059,522 bp / 37.93% GC |
| No. of predicted CDSs | 957 |
| Chlamydial plasmid and size | Present: pJX-1 (7.49 Kbp) |
| nc RNAs | 39 tRNAs, two 5S rRNA, and single copy of 16S rRNA and 23S rRNA |
| Pyrimidine genes | Present (pyrG, pyrE, pyrH, ndk) |
| Biotin operon | Present (bioADFB, bioY) |
| Tryptophan operon | Absent |
| % DNA sequence identity C. gallinacea 08-1274/3 | 99.4% |
| Accession number | CP019792 |
Fig. 1.
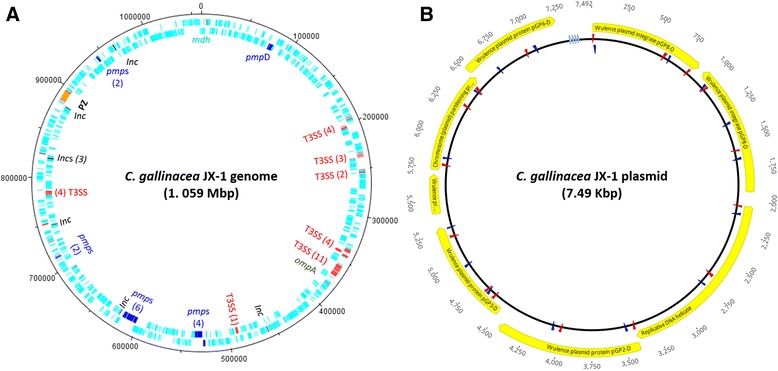
Circular representation of the C. gallinacea JX-1 genome and its cognate plasmid pJX-1. a JX-1 genome: First ring denotes CDSs (in light blue) in forward direction, while the second ring denotes CDSs in reverse direction. Genomic location of T3SS, pmp, Inc., and ompA genes, as well as the PZ are also outlined on the JX-1 genome plot. The genome start is denoted by the malate dehydrogenase gene (mdh) at 0 position. Image was generated with the DNAplotter. b Graphical representation of the pCgall JX-1 and its CDSs, including plasmid primer locations. The light blue arrows denote the 22 bp tandem repeat units
With the availability of the JX-1 genome, we were able to evaluate genome similarities and differences between C. gallinacea strains, but also to related chlamydial species (Fig. 2a). The two C. gallinacea genomes are virtually identical to each other (99.4% identity) and share the highest similarity with the C. avium genome (~78.9% sequence similarity) based on whole-genome MAFFT alignment. Comparison to the genomes of the other chlamydial species revealed more pronounced differences, mainly in the PZ and Pmp clusters, however with a remarkable overall chlamydial genome synteny (Fig. 2a). Phylogenetically, although closely related, the two C. gallinacea strains, grouped in a larger clade with their closest relative, C. avium (Fig. 2b).
Fig. 2.
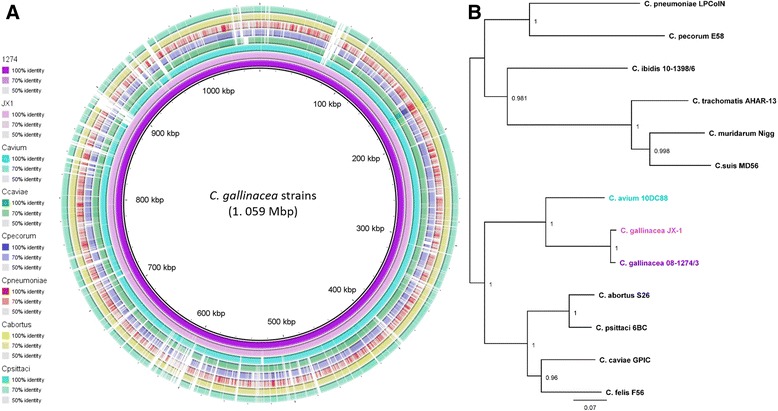
C. gallinacea genome comparisons and phylogenetic analyses. a Whole genome BLAST comparisons between the C. gallinacea and the related species based on translated nucleotide identity (tblastx algorithm). b The mid-point rooted maximum-likelihood phylogenetic analyses of the 12 conserved phylogenetic markers, resulting in 11.2 Kbp concatenated sequence alignment of the 13 chlamydial species. Bootstrap values are displayed on the tree nodes
The C. gallinacea genomes of field strain JX-1 and type strain 08-1274/3 exhibited high sequence similarity (99.4% identity) with ~6250 SNP differences between the two strains, while maintaining synteny and gene order. Both C. gallinacea genomes also contained the hallmark genomic features of chlamydiae, such as the highly conserved Type III Secretion System (T3SS), clusters of pmp genes, inclusion protein genes (incs), and a PZ (Fig. 1a) [27].
With SNPs evenly distributed along the chromosome, the major genetic differences between strains JX-1 and 08-1274/3 are presented in Table 2. In comparative genomic analysis, we have identified genes with high density of SNPs (with at least 5% total length sequence dissimilarity between the two strains), Interestingly, besides the previously recognized highly variable ompA gene, most of the remaining genes with high numbers of SNPs were annotated as metabolic genes (Table 2). Highly variable CDS GM000264, annotated as a hypothetical protein, appears to harbor a C. gallinacea-specific sequence based on BLAST searches, with only 20% similarity to a conserved hypothetical protein of C. psittaci. The putative product, however, does not seem to have a conserved domain. None of these identified genes appears to be under positive selection with the majority of accumulating SNPs being synonymous (Table 2). However, due to only two strains analyzed, at present we cannot accurately assess the selection on these genes.
Table 2.
Sequence analysis of C. gallinacea polymorphic genes
| Locus tag and length (bp) in C. gallinacea JX-1 | Predicted product | % DNA sequence similarity | Total No. of polymorphisms | No. of non-synonymous substitutions | No. of synonymous substitutions |
|---|---|---|---|---|---|
| GM000925 (1581 bp) | Lysine-tRNA ligase | 95.1% | 77 SNPs | 11 | 65 |
| GM000895 (873 bp) | Serine/threonine protein phosphatase | 93.2% | 59 SNPs | 12 | 53 |
| GM000890 (2631 bp) | Alanyl-tRNA synthetase | 92.5% | 198 SNPs | 36 | 162 |
| GM000889 (3252 bp) | Transcription-repair coupling factor | 94.2% | 190 SNPs | 37 | 153 |
| GM000888 (924 bp) | Uroporphyrinogen decarboxylase (hemE) | 92.2% | 72 SNPs | 9 | 63 |
| GM000887 (1374 bp) | Coproporphyrinogen oxidase (hemN) | 93.0% | 96 SNPs | 31 | 65 |
| GM000705 (1206 bp) | Major outer membrane protein, porin | 86.7% | 161 (104 SNPs and 57 indel) | 38 | 60 |
| GM000539 (1356 bp) | Sodium/alanine symporter family protein | 95.2% | 65 SNPs | 12 | 53 |
| GM000349 (1236 bp) | Cell wall hydrolase | 91.7% | 102 (87 SNPs and 15 indel) | 41 | 45 |
| GM000348 (1179 bp) | Phage T7 tail fiber family protein | 80.3% | 232 (133 SNPs and 99 indel) | 59 | 74 |
| GM000289 (2409 bp) | Glycogen phosphorylase | 94.1% | 142 SNPs | 20 | 122 |
| GM000288 (1290 bp) | Dihydrolipoamide acetyltransferase component | 91.1% | 115 SNPs | 40 | 75 |
| GM000264a (1765 bp) | Hypothetical protein | 88.3% | 202 (175 SNPs and 27 indel) | 67 | 108 |
aC. gallinacea-specific sequence based on BLAST search
The C. gallinacea strains possess a chlamydial T3SS comprised of a total of 36 genes encoding T3SS structural components, chaperones and secreted effectors (Additional file 6: Table S6). T3SS genes of the two C. gallinacea strains were highly conserved with 98–100% sequence similarity, in stark contrast to the previously described genetic diversity in these genes in related chlamydial species such as C. psittaci [28], C. pecorum [29] and human C. trachomatis [30].
We have also assessed the predicted Inclusion membrane proteins (Incs) for C. gallinacea, as during early infection the inclusion membrane modified by the insertion of a number of type III secreted effector proteins, and the inclusion proteins play a significant role [31]. Using open source prediction software TMHMM Server v. 2.0 using a cut-off of more than 40 amino acids in the bi-lobed hydrophobic domain, we have predicted a total of 29 putative Inc’s with two transmembrane domains, besides the two annotated IncA, and IncB and IncC; five with four transmembrane domains; and three with six transmembrane domains (Additional file 7: Table S7; Additional file 8: Figure S1). This number of predicted Incs is comparable to that observed in the related chlamydial species [31].
Variation in the pmp genes: gene truncations rather than SNP accumulation
In the C. gallinacea genomes, the pmp genes were found to form two major clusters. In our analysis, we predicted a total of 15 pmp genes, however only B, A, E1, H, G1 and G2 appear to be intact in both C. gallinacea strains (Fig. 3). The pmpD gene of strain JX-1 was found to have a premature STOP codon (Fig. 3a). The remaining four pmp genes also had a premature STOP codon in both strains, which was predicted to truncate the encoded proteins before their respective C-terminal autotransporter domains as based on BLAST and CDD (Conserved domains) BLAST analysis. BLAST and phylogenetic analyses confirmed that the majority of the predicted pmp genes in the C. gallinacea genomes are paralogs of pmpG gene lineage (Fig. 3b).
Fig. 3.
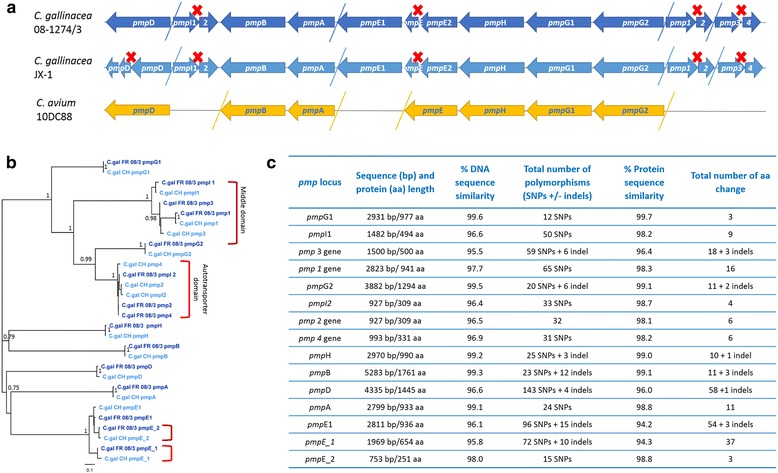
Sequence and phylogenetic analyses of C. gallinacea pmps. a Schematic representation of pmp gene order in the C. gallinacea and C. avium genomes. Red X denotes STOP codon. b A mid-point rooted maximum likelihood phylogenetic tree of the C. gallinacea pmp genes. Autotransporter and middle pmp gene domains are denoted with brackets. c Sequence analyses of the C. gallinacea pmp genes and Pmp protein
In terms of the number of intact pmp genes, C. gallinacea was closest to C. avium (n = 7), while the related chlamydial species harbor between 14 (C. abortus) and 22 pmp genes (C. psittaci). In contrast to the related chlamydial species where these genes are major contributors of SNPs [29, 32], pmp genes of the two analysed C. gallinacea strains were conserved, with overall sequence similarity ranging from 95.5 to 99.6%. Comparable levels of similarity were observed when comparing the pmp amino acid sequences (Fig. 3c).
The C. gallinacea plasticity zone (PZ)
The PZ, notoriously known for harboring key virulence genes of chlamydiae, has been considered as the region of most extensive genetic differences between chlamydial genomes [27]. In comparison to the related chlamydial species where the PZ may be up to 50 kbp in size (e.g. in C. muridarum and C. trachomatis), C. gallinacea displayed gene content reduction in this region (14 kbp), similar to C. abortus (12 kbp). In our analysis, C. avium appeared to have the most reduced PZ (4.6 kbp) (Fig. 4). The PZ of two C. gallinacea strains included three hypothetical proteins, two acetyl-co-carboxylases and a single copy of the chlamydial cytotoxin (tox) gene, but remained highly conserved with 99.2% sequence similarity. Interestingly, C. gallinacea JX-1 had a premature STOP codon in the cytotoxin (tox) gene, while tox appeared to be intact in the type strain 08-1274/3 (Fig. 4).
Fig. 4.
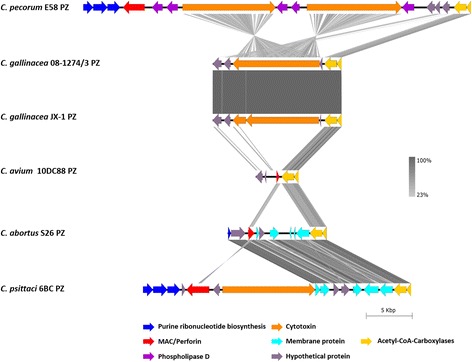
Graphical representation of the gene content in chlamydial PZs, including the two C. gallinacea strains analyzed in this study. Colored arrows in the legend denote PZ genes according to their function, while the grey shading scale denotes % sequence similarity. Image was generated with Easyfig using tblastx comparison
Molecular characterization of plasmid pJX-1 and its distribution in C. gallinacea strains
Newly characterised plasmid pJX-1 of C. gallinacea was 7.49 kbp in length, sharing an identical annotation with eight CDSs and four 22 bp tandem repeats with plasmid p1274 of the type strain 08-1274/3 (Fig. 1b). Briefly, in pJX-1, CDSs 1 (pGP8), 2 (pGP8), 3 (pGP1), and 7 (parA) were denoted as putative integrase, helicase, and a partitioning plasmid proteins, respectively, while the CDSs 4 (pGP2), 5 (pGP3), 6 (pGP4), and 8 (pGP6) were denoted as putative chlamydia-specific plasmid virulence proteins, as previously described for related chlamydial plasmids [33, 34]. The sequence of pJX-1 was 99.9% identical to p1274, except for two point mutations at positions 6573 (C changed to T) and 7170 (C changed to A) in CDS 8 (pGP6). Further PCR examination of C. gallinacea-positive clinical samples revealed that pJX-1 was detected in all the 24 oral/cloacal swab samples from chickens, while clinical samples from other avian hosts remained negative (data not shown).
MLST of C. gallinacea reveals genetic diversity among strains in chickens
In order to obtain a snapshot of the genetic diversity of this emerging pathogen, we have applied our newly developed C. gallinacea-specific MLST to a range of C. gallinacea-positive clinical samples collected from birds (Additional file 1: Table S1). Unfortunately, due to insufficient amounts of C. gallinacea DNA, MLST was only successful in 23 cloacal and oral clinical samples from 3 hens and 20 chickens.
Using a set of 25 sequences, including the MLST sequences obtained from the two sequenced C. gallinacea genomes, sequence analysis of individual as well as concatenated gene fragments confirmed the evolutionary conservation of the HK genes, as all alleles were under purifying selection with dn/ds ratios < 1(Table 3). The highest number of mutations was noted in hemN (41 SNPs) and enoA (25 SNPs), resulting mainly in synonymous substitution, whereas gatA had none. enoA and oppA3 were the most diverse loci as they both occurred in six allelic variants (Table 3). The concatenated sequences (further used for phylogenetic analysis) and its cognate MLST allelic profiles resulted in a total of 15 haplotypes or C. gallinacea STs (Table 3).
Table 3.
Sequence analysis of C. gallinacea MLST alleles for samples denoted in bold in Additional file 1: Table S1
| Allele | Total number of mutations (Δnt) | No. of non-synonymous substitutions | No. of synonymous substitutions | N alleles |
|---|---|---|---|---|
| gatA | 0 | 0 | 0 | 1 |
| oppA_3 | 5 | 2 | 3 | 6 |
| hflX | 3 | 0 | 3 | 4 |
| gidA | 2 | 1 | 1 | 2 |
| enoA | 25 | 1 | 24 | 6 |
| hemN | 41 | 13 | 28 | 3 |
| fumC | 7 | 1 | 6 | 5 |
| Concatenated (3098 bp) | 83 | 18 | 65 | 15 |
Δnt: No. of polymorphic sites; *ds and dn: the average number of synonymous substitutions per synonymous site and non- synonymous substitutions per non-synonymous site, respectively (Jukes – Cantor corrected); N Alleles: No. of unique sequences according to the gene
To examine the genetic relationships between the C. gallinacea strains typed using our MLST method, a mid-point rooted Bayesian phylogenetic tree was constructed using the concatenated HK gene sequences amplified from the 25 strains included. Using the concatenated MLST sequence of the closest relative C. avium as an out-group, the phylogenetic tree separated the C. gallinacea type strain 08-1274/3 from all Chinese C. gallinacea strains into two distinct clades (Fig. 5a). Although in the same well-supported larger clade, C. gallinacea strains from China could be further sub-divided into 14 genetically distinct lineages. The phylogenetic tree constructed from the present C. gallinacea STs also revealed that: a) The same strain can infect two different anatomical sites in a single host (e.g. A/Ch_40: oral and cloaca); b) the same strain can be found in different animals from the same area (e.g. J/Hen 31, 12 and 4 strains) or geographically distinct areas (e.g. JX-1, J/ChA2432, J/ChA2360 and A/Ch29-1 strains); and c) closely related strains can also be found in geographically distinct areas (e.g. Ha/ChA3274 and Gx/ChA612) (Fig. 5).
Fig. 5.
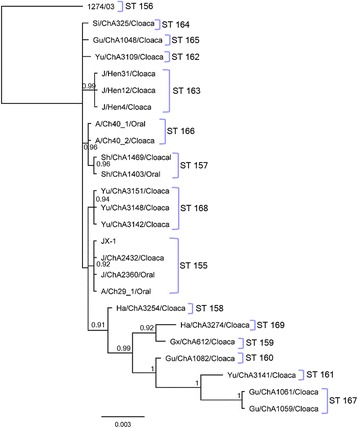
Phylogenetic and cluster analyses of C. gallinacea STs. A: Bayesian phylogenetic analysis of the concatenated sequences of seven MLST fragments of 25 C. gallinacea genotypes. Posterior probabilities > 0.75 are displayed on tree nodes, and STs in a clade are indicated by brackets
Discussion
In this study, we present the first detailed analysis of C. gallinacea genomes. The two C. gallinacea genomes studied are compact, syntenic and highly conserved between them, while sharing some of the classical genomic features of Chlamydia spp., such as the highly conserved T3SS, pmp gene clusters, and a potentially virulence-associated plasmid. The two strains analyzed in this study, JX-1 from China and type strain 08-1274/3 from France form separate phylogenetic lineages within a clade with its closest relative C. avium. Using information derived from these genomes and previously described Chlamydiales MLST scheme [13], we also adapted a complete C. gallinacea MLST scheme and applied it to clinical strains from chickens of various Chinese provinces. The latter analysis revealed that this organism is genetically diverse, indicating the potential for a complex epidemiology similar to other chlamydial species found in animals.
The previously described typical synteny and gene order for chlamydial organisms [9] was also observed in the C. gallinacea genomes. The two strains differed in approximately 6250 SNPs, mostly synonymous, which were distributed evenly along the chromosome. The extent of genetic variation in the ompA locus was not surprising, as a previous study on C. gallinacea ompA typing identified at least 13 genotypes [4]. The ompA variation at the variable domains in this species is also consistent with ompA diversity seen in other chlamydial pathogens [35–37].
As outlined in Table 2, the majority of SNPs accumulated in genes associated with metabolism, most notably in hemE and hemN genes that are involved in heme metabolism [38]. In prokaryotes, heme is an integral part of proteins involved in multiple electron transport chains for respiration, and a cofactor of many enzymes including catalases, peroxidases, and P(450) class cytochromes [39]. Further analysis including genome sequences of more strains from different hosts and regions will be necessary to elucidate possible functional consequences of the present observation. Similarly, the present dataset cannot provide an explanation why gene GM000264, which is coding for a hypothetical protein of unknown function, was found to accumulate SNPs and have the highest dn/ds ratio (0.526) among all analyzed genes.
In contrast to the high number of SNPs observed in the metabolic genes outlined in Table 2, the T3SS and pmp genes were unexpectedly conserved between the two C. gallinacea strains with less than 1% sequence dissimilarity. Such congruence and conservation in these genomic regions is in stark contrast to the high genetic variation (up to 15%) in these genes of other chlamydial species [28, 29]. The chlamydial T3SS, a system of structural, chaperone and secreted effector proteins, is considered as “virulence machinery” with a function to deliver effector proteins in order to subvert host cellular processes [40, 41]. As such, genetic variation and polymorphisms in these genes are probably associated with differences in virulence and host and/or tissue tropism, as previously observed for C. pecorum, C. trachomatis, C. psittaci and other chlamydial species [28, 42, 43]. Whether the sequence conservation in T3SS genes is present throughout the C. gallinacea taxon remains to be investigated, as well as their role in virulence of this organism. Equally, we further need to investigate the role in infection of the 37 putative C. gallinacea Incs identified in this study, and how genetically diverse they will be throughout the taxon.
Highly polymorphic chlamydial pmp gene families account for the majority of chromosomal SNPs in other related species. Generally speaking, the pmp loci constitute almost 4% of the chlamydial genome, thus suggesting an important biological function due to their adhesive as well as antigenic properties [32]. In this study, the pmp genes of C. gallinacea somewhat surprisingly displayed sequence conservation, although we observed non-synonymous changes and indels between the two strains. Proteins PmpB, A, E1, H, G1 and G2 appear to be intact in both C. gallinacea strains, while strain JX-1 harbored a premature STOP codon in pmpD, which will require further investigations to assess whether the protein’s function has been impaired. The remaining pmp genes, although truncated, appear to be PmpG paralogs based on our blast and phylogenetic analyses. Based on these (early) observations, our current hypothesis is that pmpG expansion may not be necessary for virulence or biology of C. gallinacea. Studies on C. pneumoniae, where Pmps (including at least 13 pmpG subtypes) represent major proteins in the outer membrane, showed that all pmp genes are transcribed and expressed during the infectious cycle [32, 44]. In C. psittaci, several pmpG genes that may be related to host tropism and virulence were identified. It is possible that the pmpG group plays a major role in host range, tissue tropism and virulence for different C. psittaci strains due to their high diversity and rapid evolution [45, 46]. Considering the importance of pmps in chlamydial genomes, the observed non-synonymous changes and indels between the two analyzed strains render future investigations on pmp diversity within the species of C. gallinacea highly pertinent.
The PZs of the two C. gallinacea strains proved compact and highly conserved, with 99.2% sequence similarity, with Mac/Perforin and Phospholipase D genes missing, but harboring the full-length chlamydial cytotoxin (tox) gene [47]. C. gallinacea JX-1 had a premature STOP codon in the N-terminal regions of the tox gene, a region that contain a catalytic glycosyltransferase domain, while tox was intact in C. gallinacea type strain 08-1274/3. However, whether this has an impact on the function of the tox gene remains to be elucidated. Chlamydial tox genes are considered important virulence factors and were associated with acute infection and disease [48]. They appear to be a species and niche-specific feature due to their full-length presence in only five related species, including C. psittaci, C. felis, C. caviae with a single gene copy, and C. pecorum and C. muridarum with two and three copies, respectively [9, 27]. In contrast, tox is partially truncated in C. trachomatis, while C. avium, C. abortus and C. pneumoniae do not have a tox gene. It will be interesting to see how the presence or absence of tox gene(s) in chlamydial pathogens can be correlated with virulence properties in the future.
Both C. gallinacea strains were found to carry the highly conserved plasmid. In the present study, the plasmid was only detected in 24 C. gallinacea-positive samples from chickens out of total 45 samples tested from different avian. Its absence in strains infecting other hosts could be due to a) the plasmid being host specific; and b) more likely, failure to detect it because of low DNA concentration and rapid degradation, considering that chlamydial plasmids are present in low copy numbers (1–10 per chromosome) [49]. Another possibility leading to failure of plasmid detection in clinical samples might be associated with high temperature during DNA extraction. In order to increase the efficiency of DNA extraction from clinical samples, we usually give high temperature (72 °C) and a long time for proteinase K incubation and for the multiple elution steps. This approach increases sensitivity for PCR-based clinical diagnostics, but may induce DNA breakage which probably result in failure of detecting the 7.49 kbp plasmid. The presence of a plasmid is a frequent feature of Chlamydia spp. genomes, but naturally occurring plasmid-less strains can also be found [29]. Species like C. abortus and (human) C. pneumoniae do not carry plasmids at all [50]. Both the role and distribution of the plasmid in C. gallinacea strains warrant further investigations.
Previous data from ompA genotyping indicated considerable genetic diversity of C. gallinacea field strains [4]. The present MLST confirmed this by identifying 15 novel STs among 25 strains.
An analysis of the C. gallinacea MLST data allows us to make some speculation on the epidemiology of this pathogen. Identification of the same sequence type in cloacal and oral sites (e.g. A/Ch_40: oral and cloaca, both ST 166) indicates that at least one possible transmission route could be fecal-oral (cloacal). The observations above also indicated transmission among different avian hosts in the same area (e.g. J/Hen 31, 12 and 4 strains, all ST 163) or geographically distinct areas (e.g. JX-1, J/ChA2432, J/ChA2360 and A/Ch29-1 strains, all ST 155). Moreover, there is probably transmission across different hosts, as C. gallinacea strains have been frequently isolated from birds and were also found in livestock that have been in contact [2].
While we could not apply MLST on C. gallinacea strains from ducks, geese and pigeons, nor to European and livestock strains, we do anticipate that future typing studies will shed more light on the complex epidemiology and genetic diversity of this chlamydial agent.
Conclusions
Awareness is growing that C. gallinacea infections in avian hosts are globally highly prevalent. In the present study, we have provided new evidence that this pathogen is genetically diverse, even though it is still open how these genetic differences among strains infecting chickens and other hosts translate into its biology and pathogenicity. Future studies should include analysis of strains from a variety of avian and other livestock hosts to enable us to understand the evolution and host adaption of this enigmatic pathogen.
Additional files
C. gallinacea-positive samples used for MLST and plasmid detection. (DOCX 13 kb)
Primers used for generating C. gallinacea plasmid fragments in this study. (DOCX 14 kb)
Primers used for MLST of C. gallinacea in this study. (DOCX 14 kb)
PCR conditions for amplifying seven HK genes in this study. (DOCX 12 kb)
Original data for MLST analysis of the C. gallinacea-positive samples. (XLSX 10 kb)
Original data for T3SS of C. gallinacea, including the locus-tags, and their annotation/putative function, length and direction. (XLS 36 kb)
List of the predicted transmembrane helices in the analyzed putative C. gallinacea inclusion (Inc) proteins from this study. (XLSX 10 kb)
Graphical representation of the predicted transmembrane helices in the analyzed putative C. gallinacea inclusion (Inc) proteins from this study. (DOCX 423 kb)
Acknowledgements
This publication made use of the PubMLST website (http://pubmlst.org/) developed by Keith Jolley (24) and hosted at the University of Oxford. The development of this website was funded by the Wellcome Trust.
Funding
This project is funded by the National Key R & D Program of China (2016YFD0500804), A Key Project of Anhui Province Education Department (No: KJ2016A822) and the Program of Anhui Science and Technology University (No: ZRC2016497).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files. The data from this study is also available from Treebase: http://purl.org/phylo/treebase/phylows/study/TB2:S21854.
Abbreviations
- MLST
Multilocus sequence typing
- pmp
Polymorphic membrane protein
- PZ
Plasticity zone
- tox
Cytotoxin
Authors’ contributions
CW, WG, MJ, KS, AP and BK designed the study and interpreted the data. WG, JL, JG, and JY performed the DNA extraction, MLST and DNA sequencing. WG, MJ and YP analyzed the MLST data and drew the figure. CW, WG, MJ, KS, AP, BK and YP were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Ethics approval was not needed as the DNA used in this study was extracted from the chickens in a previous study.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4343-9) contains supplementary material, which is available to authorized users.
Contributor Information
Weina Guo, Email: weina0925@126.com.
Martina Jelocnik, Email: martina.jelocnik@research.usc.edu.au.
Jing Li, Email: lijing900401@163.com.
Konrad Sachse, Email: konrad.sachse@gmx.net.
Adam Polkinghorne, Email: apolking@usc.edu.au.
Yvonne Pannekoek, Email: y.pannekoek@amc.uva.nl.
Bernhard Kaltenboeck, Email: kaltebe@auburn.edu.
Jiansen Gong, Email: jjsensen@163.com.
Jinfeng You, Email: chijichi@qq.com.
Chengming Wang, Email: wangche@auburn.edu.
References
- 1.Sachse K, Laroucau K. Avian chlamydiosis: two more bacterial players discovered. Vet J. 2014;200(3):347–348. doi: 10.1016/j.tvjl.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Guo W, Kaltenboeck B, Sachse K, Yang Y, Lu G, et al. Chlamydia pecorum is the endemic intestinal species in cattle while C. gallinacea, C. psittaci and C. pneumoniae associate with sporadic systemic infection. Vet Microbiol. 2016;193:93–99. doi: 10.1016/j.vetmic.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Luther M, Macklin K, Pugh D, Li J, Zhang J, et al. Chlamydia gallinacea: a widespread emerging Chlamydia agent with zoonotic potential in backyard poultry. Epidemiol Infect. 2017;145(13):2701–3. [DOI] [PMC free article] [PubMed]
- 4.Guo W, Li J, Kaltenboeck B, Gong J, Fan W, Wang C. Chlamydia gallinacea, not C. psittaci, is the endemic chlamydial species in chicken (Gallus gallus) Sci Rep. 2016;6:19638. doi: 10.1038/srep19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zocevic A, Vorimore F, Marhold C, Horvatek D, Wang D, Slavec B, et al. Molecular characterization of atypical Chlamydia and evidence of their dissemination in different European and Asian chicken flocks by specific real-time PCR. Environ Microbiol. 2012;14(8):2212–2222. doi: 10.1111/j.1462-2920.2012.02800.x. [DOI] [PubMed] [Google Scholar]
- 6.Krautwald-Junghanns ME, Stolze J, Schmidt V, Bohme J, Sachse K, Cramer K. Efficacy of doxycycline for treatment of chlamydiosis in flocks of racing and fancy pigeons. Tierarztl Prax Ausg K Kleintiere Heimtiere. 2013;41(6):392–398. [PubMed] [Google Scholar]
- 7.Laroucau K, Aaziz R, Meurice L, Servas V, Chossat I, Royer H, et al. Outbreak of psittacosis in a group of women exposed to Chlamydia psittaci-infected chickens. Euro Surveill. 2015;20(24) [DOI] [PubMed]
- 8.Laroucau K, Vorimore F, Aaziz R, Berndt A, Schubert E, Sachse K. Isolation of a new chlamydial agent from infected domestic poultry coincided with cases of atypical pneumonia among slaughterhouse workers in France. Infect Genet Evol. 2009;9(6):1240–1247. doi: 10.1016/j.meegid.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Bachmann NL, Polkinghorne A, Timms P. Chlamydia genomics: providing novel insights into chlamydial biology. Trends Microbiol. 2014;22(8):464–472. doi: 10.1016/j.tim.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 10.Taylor-Brown A, Vaughan L, Greub G, Timms P, Polkinghorne A. Twenty years of research into Chlamydia-like organisms: a revolution in our understanding of the biology and pathogenicity of members of the phylum Chlamydiae. Pathog Dis. 2015;73(1):1–15. doi: 10.1093/femspd/ftu009. [DOI] [PubMed] [Google Scholar]
- 11.Holzer M, Laroucau K, Creasy HH, Ott S, Vorimore F, Bavoil PM, et al. Whole-Genome Sequence of Chlamydia gallinacea Type Strain 08-1274/3. Genome Announc. 2016;4(4) [DOI] [PMC free article] [PubMed]
- 12.Sachse K, Laroucau K, Riege K, Wehner S, Dilcher M, Creasy HH, et al. Evidence for the existence of two new members of the family Chlamydiaceae and proposal of Chlamydia avium sp. nov. and Chlamydia gallinacea sp. nov. Syst Appl Microbiol. 2014;37(2):79–88. doi: 10.1016/j.syapm.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Pannekoek Y, Morelli G, Kusecek B, Morre SA, Ossewaarde JM, Langerak AA, et al. Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis. BMC Microbiol. 2008;8:42. doi: 10.1186/1471-2180-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24(23):2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darling AE, Mau B, Perna NT. Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katoh K, Standley DM. MAFFT: iterative refinement and additional methods. Methods Mol Biol. 2014;1079:131–146. doi: 10.1007/978-1-62703-646-7_8. [DOI] [PubMed] [Google Scholar]
- 19.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 20.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 21.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27(7):1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25(1):119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 27.Nunes A, Gomes JP. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol. 2014;23:49–64. doi: 10.1016/j.meegid.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Wolff BJ, Morrison SS, Pesti D, Ganakammal SR, Srinivasamoorthy G, Changayil S, et al. Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type III secretion system genes. Microbiology. 2015;161(7):1378–1391. doi: 10.1099/mic.0.000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jelocnik M, Bachmann NL, Kaltenboeck B, Waugh C, Woolford L, Speight KN, et al. Genetic diversity in the plasticity zone and the presence of the chlamydial plasmid differentiates Chlamydia pecorum strains from pigs, sheep, cattle, and koalas. BMC Genomics. 2015;16:893. doi: 10.1186/s12864-015-2053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdelsamed H, Peters J, Byrne GI. Genetic variation in Chlamydia trachomatis and their hosts: impact on disease severity and tissue tropism. Future Microbiol. 2013;8(9):1129–1146. doi: 10.2217/fmb.13.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mital JMN, Dorward DW, Dooley CA, Hackstadt T. Role for chlamydial inclusion membrane proteins in inclusion membrane structure and biogenesis. PLoS One. 2013;8(5):e63426. doi: 10.1371/journal.pone.0063426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasilevsky S, Stojanov M, Greub G, Baud D. Chlamydial polymorphic membrane proteins: regulation, function and potential vaccine candidates. Virulence. 2016;7(1):11–22. doi: 10.1080/21505594.2015.1111509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong S, Yang Z, Lei L, Shen L, Zhong G. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J Bacteriol. 2013;195(17):3819–3826. doi: 10.1128/JB.00511-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jelocnik M, Bachmann NL, Seth-Smith H, Thomson NR, Timms P, Polkinghorne AM. Molecular characterisation of the Chlamydia pecorum plasmid from porcine, ovine, bovine, and koala strains indicates plasmid-strain co-evolution. PeerJ. 2016;4:e1661. doi: 10.7717/peerj.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Legione AR, Patterson JL, Whiteley PL, Amery-Gale J, Lynch M, Haynes L, et al. Identification of unusual Chlamydia pecorum genotypes in Victorian koalas (Phascolarctos cinereus) and clinical variables associated with infection. J Med Microbiol. 2016;65(5):420–428. doi: 10.1099/jmm.0.000241. [DOI] [PubMed] [Google Scholar]
- 36.Sachse K, Laroucau K, Hotzel H, Schubert E, Ehricht R, Slickers P. Genotyping of Chlamydophila psittaci using a new DNA microarray assay based on sequence analysis of ompA genes. BMC Microbiol. 2008;8:63. doi: 10.1186/1471-2180-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlson JH, Porcella SF, McClarty G, Caldwell HD. Comparative genomic analysis of Chlamydia trachomatis oculotropic and genitotropic strains. Infect Immun. 2005;73(10):6407–6418. doi: 10.1128/IAI.73.10.6407-6418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinemann IU, Jahn M, Jahn D. The biochemistry of heme biosynthesis. Arch Biochem Biophys. 2008;474(2):238–251. doi: 10.1016/j.abb.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Panek H, O’Brian MR. A whole genome view of prokaryotic haem biosynthesis. Microbiology. 2002;148(Pt 8):2273–2282. doi: 10.1099/00221287-148-8-2273. [DOI] [PubMed] [Google Scholar]
- 40.Nans A, Kudryashev M, Saibil HR, Hayward RD. Structure of a bacterial type III secretion system in contact with a host membrane in situ. Nat Commun. 2015;6:10114. doi: 10.1038/ncomms10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. Type III secretion a la Chlamydia. Trends Microbiol. 2007;15(6):241–251. doi: 10.1016/j.tim.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Bachmann NL, Fraser TA, Bertelli C, Jelocnik M, Gillett A, Funnell O, et al. Comparative genomics of koala, cattle and sheep strains of Chlamydia pecorum. BMC Genomics. 2014;15:667. doi: 10.1186/1471-2164-15-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borges V, Gomes JP. Deep comparative genomics among Chlamydia trachomatis lymphogranuloma venereum isolates highlights genes potentially involved in pathoadaptation. Infect Genet Evol. 2015;32:74–88. doi: 10.1016/j.meegid.2015.02.026. [DOI] [PubMed] [Google Scholar]
- 44.Grimwood J, Olinger L, Stephens RS. Expression of Chlamydia pneumoniae polymorphic membrane protein family genes. Infect Immun. 2001;69(4):2383–2389. doi: 10.1128/IAI.69.4.2383-2389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Read TD, Joseph SJ, Didelot X, Liang B, Patel L, Dean D. Comparative analysis of Chlamydia psittaci genomes reveals the recent emergence of a pathogenic lineage with a broad host range. MBio. 2013;4(2) [DOI] [PMC free article] [PubMed]
- 46.Voigt A, Schofl G, Saluz HP. The Chlamydia psittaci genome: a comparative analysis of intracellular pathogens. PLoS One. 2012;7(4):e35097. doi: 10.1371/journal.pone.0035097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28(6):1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajaram K, Giebel AM, Toh E, Hu S, Newman JH, Morrison SG, et al. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun. 2015;83(7):2870–2881. doi: 10.1128/IAI.00106-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pickett MA, Everson JS, Pead PJ, Clarke IN. The plasmids of Chlamydia trachomatis and Chlamydophila pneumoniae (N16): accurate determination of copy number and the paradoxical effect of plasmid-curing agents. Microbiology. 2005;151(Pt 3):893–903. doi: 10.1099/mic.0.27625-0. [DOI] [PubMed] [Google Scholar]
- 50.Rockey DD. Unraveling the basic biology and clinical significance of the chlamydial plasmid. J Exp Med. 2011;208(11):2159–2162. doi: 10.1084/jem.20112088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C. gallinacea-positive samples used for MLST and plasmid detection. (DOCX 13 kb)
Primers used for generating C. gallinacea plasmid fragments in this study. (DOCX 14 kb)
Primers used for MLST of C. gallinacea in this study. (DOCX 14 kb)
PCR conditions for amplifying seven HK genes in this study. (DOCX 12 kb)
Original data for MLST analysis of the C. gallinacea-positive samples. (XLSX 10 kb)
Original data for T3SS of C. gallinacea, including the locus-tags, and their annotation/putative function, length and direction. (XLS 36 kb)
List of the predicted transmembrane helices in the analyzed putative C. gallinacea inclusion (Inc) proteins from this study. (XLSX 10 kb)
Graphical representation of the predicted transmembrane helices in the analyzed putative C. gallinacea inclusion (Inc) proteins from this study. (DOCX 423 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The data from this study is also available from Treebase: http://purl.org/phylo/treebase/phylows/study/TB2:S21854.