

Abstract
BACKGROUND
Thyroid-associated ophthalmopathy, a condition commonly associated with Graves’ disease, remains inadequately treated. Current medical therapies, which primarily consist of glucocorticoids, have limited efficacy and present safety concerns. Inhibition of the insulin-like growth factor I receptor (IGF-IR) is a new therapeutic strategy to attenuate the underlying autoimmune pathogenesis of ophthalmopathy.
METHODS
We conducted a multicenter, double-masked, randomized, placebo-controlled trial to determine the efficacy and safety of teprotumumab, a human monoclonal antibody inhibitor of IGF-IR, in patients with active, moderate-to-severe ophthalmopathy. A total of 88 patients were randomly assigned to receive placebo or active drug administered intravenously once every 3 weeks for a total of eight infusions. The primary end point was the response in the study eye. This response was defined as a reduction of 2 points or more in the Clinical Activity Score (scores range from 0 to 7, with a score of ≥3 indicating active thyroid-associated ophthalmopathy) and a reduction of 2 mm or more in proptosis at week 24. Secondary end points, measured as continuous variables, included proptosis, the Clinical Activity Score, and results on the Graves’ ophthalmopathy–specific quality-of-life questionnaire. Adverse events were assessed.
RESULTS
In the intention-to-treat population, 29 of 42 patients who received teprotumumab (69%), as compared with 9 of 45 patients who received placebo (20%), had a response at week 24 (P<0.001). Therapeutic effects were rapid; at week 6, a total of 18 of 42 patients in the teprotumumab group (43%) and 2 of 45 patients in the placebo group (4%) had a response (P<0.001). Differences between the groups increased at subsequent time points. The only drug-related adverse event was hyperglycemia in patients with diabetes; this event was controlled by adjusting medication for diabetes.
CONCLUSIONS
In patients with active ophthalmopathy, teprotumumab was more effective than placebo in reducing proptosis and the Clinical Activity Score. (Funded by River Vision Development and others; ClinicalTrials.gov number, NCT01868997.)
Medical therapies for moderate-to-severe thyroid-associated ophthalmopathy (Graves’ orbitopathy) that have proved to be effective and safe in adequately powered, prospective, placebo-controlled trials are lacking. This unmet need is due to the incompletely understood pathogenesis of the disease.1 Current treatments are inconsistently beneficial and often associated with side effects, and their modification of the ultimate disease outcome is uncertain.1-3 Previous clinical trials, which were rarely placebo-controlled, suggest that high-dose glucocorticoids, alone3-5 or with radiotherapy,6,7 can reduce inflammation-related signs and symptoms in patients with active ophthalmopathy. However, glucocorticoids and orbital radiotherapy minimally affect proptosis and can cause dose-limiting adverse reactions.5 In many patients, the condition does not improve, and in some patients it progresses to dysthyroid optic neuropathy.
The thyrotropin receptor is uniquely targeted in Graves’ disease by pathogenic autoantibodies known as thyroid-stimulating immunoglobulins.8 These autoantibodies can be detected in most persons who have Graves’ disease with or without ophthalmopathy.9 The expression of the thyrotropin receptor in orbital tissues10,11 and by orbit-infiltrating fibrocytes12 suggests that it contributes to ophthalmopathy. However, the fact that thyroid-stimulating immunoglobulins are not detectable in some persons with ophthalmopathy13 suggests that additional autoantigens may be involved.
Immunoglobulins that activate insulin-like growth factor I (IGF-I) receptor (IGF-IR) signaling have been detected in patients with Graves’ disease,14 and IGF-I synergistically enhances the actions of thyrotropin.15 IGF-IR is a membrane-spanning tyrosine kinase receptor with roles in development and metabolism.16 It regulates immune function and thus might be targeted therapeutically in autoimmune diseases.17 IGF-IR is overexpressed by orbital fibroblasts18 and by T cells and B cells in persons with Graves’ disease.19,20 It forms a signaling complex with the thyrotropin receptor through which it is transac-tivated.18 In vitro studies of orbital fibroblasts and fibrocytes show that IGF-IR–inhibitory antibodies can attenuate the actions of IGF-I, thyrotropin, thyroid-stimulating immunoglobulins, and immunoglobulins isolated from patients with Graves’ disease.18,21 These observations prompted a trial of teprotumumab, a fully human IGF-IR–inhibitory monoclonal antibody formerly known as R1507,22 in patients with active, moderate-to-severe ophthalmopathy. In August 2016, after a review of the data from this trial, teprotumumab received a “breakthrough therapy” designation from the Food and Drug Administration.
METHODS
TRIAL SITES AND PARTICIPANTS
The trial was conducted at 15 sites. Patients were recruited between July 2, 2013, and September 23, 2015. Major inclusion criteria were the following: patients were 18 to 75 years of age, with ophthalmopathy that had been diagnosed no more than 9 months after the onset of symptoms, had a Clinical Activity Score of 4 or more on a 7-point scale (with a score of ≥3 indicating active thyroid-associated ophthalmopathy) in the more severely affected (study) eye, and had not received surgical or medical treatment, with the exception of oral glucocorticoids (a cumulative dose of ≤1 g of methylprednisolone or equivalent, with a 6-week washout period).
Serum glucose levels in patients with diabetes were well controlled. Female patients had negative pregnancy tests and used approved contraception. Patients with optic neuropathy, severe ocular surface damage, or an improved Clinical Activity Score of 2 points or more between screening and baseline visits were excluded.
TRIAL DESIGN
The trial comprised three phases: screening (2 to 6 weeks), intervention (24 weeks), and follow-up (48 weeks). A schedule of assessments is provided in Table S1 in the Supplementary Appendix, which is available, along with the trial protocol, with the full text of this article at NEJM.org.
Screening involved one to three visits. During the intervention phase, patients were assessed at baseline and every 3 weeks for 24 weeks; efficacy was assessed at weeks 6, 12, 18, and 24. Data from week 24 were used to assess the primary and secondary end points. With the exception of rare instances, at every assessment, patients were evaluated by the same ophthalmologist, who was unaware of the trial-group assignments. A change of 2 points in the 7-component Clinical Activity Score was considered to be clinically relevant.23 Proptosis was assessed with the use of a Hertel exophthalmometer. Quality of life was evaluated with the use of the Graves’ ophthalmopathy–specific quality-of-life questionnaire (GO-QOL),24 comprising two subscales assessed separately or in combination; scores on each subscale as well as the score on the overall GO-QOL scale have a range of 0 to 100 points. A change of 8 points was considered to be clinically relevant. Subjective diplopia was assessed by categorizing patients according to four grades. A change of one grade was considered to be clinically relevant (see the Supplementary Appendix).
TRIAL OVERSIGHT
The trial was designed by the academic investigators in collaboration with the manufacturer of teprotumumab, River Vision Development, which provided primary financial support for the trial. River Vision Development provided teprotumumab free of charge and was responsible for trial oversight.
Institutional review and ethics committees of the participating centers and the investigators approved the research protocol. An independent data and safety monitoring board oversaw the safety aspects of the trial. Witnessed written informed consent was obtained from all patients. Data were obtained by the investigators and their staff.
The investigators vouch for the accuracy and completeness of the data generated at their respective institutions. The investigators and River Vision Development vouch for the fidelity of the trial to the protocol.
INTERVENTIONS
Teprotumumab was provided as freeze-dried powder in glass vials. The placebo was 0.9% sodium chloride solution supplied by the research pharmacy. Patients received eight intravenous infusions, one every 3 weeks starting with an initial dose of 10 mg per kilogram of body weight, followed by 20 mg per kilogram for the remaining seven infusions.
RANDOMIZATION AND MASKING
This randomized trial was designed to assess efficacy and safety. Patients were randomly assigned in the double-masked intervention phase to either of two intervention groups in a 1:1 ratio in blocks of two, stratified within each clinical center according to smoking status with the use of an interactive Web-response system. Study pharmacists who were aware of the trial-group assignments prepared the masked infusion. The on-site principal investigators could identify a patient’s intervention only in the case of an emergency.
OUTCOMES
Patients who had a response were defined as those who met the composite primary end point at week 24. This end point comprised a reduction of 2 points or more in the Clinical Activity Score and a reduction of 2 mm or more in proptosis in the study eye in the absence of a corresponding amount of worsening in the nonstudy eye.
Secondary end points were proptosis and the Clinical Activity Score (both measured as continuous variables over time) and assessment of the patient’s quality of life with the use of the GO-QOL instrument (which includes two subscales that measure limitations in visual functioning and psychosocial functioning as a consequence of changed physical appearance). Patients were also categorized according to their level of response. Safety was assessed according to the incidence of adverse events, serious adverse events, and withdrawals due to adverse events.
STATISTICAL ANALYSIS
We estimated that a sample of 84 patients would provide the trial with a power of 80% or more to detect a between-group difference if 42 patients per trial group had data that could be evaluated. On the basis of published trial results, it was expected that 30% of the patients in the placebo group and 60% of the patients in the teprotumumab group would have a response. All statistical tests were two-sided and performed at the 5% significance level. No interim data analysis was performed. Analyses were performed with the use of SAS software, version 9.1 (SAS Institute), after database lock.
The intention-to-treat population, which included all randomly assigned patients who received at least one infusion, was used to analyze the primary and secondary efficacy outcomes. The safety population, which included all patients who received teprotumumab or placebo, was used for all safety analyses.
In the primary analysis, we used a logistic-regression model with the intervention group as the model effect and smoking as a covariate. Patients who did not have week 24 data for any reason were considered to have treatment failure. Results of a stratified Cochran–Mantel–Haenszel chi-square test, with smoking status as the stratification factor, enabled confirmation of the logistic analysis results (see Table S5 in the Supplementary Appendix).
For assessment of the secondary end points (proptosis, Clinical Activity Score, and GO-QOL scores), a mixed model of repeated measurements was fit to the individual change from baseline with the use of the PROC MIXED procedure in SAS software. Each baseline score (including smoking status, intervention group, time, time according to intervention, and time according to baseline interaction) was incorporated into the analysis. Responses in the nonstudy eye were assessed with the use of identical analyses.
RESULTS
PATIENTS
A total of 88 eligible patients underwent randomization. Of these patients, the intention-to-treat population of 87 patients (45 in the placebo group and 42 in the teprotumumab group) had more than one infusion. A total of 39 patients in the placebo group (87%) and 37 patients in the teprotumumab group (88%) completed the intervention (Fig. 1A).
Figure 1. Screening, Randomization, Response, and Follow-up of Trial Patients.

As shown in Panel A, patients who met the primary inclusion criteria for disease that was diagnosed 9 months or less after the onset of symptoms and who had a Clinical Activity Score of 4 points or more (on a scale from 0 to 7, with a score of ≥3 indicating active thyroid-associated ophthalmopathy) were entered into the screening phase of the trial. One patient who did not meet the screening criteria for an “administrative reason” was screened after the screening period was closed. At the baseline visit (week 0), patients who met all inclusion and exclusion criteria were randomly assigned to receive teprotumumab or placebo in a 24-week intervention phase of the trial. Patients then entered a 1-year follow-up phase, which is ongoing. The intention-to-treat population was defined as all patients who received one infusion of teprotumumab or placebo and excluded one patient who was randomly assigned to teprotumumab but withdrew consent before the drug was administered. One patient discontinued the intervention during the intervention phase but returned for the week 24 assessment. As shown in Panels B through D, the primary end point was a logistic regression of response status according to trial-drug group at week 24. A response was defined as a reduction of 2 mm or more in proptosis and a reduction of 2 points or more in the Clinical Activity Score in the study eye, without an equivalent increase in proptosis or in the Clinical Activity Score in the nonstudy eye. The Clinical Activity Score, which comprises seven components, ranges from 0 to 7, and a change of 2 points is considered to be clinically relevant.23 As shown in Panel B, in the analysis of the time to first response, data are expressed as means ±SE. As shown in Panel C, in the analysis of the time course in patients who met the response criteria, P values were calculated with the use of a logistic-regression model. As shown in Panel D, in the grading of a response at week 24, P<0.001 was calculated with the use of a logistic-regression model. A high response indicates that proptosis was reduced by 3 mm or more and the Clinical Activity Score was reduced by 3 points or more. A response indicates a reduction of 2 mm or more but less than 3 mm in proptosis and 2 points or more but less than 3 points in the Clinical Activity Score. A low response indicates reductions of 1 mm or more but less than 2 mm in proptosis and 1 point or more but less than 2 points in the Clinical Activity Score. No response indicates that the patient did not meet any response criteria or had missing evaluations at week 24.
Baseline characteristics of the patients are shown in Table 1, and in Table S2 in the Supplementary Appendix. A breakdown of randomization according to clinical center is provided in Table S3 in the Supplementary Appendix. All patients met criteria for euthyroid status (within ±50% of the reference range) at trial entry; 25 of 87 patients (29%) had minor adjustments in the dose of levothyroxine or an antithyroid drug.
Table 1.
Baseline Characteristics of the Patients.*
| Characteristic | Teprotumumab (N = 43) | Placebo (N = 44) |
|---|---|---|
| Demographic and clinical characteristics | ||
| Age — yr | 51.6±10.6 | 54.2±13.0 |
| Female sex — no. of patients (%) | 28 (65) | 36 (82) |
| Time since initiation of treatment for thyroid disease — mo† | ||
| Median | 8 | 15 |
| Range | 1–134 | 3–189 |
| Current treatment for thyroid disease — no. of patients (%) | ||
| Antithyroid drug‡ | 15 (35) | 20 (45) |
| Levothyroxine | 26 (60) | 23 (52) |
| Thyroid extract | 1 (2) | 3 (7) |
| Adjustment of medication at trial entry — no. of patients (%) | ||
| Levothyroxine | 8 (19) | 5 (11) |
| Antithyroid drug | 5 (12) | 7 (16) |
| Duration of eye symptoms or signs — mo | 4.7±2.1 | 5.2±2.3 |
| Duration of Graves’ disease — mo | ||
| Median | 10.7 | 10.8 |
| Range | 1.2–228.0 | 1.2–299.0 |
| Smoking status — no. of patients (%)§ | ||
| Nonsmoker | 32 (74) | 26 (59) |
| Smoker | 11 (26) | 18 (41) |
| Biochemical characteristics | ||
| Thyrotropin-binding inhibitory immunoglobulins — % | 51.6±26.9 | 48.7±25.4 |
| Thyroid-stimulating immunoglobulins — % | 422.9±118.1 | 435.1±105.2 |
| Mean thyroid hormone levels — pmol/liter¶ | ||
| Free triiodothyronine | 4.8±1.4 | 4.9±1.7 |
| Free thyroxine | 16.3±4.8 | 16.3±3.6 |
| Levels of free triiodothyronine and free thyroxine — no. of patients (%) | ||
| Euthyroid at baseline and through intervention phase | 20 (46) | 13 (30) |
| Values occasionally outside normal range during intervention phase | 18 (42) | 25 (57) |
| Sustained out-of-range values during intervention phase | 5 (12) | 6 (14) |
Plus–minus values are means ±SD. Additional patient characteristics are provided in Table S2 in the Supplementary Appendix. Patients were recruited in the following countries: Germany (19 patients), Italy (6), the United Kingdom (10), and the United States (53). A breakdown of randomization according to clinical center is provided in Table S3 in the Supplementary Appendix. Unless otherwise stated, baseline characteristics shown are from the safety population (44 patients in the placebo group and 43 patients in the teprotumumab group), which differs from the intention-to-treat population (45 patients in the placebo group and 42 patients in the teprotumumab group), since 1 patient in the placebo group received teprotumumab in error.
Data shown are for 10 patients in the placebo group and 10 patients in the teprotumumab group.
The antithyroid drugs were carbimazole, methimazole, and propylthiouracil.
Data for smoking status were based on patients who were current smokers at the time of the screening visit. Patients were stratified at randomization according to smoking status to balance the trial groups with respect to this known risk factor for thyroid-associated ophthalmopathy. However, there were imbalances in randomization blocks and discrepancies between patients who were randomly assigned to an intervention as nonsmokers at week 0 and those recorded as being smokers in their case-report forms. Data from the case-report forms were considered to be more accurate and were used to define smoking status at baseline.
The normal ranges of free triiodothyronine are 2.3 to 4.2 pg per milliliter (3.5 to 6.5 pmol per liter), and of free thyroxine, 0.9 to 1.8 ng per deciliter (11.6 to 23.2 pmol per liter).
Although patients were stratified according to smoking status, there was an imbalance between the two groups with respect to this variable (Table 1). The P values associated with the adjusted odds ratios presented in the next section, as well as the P values for the reduced proptosis and the reduction in the Clinical Activity Score (both measured as continuous variables) and for the GO-QOL score, were calculated with the use of analytic methods that adjusted for a potential imbalance in smoking status by including smoking as a covariate.
PRIMARY AND SECONDARY END POINTS
Proptosis, the Clinical Activity Score, and the GO-QOL score were nearly identical at baseline in the two groups (Table 2). At baseline, there was an imbalance between the groups with respect to diplopia, with a higher occurrence in the teprotumumab group.
Table 2.
Results of Efficacy Assessment before and after Administration of Trial Drug.*
| Variable | Teprotumumab (N = 42) | Placebo (N = 45) | Odds Ratio (95% CI) | P Value |
|---|---|---|---|---|
| Response analysis | ||||
| Primary outcome measure: response in study eye at wk 24 — no. of patients/total no. (%)† | ||||
| Intention-to-treat population | 29/42 (69) | 9/45 (20) | 8.86 (3.29–23.8) | <0.001 |
| Per-protocol population | 26/33 (79) | 8/36 (22) | 12.73 (4.01–40.4) | <0.001 |
| Time to first response — wk | 11.2±6.6 | 18.7±7.6 | NC | |
| Graded response — no. of patients (%)‡ | 11.80 (4.72–29.5) | <0.001 | ||
| High response | 21 (50) | 4 (9) | ||
| Response | 8 (19) | 5 (11) | ||
| Low response | 9 (21) | 8 (18) | ||
| No response or missing data | 4 (10) | 28 (62) | ||
| Proptosis — mm | ||||
| Baseline | 23.4±3.2 | 23.1±2.9 | ||
| Change from baseline | -2.46±0.20 | -0.15±0.19 | <0.001 | |
| Clinical Activity Score§ | ||||
| Baseline | 5.1±0.97 | 5.2±0.74 | ||
| Change from baseline | -3.43±0.18 | -1.85±0.17 | <0.001 | |
| GO-QOL score¶ | ||||
| Combined visual-functioning and appearance subscales | ||||
| Baseline | 34.5±7.3 | 34.5±6.8 | ||
| Change from baseline | 17.7±2.4 | 6.8±2.3 | <0.01 | |
| Visual-functioning subscale | ||||
| Baseline | 16.9±4.4 | 17.8±4.3 | ||
| Change from baseline | 21.7±2.9 | 7.5±2.7 | <0.001 | |
| Appearance subscale | ||||
| Baseline | 17.6±4.5 | 16.7±3.8 | ||
| Change from baseline | 12.9±2.8 | 6.6±2.7 | 0.10 | |
| Subjective diplopia║ | ||||
| Baseline according to grade — no. of patients (%) | 3.78 (1.68–8.54)** | 0.001 | ||
| No diplopia | 4 (10) | 14 (31) | ||
| Intermittent | 16 (38) | 19 (42) | ||
| Inconstant | 7 (17) | 8 (18) | ||
| Constant | 15 (35) | 4 (9) | ||
| Wk 24 according to grade — no. of patients (%) | ||||
| No diplopia | 21 (50) | 18 (40) | ||
| Intermittent | 4 (10) | 8 (18) | ||
| Inconstant | 9 (21) | 7 (16) | ||
| Constant | 4 (10) | 6 (13) | ||
| Wk 24 response — no. of patients/total no. (%) | 26/38 (68) | 10/39 (26) | <0.001 |
Plus–minus values are means ±SD, except for the change from baseline in proptosis, which is expressed as means ±SE. CI denotes confidence interval, and NC not calculated.
The primary outcome measure was analyzed with the use of a logistic-regression model that included smoking as a covariate, with a supporting sensitivity analysis conducted in the per-protocol population. The intention-to-treat population, defined as all patients who received an infusion of placebo or teprotumumab, excluded one randomly assigned patient who withdrew consent before drug administration. Secondary end points, analyzed hierarchically, were the Graves’ ophthalmopathy–specific quality-of-life (GO-QOL) combined score, proptosis, the Clinical Activity Score, the GO-QOL visual-functioning score, and the GO-QOL appearance score. These continuous variables were analyzed with the use of a mixed model of repeated measurements with smoking as a covariate. Any apparent discrepancies between the tabulated mixed model of repeated-measurements calculations for change from baseline and data plotted in the figures are because tabulated data are an average difference between the trial groups over all post-baseline time points up to week 24. Values plotted in the figures are calculations of the mean change from baseline for specific time points. Prespecified exploratory end points included the time to first response, graded response, and subjective diplopia.
A high response indicates that proptosis was reduced by 3 mm or more and the Clinical Activity Score was reduced by 3 points or more. A response indicates a reduction of 2 mm or more but less than 3 mm in proptosis and 2 points or more but less than 3 points in the Clinical Activity Score. A low response indicates reductions of 1 mm or more but less than 2 mm in proptosis and 1 point or more but less than 2 points in the Clinical Activity Score. No response indicates that the patient did not meet any response criteria or had missing evaluations at week 24. The P value for the comparison of placebo with teprotumumab was calculated for the overall distribution of graded responses, with the use of a logistic-regression model for cumulative logits.
The Clinical Activity Score comprised seven components. Scores range from 0 to 7, and a change of 2 points is considered to be clinically relevant.23
Quality of life was evaluated with the use of the GO-QOL questionnaire.24 The GO-QOL comprises two subscales, and scores on each subscale as well as the score on the overall GO-QOL scale have a range of 0 to 100 points. A change of 8 points is considered to be clinically relevant.
Subjective diplopia was assessed on the basis of four grades, and a change of one grade or more is considered to be clinically relevant. A response in patients with diplopia was defined as a decrease of one grade or more. The chi-square test was used to compare data from patients who had a response with data from those who did not at week 24.
The estimated odds ratio shown was calculated with the use of logistic regression for cumulative logits. This imbalance should be viewed in light of the large number of characteristics assessed at baseline. A sensitivity post hoc logistic-regression analysis of the week 24 response, which included adjustment for baseline smoking status, sex, study eye, and diplopia, did not show a substantial difference from the primary analysis (adjusted odds ratio, 8.16; P<0.001).
In the primary outcome measure in the intention-to-treat population, 9 of 45 patients who received placebo (20%) and 29 of 42 patients who received teprotumumab (69%) had a response at week 24 (adjusted odds ratio, 8.86; P<0.001). Similarly, in the per-protocol population, 8 of 36 patients who received placebo (22%) and 26 of 33 patients who received teprotumumab (79%) had a response at week 24 (adjusted odds ratio, 12.73; P<0.001). The time to the first response was markedly shorter in the teprotumumab group than in the placebo group (Fig. 1B). The onset of the response was rapid. The proportion of patients who had a response was greater in the teprotumumab group than in the placebo group at weeks 6, 12, and 18 (P<0.001 for all comparisons) (Fig. 1C). In a separate analysis that graded the level of response, more patients in the teprotumumab group than in the placebo group had reductions of 3 points or more in the Clinical Activity Score and reductions of 3 mm or more in proptosis (P<0.001 for the comparisons at every level of response) (Fig. 1D).
At weeks 6, 12, 18, and 24, the reduction in proptosis from baseline, measured as a continuous variable, was significantly greater in patients who received teprotumumab than in those who received placebo (P<0.001 for all comparisons) (Fig. 2A). At week 24, a total of 17 of 42 patients (40%) who received teprotumumab had a reduction of 4 mm or more in proptosis, as compared with 0% of patients who received placebo.
Figure 2. Secondary Efficacy End Points.
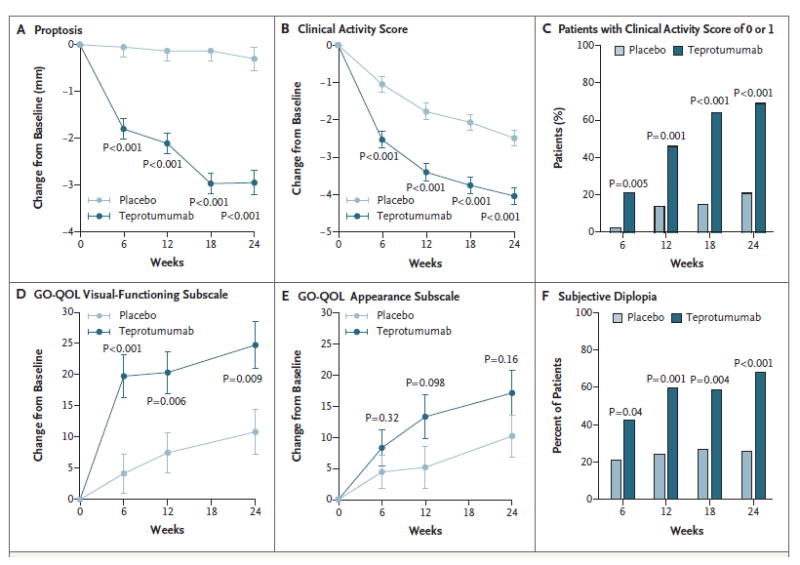
Panel A shows the change in proptosis from baseline. Panel B shows the change in the Clinical Activity Score from baseline. Panel C shows the results of the post hoc analysis of the percentage of patients with a Clinical Activity Score of 0 or 1 through week 24. Panel D shows the change in the visual-functioning subscale of the Graves’ ophthalmopathy–specific quality-of-life scale (GO-QOL) from baseline. Scores on the visual-functioning subscale range from 0 to 100, and a change of 8 points is considered to be clinically relevant. Panel E shows the change in the GO-QOL appearance subscale from baseline. Scores on the appearance subscale range from 0 to 100, and a change of 8 points is considered to be clinically relevant. In Panels A, B, D, and E, means ±SE are shown, and P values were calculated with the use of a mixed model of repeated-measurements analysis involving the intention-to-treat population (45 patients in the placebo group and 42 patients in the teprotumumab group). Panel F shows the response with respect to subjective diplopia. In this assessment, patients are categorized according to four grades, and a change of one grade or more is considered to be clinically relevant. P values shown in Panels C and F were calculated with the use of the chi-square test comparing data from patients who had a response with data from those who did not.
At weeks 6, 12, 18, and 24, the reduction in the Clinical Activity Score in the teprotumumab group was also significantly greater than that in the placebo group (P<0.001 for all comparisons) (Fig. 2B), although this score also decreased markedly and progressively in the placebo group. The baseline Clinical Activity Score in the teprotumumab group was 5.1 points, and the mean reduction at week 24 was 4 points; this indicates that some patients had a near-maximal therapeutic effect. This finding was confirmed by a post hoc categorical analysis involving patients who had a Clinical Activity Score of 0 points or 1 point; this analysis showed that 69% of the patients who received teprotumumab had a Clinical Activity Score of 0 or 1 at week 24, as compared with 21% of the patients who received placebo (adjusted odds ratio, 8.97; P<0.001) (Fig. 2C). A post hoc analysis showed that the reductions in the Clinical Activity Score in the teprotumumab group were broad-based (i.e., they were not driven by large decreases in subsets of the Clinical Activity Score components). Reductions in the Clinical Activity Score seen in the placebo group were similarly broad-based.
The GO-QOL visual-functioning score increased significantly in the teprotumumab group at all time points (Fig. 2D). This increase ranged from 12.8 to 15.6 points greater than the increase in the placebo group. On the GO-QOL appearance subscale, a consistent trend toward improvement emerged but did not achieve significance (Fig. 2E). When the two scales were combined, significance was seen at weeks 6, 12, and 24 (P = 0.003, P = 0.007, and P = 0.012, respectively) (Table 2). Response rates with respect to subjective diplopia were also significantly higher in the teprotumumab group than in the placebo group (Fig. 2F).
Efficacy was also assessed at week 28 (7 weeks after the final dose was administered), and this assessment showed no evidence of diminution (i.e., no “rebound” phenomenon). Indeed, the numbers of patients with a response increased and proptosis and the Clinical Activity Score were further reduced at week 28 as compared with week 24. The efficacy of teprotumumab in the nonstudy eye was also similar to the efficacy in the study eye with respect to response rates, proptosis, and the Clinical Activity Score. Serum assays showed no drug-induced changes in interleukin-6, interleukin-16, and RANTES (regulated on activation, normal T-cell expressed and secreted) levels. During the intervention phase, levels of thyroid-stimulating immunoglobulins and thyrotropin-binding inhibitory immunoglobulins decreased progressively (by 15 to 30%) in the two groups.
SAFETY
Adverse events that emerged during the intervention phase, that occurred in more than 5% of patients in the teprotumumab group, and that were greater in number in the teprotumumab group than in the placebo group are listed in Table 3. The majority of adverse events were mild, involved no treatment, and resolved while the patients continued to receive the intervention. Hyperglycemia, the only adverse event clearly identified by the investigators as being related to teprotumumab, was monitored by assessing blood glucose and glycated hemoglobin levels. Hyperglycemia in patients who did not have diabetes was uniformly grade 1, was intermittent, and occurred at similar rates in the two groups. Grade 2 or 3 hyperglycemia occurred in some patients with diabetes who received teprotumumab, and it was well controlled after adjustment of the medication for diabetes. Glycemic control, assessed according to the glycated hemoglobin level, was similar to that at baseline levels after the intervention phase in all patients who received teprotumumab (Table S4 in the Supplementary Appendix).
Table 3.
Adverse Events and Serious Adverse Events.
| Variable | Teprotumumab (N = 43)* | Placebo (N = 44)* | Summary Details of Adverse Events in Teprotumumab Group |
|---|---|---|---|
| number of patients (percent) | |||
| Adverse event during intervention† | |||
| Nausea | 8 (19) | 4 (9) | Generally mild and reported after first and second infusions |
| Muscle spasms | 8 (19) | 2 (5) | Intermittent, 2 of 8 patients had muscle spasms for >1 wk and received muscle relaxants |
| Diarrhea | 6 (14) | 2 (5) | Treatment occurred in 2 of 6 patients, 1 case designated as a serious adverse event (see below) |
| Hyperglycemia | 5 (12) | 2 (5) | Mechanism-based adverse event |
| Alopecia | 3 (7) | 2 (5) | All mild and no treatment necessary |
| Dry skin | 3 (7) | 0 | All mild, 1 patient used topical dry-skin cream |
| Dysgeusia | 3 (7) | 0 | In 2 of 3 patients, a transient “metallic” taste reported on days 1–2 |
| Headache | 3 (7) | 2 (5) | Generally mild, 1 patient took paracetamol |
| Paresthesia | 3 (7) | 0 | “Tingling” reported in nose, feet, or chest; variable onset and in 2 of 3 patients occurred on 1 day |
| Hearing impairment | 3 (7) | 0 | Disparate symptoms, onset, and duration (i.e., one case of unilateral hearing impairment with onset 16 wk after end of therapy, ‡ one case of mild bilateral hearing impairment that resolved, and one case of tinnitus in a patient with a history of tinnitus) |
| Weight loss | 3 (7) | 0 | Variable timing; decreases ranged from 5–9 lb (11–20 kg) |
| Any adverse event during intervention | 32 (74) | 32 (73) | |
| Serious adverse event§ | |||
| Optic neuropathy¶ | 0 | 1 (2) | |
| Diarrhea | 1 (2) | 0 | Severe diarrhea in 1 patient with a 6-mo history of ulcerative colitis |
| Inflammatory bowel disease | 1 (2) | 0 | In 1 patient with recent diagnosis of ileitis and colitis, inflammatory bowel disease diagnosed and treated while patient received trial drug |
| Escherichia sepsis║ | 1 (2) | 0 | Escherichia coli infection of unknown origin treated with intravenous antibiotics |
| Hashimoto’s encephalopathy | 1 (2) | 0 | Provisional diagnosis after episodic mental confusion with no other neurologic symptoms |
| Urinary retention | 1 (2) | 0 | Diagnosed after patient had an inguinal herniorrhaphy |
| Any serious adverse event | 5 (12) | 1 (2) | |
One patient in the placebo group received a single dose of teprotumumab in error at week 15. That patient is included here in the teprotumumab group.
Adverse events of any cause were defined as those that occurred between the administration of the first dose and 30 days after the administration of the final dose. Listed adverse events that emerged during the intervention phase are those that occurred in more than 5% of patients in the teprotumumab group and that occurred in greater numbers in the teprotumumab group than in the placebo group. Patients may have had more than one adverse event.
This case is included because the patient had unrelated, transient eustachian-tube dysfunction while receiving teprotumumab.
Listed are all serious adverse events reported in the trial, including any adverse event involving hospitalization. In the teprotumumab group, four patients discontinued the intervention because of the following serious adverse events: diarrhea that occurred after six infusions, inflammatory bowel disease after seven, Escherichia coli sepsis after three, and Hashimoto’s encephalopathy after six.
A total of three patients in the placebo group withdrew from the trial because of worsening eye symptoms or a lack of response. One case of dysthyroid optic neuropathy designated by an investigator as a serious adverse event occurred 3 days after the week 24 evaluation.
This patient was the only one whose intervention was unmasked during the course of the trial.
No deaths occurred during the trial. A total of 6 patients in each group discontinued the intervention. Serious adverse events occurred in 5 of 43 patients in the teprotumumab group (12%) and in 1 of 45 patients in the placebo group (2%) (Table 3). Two serious adverse events (diarrhea and mental confusion) in patients who received teprotumumab were categorized by the investigators as “possibly related” to the drug. Other serious adverse events were categorized as “unrelated.” Although designated as having a nonresponse treatment failure in the analyses, these 5 patients all met response criteria at their early withdrawal visit. Antidrug antibodies were detected in 1 patient in the teprotumumab group at baseline and in 1 patient during the intervention phase (week 3). Both patients tested negative on subsequent visits (at weeks 9 and 24), and neither patient had neutralizing antibodies. Four patients in the placebo group tested positive for antidrug antibodies at baseline; all were graded as having low-level antibody responses.
DISCUSSION
Patients who received teprotumumab had reductions in proptosis, the Clinical Activity Score, the GO-QOL (both the visual-functioning subscale and combined scales), and subjective diplopia that were clinically meaningful and significant (P≤0.001 for all comparisons with placebo).25 The reductions observed across all components of the Clinical Activity Score suggest a therapeutic mechanism upstream from the inflammation in orbital tissues. The marked reduction in proptosis is similar to that reported after decompression surgery.26,27 Moreover, orbital surgery can provoke reactivation of ophthalmopathy and can cause or exacerbate strabismus.26,28 Taken together, these findings suggest that inhibition of IGF-IR in patients with ophthalmopathy may result in a disease-modifying reduction in the volume of orbital fat, muscle, or both. Data from studies to determine the mechanism underlying the drug action are lacking.
The rationale for examining the clinical benefit of inhibiting the IGF-IR pathway in patients with ophthalmopathy derives from previous studies conducted in vitro. These studies showed the presence of autoantibodies recognizing IGF-IR and activating IGF-IR signaling in patients with Graves’ disease 14,18,29-31 and widespread IGF-IR overexpression in patients with Graves’ disease.14,18-20 They also showed that thyrotropin receptor and IGF-IR function interdependently.18 The presence of specific anti–IGF-IR antibodies in patients with Graves’ disease remains controversial.32 Some studies have shown their detection,14,29-31 whereas others have not.33,34 Given the evidence that actions of thyrotropin and thyroid-stimulating immunoglobulins are in part dependent on IGF-IR activity,18,21 the clinical benefits reported here may result from attenuation of pathogenic signaling mediated through both IGF-IR and the thyrotropin receptor.
The encouraging safety profile of teprotumumab in patients with ophthalmopathy is consistent with that in previous oncology studies35,36 and studies of other anti–IGF-IR antibodies.37 Teprotumumab shows no detectable affinity for the insulin receptor; thus, the hyperglycemia that was observed in some patients with diabetes probably resulted from IGF-IR inhibition. There was no evidence of residual worsening of glycemic control after the course of therapy. These findings indicate that patients with diabetes who receive teprotumumab will probably need glucose monitoring and potential adjustment of medication. Teprotumumab could also be associated with muscle spasms and diarrhea, particularly in patients with gastrointestinal disease; however, no mechanistic link has been established. Involvement of teprotumumab in the serious adverse events of Escherichia coli infection, mental confusion, and urinary retention (after herniorrhaphy) appears to be unlikely. Thrombocytopenia, anemia, and fatigue that have been reported in previous cancer studies22,37 were not observed in patients with ophthalmopathy.
As of this writing, the most informative trial of glucocorticoids in ophthalmopathy compared three doses of intravenous methylprednisolone, and the highest cumulative dose (7.47 g) produced mean reductions of 2.7 points from baseline in the Clinical Activity Score and 0.6 mm in proptosis.3 In a recent trial, rituximab did not result in greater reductions from baseline in the Clinical Activity Score or proptosis than placebo,38 whereas in a second trial, rituximab, as compared with intravenous methylprednisolone, was shown to reduce the Clinical Activity Score from baseline with no clinically meaningful effect on proptosis.39 Our trial, which showed appreciable responses across multiple end points in the placebo group, emphasizes the importance of conducting double-masked, placebo-controlled trials in ophthalmopathy.
Our trial has limitations. We enrolled only patients with active disease of recent onset, with a Clinical Activity Score of 4 or more. Thus, the potential of teprotumumab in benefiting patients with milder, less active, or stable disease was not assessed. Longer-term observation in the ongoing 1-year follow-up trial phase is necessary for assessing the durability of the response. No orbital imaging was performed; thus, it remains uncertain which orbital tissues were primarily affected by teprotumumab therapy. Our findings may have implications for other autoimmune diseases, such as rheumatoid arthritis, in which IGF-IR is involved and activating anti–IGF-IR antibodies also have been detected.17,40
In conclusion, a 24-week course of teprotumumab therapy provided clinical benefit in patients with active, moderate-to-severe thyroid-associated ophthalmopathy by reducing proptosis and the Clinical Activity Score and by improving the patients’ quality of life.
Supplementary Material
Acknowledgments
Supported by River Vision Development; a grant (1R01FD004792-01A1, to River Vision Development) from the Office of Orphan Products Development, Food and Drug Administration; grants (EY008976, EY0011708, DK063121, and 5UM1AI110557, to Dr. Smith) from the National Institutes of Health; a Center for Vision grant (EY007003, to Dr. Smith) and a grant (EY021197, to Dr. Douglas) from the National Eye Institute; an unrestricted grant (to Dr. Smith) from Research to Prevent Blindness; a grant (to Dr. Smith) from the Bell Charitable Foundation; a grant (to Dr. Ezra) from the Department of Health’s National Institute for Health Research Biomedical Research Centre for Ophthalmology at Moorfields Eye Hospital and University College London Institute of Ophthalmology; an unrestricted grant (to Dr. Fleming) from Research to Prevent Blindness to the Hamilton Eye Institute, University of Tennessee; and an unrestricted grant (to Dr. Harris) from Research to Prevent Blindness.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank the following persons for their contributions to the trial: Dave Madden, B.S., M.B.A., chief executive officer of River Vision Development, whose leadership and industry made this trial possible; Michael Friedman, Ph.D., for statistical support; Darla Kroft, B.A., who helped prepare an earlier version of the manuscript; and Sonal Trivedi, M.S., C.C.R.P., and Shivani Gupta, M.D. (Ann Arbor, MI); Elisa Kolbe, R.N., and Michaela Riedl, R.Ph. (Mainz, Germany); Dov Hersh, M.D., and Daniele Lorenzano, M.D. (London); Brian T. Fowler, M.D., and Moana Mosby, R.N. (Memphis, TN); Eric Ahn, M.D., and Chad Sorenson, B.S. (Portland, OR); Alonso Prusmack, B.S., and Alejandro Cruz Saldana, M.D. (Houston); Sang H. Hong, M.D. (Milwaukee); Poupak Fallahi, M.D. (Pisa, Italy); Nicola Currò, M.D., and Irene Campi, M.D. (Milan); Catherine Hwang, M.D.; Daniel Rootman, M.D. (Los Angeles); Shannon C. Lynch, M.D., and Donna G. Neely, M.B.A., C.O.M.T. (Omaha, NE); Ellen Fischbach, B.S., C.C.R.P. (St. Louis); Christine A. Sinkey, B.S.N. (Iowa City, IA); Ted Wojno, M.D., and Judy Brower, C.O.M.T. (Atlanta); and Mary Preston, C.O.M.T. (Aurora, CO) for executing the trial.
APPENDIX
The authors’ affiliations are as follows: the Department of Ophthalmology and Visual Sciences, Kellogg Eye Center (T.J.S., R.S.D.), and the Division of Metabolism, Endocrinology, and Diabetes, Department of Internal Medicine (T.J.S.), University of Michigan Medical School, Ann Arbor; the Department of Medicine, Johannes Gutenberg University Medical Center, Mainz, Germany (G.J.K.); Moorfields Eye Hospital, London (D.G.E.); the University of Tennessee Health Science Center, Memphis (J.C.F.); the Oculofacial Plastic Surgery Division, Oregon Health and Science University, Portland (R.A.D.); Eye Wellness Center, Neuro-Ophthalmology of Texas, Houston (R.A.T.); the Department of Ophthalmology, Medical College of Wisconsin, Milwaukee (G.J.H.); the Department of Clinical and Experimental Medicine, University of Pisa, Pisa (A.A.), and the Endocrinology and Diabetology Unit, Fondazione IRCCS Ca’ Granda, University of Milan, Milan (M.S.) — both in Italy; the Jules Stein Eye Institute, University of California, Los Angeles, Los Angeles (R.A.G.); the University of Nebraska Medical Center, Omaha (J.W.G.); Barnes–Jewish Hospital, Washington University, St. Louis (S.M.C.); the Department of Ophthalmology, University of Iowa Hospitals and Clinics, Iowa City (E.M.S.); the Department of Ophthalmology, Emory University, Atlanta (B.R.H.); the Department of Ophthalmology, University of Colorado, Aurora (E.M.H.); and River Vision Development, New York (R.M.W., K.G., G.M.).
References
- 1.Smith TJ, Hegedüs L. Graves’ disease. N Engl J Med. 2016;375:1552–65. doi: 10.1056/NEJMra1510030. [DOI] [PubMed] [Google Scholar]
- 2.Sandler HM, Rubenstein JH, Fowble BL, Sergott RC, Savino PJ, Bosley TM. Results of radiotherapy for thyroid ophthalmopathy. Int J Radiat Oncol Biol Phys. 1989;17:823–7. doi: 10.1016/0360-3016(89)90073-4. [DOI] [PubMed] [Google Scholar]
- 3.Bartalena L, Krassas GE, Wiersinga W, et al. Efficacy and safety of three different cumulative doses of intravenous methylprednisolone for moderate to severe and active Graves’ orbitopathy. J Clin Endocrinol Metab. 2012;97:4454–63. doi: 10.1210/jc.2012-2389. [DOI] [PubMed] [Google Scholar]
- 4.Zang S, Ponto KA, Kahaly GJ. Clinical review: intravenous glucocorticoids for Graves’ orbitopathy: efficacy and morbidity. J Clin Endocrinol Metab. 2011;96:320–32. doi: 10.1210/jc.2010-1962. [DOI] [PubMed] [Google Scholar]
- 5.Sisti E, Coco B, Menconi F, et al. Intravenous glucocorticoid therapy for Graves’ ophthalmopathy and acute liver damage: an epidemiological study. Eur J Endocrinol. 2015;172:269–76. doi: 10.1530/EJE-14-0712. [DOI] [PubMed] [Google Scholar]
- 6.Tanda ML, Bartalena L. Efficacy and safety of orbital radiotherapy for Graves’ orbitopathy. J Clin Endocrinol Metab. 2012;97:3857–65. doi: 10.1210/jc.2012-2758. [DOI] [PubMed] [Google Scholar]
- 7.Marcocci C, Bartalena L, Tanda ML, et al. Comparison of the effectiveness and tolerability of intravenous or oral glucocorticoids associated with orbital radiotherapy in the management of severe Graves’ ophthalmopathy: results of a prospective, single-blind, randomized study. J Clin Endocrinol Metab. 2001;86:3562–7. doi: 10.1210/jcem.86.8.7737. [DOI] [PubMed] [Google Scholar]
- 8.Morshed SA, Davies TF. Graves’ disease mechanisms: the role of stimulating, blocking, and cleavage region TSH receptor antibodies. Horm Metab Res. 2015;47:727–34. doi: 10.1055/s-0035-1559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diana T, Wüster C, Kanitz M, Kahaly GJ. Highly variable sensitivity of five binding and two bio-assays for TSH-receptor antibodies. J Endocrinol Invest. 2016;39:1159–65. doi: 10.1007/s40618-016-0478-9. [DOI] [PubMed] [Google Scholar]
- 10.Feliciello A, Porcellini A, Ciullo I, Bonavolontà G, Avvedimento EV, Fenzi G. Expression of thyrotropin-receptor mRNA in healthy and Graves’ disease retro-orbital tissue. Lancet. 1993;342:337–8. doi: 10.1016/0140-6736(93)91475-2. [DOI] [PubMed] [Google Scholar]
- 11.Bahn RS, Dutton CM, Natt N, Joba W, Spitzweg C, Heufelder AE. Thyrotropin receptor expression in Graves’ orbital adipose/connective tissues: potential autoantigen in Graves’ ophthalmopathy. J Clin Endocrinol Metab. 1998;83:998–1002. doi: 10.1210/jcem.83.3.4676. [DOI] [PubMed] [Google Scholar]
- 12.Douglas RS, Afifiyan NF, Hwang CJ, et al. Increased generation of fibrocytes in thyroid-associated ophthalmopathy. J Clin Endocrinol Metab. 2010;95:430–8. doi: 10.1210/jc.2009-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabasum A, Khan I, Taylor P, Das G, Okosieme OE. Thyroid antibody-negative euthyroid Graves’ ophthalmopathy. Endocrinol Diabetes Metab Case Rep 2016. 2016:160008. doi: 10.1530/EDM-16-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pritchard J, Han R, Horst N, Cruikshank WW, Smith TJ. Immunoglobulin activation of T cell chemoattractant expression in fibroblasts from patients with Graves’ disease is mediated through the insulin-like growth factor I receptor pathway. J Immunol. 2003;170:6348–54. doi: 10.4049/jimmunol.170.12.6348. [DOI] [PubMed] [Google Scholar]
- 15.Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and Graves’-IgG. Endocrinology. 1986;119:940–2. doi: 10.1210/endo-119-2-940. [DOI] [PubMed] [Google Scholar]
- 16.Gallagher EJ, LeRoith D. Minireview: IGF, insulin, and cancer. Endocrinology. 2011;152:2546–51. doi: 10.1210/en.2011-0231. [DOI] [PubMed] [Google Scholar]
- 17.Smith TJ. Insulin-like growth factor-I regulation of immune function: a potential therapeutic target in autoimmune diseases? Pharmacol Rev. 2010;62:199–236. doi: 10.1124/pr.109.002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsui S, Naik V, Hoa N, et al. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in Graves’ disease. J Immunol. 2008;181:4397–405. doi: 10.4049/jimmunol.181.6.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas RS, Gianoukakis AG, Kamat S, Smith TJ. Aberrant expression of the insulin-like growth factor-1 receptor by T cells from patients with Graves’ disease may carry functional consequences for disease pathogenesis. J Immunol. 2007;178:3281–7. doi: 10.4049/jimmunol.178.5.3281. [DOI] [PubMed] [Google Scholar]
- 20.Douglas RS, Naik V, Hwang CJ, et al. B cells from patients with Graves’ disease aberrantly express the IGF-1 receptor: implications for disease pathogenesis. J Immunol. 2008;181:5768–74. doi: 10.4049/jimmunol.181.8.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Mester T, Raychaudhuri N, et al. Teprotumumab, an IGF-1R blocking monoclonal antibody inhibits TSH and IGF-1 action in fibrocytes. J Clin Endocrinol Metab. 2014;99:E1635–E1640. doi: 10.1210/jc.2014-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurzrock R, Patnaik A, Aisner J, et al. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin Cancer Res. 2010;16:2458–65. doi: 10.1158/1078-0432.CCR-09-3220. [DOI] [PubMed] [Google Scholar]
- 23.Wiersinga WM, Perros P, Kahaly GJ, et al. Clinical assessment of patients with Graves’ orbitopathy: the European Group on Graves’ Orbitopathy recommendations to generalists, specialists and clinical researchers. Eur J Endocrinol. 2006;155:387–9. doi: 10.1530/eje.1.02230. [DOI] [PubMed] [Google Scholar]
- 24.Terwee CB, Gerding MN, Dekker FW, Prummel MF, Wiersinga WM. Development of a disease specific quality of life questionnaire for patients with Graves’ ophthalmopathy: the GO-QOL. Br J Ophthalmol. 1998;82:773–9. doi: 10.1136/bjo.82.7.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rootman DB, Golan S, Pavlovich P, Rootman J. Postoperative changes in strabismus, ductions, exophthalmometry, and eyelid retraction after orbital decompression for thyroid orbitopathy. Ophthal Plast Reconstr Surg. 2016 Aug 1; doi: 10.1097/IOP.0000000000000758. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 26.Wu CY, Niziol LM, Musch DC, Kahana A. Thyroid-related orbital decompression surgery: a multivariate analysis of risk factors and outcomes. Ophthal Plast Reconstr Surg. 2016 Apr 19; doi: 10.1097/IOP.0000000000000699. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terwee CB, Dekker FW, Mourits MP, et al. Interpretation and validity of changes in scores on the Graves’ ophthalmopathy quality of life questionnaire (GO-QOL) after different treatments. Clin Endocrinol (Oxf) 2001;54:391–8. doi: 10.1046/j.1365-2265.2001.01241.x. [DOI] [PubMed] [Google Scholar]
- 28.Baldeschi L, Lupetti A, Vu P, Wakelkamp IM, Prummel MF, Wiersinga WM. Reactivation of Graves’ orbitopathy after rehabilitative orbital decompression. Ophthalmology. 2007;114:1395–402. doi: 10.1016/j.ophtha.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 29.Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. Autoantibodies to IGF-1 binding sites in thyroid associated ophthalmopathy. Autoimmunity. 1993;16:251–7. doi: 10.3109/08916939309014643. [DOI] [PubMed] [Google Scholar]
- 30.Smith TJ, Hoa N. Immunoglobulins from patients with Graves’ disease induce hyaluronan synthesis in their orbital fibroblasts through the self-antigen, IGF-1 receptor. J Clin Endocrinol Metab. 2004;89:5076–80. doi: 10.1210/jc.2004-0716. [DOI] [PubMed] [Google Scholar]
- 31.Varewijck AJ, Boelen A, Lamberts SW, et al. Circulating IgGs may modulate IGF-I receptor stimulating activity in a subset of patients with Graves’ ophthalmopathy. J Clin Endocrinol Metab. 2013;98:769–76. doi: 10.1210/jc.2012-2270. [DOI] [PubMed] [Google Scholar]
- 32.Smith TJ. Is IGF-I receptor a target for autoantibody generation in Graves’ disease? J Clin Endocrinol Metab. 2013;98:515–8. doi: 10.1210/jc.2013-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minich WB, Dehina N, Welsink T, et al. Autoantibodies to the IGF1 receptor in Graves’ orbitopathy. J Clin Endocrinol Metab. 2013;98:752–60. doi: 10.1210/jc.2012-1771. [DOI] [PubMed] [Google Scholar]
- 34.Krieger CC, Place RF, Bevilacqua C, et al. TSH/IGF-1 receptor cross talk in Graves’ ophthalmopathy pathogenesis. J Clin Endocrinol Metab. 2016;101:2340–7. doi: 10.1210/jc.2016-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pappo AS, Patel SR, Crowley J, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2011;29:4541–7. doi: 10.1200/JCO.2010.34.0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramalingam SS, Spigel DR, Chen D, et al. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2011;29:4574–80. doi: 10.1200/JCO.2011.36.6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma H, Zhang T, Shen H, Cao H, Du J. The adverse events profile of anti-IGF-1R monoclonal antibodies in cancer therapy. Br J Clin Pharmacol. 2014;77:917–28. doi: 10.1111/bcp.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stan MN, Garrity JA, Carranza Leon BG, Prabin T, Bradley EA, Bahn RS. Randomized controlled trial of rituximab in patients with Graves’ orbitopathy. J Clin Endocrinol Metab. 2015;100:432–41. doi: 10.1210/jc.2014-2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salvi M, Vannucchi G, Currò N, et al. Efficacy of B-cell targeted therapy with rituximab in patients with active moderate to severe Graves’ orbitopathy: a randomized controlled study. J Clin Endocrinol Metab. 2015;100:422–31. doi: 10.1210/jc.2014-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pritchard J, Tsui S, Horst N, Cruikshank WW, Smith TJ. Synovial fibroblasts from patients with rheumatoid arthritis, like fibroblasts from Graves’ disease, express high levels of IL-16 when treated with Igs against insulin-like growth factor-1 receptor. J Immunol. 2004;173:3564–9. doi: 10.4049/jimmunol.173.5.3564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.