

Abstract
Polypoidal choroidal vasculopathy (PCV) is a common subtype of wet age-related macular degeneration in Asian populations, whereas choroidal neovascularization is the typical subtype in Western populations. The cause of PCV is unknown. By comparing the phenotype of a PCV mouse model expressing protease high temperature requirement factor A1 (HTRA1) in retinal pigment epithelium with transgenic mice expressing the inactive HTRA1S328A, we showed that HTRA1-mediated degradation of elastin in choroidal vessels is critical for the development of PCV, which exhibited destructive extracellular matrix remodeling and vascular smooth muscle cell loss. Compared with weak PCV, severe PCV exhibited prominent immune complex deposition, complement activation, and infiltration of inflammatory cells, suggesting inflammation plays a key role in PCV progression. More important, we validated these findings in human PCV specimens. Intravitreal delivery of an HTRA1 inhibitor (DPMFKLboroV) was effective (36% lesion reduction; P = 0.009) in preventing PCV initiation but ineffective in treating existing lesions. Anti-inflammatory glucocorticoid was effective in preventing PCV progression but ineffective in preventing PCV initiation. These results suggest that PCV pathogenesis occurs through two stages. The initiation stage is mediated by proteolytic degradation of extracellular matrix proteins attributable to increased HTRA1 activity, whereas the progression stage is driven by inflammatory cascades. This study provides a basis for understanding the differences between PCV and choroidal neovascularization, and helps guide the design of effective therapies for PCV.
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in elderly people globally. The projected number of people with AMD is 196 million in 2020 and 288 million in 2040, representing a substantial global burden on the health care system.1 Wet AMD includes choroidal neovascularization (CNV) and polypoidal choroidal vasculopathy (PCV). CNV describes the growth of new blood vessels from the choroid into the subretinal space, whereas PCV refers to choroidal vessel abnormalities (eg, polypoidal dilations).2 PCV is a common subtype of wet AMD in Asian populations, whereas CNV is the typical subtype in Western populations. Although the two subtypes share genetic and risk factors, they have distinct clinical characteristics, natural histories, and treatment outcomes.2 The reasons for these differences are unknown.
PCV is frequently associated with recurrent hemorrhagic or exudative pigment epithelium detachment (PED), with leakage and bleeding from the polypoidal components.2, 3, 4 The cause and pathogenesis of PCV are largely unknown. Moreover, the current anti–vascular endothelial growth factor (VEGF) therapy is less effective in treating PCV compared with classic CNV.5 The visual prognosis in human PCV is correlated with lesion size.6, 7, 8 Patients with larger lesions often have higher rates of lesion progression, severe complications (eg, PED, subretinal hemorrhage, and classic CNV), and poor visual outcome. In contrast, patients with small lesions have minimal lesion progression, limited exudative changes, and more stable vision performance. Therefore, understanding the pathophysiological mechanism for PCV initiation and progression is critical to design both prevention and treatment strategies.
On the basis of genome-wide association studies implicating the involvement of high temperature requirement factor A1 (HTRA1), a multifunctional secreted serine protease that is ubiquitously expressed in mammalian tissues,9, 10 in AMD, including PCV,11, 12, 13, 14, 15 we generated the first PCV model by transgenically expressing human HTRA1 in mouse retinal pigment epithelium (RPE).16, 17 Increased expression of HTRA1 induced two key features of PCV, polypoidal dilations (polyps) and branching vascular network (BVN), in transgenic hHTRA1+ mice. Furthermore, hHTRA1+ mice exhibit additional features of PCV, such as late geographic hyperfluorescence, PED, and hyperfluorescent plaque.17 On funduscopy, polypoidal lesions appear as reddish orange nodules. These phenotypes share remarkable similarities to the well-established clinical features of human PCV. Thus, this animal model provides an unprecedented opportunity for us to investigate the underlying pathophysiological mechanism of PCV.
In this study, we performed comprehensive genetic, histopathological, imaging, and molecular biological studies on the hHTRA1+ mouse model in combination with analysis on human PCV specimens. We demonstrated that PCV is caused by a multitude of degenerative processes, including destructive extracellular matrix (ECM) remodeling, vascular smooth muscle cell (SMC) loss, immune complex deposition, complement activation, and infiltration of inflammatory cells. Our results provide support for a two-stage process for PCV pathogenesis. PCV initiation is mediated by increased HTRA1 activity, whereas progression is driven by chronic inflammation.
Materials and Methods
Generation of Tg44hHTRA1+, Tg26hHTRA1+S328A, and Tg33hHTRA1+S328A Transgenic Mice
To generate Tg44 mice, a 1.5-kb human HTRA1 cDNA (TAG stop codon was removed) was amplified using primers with BamHI and XhoI overhangs. The myc-his6 fragment was amplified using primers with XhoI and EcoRI overhangs. For both PCRs, pcDNA3.1-CMV-VMD-hHtra1-myc-His vector was used as a template.16 The destination plasmid, pAAV-L-CAG-Shuttle-WPRE, was digested with BamHI and EcoRI and ligated with human HTRA1 cDNA and myc-his fragments in a three-piece ligation reaction. Subsequently, the human vitelliform macular dystrophy 2 promoter (−585 to 38 bp)18 was inserted as a KpnI-BamHI fragment before human HTRA1 cDNA. The regions containing the vitelliform macular dystrophy 2 promoter, hHTRA1-myc-His6, woodchuck post-transcriptional regulatory element, and human growth hormone poly(A) were sequence verified. The transgene construct was excised by KpnI-RsrII digestion, gel purified, and injected into C57BL/6 × CBA embryos at the University of Utah Transgenic/Gene Targeting Core Facility (Salt Lake City, UT). The same strategy was used to generate Tg26 and Tg33 mice, except that the catalytic residue S328 of hHTRA1 cDNA was mutated by site-directed mutagenesis. All three mouse lines were crossed 10 times with CD1 mice to facilitate indocyanine green angiography (ICGA).19 Wild-type CD1 mice were ordered from Charles River (San Diego, CA). All animal experiments were approved by the Institutional Animal Care and Use Committees at the University of Utah and Baylor College of Medicine (Houston, TX). All of the experiments were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Human Specimens
Human PCV specimens were obtained from three patients (one 74-year–old man, one 77-year–old woman, and one 60-year–old woman) (Supplemental Table S1). Control eye specimens were obtained from 60- to 80-year–old donors without AMD and PCV. Informed consent for the surgical procedure and for the use of excised tissue was obtained from all patients, in accordance with the Declaration of Helsinki. All human experiments were approved by the Institutional Review Board at Nihon University School of Medicine (Tokyo, Japan), Johns Hopkins University School of Medicine (Baltimore, MD), and Baylor College of Medicine.
Immunohistochemistry
Except for the detection of human HTRA1 or HTRA1S328A in Tg44, Tg33, or Tg26, which was performed on frozen sections, all other staining was performed on paraffin sections. A standard deparaffinization procedure was used for paraffin-embedded sections. An antigen retrieval step (10 mmol/L sodium citrate, pH 6.0, pressure cooker, 3 minutes) was added before the labeling step to unmask epitopes on tissues. Either immunoperoxidase (Avidin/Biotin Complex Kit; Vector Laboratories, Burlingame, CA) or immunofluorescence staining was used for detection. Immunohistochemistry was performed, as previously described.16, 17 Isotype controls or secondary antibodies alone (Supplemental Figures S1, S2, S3, and S4) were used as negative controls. Mouse (G3A1) monoclonal antibody IgG1 (catalog number 5415; Cell Signaling, Danvers, MA) was used as an isotype control for anti-C3 monoclonal antibody (sc-28294; Santa Cruz Biotechnology, Dallas, TX), whereas normal rabbit serum (011-000-002; Jackson ImmunoResearch, West Grove, PA) was used as an isotype control for anti-HTRA1 rabbit polyclonal antibody (55011-1-AP; Proteintech, Chicago, IL). The primary antibodies were listed in Table 1. The following secondary antibodies were used: Alexa 488 donkey anti-mouse IgG (715-545-150), Cy3 goat anti-rabbit IgG (111-165-144), Cy3 goat anti-rat IgG (112-165-167), Cy3 bovine anti-goat IgG (805-165-180), and DyLight 549 goat anti-chicken IgY (103-505-1549), from Jackson ImmunoResearch Laboratories (West Grove, PA); Alexa 488 goat anti-chicken IgG (A11039), from Invitrogen (Waltham, MA); and Alexa 488 donkey anti-goat IgG (ab150129) from Abcam (Cambridge, MA).
Table 1.
List of Primary Antibodies Used in Immunohistochemistry
| Antibody name | Antibody type | Manufacturer | Catalog no. | Dilution |
|---|---|---|---|---|
| HTRA1 | Mouse monoclonal | R&D Systems (Minneapolis, MN) | MAB2916 | 1:1000 |
| HTRA1 | Rabbit polyclonal | Proteintech (Chicago, IL) | 55011-1-AP | 1:200 |
| MMP2 | Rabbit polyclonal | Proteintech | 10373-2-AP | 1:100 |
| MMP9 | Rabbit polyclonal | Abcam (Cambridge, MA) | ab38898 | 1:100 |
| C3 | Goat polyclonal | MP Cappel (Santa Ana, CA) | 55444 | 1:100 |
| C3 | Mouse monoclonal | Santa Cruz Biotechnology | sc-28294 | 1:50 |
| Sc5b-9 | Chicken IgY | Quidel (San Diego, CA) | A802 | 1:100 |
| α-SMA | Rabbit polyclonal | Abcam | Ab5694 | 1:200 |
| F4/80 | Rat monoclonal | Abcam | Ab6640 | 1:100 |
| CD68 | Mouse monoclonal | Dako North America, Inc. (Carpinteria, CA) | PG-M1 | 1:80 |
| Myc-Tag | Rabbit monoclonal | Cell Signaling (Danvers, MA) | 71D10 | 1:500 |
MMP, matrix metalloproteinase; α-SMA, α-smooth muscle actin.
Western Blotting
Western blotting was performed as described previously.16, 17 Proteins were separated under nonreducing conditions for the detection of human HTRA1, and were run under reducing condition for others. The following antibodies were used: anti-human HTRA1 (MAB2916; R&D Systems, Minneapolis, MN), anti–α-smooth muscle actin (ab5694; Abcam), anti–Myc-tag rabbit monoclonal antibody (71D10; Cell Signaling) (Supplemental Figure S5A) anti–fibulin-5 (AB6065; Millipore, Billerica, MA) (Supplemental Figure S6), anti–β-actin (AC-15; Sigma, St. Louis, MO) (Supplemental Figure S6A), and anti–glyceraldehyde-3-phosphate dehydrogenase (G8795; Sigma) (Supplemental Figure S6B). For quantification, gel images were captured, and the pixel value of each protein band was obtained by ImageJ software version 1.51J8 (NIH, Bethesda, MD; http://imagej.nih.gov/ij).
Isolation of PCV Lesions from Tg44 Mice
Tg44 mice were anesthetized by i.p. injection of a combination of ketamine (65 to 100 mg/kg) and xylazine (10 to 20 mg/kg). Pupils were dilated with 1% tropicamide (Bausch & Lomb, Rochester, NY). Anesthetized mice were injected with high–mol. wt. fluorescein isothiocyanate (FITC)–dextran (80 mg/kg; mol. wt., 2 × 106; Sigma) and ICG (2 mg/kg; Pfaltz & Bauer, Waterbury, CT) via tail vein injection. ICGA and fluorescein angiography were captured with the Heidelberg Retina Angiograph—Optical Coherence Tomography device (Heidelberg Engineering, Franklin, MA). ICG signal allowed us to identify PCV lesions, whereas FITC-dextran allowed us to image the retinal vasculature as landmarks for lesion locations (see below). It was important to use the high–mol. wt. FITC-dextran because low–mol. wt. FITC-dextran and ICG tended to leak out of the vasculature rapidly, which made the subsequent identification by confocal microscopy difficult.20 The geographic location of the lesion was noted (ie, superior nasal quadrant, distance from optical disk, and relative location to nearby vasculature). After euthanasia and dissection, eyes were fixed for 15 minutes in either 2.5% glutaraldehyde/1% formaldehyde for ultrastructure analysis or 4% paraformaldehyde for paraffin processing. Cornea, iris, and lens were removed. The remaining eye cup was flat mounted and coverslipped. Images were acquired from both the bright-field and FITC channels with an Olympus Fluoview FV1000 confocal microscope (Olympus Corporation, Tokyo, Japan). Lesions were identified by their geographic location relative to nearby vasculature landmarks revealed by the FITC-dextran signal. Guided by images from ICGA/fluorescein angiography and confocal microscopy, lesions were dissected out for further analysis under a Leica fluorescence stereomicroscope (MZ16F; Leica Microsystems GmbH, Wetzlar, Germany).
Fundus Photography and Angiography
In vivo imaging was performed, as described previously.17, 19 Small and large lesions were defined as the largest linear dimension ≤100 and >100 μm, respectively.
Histology and Electron Microscopy
Procedures for Richardson staining and electron microscopy were described in our previous publication.16 Elastica van Gieson stain (26369; Electron Microscopy Sciences, Hatfield, PA) was used for elastic fiber staining on paraffin sections.
Elastase Assays
The elastase activity of recombinant human wild-type HTRA1 and HTRA1S328A was measured using the EnzChek Elastase Assay kit (Invitrogen), according to the manufacturer's instructions.
IgG-IgM Deposition in Human and Mouse PCV Lesions
To minimize nonspecific labeling of IgG-IgM, a perfusion step was performed to flush out remaining serum proteins in mouse vasculature. Under deep anesthesia, mice underwent a thoracotomy operation. The right atrium was cut, and a heparinized catheter was inserted into the left ventricle. First, mice were perfused with 1× phosphate-buffered saline at 37°C for 5 minutes, followed by 15-minute perfusion with 4% paraformaldehyde. Then, lesions were isolated and embedded in paraffin. The following antibodies from Jackson ImmunoResearch Laboratories were used to detect IgG and IgM deposition in human and mouse tissues: Alexa 647 donkey anti-mouse IgM (μ chain specific, 715-605-020), Alexa 488 goat anti-mouse IgG (Fcγ fragment specific, 115-545-008), Alexa 647 goat anti-human IgM (Fc5μ fragment specific, 109-605-043), and Alexa 488 goat anti-human IgG (Fcγ fragment specific, 109-545-008).
Enzyme-Linked Immunosorbent Assay
RPE/choroid lysates were prepared in lysis buffer (AA-LYS; Raybiotech Inc., Norcross, GA) containing protease (M250; Amresco, Solon, OH) and phosphatase (PhosSTOP; Roche, Basel, Switzerland) inhibitors. Protein concentrations were quantified by bicinchoninic acid assay (Pierce, Rockford, IL). An enzyme-linked immunosorbent assay to measure the VEGF level in Tg44 and control mice was performed by Raybiotech, Inc.
HTRA1 Inhibitor Synthesis and Delivery
The HTRA1 peptide inhibitor (DPMFKLboroV) was synthesized and purified, as described.21 Nanoparticles (NPs) were fabricated using the double emulsion (water/oil/water) solvent evaporation technique, as described,22 except that 0.2 mg coumarin 6 in 2 mL of CH2Cl2 and 0.3 mL of HTRA1 inhibitor (17.8 mg/mL) in formamide were used. For intravitreal delivery, mice were anesthetized by i.p. injection of ketamine/xylazine (70 to 100 mg/10 to 20 mg/kg body weight). Using a 30.5-gauge needle (Becton-Dickinson, Houston, TX), an initial puncture was made through the conjunctiva and sclera immediately posterior to the superonasal limbus. A 32-gauge blunt needle attached to a 5-μL syringe (Hamilton, Reno, NV) was introduced to the vitreous cavity through this opening. Mice were injected with 0.8 μL (for postnatal day 16 to postnatal day 21) or 1.5 μL (for 2 to 3 months) of NPs (1.5 μg/μL) in NP buffer (15 mmol/L 3-Morpholinopropane-1-sulfonic acid, pH 6.0, and 0.9% NaCl). For the preventive experiment, Tg44 mice were injected at P16 to P22. For the treatment experiment, Tg44 mice were injected at approximately 3 months. Injected mice were monitored by ICGA.
TAA Delivery
Mice were anesthetized by i.p. injection of ketamine/xylazine (70 to 100 mg/10 to 20 mg/kg body weight). Local anesthesia, proparacaine hydrochloride ophthalmic solution USP, 0.5% (Akorn, Lake Forest, IL), was applied to the mouse eyes. For triamcinolone acetonide (TAA) delivery, suprachoroidal injection was performed at P22. In brief, a bevel-type 30.5-gauge needle, bevel facing inward, was inserted at least 0.25 to 0.50 mm posterior from the limbus at an angle of 45 degrees through the conjunctiva and sclera. TAA was delivered to the suprachoroidal space once minimum resistance occurred to the injection. One eye was injected with 1000 μg (25 μL) of TAA (Kenalog-40; 40 mg/mL; NDC 0003-0293-05; Bristol-Myers Squibb Company, New York, NY), and the fellow eye was injected with sterile phosphate-buffered saline. Injected mice were monitored by ICGA. The preventive effect was evaluated by comparing the PCV severity between the control eye and TAA injected eye.
Statistical Analysis
All group results are expressed as means ± SEM. We performed each mouse experiment at least three times and used representative data in the calculations. Comparisons between groups were made using the two-tailed t-test or one-way analysis of variance and Tukey post hoc tests for multiple groups. Values of n are detailed in figure legends. Statistical analysis was performed in OriginPro version b9.3.2.303 (OriginLab Corporation, Northampton, MA). No statistical methods were used to predetermine the sample size, but our sample sizes are consistent with those generally used within the field. The mice were not randomized.
Results
The Proteolytic Activity of HTRA1 Is Critical for PCV Development
To test whether the proteolytic activity of HTRA1 is important for PCV development, we generated two transgenic lines (Tg33 and Tg26) expressing the protease-inactive mutant human HTRA1S328A in mouse RPE using the human vitelliform macular dystrophy 2 promoter.18 As expected, HTRA1S328A has negligible elastolytic activity in vitro (Figure 1A) in contrast to wild-type (WT) HTRA1.16 The protein level of HTRA1S328A in RPE-choroid of Tg33 was 1.3 and 5.2 times that of WT HTRA1 in Tg44 (a previously generated transgenic line expressing human HTRA1 in mouse RPE17) and human donor RPE, respectively (Figure 1, B and C). The high expression line Tg26 expressed 2.5 times more HTRA1S328A than Tg33. Both WT HTRA1 and HTRA1S328A were specifically expressed in RPE, except that weak expression was detected in the brain in the high expression line Tg26 (Supplemental Figure S5A). The expression pattern of HTRA1S328A in both Tg33 and Tg26 was similar to that of WT HTRA1 in Tg44 in the eye. Both proteins were expressed in RPE (Supplemental Figure S5B) and were secreted into the choroid, consistent with previous results.16, 17 Intense signals were detected throughout RPE and choroid in the high expression line Tg26.
Figure 1.

Transgenic mice Tg33 and Tg26 expressing HTRA1S328A did not develop polypoidal choroidal vasculopathy (PCV). A: Elastase assay of recombinant wild-type (WT) human HTRA1 and mutant human HTRA1S328A. Statistical significance was assessed using t-test. B and C: Western blot and quantification of human HTRA1 expression in retinal pigment epithelium (RPE) and choroid of Tg44 and Tg33 mice. Data showed the relative protein levels of human HTRA1 (or HTRA1S328A) signal from 4 μg of RPE/choroid lysates of Tg44 and Tg33, and 15 μg lysate of human RPE/choroid. C: The myc-His6 tagged transgenic HTRA1 and HTRA1S328A ran at 55 kDa, whereas the native HTRA1 [from human RPE (hRPE)] ran at 50 kDa. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was included as the loading control. Statistical significance was assessed using one-way analysis of variance with post-hoc Tukey test. HTRA1 in hRPE is significantly different from all three transgenic lines (Tg44, Tg33, and Tg26; P < 0.0001). D: Early-, middle-, and late-phase indocyanine green angiography (ICGA) showed that Tg33 and Tg26 mice had normal choroidal vasculature, which was similar to WT. However, Tg44 mice developed two cardinal features of PCV, polyp dilations (blue arrows) and branching vascular network (red circles). E: Funduscopy (left column) and paired ICGA (right column). Tg44 mice exhibited reddish orange nodules corresponding to PCV lesion structures (red circles), hemorrhage (asterisks), and RPE degeneration (green arrows). Tg33 and WT controls were normal. Data are expressed as means ± SEM (A and B). n = 4 (A); n = 3 (B). ∗∗P < 0.01, ∗∗∗∗P < 0.0001. Scale bar = 1000 μm (D and E). u, units.
We examined 45 Tg33 mice and 17 Tg26 mice (1 to 14 months old) by fluorescein angiography, ICGA, and funduscopy. Neither line exhibited any PCV phenotype (Figure 1D). Consistent with our previous studies,16, 17 normal fluorescein angiography was observed in all transgenic lines (Supplemental Figure S7), suggesting normal retinal vasculature (see more below). This is expected because transgenic HTRA1 is mainly expressed in RPE and is secreted toward the choroid (Supplemental Figure S5). Because the expression level of HTRA1S328A in the Tg33 line was more similar to WT HTRA1 in Tg44, Tg33 was selected for further comparison study with Tg44. Tg33 mice had normal-looking ICGA and fundus, similar to WT controls (Figure 1E). This was in contrast to the cardinal features of PCV exhibited by Tg44 mice, including BVN (Figure 1D) and polypoidal lesions (Figure 1D) on ICGA, reddish orange nodules on funduscopy (Figure 1E), sub-RPE hemorrhage (Figure 1E), and RPE degeneration (Figure 1E). Our data suggest that the proteolytic activity of HTRA1 plays a critical role in PCV pathogenesis.
Elastic Fiber Degradation and Loss of Vascular Smooth Muscle Cells in PCV Lesions
We examined the choroidal vasculature and Bruch membrane in PCV lesions. The internal elastic laminae (IELs) of Tg44 choroidal arteries in the lesion area were either degenerating (Figure 2A) or missing (Figure 2, A and B). In fact, the artery was collapsing in Tg44, probably because of the combination of IEL degradation, degeneration of endothelial cells (Figure 2B), and atrophic changes of SMCs (Figure 2B). In addition to vascular defects, the EL of the Bruch membrane in Tg44 mice was fragmented, and interrupted by gaps of varying sizes, as we had shown previously (Supplemental Figure S8A).16 In sharp contrast, Tg33 mice expressing mutant HTRA1S328A had intact IELs in arteries (Figure 2, A and B) and intact elastic lamina in Bruch membrane (Supplemental Figure S8A), similar to WT mice (Figure 2, A and B, and Supplemental Figure S8A). These results suggest that HTRA1 is involved in elastin fiber degradation in both choroidal vessels and Bruch membrane.
Figure 2.

Elastic fiber degradation and loss of smooth muscle cells (SMCs) in choroidal vessels of both Tg44 and human polypoidal choroidal vasculopathy (PCV). A: Van Gieson staining showed the loss of internal elastic laminae (IELs) of a choroidal artery in Tg44 PCV lesions. Black brackets and black arrows in Tg44 indicated missing and partial loss of IEL, respectively. Green arrows indicated normal IEL in WT and Tg33. B: Transmission electron microscopy image showed a collapsed artery in Tg44 PCV lesion. Endothelial cell degeneration (red arrows), SMC atrophy (asterisks), and degeneration of both IEL (blue arrow) and external elastic layer (EEL; yellow arrows) were evident. Part of the IELs was missing (red bracket). The EEL (yellow arrows) drifted away through the tunica media and also merged with IEL (blue arrow). Tg33 (red arrowheads) and wild type (WT; orange arrows) had normal IEL. EEL (green arrow) and endothelial cells (black arrows) were normal in WT. C: Van Gieson staining showed stretches of IELs of a collapsed choroidal vessel in human PCV lesion were missing (red brackets), degenerating (black bracket), or broken (red arrows). IEL was normal in a control artery (black arrows). D and E: Immunostaining of α-smooth muscle actin (α-SMA) in human (D) and mouse (E) PCV lesions. The choroidal medial SMCs were partially lost in the bottom left panel (white brackets), and were completely lost in the bottom right panel (D). SMC loss (white brackets) and degeneration (yellow bracket) in choroidal vessel wall of Tg44 were shown (E). SMCs were intact in WT and Tg33. Nuclei were counterstained with DAPI (blue). F: Western blot analysis on retinal pigment epithelium/choroid lysates showed that the α-SMA level was significantly decreased in Tg44 when compared with both Tg33 and WT controls. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was included as the loading control. Statistical significance was assessed using one-way analysis of variance with post-hoc Tukey test. Data are expressed as means ± SEM (F). n = 4 (F). ∗P < 0.05. Scale bars: 10 μm (A and E); 1 μm (B); 20 μm (C and D).
In human PCV lesion specimens, stretches of IELs of choroidal vessels were missing (Figure 2C), degenerating (Figure 2C), or broken (Figure 2C), consistent with previous histopathological studies.23, 24 This was in contrast to the wave-shaped IELs in control arteries (Figure 2C). The Bruch membrane in PCV lesions was variably degenerating (Supplemental Figure S8B), which was in agreement with previous ultrastructural studies on human PCV specimens showing that the elastic lamina of Bruch membrane in PCV was frequently missing.23, 24 Collectively, our results suggest that elastin fiber degradation in choroidal vasculature and Bruch membrane is a common pathological feature of PCV.
Another prominent feature of PCV was the significant loss of medial SMCs in the choroidal vessel wall in both mouse and human specimens. In human PCV lesions, the degeneration of choroidal medial SMCs ranged from partial loss (Figure 2D) to complete loss (Figure 2D). In Tg44 mice, SMCs were either missing (Figure 2E) or degenerating (Figure 2E) in PCV lesions in contrast to intact SMCs in WT (Figure 2E) and Tg33 (Figure 2E). By Western blotting of RPE-choroid lysates using an antibody against α-smooth muscle actin, SMC contents were decreased 1.8 times (P < 0.05) in Tg44 compared with those of WT and Tg33 mice (Figure 2F). We also examined SMCs in retinal vessels by immunohistochemistry. All transgenic lines exhibited intact SMCs (Supplemental Figure S9), which provided further support that their retinal vasculatures were normal (Supplemental Figure S7).
Immune Complex Deposition and Complement Activation in PCV Lesions
Complement activation and immune complex deposition play an important role in drusen formation of AMD and other age-related diseases (eg, Alzheimer disease and atherosclerosis) by eliciting local chronic inflammation.25, 26, 27 In Tg44 mice, we found abundant IgG and IgM deposition in the choroidal vessel wall of severe PCV lesions (Figure 3, C and F). Weak IgG-IgM deposition was also present in the choroidal vessel wall of weak PCV lesions (Figure 3, B and E), whereas no IgG-IgM deposition was detected in WT (Figure 3, A and D). Both the complement component C3 (Figure 3I) and membrane attack complex (MAC) (Figure 3L) were drastically increased in the choroidal vessel wall of severe PCV lesions, but absent in the WT (Figure 3, G and J). C3 and MAC were also detected in the choroidal vessel wall of weak PCV lesions (Figure 3, H and K).
Figure 3.

Immune complex deposition and complement activation in both Tg44 and human polypoidal choroidal vasculopathy (PCV) lesions. IgG (A–C) and IgM (D–F) staining in the choroidal vessel wall of wild-type (WT) and Tg44 PCV lesions (C and F). White arrows indicated strong signals in severe PCV lesions, and yellow arrowheads indicated weak signals in weak PCV lesions (B and E). C3 (G–I) and membrane attack complex (MAC; Sc5b9) (J–L) staining in the choroidal vessel wall of WT and Tg44 PCV lesions. White arrows indicated signals in severe PCV lesions, and white arrowheads indicated signals in weak PCV lesions. No signal was detected in WT. IgG (M and N), IgM (O and P), C3 (Q and R), and MAC (S and T) staining in the choroidal vessel wall of human PCV lesions. White arrows indicated strong signals in lesions (N, P, R, and T). Nuclei were counterstained with DAPI (blue). Scale bars = 10 μm (A–T).
Similar to the findings in mouse PCV lesions, prominent IgG and IgM deposition was present in the choroidal vessel wall of human PCV lesions (Figure 3, N and P). Both C3 (Figure 3R and Supplemental Figure S10) and MAC (Figure 3T and Supplemental Figure S10) were drastically increased in the choroidal vessel wall compared with those in controls. Both C3 (Supplemental Figure S10) and MAC (Supplemental Figure S10) were also present outside vessels, whereas C3 was strongly expressed in the lumen (Supplemental Figure S10). No signal was detected on control tissues (Figure 3, M, O, Q, and S). These results suggest that immune complex deposition and complement activation may play a role in PCV progression (see more below). Staining using the isotype IgG or secondary antibody alone on PCV and control tissues showed no signal (Supplemental Figure S1).
Presence of Inflammatory Cells and Degeneration in Photoreceptor-RPE-Choroid of Large PCV Lesions
We surgically excised small (≤100 μm) and large (>100 μm) PCV lesions from Tg44 mice (Figure 4, A and B). Thin plastic sections were prepared and stained with Richardson stain. In the small lesion, a cluster of dilated thin-wall vessels (Figure 4, C and E) with a broken vessel wall (Figure 4, C and E) were observed. The lesion pushed RPE and retinal layers upward, likely because of the buildup of pressure from leaked fluid. PEDs were frequently observed near the dilated thin-wall vessels (Figure 4, C and E). Choriocapillaris was completely missing in large lesions (Figure 4, D and F) but was present in small lesions (Figure 4, C and E). When compared with WT (Figure 4G) and small lesions (Figure 4, C and E), large PCV lesions caused severe and extensive RPE degeneration (Figure 4D) accompanied by photoreceptor degeneration (Figure 4F). In fact, the RPE layer and the tip of outer segment were disintegrated into a debris zone (Figure 4, D and F) containing large undigested outer segment phagosomes as a result of RPE degeneration (Figure 4F). The outer segment was ruffled, and the outer nuclear layer was shortened (Figure 4F). In contrast, the small PCV lesion caused RPE dysfunction with the presence of numerous empty vacuoles (Figure 4, C and E). There were no obvious signs of photoreceptor degeneration (Figure 4E). This observation is consistent with human studies showing that patients with larger lesions often have higher rates of lesion progression and severe complications.6, 7, 8
Figure 4.

Histopathological features of mouse and human polypoidal choroidal vasculopathy (PCV) lesions. A–F: Representative indocyanine green angiography images of small (red arrow; A) and large (red arrow; B) PCV lesions that were isolated from a 9-month–old Tg44 for histopathology analysis (Richardson stain; C–F). C and E: Small lesions. D and F: Large lesions. G: Wild-type control. C–E: Dilated thin-wall vessels (black dashed ovals; C and E) with broken vessel wall (red arrowheads; C and E) were present in small lesions, whereas hyalinized vessels with thick wall were present in both small and large lesions (red dashed circle; C–E). Yellow arrows indicated pigment epithelium detachments frequently observed near the dilated thin-wall vessels (C and E). Red arrows and black arrows indicate small and large vacuoles, respectively, in the small PCV lesion (C and E). Red arrows indicated severe and extensive retinal pigment epithelium (RPE) degeneration in large PCV lesions (D). Yellow arrows, black arrowhead, and orange arrowheads indicated macrophages, neutrophil, and monocytes, respectively (D). Red brackets indicated that the RPE layer and the tip of outer segment (OS) were disintegrated into a debris zone (D and F). Red arrows indicated large undigested OS phagosomes as a result of RPE degeneration (F). H and I: Immunopositive cells for macrophage markers F4/80 (yellow arrows; H) and CD68 (black arrows; I) were present in severe Tg44 and human PCV lesions, respectively. J–M: Transmission electron microscopic images showed the presence of mast cells (purple asterisks; J), platelets (blue asterisks; K), macrophages (red oval and red cap; L and M), and neutrophils (yellow oval; M) in severe PCV lesions of Tg44. L: Red arrowheads mark the crossing points in Bruch membrane (BM) of a macrophage en route to RPE. The red cap indicates another macrophage that was already invaded into the sub-RPE space in a severe PCV lesion. The red arrow and yellow arrow indicate degenerating endothelial cells and pericytes, respectively, in the large lesion. Scale bars: 100 μm (A and B); 10 μm (C, D, H, and I); 20 μm (E–G); 2 μm (J–M). Cho, choroid; IS, inner segment; ONL, outer nuclear layer.
We frequently observed large numbers of inflammatory cells in large PCV lesions. Numerous macrophages infiltrated the lesion area (Figure 4, D, H, L, and M), as in the human PCV lesion specimen (Figure 4I).28 Besides macrophages, neutrophils (Figure 4, D and M), monocytes (Figure 4D), mast cells (Figure 4J), and platelets (Figure 4K) were also frequently present. Interestingly, a macrophage was caught in action crossing into the RPE (Figure 4L), whereas another macrophage was already invaded into the sub-RPE space (Figure 4L) in a severe PCV lesion. Endothelial cells (Figure 4L) and pericytes (Figure 4L) were degenerating in the large lesion. The artery wall was thick and hyalinized in both small and large lesions (Figure 4, C–E). Obstruction of the hyalinized vessels was apparent. Similar arteriosclerotic changes in the choroid have been reported in human PCV.24, 28 Thus, both thin-wall vessels and hyalinized vessels seem to be involved in the complex pathology of PCV.
MMP2 and MMP9 Were Increased in PCV Lesions
Large numbers of inflammatory cells (neutrophils and macrophages) in PCV lesions can produce matrix-degrading proteases [eg, matrix metalloproteinases (MMPs)] and may cause further structural damages to the choroidal vasculature. We examined the expression of MMP2 and MMP9 in mouse and human PCV lesions (Figure 5, A and B). MMP9 was markedly increased in the choroidal vessel wall (Figure 5A) and near the vessel (Figure 5A) of severe PCV lesions comparing with that in WT and weak PCV lesions (Figure 5A). On the other hand, MMP2 was markedly increased in both the vessel wall (Figure 5A) and inside lumen (Figure 5A) in severe PCV lesions, whereas it was slightly increased in the vessel wall of weak PCV lesions (Figure 5A). In human PCV specimens, both MMP9 (Figure 5B) and MMP2 (Figure 5B) were prominently increased in the vessel wall. MMP9 was also increased near the vessel (Figure 5B), whereas MMP2 was increased inside the lumen (Figure 5B). The expression of both matrix-degrading enzymes in the choroid of control human eyes was minimal. Staining using the secondary antibody alone showed no signal (Supplemental Figure S2).
Figure 5.
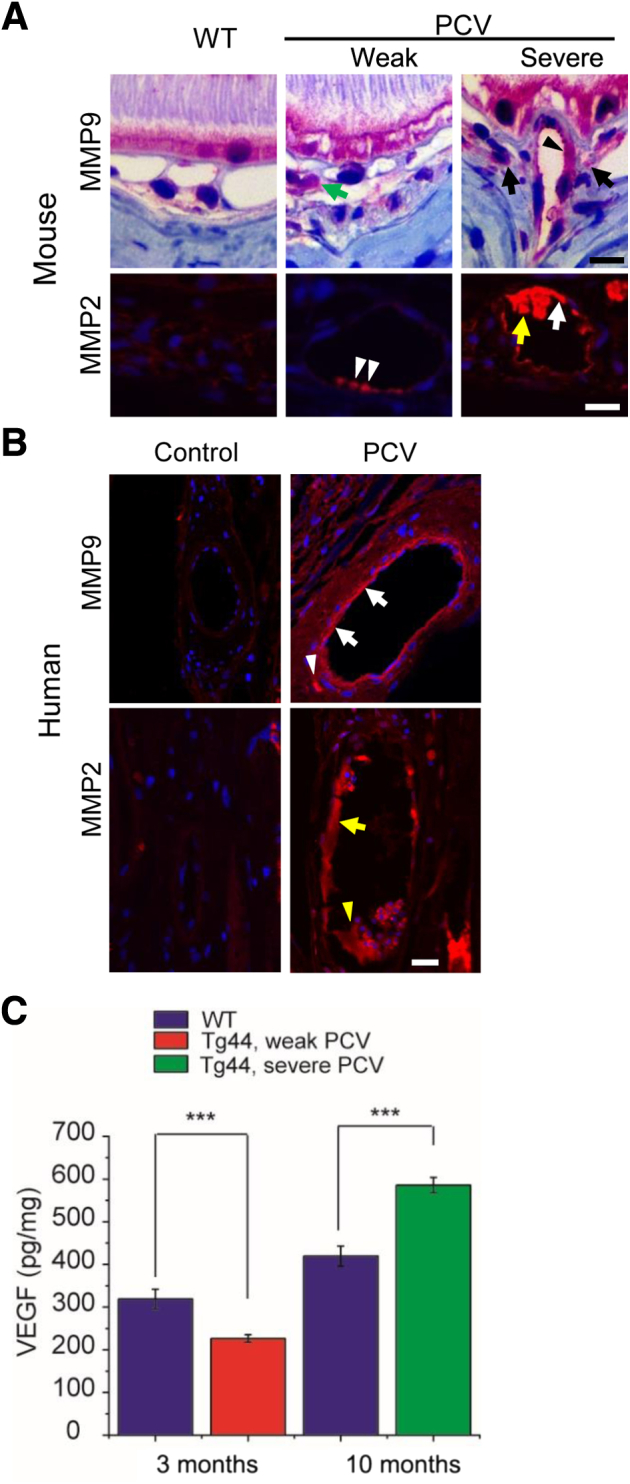
Analysis of matrix metalloproteinase (MMP) 2, MMP9, and vascular endothelial growth factor (VEGF) in polypoidal choroidal vasculopathy (PCV) lesions. A: Immunostaining of MMP9 and MMP2 in mouse PCV lesions. MMP9 was markedly increased in an artery wall (black arrowhead) and near the vessel (black arrows) of a severe PCV lesion. MMP9 was also increased near the vessel in a weak lesion (green arrow). MMP2 was robustly expressed in both the artery wall (white arrow) and lumen (yellow arrow) of a severe PCV lesion, whereas it was weakly expressed in the vessel wall of a weak PCV lesion (white arrowheads). B: Immunostaining of MMP9 and MMP2 in human PCV lesions. MMP9 was increased in the choroidal vessel wall (white arrows) and near the vessel (white arrowhead), whereas MMP2 was increased in the choroidal vessel wall (yellow arrow) and lumen (yellow arrowhead). Nuclei were counterstained with DAPI (blue). C: The VEGF levels in retinal pigment epithelium (RPE)/choroid lysates of Tg44 and wild-type (WT) littermates were detected with enzyme-linked immunosorbent assay; 3-month–old Tg44 with weak PCV, 10-month–old Tg44 with severe PCV, and their respective age-matched WT controls were used in the assay. Data are expressed as means ± SEM (C). n = 16 3-month–old Tg44 and WT (C); n = 12 10-month–old Tg44 and WT (C). ∗∗∗P < 0.001 (t-test). Scale bars: 10 μm (A); 20 μm (B).
Role of VEGF in PCV Pathogenesis
VEGF is important in the pathogenesis of choroidal neovascularization through its potent proangiogenic activity. To examine the role of VEGF in PCV pathogenesis, we measured the VEGF level in the RPE/choroid of both 3-month–old Tg44 with weak PCV and 10-month–old Tg44 with severe PCV (Figure 5C). Surprisingly, the VEGF level was decreased approximately 30% (P < 0.001) in weak PCV compared with 3-month–old WT littermates, suggesting that, unlike in the case of CNV pathogenesis, VEGF was unlikely involved in PCV initiation via its proangiogenic activity. In 10-month–old Tg44 with severe PCV, the VEGF level was increased approximately 40% (P < 0.001) compared with 10-month–old WT littermates, suggesting that VEGF might contribute to the progression and development of complications (eg, classic CNV, as described in Discussion).
HTRA1 Was Increased in Human PCV Lesions
We examined the HTRA1 expression level in human donor eyes from three PCV cases. HTRA1 was markedly increased in the inner wall of degenerating choroidal vessels (Figure 6, A, E, and G) compared with the weak signal in the choroidal vessel wall of control human eyes (Figure 6, C and H). HTRA1 was highly expressed in plasma proteins inside the lumen of hyalinized and obstructed vessels (Figure 6A, E, and F) and in fibrin deposit (Figure 6A). In addition, HTRA1 was also increased in RPE of the lesion area (Figure 6G and Supplemental Figure S3A) and in collapsed choriocapillaris (Figure 6, F and G) compared with those of control eyes (Figure 6H and Supplemental Figure S3B). Vessels with higher HTRA1 levels in both the inner wall and the lumen appeared to have more extensive degeneration and leakage (Figure 6A, E, and G). In control human eyes, HTRA1 was weakly expressed in the RPE (Figure 6H and Supplemental Figure S3B), Bruch membrane (Figure 6, C and H and Supplemental Figure S3B), choriocapillaris (Figure 6, C and H, and Supplemental Figure S3B), and choroid vessel wall (Figure 6, C and H). Staining using the isotype IgG or secondary antibody alone on PCV and control tissues showed no signal (Figure 6, B and D, and Supplemental Figure S3).
Figure 6.

HTRA1 expression in human polypoidal choroidal vasculopathy (PCV) lesions. The expression level of HTRA1 was examined by immunohistochemistry in PCV specimens (A, C, and E–G) and in controls (B, D, and H). Immunoperoxidase method was used in A–D, whereas immunofluorescence method was used in E–H. Nuclei were counterstained with hematoxylin (blue; A–D) or DAPI (blue; E–H). A: Red dashed line surrounded area indicated a severely degenerating vessel with extensive plasma protein leakage. Green arrows indicated a less degenerating vessel. The black dashed line surrounded area indicated the increased expression of HTRA1 in fibrin deposit. C and D: Isotype IgG staining showed no positive signal. E and G: Yellow dashed line surrounded area indicated a severely degenerating vessel (E and G), whereas the green dashed line surrounded area indicated a less degenerating vessel (G). A, E, and G: Red arrows and yellow arrows indicate the marked increase of HTRA1 in the inner wall of degenerating choroidal vessels. C and H: Red arrows (C) and white arrows (H) indicated the weak signal in choroidal vessel wall of control human eyes. A, E, and F: Red arrowheads (A), yellow arrowheads (E), and white dashed ovals (F) indicated highly expressed HTRA1 in plasma proteins inside the lumen of hyalinized and obstructed vessels. F and G: White arrows (G) indicated increased HTRA1 in retinal pigment epithelium (RPE) of the lesion area, whereas white arrowheads (F and G) indicated increased HTRA1 in collapsed choriocapillaris. C and H: In control human eyes, HTRA1 was weakly expressed in the RPE (red signal in H, indicated by white arrowheads; RPE autofluorescence was in pink), Bruch membrane (black arrows in C and yellow arrow in H), choriocapillaris (black arrowheads in C and yellow arrowhead in H), and choroid vessel wall (red arrows in C and white arrows in H). Scale bars: 50 μm (A, C, and E–H); 20 μm (B and D). Cho, choroid.
HTRA1 Inhibitor Was Effective in Preventing PCV Initiation but Ineffective in Treating Existing PCV Lesions
An HTRA1 inhibitor, DPMFKLboroV, was shown to bind covalently to the active-site serine of HTRA1 via its C-terminal boronic acid group, with an inhibitory concentration of 50% of 2.6 μmol/L.21 To test whether this inhibitor was effective in preventing or treating PCV, we used biodegradable polymer-based NPs to deliver the HTRA1 inhibitor to the posterior segment of the eye of Tg44 mice to achieve long-term sustained release. Previous work demonstrated that intravitreally administered NPs transiently moved across the retina and preferentially localized to the RPE.22, 29, 30 We prepared two types of NPs using polylactic acid/polylactic acid–polyethylene oxide, following a published procedure22: NPs encapsulating a lipophilic fluorescent marker, coumarin 6; and NPs encapsulating coumarin 6 and HTRA1 inhibitor. We first injected adult WT mice intravitreally with NPs containing coumarin 6. Four days after injection, NPs were widely distributed in the eye (Supplemental Figure S11A), whereas confocal microscopy showed that NPs were concentrated in the RPE and outer segment (Supplemental Figure S11B). NPs were mainly located in the RPE afterward and were detectable at least 21 days after injection. Consistent with previous work,22 NP-injected eyes had normal retinal architecture, with no signs of toxicity compared with the control eye. We then injected Tg44 mice before they developed PCV at either P16 or P22. Because PCV was bilateral in Tg44 mice, we injected one eye with NPs containing coumarin 6 as the control and the other eye with NPs containing HTRA1 inhibitor plus coumarin 6. We analyzed the preventive effect 21 to 32 days after injection (Figure 7, A and B). The number of lesions was reduced by 36% on average (P = 0.009) in inhibitor-injected eyes (Figure 7B). In the example shown in Figure 7A, the control eye displayed a large polypoidal lesion and a cluster of small lesions 13 days after injection. In contrast, there was only one small lesion visible in the inhibitor-injected eye. Twenty-seven days after injection, only a few small lesions could be seen in the inhibitor-injected eye compared with much more severe PCV lesions (in both number and size) (Figure 7A) in the control eye. Two early-stage lesions (Figure 7A) started to appear, probably attributable to the waning of inhibitor at this time.
Figure 7.

Treatment of Tg44 mice with HTRA1 inhibitor or triamcinolone acetonide (TAA). A: Representative indocyanine green angiography (ICGA) images showed that HTRA1 inhibitor was effective in preventing polypoidal choroidal vasculopathy (PCV) when delivered before its onset. PCV lesions were imaged by ICGA at 13 and 27 days after injection. Red arrows and circles indicated large polypoidal lesions and cluster-type lesions, respectively, in nanoparticle-injected eye. Yellow arrow indicated an intermediate lesion. Red arrowheads indicated small lesions in both injected eyes. Two black arrows indicated two early-stage lesions in the inhibitor-injected eye. B: Quantitative results on the efficacy of HTRA1 inhibitor in preventing PCV initiation in Tg44. Lesion numbers were determined by ICGA. C and D: Intravitreal injection of HTRA1 inhibitor into Tg44 after they had developed PCV (approximately 3 months). Lesion numbers were determined by ICGA before and after injection. E: Representative ICGA images showing the efficacy of suprachoroidal injection of TAA treatment. PCV lesions were imaged by ICGA at 21 days after injection. The black dashed circle, red dashed circle, and red arrowheads indicated a cluster of polyps, a branching vascular network, and small lesions in vehicle-injected eyes (control), respectively. Black arrowheads indicated small lesions in TAA-injected eyes. F: Collective results on the efficacy of TAA. B, D, and F: Statistical significance was assessed using t-test. Data are expressed as means ± SEM (B, D, and F). n = 15 (B); n = 12 before treatment, control and inhibitor-injected groups (D); n = 9 after treatment, control and inhibitor-injected groups (D); n = 12 (F). ∗P < 0.05, ∗∗P < 0.01. Scale bars = 2000 μm (A, C, and E).
We also injected Tg44 mice with HTRA1 inhibitor after they developed PCV (approximately 3 months old). The treatment effect was analyzed 3 to 4 weeks after injection. There was no significant difference when comparing lesion numbers before and after injection (Figure 7, C and D). This result suggests that HTRA1 inhibitor is effective in preventing PCV but ineffective in treating existing PCV lesions.
Anti-Inflammatory Glucocorticoid Was Effective in Preventing PCV Progression
Because our results suggest that inflammation plays a critical role in PCV progression, we reasoned that treatment of Tg44 mice early with anti-inflammatory glucocorticoid TAA might be able to prevent the formation of severe PCV lesions. To deliver TAA effectively to the choroidal vasculature, we performed suprachoroidal injection on P22 Tg44 mice. One eye was injected with TAA, and the contralateral eye was injected with phosphate-buffered saline as a control. After 21 to 28 days, we assessed the efficacy of TAA by ICGA. TAA treatment was highly effective in preventing the formation of large lesions (5.8 times reduction; P = 0.017), whereas it was ineffective in preventing small lesion formation when compared with the controls (Figure 7, E and F). An example was shown in Figure 7E. Control eyes exhibited a cluster of polyps and a BVN in addition to small lesions. In contrast, only small lesions could be seen on TAA-treated eyes.
Discussion
The key finding of this work was that PCV was caused by proteolytic degradation of ECM of the choroid–Bruch membrane–RPE region and the resulting chronic inflammation. Specifically, PCV was caused by a multitude of degenerative processes, including destructive ECM remodeling, vascular SMC loss, immune complex deposition, complement activation, and infiltration of inflammatory cells. We found that the proteolytic activity of HTRA1 was responsible for PCV initiation; inflammatory processes played a key role in PCV progression. The first conclusion was supported by both genetic (ie, expression of mutant HTRA1S328A in mouse RPE) and pharmacological (delivery of an HTRA1 inhibitor) experiments, which prevented PCV initiation. The second conclusion was supported by complement activation, infiltration of inflammatory cells in severe PCV lesions, and especially the prevention of large lesion formation by anti-inflammatory TAA treatment in Tg44 mice. Collectively, our results provided strong support for a two-stage hypothesis for PCV initiation and progression (Figure 8). In stage 1 (initiation stage), increased HTRA1 degrades ECM proteins (eg, elastin) in the RPE/choroid region through its proteolytic activity, which leads to the limited degradation of Bruch membrane and choroidal vessel wall, producing weak PCV. In stage 2 (progression stage), tissue injury in RPE/choroid and leakage of plasma proteins in the lesion area trigger chronic inflammatory responses, which promote progression to severe PCV and complications (eg, PED and hemorrhage).
Figure 8.

Schematic drawing of a two-stage model for polypoidal choroidal vasculopathy (PCV) initiation and progression. In stage 1, increased HTRA1 degrades extracellular matrix (ECM) proteins (eg, elastin). This leads to the limited degradation of the Bruch membrane (BM) and the choroidal vessel wall, which initiates PCV. In stage 2, HTRA1-mediated proteolytic degradation unmasks hidden neoepitopes in choroidal vessel wall, which leads to IgG-IgM deposition and complement activation. This serves as an inflammatory focus to initiate chronic local inflammatory responses (infiltration of inflammatory cells, such as macrophages and neutrophils; release of proinflammatory cytokines; and further proteolysis), which, in turn, leads to PCV progression and disease complications. Either genetic mutation (S328A) or the HTRA1 inhibitor can abolish the protease activity of HTRA1 and prevents PCV. Triamcinolone acetonide (TAA) treatment blocks the inflammatory processes and prevented the development of severe PCV and complications. MMP, matrix metalloproteinase; RPE, retinal pigment epithelium.
We found abundant deposition of both IgG and IgM as well as complement activation in mouse and human PCV lesions, suggesting the involvement of both innate and adaptive immunity in PCV pathogenesis. It is likely that HTRA1-mediated proteolytic degradation unmasks hidden neoepitopes (self-antigens) in the choroidal vessel wall, which are recognized by IgG and IgM antibodies, providing a platform for complement activation. This, in turn, serves as an inflammatory focus to initiate chronic local inflammatory responses that lead to the infiltration of inflammatory cells, such as neutrophils and macrophages, and the production of proinflammatory cytokines and matrix-degrading proteases (eg, MMPs). These cytokines and proteases cause further structural damage and weakening of the choroidal vessel wall, leading to PCV progression and complication. The presence of IgG, IgM, MMPs, C3, and MAC in weak PCV lesions (ie, before severe PCV lesions), and the fact that anti-inflammatory TAA treatment prevented large, but not small, lesion formation, suggests a causative role of inflammatory processes in the progression, rather than the initiation, of PCV. Corticosteroids (ie, triamcinolone) may exert several effects that are beneficial in treating PCV lesions (eg, inhibiting the release of proinflammatory cytokines on endothelial and inflammatory cells; inhibiting the migration and activation of inflammatory cells, including macrophages, monocytes, and leukocytes; and inhibiting the activation of MMPs).31
There is an ongoing debate on whether PCV represents a subtype of neovascular AMD (ie, CNV) or a distinct vascular abnormality of the choroidal vessels.32, 33, 34, 35, 36 Because VEGF plays an important role in CNV, we analyzed the VEGF level in Tg44. Surprisingly, there was actually a decrease of VEGF in the RPE/choroid of weak PCV when compared with the age-matched WT control (Figure 5C), which may contribute to endothelial cell degeneration,37 vascular injury, and hypoxic condition in the early stage of PCV. In 10-month–old Tg44 with severe PCV, there was a 40% increase of VEGF in the RPE/choroid when compared with the 10-month–old WT controls, probably as a result of hypoxic and inflammatory conditions. This suggests that VEGF-associated neovascularization may play a role in PCV progression and the development of complications (eg, classic CNV and hemorrhage). Our data may explain the inconsistent results in the literature regarding the role of VEGF in PCV28, 38, 39, 40 because its expression and role depends on the PCV progression stage and condition. PCV may manifest features of both choroid abnormality and neovascular AMD at different disease stages. Thus, reducing VEGF may be useful to help control the progression of PCV, but it is ineffective to treat the underlying degenerative processes. According to our results, it may even be detrimental to treat early-stage PCV with anti-VEGF therapy. Overall, our proposed mechanism may provide a basis to explain the different pathological features and treatment outcomes between PCV and CNV.
Numerous studies have shown that genetic loci in chromosome 10q26, which surrounds two genes, HTRA1 and ARMS2, are strongly associated with AMD, including PCV.12, 13, 14, 15, 41, 42, 43, 44 A series of studies on the influence of AMD-associated polymorphisms on the expression of ARMS2 and/or HTRA1 have yielded conflicting results.15, 45, 46, 47, 48, 49, 50, 51, 52 However, recent studies started to provide evidence that variants in the promoter region of HTRA1 can transcriptionally up-regulate HTRA1.53, 54 Transgenic expression of HTRA1 or ARMS2 in mouse has shown that overexpression of HTRA1, but not ARMS2, induced PCV and CNV.16, 17, 53, 54 To our knowledge, this is the first study examining the expression of HTRA1 in human PCV lesions. We showed that HTRA1 protein was significantly increased in RPE and degenerating choroidal vessels of PCV lesions (Figure 6 and Supplemental Figure S3, A and B). Moreover, loss-of-function studies, by expressing the catalytically inactive HTRA1 and delivering HTRA1 inhibitor, strongly suggest that HTRA1 plays a key role in PCV initiation.
Because HTRA1 is genetically associated with both PCV and CNV, why did the Tg44 mice only exhibit the PCV phenotype? One possibility is that additional factors are required to develop typical CNV. For example, drusen, a hallmark of AMD that is implicated in the pathology of both neovascular and atrophic AMD, is more common in CNV than in PCV patients.2, 34, 55 There are no drusen in Tg44 mice, which may partly contribute to the lack of CNV. Nevertheless, increased expression of HTRA1 leads to the degradation of EL of Bruch membrane and up-regulation of VEGF at the PCV progression stage, both of which are risk factors of CNV. By subjecting Tg44 mice to additional AMD-associated stress factors, it is likely that they will have increased susceptibility of developing CNV. It is actually surprising that overexpressing a single genetic risk factor (ie, HTRA1) in mouse can induce robust PCV phenotypes. It may imply that PCV pathogenesis involves simpler steps than CNV. Indeed, the onset of PCV is generally much younger than CNV,2 which is consistent with the idea that more age-dependent stresses (eg, oxidative stress) are required to develop CNV.
Elastin is an important structural protein of ECM in the choroidal vessel that maintains the integrity of the wall. Our results showed that proteolytic degradation of elastin in choroidal vessel wall attributable to both HTRA1 and MMPs was a key pathological feature of PCV, which led to weakening and dilation of the vessel wall. Because elastin is important in stabilizing the vasculature by keeping vascular SMCs in a quiescent contractile state,56, 57 loss of elastin in choroidal vessels can contribute to the loss of SMCs in PCV lesions. Our findings that both MMP2 and MMP9 are increased in PCV lesions are consistent with previous work showing that both MMPs were increased in the serum of PCV patients.58 Increased MMPs can degrade collagen, another major component of choroidal vessel, leading to further damage of the aneurysmal wall. Polypoidal dilations (polyps) and BVN, two key features of PCV, could be the direct result of this destructive vessel remodeling.
In addition to the degradation of elastin in choroidal vessels, HTRA1 also degrades the EL of the Bruch membrane in Tg44 mice, leading to EL fragmentation in the Bruch membrane. This generates a favorable environment for the invasion of polyps into the sub-RPE space. Our histopathology data showed that PCV lesions frequently pushed RPE and Bruch membrane to the retinal side, likely because of the buildup of pressure of leaked fluid from compromised vessels. The leakage and pressure buildup may explain the high frequency occurrence of PEDs and hemorrhage in human PCV. In conjunction with compromised Bruch membrane, it may explain why polyps are often located in the sub-RPE space in high-resolution optical coherence tomography studies of human PCV.59
A previous study attributed HTRA1-mediated cleavage of fibulin-5 (an essential protein for elastogenesis by promoting elastic fiber assembly) as the reason for elastic lamina degradation in Bruch membrane in HTRA1 transgenic mice.60 However, we found no difference in the protein level of fibulin-5 in the RPE/choroid of two different hHTRA1+ mouse lines (a previously generated Vh7316 and the current Tg44) (Supplemental Figure S6). Moreover, there was no correlation between the severity of PCV phenotype and the level of fibulin-5, ruling out a role of fibulin-5 in PCV pathogenesis. Missense mutations of fibulin-5 were shown to be associated with cuticular drusen formation,61 whereas we found no evidence of drusen in hHTRA1+ mice, consistent with the lack of drusen in PCV patients.2, 3 The reasons for these differences were unclear and were unlikely attributable to the difference between human HTRA1 used in our animal model and mouse HTRA1 in the previous study, because the two proteins were 91% identical at the amino acid level.
The principal therapies for PCV are laser photocoagulation, photodynamic therapy, and anti-VEGF drugs (ranibizumab, bevacizumab, and aflibercept). The best reported treatment combines photodynamic therapy with anti-VEGF drugs.62 However, photodynamic therapy can cause subretinal hemorrhage, resulting in a poor visual prognosis.63, 64 Another major concern regarding photodynamic therapy is the high rate of recurrence or the development of new polypoidal lesions.65 Ranibizumab and bevacizumab were reported to be less successful in treating PCV than CNV. Recent studies showed that the latest anti-VEGF agent, aflibercept, was effective in reducing polyps; however, long-term study is needed to fully assess the efficacy of this treatment.66, 67, 68, 69 This is important because long-term use of anti-VEGF therapy (eg, ranibizumab or bevacizumab) can lead to anti-VEGF resistance.70, 71, 72 Aflibercept is less effective in reducing BVN, which may lead to the recurrence of polyps. Moreover, RPE-derived VEGF is essential in maintaining the choriocapillaris, suggesting that long-term blockade of VEGF signaling in retinal diseases may have detrimental adverse effects.73, 74 Therefore, the development of novel drugs that prevent or reduce both BVN and polypoidal lesions could have a considerable impact on current therapeutic strategy. Our result showing that intravitreally delivering an NP-encapsulated HTRA1 inhibitor is effective in preventing PCV but ineffective in treating existing lesions suggests that successful treatment of PCV requires the blocking of both HTRA1 activity and the underlying multiple degenerative processes (eg, attributable to MMPs). Combination therapy using both HTRA1 and MMP inhibitors as well as anti-inflammation medicine could be a potential effective treatment for PCV.
Acknowledgments
We thank Noriko Esumi for providing the vitelliform macular dystrophy 2 promoter, Carl B. Watt for assistance with electron microscopy, Balamurali K. Ambati for assistance with the Heidelberg Retina Angiograph-Optical Coherence Tomography device (Spectralis), Mary E. Hartnett for assistance with MICRON (Phoenix Research Laboratories Inc.), and Peter Westenskow, Benjamin Frankfort, Nduka Enemchukwu, and Hui Xu for comments on the manuscript.
S.K. performed most studies related to Tg44, Tg33, Tg26, and human polypoidal choroidal vasculopathy (PCV) specimens, was involved in experimental design and data analysis, and helped write the manuscript; H.N. provided human PCV specimens and performed histopathological studies on them; M.T. provided human PCV specimens; P.C.-B. provided human specimens; A.J. and S.W. performed histological experiments on mouse specimens; A.L., X.Z., and J.R. prepared HTRA1 inhibitor and nanoparticles; M.S. performed α-smooth muscle actin labeling of retinal vessels of all transgenic lines and some indocyanine green angiography (ICGA) and fluorescein angiography (FA); M.P. performed some ICGA, FA, and genotyping; Z.B., A.F., and S.V. assisted in genotyping; Y.F. developed the overall concept, designed experiments, analyzed data, supervised the study, and wrote the manuscript.
Footnotes
Supported by NIH grants 5R01EY022901 (Y.F.), 2P30EY002520 (Baylor College of Medicine), and 5P30EY014800 (University of Utah); The Sarah Campbell Blaffer Endowment in Ophthalmology (Y.F.); the Research to Prevent Blindness Career Development Award (Y.F.) and Research to Prevent Blindness unrestricted grants to the Department of Ophthalmology, University of Utah and Department of Ophthalmology, Baylor College of Medicine; the C.M. Reeves and M.A. Reeves Foundation (Y.F.); and the Moran Center for Translational Medicine (Y.F.).
Disclosures: Y.F. has received royalty from the University of Utah and has a patent pending relevant to a University of Utah research foundation study (application 20140323413).
Supplemental material for this article can be found at https://doi.org/10.1016/j.ajpath.2017.08.025.
Supplemental Data
Secondary or isotype controls. A: Secondary antibody control for C3 and membrane attack complex (MAC) staining in mouse tissues. B: Isotype IgG1 control for C3 staining in human tissues. C: Secondary antibody control for MAC staining in human tissues. Scale bars: 10 μm (A); 50 μm (B); 20 μm (C). PCV, polypoidal choroidal vasculopathy.
Secondary antibody controls for matrix metalloproteinase (MMP) 2, MMP9, and α-smooth muscle actin staining in mouse (A) and human (B) tissues. Scale bars: 10 μm (A); 20 μm (B).
HTRA1 expression in human polypoidal choroidal vasculopathy (PCV) lesions. A and B: HTRA1 level was markedly increased in retinal pigment epithelium (RPE; red arrows) of the PCV lesion (A) compared with that in the control eye (B). B: Weak expression of HTRA1 was detected in RPE (red arrows), Bruch membrane (black arrows), and choriocapillaris (black arrowhead) in the control eye. A–D: Isotype IgG (A–C) and secondary antibody (D) staining showed minimal background signals. C and D: RPE autofluorescence can be seen. Scale bars: 50 μm (A); 20 μm (B–D). Cho, choroid.
Secondary antibody controls for CD68 staining in human tissues (A) and F4/80 staining in mouse tissues (B). Scale bars: 20 μm (A); 10 μm (B). PCV, polypoidal choroidal vasculopathy.
Expression of human HTRA1S328A (or HTRA1) in transgenic mouse lines. A: Western blot analysis on tissue-type specificity of HTRA1S328A (or HTRA1) expression. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as a loading control. B: Retinal sections from wild-type (WT), Tg26, Tg33, and Tg44 mice were labeled with a rabbit anti–Myc-tag monoclonal antibody (green) or the isotype control. Nuclei were stained with DAPI (blue). Transgenic HTRA1 and HTRA1S328A were detected in the retinal pigment epithelium (RPE; red arrows) and the choroid (Cho; red arrowheads). Scale bar = 10 μm (B).
The expression level of fibulin-5 was similar in the retinal pigment epithelium (RPE)/choroid of wild-type (WT) and two hHTRA1+ mouse lines, VH73 (A) and Tg44 (B). Western blot analysis was performed on RPE/choroid lysate from two transgenic lines, VH73 and Tg44, and their respective WT controls. Equal loading was indicated by β-actin (A) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh; B).
Early-, middle-, and late-phase fluorescein angiography showed that all transgenic lines (Tg26, Tg33, and Tg44) had normal retinal vasculature, which was similar to wild type. Scale bar = 200 μm.
Elastic fiber degradation in Bruch membrane (BM) of Tg44 and human polypoidal choroidal vasculopathy (PCV). A: Elastic lamina (EL) of the Bruch membrane in Tg44 was fragmented and interrupted by gaps (red brackets). EL was normal in both wild type (WT) and Tg33. B: The Bruch membrane in human PCV lesions was degenerating (red brackets), whereas it was normal in the control (black arrows). Scale bars: 1 μm (A); 10 μm (B). Cho, choroid; RPE, retinal pigment epithelium.
Immunostaining of α-smooth muscle actin in retinal vessels in different transgenic lines. Staining was performed on retinal frozen sections. Smooth muscle cells were intact in wild-type (WT) and transgenic lines (Tg26, Tg33, and Tg44). Nuclei were counterstained with DAPI (blue). Scale bar = 10 μm.
Complement deposition in human polypoidal choroidal vasculopathy (PCV) lesions. C3 and membrane attack complex (MAC; Sc5b9) staining in the choroid of human PCV lesions. White arrows and yellow arrows indicated increased C3 and MAC, respectively, in the choroidal vessel wall of PCV. The red arrow and red arrowhead indicated the increase of C3 and MAC, respectively, outside of choroid vessels in PCV. The white arrowheads indicated the strong increase of C3 in the lumen of choroid vessels in PCV. Nuclei were counterstained with DAPI (blue). Scale bar = 10 μm.
Nanoparticle (NP) distribution in injected eyes. A: Widespread NPs in injected Tg44 eyes determined by coumarin fluorescence using fluorescein angiography. B: Localization of NPs was analyzed by confocal microscopy 4 days after injection in wild-type (WT) mice. NPs were concentrated in the retinal pigment epithelium and outer segment (white arrows). WT mice were injected with NPs containing coumarin or with NP buffer control. Scale bars: 1000 μm (A); 100 μm (B).
References
- 1.Wong W.L., Su X., Li X., Cheung C.M.G., Klein R., Cheng C.-Y., Wong T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2:e106–e116. doi: 10.1016/S2214-109X(13)70145-1. [DOI] [PubMed] [Google Scholar]
- 2.Imamura Y., Engelbert M., Iida T., Freund K.B., Yannuzzi L.A. Polypoidal choroidal vasculopathy: a review. Surv Ophthalmol. 2010;55:501–515. doi: 10.1016/j.survophthal.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Coppens G., Spielberg L., Leys A. Polypoidal choroidal vasculopathy, diagnosis and management. Bull Soc Belge Ophtalmol. 2011:39–44. [PubMed] [Google Scholar]
- 4.Tsujikawa A., Sasahara M., Otani A., Gotoh N., Kameda T., Iwama D., Yodoi Y., Tamura H., Mandai M., Yoshimura N. Pigment epithelial detachment in polypoidal choroidal vasculopathy. Am J Ophthalmol. 2007;143:102–111. doi: 10.1016/j.ajo.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Wong C.W., Yanagi Y., Lee W.-K., Ogura Y., Yeo I., Wong T.Y., Cheung C.M.G. Age-related macular degeneration and polypoidal choroidal vasculopathy in Asians. Prog Retin Eye Res. 2016;53:107–139. doi: 10.1016/j.preteyeres.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Tsujikawa A., Ojima Y., Yamashiro K., Nakata I., Ooto S., Tamura H., Nakanishi H., Hayashi H., Otani A., Yoshimura N. Association of lesion size and visual prognosis to polypoidal choroidal vasculopathy. Am J Ophthalmol. 2011;151:961–972.e1. doi: 10.1016/j.ajo.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Okubo A., Hirakawa M., Ito M., Sameshima M., Sakamoto T. Clinical features of early and late stage polypoidal choroidal vasculopathy characterized by lesion size and disease duration. Graefes Arch Clin Exp Ophthalmol. 2008;246:491–499. doi: 10.1007/s00417-007-0680-8. [DOI] [PubMed] [Google Scholar]
- 8.Uyama M., Wada M., Nagai Y., Matsubara T., Matsunaga H., Fukushima I., Takahashi K., Matsumura M. Polypoidal choroidal vasculopathy: natural history. Am J Ophthalmol. 2002;133:639–648. doi: 10.1016/s0002-9394(02)01404-6. [DOI] [PubMed] [Google Scholar]
- 9.Nie G.Y., Hampton A., Li Y., Findlay J.K., Salamonsen L.A. Identification and cloning of two isoforms of human high-temperature requirement factor A3 (HtrA3), characterization of its genomic structure and comparison of its tissue distribution with HtrA1 and HtrA2. Biochem J. 2003;371:39–48. doi: 10.1042/BJ20021569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zurawa-Janicka D., Skorko-Glonek J., Lipinska B. HtrA proteins as targets in therapy of cancer and other diseases. Expert Opin Ther Targets. 2010;14:665–679. doi: 10.1517/14728222.2010.487867. [DOI] [PubMed] [Google Scholar]
- 11.Kondo N., Honda S., Ishibashi K., Tsukahara Y., Negi A. LOC387715/HTRA1 variants in polypoidal choroidal vasculopathy and age-related macular degeneration in a Japanese population. Am J Ophthalmol. 2007;144:608–612. doi: 10.1016/j.ajo.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Lee K.Y., Vithana E.N., Mathur R., Yong V.H., Yeo I.Y., Thalamuthu A., Lee M.W., Koh A.H., Lim M.C., How A.C., Wong D.W., Aung T. Association analysis of CFH, C2, BF, and HTRA1 gene polymorphisms in Chinese patients with polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci. 2008;49:2613–2619. doi: 10.1167/iovs.07-0860. [DOI] [PubMed] [Google Scholar]
- 13.Lima L.H., Schubert C., Ferrara D.C., Merriam J.E., Imamura Y., Freund K.B., Spaide R.F., Yannuzzi L.A., Allikmets R. Three major loci involved in age-related macular degeneration are also associated with polypoidal choroidal vasculopathy. Ophthalmology. 2010;117:1567–1570. doi: 10.1016/j.ophtha.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewan A., Liu M., Hartman S., Zhang S.S., Liu D.T., Zhao C., Tam P.O., Chan W.M., Lam D.S., Snyder M., Barnstable C., Pang C.P., Hoh J. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 15.Yang Z., Camp N.J., Sun H., Tong Z., Gibbs D., Cameron D.J., Chen H., Zhao Y., Pearson E., Li X., Chien J., Dewan A., Harmon J., Bernstein P.S., Shridhar V., Zabriskie N.A., Hoh J., Howes K., Zhang K. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–993. doi: 10.1126/science.1133811. [DOI] [PubMed] [Google Scholar]
- 16.Jones A., Kumar S., Zhang N., Tong Z., Yang J.H., Watt C., Anderson J., Amrita, Fillerup H., McCloskey M., Luo L., Yang Z., Ambati B., Marc R., Oka C., Zhang K., Fu Y. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci U S A. 2011;108:14578–14583. doi: 10.1073/pnas.1102853108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S., Berriochoa Z., Ambati B.K., Fu Y. Angiographic features of transgenic mice with increased expression of human serine protease HTRA1 in retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2014;55:3842–3850. doi: 10.1167/iovs.13-13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esumi N., Oshima Y., Li Y., Campochiaro P.A., Zack D.J. Analysis of the VMD2 promoter and implication of E-box binding factors in its regulation. J Biol Chem. 2004;279:19064–19073. doi: 10.1074/jbc.M309881200. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S., Berriochoa Z., Jones A.D., Fu Y. Detecting abnormalities in choroidal vasculature in a mouse model of age-related macular degeneration by time-course indocyanine green angiography. J Vis Exp. 2014:e51061. doi: 10.3791/51061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D'Amato R., Wesolowski E., Smith L.E. Microscopic visualization of the retina by angiography with high-molecular-weight fluorescein-labeled dextrans in the mouse. Microvasc Res. 1993;46:135–142. doi: 10.1006/mvre.1993.1042. [DOI] [PubMed] [Google Scholar]
- 21.Truebestein L., Tennstaedt A., Monig T., Krojer T., Canellas F., Kaiser M., Clausen T., Ehrmann M. Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat Struct Mol Biol. 2011;18:386–388. doi: 10.1038/nsmb.2013. [DOI] [PubMed] [Google Scholar]
- 22.Kim H., Csaky K.G. Nanoparticle-integrin antagonist C16Y peptide treatment of choroidal neovascularization in rats. J Control Release. 2010;142:286–293. doi: 10.1016/j.jconrel.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 23.Kuroiwa S., Tateiwa H., Hisatomi T., Ishibashi T., Yoshimura N. Pathological features of surgically excised polypoidal choroidal vasculopathy membranes. Clin Exp Ophthalmol. 2004;32:297–302. doi: 10.1111/j.1442-9071.2004.00827.x. [DOI] [PubMed] [Google Scholar]
- 24.Okubo A., Sameshima M., Uemura A., Kanda S., Ohba N. Clinicopathological correlation of polypoidal choroidal vasculopathy revealed by ultrastructural study. Br J Ophthalmol. 2002;86:1093–1098. doi: 10.1136/bjo.86.10.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson D.H., Mullins R.F., Hageman G.S., Johnson L.V. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 26.Mullins R.F., Russell S.R., Anderson D.H., Hageman G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846. [PubMed] [Google Scholar]
- 27.Mullins R.F., Aptsiauri N., Hageman G.S. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye (Lond) 2001;15:390–395. doi: 10.1038/eye.2001.142. [DOI] [PubMed] [Google Scholar]
- 28.Nakashizuka H., Mitsumata M., Okisaka S., Shimada H., Kawamura A., Mori R., Yuzawa M. Clinicopathologic findings in polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci. 2008;49:4729–4737. doi: 10.1167/iovs.08-2134. [DOI] [PubMed] [Google Scholar]
- 29.Li H., Tran V.V., Hu Y., Mark Saltzman W., Barnstable C.J., Tombran-Tink J. A PEDF N-terminal peptide protects the retina from ischemic injury when delivered in PLGA nanospheres. Exp Eye Res. 2006;83:824–833. doi: 10.1016/j.exer.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Bourges J.L., Gautier S.E., Delie F., Bejjani R.A., Jeanny J.C., Gurny R., BenEzra D., Behar-Cohen F.F. Ocular drug delivery targeting the retina and retinal pigment epithelium using polylactide nanoparticles. Invest Ophthalmol Vis Sci. 2003;44:3562–3569. doi: 10.1167/iovs.02-1068. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y., Wang V.M., Chan C.C. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye (Lond) 2011;25:127–139. doi: 10.1038/eye.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim T.H., Laude A., Tan C.S. Polypoidal choroidal vasculopathy: an angiographic discussion. Eye (Lond) 2010;24:483–490. doi: 10.1038/eye.2009.323. [DOI] [PubMed] [Google Scholar]
- 33.Laude A., Cackett P.D., Vithana E.N., Yeo I.Y., Wong D., Koh A.H., Wong T.Y., Aung T. Polypoidal choroidal vasculopathy and neovascular age-related macular degeneration: same or different disease? Prog Retin Eye Res. 2010;29:19–29. doi: 10.1016/j.preteyeres.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Coscas G., Lupidi M., Coscas F., Benjelloun F., Zerbib J., Dirani A., Semoun O., Souied E.H. Toward a specific classification of polypoidal choroidal vasculopathy: idiopathic disease or subtype of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2015;56:3187–3195. doi: 10.1167/iovs.14-16236. [DOI] [PubMed] [Google Scholar]
- 35.Kawamura A., Yuzawa M., Mori R., Haruyama M., Tanaka K. Indocyanine green angiographic and optical coherence tomographic findings support classification of polypoidal choroidal vasculopathy into two types. Acta Ophthalmol. 2013;91:e474–e481. doi: 10.1111/aos.12110. [DOI] [PubMed] [Google Scholar]
- 36.Yuzawa M., Mori R., Kawamura A. The origins of polypoidal choroidal vasculopathy. Br J Ophthalmol. 2005;89:602–607. doi: 10.1136/bjo.2004.049296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerber H.-P., Dixit V., Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 38.Matsuoka M., Ogata N., Otsuji T., Nishimura T., Takahashi K., Matsumura M. Expression of pigment epithelium derived factor and vascular endothelial growth factor in choroidal neovascular membranes and polypoidal choroidal vasculopathy. Br J Ophthalmol. 2004;88:809–815. doi: 10.1136/bjo.2003.032466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terasaki H., Miyake Y., Suzuki T., Nakamura M., Nagasaka T. Polypoidal choroidal vasculopathy treated with macular translocation: clinical pathological correlation. Br J Ophthalmol. 2002;86:321–327. doi: 10.1136/bjo.86.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong J.P., Chan W.M., Liu D.T., Lai T.Y., Choy K.W., Pang C.P., Lam D.S. Aqueous humor levels of vascular endothelial growth factor and pigment epithelium-derived factor in polypoidal choroidal vasculopathy and choroidal neovascularization. Am J Ophthalmol. 2006;141:456–462. doi: 10.1016/j.ajo.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 41.Cheung C.M.G., Laude A., Yeo I., Tan S.-P., Fan Q., Mathur R., Lee S.Y., Chan C.M., Tan G., Lim T.H., Cheng C.-Y., Wong T.Y. Systemic, ocular and genetic risk factors for age-related macular degeneration and polypoidal choroidal vasculopathy in Singaporeans. Sci Rep. 2017;7:41386. doi: 10.1038/srep41386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.AMD Gene Consortium Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45:433–439. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goto A., Akahori M., Okamoto H., Minami M., Terauchi N., Haruhata Y., Obazawa M., Noda T., Honda M., Mizota A., Tanaka M., Hayashi T., Tanito M., Ogata N., Iwata T. Genetic analysis of typical wet-type age-related macular degeneration and polypoidal choroidal vasculopathy in Japanese population. J Ocul Biol Dis Infor. 2009;2:164–175. doi: 10.1007/s12177-009-9047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park D.H., Kim I.T. Association of ARMS2/HTRA1 variants with polypoidal choroidal vasculopathy phenotype in a Korean population. Jpn J Ophthalmol. 2012;56:60–67. doi: 10.1007/s10384-011-0089-0. [DOI] [PubMed] [Google Scholar]
- 45.Fritsche L.G., Loenhardt T., Janssen A., Fisher S.A., Rivera A., Keilhauer C.N., Weber B.H. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008;40:892–896. doi: 10.1038/ng.170. [DOI] [PubMed] [Google Scholar]
- 46.Friedrich U., Myers C.A., Fritsche L.G., Milenkovich A., Wolf A., Corbo J.C., Weber B.H. Risk- and non-risk-associated variants at the 10q26 AMD locus influence ARMS2 mRNA expression but exclude pathogenic effects due to protein deficiency. Hum Mol Genet. 2011;20:1387–1399. doi: 10.1093/hmg/ddr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z., Tong Z., Chen Y., Zeng J., Lu F., Sun X., Zhao C., Wang K., Davey L., Chen H., London N., Muramatsu D., Salasar F., Carmona R., Kasuga D., Wang X., Bedell M., Dixie M., Zhao P., Yang R., Gibbs D., Liu X., Li Y., Li C., Campochiaro B., Constantine R., Zack D.J., Campochiaro P., Fu Y., Li D.Y., Katsanis N., Zhang K. Genetic and functional dissection of HTRA1 and LOC387715 in age-related macular degeneration. PLoS Genet. 2010;6:e1000836. doi: 10.1371/journal.pgen.1000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan C.C., Shen D., Zhou M., Ross R.J., Ding X., Zhang K., Green W.R., Tuo J. Human HtrA1 in the archived eyes with age-related macular degeneration. Trans Am Ophthalmol Soc. 2007;105:92–97. discussion 97–98. [PMC free article] [PubMed] [Google Scholar]
- 49.Tuo J., Ross R.J., Reed G.F., Yan Q., Wang J.J., Bojanowski C.M., Chew E.Y., Feng X., Olsen T.W., Ferris F.L., 3rd, Mitchell P., Chan C.C. The HtrA1 promoter polymorphism, smoking, and age-related macular degeneration in multiple case-control samples. Ophthalmology. 2008;115:1891–1898. doi: 10.1016/j.ophtha.2008.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanda A., Stambolian D., Chen W., Curcio C.A., Abecasis G.R., Swaroop A. Age-related macular degeneration-associated variants at chromosome 10q26 do not significantly alter ARMS2 and HTRA1 transcript levels in the human retina. Mol Vis. 2010;16:1317–1323. [PMC free article] [PubMed] [Google Scholar]
- 51.Wang G., Spencer K.L., Scott W.K., Whitehead P., Court B.L., Ayala-Haedo J., Mayo P., Schwartz S.G., Kovach J.L., Gallins P., Polk M., Agarwal A., Postel E.A., Haines J.L., Pericak-Vance M.A. Analysis of the indel at the ARMS2 3′UTR in age-related macular degeneration. Hum Genet. 2010;127:595–602. doi: 10.1007/s00439-010-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.An E., Sen S., Park S.K., Gordish-Dressman H., Hathout Y. Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis Sci. 2010;51:3379–3386. doi: 10.1167/iovs.09-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakayama M., Iejima D., Akahori M., Kamei J., Goto A., Iwata T. Overexpression of HtrA1 and exposure to mainstream cigarette smoke leads to choroidal neovascularization and subretinal deposits in aged mice. Invest Ophthalmol Vis Sci. 2014;55:6514–6523. doi: 10.1167/iovs.14-14453. [DOI] [PubMed] [Google Scholar]
- 54.Iejima D., Itabashi T., Kawamura Y., Noda T., Yuasa S., Fukuda K., Oka C., Iwata T. HTRA1 (high temperature requirement A serine peptidase 1) gene is transcriptionally regulated by insertion/deletion nucleotides located at the 3′ end of the ARMS2 (age-related maculopathy susceptibility 2) gene in patients with age-related macular degeneration. J Biol Chem. 2015;290:2784–2797. doi: 10.1074/jbc.M114.593384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maruko I., Iida T., Saito M., Nagayama D., Saito K. Clinical characteristics of exudative age-related macular degeneration in Japanese patients. Am J Ophthalmol. 2007;144:15–22. doi: 10.1016/j.ajo.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 56.Karnik S.K., Brooke B.S., Bayes-Genis A., Sorensen L., Wythe J.D., Schwartz R.S., Keating M.T., Li D.Y. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 57.Duca L., Blaise S., Romier B., Laffargue M., Gayral S., El Btaouri H., Kawecki C., Guillot A., Martiny L., Debelle L., Maurice P. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res. 2016;110:298–308. doi: 10.1093/cvr/cvw061. [DOI] [PubMed] [Google Scholar]
- 58.Zeng R., Wen F., Zhang X., Su Y. Serum levels of matrix metalloproteinase 2 and matrix metalloproteinase 9 elevated in polypoidal choroidal vasculopathy but not in age-related macular degeneration. Mol Vis. 2013;19:729–736. [PMC free article] [PubMed] [Google Scholar]
- 59.De Salvo G., Vaz-Pereira S., Keane P.A., Tufail A., Liew G. Sensitivity and specificity of spectral-domain optical coherence tomography in detecting idiopathic polypoidal choroidal vasculopathy. Am J Ophthalmol. 2014;158:1228–1238.e1. doi: 10.1016/j.ajo.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 60.Vierkotten S., Muether P.S., Fauser S. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch's membrane via cleavage of extracellular matrix components. PLoS One. 2011;6:e22959. doi: 10.1371/journal.pone.0022959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stone E.M., Braun T.A., Russell S.R., Kuehn M.H., Lotery A.J., Moore P.A., Eastman C.G., Casavant T.L., Sheffield V.C. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–353. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- 62.Nowak-Sliwinska P., van den Bergh H., Sickenberg M., Koh A.H.C. Photodynamic therapy for polypoidal choroidal vasculopathy. Prog Retin Eye Res. 2013;37:182–199. doi: 10.1016/j.preteyeres.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 63.Hirami Y., Tsujikawa A., Otani A., Yodoi Y., Aikawa H., Mandai M., Yoshimura N. Hemorrhagic complications after photodynamic therapy for polypoidal choroidal vasculopathy. Retina. 2007;27:335–341. doi: 10.1097/01.iae.0000233647.78726.46. [DOI] [PubMed] [Google Scholar]
- 64.Ojima Y., Tsujikawa A., Otani A., Hirami Y., Aikawa H., Yoshimura N. Recurrent bleeding after photodynamic therapy in polypoidal choroidal vasculopathy. Am J Ophthalmol. 2006;141:958–960. doi: 10.1016/j.ajo.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 65.Kang H.M., Kim Y.M., Koh H.J. Five-year follow-up results of photodynamic therapy for polypoidal choroidal vasculopathy. Am J Ophthalmol. 2013;155:438–447.e1. doi: 10.1016/j.ajo.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 66.Hosokawa M., Morizane Y., Hirano M., Kimura S., Kumase F., Shiode Y., Doi S., Toshima S., Hosogi M., Fujiwara A., Mitsuhashi T., Shiraga F. One-year outcomes of a treat-and-extend regimen of intravitreal aflibercept for polypoidal choroidal vasculopathy. Jpn J Ophthalmol. 2017;61:150–158. doi: 10.1007/s10384-016-0492-7. [DOI] [PubMed] [Google Scholar]
- 67.Kikushima W., Sakurada Y., Sugiyama A., Tanabe N., Kume A., Iijima H. Comparison of initial treatment between 3-monthly intravitreal aflibercept monotherapy and combined photodynamic therapy with single intravitreal aflibercept for polypoidal choroidal vasculopathy. Graefes Arch Clin Exp Ophthalmol. 2017;255:311–316. doi: 10.1007/s00417-016-3467-y. [DOI] [PubMed] [Google Scholar]
- 68.Lee J.E., Shin J.P., Kim H.W., Chang W., Kim Y.C., Lee S.J., Chung I.Y., Lee J.E., VAULT Study Group Efficacy of fixed-dosing aflibercept for treating polypoidal choroidal vasculopathy: 1-year results of the VAULT study. Graefes Arch Clin Exp Ophthalmol. 2017;255:493–502. doi: 10.1007/s00417-016-3489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamamoto A., Okada A.A., Kano M., Koizumi H., Saito M., Maruko I., Sekiryu T., Iida T. One-year results of intravitreal aflibercept for polypoidal choroidal vasculopathy. Ophthalmology. 2015;122:1866–1872. doi: 10.1016/j.ophtha.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 70.Rofagha S., Bhisitkul R.B., Boyer D.S., Sadda S.R., Zhang K., SEVEN-UP Study Group Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP) Ophthalmology. 2013;120:2292–2299. doi: 10.1016/j.ophtha.2013.03.046. [DOI] [PubMed] [Google Scholar]
- 71.Silva R., Axer-Siegel R., Eldem B., Guymer R., Kirchhof B., Papp A., Seres A., Gekkieva M., Nieweg A., Pilz S., SECURE Study Group The SECURE study: long-term safety of ranibizumab 0.5 mg in neovascular age-related macular degeneration. Ophthalmology. 2013;120:130–139. doi: 10.1016/j.ophtha.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 72.Singer M.A., Awh C.C., Sadda S., Freeman W.R., Antoszyk A.N., Wong P., Tuomi L. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119:1175–1183. doi: 10.1016/j.ophtha.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 73.Kurihara T., Westenskow P.D., Bravo S., Aguilar E., Friedlander M. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122:4213–4217. doi: 10.1172/JCI65157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saint-Geniez M., Kurihara T., Sekiyama E., Maldonado A.E., D'Amore P.A. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106:18751–18756. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Secondary or isotype controls. A: Secondary antibody control for C3 and membrane attack complex (MAC) staining in mouse tissues. B: Isotype IgG1 control for C3 staining in human tissues. C: Secondary antibody control for MAC staining in human tissues. Scale bars: 10 μm (A); 50 μm (B); 20 μm (C). PCV, polypoidal choroidal vasculopathy.
Secondary antibody controls for matrix metalloproteinase (MMP) 2, MMP9, and α-smooth muscle actin staining in mouse (A) and human (B) tissues. Scale bars: 10 μm (A); 20 μm (B).
HTRA1 expression in human polypoidal choroidal vasculopathy (PCV) lesions. A and B: HTRA1 level was markedly increased in retinal pigment epithelium (RPE; red arrows) of the PCV lesion (A) compared with that in the control eye (B). B: Weak expression of HTRA1 was detected in RPE (red arrows), Bruch membrane (black arrows), and choriocapillaris (black arrowhead) in the control eye. A–D: Isotype IgG (A–C) and secondary antibody (D) staining showed minimal background signals. C and D: RPE autofluorescence can be seen. Scale bars: 50 μm (A); 20 μm (B–D). Cho, choroid.
Secondary antibody controls for CD68 staining in human tissues (A) and F4/80 staining in mouse tissues (B). Scale bars: 20 μm (A); 10 μm (B). PCV, polypoidal choroidal vasculopathy.
Expression of human HTRA1S328A (or HTRA1) in transgenic mouse lines. A: Western blot analysis on tissue-type specificity of HTRA1S328A (or HTRA1) expression. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as a loading control. B: Retinal sections from wild-type (WT), Tg26, Tg33, and Tg44 mice were labeled with a rabbit anti–Myc-tag monoclonal antibody (green) or the isotype control. Nuclei were stained with DAPI (blue). Transgenic HTRA1 and HTRA1S328A were detected in the retinal pigment epithelium (RPE; red arrows) and the choroid (Cho; red arrowheads). Scale bar = 10 μm (B).
The expression level of fibulin-5 was similar in the retinal pigment epithelium (RPE)/choroid of wild-type (WT) and two hHTRA1+ mouse lines, VH73 (A) and Tg44 (B). Western blot analysis was performed on RPE/choroid lysate from two transgenic lines, VH73 and Tg44, and their respective WT controls. Equal loading was indicated by β-actin (A) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh; B).
Early-, middle-, and late-phase fluorescein angiography showed that all transgenic lines (Tg26, Tg33, and Tg44) had normal retinal vasculature, which was similar to wild type. Scale bar = 200 μm.
Elastic fiber degradation in Bruch membrane (BM) of Tg44 and human polypoidal choroidal vasculopathy (PCV). A: Elastic lamina (EL) of the Bruch membrane in Tg44 was fragmented and interrupted by gaps (red brackets). EL was normal in both wild type (WT) and Tg33. B: The Bruch membrane in human PCV lesions was degenerating (red brackets), whereas it was normal in the control (black arrows). Scale bars: 1 μm (A); 10 μm (B). Cho, choroid; RPE, retinal pigment epithelium.
Immunostaining of α-smooth muscle actin in retinal vessels in different transgenic lines. Staining was performed on retinal frozen sections. Smooth muscle cells were intact in wild-type (WT) and transgenic lines (Tg26, Tg33, and Tg44). Nuclei were counterstained with DAPI (blue). Scale bar = 10 μm.
Complement deposition in human polypoidal choroidal vasculopathy (PCV) lesions. C3 and membrane attack complex (MAC; Sc5b9) staining in the choroid of human PCV lesions. White arrows and yellow arrows indicated increased C3 and MAC, respectively, in the choroidal vessel wall of PCV. The red arrow and red arrowhead indicated the increase of C3 and MAC, respectively, outside of choroid vessels in PCV. The white arrowheads indicated the strong increase of C3 in the lumen of choroid vessels in PCV. Nuclei were counterstained with DAPI (blue). Scale bar = 10 μm.
Nanoparticle (NP) distribution in injected eyes. A: Widespread NPs in injected Tg44 eyes determined by coumarin fluorescence using fluorescein angiography. B: Localization of NPs was analyzed by confocal microscopy 4 days after injection in wild-type (WT) mice. NPs were concentrated in the retinal pigment epithelium and outer segment (white arrows). WT mice were injected with NPs containing coumarin or with NP buffer control. Scale bars: 1000 μm (A); 100 μm (B).