

Abstract
Our goal was to measure the association of CXCL5 and molecular phenotypes associated with coronary atherosclerosis severity in patients at least 65 years old. CXCL5 is classically defined as a proinflammatory chemokine, but its role in chronic inflammatory diseases, such as coronary atherosclerosis, is not well defined. We enrolled individuals who were at least 65 years old and undergoing diagnostic cardiac catheterization. Coronary artery disease (CAD) severity was quantified in each subject via coronary angiography by calculating a CAD score. Circulating CXCL5 levels were measured from plasma, and both DNA genotyping and mRNA expression levels in peripheral blood mononuclear cells were quantified via microarray gene chips. We observed a negative association of CXCL5 levels with CAD at an odds ratio (OR) of 0.46 (95% CI, 0.27–0.75). Controlling for covariates, including sex, statin use, hypertension, hyperlipidemia, obesity, self-reported race, smoking, and diabetes, the OR was not significantly affected [OR, 0.54 (95% CI, 0.31–0.96)], consistent with a protective role for CXCL5 in coronary atherosclerosis. We also identified 18 genomic regions with expression quantitative trait loci of genes correlated with both CAD severity and circulating CXCL5 levels. Our clinical findings are consistent with the emerging link between chemokines and atherosclerosis and suggest new therapeutic targets for CAD.
It is well established that age is associated with cardiovascular disease (CVD) risk. Epidemiological studies demonstrated that the lifetime risk for CVD remains high in older individuals and increases dramatically in the presence of multiple risk factors.1 In addition, older individuals who experience an acute coronary syndrome have a poorer prognosis compared with the general population. For example, in patients aged 65 years or older, the 5-year mortality rate after a myocardial infarction is 46% to 58%, and all-cause mortality, death from cardiovascular causes, recurrent myocardial infarction, and stroke are all higher compared with younger individuals.2 However, older patients are frequently underrepresented in clinical trials, and the pathophysiology underlying the development and progression of atherosclerotic coronary artery disease (CAD) has not been rigorously evaluated in this population.
Increasing age is associated with pathophysiological changes in the vasculature, including increased carotid intima-media thickness, arterial stiffening, endothelial dysfunction, and elevated systolic blood pressure.3 Although it is well established that systemic and vascular inflammation are important in the development and progression of CAD in the general population,4, 5, 6 differing reports on inflammatory markers and their association with CAD are found throughout the literature. It remains unclear how age modifies the disease process. Although studies have looked at the use of statins and treatment of hypertension in geriatric cohorts,7, 8 further analyses are needed to ascertain the inflammatory processes that are involved in the pathophysiology of CVD in older adults. More important, identification of the key biological processes underlying CAD in the older high-risk geriatric patient population may facilitate the development of novel therapeutic strategies to improve prognosis.
Systems biology studies identified global gene expression signatures in peripheral blood mononuclear cells (PBMCs) that are predictive of the presence and extent of CAD.9, 10 In addition, changes in gene expression in peripheral blood mirror changes in gene expression in atherosclerotic arteries.9 Therefore, evaluation of global gene expression changes in PBMCs represents a nonbiased approach to elucidating the key mechanisms underlying CAD. Likewise, genomic data and quantification of circulating factors that are thought to contribute to inflammation can be combined with gene expression data to identify molecular phenotypes and elucidate the potential dysregulation of biological pathways that are associated with the presence and severity of obstructive CAD, potentially uncovering new therapeutic targets.
The ELR+-CXCL chemokines contain a glutamic acid-leucine-arginine motif (ELR) and are classically defined as chemoattractants and are involved in neutrophil activation in models of acute inflammation. In chronic inflammatory disease, such as atherosclerosis, ELR+-CXC chemokines are proposed to be involved throughout the pathophysiological response. Of particular interest to our study was the leukocyte-derived CXCL5 (initially described as epithelial neutrophil-activating peptide-78), because of the observed increases in CXCL5 with subclinical atherosclerosis in type 2 diabetics and CAD (as defined as 70% narrowing of one epicardial coronary artery) in Chinese populations.11, 12 These results seem to be at odds with the protective role for CXCL5 in a mouse model of atherosclerosis.13 Therefore, we investigated the relationship between circulating CXCL5 and CAD in a clinical cohort from the southeastern United States and used a systems biology approach to understand the genetic influences of CXCL5 regulation and molecular phenotypes associated with CAD.
Materials and Methods
Study Population
Phase 2 of the Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARA) study enrolled individuals who were at least 65 years old and undergoing clinically indicated diagnostic cardiac catheterization. Between March 2007 and February 2009, 200 individuals from The University of North Carolina Cardiac Catheterization Laboratory were enrolled. Individuals with a history of any of the following conditions were not eligible to participate in this study: HIV or hepatitis C virus infection, lymphoma, leukemia, anemia, rheumatoid arthritis, lupus, solid organ transplantation, active cancer treatment, or chronic immunosuppressive therapy. Participants were interviewed to obtain pertinent medical information, including medical history, current medication use, family medical history, and tobacco use. This study was approved by The University of North Carolina Institutional Review Board (number 05-1798) and conducted according to institutional guidelines; all participants provided written informed consent.
CAD Severity
Subjects underwent clinically indicated diagnostic left heart catheterization with coronary angiography. After the procedure, angiograms were reviewed independently by two cardiologists and CAD severity was quantified in each subject on either an ordinal scale (range, 0 to 4) or binary classification (obstructive CAD, yes/no). The ordinal CAD score was calculated as follows: 0 (<10% stenosis in all major coronary arteries), 1 (10% to 70% stenosis in one vessel, left main coronary artery excluded), 2 (>70% stenosis in one vessel), 3 (>70% stenosis in two vessels), and 4 (>70% stenosis in 3 vessels or >50% stenosis in the left main coronary artery). Obstructive CAD was defined as >70% stenosis in one or more major epicardial coronary arteries or >50% left main coronary artery stenosis. Subjects with ≤70% occlusion in any vessel or <50% in the left main coronary artery were considered free of obstructive CAD.14, 15
Sample Collection and Processing
All catheterizations took place in the morning after an overnight fast. A complete blood cell count (without differential) was performed as routine care on the day of the catheterization. Lipid panel data were obtained from a previous office visit (within 6 months before catheterization). At the start of the procedure, 30 mL of blood was drawn from the femoral artery into Na-EDTA Vacutainer tubes (Becton, Dickinson and Co, Cockeysville, MD). After blood collection, RNA and DNA were isolated from PBMCs, as described.16, 17 RNA quantity, purity, and integrity were assessed by spectrophotometry and microcapillary electrophoresis on an Agilent BioAnalyzer 2100 (Agilent Technologies, Inc., Santa Clara, CA). Only RNA with an A260/280 ratio of ≥1.8 and an electrophoretic profile consistent with only minimal degradation was considered acceptable for use. An additional 8 mL of blood collected in tubes containing EDTA and proteinase inhibitor was processed to plasma, aliquoted, and stored at −80°C, pending analysis.
Quantification of Circulating Markers of Inflammation
Plasma samples from 143 of the 200 subjects were used to measure circulating inflammatory mediators. We limited these analyses (n = 143) to only self-reported blacks and whites and avoided potential inflammatory bias by excluding subjects who were diagnosed as having acute infections. Plasma concentrations of cellular adhesion molecules (CAMs; E-selectin, P-selectin, intracellular adhesion molecule-1, and vascular CAM-1) and chemokines [monocyte chemoattractant protein (MCP)-1 and CXCL5] were quantified using the Human Adhesion Molecule and Human Cytokine Fluorokine Multi-Analyte Profiling Kits (R&D Systems, Minneapolis, MN), respectively.18 High-sensitivity C-reactive protein (hsCRP) was quantified using the Human C-Reactive Protein Fluorokine MAP Kit for Use in Cardiac Panel B (R&D Systems, Minneapolis, MN). All analytes were quantified with fluorescence detection on the Bio-Plex 200 System (Bio-Rad, Hercules, CA), according to the manufacturer's instructions, at the Bioanalytical Core Laboratory at The University of North Carolina at Chapel Hill. Because hsCRP was higher than the limit of detection in 53 (37%) of the samples analyzed, a value of 15.2 mg/L (1.5-fold higher than the highest standard) was imputed for each sample above the upper limit of detection; in addition, we stratified subjects by hsCRP level higher or lower than the median. Pearson and Spearman correlation analyses were used to determine the correlation among circulating biomarkers of inflammation. Multiple CAMs were correlated at P < 0.05 and correlation coefficients at >0.2 (Supplemental Table S1). To minimize redundancy in our analysis, a consolidated CAM score phenotype was calculated for each individual by summing the z-scores of E-selectin, P-selectin, intracellular adhesion molecule-1, and vascular CAM-1.18, 19 The association between plasma levels of the selected inflammatory biomarkers and obstructive CAD status or ordinal CAD score was evaluated using nominal or ordinal logistic regression, respectively, using an unadjusted model and a model that adjusted for the indicated variables (JMP Pro 12; SAS, Cary, NC).
Gene Chip Analysis
RNA labeling, hybridization, and data extraction were performed in our laboratory. RNA was cohybridized to Agilent G4112F Whole Human Genome 4 × 44K oligonucleotide arrays (Agilent Technologies, Inc.) in the presence of cyanine-3–labeled Universal Human Reference RNA (Stratagene, La Jolla, CA) for array normalization, as described.16 Slides were hybridized and washed, then scanned on an Axon 4000b microarray scanner; data were processed using Feature Extraction version 9.5.1.1 (Agilent Technologies, Inc.). Probes were filtered by only including probes flagged as detected in at least 85% of all SAMARA phase 2 samples, resulting in 18,411 probes available for analysis. In addition, samples were filtered by only including RNA samples with at least 90% of the 18,411 probes flagged as detected. Missing data were imputed using the k-nearest neighbor algorithm (k = 10). Data sets were deposited in the Gene Expression Omnibus (GEO) of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/geo; accession number GSE90074) containing the 143 samples used in the biomarker analyses. The processing date (batch effect) and sex were controlled for using an analysis of variance linear model using Partek Genomics Suite version 6.6 (Partek Inc., Chesterfield, MO). Probes were updated using the annotations updated by Agilent on June 12, 2015. We used BioMart version 0.9 identification conversion (http://www.biomart.org)20 for all significant probes that contained missing associated gene name annotations. Spearman rank correlation was used to generate a list of genes that correlated with the CAD score (range, 0 to 4) with a predefined cutoff of |rs| ≥ 0.25 using Partek Genomics Suite v6.6, as described.9 SAMARA phase 1 microarray data were downloaded from GEO (accession number GSE12959), as previously described.17
DNA labeling, hybridization, and data extraction were performed by the DNA Array Core Facility at The Scripps Research Institute (Jupiter, FL). The Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA) was used for hybridizations. We obtained high-purity DNA samples from 126 of the 143 subjects with gene expression and biomarker analyses that were analyzed via single-nucleotide polymorphism (SNP) array. Subsequently, 20 of the 126 arrays had no call rates of >20% and were dropped from further analysis, leaving 106 samples with 909,624 probes. Data sets were deposited in the GEO of the National Center for Biotechnology Information (GEO series accession number GSE90073). SNP probes were filtered to include only those with minor allele frequencies >0.05 and Hardy-Weinberg equilibrium >0.01, leaving 398,997 available for analysis. Principal components analysis was used to classify subjects as Americans with either African or European ancestry. Missing genotypes were imputed using MaCH version 1.0.6 (http://csg.sph.umich.edu/abecasis/mach/index.html)21 with estimated mismatch rates in the Markov model of 0.1256 and 0.00481 in each group, respectively.
eQTL Analysis
Identification of local elements associated with expression [expression quantitative trait loci (eQTL)] was performed with Matrix eQTL version 2.1.022 in the software package R version 3.2.1 (https://www.r-project.org).23 We used the linear additive model to identify cis-acting eQTLs (distance between SNP and expression probe, <1e6 bp) accounting for ancestry covariance and population structure. This resulted in testing 4,600,673 SNP-gene pairs in the 106 subjects with both DNA and RNA analyses. The DNA and RNA data sets are collated as a SuperSeries in the GEO of the National Center for Biotechnology Information (GEO series accession number GSE90076). Where indicated, additional data used for the analyses described herein were obtained from the GTEx Portal (https://www.ncbi.nlm.nih.gov/gap, accession number phs000424.v6.p1).
Overlap of Two Groups of Genes
The probability of overlap of two groups of genes was determined by measuring the hypergeometric distribution using the number of probes that passed quality control measures (18,411). To compare the gene expression signatures identified in our analysis with those previously identified in CAD patients, we accessed the published list of 655 genes24 that were associated with CAD in the case-control analyses from both the CATHeterization GENetics (CATHGEN)10 and Personalized Risk Evaluation and Diagnosis in the Coronary Tree cohorts.25
Results
Study Population
The study population characteristics of the 143 subjects stratified by obstructive CAD status are shown in Table 1. A total of 93 subjects (65%) had obstructive CAD, whereas 50 subjects (35%) did not have obstructive CAD. In both groups, most subjects were white and had multiple comorbidities, including hyperlipidemia and hypertension. More of those diagnosed as having obstructive CAD were men, had a previous diagnosis of hyperlipidemia or myocardial infarction (MI), and were taking medications for the treatment of CVD, including statins, aspirin, and clopidogrel. There was no difference in lipid profiles, white blood cell count, or platelet count between the two groups at P < 0.05 (Supplemental Table S1).
Table 1.
Study Population Characteristics by Obstructive CAD Status
| Characteristic | Obstructive CAD (n = 93) | No obstructive CAD (n = 50) | P value |
|---|---|---|---|
| Age, years | 74 ± 6.3 | 73 ± 6.2 | 0.220 |
| Male sex∗ | 57 (61) | 18 (36) | 0.004 |
| White race | 74 (80) | 37 (74) | 0.446 |
| Body mass index, kg/m2 | 29 ± 6.3 | 30 ± 7.1 | 0.323 |
| Current smoker | 4 (4) | 3 (6) | 0.653 |
| Diabetes | 38 (41) | 17 (34) | 0.421 |
| Hyperlipidemia∗ | 74 (80) | 28 (56) | 0.003 |
| Hypertension | 82 (88) | 44 (88) | 0.976 |
| Previous MI∗ | 41 (44) | 3 (6) | <0.001 |
| Multivessel disease∗ | 62 (67) | 0 (0) | <0.001 |
| Systolic blood pressure, mmHg | 143 ± 20 | 141 ± 20 | 0.573 |
| Diastolic blood pressure, mmHg | 77 ± 13 | 78 ± 12 | 0.588 |
| Statin use∗ | 74 (80) | 23 (46) | <0.001 |
| Aspirin use∗ | 78 (84) | 33 (66) | 0.015 |
| Clopidogrel use∗ | 33 (36) | 6 (12) | 0.003 |
Data presented as means ± SD or n (%). Age, body mass index, and diastolic blood pressure were not normally distributed and were log transformed before analysis.
CAD, coronary artery disease; MI, myocardial infarction.
P < 0.05 (t-test).
Circulating Biomarkers of Inflammation
The Protective Chemokine CXCL5 Inversely Correlates with CAD Severity
Given the proposed protective role for CXCL5 in atherosclerosis, we hypothesized that CXCL5 levels would vary inversely with disease severity in our cohort. To test our hypothesis, we measured levels of CXCL5 in plasma and used logistic regression to measure the association between CAD severity and circulating levels of CXCL5. We observed a negative association between CXCL5 levels and CAD severity using either ordinal (Figure 1A) or nominal (Figure 1B) classification, with odds ratios (ORs) of 0.52 and 0.46, respectively (Table 2 and Figure 1). To control for other potential confounding variables, we included sex, prior statin use, hyperlipidemia, self-reported race, obesity, hypertension, smoking, and diabetes in an adjusted model. When we controlled for these variables, the ORs for CXCL5 and CAD severity were similar to the unadjusted model, 0.59 and 0.54, in both the ordinal and nominal models, respectively, and within the 95% CI of the unadjusted ORs (Table 2). Of the variables included in the model, only prior statin use, sex, and CXCL5 associated with CAD classification (P < 0.05); the other variables had no association with CAD score (Supplemental Table S2).26 In addition, the fit of both ordinal and logistic models with the above risk factors was improved when circulating CXCL5 levels were included in the model (Supplemental Table S2).
Figure 1.

Plasma CXCL5 levels negatively associated with coronary artery disease (CAD) severity. Plasma levels of CXCL5 represented by dot plot and summarized by the means and 95% CIs of the log values. A: CXCL5 levels in subjects with increasing CAD severity represented by dot plot, with the corresponding P value of the logistic regression analysis. B: CXCL5 levels stratified by obstructive CAD status for the entire cohort (left of dashed line) or within each sex (right of dashed line). ∗P < 0.05 on CAD term testing the negative association between CAD and CXCL5 levels within each sex (t-test); ††P < 0.01 on CAD term from two-way analysis of variance of CXCL5, factoring sex and CAD. N, no; Y, yes.
Table 2.
Regression Analysis of Plasma CXCL5 Levels and CAD Score
| Analyte | P value | OR (95% CI) | Reciprocal OR | CAD Class 0–4 |
|---|---|---|---|---|
| Ordinal | ||||
| CXCL5 unit odds | 0.0013 | 0.52 (0.36–0.78) | 1.91 | 1570, 1638, 1048, 1324, 886 |
| Adjusted model | ||||
| CXCL5 unit odds adj | 0.0148 | 0.59 (0.39–0.90) | 1.69 | |
| Prior statin use (Y) adj | 0.0023 | 1.75 (1.15–2.67) | 5%, 19%, 25%, 21%, 31% | |
| Sex (M) adj | 0.0003 | 1.78 (1.27–2.48) | 7%, 17%, 18%, 24%, 33% | |
| Analyte | P | OR (95% CI) | Recip | Obstructive CAD-no | Obstructive CAD-yes |
|---|---|---|---|---|---|
| Nominal | |||||
| CXCL5 unit odds | 0.0014 | 0.46 (0.27–0.75) | 2.15 | 1638 (1439) | 998 (943) |
| CXCL5 range odds | 0.03 (3.2E-3–0.30) | 32.06 | |||
| Adjusted model | |||||
| CXCL5 unit odds adj | 0.0309 | 0.54 (0.31–0.96) | 1.84 | ||
| CXCL5 range odds adj | 0.06 (4.7E-3 - 0.84) | 57.33 | |||
| Prior statin usage (Y) adj | 0.0100 | 4.02 (1.39–11.58) | 46% | 80% | |
| Sex (M) adj | 0.0140 | 3.03 (1.25–7.33) | 36% | 61% | |
We measured the associations between each CXCL5 and CAD score using nominal and ordinal logistic regression. The adj model controlled for sex, statin use, hypertension, hyperlipidemia, obesity, self-reported race, smoking status, and diabetes. The P value of the effect likelihood test for each analyte in the model at P < 0.05 and the OR (95% CI) are provided. The reciprocal OR is provided for ratios <1. Summary CXCL5 data are presented as median (interquartile range), with units of pg/mL or percentage of subjects in the indicated category for statin use and sex.
M, male; Adj, adjusted model; CAD, coronary artery disease; OR, odds ratio; Y, yes.
Both sex and prior statin use covariates were of particular interest to the CAD-CXCL5 association. First, studies in younger populations identified higher circulating levels of CXCL5 in women compared with men,27 and our geriatric cohort reflected a similar observation (1.8 ± 1.7 and 1.3 ± 0.9 ng/mL in women and men, respectively, represented by the means ± SD; P = 0.019). To explore the possible confounding of sex on the CXCL5-CAD association (Table 1), we measured the association of CXCL5 levels and the interaction between sex and obstructive CAD class using a two-way analysis of variance. Only CAD status had a significant association with CXCL5 (P = 0.007), whereas sex alone or the interaction between sex and CAD status did not associate with CXCL5 levels (P = 0.139 and P = 0.708, respectively). Furthermore, we tested for a negative association between CAD and CXCL5 levels after stratifying the data set by sex and found that CXCL5 levels decreased with obstructive CAD, irrespective of sex (P = 0.044 and P = 0.018 in women and men, respectively) (Figure 1B).
Statins decreased CXCL5 levels in an in vitro model of inflammation using endothelial cells,28 suggesting that statin use might confound our analysis of the CXCL5-CAD association. Furthermore, CXCL5 is expressed in other cell types, such as platelets, enterocytes, fibroblasts, microglia, endometrial stromal cells, and various white blood cells.29, 30, 31, 32, 33, 34, 35, 36 Given the influence of statins on both CXCL5 and circulating lipids, as well as the various potential sources of CXCL5 found in the blood, we performed pairwise association analyses of CXCL5 with lipid variables, white blood cell count, and platelet count in our study subjects. Interestingly, CXCL5 did not correlate with any of the lipid molecules, but it did have a small positive correlation with platelet count (Figure 2A). However, when included in our adjusted model, platelet count did not associate with CAD (P = 0.74 and P = 0.16 in the ordinal and nominal models, respectively); inclusion of platelet count did not affect the impact of CXCL5, statins, or sex on CAD associations (data not shown). Similar to our two-way analysis of variance analysis with sex and CAD status, we observed an association between CXCL5 and CAD status (P = 0.005); however, there was no association between CXCL5 levels and statin use (P = 0.699) and no interaction between statin use and CAD status with CXCL5 levels (P = 0.824). When we further stratified CXCL5 levels by sex and obstructive CAD, we identified an association of statin use and decreased CXCL5 only in women with obstructive CAD (P = 0.0098) (Figure 2B).
Figure 2.

Association of plasma CXCL5 levels with lipids, cell counts, and statin use. A: Pairwise association analysis of the indicated variables summarized by the regression line and means (95% CIs). The correlation coefficient for each association is indicated and colored to reflect the P value. B–D: CXCL5 levels represented by dot plot and summarized by the means and 95% CIs of the log values. B: CXCL5 levels stratified by obstructive CAD status and statin use. C: CXCL5 levels stratified by statin type; the P value from one-way analysis of variance is indicated. D: CXCL5 levels stratified by statin intensity; the P value from one-way analysis of variance is indicated. ∗∗P < 0.01 for statin use with CXCL5 levels (t-test); †P < 0.05, ††P < 0.01 versus rosuvastatin (Tukey post-test). CAD, coronary artery disease; CHOL, cholesterol; HDL, high-density lipoprotein; LDL, low-density lipoprotein; N, no; PC, platelet counts; TG, triglyceride; WBC, white blood cell; Y, yes.
Statins vary in their low-density lipoprotein–lowering intensity and, hence, could differentially affect CXCL5 levels.37, 38, 39 To determine whether differences in statin intensity correlated with CXCL5 levels, we examined the association of the five prescribed statins in our cohort to circulating CXCL5 levels using analysis of variance. Subjects prescribed the high-intensity statin rosuvastatin39 had lower levels of circulating CXCL5 compared with the other statins. However, only three subjects were prescribed rosuvastatin, and there were no other differences observed across other statins when analyzed individually (Figure 2C) or when grouped into high versus low intensity (Figure 2D).
Given the age of our cohort (>65 years), it was not surprising that 31% of the subjects had a previous MI; likewise, a disproportionate number of subjects who were diagnosed as having obstructive CAD in our study also had a previous MI (Table 1). We found that CXCL5 levels were lower in subjects who had a previous MI (1.1 ± 0.8 versus 1.7 ± 1.5 ng/mL, represented by the means ± SD in subjects with or without a previous MI, respectively; P = 0.025). Therefore, we stratified our data set on subjects with or without a previous MI as well as CAD status to compare circulating CXCL5 levels using analysis of variance. As with our previous analyses (Figure 1), we observed that subjects with CAD had lower levels of CXCL5, and that CXCL5 levels were independent of a history of previous MI (Figure 3).
Figure 3.
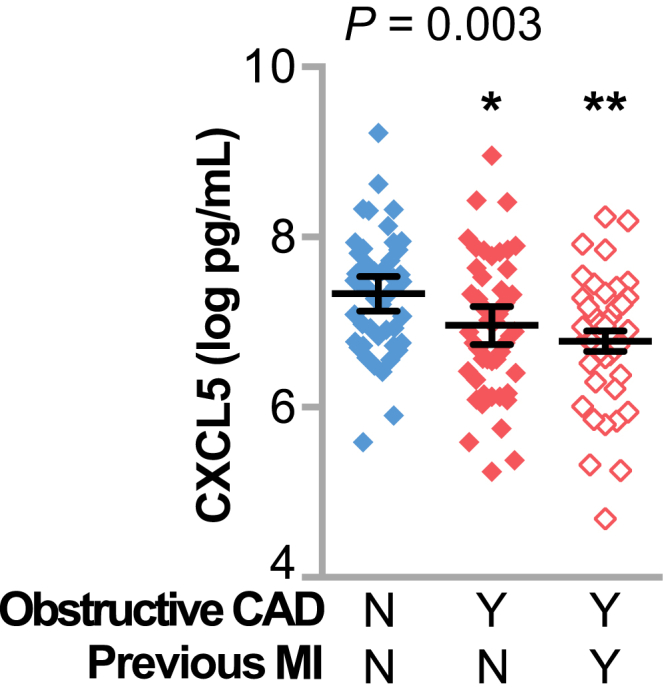
Plasma CXCL5 levels negatively associated with coronary artery disease (CAD) severity, irrespective of a previous myocardial infarction (MI). Plasma levels of CXCL5 represented by dot plot and summarized by the means and 95% CIs of the log values in subjects stratified by previous MI and the presence of obstructive CAD; the P value from one-way analysis of variance is indicated. ∗P < 0.05, ∗∗P < 0.01 versus CAD [no (N)] and previous MI (N; Tukey post-test). Y, yes.
hsCRP, but Not Other Circulating Inflammatory Biomarkers, Correlates with CAD Severity
Previous studies indicate that certain mediators of inflammation and leukocyte mobilization, including hsCRP, CAMs, selectins, and MCP-1, associate with CAD incidence in the general population and predict higher mortality in patients with established CAD40, 41, 42, 43; however, the association between these inflammatory biomarkers and disease severity, particularly in geriatric populations, is less clear. We measured these additional mediators and performed the identical analyses, as we did for CXCL5. Surprisingly, only hsCRP associated with CAD classified either nominally (P = 0.083) or on the ordinal scale (P = 0.021) in the adjusted model at an OR of 1.1 (Table 3). The association between CRP and severity of CAD is inconsistent and contradictory.44, 45 Although hsCRP is a common marker of CVD risk, our results are similar to others that find hsCRP less effective at distinguishing CAD severity.46, 47, 48, 49 Not surprisingly, hsCRP had small-to-moderate positive correlations with the CAM score and MCP-1, but no correlation with CXCL5 (Supplemental Table S3). Likewise, the addition of hsCRP with CXCL5 to the adjusted model further increased the fitness of both the ordinal and nominal models of CAD, whereas addition of the CAM score and MCP-1 did not (Supplemental Table S2), suggesting that changes in hsCRP and CXCL5 reflect different pathophysiological processes.
Table 3.
Regression Analysis of Plasma Inflammatory Biomarker Levels and CAD Score
| Analyte | P value | OR (95% CI) | CAD Class 0–4 |
|---|---|---|---|
| Ordinal | |||
| MCP1 | 0.706 | 0.90 (0.51–1.59) | 106, 125, 132, 127, 109 |
| MCP1 adj | 0.586 | 0.99 (0.54–1.84) | |
| hsCRP | 0.105 | 1.04 (0.99–1.09) | 4.9, 4.4, 4.3, 5.8, 8.1 |
| hsCRP adj∗ | 0.021 | 1.06 (1.01–1.12) | |
| hsCRPmed [1] | 0.137 | 1.55 (0.87–2.80) | 11%, 20%, 18%, 21%, 30% [1] |
| hsCRPmed adj [1] | 0.097 | 1.68 (1.59–1.76) | 14%, 25%, 25%, 15%, 21% [0] |
| CAM score | 0.285 | 1.08 (0.94–1.23) | −0.12, −0.07, −0.044, 0.60, −0.16 |
| CAM score adj | 0.548 | 1.04 (0.91–1.20) |
| Analyte | P | OR (95% CI) | Obstructive CAD-no | Obstructive CAD-yes |
|---|---|---|---|---|
| Nominal | ||||
| MCP1 unit | 0.661 | 1.16 (0.59–2.29) | 115 (46) | 122 (58) |
| MCP1 range | 1.77 (0.14–22.5) | |||
| MCP1 unit adj | 0.521 | 1.30 (0.59–2.87) | ||
| MCP1 range adj | 2.68 (0.13–54.5) | |||
| hsCRP unit | 0.212 | 1.04 (0.98–1.10) | 4.6 (13.8) | 5.7 (13.1) |
| hsCRP range | 1.72 (0.73–4.04) | |||
| hsCRP unit adj | 0.083 | 1.06 (0.99–1.14) | ||
| hsCRP range adj | 2.41 (0.88–6.63) | |||
| hsCRPmed [1] | 0.321 | 1.42 (0.71–2.85) | 15%/20% [1/0] | 34%/31% [1/0] |
| hsCRPmed adj [1] | 0.201 | 1.69 (0.75–3.80) | ||
| CAM score unit | 0.485 | 1.06 (0.90–1.25) | −0.07 (2.86) | 0.10 (2.68) |
| CAM score range | 1.92 (0.90–1.25) | |||
| CAM score unit adj | 0.665 | 1.04 (0.87–1.25) | ||
| CAM score range adj | 1.58 (0.20–12.6) |
We measured the associations between each analyte and CAD score using nominal and ordinal logistic regression. The adj model controlled for sex, statin use, hypertension, hyperlipidemia, obesity, self-reported race, smoking status, and diabetes. In addition, for hsCRP, the data were stratified higher than [1] or lower than [0] hsCRPmed. The P value of the effect likelihood test for each analyte and the OR (95% CI) are provided. Summary data for continuous variables are presented as the median value from each CAD class for ordinal classification of CAD or as the median (interquartile range) for nominal classification of CAD, with units of pg/mL and mg/L for MCP-1 and hsCRP, respectively, or the median CAM score (normalized distribution value). For hsCRPmed, summary data are presented as the percentage of subjects higher [1] or lower [0] than the median value for the indicated CAD class.
Adj, adjusted model; CAD, coronary artery disease; CAM, cellular adhesion molecule; hsCRP, high-sensitivity C-reactive protein; hsCRPmed, median hsCRP; MCP, monocyte chemoattractant protein; OR, odds ratio.
P < 0.05.
Global Gene Expression and Genotype Profiling
PBMCs are integral mediators of vascular inflammation and express signaling molecules that drive monocyte recruitment and infiltration into the vessel, including cytokines, chemokines, and CAMs, ultimately leading to plaque development and obstructive CAD.50 The heritable component of gene expression variance is a combination of epigenetic and genetic factors, the latter of which we can measure by combining measures of DNA variation with CAD gene expression data to identify eQTL. We used gene expression and genotype microarray data from the SAMARA2 study to explore pathways and targets involved in CXCL5-associated protection.
Polymorphisms Associated with CXCL5 Expression and Circulating CXCL5 Levels
To identify genes and pathways that associate with circulating CXCL5 and CAD severity, we first identified gene expression changes that correlated with plasma CXCL5, resulting in 1041 probes [absolute correlation coefficient (rs) ≥ 0.25, corresponding to P ≤ 0.0026; false-discovery rate, <7%], including the probe for CXCL5 (A_24_P277367). Previous studies identified polymorphisms at the CXCL5 locus that associate with changes in PBMC CXCL5 mRNA and plasma CXCL5 levels,51, 52 suggesting that genetic variance at the CXCL5 locus can affect the expression and production of CXCL5 and that these differences are detectable in PBMCs. Likewise, we identified an eQTL for CXCL5 expression in our cohort (rs394408|CXCL5) (Figure 4A). The expression regulatory SNP (eSNP) rs394408 is not a rare variant in our population (Supplemental Figure S1)54 and is in high linkage disequilibrium (Supplemental Table S4) with these two other known variants (rs425535 and rs352046) that correlated with CXCL5 expression and plasma levels in both black and white populations.55 To validate our findings on rs394408, we analyzed the same eQTL in whole blood from 338 independent samples from the Genotype-Tissue Expression project, consisting of 66% men and 84% whites (16% blacks); their mean age was 53 years [The Database of Genotypes and Phenotypes (dbGaP) phs000424.v6.p1]. This analysis confirmed our initial observation of the minor allele of rs394408 associating with increased CXCL5 expression (Figure 4B).
Figure 4.

Genetic association with CXCL5 expression and circulating CXCL5 levels. A and B: Expression quantitative trait loci (eQTL) analysis of the expression regulatory single-nucleotide polymorphism (eSNP) rs394408 with the number of subjects, genotype, and CXCL5 expression levels from either the Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARA) cohort (A) or dbGaP phs000424.v6.p1 (B), represented by box plots (5% to 95% CI) with outliers identified (open circles). The P value and β coefficient of the linear regression are noted. C: The genetic locus of rs394408 on chromosome 4 with the physical position (in million bp; top) and known genes; several eQTLs (green vertical lines; middle) for CXCL5 identified in monocytes in the region containing rs394408 (dark green line) and further represented in the corresponding dot plot of the −log10 of the false-discovery rate (q; bottom) were measured in a cohort of 1490 participants.53D: Pairwise association analysis of the indicated variables summarized by the regression line and means (95% CIs). The correlation coefficient for each association is indicated and colored to reflect the P value. E–G: eQTL analysis of the eSNP rs394408 with the genotype and either PF4 (E) or PPBP (F) expression levels or QTL analysis of rs394408 and plasma levels of SAMARA subjects represented by box plots (5% to 95% CI) with outliers identified (open circles; G). The P value and β coefficient of the linear regression are noted. PF4, platelet factor 4; PPBP, pro-platelet basic protein.
In addition, we looked at eQTLs specifically in monocytes at the locus containing rs394408 from 1490 Western Europeans enrolled in the Gutenberg Heart Study, consisting of 51% men (mean age, 55 years). Within this region on chromosome 4, we identified a hotspot for CXCL5 cis-eQTLs (Figure 4C) in a region that also associated with sex-based disease risk in immunity-based diseases and atherosclerosis.53 We also confirmed this eQTL in the most comprehensive circulating monocyte data set on >2000 Western Europeans from the Leipzig LIFE Heart Study (Supplemental Table S5).56 Given that rs394408 is located in a region containing two other chemokine genes, PF4 (CXCL4) and PPBP (CXCL7) (Figure 4C), we examined the pairwise correlation between the expression levels of these three chemokine genes and their association with rs394408 to determine the relationship between the expression of these genes and the potential influence of DNA variation at rs394408. We observed a robust positive correlation between PF4 and PPBP expression (Figure 4D) compared with a much smaller correlation between CXCL5 and PF4 or PPBP expression. Furthermore, we did not observe the same association of rs394408 with either PF4 (Figure 4E) or PPBP (Figure 4F) expression as we did with CXCL5 (Figure 4A).
Collectively, these data suggest PF4 and PPBP are coregulated and under distinct transcriptional regulation compared with CXCL5 in PMBCs in our cohort. More important, we extended the rs394408-CXCL5 eQTL to the protein level by measuring the genetic association of rs394408 with circulating levels of CXCL5 (Figure 4G), demonstrating that this eQTL extends to similar changes in the circulating amount of CXCL5 peptide.
Humans have two homologs of the mouse CXCL5 gene, CXCL5 and CXLC6. We examined the correlations between CXCL5 levels in plasma and CXCL5 and CXCL6 expression in PMBCs. As expected, we identified a positive correlation between the circulating levels and expression of CXCL5 (r = 0.4842, P = 0.0042); however, there was no correlation between CXCL6 and CXCL5 expression (r = −0.1134, P = 0.1855) or between CXCL6 expression and circulating CXCL5 levels (r = 0.0567, P = 0.5089), as calculated by Pearson correlation analysis.
CXCL6 associated with diabetes in other clinical cohorts.57 We also observed an association with CXCL6 expression in PMBCs with diabetes [β = 0.38 (no CAD), P = 0.0312), but not with CAD class [β = 0.06138 (no CAD), P = 0.4795], as calculated by t-test. A report on a Han Chinese cohort in 201511 reported that CAD subjects (n = 77) had higher plasma levels of CXCL5 compared with control participants (n = 64), in stark contrast with our observations (Figure 1B). However, diabetes, hypertension, and smoking were all more prevalent in their CAD versus control participants, and circulating CXCL5 appears to be modified by diabetes in Chinese populations.12, 58 These additional inflammatory phenotypes were not distinguishing factors in our data set (Table 1) nor did they have any predictive power in our regression models (Supplemental Table S2), perhaps allowing us to uncover a link between CXCL5 and CAD.
CXCL5 Polymorphisms Associated with CAD in a Larger Cohort
Given the link between rs394408 and circulating levels of CXCL5 (Figure 4G), we used a dominant model with the minor allele that corresponded to increased CXCL5 levels. We found that the protective T allele associated with less severe CAD (Figure 5A), with an OR of 0.40 (95% CI, 0.17–0.93) using a nominal CAD classification (Table 4), similar to the inverse relationship between circulating CXCL5 and disease severity (Table 2).
Figure 5.

CXCL5 genetic association with coronary artery disease (CAD) in additional cohorts. A and C: Contingency plot and P values of CAD status and the minor allele of rs394408 in phase 1 (S1) or phase 2 (S2) of Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARA; A) or in the combined SAMARA cohort (C). B and D: Expression quantitative trait loci analysis of the single-nucleotide polymorphism rs394408 with the number of subjects, genotype, and relative CXCL5 expression levels from either phase 1 of SAMARA (B) or the combined SAMARA cohort (D), represented by box plots (5% to 95% CI) with outliers identified (open circles). The P value and β coefficient of the linear regression are noted. E and F: Plasma levels of CXCL5 represented by dot plot and summarized by the means and 95% CIs of the log values (pg/mL). E: Phase 1 (left), phase 2 (middle), or combined (right) SAMARA subjects were stratified by obstructive CAD status. F: Plasma levels of CXCL5 in subjects with increasing CAD severity, with the corresponding P value of the logistic regression analysis. D–F: The combined data sets used the z-scores of relative CXCL5 expression within each cohort. ∗P < 0.05, ∗∗P < 0.01 versus the decreased CXCL5 levels in subjects with obstructive CAD (t-test).
Table 4.
Association Between the Minor Allele of rs394408 and No Obstructive CAD
| Cohort | Participants, n | P value | OR (95% CI) | Reciprocal OR |
|---|---|---|---|---|
| SAMARA 1 | 116 | 0.0612 | 0.48 (0.22–1.05) | 2.15 |
| SAMARA 2 | 106 | 0.0330 | 0.40 (0.17–0.93) | 2.51 |
| SAMARA 1 and 2 | 222 | 0.0037 | 0.43 (0.24–0.77) | 2.31 |
We measured the association between the minor allele of rs394408 (TT + TC ↔ CC) and CAD status (nominal) using contingency analysis. The P value of the effect likelihood test for each analysis and the OR (95% CI) are provided as well as the reciprocal OR.
CAD, coronary artery disease; OR, odds ratio; SAMARA, Supporting a Multidisciplinary Approach to Researching Atherosclerosis.
To test whether our CAD association findings could be replicated in other data sets, we used the genetic data from phase 1 of the SAMARA study (n = 116; median age, 47 years; interquartile range, 43–49 years of age)17 and observed the same protective trend (Figure 5A) and eQTL (Figure 5B), finding the minor allele more prevalent in subjects without obstructive CAD (Table 4), with an OR of 0.48 (95% CI, 0.22–1.05). We leveraged the data from phase 1 participants to allow us to more than double our study size (n = 222), with which we calculated the OR of the minor allele and no obstructive CAD as 0.43 (95% CI, 0.24–0.77). This larger cohort allowed us to examine all three genotypes of rs394408, and we found a dose-dependent association of the minor allele with less severe CAD (Figure 5C), as expected given the presence of the linear eQTL (Figure 5D). Consistent with our hypothesis, we found that decreased CXCL5 expression associated with obstructive CAD from phase 1, phase 2, and the combined cohort via t-test (Figure 5E) and across all stages of CAD via logistic regression in the combined cohort (Figure 5F). These data suggest CXCL5 may modify disease risk in younger populations as well.
Identifying Molecular Phenotypes of CXCL5-Related CAD in a Geriatric Cohort
To identify molecular phenotypes associated with both circulating CXCL5 and CAD severity, we analyzed whether gene expression changes correlated with CAD severity (ordinal), resulting in 681 probes (618 unique genes; absolute rs ≥ 0.25, corresponding to P ≤ 0.0026; false-discovery rate, <7%). We next compared the SAMARA CAD expression signature with independent analyses of case-control studies performed in younger populations (all mean ages, <65 years).24 We compared our list of 618 unique genes with the 655 genes that significantly associated with CAD in analyses from both the CATHGEN10 and Personalized Risk Evaluation and Diagnosis in the Coronary Tree25 studies and identified 43 genes that overlapped (Table 5 and Supplemental Table S6),56, 59 threefold greater than the number of overlapping genes expected by chance (Table 5). These results indicate that, although some of the gene expression changes in our cohort of older individuals are similar to previously validated signatures in younger populations, there are likely age-dependent CAD changes in gene expression reflected in our cohort.
Table 5.
Over-Representation Analysis of Gene Lists
| Comparison | Set 1 | Set 2 | Overlap | Total | Representation factor | P value |
|---|---|---|---|---|---|---|
| SAMARA vs CATHGEN/PREDICT∗ | 618 | 655 | 43 | 29,281† | 3.1 | 6.70 × 10−11 |
| CXCL5 vs CAD‡ | 1041 | 681 | 253 | 18,425§ | 6.6 | 6.01 × 10−145 |
Two sets of genes and their overlap were compared, and the representation factor and probability of finding an overlap of genes were calculated. The representation factor is the number of overlapping genes/the expected number of overlapping genes drawn from two independent groups. A representation factor >1 indicates more overlap than expected of two independent groups, a representation factor <1 indicates less overlap than expected, and a representation factor of 1 indicates two groups by the number of genes expected for independent groups of genes. The probability (P) of finding more than the number of identified overlapping genes was calculated using the hypergeometric probability formula.
CAD, coronary artery disease; CATHGEN, CATHeterization GENetics; PREDICT, Personalized Risk Evaluation and Diagnosis in the Coronary Tree; SAMARA, Supporting A Multidisciplinary Approach to Researching Atherosclerosis.
Indicates gene-level comparison (unique genes only).
Indicates the number of unique genes (GenBank accessions) represented on the Agilent arrays used in the SAMARA, CATHGEN, and PREDICT studies.
Indicates probe-level comparison.
Indicates the number of probes that passed quality control in the SAMARA microarray data set used to identify CXCL5- and CAD-associated gene expression.
We then overlapped our CXCL5 and CAD gene lists and identified 253 probes associated with both circulating CXCL5 levels and CAD severity (Figure 6A). The size of the probe overlap was more than six times larger than expected by chance (expected by chance, 36 probes) (Table 5), suggesting that the inverse correlation between circulating CXCL5 levels and CAD severity extends to the molecular phenotype of gene expression. This is evident in the inverse correlations between the 253 overlapping probes (Figure 6B). For a broader overview of functional themes represented in the overlapping gene set, we performed overrepresentation analysis of immunological signatures and curated gene sets60 within these two clusters of genes (Figure 6B). Genes negatively associated with plasma levels of CXCL5 (and positively associated with CAD severity) were enriched for genes previously shown to be increased in systolic heart failure61 and aging62 (Supplemental Table S7). They were also enriched for several sets of genes that are down-regulated in B cells compared with monocytes, dendritic cells, or neutrophils,63 suggesting changes in immune cell gene expression. Likewise, healthy immune response gene expression changes64, 65 and T-cell signatures66, 67, 68 were associated with increased CXCL5 levels (Supplemental Table S7).
Figure 6.

Expression quantitative trait loci (eQTL) analysis associated with circulating CXCL5 levels and coronary artery disease (CAD) severity. A: Venn diagram of gene expression that correlated with either circulating CXCL5 levels (red), CAD severity (green), or both (yellow) in the Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARA) cohort. B: The overlapping 253 gene probes are represented by a scatterplot of the Spearman correlation coefficients (rs) of each probe to circulating CXCL5 levels (y axis) versus CAD severity (x axis). The two clusters of genes (dashed circles) were enriched for distinct gene sets, as indicated (Supplemental Table S4 provides a complete list). C: The heat map represents the relative gene expression of 18 genes with identified eQTLs in the SAMARA cohort. Each column represents one subject; the first and second rows display the CAD score and CXCL5 rank, respectively, followed by the individual gene expression profile.
We used eQTL analyses and gene expression levels as intermediate molecular phenotypes to understand how variation in cis regulatory regions contribute to disease-associated gene expression. Therefore, we further filtered the CXCL5-CAD genes that also had an eSNP within 1E6 bases of the gene probe. We used associative statistical methods using a linear model and identified 30 SNP-gene associations (cis eQTLs) (Table 6) at 18 loci (Figure 6C). One gene, LPCAT2, accounted for six eQTLs from two high linkage disequilibrium clusters of eSNPs (Figure 7, A and B). LPCAT2 expression was positively associated with CAD severity when CAD classified as either an ordinal or a nominal variable (Figure 7C). We validated these LPCAT2 eQTLs in the Kirsten data set on circulating monocytes from >2000 individuals56 and found a similar pattern of lower false-discovery rate in the linkage disequilibrium cluster ii (1e-304) compared with cluster i (2e-150), consistent with a strong link between genetic variance and LPCAT2 expression (Supplemental Table S5).
Table 6.
Local Cis eQTL Identified in CAD Genes
| rs Identification | Gene symbol | Gene description | P value | FDR, % | β Value |
|---|---|---|---|---|---|
| rs12708952 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 3 × 10−7 | 0.1 | −0.52 |
| rs4402561 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 4 × 10−7 | 0.1 | 0.49 |
| rs1891241 | LGALS8 | Lectin, galactoside-binding, soluble, 8 | 2 × 10−6 | 0.4 | −0.33 |
| rs1796415 | RNF10 | Ring finger protein 10 | 4 × 10−6 | 0.8 | 0.60 |
| rs10158939 | RP11-553N16.1∗ | Known processed pseudogene | 5 × 10−6 | 0.9 | −0.24 |
| rs11254468 | VIM | Vimentin | 7 × 10−6 | 1.3 | 0.43 |
| rs6599244 | RPL18AP7∗ | Ribosomal protein L18a pseudogene 7 | 1 × 10−5 | 1.9 | 0.44 |
| rs9511357 | ZMYM5 | Zinc finger, MYM-type 5 | 1 × 10−5 | 2.2 | −0.18 |
| rs831757 | ARL8A | ADP-ribosylation factor-like 8A | 2 × 10−5 | 2.5 | −0.23 |
| rs2799428 | LGALS8 | Lectin, galactoside-binding, soluble, 8 | 2 × 10−5 | 2.8 | −0.29 |
| rs244836 | COTL1 | Coactosin-like F-actin binding protein 1 | 2 × 10−5 | 2.9 | 0.23 |
| rs1583585 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 2 × 10−5 | 3.2 | −0.41 |
| rs1963773 | RNF10 | Ring finger protein 10 | 2 × 10−5 | 3.4 | −0.45 |
| rs41416850 | RNF10 | Ring finger protein 10 | 2 × 10−5 | 3.4 | −0.63 |
| rs2737706 | LGALS8 | Lectin, galactoside-binding, soluble, 8 | 3 × 10−5 | 3.7 | 0.30 |
| rs1583587 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 3 × 10−5 | 3.9 | 0.42 |
| rs3779647 | GSR | Glutathione reductase | 3 × 10−5 | 4.0 | −0.19 |
| rs913863 | RRAGC | Ras-related GTP binding C | 4 × 10−5 | 4.8 | −0.10 |
| rs11646643 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 5 × 10−05 | 6.0 | −0.41 |
| rs9989481 | UBALD2 | UBA-like domain containing 2 | 6 × 10−5 | 6.3 | 0.24 |
| rs11206237 | TALDO1 | Transaldolase 1 | 6 × 10−5 | 6.8 | −0.44 |
| rs913863 | RRAGC | Ras-related GTP binding C | 7 × 10−5 | 7.3 | −0.10 |
| rs1477017 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 7 × 10−5 | 7.6 | 0.40 |
| rs1799917 | LPCAT2 | Lysophosphatidylcholine acyltransferase 2 | 7 × 10−5 | 7.6 | 0.41 |
| rs2715143 | GRB10 | Growth factor receptor-bound protein 10 | 7 × 10−5 | 7.6 | −0.84 |
| rs9553323 | ZMYM5 | Zinc finger, MYM-type 5 | 7 × 10−5 | 7.7 | −0.17 |
| rs4415303 | GYG1P1∗ | Glycogenin 1 pseudogene 1 | 8 × 10−5 | 7.8 | 0.49 |
| rs1611699 | HLA-K∗ | Major histocompatibility complex, class I, K | 9 × 10−5 | 8.5 | −0.18 |
| rs3013777 | YIPF1 | Yip1 domain family, member 1 | 1 × 10−4 | 9.5 | −0.21 |
| rs11185517 | NCBP2-AS2 | NCBP2 antisense RNA 2 (head to head) | 1 × 10−4 | 9.7 | 0.16 |
The single-nucleotide polymorphism identifier (rs identification), official gene symbol and description, and the P values, FDRs, and β values from the linear model are provided.
CAD, coronary artery disease; eQTL, expression quantitative trait loci; FDR, false-discovery rate; HLA, human leukocyte antigen; MYM, myeloproliferative and mental retardation motif; NCBP, Nuclear Cap Binding Protein Subunit; UBA, ubiquitin-associated domain; Yip, Ypt-interacting proteins.
Gene symbols and descriptions updated in BioMart that were not initially annotated.
Figure 7.

Expression quantitative trait loci (eQTL) analysis associated with circulating CXCL5 levels and coronary artery disease (CAD) severity. A: Linkage disequilibrium analysis (top) of six expression regulatory single-nucleotide polymorphisms (eSNPs) at the locus of LPCAT2 on chromosome 16 from black and white American 1000 genomes population data (represented in million bp; middle) identified two clusters of correlated SNPs (i and ii). Tissues where four of the LPCAT2 eQTLs were also present are indicated (bottom), with the effect size and significance level indicated by color intensity and size, respectively, from dbGaP phs000424.v6.p1. B: eQTL analysis of the eSNP rs12708952 (left panel) and rs11646643 (right panel) with the number of subjects per genotype from the Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARA) cohort represented by box plots (5% to 95% CI) with outliers identified (open circles). The P value and β coefficient of the linear regression are noted. C:LPCAT2 expression versus CAD severity in the SAMARA cohort is represented by dot plot, summarized by the means and 95% CIs, classified as a nominal (left panel) or an ordinal (right panel) variable, with the corresponding P value from either a t-test or logistic regression, respectively. D: QTL analysis of rs12708952 and platelet counts (left panel) or PF4 expression (right panel), represented by box plots (5% to 95% CI) with outliers identified (open circles). The P value and β coefficient of the linear regression are noted.
LPCAT2 encodes an enzyme that synthesizes glycerophospholipid precursors used in membrane biogenesis and the inflammatory lipid platelet-activating factor. Because LPCAT2 is expressed in circulating blood cells, we extended our QTL and correlation analyses to platelet counts and PF4 expression levels to determine whether variation at rs12708952 associates with altered platelet levels or platelet activation. The correlation between LPCAT2 and either platelet counts or PF4 expression was weak and opposed (Supplemental Figure S2). Furthermore, there was no association between variation at rs12708952 and either platelet counts or PF4 expression (Figure 7D), suggesting that the repercussions of LPCAT2 expression do not extend to these measures of platelet function and that perhaps other pathways or signaling affected by LPCAT2 expression may be important to the phenotypes observed in our study. In addition to being expressed in circulating blood cells, LPCAT2 is expressed throughout the body. Given the complex nature of CVD, we looked for tissue specific for the LPCAT2 eQTLs from cluster ii from dbGAP. Interestingly, we found the presence of tissue-specific eQTLs, including adipose tissue and various blood vessels, suggesting that LPCAT2 biology in these tissues, in addition to circulating blood cells, may be important in human CAD (Figure 7A).
Discussion
As the percentage of our population that is 65 years and older increases annually, it is important to understand the genetic and molecular underpinnings of CAD in geriatric patients. To assess the relationship between CXCL5, the severity of CAD, and the corresponding molecular phenotypes, we characterized the circulating levels of CXCL5 and other proinflammatory markers, gene expression patterns, and genetic variation in patients 65 years and older who were undergoing cardiac catheterization (Table 1). Interestingly, we identified a negative association between circulating CXCL5 levels and CAD severity (Table 2). Similar to a prior report of an inverse relationship between CXCL5 activity and atherosclerosis in a mouse model,13 our clinical data are consistent with a protective role of CXCL5 in CAD (Figure 1). To ensure our observation was not confounded by other known factors that could influence CXCL5 levels, we extensively examined several covariates, such as sex (Figures 1B and 2B), circulating lipid levels (Figure 2A), inflammatory biomarkers (Table 3 and Supplemental Tables S2 and S3), prior MI (Figure 3), and statin use (Figure 2, B–D). Interestingly, there was no association between prior statin use and CXCL5 levels, but by using various stratifications, we identified a decrease in CXCL5 levels in female statin users with obstructive CAD (Figure 2B). The high-intensity statin, rosuvastatin, had a differential effect on CXCL5 levels (Figure 2C), suggesting that statin intensity and sex-specific effects of statins on CXCL5 biology should be considered in future clinical studies. Although white blood cell counts did not correlate with plasma levels of CXCL5, we identified a positive correlation between platelet counts and CXCL5 (Figure 2A). These data suggest that platelets could be a physiologically important source of CXCL5 in the context of CAD. Previous studies in mice demonstrated that aortic endothelial cells are an important source of CXCL5 in proatherogenic conditions,13 highlighting the need to better understand the production and turnover of CXCL5 in physiological and pathophysiological conditions.
Genomic analyses of our cohort revealed a genetic contribution to CXCL5 expression (Figures 4A and 5, B and D) that ultimately extended to both circulating plasma CXCL5 levels (Figure 4G) and CAD severity (Figure 5, A, C, E, and F). Furthermore, the eSNP rs394408 corresponds specifically with CXCL5 expression levels and not PF4 or PPBP, which are in close proximity (Figure 4C). Furthermore, gene expression changes that were common to both plasma CXCL5 levels and CAD severity (Figure 6A) also had an inverse relationship, suggesting that genomic analyses provide a unique molecular insight into the relationship between CXCL5 and CAD severity (Figure 6B). This approach revealed the dysregulation of LPCAT2 (Figures 6C and 7, B and C), an enzyme involved in both glycerophospholipid precursor synthesis to help maintain inflammatory cell membrane integrity and synthesis of platelet-activating factor. This factor is a potent phospholipid activator that affects vascular permeability and chemotaxis of leukocytes, platelet aggregation and degranulation, and inflammation; it also causes a negative inotropic effect.69 In addition to the presence of LPACT2 eQTLs in PBMCs (Figure 7B) and isolated monocytes (Supplemental Table S5), multitissue analysis (Figure 7A) identified these same eQTLs in vascular tissues, such as the aorta and tibial arteries, and adipose tissue. The presence of tissue-specific eQTLs suggests that LPCAT2 expression in these tissues may play a role in the disease susceptibility identified in our study (Figure 7C).
How is CXCL5 regulated during the development of CAD? In ApoE−/− mice, plasma CXCL5 levels increased threefold after 6 weeks of a Western diet and remained elevated throughout atherogenesis.13 In a Chinese Han cohort, higher plasma CXCL5 levels were found in CAD patients compared with subjects without CAD.11 However, CAD severity was not considered, and other chronic inflammatory conditions, including diabetes and hypertension, were more commonly comorbid with CAD in these subjects. These reports, combined with the data presented herein, suggest that CXCL5 increases with the onset of atherosclerosis and maintaining (or further increasing) CXCL5 levels may protect against obstructive CAD. This provides a clinical extension of mouse studies in which CXCL5 was directly manipulated to demonstrate the atheroprotective effect of this chemokine.13
Other studies demonstrated that markers, such as MCP-1, selectins, and CAMs, positively associate with CAD41, 70; however, our study did not reveal any associations between CAD severity and these inflammatory markers. The only other marker in our analyses that positively associated with CAD was hsCRP (Table 3). Numerous studies reported positive associations of hsCRP with CAD,44, 45 although studies using a mixed population of older and younger patients reported no association between hsCRP and CAD.71, 72, 73 Further studies that evaluate younger and geriatric populations separately, and analysis of longitudinal studies over age, are necessary to identify the distinct pathophysiological processes in CAD that are likely modified by aging. For example, we observed a similar, but weaker, genetic association between CXCL5 and CAD in a younger cohort (Figure 5, A and B), suggesting that CXCL5 may play a role, even in younger patients with CAD.
It is important to identify the limitations of our study. First, our patient population consists of subjects referred to the cardiac catheterization laboratory for clinically indicated coronary angiography; therefore, we lack a true age-matched, healthy control group, a known challenge in the genetic characterization of CAD.74 However, our cohort did include patients with little or no angiographically detectable CAD. Second, our sample size for genomic analyses (n = 106) prevented us from performing additional analyses, such as genome-wide associations or trans-eQTL identification. Therefore, we leveraged our data by combining genetic and transcriptional data sets, and we used public data sets to serve as validation cohorts for our genomic findings. In addition, we validated our primary findings from phase 1 of SAMARA to increase the power of our contingency analysis looking at disease risk and the protective allele at rs394408. Although our study demonstrates that CXCL5 levels are diminished with increasing CAD severity, future longitudinal studies tracking CXCL5 expression over age are necessary to elucidate the role of CXCL5 in disease initiation and progression. In addition, cell and animal studies can be used to elucidate the role of the biological interaction of CXCL5 and LPCAT2 in both the pathology of atherosclerosis and the design of new therapeutic interventions to modify disease risk.
Acknowledgments
We thank members of the Schisler Laboratory for critical review of the manuscript; the Willis, Jensen, Mclean, and Stouffer Laboratories for support; previous collaborators of the SAMARA project, including Peter C. Charles and Robert E. Lineberger; and The McAllister Heart Institute and Cardiology administration teams.
S.R., R.N.S., and J.C.S. contributed to conception and design, analysis and interpretation of, data, or both; drafting of the manuscript or revising it critically for important intellectual content; final approval of the manuscript submitted; E.H., C.R.L., X.D., G.A.S., and C.P. contributed to conception and design, analysis and interpretation of data, or both; and final approval of the manuscript submitted; K.L., M.S.W., and B.C.J. contributed to drafting of the manuscript or revising it critically for important intellectual content and final approval of the manuscript submitted; J.C.S. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Supported by a Le Fondation Leducq Trans-Atlantic Network of Excellence grant, The University of North Carolina at Chapel Hill grant 6066230 (Investments in the Future: Understanding Clinical Cardiovascular Disease and Health Disparities in North Carolina Using a Systems Biology Approach), American Heart Association grant 16GRNT29300003 (C.R.L.) and predoctoral fellowship 11PRE7240059 (R.N.S.), NIH/National Heart, Lung, and Blood Institute (NHLBI) predoctoral training program in Integrative Vascular Biology T32 HL069768, and NIH/NHLBI grant 4R37HL065619 (C.P.). The Genotype-Tissue Expression Project was supported by the Common Fund of the Office of the Director of the NIH and by National Cancer Institute, National Human Genome Research Institute, NHLBI, National Institute of Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Diseases and Stroke.
Disclosures: None declared.
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2017.08.006.
Supplemental Data
Allele frequency of rs394408 in different ancestral populations. The allele frequency of the minor (T) and major (C) alleles represented by a stacked bar plot. For reference, the 5% minor allele frequency (MAF) cutoff is indicated by a green arrow, and frequencies <5% are usually considered rare within a population. Data from the following ancestral populations are provided: Han Chinese in Beijing, China (CHB); Japanese in Tokyo, Japan (JPT); Mexican ancestry in Los Angeles, CA (MXL); British in England and Scotland (GRB); Utah residents with Northern and Western European ancestry (CEU); Puerto Ricans from Puerto Rico (PUR); African ancestry in Southwest United States (ASW); and Yoruba in Ibadan, Nigeria (YRI).54 The expected allele frequency in Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARAex) for rs394408 within the SAMARA cohort (phase 1 and phase 2) was calculated using the corresponding frequencies found in the black (ASW) and white (CEU) American F1000 population data. The observed allele frequency (SAMARAobs) is also provided.
Association of lysophosphatidylcholine acyltransferase 2 (LPCAT2) expression, platelet factor 4 (PF4) expression, and platelet counts. Pairwise association analysis (Pearson) of the indicated variables summarized by scatterplot, regression line, and means (95% CIs). The histogram of the variable distribution is provided. The unit of mRNA is relative expression, and platelets are log transformed. The correlation coefficient for each association is indicated. ∗P < 0.05 LPCAT2 versus platelet counts; ∗∗P < 0.01 LPCAT2 versus PF4.
References
- 1.Berry J.D., Dyer A., Cai X., Garside D.B., Ning H., Thomas A., Greenland P., Van Horn L., Tracy R.P., Lloyd-Jones D.M. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366:321–329. doi: 10.1056/NEJMoa1012848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roger V.L., Go A.S., Lloyd-Jones D.M., Benjamin E.J., Berry J.D., Borden W.B., American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakatta E.G., Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises, part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 4.Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 5.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 6.Libby P., Ridker P.M., Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 7.Beckett N.S., Peters R., Fletcher A.E., Staessen J.A., Liu L., Dumitrascu D., Stoyanovsky V., Antikainen R.L., Nikitin Y., Anderson C., Belhani A., Forette F., Rajkumar C., Thijs L., Banya W., Bulpitt C.J. Treatment of hypertension in patients 80 years of age or older. N Engl J Med. 2008;358:1887–1898. doi: 10.1056/NEJMoa0801369. [DOI] [PubMed] [Google Scholar]
- 8.Shepherd J., Blauw G.J., Murphy M.B., Bollen E.L.E.M., Buckley B.M., Cobbe S.M., Ford I., Gaw A., Hyland M., Jukema J.W., Kamper A.M., Macfarlane P.W., Meinders A.E., Norrie J., Packard C.J., Perry I.J., Stott D.J., Sweeney B.J., Twomey C., Westendorp R.G.J., PROSPER Study Group. PROspective Study of Pravastatin in the Elderly at Risk Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 9.Sinnaeve P.R., Donahue M.P., Grass P., Seo D., Vonderscher J., Chibout S.-D., Kraus W.E., Sketch M., Nelson C., Ginsburg G.S., Goldschmidt-Clermont P.J., Granger C.B. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS One. 2009;4:e7037. doi: 10.1371/journal.pone.0007037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wingrove J.A., Daniels S.E., Sehnert A.J., Tingley W., Elashoff M.R., Rosenberg S., Buellesfeld L., Grube E., Newby L.K., Ginsburg G.S., Kraus W.E. Correlation of peripheral-blood gene expression with the extent of coronary artery stenosis. Circ Cardiovasc Genet. 2008;1:31–38. doi: 10.1161/CIRCGENETICS.108.782730. [DOI] [PubMed] [Google Scholar]
- 11.Wang X.Z., Liu L.W., Du X.M., Gu R.X., Sun Z.J. CXCL5 is associated with the increased risk of coronary artery disease. Coron Artery Dis. 2015;26:612–619. doi: 10.1097/MCA.0000000000000292. [DOI] [PubMed] [Google Scholar]
- 12.Chen L., Yang Z., Lu B., Li Q., Ye Z., He M., Huang Y., Wang X., Zhang Z., Wen J., Liu C., Qu S., Hu R. Serum CXC ligand 5 is a new marker of subclinical atherosclerosis in type 2 diabetes. Clin Endocrinol (Oxf) 2011;75:766–770. doi: 10.1111/j.1365-2265.2011.04119.x. [DOI] [PubMed] [Google Scholar]
- 13.Rousselle A., Qadri F., Leukel L., Yilmaz R., Fontaine J.-F., Sihn G., Bader M., Ahluwalia A., Duchene J. CXCL5 limits macrophage foam cell formation in atherosclerosis. J Clin Invest. 2013;123:1343–1347. doi: 10.1172/JCI66580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maddox T.M., Stanislawski M.A., Grunwald G.K., Bradley S.M., Ho P.M., Tsai T.T., Patel M.R., Sandhu A., Valle J., Magid D.J., Leon B., Bhatt D.L., Fihn S.D., Rumsfeld J.S. Nonobstructive coronary artery disease and risk of myocardial infarction. JAMA. 2014;312:1754–1763. doi: 10.1001/jama.2014.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel M.R., Dai D., Hernandez A.F., Douglas P.S., Messenger J., Garratt K.N., Maddox T.M., Peterson E.D., Roe M.T. Prevalence and predictors of nonobstructive coronary artery disease identified with coronary angiography in contemporary clinical practice. Am Heart J. 2014;167:846–852. doi: 10.1016/j.ahj.2014.03.001. e2. [DOI] [PubMed] [Google Scholar]
- 16.Charles P.C., Alder B.D., Hilliard E.G., Schisler J.C., Lineberger R.E., Parker J.S., Mapara S., Wu S.S., Portbury A., Patterson C., Stouffer G.A. Tobacco use induces anti-apoptotic, proliferative patterns of gene expression in circulating leukocytes of Caucasian males. BMC Med Genomics. 2008;1:38. doi: 10.1186/1755-8794-1-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schisler J.C., Charles P.C., Parker J.S., Hilliard E.G., Mapara S., Meredith D., Lineberger R.E., Wu S.S., Alder B.D., Stouffer G.A., Patterson C. Stable patterns of gene expression regulating carbohydrate metabolism determined by geographic ancestry. PLoS One. 2009;4:e8183. doi: 10.1371/journal.pone.0008183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuck R.N., Theken K.N., Edin M.L., Caughey M., Bass A., Ellis K., Tran B., Steele S., Simmons B.P., Lih F.B., Tomer K.B., Wu M.C., Hinderliter A.L., Stouffer G.A., Zeldin D.C., Lee C.R. Cytochrome P450-derived eicosanoids and vascular dysfunction in coronary artery disease patients. Atherosclerosis. 2013;227:442–448. doi: 10.1016/j.atherosclerosis.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anuurad E., Enkhmaa B., Gungor Z., Zhang W., Tracy R.P., Pearson T.A., Kim K., Berglund L. Age as a modulator of inflammatory cardiovascular risk factors. Arterioscler Thromb Vasc Biol. 2011;31:2151–2156. doi: 10.1161/ATVBAHA.111.232348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smedley D., Haider S., Durinck S., Pandini L., Provero P., Allen J. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43:W589–W598. doi: 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R Core Team (2013). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available at http://www.R-project.org
- 24.Elashoff M.R., Wingrove J.A., Beineke P., Daniels S.E., Tingley W.G., Rosenberg S., Voros S., Kraus W.E., Ginsburg G.S., Schwartz R.S., Ellis S.G., Tahirkheli N., Waksman R., McPherson J., Lansky A.J., Topol E.J. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Med Genomics. 2011;4:26. doi: 10.1186/1755-8794-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg S., Elashoff M.R., Beineke P., Daniels S.E., Wingrove J.A., Tingley W.G., Sager P.T., Sehnert A.J., Yau M., Kraus W.E., Newby L.K., Schwartz R.S., Voros S., Ellis S.G., Tahirkheli N., Waksman R., McPherson J., Lansky A., Winn M.E., Schork N.J., Topol E.J. Multicenter validation of the diagnostic accuracy of a blood-based gene expression test for assessing obstructive coronary artery disease in nondiabetic patients. Ann Intern Med. 2010;153:425–434. doi: 10.7326/0003-4819-153-7-201010050-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akaike H. A new look at the statistical model identification. IEEE Trans Autom Control. 1974;19:716–723. [Google Scholar]
- 27.Ramsey J.M., Schwarz E., Guest P.C., van Beveren N.J.M., Leweke F.M., Rothermundt M., Bogerts B., Steiner J., Ruta L., Baron-Cohen S., Bahn S. Molecular sex differences in human serum. PLoS One. 2012;7:e51504. doi: 10.1371/journal.pone.0051504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zineh I., Beitelshees A.L., Welder G.J., Hou W., Chegini N., Wu J., Cresci S., Province M.A., Spertus J.A. Epithelial neutrophil-activating peptide (ENA-78), acute coronary syndrome prognosis, and modulatory effect of statins. PLoS One. 2008;3:e3117. doi: 10.1371/journal.pone.0003117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chegini N., Luo X., Pan Q., Rhoton-Vlasak A., Archer D.F. Endometrial expression of epithelial neutrophil-activating peptide-78 during the menstrual cycle or in progestin-only contraceptive users with breakthrough bleeding and the influence of doxycycline therapy. Hum Reprod. 2007;22:427–433. doi: 10.1093/humrep/del398. [DOI] [PubMed] [Google Scholar]
- 30.Imaizumi T., Albertine K.H., Jicha D.L., McIntyre T.M., Prescott S.M., Zimmerman G.A. Human endothelial cells synthesize ENA-78: relationship to IL-8 and to signaling of PMN adhesion. Am J Respir Cell Mol Biol. 1997;17:181–192. doi: 10.1165/ajrcmb.17.2.2818. [DOI] [PubMed] [Google Scholar]
- 31.Jeyaseelan S., Manzer R., Young S.K., Yamamoto M., Akira S., Mason R.J., Worthen G.S. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am J Respir Cell Mol Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch A.E., Kunkel S.L., Harlow L.A., Mazarakis D.D., Haines G.K., Burdick M.D., Pope R.M., Walz A., Strieter R.M. Epithelial neutrophil activating peptide-78: a novel chemotactic cytokine for neutrophils in arthritis. J Clin Invest. 1994;94:1012–1018. doi: 10.1172/JCI117414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin M., Carlson E., Diaconu E., Pearlman E. CXCL1/KC and CXCL5/LIX are selectively produced by corneal fibroblasts and mediate neutrophil infiltration to the corneal stroma in LPS keratitis. J Leukoc Biol. 2007;81:786–792. doi: 10.1189/jlb.0806502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mei J., Liu Y., Dai N., Favara M., Greene T., Jeyaseelan S., Poncz M., Lee J.S., Worthen G.S. CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity. 2010;33:106–117. doi: 10.1016/j.immuni.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mei J., Liu Y., Dai N., Hoffmann C., Hudock K.M., Zhang P., Guttentag S.H., Kolls J.K., Oliver P.M., Bushman F.D., Worthen G.S. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J Clin Invest. 2012;122:974–986. doi: 10.1172/JCI60588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L.-Y., Tu Y.-F., Lin Y.-C., Huang C.-C. CXCL5 signaling is a shared pathway of neuroinflammation and blood-brain barrier injury contributing to white matter injury in the immature brain. J Neuroinflammation. 2016;13:6. doi: 10.1186/s12974-015-0474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anand S.S. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. Vasc Med. 2003;8:289–290. doi: 10.1191/1358863x03vm518xx. [DOI] [PubMed] [Google Scholar]
- 38.Jones P., Kafonek S., Laurora I., Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study) Am J Cardiol. 1998;81:582–587. doi: 10.1016/s0002-9149(97)00965-x. [DOI] [PubMed] [Google Scholar]
- 39.Jones P.H., Davidson M.H., Stein E.A., Bays H.E., McKenney J.M., Miller E., Cain V.A., Blasetto J.W., STELLAR Study Group Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial) Am J Cardiol. 2003;92:152–160. doi: 10.1016/s0002-9149(03)00530-7. [DOI] [PubMed] [Google Scholar]
- 40.Blankenberg S., Rupprecht H.J., Bickel C., Peetz D., Hafner G., Tiret L., Meyer J. Circulating cell adhesion molecules and death in patients with coronary artery disease. Circulation. 2001;104:1336–1342. doi: 10.1161/hc3701.095949. [DOI] [PubMed] [Google Scholar]
- 41.de Lemos J.A., Morrow D.A., Blazing M.A., Jarolim P., Wiviott S.D., Sabatine M.S., Califf R.M., Braunwald E. Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: results from the A to Z trial. J Am Coll Cardiol. 2007;50:2117–2124. doi: 10.1016/j.jacc.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 42.Ridker P.M., Cushman M., Stampfer M.J., Tracy R.P., Hennekens C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- 43.Ridker P.M., Cannon C.P., Morrow D., Rifai N., Rose L.M., McCabe C.H., Pfeffer M.A., Braunwald E., Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 44.Bajpai A., Goyal A., Sperling L. Should we measure C-reactive protein on Earth or just on JUPITER? Clin Cardiol. 2010;33:190–198. doi: 10.1002/clc.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva D., Pais de Lacerda A. High-sensitivity C-reactive protein as a biomarker of risk in coronary artery diseaseRev Port Cardiol. 2012;31:733–745. doi: 10.1016/j.repc.2012.02.018. Portuguese. [DOI] [PubMed] [Google Scholar]
- 46.Chang T.-Y., Hsu C.-Y., Huang P.-H., Chiang C.-H., Leu H.-B., Huang C.-C., Chen J.-W., Lin S.-J. Usefulness of circulating decoy receptor 3 in predicting coronary artery disease severity and future major adverse cardiovascular events in patients with multivessel coronary artery disease. Am J Cardiol. 2015;116:1028–1033. doi: 10.1016/j.amjcard.2015.06.041. [DOI] [PubMed] [Google Scholar]
- 47.Lin T., Liu J.-C., Chang L.-Y., Shen C.-W. Association of C-reactive protein and homocysteine with subclinical coronary plaque subtype and stenosis using low-dose MDCT coronary angiography. Atherosclerosis. 2010;212:501–506. doi: 10.1016/j.atherosclerosis.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 48.Peer A., Falkensammer G., Alber H., Kroiss A., Griesmacher A., Ulmer H., Pachinger O., Mair J. Limited utilities of N-terminal pro B-type natriuretic peptide and other newer risk markers compared with traditional risk factors for prediction of significant angiographic lesions in stable coronary artery disease. Heart. 2009;95:297–303. doi: 10.1136/hrt.2008.145425. [DOI] [PubMed] [Google Scholar]
- 49.Potekhina A.V., Pylaeva E., Provatorov S., Ruleva N., Masenko V., Noeva E., Krasnikova T., Arefieva T. Treg/Th17 balance in stable CAD patients with different stages of coronary atherosclerosis. Atherosclerosis. 2015;238:17–21. doi: 10.1016/j.atherosclerosis.2014.10.088. [DOI] [PubMed] [Google Scholar]
- 50.Mestas J., Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beitelshees A.L., Aquilante C.L., Allayee H., Langaee T.Y., Welder G.J., Schofield R.S., Zineh I. CXCL5 polymorphisms are associated with variable blood pressure in cardiovascular disease-free adults. Hum Genomics. 2012;6:9. doi: 10.1186/1479-7364-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zineh I., Aquilante C.L., Langaee T.Y., Beitelshees A.L., Arant C.B., Wessel T.R., Schofield R.S. CXCL5 gene polymorphisms are related to systemic concentrations and leukocyte production of epithelial neutrophil-activating peptide (ENA-78) Cytokine. 2006;33:258–263. doi: 10.1016/j.cyto.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 53.Zeller T., Wild P., Szymczak S., Rotival M., Schillert A., Castagne R., Maouche S., Germain M., Lackner K., Rossmann H., Eleftheriadis M., Sinning C.R., Schnabel R.B., Lubos E., Mennerich D., Rust W., Perret C., Proust C., Nicaud V., Loscalzo J., Hübner N., Tregouet D., Münzel T., Ziegler A., Tiret L., Blankenberg S., Cambien F. Genetics and beyond: the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Machiela M.J., Chanock S.J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirsten H., Al-Hasani H., Holdt L., Gross A., Beutner F., Krohn K., Horn K., Ahnert P., Burkhardt R., Reiche K., Hackermüller J., Löffler M., Teupser D., Thiery J., Scholz M. Dissecting the genetics of the human transcriptome identifies novel trait-related trans-eQTLs and corroborates the regulatory relevance of non-protein coding loci†. Hum Mol Genet. 2015;24:4746–4763. doi: 10.1093/hmg/ddv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alfadda A.A., Alzoghaibi M.A. Circulatory neutrophil chemokines in statin-treated diabetic patients. Saudi Med J. 2008;29:584–588. [PubMed] [Google Scholar]
- 58.Yang Z., Zhang Z., Wen J., Wang X., Lu B., Yang Z., Zhang W., Wang M., Feng X., Ling C., Wu S., Hu R. Elevated serum chemokine CXC ligand 5 levels are associated with hypercholesterolemia but not a worsening of insulin resistance in Chinese people. J Clin Endocrinol Metab. 2010;95:3926–3932. doi: 10.1210/jc.2009-2194. [DOI] [PubMed] [Google Scholar]
- 59.Fehrmann R.S.N., Jansen R.C., Veldink J.H., Westra H.-J., Arends D., Bonder M.J., Fu J., Deelen P., Groen H.J.M., Smolonska A., Weersma R.K., Hofstra R.M.W., Buurman W.A., Rensen S., Wolfs M.G.M., Platteel M., Zhernakova A., Elbers C.C., Festen E.M., Trynka G., Hofker M.H., Saris C.G.J., Ophoff R.A., van den Berg L.H., van Heel D.A., Wijmenga C., te Meerman G.J., Franke L. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thum T., Galuppo P., Wolf C., Fiedler J., Kneitz S., van Laake L.W., Doevendans P.A., Mummery C.L., Borlak J., Haverich A., Gross C., Engelhardt S., Ertl G., Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 62.Rodwell G.E.J., Sonu R., Zahn J.M., Lund J., Wilhelmy J., Wang L., Xiao W., Mindrinos M., Crane E., Segal E., Myers B.D., Brooks J.D., Davis R.W., Higgins J., Owen A.B., Kim S.K. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004;2:e427. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abbas A.R., Baldwin D., Ma Y., Ouyang W., Gurney A., Martin F., Fong S., van Lookeren Campagne M., Godowski P., Williams P.M., Chan A.C., Clark H.F. Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 2005;6:319–331. doi: 10.1038/sj.gene.6364173. [DOI] [PubMed] [Google Scholar]
- 64.Hutcheson J., Scatizzi J.C., Siddiqui A.M., Haines G.K., Wu T., Li Q.-Z., Davis L.S., Mohan C., Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 65.Ramilo O., Allman W., Chung W., Mejias A., Ardura M., Glaser C., Wittkowski K.M., Piqueras B., Banchereau J., Palucka A.K., Chaussabel D. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109:2066–2077. doi: 10.1182/blood-2006-02-002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doering T.A., Crawford A., Angelosanto J.M., Paley M.A., Ziegler C.G., Wherry E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee M.S., Hanspers K., Barker C.S., Korn A.P., McCune J.M. Gene expression profiles during human CD4+ T cell differentiation. Int Immunol. 2004;16:1109–1124. doi: 10.1093/intimm/dxh112. [DOI] [PubMed] [Google Scholar]
- 68.Lund R., Aittokallio T., Nevalainen O., Lahesmaa R. Identification of novel genes regulated by IL-12, IL-4, or TGF-beta during the early polarization of CD4+ lymphocytes. J Immunol. 2003;171:5328–5336. doi: 10.4049/jimmunol.171.10.5328. [DOI] [PubMed] [Google Scholar]
- 69.Detopoulou P., Nomikos T., Fragopoulou E., Chrysohoou C., Antonopoulou S. Platelet activating factor in heart failure: potential role in disease progression and novel target for therapy. Curr Heart Fail Rep. 2013;10:122–129. doi: 10.1007/s11897-013-0131-2. [DOI] [PubMed] [Google Scholar]
- 70.Hwang S.J., Ballantyne C.M., Sharrett A.R., Smith L.C., Davis C.E., Gotto A.M., Boerwinkle E. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation. 1997;96:4219–4225. doi: 10.1161/01.cir.96.12.4219. [DOI] [PubMed] [Google Scholar]
- 71.Folsom A.R., Chambless L.E., Ballantyne C.M., Coresh J., Heiss G., Wu K.K., Boerwinkle E., Mosley T.H., Sorlie P., Diao G., Sharrett A.R. An assessment of incremental coronary risk prediction using C-reactive protein and other novel risk markers: the Atherosclerosis Risk in Communities Study. Arch Intern Med. 2006;166:1368–1373. doi: 10.1001/archinte.166.13.1368. [DOI] [PubMed] [Google Scholar]
- 72.Ridker P.M., Rifai N., Rose L., Buring J.E., Cook N.R. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 73.Rodondi N., Marques-Vidal P., Butler J., Sutton-Tyrrell K., Cornuz J., Satterfield S., Harris T., Bauer D.C., Ferrucci L., Vittinghoff E., Newman A.B., Health, Aging, and Body Composition Study Markers of atherosclerosis and inflammation for prediction of coronary heart disease in older adults. Am J Epidemiol. 2010;171:540–549. doi: 10.1093/aje/kwp428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo A.K., Jefferson B.K., Garcia M.J., Ginsburg G.S., Topol E.J. Challenges in the phenotypic characterisation of patients in genetic studies of coronary artery disease. J Med Genet. 2007;44:161–165. doi: 10.1136/jmg.2006.045732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Allele frequency of rs394408 in different ancestral populations. The allele frequency of the minor (T) and major (C) alleles represented by a stacked bar plot. For reference, the 5% minor allele frequency (MAF) cutoff is indicated by a green arrow, and frequencies <5% are usually considered rare within a population. Data from the following ancestral populations are provided: Han Chinese in Beijing, China (CHB); Japanese in Tokyo, Japan (JPT); Mexican ancestry in Los Angeles, CA (MXL); British in England and Scotland (GRB); Utah residents with Northern and Western European ancestry (CEU); Puerto Ricans from Puerto Rico (PUR); African ancestry in Southwest United States (ASW); and Yoruba in Ibadan, Nigeria (YRI).54 The expected allele frequency in Supporting a Multidisciplinary Approach to Researching Atherosclerosis (SAMARAex) for rs394408 within the SAMARA cohort (phase 1 and phase 2) was calculated using the corresponding frequencies found in the black (ASW) and white (CEU) American F1000 population data. The observed allele frequency (SAMARAobs) is also provided.
Association of lysophosphatidylcholine acyltransferase 2 (LPCAT2) expression, platelet factor 4 (PF4) expression, and platelet counts. Pairwise association analysis (Pearson) of the indicated variables summarized by scatterplot, regression line, and means (95% CIs). The histogram of the variable distribution is provided. The unit of mRNA is relative expression, and platelets are log transformed. The correlation coefficient for each association is indicated. ∗P < 0.05 LPCAT2 versus platelet counts; ∗∗P < 0.01 LPCAT2 versus PF4.