

Abstract
The type strain of the prospective 10.1601/nm.30737 sp. nov. ERR11T, was isolated from a nodule of the leguminous tree Erythrina brucei native to Ethiopia. The type strain 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T, was isolated from the nodules of Lespedeza cuneata in Beijing, China. The genomes of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T were sequenced by DOE–JGI and deposited at the DOE–JGI genome portal as well as at the European Nucleotide Archive. The genome of ERR11T is 9,163,226 bp in length and has 102 scaffolds, containing 8548 protein–coding and 86 RNA genes. The 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T genome is arranged in 108 scaffolds and consists of 8,201,522 bp long and 7776 protein–coding and 85 RNA genes. Both genomes contain symbiotic genes, which are homologous to the genes found in the complete genome sequence of 10.1601/nm.24498 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T. The genes encoding for nodulation and nitrogen fixation in ERR11T showed high sequence similarity with homologous genes found in the draft genome of peanut–nodulating 10.1601/nm.27386 10.1601/strainfinder?urlappend=%3Fid%3DLMG+26795 T. The nodulation genes nolYA-nodD2D1YABCSUIJ-nolO-nodZ of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T are organized in a similar way to the homologous genes identified in the genomes of 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T , 10.1601/nm.25806 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/nm.1462 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525. The genomes harbor hupSLCFHK and hypBFDE genes that code the expression of hydrogenase, an enzyme that helps rhizobia to uptake hydrogen released by the N2-fixation process and genes encoding denitrification functions napEDABC and norCBQD for nitrate and nitric oxide reduction, respectively. The genome of ERR11T also contains nosRZDFYLX genes encoding nitrous oxide reductase. Based on multilocus sequence analysis of housekeeping genes, the novel species, which contains eight strains formed a unique group close to the 10.1601/nm.25806 branch. Genome Average Nucleotide Identity (ANI) calculated between the genome sequences of ERR11T and closely related sequences revealed that strains belonging to 10.1601/nm.25806 branch (10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615), were the closest strains to the strain ERR11T with 95.2% ANI. Type strain ERR11T showed the highest DDH predicted value with 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 (58.5%), followed by 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 (53.1%). Nevertheless, the ANI and DDH values obtained between ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 or 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 were below the cutoff values (ANI ≥ 96.5%; DDH ≥ 70%) for strains belonging to the same species, suggesting that ERR11T is a new species. Therefore, based on the phylogenetic analysis, ANI and DDH values, we formally propose the creation of 10.1601/nm.30737 sp. nov. with strain ERR11T (10.1601/strainfinder?urlappend=%3Fid%3DHAMBI+3532 T=10.1601/strainfinder?urlappend=%3Fid%3DLMG+30162 T) as the type strain.
Electronic supplementary material
The online version of this article (10.1186/s40793-017-0283-x) contains supplementary material, which is available to authorized users.
Keywords: Bradyrhizobium shewense sp. nov. ERR11T, Erythrina brucei, Bradyrhizobium yuanmingense CCBAU 10071T, Symbiotic, Ethiopia, Genome average nucleotide identity, Digital DNA-DNA hybridization
Introduction
Biological nitrogen fixation is a vital process in ecosystem functioning, offering a nitrogen for plant growth. Legume plants form a nitrogen–fixing symbiotic association with soil bacteria known as rhizobia. The symbiotic association results in the formation of nodules, shelter and powerhouse of nitrogen fixation for the rhizobia, on the roots or stems of host legumes [1]. The rhizobia belong to 10.1601/nm.809 and 10.1601/nm.1616 [2]. alphaproteobacterial 10.1601/nm.1459 was first described as slow–growing rhizobia by Jordan [3]. Since then, 33 distinct rhizobial species belonging to the genus 10.1601/nm.1459 were formally described [4]. In addition, unique 10.1601/nm.1459 groups isolated from diverse legume species might represent new species [5–11].
In rhizobial taxonomic studies, polyphasic approaches such as phenotypic features, analysis of the 16S rRNA genetic marker, and DDH were for years used as standard criteria for the description of new bacterial species. Nevertheless, the 16S rRNA gene sequence difference between closely related species, particularly in the genus 10.1601/nm.1459 is low for differentiation of closely related species [5, 12, 13]. Bacterial strains in the same species could be delineated at ≥70% DDH relatedness [14, 15], but yet this method is vulnerable to variable laboratory results that lead to an inconsistent classification of the same species [16]. To resolve the issues related to the traditional wet–lab DDH technique, a digital DDH method was proposed for calculation of the DDH from genome sequences for bacterial classification study [17–19].
Multilocus sequence analysis (MLSA) of housekeeping protein–coding genes has become a common practice in bacterial taxonomic studies. The method offers high resolution and hence, has been used in rhizobial taxonomic studies for species identification and differentiating strains at the species level [5, 13, 20, 21]. Recently, the genome–wide average nucleotide Identity (ANI) method has successfully been used for classification of various bacterial species [22–24]. According to Richter and Rosselló-Móra [25] and Kim et al. [23], the ANI cutoff value that corresponds to the traditional 70% DNA–DNA relatedness cutoff value for species delineation was in the range 95–96%, depending on the nature of bacterial genome sequences. A more advanced ANI calculation was carried–out by Varghese et al. [24] by including a large number of genome sequences. Based on this study, a 96.5% ANI value is the minimum threshold that corresponds to 70% DNA–DNA relatedness cutoff value for strains (genomes) belong to the same species. To set the 96.5% ANI cutoff value for species description, the alignment fraction (AF) between the genomes should be 0.6 or above (i.e. AF covering at least 60% of the gene content of a pair of genomes) [24].
In Ethiopia, an endemic multipurpose legume tree E. brucei [26] is used for the production of firewood and a shade for coffee plantations [27] and it also improves soil fertility [28]. Crotalaria spp. [29] and Indigofera spp. [30] are among the diverse perennial herb and shrub legumes found in Ethiopia [31]. Crotalaria spp. [29] are used for green manuring, as a fallow before the main crop or for intercropping with cereal plants in order to amend soil nitrogen fertility. Some Crotalaria spp. [29] can be used as food and feed [32–34]. Indigofera spp. [30] are used for fodder for livestock, particularly in dryland areas as the species are resistant to water stress [35]. A group of rhizobial strains belonging to the genus 10.1601/nm.1459 was isolated from nodules of the legume tree E. brucei [26] and the shrub legumes Crotalaria spp. [29] and Indigofera spp. [30] growing in Ethiopia. These bacteria formed a unique branch which was distinct from other known species of the genus 10.1601/nm.1459 in phylogenetic trees constructed based on sequence analysis of housekeeping genes [5]. To describe this group as a new 10.1601/nm.1459 species using the genome–wide ANI and digital DDH methods, a representative strain 10.1601/nm.1459 sp. ERR11 (hereafter 10.1601/nm.30737 sp. nov. ERR11T) was selected for genome sequencing. The sequencing was done under the DOE–JGI 2014 Genomic Encyclopedia of Type Strains, Phase III, a project designed for sequencing of soil and plant–associated and newly described type strains [36]. Therefore, the main purpose of this study was 1) to present classification and general features of 10.1601/nm.30737 sp. nov., 2) to report the genome sequence and annotation of the type strain ERR11T. In addition, the genome sequence and annotation of reference type strain 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T [37] sequenced for this study will be reported.
Organism information
Classification and features
The strain ERR11T is the type strain of newly proposed 10.1601/nm.30737 sp. nov. This novel species includes strains isolated from nodules of E. brucei [26], Indigofera spp. [30] and Crotalaria spp. [29] growing in Ethiopia. Previously, the strains were identified as a unique group using recA, glnII, and rpoB single gene sequence analysis and on the phylogenetic tree constructed based concatenated recA–glnII–rpoB gene sequences. On the phylogenetic tree, the strains in the novel group formed their own cluster exclusive of validly published species, and consequently, this group were designated as 10.1601/nm.1459 genosp ETH1 [5]. To define the current taxonomic position of the novel rhizobial species, we reconstructed a phylogenetic tree from concatenated recA–glnII–rpoB sequences by including more and recently published reference sequences from the public database. In this phylogenetic tree, the bacterial grouping was consistent with our previous tree produced from concatenated recA, rpoB and glnII gene sequences [5]. The novel species formed a distinct group close to a 10.1601/nm.25806 branch that contains strains isolated from the nodules of soybean (Glycine max) [38] grown in Ottawa, Canada [39] (Fig. 1). The average recA–glnII–rpoB gene sequences (1411 bp) similarity between the type strain ERR11T and other strains in the novel species was in the range 99–100% (data not shown). The closest species was 10.1601/nm.25806 [39] followed by 10.1601/nm.1462 [40]. The similarity between strains in the novel group and strains in the closest species was 96% and they all showed 95% average gene sequence similarity with strains in 10.1601/nm.1462 (Table 5). The type strain ERR11T showed 94–95% similarity of recA–glnII–rpoB gene sequence with the type strains of neighbor branches; 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T [37], 10.1601/nm.23808 10.1601/strainfinder?urlappend=%3Fid%3DCGMCC+1.10947 T [41], 10.1601/nm.27386 10.1601/strainfinder?urlappend=%3Fid%3DLMG+26795 T [42, 43] and 10.1601/nm.27430 58 2–1T [44].
Fig. 1.

Maximum Likelihood phylogenetic tree reconstructed based on recA-glnII-rpoB concatenated nucleotide sequences, showing the relationships between 10.1601/nm.30737 sp. nov. (in green) and recognized species of the genus 10.1601/nm.1459 as well as the position of type strain 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+1007 T.The tree was constructed by using General Time Reversible model using MEGA version 7. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.2999). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 31.7544% sites). Bootstrap values (100 replicates) are indicated at the branching points. Reference type strains are indicated with superscript ‘T’. Bar, % estimated substitutions. GenBank accession numbers of the sequences (recA, glnII, rpoB in order) are listed in parentheses next to the strains codes. The accession numbers of whole genome sequenced strains are indicated with bold*. Abbreviations: B, 10.1601/nm.1459; R, 10.1601/nm.1279; sp., species
Table 5.
ANI and DDH Genomic comparison between 10.1601/nm.30737 sp. nov. ERR11T and reference 10.1601/nm.1459 species
| Genome name | NCBI/ENA accession number | MSLA | ANI was computed from protein-coding genes of the genomes using the MiSI program | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |||
| 1 | B. shewense sp.nov. ERR11T | FMAI01000000 | 95.2 | 95.2 | 89.6 | 89.3 | 89.3 | 89.2 | 89.1 | 89.0 | 89.0 | 89.0 | 89.0 | 86.9 | 89.6 | ||
| 2 | B. ottawaense USDA 4 | AXAF00000000 | 96.0 | 53.1 | 99.9 | 90.0 | 90.3 | 90.3 | 89.1 | 90.2 | 89.1 | 89.1 | 90.0 | 89.1 | 87.1 | 90.1 | |
| 3 | B. ottawaense CCBAU15615 | AJQG0100000 | 96.0 | 58.3 | 99.0 | 90.2 | 90.3 | 90.4 | 89.2 | 90.3 | 89.2 | 89.1 | 90.0 | 89.2 | 87.1 | 90.3 | |
| 4 | B. liaoningense CCBAU 83689 | AJQD00000000 | 95.0 | 36.6 | 38.0 | 38.7 | 89.7 | 89.6 | 88.8 | 89.2 | 90.4 | 90.4 | 89.6 | 90.4 | 87.0 | 99.9 | |
| 5 | B. huanghuaihaiense CCBAU 23303T | PRJNA255602 | 94.0 | 35.6 | 39.2 | 39.4 | 37.4 | 91.2 | 90.0 | 90.3 | 89.3 | 89.2 | 89.8 | 89.2 | 87.6 | 89.7 | |
| 6 | B. diazoefficiens USDA 110T | CP011360 | 94.0 | 35.7 | 39.3 | 39.6 | 37.2 | 42.1 | 89.4 | 91.0 | 88.8 | 88.8 | 89.7 | 88.8 | 87.7 | 89.6 | |
| 7 | B.arachidis LMG 26795T | FPBQ01000000 | 94.0 | 35.4 | 35.1 | 35.2 | 34.8 | 37.3 | 35.9 | 89.5 | 88.6 | 88.6 | 89.6 | 88.6 | 87.7 | 88.8 | |
| 8 | B.japonicum USDA 6T | AP012206 | 94.0 | 35.4 | 39.0 | 39.3 | 36.3 | 39.3 | 41.0 | 36.4 | 88.4 | 88.4 | 89.3 | 88.5 | 87.5 | 89.2 | |
| 9 | B.yuanmingense CCBAU 10071T | FMAE00000000 | 94.0 | 34.7 | 34.7 | 35.1 | 38.3 | 35.3 | 34.5 | 34.0 | 33.6 | 100.0 | 89.1 | 98.2 | 86.8 | 90.3 | |
| 10 | B.yuanmingense CGMCC1.3531 | PRJNA255601 | 94.0 | 34.7 | 34.7 | 35.0 | 38.3 | 35.5 | 34.5 | 33.8 | 33.6 | 100.0 | 90.0 | 98.2 | 86.8 | 90.3 | |
| 11 | B.daqingense CGMCC 1.10947T | PRJNA255603 | 94.0 | 34.9 | 38.3 | 38.5 | 37.0 | 37.8 | 37.7 | 34.0 | 36.7 | 35.1 | 35.1 | 89.1 | 86.8 | 89.7 | |
| 12 | B.yuanmingense CCBAU 35157 | AJQL00000000 | 94.0 | 34.7 | 35.0 | 35.1 | 38.4 | 35.5 | 34.5 | 34.0 | 33.7 | 82.5 | 82.5 | 35.0 | 86.8 | 90.4 | |
| 13 | B. manausense BR3351T | LJYG00000000 | 94.0 | 31.1 | 58.1 | 31.6 | 31.5 | 32.4 | 32.3 | 32.5 | 32.2 | 31.1 | 31.1 | 30.9 | 31.0 | 87.0 | |
| 14 | B.liaoningense CCBAU 05525 | AJQC00000000 | 95.0 | 36.6 | 38.3 | 39.1 | 99.5 | 37.7 | 37.6 | 34.8 | 36.7 | 38.2 | 38.2 | 37.3 | 38.3 | 31.6 | |
| DDH values were predicted by the Genome-to-Genome Distance calculator 2.0, formula 2 | |||||||||||||||||
The numbers in MLSA column indicate recA, glnII, rpoB concatenated gene sequence similarities between ERR11T and reference strains. The numbers below the diagonal are DDH values predicted between pairwise genomes. The numbers above the diagonal are ANI values between genomes; in all ANI calculations AF was > = 60%. Reference type strains are indicated with superscript ‘T’; B, Bradyrhizobium
Minimum Information about the Genome Sequence is provided in Table 1 and the Additional file 1: Table S1. The type strain ERR11T is a rod–shaped Gram–negative strain and has a dimension of 1.0–2.3 μm length and 0.7–1.0 μm width (Fig. 2). The species includes slow–growing bacteria, forming creamy, raised, smooth margin colonies of 1–2 mm in diameter after 7–10 days of incubation on YEM agar plates at 28 °C. The bacteria are able to grow at 15 °C–30 °C temperature, in 0.0–0.5% NaCl concentrations and in the pH range 5–10. The type strain ERR11T and all other strains in the novel group were not able to grow at pH 4, at 4 °C and 35 °C, and in the 1–5% NaCl range (Additional file 1: Table S1). The carbon source utilization pattern of the type strain ERR11T and other strains was tested as previously described [22] using Biolog GN2 plates with 95 carbon sources, following the manufacturer’s guideline [45]. Concisely, bacterial colonies grown on YEM agar were transferred and incubated on R2A media. Bacterial suspension was made by transferring colonies from R2A media into 0.5% (w/v) saline solution. Then, each of the wells of the Biolog GN2 Microplate was filled with 150 μl of the suspension. The results were recorded as positive when the wells turned purple after 4, 24, 48 h or 96 h incubation at 28 °C [46]. The carbon source utilization characteristics are presented in Additional file 2: Table S2. In general, the test strains showed a positive reaction for 66 of the carbon sources and negative reaction for 29 of the carbon sources (Additional file 2: Table S2). Despite that the diversity in carbon utilization patterns was minimal among test strains and between reference strain 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T [37], only the test strains responded positively for adonitol, xylitol, and cis–aconitic acid carbon sources.
Table 1.
Classification and general features of Bradyrhizobium shewense sp. nov. ERR11T and B. yuanmingense CCBAU 10071T [94]
| MIGS ID | Property | ERR11T | CCBAU 10071T | ||
|---|---|---|---|---|---|
| Term | Evidence code | Term | Evidence code | ||
| Domain Bacteria | TAS [95] | Domain Bacteria | TAS [95] | ||
| Phylum 10.1601/nm.808 | TAS [96] | Phylum 10.1601/nm.808 | TAS [96] | ||
| Class 10.1601/nm.809 | TAS [97, 98] | Class 10.1601/nm.809 | TAS [97, 98] | ||
| Classification | Order 10.1601/nm.1277 | TAS [98, 99] | Order 10.1601/nm.1277 | TAS [98, 99] | |
| Family 10.1601/nm.1458 | TAS [98, 100] | Family 10.1601/nm.1458 | TAS [98, 100] | ||
| Genus 10.1601/nm.1459 | TAS [3] | Genus 10.1601/nm.1459 | TAS [3] | ||
| Species 10.1601/nm.30737 sp. nov. | IDA | Species 10.1601/nm.1463 | TAS [37] | ||
| Type strain ERR11T | IDA | Type strain CCBAU 10071T | TAS [37] | ||
| Gram stain | Negative | IDA | Negative | IDA | |
| Cell shape | Rod | IDA | Rod | IDA | |
| Motility | Motile | IDA | Motile | IDA | |
| Sporulation | Non-sporulating | IDA | Non-sporulating | IDA | |
| Temperature range | Mesophile | IDA | Mesophile | TAS [37] | |
| Optimum temperature | 28 °C | IDA | 28 °C | TAS [37] | |
| pH range; Optimum | 5–10; 7 | IDA | 6.5–7.5; 7 | TAS [37] | |
| Carbon source | Varied (Additional file 2) | IDA | Varied | TAS [37] | |
| MIGS-6 | Habitat | Soil, root nodule | [4] | Soil, root nodule | TAS [37] |
| MIGS-6.3 | Salinity | Non-halophile | IDA | Non-halophile | TAS [37] |
| MIGS-22 | Oxygen requirement | Aerobic | IDA | Aerobic | TAS [37] |
| MIGS-15 | Biotic relationship | Free living, symbiotic | IDA | Free living, symbiotic | TAS [37] |
| MIGS-14 | Pathogenicity | Non-pathogenic | NAS | Non-pathogenic | NAS |
| MIGS-4 | Geographic location | Central Ethiopia | [4] | Beijing, China | TAS [37] |
| MIGS-5 | Sample collection | September, 2007 | [4] | 1995 | TAS [37] |
| MIGS-4.1 | Latitude | 08o 59' 38" | [4] | Not reported | TAS [37] |
| MIGS-4.2 | Longitude | 038o 4' 18.5" | [4] | Not reported | TAS [37] |
| MIGS-4.4 | Altitude | 2327 | [4] | Not reported | TAS [37] |
Evidence codes – IDA Inferred from Direct Assay, TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e.,not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [101]
Fig. 2.

Gram stain and dimensions of 10.1601/nm.30737 sp. nov. ERR11T and 10.1601/nm.1463 CBAU1007T
Type strain 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T and other strains in 10.1601/nm.1463 were first described as distinct species using phenotypic features, SDS–PAGE analysis of whole–cell proteins, DNA–DNA hybridization and16S rRNA gene sequence analyses [37]. In agreement with the previous study, in this study based on recA–glnII–rpoB sequence analysis, the strains belonging to 10.1601/nm.1463 formed a district branch in Fig. 1. 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T is motile and Gram–negative. The rod–shaped form (Fig. 2) has dimensions of approximately 0.5 μm in width and 1.5–2.0 μm in length. It is slow–growing, forming colonies with about 1–2 mm diameter after 7 days incubation at 28 °C on YMA. The optimum growth temperature reported was between 25 °C and 30 °C [37]. The organism grows best at pH 6.5–7.5 and growth recorded negative at pH 5.0 and pH 10.0, 10 °C or 40 °C and with 1.0% NaCl in YEMA [37]. Minimum Information about the Genome Sequence (MIGS) of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T is provided in Table 1.
Symbiotaxonomy
The symbiotic properties of the strains in 10.1601/nm.30737 sp. nov. was studied in our previous study [5]. The strains recovered from nodules of Indigofera spp. [30] and Crotalaria spp. [29] formed an effective symbiotic association with the original host plants and also on soybean plants [5]. The type strain ERR11T and other strains were again tested in this study for nodulation and nitrogen fixation ability on E. brucei [26], Indigofera arrecta [47] and Crotalaria juncea [48] as well as on food legumes soybean and peanut (Arachis hypogaea) [49]. All the sterilization and germination methods for I. arrecta [47] and C. juncea [48] seeds were as described previously [5]. Seeds from E. brucei [26], soybean and peanut were sterilized by soaking in 70% alcohol for 3 min and a sodium hypochlorite solution for 3 min followed by rinsing with 5–6 changes of sterilized water. E. brucei [26] seeds were germinated at room temperature (at about 25 °C) on 0.75% water agar or by wrapping with a sterilized paper towel. The soybean and peanut were germinated at 28 °C on 0.75% water agar. The symbiotic characteristics of 10.1601/nm.30737 sp. nov. strains are presented in Additional file 1: Table S1. The results show that the type strain ERR11T and other strains obtained from E. brucei [26], Crotalaria spp. [29] and Indigofera spp. [30] formed an effective symbiosis with E. brucei [26], I. arrecta [47], soybean or peanut plants, suggesting the same origin of symbiotic genes among the rhizobia nodulating the legume plants E. brucei, I. arrecta, soybean or peanut. Only the strains from Crotalaria spp. [29] and Indigofera spp. [30] were able to form effective nodules on C. juncea plants [48]. Strains from E. brucei [26] including the type strain were unable to form effective symbiotic associations with soybean plants. 10.1601/nm.1463 is 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T was isolated from the nodules of the Lespedeza cuneata [50] legume in Beijing, China. In addition to its original host, the strain was also able to form an ineffective symbiotic association with Medicago sativa [51] and Melilotus albus [37, 52].
Genome sequencing information
Genome project history
Type strains ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T were sequenced at the DOE–JGI as part of the Genomic Encyclopedia of Bacterial and Archaeal Type Strains, Phase III: the genomes of soil and plant–associated and newly described type strains sequencing project. The plant and soil associated bacteria were considered for sequencing to understand better their environmental and agricultural importance from the sequence information. The sequencing project was also designed to produce genome sequence data that can be used for bacterial classification studies and for a description of a new species using ANI and Genome–to–Genome–Distance values [36]. Based on our previous MLSA, the type strain ERR11T together with other strains formed a distinctive phylogenetic group without including any known 10.1601/nm.1459 species, and this group representing most likely a new species [5]. Therefore, the aim of the genome sequencing of ERR11T was to describe the group as a new species by comparing the genome sequence data of ERR11T with the genome sequences of other 10.1601/nm.1459 species present in public databases. For this purpose, the type strain 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T [37] was also sequenced in this study to be used as a reference for our genome sequence comparison analysis. The ERR11T genome project is deposited at the DOE–JGI genome portal [53] as well as at European Nucleotide Archive [54] under accession numbers FMAI01000001–FMAI01000102. The genome sequence of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T is also available at DOE–JGI genome portal [53] and at European Nucleotide Archive [55] under accession numbers FMAE01000001–FMAE01000108. The Sequencing, assembling, finishing, and annotation were performed by the DOE–JGI [53]. The genome projects information is depicted in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term, ERR11T | Term, CCBAU 10071T |
|---|---|---|---|
| MIGS 31 | Finishing quality | High-quality draft | High-quality draft |
| MIGS-28 | Libraries used | Illumina std. shotgun library | Illumina std. shotgun library |
| MIGS 29 | Sequencing platforms | Illumina HiSeq 2500, Illumina HiSeq 2500–1 TB | Illumina HiSeq 2500, Illumina HiSeq 2500–1 TB |
| MIGS 31.2 | Fold coverage | 225.2X | 279.9× |
| MIGS 30 | Assemblers | Velvet (version 1.2.07), Allpaths–LG (version r46652) | Velvet (version 1.2.07), Allpaths–LG (version r46652) |
| MIGS 32 | Gene calling method | Prodigal | Prodigal |
| Locus Tag | ATF67 | ATF66 | |
| GenBank ID | FMAI01000000 | FMAE01000000 | |
| GenBank Date of Release | 01-AUG-2016 | 01-10.1601/strainfinder?urlappend=%3Fid%3DAUG+2016 | |
| GOLD ID | Gp0108279 | Gp0108280 | |
| BIOPROJECT | PRJNA303280 | PRJNA303279 | |
| MIGS 13 | Source Material Identifier | ERR11 | CCBAU 10071 |
| Project relevance | Symbiotic N2 fixation, agriculture | Symbiotic N2 fixation, agriculture |
Growth conditions and genomic DNA preparation
The growth conditions and DNA isolation methods were as previously described [22]. In brief, the strains ERR11T =10.1601/strainfinder?urlappend=%3Fid%3DHAMBI+3532 T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T = 10.1601/strainfinder?urlappend=%3Fid%3DLMG+21827 T were grown on YEM agar plates at 28 °C for 7–10 days and a pure colony of the cultures was transferred and grown in YEM broth till the culture reached late–logarithmic phase. The genomic DNAs were extracted from cell pellets following the CTAB DNA extraction protocol of the DOE–JGI [56].
Genome sequencing and assembly
Strains ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T were sequenced at the DOE–JGI by using the Illumina technology [57]. An Illumina std. shotgun library was constructed and sequenced using the Illumina HiSeq 2000 platform which produced 7,620,202 reads totaling 1150.7 Mb for ERR11T and 9,923,442 reads counting 1498.4 Mb of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T. Details regarding the general aspects of library construction and sequencing methods can be found at the DOE–JGI website [53]. Artifacts from Illumina sequencing and library preparation were removed by passing all raw Illumina sequence data through DUK, filtering program developed by DOE–JGI [58]. The filtered Illumina reads were assembled first using Velvet (version 1.2.07) [59] and 1–3 kb simulated paired–end reads were created from Velvet contigs using wgsim (version 0.3.0) [60]. The Illumina reads were then assembled with the simulated read pairs using Allpaths–LG (version r46652) [61]. The final draft genome assembly comprises 9.2 Mb genome size containing 107 contigs in 102 scaffolds for strain ERR11T; 109 contigs in 108 scaffolds with a total size of 8.2 Mb for 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T. The final assembly was based on 1399.7 Mb Illumina data and 225.2X input read coverage for the strain ERR11T; 1399.7 Mb Illumina data and 279.9× input read coverage for the strain 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T.
Genome annotation
Genes were first predicted by the Prodigal [62] program at the DOE–JGJ annotation pipeline [63], followed by a round of manual curation using GenePRIMP [64]. The predicted CDSs were translated and functionally annotated by searching against the NCBI non redundant database, UniProt, TIGRFam, Pfam, KEGG, COG, and InterPro databases. The tRNAScanSE tool [65] was used to identify tRNA genes and ribosomal RNA genes were predicted by searches against the ribosomal RNA genes in the SILVA database [66]. Other non–coding RNAs such as the RNA components of the protein secretion complex and the RNase P were identified by searching the genomes for the corresponding Rfam profiles using INFERNAL [67]. Additional gene prediction and functional annotation of the predicted genes were accomplished by using the Integrated Microbial Genomes (IMG) platform [68] developed by DOE–JGI [69].
Genome properties
The genome of ERR11T consists 102 scaffolds with a total size of 9,163,226 bp and a 63.2% G + C content. From a total 8634 genes, 8548 were protein–coding genes and 86 RNA encoding genes. The genome of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T is arranged in 108 scaffolds and has a size of 6,928,453 bp with a 63.8% G + C content and of the 7861 predicted genes 7776 were protein–coding genes and 85 were RNAs–coding genes (Table 3). The majority of the protein–coding genes of ERR11T (72.8%) and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T (72.6%) were annotated to functions and the remaining 2266 (26.3%) and 2073 (26.4%) genes were without a functional prediction for ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T, respectively. About 62% CDSs of ERR11T and 63% CDSs of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T were assigned to COG functional categories. The distribution of the genes assigned into COGs functional categories is presented in Table 4.
Table 3.
Genome statistics
| Attribute | ERR11T | CCBAU 10071T | ||
|---|---|---|---|---|
| Value | % of Total | Value | % of Total | |
| Genome size (bp) | 9,163,226 | 100% | 8,201,522 | 100% |
| DNA coding (bp) | 8548 | 99% | 6,928,453 | 84.48% |
| DNA G + C (bp) | 5,792,812 | 63.22% | 5,230,108 | 63.77% |
| DNA scaffolds | 102 | 100% | 108 | 100% |
| Total genes | 8634 | 100% | 7861 | 100% |
| Protein coding genes | 8548 | 99% | 7776 | 98.92% |
| RNA genes | 86 | 1% | 85 | 1.08% |
| Pseudo genes | not determined | not determined | ||
| Genes in internal clusters | 1889 | 21.88% | 1457 | 18.53% |
| Genes with function prediction | 6282 | 72.76% | 5703 | 72.55% |
| Genes assigned to COGs | 5346 | 61.92% | 4913 | 62.50% |
| Genes with Pfam domains | 6555 | 75.92% | 6014 | 76.50% |
| Genes with signal peptides | 924 | 10.70% | 812 | 10.33% |
| Genes with transmembrane helices | 1956 | 22.65% | 1772 | 22.54% |
| CRISPR repeats | 3 | 1 | ||
Table 4.
Number of genes associated with general COG functional categories
| Code | ERR11T | CCBAU 10071T | Description | ||
|---|---|---|---|---|---|
| Value | %age | Value | %age | ||
| J | 225 | 3.65% | 231 | 4.08% | Translation, ribosomal structure and biogenesis |
| A | 0 | 00% | 0 | 00% | RNA processing and modification |
| K | 458 | 7.44% | 392 | 6.93% | Transcription |
| L | 135 | 2.19% | 143 | 2.53% | Replication, recombination and repair |
| B | 2 | 0.03% | 2 | 0.04% | Chromatin structure and dynamics |
| D | 36 | 0.58% | 39 | 0.69% | Cell cycle control, Cell division, chromosome partitioning |
| V | 162 | 2.63% | 134 | 2.37% | Defense mechanisms |
| T | 288 | 4.68% | 263 | 4.47% | Signal transduction mechanisms |
| M | 316 | 5.13% | 300 | 5.3% | Cell wall/membrane biogenesis |
| N | 106 | 1.72% | 109 | 1.93% | Cell motility |
| U | 85 | 1.38% | 113 | 2% | Intracellular trafficking and secretion |
| O | 245 | 3.98% | 221 | 3.9% | Posttranslational modification, protein turnover, chaperones |
| C | 440 | 7.15% | 378 | 6.68% | Energy production and conversion |
| G | 438 | 7.11% | 339 | 5.97% | Carbohydrate transport and metabolism |
| E | 665 | 10.8% | 623 | 11.01% | Amino acid transport and metabolism |
| F | 98 | 1.59% | 94 | 1.66% | Nucleotide transport and metabolism |
| H | 309 | 5.02% | 271 | 4.79% | Coenzyme transport and metabolism |
| I | 413 | 6.71% | 398 | 7.03% | Lipid transport and metabolism |
| P | 358 | 5.81% | 311 | 5.49% | Inorganic ion transport and metabolism |
| Q | 266 | 4.32% | 278 | 4.91% | Secondary metabolites biosynthesis, transport and catabolism |
| R | 684 | 11.11% | 626 | 11.06% | General function prediction only |
| S | 353 | 5.73% | 333 | 5.83% | Function unknown |
| – | 3288 | 38.08% | 2948 | 37.5% | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Insights from the genome sequence
Genome wide comparative analysis
The strains belonging to 10.1601/nm.30737 sp. nov. formed their own group close to 10.1601/nm.25806 branch on the phylogenetic tree reconstructed based on recA–glnII–rpoB concatenated gene sequences (Fig. 1). Comparative analysis of the genome sequences between type strain ERR11T and relatively close references was thus done for detail taxonomic study of the unique group and to describe it as a novel species. Among the reference genomes presented in the Fig. 1, completely sequenced 10.1601/nm.1460 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+6 T [3, 70] and 10.1601/nm.24498 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T [71, 72] and draft sequences of 10.1601/nm.27386 26795T [42, 43], 10.1601/nm.1460 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 [73], 10.1601/nm.1459 sp. 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 [74], 10.1601/nm.1462 strains (10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689, 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525) [74], 10.1601/nm.1463 strains (10.1601/strainfinder?urlappend=%3Fid%3DCGMCC+1.3531, 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+35157) [37, 74], 10.1601/nm.23808 10.1601/strainfinder?urlappend=%3Fid%3DCGMCC+1.10947 T [41], 10.1601/nm.23259 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+23303 T [75] and 10.1601/nm.25639 BR3351T [76] were collected from the DOE–JGI genome portal [53] as well as from the GenBank database [77]. The type strain 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T [37] sequenced in this study was also included in the comparative analyses.
To evaluate the similarity between the genomes, we calculated genome–wide ANI by averaging the nucleotide identity of orthologous genes identified as bidirectional best hits as previously described [22, 24]. Based on this method, 96.5% ANI and 0.6 AF were set as the threshold values between strains in the same species [24]. In addition, DDH values were predicted between the genomes by using Genome–to–Genome Distance Calculator (GGDC) [78, 79]. This program computes the distance between genomes using three different formals: 1, high–scoring segment pairs (HSPs) /total length; 2, identities /HSP length; 3, identities/total length. The formula 2 proved to be a robust and recommended method for draft genome distance comparison [80].
The ANI values and DDH estimated results are presented in Table 5. The soybean–nodulating strain 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 was previously classified as 10.1601/nm.1460 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 based on sequence analysis of 16S rRNA gene and the internally transcribed spacer region of the 5′–23S rRNA gene [73]. However, the ANI value between type the strain 10.1601/nm.1460 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+6 T and USD 4 was 90.2% and the DDH value between the two was 39.0%, suggesting that 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 does not belong to the 10.1601/nm.1460 species. The strains 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 were tightly grouped with strains in 10.1601/nm.25806 on the phylogenetic tree in Fig. 1. Both 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 shared 99% recA–glnII–rpoB sequence identity with 10.1601/nm.25806 OO99T and 10.1601/nm.25806 OO100 [39]. Even though the reference strains OO99T and OO100 were not sequenced and not included in our ANI calculation, the recA–glnII–rpoB sequence analysis result strongly indicates that both 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 belong to 10.1601/nm.25806. The ANI values between type strain ERR11T and references ranged from 86.8 to 95.2%, which is below 96.5%, the value for strains belong to the same species [24]. The closest strains were 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615, with 95.2% ANI values followed by 10.1601/nm.1462 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525, both sharing 89.6% ANI with strain ERR11T. The DDH predicted between strain ERR11T and references were in the range of 31.1_58.5%. The highest DDH value obtained between ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 (58.5%) followed by 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 (53.1%) is below the threshold of 70%, which is commonly used value for species delineation [78, 79]. In agreement with recA–glnII–rpoB gene sequence analysis, both ANI and DDH results revealed that the closest strains for ERR11T were strains belong to 10.1601/nm.25806 group (10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 and 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4). Nevertheless, both the ANI and DDH values between ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+15615 or 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 were below the cutoff values of the strains of the same species, suggesting that ERR11T belong to the novel group.
Shared orthologous protein clusters between the genomes of ERR11T and the closest reference strains 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689 were identified using an OrthoVenn program [81] as described previously [22]. The orthologous clusters are shown in a Venn diagram (Fig. 3). The number of protein clusters identified in each of ERR11T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689 was 6850, 5897 and 6923, respectively. In the genome of ERR11T, 99 of the clusters were identified as unique protein clusters without homologs in the other genomes. In 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689, 44 and 77 protein clusters respectively, were also identified as unique clusters with no detectable homologous with other genomes. Of the total proteins used in the analysis 1456, 2028, 1102 were single copy gene clusters in ERR11T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689, respectively. Of the clusters, in total 5310 homologous protein clusters were shared in common by all of the three genomes. Strain ERR11T shares about 76.7% (6560) of its proteins with 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 64.4% (5501) clusters with 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689. Based on the pairwise comparison, ERR11T shared the highest number with strain 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 with 1250 protein clusters and ERR11T shared only 191 protein clusters with 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689. This result is in accordance with the phylogenetic tree (Fig. 1), ANI and DDH results (Table 5), supporting that strain 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 (in the 10.1601/nm.25806 species group) is more closely related to ERR11T compared to strain 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689 (in 10.1601/nm.1462).
Fig. 3.
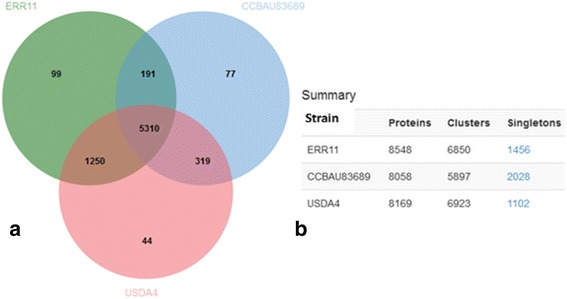
Venn diagram (panel a) plotted by OrthoVenn program shows shared orthologous protein clusters between three genomes (in the center): 10.1601/nm.30737 sp.nov. ERR11T, 10.1601/nm.25806 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4 and 10.1601/nm.1462 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+83689. The total number of protein sequences, the number of protein clusters comprising multiple protein families and also the number of singletons i.e. protein with no orthologous are summarized in (panel b) for each genome
Comparative analysis of genes linked to symbiosis and denitrification
Symbiotic genes
The nodulation genes (nod, nol, noe) for the synthesis of the backbone of LCO Nod factors and substituent groups and genes coding for nitrogen fixation (nif, fix) are required in rhizobia–legume symbiosis [70, 72]. In order to search the symbiotic genes in ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T, the genomes were assembled against completely sequenced 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T and 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+6 T using the Genome Gene Best Homologs package from program IMG–ER [69]. In addition, the symbiotic genes were also compared against other draft 10.1601/nm.1459 genomes: 10.1601/strainfinder?urlappend=%3Fid%3DLMG+26795 T, 10.1601/strainfinder?urlappend=%3Fid%3DCGMCC+1.10947 T, 10.1601/strainfinder?urlappend=%3Fid%3DCGMCC+1.10948 T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4, and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525. To see the arrangement of symbiotic genes, the genome of ERR11T and references were aligned using the progressive Mauve alignment method [82]. Summary of the symbiotic genes identified in ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T and their locations in the genomes and resemblance with genes in the reference genomes are shown in Additional file 3: Table S3. The main nodulation genes; nolY–nolA–nodD2–nodD1YABCSUIJ–nolO–nodZ were identified in scaffolds Ga0061098_1039 and Ga0061099_1014, in the genome of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T, respectively. The result of the Mauve alignment (Fig. 4) shows that these genes are homologous and organized in the same region (module) similarly as the genes found in the genome of 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4, and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525. Additional nodulation genes of ERR11T are scattered in scaffolds Ga0061098_1005 (nodWV, nodM, noeL, nolXWTUV), Ga0061098_1016 (nodU), Ga0061098_1006 (nodT) and Ga0061098_1031 (noeE, noeI). These genes are also identified in the genome of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T in Ga0061099_1013 and Ga0061099_1018, Ga0061099_1014, Ga0061099_1005 and Ga0061099_1022, respectively.
Fig. 4.
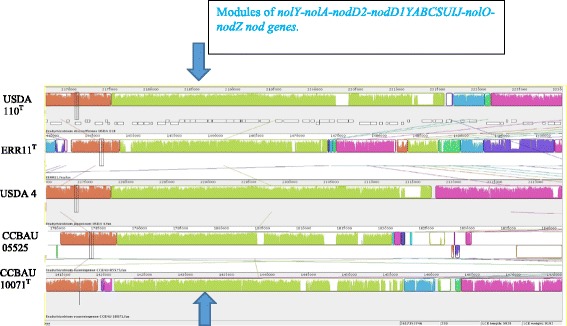
Mauve alignment comparing the genome of ERR11T with the genome of 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4, 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525 and CCBAU 10071T. The nod genes: nolY-nolA-nodD2-nodD1YABCSUIJ-nolO-nodZ indicated by the arrows are homologous and organized similarly between the genomes
In the genome of ERR11T, the genes coding for the nitrogen–fixing nitrogenase complex [83] are mainly located in scaffolds Ga0061098_1005 (nifDKENX–nifT–nifB–nifZ–nifHQV–fixBCX), and Ga0061098_1039 (fixR–nifA–fixA). The nif/fix genes in the genome of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T are distributed in scaffolds Ga0061099_1013 (nifDKENX), Ga0061099_1041 (nifT–nifB–nifZ), Ga0061099_1036 (nifHQV–fixBCX), and Ga0061099_1014 (fixR–nifA–fixA). The fix genes (fixK2–fixJL–fixNOPGHIS), which are required for creating microoxic respiration for the rhizobia during symbiosis, are also conserved in the genomes of ERR11T (in scaffold Ga0061098_1024) and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T (in scaffold Ga0061099_10014) in a similar fashion as the homologous genes found in 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T. Generally, the nodulation and nitrogen fixation genes of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T showed 70.0–100% sequence similarity with homologous genes found in the reference genomes of 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+6 T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4, 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+23303 T, 10.1601/strainfinder?urlappend=%3Fid%3DCGMCC+1.10947 T, and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525 (Additional file 3: Table S3). The nodulation and nitrogen fixation genes of ERR11T mostly showed the highest sequence similarities (>90%) specifically with homologous genes found in the genome of peanut–nodulating strain 10.1601/strainfinder?urlappend=%3Fid%3DLMG+26795 T, suggesting that these strains may have a similar origin of symbiotic genes.
Nitrogen fixation in symbiosis is an ATP–dependent energy intensive reaction, where energy is released in the form of H2 as a result of the reduction of N2 by nitrogenase. The rhizobia which have hydrogen–uptake systems are capable of recycling the released H2 in the rhizobia–legume symbiosis [84]. This way some rhizobia increase the energy efficiency in symbiosis and consequently the nitrogen–fixation and legume productivity. The hydrogenase uptake complex is coded by clusters of hupNCUVSLCDFGHIJK, hypABFCDE, and hoxXA genes [70, 72, 85, 86]. Clusters of hupSLCFHK and hypBFDE genes were identified in the genomes of ERR11T in scaffold Ga0061098_1005 and in 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T in scaffold Ga0061099_1013 (Additional file 3: Table S3). The composition of hydrogenase genes in the clusters hup, hyp and hox and their expression can be different between rhizobial species and are also missing in some rhizobia [70, 72, 84]. Rhizobia with the functional hydrogenase uptake system, such as strain 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T contained a complete set of hup–hyp–hox genes [72]. In the genomes of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T, some of the genes are missing or incomplete. Therefore, further study and complete sequencing may confirm if the hydrogenase uptake system is functional in these strains.
Denitrifying genes
Denitrification is a process by which NO3 − and NO2 − are reduced to N2 when NO3 − or NO2 − is used by microorganisms as a final electron acceptor for respiration as an alternative in oxygen limitation. NO and N2O are produced as intermediate products during this process [87]. Thus, denitrification result in nitrogen losses from terrestrial and aquatic ecosystems and also contribute to the production of a potent greenhouse gas, N2O. The denitrification is common among the bacteria in the 10.1601/nm.808 class and also in Archaea [88]. Symbiotic nitrogen–fixing rhizobia, particularly species belonging to 10.1601/nm.1459 were reported to be involved in the denitrification process in low oxygen environments [89]. All or some of the genes for NO3 −, NO2 −, NO and N2O reductions were found in several rhizobial species investigated thus far [90] and emission of N2O by symbiotic rhizobia inside the root nodules was reported [91, 92]. Nitrogen–fixing 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T is known to denitrify as free living and also in the symbiotic condition in root nodules of soybean [89, 93]. Strain 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T requires napEDABC, nirK, norCBQD, and nosRZDFYLX gene clusters for NO3 −, NO2 −, NO and N2O reductase, respectively [90]. In the genome of ERR11T, the napCBADE, norDQBCE, nosRZDYL cluster of genes are present in the scaffolds Ga0061098_1005, Ga0061098_1001, and Ga0061098_1006, respectively. The gene for nitrite reductase (nir) was not found in ERR11T. Therefore, the nitrite reductase activity may be lacking in ERR11T and denitrification in this strain may depend only on nitrate, nitric oxide, and nitrous oxide reductase reactions. The genome of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T harbors only denitrifying genes napEDABC and norCBQD for nitrate and nitric oxide reduction, respectively (Additional file 3: Table S3). Further experimental study with appropriate methods and techniques can help to understand better the presence of denitrification enzyme activities in the type strains ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T and to confirm if the type strains are involved in the denitrification process and N2O emission.
Conclusion
In this study, we present the genome sequences of 10.1601/nm.30737 sp. nov. strain ERR11T and the type strain 10.1601/nm.1463 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T. The draft genome size of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T is about 9.2Mbp and 8.2Mbp, respectively. Type strain 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T was selected for sequencing to be used as a reference for our comparative genomic analysis. The genomes of the type strains ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T carry genes for nodulation, nitrogen fixation, the hydrogen–uptake system as well as genes for denitrification. The nod genes nolY–nolA–nodD2–nodD1YABCSUIJ–nolO–nodZ in the genomes of ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T are organized similarly as homologous genes identified in the genomes of 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T, 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4, and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+05525. The nodulation and nitrogen fixation genes of ERR11T share high sequence similarity with peanut–nodulating type strain 10.1601/nm.27386 10.1601/strainfinder?urlappend=%3Fid%3DLMG+26795 T [42, 43] The denitrification genes nap, nor and nos of ERR11T and nap and nor of 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T are homologous to the genes in found in the genome of 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+110 T, a known denitrifying rhizobium, indicating that ERR11T and 10.1601/strainfinder?urlappend=%3Fid%3DCCBAU+10071 T may involve in reduction of nitrate, nitric oxide, or nitrous oxide. Based on the phylogenetic analyses of recA–glnII–rpoB sequences, the strains (ERR2A, ERR2B, ERR11, ERR13, 10.1601/strainfinder?urlappend=%3Fid%3DCIR+42, 10.1601/strainfinder?urlappend=%3Fid%3DCSR+10B, IAR8 and AURI6) belonging to the novel species formed a unique group within the genus 10.1601/nm.1459 . In order to verdict this result, comparative genomic analyses based on ANI calculation and DDH methods were done. The results from both ANI and DDH supported the result from the phylogenetic analysis, in which the genome of the type strain ERR11T showed 95.2% ANI and 53.1 DDH similarity with the closest reference strain 10.1601/strainfinder?urlappend=%3Fid%3DUSDA+4. These values are lower than the 96.5% ANI and 70% DDH cutoff values designed for strains of the same species. These results confirm that 10.1601/nm.30737 sp. nov. should be considered as a new 10.1601/nm.1459 species. Therefore, based on the phylogenetic analysis, ANI and DDH results and by including phenotypic characteristics, we formally propose the creation of 10.1601/nm.30737 sp. nov. that contains the strain ERR11T (= 10.1601/strainfinder?urlappend=%3Fid%3DHAMBI+3532 T=10.1601/strainfinder?urlappend=%3Fid%3DLMG+30162 T). The type strain forms an effective nitrogen–fixing symbiosis with E. brucei [26], I. arrecta (47) and peanut.
Description of 10.1601/nm.30737 sp. nov.
10.1601/nm.30737 (she.wen’se. L. neut. Adj. shewense of Shewa, pertaining to Shewa, the region in Ethiopia, where the type strain was obtained). The bacteria are non–spore–forming, Gram–negative rods with a size of 1.0–2.4 μM in length and 0.7–1.0 μm width. Strains included in species are slow–growing, forming creamy, raised and smooth margin colonies of 1–2 mm in diameter after 7 to 10 days of incubation on YEMA plate containing Congo red at 28 °C and pH 7 optimal growth conditions. The strains are able to grow at 15 °C–30 °C, in 0.0–0.5 NaCl and at 5–10 pH ranges. They do not grow at pH 4, at 4 °C and at 35 °C and in 1–5% NaCl. In general, the type and the other strains in this species could ferment the following substrates as carbon sources in Biolog GN2 microplates; Tween 40, Tween 80, adonitol, L–arabinose, D–arabitol, glycogen, N–acetyl–D–glucosamine, adonitol, L–arabinose, D–arabitol, D–cellobiose, I–erythritol, D–fructose, L–fucose, D–galactose, α–D–glucose, α–D–lactose, lactulose, D–mannitol, D–mannose, D–melibiose, β–methyl–D–glucoside, D–raffinose, L–rhamnose, D–sorbitol, sucrose, turanose, xylitol, pyruvic acid methyl ester, acetic acid, succinic acid mono–methyl–ester, cis–aconitic acid, citric acid, formic acid, D–galactonic acid lactone, D–galacturonic acid, D–gluconic acid, D–glucosaminic acid, D–glucuronic acid, α–Hydroxybutyric acid, β–hydroxybutyric acid, γ–hydroxybutyric acid, p–hydroxy phenylacetic acid, itaconic acid, α–keto butyric acid, α–keto glutaric acid, α–keto valeric acid, D,L–lactic acid, propionic acid, quinic acid, D–saccharic acid, sebacic acid, succinic acid, bromosuccinic acid, succinamic acid, glucuronamide, L–alaninamide, D–alanine, L–alanine, L–alanyl–glycine, L–asparagine, L–aspartic Acid, L–glutamic acid, glycyl–L–aspartic acid, glycyl–L–glutamic acid, L–leucine, L–phenylalanine, L–proline, L–pyroglutamic acid, D–serine, L–threonine, urocanic acid, and glycerol. However, all the strains included in this test failed to oxidize α–cyclodextrin, glycogen, N–acetyl–D–galactosamine, N–acetyl–D–glucosamine, D–cellobiose, I–erythritol, gentiobiose, M–inositol, α–D–lactose, lactulose, D–melibiose, β–methyl–D–glucoside, D–raffinose, melonic acid, L–histidine, hydroxy–L–proline, L–ornithine, D,L–carnitine, γ–amino butyric acid, uridine, thymidine, phenyethyl–amine, putrescine, 2–aminoethanol, 2,3–butanediol, D,L–α–glycerol phosphate, α–D–glucose–1–phosphate, and D–glucose–6–phosphate. The type strain ERR11T was obtained from root nodules of E. brucei [25] growing in Ethiopia. The genome size of the type strain is 9.2Mbp and the genome G + C content is 63.2%. The genome sequence of ERR11T is available at the DOE–JGI genome portal [47] as well as at European Nucleotide Archive [48] from accession number FMAI01000001 to FMAI01000102. The type strain has been deposited in the HAMBI (10.1601/strainfinder?urlappend=%3Fid%3DHAMBI+3532 T) and LMG (10.1601/strainfinder?urlappend=%3Fid%3DLMG+30162 T) culture collections.
Additional files
Phenotypic characteristics of Bradyrhizobium shewense sp. nov. strains. [102, 103] (DOCX 28 kb)
Carbon sources utilization response between Bradyrhizobium shewense sp. nov. strains and reference type strain B. yuanmingense CCBAU 10071T. (DOCX 29 kb)
Symbiotic and denitrifying genes identified in the genomes of Bradyrhizobium shewense. sp. nov. ERR11T and B. yuanmingense CCBAU 10071T. (XLSX 121 kb)
Acknowledgements
Microbiological and molecular activities, and manuscript preparation were supported by the SOILMAN project funded by the Academy of Finland, University of Helsinki. The genome sequencing was performed by DOE–JGI as a part of the DOE–JGI 2014 Genomic Encyclopedia project of Type Strains, Phase III the genomes of soil and plant–associated and newly described type strains. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231. Crotalaria juncea [48] seeds for the nodulation test was kindly provided by Philippe Lajudie from LSTM, Campus de Baillarguet TA A-82/J, IRD, Montpellier Cedex 5, 34398, France. We also thank Belay Berza and Tassew Sirage, postgraduate students at Addis Ababa University in Ethiopia, for providing and delivering the seeds of Erythrina brucei [26] for our nodulation experiment.
Abbreviations
- AF
Alignment fraction
- ANI
Average nucleotide identity
- CTAB
Cetyl Trimethyl Ammonium Bromide
- DDH
DNA-DNA Hybridization
- DOE
Department of Energy
- GOLD
Genomes online database
- H2
Dihydrogen
- IMG
Integrated Microbial Genomes
- IMG-ER
Integrated microbial genomes – expert review
- JGI
Joint Genome Institute
- LCO
Lipochito-Oligosaccharide
- MIGS
Minimum information about a genome sequence
- MiSI
Microbial species identifier
- MLSA
Multilocus sequence analysis
- N2
Dinitrogen
- N2O
Nitrous oxide
- NO
Nitric oxide
- NO2
Nitrite
- NO3
Nitrate
- R2A
Reasoner’s 2A Agar
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- YEMA
Yeast Extract Mannitol Agara
Authors’ contributions
The genome sequencing project was initiated by AAA, KL and WBW. KL and WBW conceived and coordinated the study. AAA isolated the newly described 10.1601/nm.30737 sp. nov. strains, carried out all the microbiological laboratory works, genomic DNA extraction, PCR, phylogenetic analyses, genomic data analyses and drafted the manuscript. TW and NCK were responsible for the genome sequencing, assembly and genome annotation tasks. All authors read, reviewed and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40793-017-0283-x) contains supplementary material, which is available to authorized users.
References
- 1.Masson-Boivin C, Giraud E, Perret X, Batut J. Establishing nitrogen–fixing symbiosis with legumes: how many rhizobium recipes? Trends Microbiol. 2009;17:458–466. doi: 10.1016/j.tim.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Lindström K, Aserse AA, Mousavi SA. Evolution and Taxonomy of Nitrogen-Fixing Organisms with Emphasis on Rhizobia. In: de Bruijn FJ, editor. Biological Nitrogen Fixation. Hoboken, NJ, USA: John Wiley and Sons, Inc; 2015. pp. 21–38. [Google Scholar]
- 3.Jordan DC. Transfer of Rhizobium japonicum Buchanan 1980 to Bradyrhizobium gen. Nov., a genus of slow–growing, root nodule bacteria from leguminous plants. Int J Syst Bacteriol. 1982;32:136–139. doi: 10.1099/00207713-32-1-136. [DOI] [Google Scholar]
- 4.List of described Bradyrhizobium species. http://www.bacterio.net/bradyrhizobium.html.
- 5.Aserse AA, Räsänen LA, Aseffa F, Hailemariam A, Lindström K. Phylogenetically diverse groups of Bradyrhizobium isolated from nodules of Crotalaria spp., Indigofera spp., Erythrina brucei and Glycine max growing in Ethiopia. Mol Phylogenet Evol. 2012;65:595–609. doi: 10.1016/j.ympev.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 6.Beukes CW, Stępkowski T, Venter SN, Cłapa T, Phalane FL, le Roux MM, Steenkamp ET. Crotalaria and Genisteae of the south African great escarpment are nodulated by novel Bradyrhizobium species with unique and diverse symbiotic loci. Mol Phylogenet Evol. 2016:206–18. [DOI] [PubMed]
- 7.Horn K, Parker IM, Malek W, Rodríguez, Echeverria S, Matthew AP. Disparate origins of Bradyrhizobium symbionts for invasive populations of Cytisus scoparius (Leguminosae) in North America. FEMS Microbiol Ecol. 2014:89–98. [DOI] [PubMed]
- 8.Koppell JH, Parker MA. Phylogenetic clustering of Bradyrhizobium symbionts on legumes indigenous to North America. Microbiology. 2012;158:2050–2059. doi: 10.1099/mic.0.059238-0. [DOI] [PubMed] [Google Scholar]
- 9.Parker MA, Rousteau A. Mosaic origins of Bradyrhizobium legume symbionts on the Caribbean island of Guadeloupe. Mol Phylogenet Evol. 2014;77:110–115. doi: 10.1016/j.ympev.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Steenkamp ET, Stepkowski T, Przymusiak A, Botha WJ, Law IJ. Cowpea and peanut in southern Africa are nodulated by diverse Bradyrhizobium strains harbouring nodulation genes that belong to the large pantropical clade common in Africa. Mol Phylogenet Evol. 2008;48:1131–1144. doi: 10.1016/j.ympev.2008.04.032. [DOI] [PubMed] [Google Scholar]
- 11.Stepkowski T, Watkin E, McInnes A, Gurda D, Gracz J, Steenkamp ET. Distinct Bradyrhizobium communities nodulate legumes native to temperate and tropical monsoon Australia. Mol Phylogenet Evol. 2012;63:265–277. doi: 10.1016/j.ympev.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Amann RI, Lin CH, Key R, Montgomery L, Stahl DA. Diversity among fibrobacter strains: towards a phylogenetic classification. Syst Appl Microbiol. 1992;15:23–32. doi: 10.1016/S0723-2020(11)80133-5. [DOI] [Google Scholar]
- 13.Martens M, Dawyndt P, Coopman R, Gillis M, De Vos P, Willems A. Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer. Int J Syst Evol Microbiol. 2008;58:200–214. doi: 10.1099/ijs.0.65392-0. [DOI] [PubMed] [Google Scholar]
- 14.Stackebrandt E, Frederiksen W, Garrity GM, Grimont PAD, Kämpfer P, Maiden MCJ, Nesme X, Rosselló–Mora, Swings R, et al. Report of the ad hoc committee for the reevaluation of the species definition in bacteriology. Int J Syst Evol Microbiol. 2002;52:1043–1047. doi: 10.1099/00207713-52-3-1043. [DOI] [PubMed] [Google Scholar]
- 15.Willems A, Munive A, de Lajudie P, Gillis M. Most Bradyrhizobium groups sequence comparison of 16S–23S rDNA internal transcribed spacer regions corroborates DNA–DNA hybridizations. Syst Appl Microbiol. 2003;26:203–210. doi: 10.1078/072320203322346056. [DOI] [PubMed] [Google Scholar]
- 16.Rosselló–Mora R. DNA–DNA re–association methods applied to microbial taxonomy and their critical evaluation. In: Stackebrandt E, editor. Molecular identification, systematics and population structure of prokaryotes. Berlin: Springer; 2006. pp. 23–50. [Google Scholar]
- 17.Blom J, Kreis J, Spanig S, Juhre T, Bertelli C, Ernst C, Goesmann A. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016; 10.1093/nar/gkw255. [DOI] [PMC free article] [PubMed]
- 18.Scheuner C, Tindall BJ, Lu M, Nolan M, Lapidus A, et al. Complete genome sequence of Planctomyces brasiliensis type strain (DSM 5305T), phylogenomic analysis and reclassification of Planctomycetes including the descriptions of Gimesia gen. Nov., Planctopirus gen. Nov. and Rubinisphaera gen. Nov. and emended descriptions of the order Planctomycetales and the family Planctomycetaceae. Stand Genomic Sci. 2014;9:10. doi: 10.1186/1944-3277-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sizova MV, Chilaka A, Earl AM, Doerfert SN, Muller PA, Torralba M, McCorrison JM, Durkin AS, Nelson KE. E t c. High–quality draft genome sequences of five anaerobic oral bacteria and description of Peptoanaerobacter stomatis gen. Nov., sp. nov., a new member of the family Peptostreptococcaceae. Stand Genomic Sci. 2015;10:37. doi: 10.1186/s40793-015-0027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mousavi SA, Österman J, Wahlberg N, Nesme X, Lavire C, Vial L, Paulin L, de Lajudiee P, Lindström K. Phylogeny of the Rhizobium–Allorhizobium: Agrobacterium clade supports the delineation of Neorhizobium gen. Nov. Syst Appl Microbiol. 2014;37(3):208–215. doi: 10.1016/j.syapm.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Mousavi SA, Willems A, Nesme X, de Lajudie P, Lindström K. Revised phylogeny of Rhizobiaceae: proposal of the delineation of Pararhizobium gen. Nov., and 13 new species combinations. Syst Appl Microbiol. 2015;38(2):84–90. doi: 10.1016/j.syapm.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Aserse AA, Woyke T, Kyrpides NC, Whitman WB, Lindström K. Draft genome sequence of type strain HBR26T and description of Rhizobium aethiopicum sp. nov. Stand Genomic Sci. 2017;12:1–16. doi: 10.1186/s40793-016-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim M, HS O, Park SC, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0. [DOI] [PubMed] [Google Scholar]
- 24.Varghese JN, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, Pati A. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43(14):6761–6771. doi: 10.1093/nar/gkv657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter M. Rosselló–Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci. U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erythrina brucei.http://www.theplantlist.org/tpl1.1/record/ild-2676.
- 27.Teketay D. erythrina burana, a multipurpose tree from Ethiopia. Agrofor Today 2. 1990; 13–14.
- 28.Negash L. Erythrina brucei: propagation attributes, leaf nutrient concentration and impact on barley grain yield. Agrofor Syst. 2002;56:39–46. doi: 10.1023/A:1021182428083. [DOI] [Google Scholar]
- 29.Crotalaria spp. http://www.theplantlist.org/1.1/browse/A/Leguminosae/Crotalaria/.
- 30.Indigofera spp. http://www.theplantlist.org/1.1/browse/A/Leguminosae/Indigofera/.
- 31.Thulin M. Fabaceae. In: Hedberg M, Edwards S, editors. Flora of Ethiopia. Addis Ababa University, Ethiopia, the National Herbarium. 1989. pp. 71–96. [Google Scholar]
- 32.Bogale T, Assenga RH, Mmbaga TE, Friesen DK, Kikafunda J, Ransom JK. Legume follows for maize–based systems in eastern Africa: contribution of legumes to enhanced maize productivity and reduced nitrogen requirements. In: Seventh eastern and southern Africa regional maize conference 11–15th February, 2001; pp. 324–329.
- 33.Diabate M, Munive A, de Faria SM, Ba A, Dreyfus B, Galiana A. Occurrence of nodulation in unexplored leguminous trees native to the west African tropical rainforest and inoculation response of native species useful in reforestation. New Phytol. 2005;166:231–239. doi: 10.1111/j.1469-8137.2005.01318.x. [DOI] [PubMed] [Google Scholar]
- 34.Fischler M, Wortmann CS. Green manures for maize–bean systems in eastern Uganda: agronomic performance and farmers’ perceptions. Agrofor Syst. 1999;47:123–138. doi: 10.1023/A:1006234523163. [DOI] [Google Scholar]
- 35.Hassen A, Rethman N, Apostolides Z, van Niekerk W. Influence of moisture stress on growth, dry matter yield and allocation, water use and water–use efficiency of four Indigofera species. Afr J Range Forage Sci. 2007;24(1):25–34. doi: 10.2989/102201107780178195. [DOI] [Google Scholar]
- 36.Whitman WB, Woyke T, Klenk HP, Zhou Y, Lilburn TG, Beck BJ, De Vos P, Vandamme P, Eisen JA, Garrity G, Hugenholtz P, Kyrpides NC. Genomic encyclopedia of bacterial and Archaeal type strains, phase III: the genomes of soil and plant–associated and newly described type strains. Stand Genomic Sci. 2015;10:26. doi: 10.1186/s40793-015-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao ZY, Kan FL, Wang ET, Wei GH, Chen WX. Characterization of rhizobia that nodulate legume species of the genus Lespedeza and description of Bradyrhizobium yuanmingense sp. nov. Int J Syst Evol Microbiol. 2002;52:2219–2230. doi: 10.1099/00207713-52-6-2219. [DOI] [PubMed] [Google Scholar]
- 38.Glycine max.http://www.theplantlist.org/tpl1.1/record/ild-2760.
- 39.Yu X, Cloutier S, Tambong JT, Bromfield ESP. Bradyrhizobium ottawaense sp. nov., a symbiotic nitrogen fixing bacterium from root nodules of soybeans in Canada. Int J Syst Evol Microbiol. 2014;64:3203–3207. doi: 10.1099/ijs.0.065540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LM X, Ge C, Cui Z, Li J, Fan H. Bradyrhizobium liaoningense sp. nov., isolated from the root nodules of soybeans. Int J Syst Evol Microbiol. 1995;45:706–711. doi: 10.1099/00207713-45-4-706. [DOI] [PubMed] [Google Scholar]
- 41.Wang JY, Wang R, Zhang YM, Liu HC, Chen WF, Wang ET, Sui XH, Chen WX. Bradyrhizobium daqingense sp. nov., isolated from soybean nodules. Int J Syst Evol Microbiol. 2013;63:616–624. doi: 10.1099/ijs.0.034280-0. [DOI] [PubMed] [Google Scholar]
- 42.Wang R, Chang YL, Zheng WT, Zhang D, Zhang XX, Sui XH, Wang ET, JQ H, Zhang LY, Chen WX. Bradyrhizobium arachidis sp. nov., isolated from effective nodules of Arachis hypogaea grown in China. Syst Appl Microbiol. 2013;36:101–105. doi: 10.1016/j.syapm.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Oren A, Garrity GM. List of new names and new combinations previously effectively, but not validly published. Validation list no. 165. Int J Syst Evol Microbiol. 2015;65:2777–2783. doi: 10.1099/ijsem.0.000464. [DOI] [PubMed] [Google Scholar]
- 44.Grönemeyer JL, Chimwamurombe P, Reinhold-Hurek B. Bradyrhizobium subterraneum sp. nov., a symbiotic nitrogen-fixing bacterium from root nodules of groundnuts. Int J Syst Evol Microbiol. 2015;65:3241–3247. doi: 10.1099/ijsem.0.000403. [DOI] [PubMed] [Google Scholar]
- 45.Biolog GN2 manufacturer’s instructions. http://www.ecologiemicrobiennelyon.fr/IMG/pdf/GN2-1.pdf.
- 46.Shona GM, Campbell CD, Haukka KE, et al. Characterization of rhizobia from African acacias and other tropical woody legumes using biolog and partical 16S rRNA sequencing. FEMS Microbiol Letters. 1999;170:111–117. doi: 10.1111/j.1574-6968.1999.tb13362.x. [DOI] [PubMed] [Google Scholar]
- 47.Indigofera arrecta. http://www.theplantlist.org/tpl1.1/record/ild-3690.
- 48.Crotalaria juncea.http://www.theplantlist.org/tpl1.1/record/ild-6022.
- 49.Arachis hypogaea.http://www.theplantlist.org/tpl1.1/record/ild-2050.
- 50.Lespedeza cuneata. http://www.theplantlist.org/tpl1.1/search?q=Lespedeza+cuneata.
- 51.Medicago sativa.http://www.theplantlist.org/tpl1.1/record/ild-8536.
- 52.Melilotus albus.http://www.theplantlist.org/tpl1.1/record/ild-7888.
- 53.DOE Joint Genome Institute. http://www.jgi.doe.gov/.
- 54.European Nucleotide Archive database. http://www.ebi.ac.uk/ena/data/view/Taxon:1761772.
- 55.European Nucleotide Archive database. http://www.ebi.ac.uk/ena/data/view/PRJEB14983.
- 56.CTAB DNA extraction protocol [http://1ofdmq2n8tc36m6i46scovo2e-wpengine.netdna-ssl.com/wp-content/uploads/2014/02/JGI-Bacterial-DNA-isolation-CTAB-Protocol-2012.pdf].
- 57.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5(4):433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 58.Mingkun L, Copeland A, Han J. DUK, 2011[https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4623924/pdf/40793_2015_Article_77.pdf]. Accessed 26 Oct 2015.
- 59.Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wgsim. https://github.com/lh3/wgsim .
- 61.Gnerre S, MacCallum I. High–quality draft assemblies of mammalian genomes from massively parallel sequence data. PNAS. 2011;108(4):1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hyatt D, Chen GL, Lacascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez–Espino D, Palaniappan K, Szeto E, Pillay M, Chen IM–A, Pati A, Nielsen T, Markowitz VM, Kyrpides NC. The standard operating procedure of the DOE–JGI microbial genome annotation pipeline (MGAP v.4) Stand Genomic Sci. 2015;10:86. doi: 10.1186/s40793-015-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: A gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–457. doi: 10.1038/nmeth.1457. [DOI] [PubMed] [Google Scholar]
- 65.Lowe TM, Eddy SR. tRNAscan–SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pruesse E, Quast C, Knittel, Fuchs B, Ludwig W, Peplies J, Glckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:2188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.INFERNAL. Inference of RNA alignments. http://infernal.janelia.org.
- 68.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, Anderson I, Billis K, Varghese N, Mavromatis K, Pati A, Ivanova NN, Kyrpides NCIMG. 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42(1):D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG–ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 70.Kaneko T, Maita H, Hirakawa H, Uchiike N, Minamisawa K, Watanabe A, Sato S. Complete genome sequence of the soybean symbiont Bradyrhizobium japonicum strain USDA6T. Genes. 2011;2:763–787. doi: 10.3390/genes2040763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delamuta JRM, Ribeiro RA, Ormeño-Orrillo E, Melo IS, Martínez-Romero E, Hungria M. Polyphasic evidence supporting the reclassification of Bradyrhizobium japonicum group Ia strains as Bradyrhizobium diazoefficiens sp. nov. Int J Syst Evol Microbiol. 2013;63:3342–3351. doi: 10.1099/ijs.0.049130-0. [DOI] [PubMed] [Google Scholar]
- 72.Kaneko T, Nakamura Y, Sato S, Minamisawa K, Uchiumi T, Sasamoto S, Watanabe A, Idesawa K, Iriguchi M, Kawashima K, et al. Complete genomic sequence of nitrogen–fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 2002;9:189–197. doi: 10.1093/dnares/9.6.189. [DOI] [PubMed] [Google Scholar]
- 73.van Berkum P, Fuhrmann JJ. Characterization of soybean bradyrhizobia for which serogroup affinities have not been identified. Can J Microbiol. 2001;47(6):519–525. doi: 10.1139/w01-036. [DOI] [PubMed] [Google Scholar]
- 74.Tian CF, Zhou YJ, Zhang YM, Li QQ, Zhang YZ, Li DF, Wang S, Wang J, Gilbert LB, Li YR, Chen WX. Comparative genomics of rhizobia nodulating soybean suggests extensive recruitment of lineage-specific genes in adaptations. Proc Natl Acad Sci. 2012;109(22):8629–8634. doi: 10.1073/pnas.1120436109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang YM, Li Y, Chen WF, Wang ET, Sui XH, Li QQ, Zhang YZ, Zhou YG, Chen WX. Bradyrhizobium huanghuaihaiense sp. nov., an effective symbiotic bacterium isolated from soybean (Glycine max L.) nodules. Int J Syst Evol Microbiol. 2012;62:1951–1957. doi: 10.1099/ijs.0.034546-0. [DOI] [PubMed] [Google Scholar]
- 76.Silva FV, De Meyer SE, Simões-Araújo JL, Barbé TC, Xavier GR, O'Hara G, Ardley JK, Rumjanek NG, Willems A, Zilli JE. Bradyrhizobium manausense sp. nov., isolated from effective nodules of Vigna unguiculata grown in Brazilian Amazonian rainforest soils. Int J Syst Evol Microbiol. 2014;64:2358–2363. doi: 10.1099/ijs.0.061259-0. [DOI] [PubMed] [Google Scholar]
- 77.NCBI database. https://www.ncbi.nlm.nih.gov/.
- 78.Auch AF, von Jan M, Klenk HP, Göker M. Digital DNA–DNA hybridization for microbial species delineation by means of genome–to–genome sequence comparison. Stand Genomic Sci. 2010;2:117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Auch AF, Klenk HP, Goeker M. Standard operating procedure for calculating genome–to–genome distances based on high–scoring segment pairs. Stand Genomic Sci. 2010;2(1):142–148. doi: 10.4056/sigs.541628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meier–Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence–based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y. Coleman–Derr D, Chen G, Gu YQ. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015;43(W1):W78–W84. doi: 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Darling AE, et al. Progressive mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;87(12):4576–4579. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rubio LM, Ludden PW. Biosynthesis of the iron–molybdenum cofactor of nitrogenase. Annu Rev Microbiol. 2008;62:93–111. doi: 10.1146/annurev.micro.62.081307.162737. [DOI] [PubMed] [Google Scholar]
- 84.Baginsky C, Brito B, Imperial J, et al. Diversity and evolution of hydrogenase systems in rhizobia. Appl Environ Microb. 2002;68:4915–4924. doi: 10.1128/AEM.68.10.4915-4924.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Itakura M, Saeki K, Omori H, Yokoyama T, Kaneko T, Tabata S, Ohwada T, Tajima S, Uchiumi T, Honnma K, et al. Genomic comparison of Bradyrhizobium japonicum strains with different symbiotic nitrogen–fixing capabilities and other Bradyrhizobiaceae members. ISME J. 2009;3:326–339. doi: 10.1038/ismej.2008.88. [DOI] [PubMed] [Google Scholar]
- 86.Maier T, Böck A. Nickel incorporation into hydrogenases. In: Hausinger R, Eichlorn GL, Marzilli LG, editors. Advances in inorganic biochemistry. New York: VHC publishers Inc.; 1996. pp. 173–192. [Google Scholar]
- 87.Galloway JN. The global nitrogen cycle: changes and consequences. Environ Pollut. 1998;102:15–24. doi: 10.1016/S0269-7491(98)80010-9. [DOI] [Google Scholar]
- 88.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sanchez C, Tortosa G, Granados A, Delgado A, Bedmar EJ, Delgado MJ. Involvement of Bradyrhizobium japonicum denitrification in symbiotic nitrogen fixation by soybean plants subjected to flooding. Soil Biol Biochem. 2011;43(1):212–217. doi: 10.1016/j.soilbio.2010.09.020. [DOI] [Google Scholar]
- 90.Bedmar EJ, Robles EF, Delgado MJ. The complete denitrification pathway of the symbiotic, nitrogen–fixing bacterium Bradyrhizobium japonicum. Biochem Soc Trans. 2005;33:145–148. doi: 10.1042/BST0330141. [DOI] [PubMed] [Google Scholar]
- 91.Hirayama J, Eda S, Mitsui H, Minamisawa K. Nitrate–dependent N2O emission from intact soybean nodules via Denitrification by Bradyrhizobium japonicum Bacteroids. Applied Enviro Microbiol. 2011:8787–90. [DOI] [PMC free article] [PubMed]
- 92.Mesa S, de Dios Alché J, Bedmar EJ, Delgado MJ. Expression of the nir, nor and nos denitrification genes from Bradyrhizobium japonicum in soybean root nodules. Physiol Plant. 2004;120:205–211. doi: 10.1111/j.0031-9317.2004.0211.x. [DOI] [PubMed] [Google Scholar]
- 93.Velasco L, Mesa S, CA X, Delgado MJ, Bedmar EJ. Molecular characterization of nosRZDFYLX genes coding for denitrifying nitrous oxide reductase of Bradyrhizobium japonicum. Antonie Van Leeuwenhoek. 2004;85:229–235. doi: 10.1023/B:ANTO.0000020156.42470.db. [DOI] [PubMed] [Google Scholar]
- 94.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, bacteria, and Eucarya. PNAS USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology, volume 2, part B. Second ed. New York: Springer; 2005.
- 97.Garrity GM, Bell JA, Lilburn T. Class I. Alphaproteobacteria class. In: Garrity GM, Brenner DJ, Kreig NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. Part C: 1. 2nd ed. New York: Springer – Verlag; 2005.
- 98.Euzéby J. List of new names and new combinations previously effectively, but not validly published. Validation list no. 107. Int J Syst Evol Microbiol. 2006;56:1–6. doi: 10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- 99.Kuykendall LD, Order VI. Rhizobiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology, vol. Volume 2, Part C. 2n ed. New York: Springer; 2005. p. 324. [Google Scholar]
- 100.Garrity GM, Bell JA, Liburn T. Family VII. Bradyrhizobiaceae. In: Brenner DJ, Krieg NR, Staley JT, editors. Bergey's Manual of Systematic Bacteriology. Volume 2 part C. New York: Springer; 2005. p. 438. [Google Scholar]
- 101.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Crotalaria incana. http://www.theplantlist.org/tpl1.1/record/ild-5341.
- 103.Crotalaria spinosa. http://www.theplantlist.org/tpl1.1/record/ild-5818.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypic characteristics of Bradyrhizobium shewense sp. nov. strains. [102, 103] (DOCX 28 kb)
Carbon sources utilization response between Bradyrhizobium shewense sp. nov. strains and reference type strain B. yuanmingense CCBAU 10071T. (DOCX 29 kb)
Symbiotic and denitrifying genes identified in the genomes of Bradyrhizobium shewense. sp. nov. ERR11T and B. yuanmingense CCBAU 10071T. (XLSX 121 kb)