

Abstract
Background:
Restrictive cardiomyopathy (RCM) is the least common cardiomyopathy in which the walls are rigid and the heart is restricted from stretching and filling properly. Cardiac troponin I (cTnI) mutation-caused myofibril Ca2+ hypersensitivity has been shown to be associated with impaired diastolic function. This study aimed to investigate the linkage between the genotype and clinical therapy of RCM.
Methods:
Five sporadic pediatric RCM patients confirmed by echocardiography were enrolled in this study. Whole-exome sequencing (WES) was performed for the cohort to find out candidate causative gene variants. Sanger sequencing confirmed the WES-identified variants.
Results:
TNNI3 variants were found in all of the five patients. R192H mutation was shared in four patients while R204H mutation was found only in one patient. Structure investigation showed that the C terminus of TNNI3 was flexible and mutation on the C terminus was possible to cause the RCM. Catechins were prescribed for the five patients once genotype was confirmed. Ventricular diastolic function was improved in three patients during the follow-up.
Conclusions:
Our data demonstrated that TNNI3 mutation-induced RCM1 is the most common type of pediatric RCM in this study. In addition, WES is a reliable approach to identify likely pathogenic genes of RCM and might be useful for the guidance of clinical treatment scheme.
Keywords: Pediatric Restrictive Cardiomyopathy, Phenotype Classification, TNNI3, Whole-exome Sequencing
INTRODUCTION
Cardiomyopathy includes five original subtypes referred as hypertrophic, dilated, restrictive, arrhythmogenic right ventricular, and unclassified types.[1] Restrictive cardiomyopathy (RCM) is the least common of cardiomyopathy characterized by impaired ventricular filling.[2] RCM includes five subtypes on the basis of genotype. It is well documented that RCM1, RCM3, RCM4, and RCM5 are caused by mutations which lead to isoform of troponin I (TNNI3), troponin T, myopalladin, and filamin-C, respectively.[3,4,5,6] RCM2 mapped on mutations in the gene coding region of chromosome 10q23.3.[7] Besides these identified sites, RCM is suspected of having other pathogeneses as some RCM patients have no mutations among these identified gene locations.
Up to now, there is no effective treatment scheme for the patients with RCM due to its low prevalence and there is just few research with regard to RCM.[8,9] In this study, we present the diagnosis and treatment of five clinically diagnosed sporadic pediatric RCM patients who had taken whole-exome sequencing (WES).
METHODS
Ethical approval
This study was approved by the Ethics Committee of Beijing Anzhen Hospital. Informed consent was obtained from parents of the cohort in this study.
Patients
From October 2013 to May 2016, a total of 16 patients of unrelated pediatric RCM patients were admitted to Beijing Anzhen Hospital. A clinical diagnosis of idiopathic RCM was confirmed by transthoracic echocardiography (TTE) and other clinical characteristics. The inclusion criteria included pediatric patients without family history of RCM, patients who were diagnosed as idiopathic RCM during the first visit of hospital, patients without constrictive pericarditis caused by tuberculosis or other systemic diseases, patients who had no parasitic infection and eosinophilia, and patients who had taken WES with definite mutations. Finally, five patients (two males and three females) were enrolled in this study due to having been confirmed as idiopathic RCM by both clinical characteristics and WES. They were aged between 5 and 12 years old (mean age: 9 ± 3 years). Clinical data including medical history (onset time, age at treatment, and clinical symptoms), physical signs, results of the diagnostic examination, treatment strategy, and curative effect were collected. Clinical cardiac function improvement was defined as New York Heart Association (NYHA) functional classification increased by at least one level. The improvement of left ventricular diastolic function by TTE was defined as the reduction of velocity of early filling mitral flow/early diastolic tissue Doppler imaging mitral annular velocity (E/E’) >10%.
Physical and diagnostic examination
Physical and diagnostic examinations were performed for all patients, including routine blood test (containing eosinophil counts and percentage), liver and kidney function, myocardial enzymes, cardiac troponin I (cTnI), brain natriuretic peptide (BNP), erythrocyte sedimentation rate, C-reactive protein, antituberculosis antibody, antinuclear antibodies, antiphospholipid antibodies, coagulation function, screening of inherited metabolic diseases, serum transferrin saturation, and antiparasite antibody. All patients underwent tuberculin tests to rule out tuberculous infection. In addition, TTE, electrocardiogram (ECG), chest X-ray, and cardiac magnetic resonance imaging were carried out.
Clinical diagnosis of RCM was confirmed by two-dimensional (2D) color Doppler echocardiography. Diagnostic criteria were as follows: (1) marked enlargement of left and right atrium; (2) normal ventricular chamber size, normal or slightly thickened ventricular wall; (3) normal or less decreased ventricular systolic function; and (4) impaired ventricular diastolic function. TTE was performed according to the recommendations by American Society of Echocardiography.
Indicators for assessing diastolic function included E deceleration time (EDT), late filling mitral flow (A), ratio of E to A (E/A), E′ and mitral E/E’.[10] Left ventricular diastolic dysfunction was defined as mitral EDT <150 ms, mitral E/A >2, and average mitral E/E’>13 (average of the E/E’ from septum and LV lateral wall).
Whole-exome sequencing
DNA was extracted from 2 ml of peripheral blood following the instruction of BloodGen Midi Kit (CWBIO, Beijing, China), sheared by sonication after detected by agarose gel electrophoresis, and then hybridized with NimbleGen 2.0 probe sequence capture array (Roche, Basel, Switzerland). Captured DNA was first applied for exonic DNA enrichment. The libraries were then tested for enrichment by quantitative polymerase chain reaction (PCR), and also for size distribution and concentration by the Agilent Bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, USA). The samples were thereby sequenced by Illumina Hiseq 2500 (Illumina, Santiago, USA).
Raw image files were processed by the BclToFastq (Illumina, Santiago, USA) for base calling and raw data generating. The low-quality variations were ruled out if quality score ≥20 (Q20). The sequencing reads were aligned to the National Center for Biotechnology Information (NCBI) human reference genome (hg19) using BOADICEA Web Application. Genome Analysis Toolkit was used to analyze single-nucleotide polymorphism and insertion-deletion of the sequences.[11] The reported cardiovascular diseases-associated genes were analyzed. In addition, as mitochondria mutations could also induce cardiomyopathy, the known mitochondriopathy-related genes were assessed as well. All samples were annotated using ANNOVAR and the candidate variants were based on frequency and function.[12] A 0.5% cutoff of frequency estimated from the 1000 genomes project database was applied. Protein biological function was predicted using PROVEAN and PolyPhen-2.[13,14]
Sanger sequencing validation
Sanger sequencing was used to confirm the variants identified in both the patients and their parents. PCR primers for TNNI3 used in this study were 5’-ATAAGAAGAGAAGGAAGGA GAC-3’ and 5’-TCAATAACACAGCCAAGAGT-3’, producing a PCR product with the length of 608 bp. The PCR products were sequenced by ABI 3730XL (Thermo Fisher Scientific Inc., Waltham, MA, USA) and analyzed by DNASTAR 5.0 software (DNASTAR, Inc., Madison, WI, USA).
Medication
Once the patients were diagnosed as RCM, all of them were started with medical treatment immediately. All patients received routine treatment consisted of oral diuretics, vasodilators, and small doses of calcium antagonists. For patients with TNNI3-192 site mutation, oral administration of catechin was prescribed according to Zhang L et al.[15] The initial dose of catechin was 15 mg·kg−1·day−1 and was gradually added to 50 mg·kg−1·day−1, within 6 months if patients could tolerate.
RESULTS
General information
To understand the general condition of the five patients, clinical data were collected. Clinical symptoms included fatigue, exercise intolerance, exertional asthma, respiratory infection, tightness in the chest, and lower extremity edema [Table 1]. The initial onset age was from 3 to 6 years old, with an average of 5 years old. Time between onset and the first visit was 1–4 years (median: 2 years), and the follow-up time ranged from 3 to 35 months (mean: 17 ± 12 months). To date, one female patient (13 years old) has received heart transplant surgery, another 11-year-old girl died of sudden death after exercise, and others were still followed up continually.
Table 1.
General clinical data of five sporadic pediatric restrictive cardiomyopathy patients
| Characteristics | Patient number | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Gender | Male | Male | Female | Female | Female |
| Age (years) | 12 | 8 | 12 | 5 | 10 |
| Symptoms | |||||
| Fatigue | + | + | + | + | + |
| Exercise intolerance | + | + | + | + | + |
| Short of breath | + | + | + | – | + |
| Signs | |||||
| Edema | – | – | + | – | – |
| Jaundice | – | – | + | – | – |
| Hepatomegaly | + | + | ++ | + | + |
| Ascites | – | – | ++ | – | – |
| Heart murmur | – | – | + | – | – |
+: Positive; ++: Significant positive; –: Negative.
To gain insight into the clinical changes of the cohort, conventional diagnostic examination was performed. As displayed in Table 2, two patients were with mild elevated liver enzyme whereas renal function was normal in all patients. Abnormal myocardial enzyme was found in two cases, increased cTnI was discovered in one patient, and significantly increased BNP was detected in all five patients. Other laboratory tests were normal in all the patients. As for medical device testing, no special diagnostic information for RCM occurrence was provided by ECG, X-ray, and cardiac magnetic resonance.
Table 2.
Diagnostic examination of five sporadic pediatric restrictive cardiomyopathy patients
| Examinations | Patient number | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| CTnI (ng/ml) | 0.01 | 0.01 | 0.07 | 0.03 | 0.05 |
| CK-MB (mmol/L) | 17 | 14 | 16 | 20 | 13 |
| CRP (mg/L) | 0.23 | 0.2 | 10.93 | 0.85 | 0.25 |
| BNP (ng/L) | 1646.5 | 899 | 4869 | 525.3 | 1187 |
| ECG | |||||
| Enlargement of atrium | + | + | + | + | + |
| ST-T segment change | + | + | – | + | + |
| AF | – | – | – | – | – |
| SVT | – | – | – | – | – |
| UCG | |||||
| Enlargement of atrium | + | + | + | + | + |
| LVRF | + | + | + | + | + |
| TP | – | – | – | – | – |
| PH | – | + | + | – | + |
| CMRI | RCM | RCM | RCM | RCM | RCM |
CTnI: Cardiac troponin I; CK-MB: Isoenzyme of creatine kinase; CRP: C-reaction protein; BNP: Brain natriuretic peptide; ECG: Electrocardiograph; SVT: Supraventricular tachycardia; AF: Atrial fibrillation; UCG: Ultrasonic cardiogram; LVRF: Left ventricular restrictive filling; TP: Thickened pericardium; PH: Pulmonary hypertension; RCM: Restrictive cardiomyopathy. CMRI: Cardiac magnetic resonance imaging.
Echocardiography
2D TTE was performed to confirm the diagnosis of RCM. As illustrated in Figure 1a, dilation of left and right atriums with normal ventricular size was detected. The velocity of mitral peak E and peak A decreased significantly, E/A >2, and the variation rate of E peak flow with suction was less than 25% [Figure 1b]. Isovolumic relaxation time was shorter than 70 ms, and the deceleration time of peak E reduced significantly (<150 ms). Inferior vena cava broadened and the subsidence rate reduced. Echocardiography estimated average mitral E/E’>13.
Figure 1.
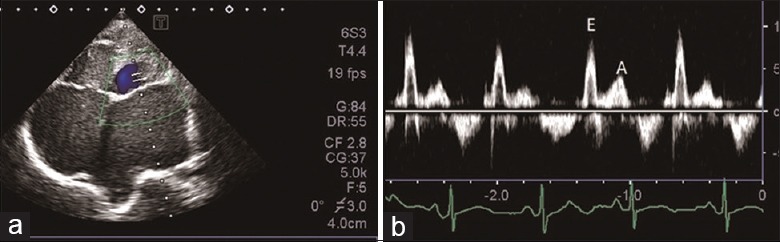
Echocardiography of restrictive cardiomyopathy. (a) Biatrial enlargement, with normal ventricular chamber size; (b) peak E wave velocity, peak A wave velocity decreased significantly, and E/A >2. E: Velocity of early filling mitral flow; A: Velocity of late filling mitral flow.
TNNI3 variants showed in whole-exome sequencing
In general, the Q20 was more than 95% and quality control files demonstrated that the data were adequate for further analysis. TNNI3 was found in all the patients. R192H mutation on TNNI3 was found in four individuals whereas R204H mutation on TNNI3 was found in one patient.
De novo mutations
Sanger sequencing was performed in the family trios using double orientation primers. The results are illustrated in Figure 2. Of note, all the five TNNI3 mutations were de novo germline mutation, clarifying that their parents are healthy.
Figure 2.

Sanger sequencing validation of the mutation sites of R192H (a) and R204H (b) in pediatric restrictive cardiomyopathy patients with their parents. NCBI: National Center for Biotechnology Information.
Clinical outcome
One patient of 13-year-old girl received heart transplant after taking catechin for 3 months due to her very serious condition. Another 11-year-old girl died of “sudden death” after taking catechin for 9 months. The other three patients survived with long-term regular follow-up. The dose of catechin was added to mean 50 mg·kg−1·day−1 in half a year for the three patients. Mean follow-up range in these patients was more than 1 year, and the longest follow-up time was 27 months. The NYHA functional classification in survived patients improved from Class III to Class II [Table 3]. TTE showed that mean E/E’ decreased from 18.4 ± 4.6 to 11.2 ± 4.6, suggesting that the TTE findings were consistent with the heart function recovery [Figure 3].
Table 3.
Changes of cardiac function and prognosis before and after administration of catechin
| Parameters | Patient number | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Weight (kg) | 42 | 25 | 32 | 16 | 30 |
| Catechin (mg/d) | 1600 | 1600 | 800 | 725 | 400 |
| NYHA | |||||
| Precatechin | III | III | VI | III | III |
| Postcatechin | II | II | I | II | II |
| Follow-up (months) | 15 | 35 | 3 | 18 | 9 |
| Prognosis | S | S | HT | S | SD |
HT: Heart transplant; S: Survival; SD: Sudden death; NYHA: New York Heart Association Functional Classification.
Figure 3.
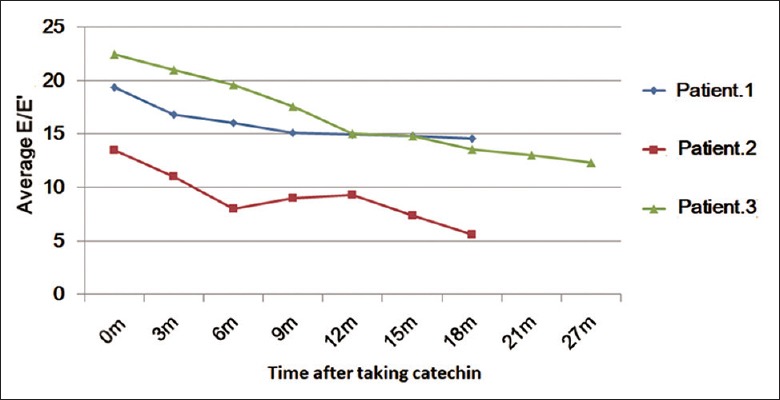
Changes of average mitral E/E’ after taking catechin in three survivors of pediatric restrictive cardiomyopathy. E/E’: Ratio of velocity of early filling mitral flow to early diastolic tissue Doppler imaging mitral annular velocity.
DISCUSSION
RCM is a rare cardiomyopathy that can occur at any age during childhood.[16,17,18,19] Progress on the treatment of the RCM was limited for the infrequent feature of the disease. A total five sporadic RCM patients from different regions around China were enrolled in the present study. In this study, WES was performed and the results showed variants of TNNI3 in the patients. The variants could induce R192H (four patients) and R204H (one patient) mutations, which all have been reported by previous studies.[20,21] In line with this, the mutations were considered as the pathogenesis. Four of the five sporadic patients had the same mutation site (R192H), indicating that R192H might be the main mutation pattern for patients around China.
To confirm the possibility that TNNI3 variants are the likely the etiology of RCM, structural investigation was performed. C terminus of TNNI3 is defined as “mobile domain” indicating that this domain is quite flexible.[22] We hypothesized that TNNI3 might play a role in conformational changes. Therefore, to further investigate the mechanism of TNNI3 for RCM occurrence, structural investigation was performed. To date, there is no structure covering the sequence of extreme C terminus of human TNNI3; therefore, the structure of Gallus gallus was applied.[23] As for human TNNI3 determination, human cardiac troponin in the Ca2+ saturated form (1j1e) was enrolled.[22] Before structure investigation, sequence conservation alignment was carried out, and as illustrated in Figure 4a, the sequence of G. gallus TNNI3 is almost identical to that of human. The result argued that it is possible to use the TNNI3 structure of G. gallus to represented human TNNI3 structure. Furthermore, structure prediction showed that the secondary structure of TNNI3 is composed by two α helixes connected by a loop, which is in accord with 1j1e. However, structure from G. gallus showed that the C terminus is composed by two α helixes that comprising two connecting short β sheets, indicating that the C terminus has different conformations under different physiological states [Figure 4b]. Together, the result demonstrated that C terminus of TNNI3 is flexible and conformational variable, which might play an important role in the function execution of protein TNNI3.
Figure 4.
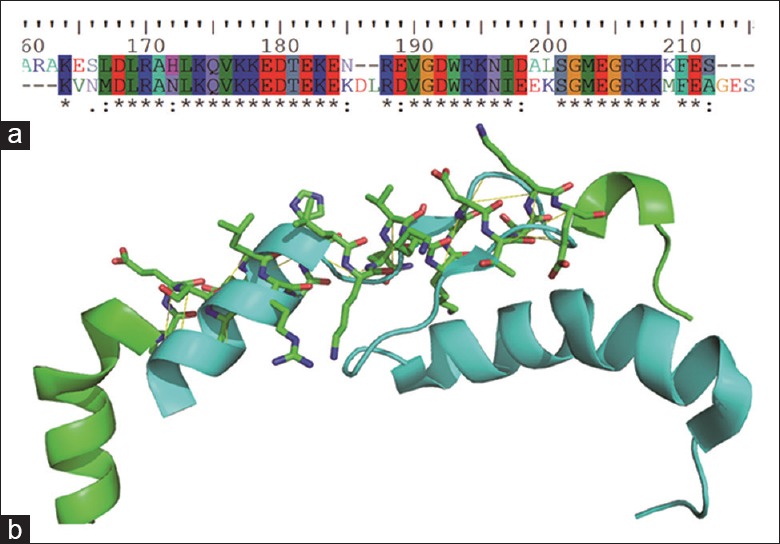
Structural investigation results of TNNI3. (a) Sequences alignment of human and Gallus gallus showed that the two sequences are mostly identical; (b) Structure alignment of human (green) and Gallus gallus (cyan) showed the structures in different physiological states. The aligned sequence of human was presented as sticks while Gallus gallus was shown as cartoon.
To date, there was no specific treatment for patients with RCM. The routine treatment contains oral diuretics, vasodilators, and calcium antagonists. However, the effect is poor. Several studies[24,25,26] have constructed RCM transgenic mouse lines expressing cTnI R193H mutation in the heart and showed that cTnI mutations could induce specific diastolic dysfunction. In addition, they argue that Ca2+ is highly linked to impaired relaxation in myocardial cells. Furthermore, they demonstrate that catechin is effective in reverse diastolic dysfunction and could be useful for the troponin mutations-induced RCM.[15] In this study, application of large doses of catechin (50 mg·kg−1·day−1) was performed and it was effective in improving diastolic function and clinical symptoms. However, catechin could neither cure RCM nor prevent sudden death. Comparing with the traditional calcium antagonists, catechin can reduce myocardial calcium hypersensitivity, which is similar to calcium antagonists. However, catechin has no obvious adverse effect such as blood pressure reduction and cardiac rhythm reduction.[15] In line with this, we consider that catechin should be deserved promotion and application in clinic.
In conclusion, five sporadic RCM pediatric patients were enrolled and had undertaken WES. TNNI3 mutation was found in all the patients and R192H mutation was shared by four individuals. Structure investigation indicated that the C terminus of TNNI3 was flexible and the mutations were possible to induce the onset of RCM. WES played an important role in the diagnosis of genotype and clinical treatment of RCM. Catechin therapy was effective in most of the patients in our group.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Ning-Ning Wang
REFERENCES
- 1.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–6. doi: 10.1093/eurheartj/ehm342. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 2.Muchtar E, Blauwet LA, Gertz MA. Restrictive cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:819–37. doi: 10.1161/CIRCRESAHA.117.310982. doi: 10.1161/CIRCRESAHA.117.310982. [DOI] [PubMed] [Google Scholar]
- 3.Huang XP, Du JF. Troponin I, cardiac diastolic dysfunction and restrictive cardiomyopathy. Acta Pharmacol Sin. 2004;25:1569–75. [PubMed] [Google Scholar]
- 4.Peddy SB, Vricella LA, Crosson JE, Oswald GL, Cohn RD, Cameron DE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117:1830–3. doi: 10.1542/peds.2005-2301. doi: 10.1542/peds.2005-2301. [DOI] [PubMed] [Google Scholar]
- 5.Meyer T, Ruppert V, Ackermann S, Richter A, Perrot A, Sperling SR, et al. Novel mutations in the sarcomeric protein myopalladin in patients with dilated cardiomyopathy. Eur J Hum Genet. 2013;21:294–300. doi: 10.1038/ejhg.2012.173. doi: 10.1038/ejhg.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W, et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum Mutat. 2016;37:269–79. doi: 10.1002/humu.22942. doi: 10.1002/humu.22942. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Kumar A, Kaplan L, Fricker FJ, Wallace MR. Genetic linkage of a novel autosomal dominant restrictive cardiomyopathy locus. J Med Genet. 2005;42:663–5. doi: 10.1136/jmg.2004.030189. doi: 10.1136/jmg.2004.030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishiwata S, Nishiyama S, Seki A, Kojima S. Restrictive cardiomyopathy with complete atrioventricular block and distal myopathy with rimmed vacuoles. Jpn Circ J. 1993;57:928–33. doi: 10.1253/jcj.57.928. [DOI] [PubMed] [Google Scholar]
- 9.Chen SC, Balfour IC, Jureidini S. Clinical spectrum of restrictive cardiomyopathy in children. J Heart Lung Transplant. 2001;20:90–2. doi: 10.1016/s1053-2498(00)00162-5. doi: 10.1016/S1053-2498(00)00162-5. [DOI] [PubMed] [Google Scholar]
- 10.Chinali M, Aurigemma GP, de Simone G, Mishra RK, Gerdts E, Wachtell K, et al. Mitral E wave deceleration time to peak E velocity ratio and cardiovascular outcome in hypertensive patients during antihypertensive treatment (from the LIFE echo-substudy) Am J Cardiol. 2009;104:1098–104. doi: 10.1016/j.amjcard.2009.05.063. doi: 10.1016/j.amjcard.2009.05.063. [DOI] [PubMed] [Google Scholar]
- 11.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi Y, Chan AP. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7. doi: 10.1093/bioinformatics/btv195. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Nan C, Chen Y, Tian J, Jean-Charles PY, Getfield C, et al. Calcium desensitizer catechin reverses diastolic dysfunction in mice with restrictive cardiomyopathy. Arch Biochem Biophys. 2015;573:69–76. doi: 10.1016/j.abb.2015.03.015. doi: 10.1016/j.abb.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Denfield SW, Rosenthal G, Gajarski RJ, Bricker JT, Schowengerdt KO, Price JK, et al. Restrictive cardiomyopathies in childhood. Etiologies and natural history. Tex Heart Inst J. 1997;24:38–44. [PMC free article] [PubMed] [Google Scholar]
- 17.Malcić I, Jelusić M, Kniewald H, Barisić N, Jelasić D, Bozikov J, et al. Epidemiology of cardiomyopathies in children and adolescents: A retrospective study over the last 10 years. Cardiol Young. 2002;12:253–9. doi: 10.1017/s1047951102000550. [DOI] [PubMed] [Google Scholar]
- 18.Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–46. doi: 10.1056/NEJMoa021737. doi: 10.1056/NEJMoa021737. [DOI] [PubMed] [Google Scholar]
- 19.Russo LM, Webber SA. Idiopathic restrictive cardiomyopathy in children. Heart. 2005;91:1199–202. doi: 10.1136/hrt.2004.043869. doi: 10.1136/hrt.2004.043869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ, et al. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–51. doi: 10.1161/01.CIR.0000080896.52003.DF. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 21.Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest. 2003;111:209–16. doi: 10.1172/JCI16336. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 23.Murakami K, Yumoto F, Ohki SY, Yasunaga T, Tanokura M, Wakabayashi T, et al. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J Mol Biol. 2005;352:178–201. doi: 10.1016/j.jmb.2005.06.067. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- 24.Du J, Liu J, Feng HZ, Hossain MM, Gobara N, Zhang C, et al. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol. 2008;294:H2604–13. doi: 10.1152/ajpheart.91506.2007. doi: 10.1152/ajpheart.91506.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Charles PY, Nan C, Pinto JR, Wang Y, Liang J, et al. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49:402–11. doi: 10.1016/j.yjmcc.2010.04.017. doi: 10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Zhang L, Jean-Charles PY, Nan C, Chen G, Tian J, et al. Dose-dependent diastolic dysfunction and early death in a mouse model with cardiac troponin mutations. J Mol Cell Cardiol. 2013;62:227–36. doi: 10.1016/j.yjmcc.2013.06.007. doi: 10.1016/j.yjmcc.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]