

Abstract
In this study, we examined early time-dependent changes in articular cartilage and synovium in response to tibial compression and sought the plausible origin of cells that respond to compression in the healer (LGXSM-6) and non-healer (LGXSM-33) recombinant inbred mouse strains. The right knee of 13-week old male mice was subjected to tibial compression using 9N axial loading. The contralateral left knee served as a control. Knees were harvested at 5, 9, and 14 days post-injury. Histological changes in cartilage and synovium, immunofluorescence pattern of CD44, aggrecan, type-II collagen, cartilage oligomeric matrix protein and the aggrecan neo-epitope NITEGE, and cell apoptosis (by TUNEL) were examined. We used a double nucleoside analog cell-labeling strategy to trace cells responsive to injury. We showed that tibial compression resulted in rupture of anterior cruciate ligament, cartilage matrix loss and chondrocyte apoptosis at the injury site. LGXSM-33 showed higher synovitis and ectopic synovial chondrogenesis than LGXSM-6 with no differences for articular cartilage lesions. With loading, an altered pattern of CD44 and NITEGE was observed: cells in the impacted area underwent apoptosis, cells closely surrounding the injured area expressed CD44, and cells in the intact area expressed NITEGE. Cells responding to injury were found in the synovium, subchondral bone marrow and the Groove of Ranvier. Taken together, we found no strain differences in chondrocytes in the early response to injury. However, the synovial response was greater in LGXSM-33 indicating that, at early time points, there is a genetic difference in synovial cell reaction to injury.
Keywords: mechanical injury, knee joint, genetics, chondrocyte apoptosis, cell source
Injury to knee is a predisposing factor for the development of post-traumatic osteoarthritis (PTOA). Individuals who suffer a knee injury are reported to have about fourfold increase in the risk of developing knee OA.1 Unlike many other joints, the knee is particularly susceptible to OA as ligamentous and meniscal damage and direct effects of injury to the joint surface and intra-articular fractures occur in the knee.2 Approximately 50% of individuals with anterior cruciate ligament (ACL) rupture or meniscus tear go on to develop PTOA within 10–20 years after injury.2 Currently, OA is diagnosed at the terminal stage at which time little can be done to slow, reverse or halt its progression and too often total knee arthroplasty is the only answer.
Studies in humans have shown that changes in chondrocytes and extracellular matrix begin in the first few weeks after injury and well before the occurrence of radiographically detectable OA.3 The timeline of these events during disease initiation and progression remains poorly understood. It has been thought that the pathogenetic events following mechanical injury to the synovial joint can temporally be divided into acute and chronic phases. The acute phase is characterized by prominent inflammation. During this phase subtle metabolic changes in cartilage and other joint structures occur that eventually may result in PTOA.3 Thus, an understanding of changes in the injured joint in the early (acute) phase of injury may offer opportunities to devise early therapeutic strategies. Joints with meniscus or ACL injuries are an ideal resource to identify early molecular changes because the time from injury is generally known. Therefore, in order to study early changes following injury, we adapted a non-invasive murine model of compressive loading to the knee joint that results in ACL rupture and injuries to both articular cartilage and synovium.4,5 Using this model, we observed chondrocyte apoptosis and altered distribution of extracellular matrix molecules in the cartilage within 5–14 days after injury.4 In the present study, we explored the timeline of cellular response to injury, attempted to unravel the plausible mechanism of the altered extracellular distribution pattern, and examined the source of cells responding to non-invasive injury (i.e., compression).
Another aspect of this study was to begin to examine the genetic differences in the susceptibility to injury. It is known that OA runs in families and has a strong hereditary component, accounting for 39–78% of OA.6,7 We have previously shown that two mouse strains (namely LGXSM-6, the healer strain and LGXSM-33, the non-healer strain), selected from a range of recombinant inbred lines of LG/J and SM/J, differ in their ability to heal full-thickness cartilage surface injuries over 16 weeks. In addition, the healer strain can heal full-thickness ear wounds.8,9 However, very little is known about how these two strains react to a non-invasive mechanical injury at early time points (i.e., up to 2 weeks). In the present study, we sought to determine whether these two mouse strains exhibit phenotypic differences in the early response to mechanical injury, in order to provide an experimental basis for our long-term studies.
MATERIALS AND METHODS
Mice and Housing
Male mice from LGXSM-6 (n=15) and LGXSM-33 (n=15) strains were used and housed under standard conditions as described previously.8 These mice are available to us through our collaborative work with Dr. James Cheverud.8–10 The Animal Studies Committee of Washington University approved all the procedures in this study. Mice were raised at our mouse facility with a constant humidity of 30–70%, temperature of 21°C, light/dark cycle of 12 h and high standards of animal husbandry. Mouse strains were bred by brother-sister mating. Offspring were housed with their mothers for 3 weeks until weaned and separated into sex-specific cages of 4–5 mice per cage with each cage ventilated individually.
Non-Invasive Mechanical Injury Model
The right knee of labeled mice (see “Labeling Procedure for Cells” below) underwent 9N of compressive loading as detailed previously.4 Briefly, under anesthesia (2.5% isoflurane in 4 L/min oxygen; Highland Medical Equipment, Temecula, CA), the right tibia was positioned vertically, with the ankle upward and the knee downward in deep flexion between custom-made cups of a materials testing machine (Instron ElectroPulse E1000, Norwood, MA) in the laboratory. Axial compressive loads were applied through the foot joint via the upper loading cup, while the lower cup holding the knee was fixed and linked to the load cell. Cyclic loads were applied for 0.34 s, with a rise and fall time each of 0.17 s and a baseline hold time of 10 s between cycles. Peak loading force was 9N with a 0.5N preload force to maintain the limb in position between periods of peak loading. Sixty cycles of this pattern were applied in the whole loading episode. The contralateral left knee served as a control. Three additional age-matched mice from each strain were used as non-loaded, non-labeled controls at day 14.
Histological Analysis for Changes in Cartilage and Synovium
Mice were sacrificed at day 5 (n=4), 9 (n=4), and 14 n=4) after loading. Knee joints were dissected and processed for histology using standard procedures with slight modifications.4 We used 12% formic acid for decalcification instead of 14% ethylene diamine tetraacetic acid and we cut the sections at 5 μm thickness instead of 10 μm thickness. We prepared serial sagittal sections entirely through the lateral femoral condyle. The average number of sections from each knee was 61. We used every 10th section to stain with Safranin-O/fast green staining. Three of seven stained sections were used to score cartilage injury length and synovitis. For immunostaining, we used at least three sections from each knee. Synovial pathology was evaluated using published methods11 and combined the synovitis score (cell layers and cell density) to report overall synovitis.
Assessment of Cell Apoptosis
In situ detection of chondrocyte apoptosis was performed using the terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) assay (In situ Cell Death Detection Kit, Roche, Indianapolis, IN). In order to compare the number of apoptotic cells, we counted the numbers of TUNEL-positive cells in non-calcified cartilage in both strains since apoptosis naturally occurs in calcified cartilage.12,13 Images taken from three TUNEL stained sections at 400X magnification were evaluated by two blinded observers as described previously.14
Pattern of Extracellular Matrix Proteins and CD44 Expression
We assessed expression pattern of extracellular matrix proteins (type-II collagen, aggrecan, and cartilage oligomeric matrix protein, that is, COMP) and CD44 (a cell surface antigen) through immunostaining as described.4 The primary antibodies used are shown in Table 1. Images were captured on an Eclipse E800 microscope (Nikon, Tokyo, Japan) with QImaging Retiga 2000R Fast 1394 camera and MetaMorph software v7.7 (Molecular Devices, Sunnyvale, CA).
Table 1.
Characteristics of Antibodies
| Antibody | Supplier | Cat. # | Dilution |
|---|---|---|---|
| Mouse anti-CIdU | Abcam (Cambridge, MA) | ab6326 | 1:100 |
| Rat anti-IdU | Abcam (Cambridge, MA) | ab8955 | 1:100 |
| Rabbit anti-COMP | Kamiya Biomedical Co. (Seattle, WA) | PC-140 | 1:100 |
| Rabbit anti-aggrecan (G2 domain) | Dr. Amanda Fosang (The University of Melbourne, Australia) | Gift | 1:100 |
| Rat anti-type-II collagen (triple helical domain) | Dr. Linda Sandell (Washington University, St. Louis, MO) | In house | 1:100 |
| Rabbit anti-NITEGE373 | Dr. Amanda Fosang (The University of Melbourne, Australia) | Gift | 1:100 |
| Alexa Fluor 594 goat anti-rat | Abcam (Cambridge, MA) | ab150168 | 1:200 |
| Alexa Fluor 488 goat anti-rabbit | Abcam (Cambridge, MA) | ab181448 | 1:200 |
| Alexa Fluor 594 goat anti-rabbit | Abcam (Cambridge, MA) | ab150092 | 1:200 |
| Alexa Fluor 488 goat anti-mouse | Abcam (Cambridge, MA) | ab150113 | 1:200 |
In Situ Hybridization Analyses of Aggrecan
To determine whether cells synthesized aggrecan after injury, in situ hybridization was performed using 35S labeled riboprobe as previously described.15,16 The aggrecan in situ probe covering the interglobular domain sequence of the mouse aggrecan has been previously described. Images for in situ hybridization analysis were captured with an OlympusBX51 microscope with a 10× objective and Olympus DP70 digital camera using the DPcontroller software (Olympus, Tokyo, Japan). For in situ analysis, pictures of hybridization signals were hued red and superimposed on toluidine blue counterstained images using Adobe Photoshop.
Labeling Procedure for Cells
To track the cells that respond to the injury, we took advantage of a previous in vivo scheme of double nucleoside analog cell-labeling model.17,18 Most stem cells are in a quiescent state and label retention is an indication of quiescence.19 Once cells have incorporated a label, rapidly dividing cells lose the label quickly due to dilution, whereas quiescent or very slow cycling cells retain the label for extended periods. Three-week old mice received 1 mg/ml iododeoxyuridine (IdU; Sigma–Aldrich, St. Louis, MO) in the drinking water for 30 days, followed by a 40-day washout period using regular drinking water. All the mice were subjected to mechanical loading as described above. To label cells responding to the injury, 10 mg/ml chlorodeoxyuridine (CIdU, Sigma–Aldrich) was subcutaneously injected respectively at 0, 5, and 9 days after loading (n=4 at each time point) followed by 1 mg/ml in drinking water until sacrificed at 5, 9, and 14 days after loading (Fig. 1A). We anticipated the following scenarios (Fig. 1B): during the initial 30-day labeling period with IdU, all dividing cells would become labeled. The IdU would be diluted and become undetectable in rapidly dividing cells after the 40-day washout period, but would be retained by slow-cycling cells. These slow-cycling cells are thought to be in a progenitor-like stage. During the second post-injury labeling period with CIdU, all dividing cells will incorporate CIdU, but only slow-cycling long-term-retaining IdU-positive cells would become double labeled. The IdU-labeled cells that did not divide after injury would not take up CIdU.
Figure 1.
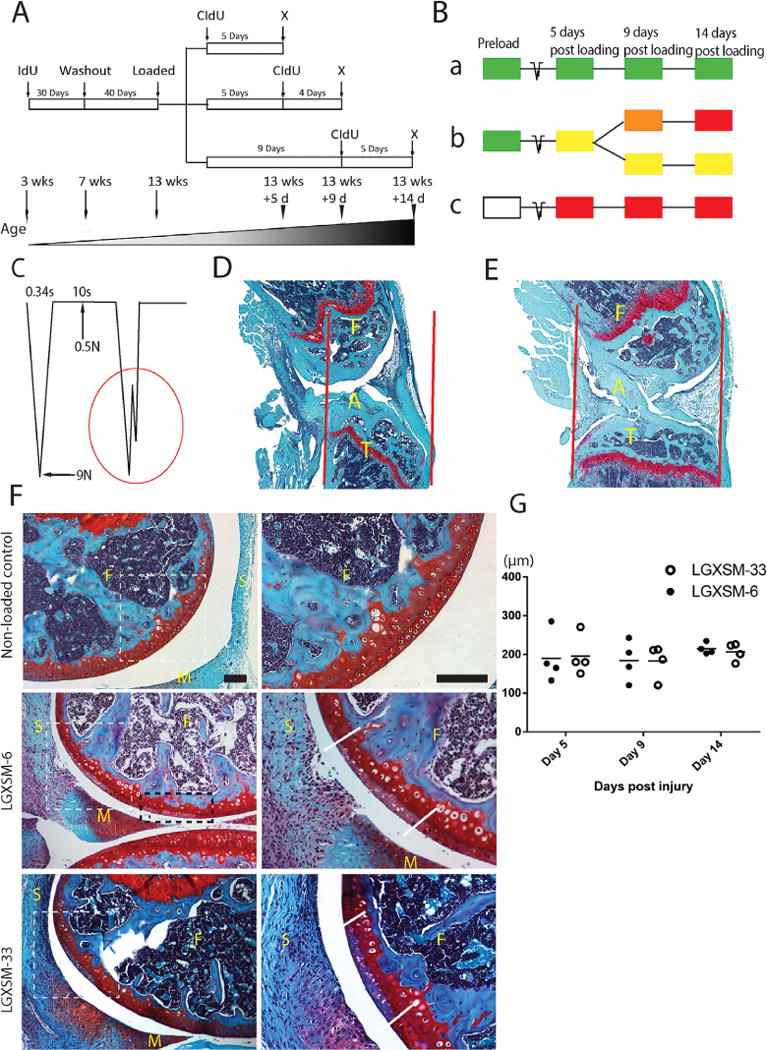
Non-invasive tibial compression and acute pathological changes in articular cartilage. (A) A sketch of double nucleoside cell-labeling procedure and (B) labeling options with three possible outcomes (a, b, and c). (a) IdU-positive cells will not proliferate and retain IdU (green); (b) IdU-positive cells will proliferate and acquire CIdU (red) and gradually lose IdU, hence appearing yellow, orange, or red; (c) Unlabeled IdU-negative cells (white) will proliferate and acquire CIdU (red); x = sacrifice. (C) A schematic diagram of loading cycle with basal maintenance force, peak force, time-interval between loading episodes and a typical indication of ACL rupture (red circle). (D) Compression-induced ACL rupture indicated by disrupted alignment of the ligament fibrils (A=ACL, F=femur, T=tibia), increased joint gap, and the dislocation of the tibia in relation to the femur; red lines indicate joint alignment. (E) Non-loaded control knee with intact ACL (A=ACL, F=femur, T=tibia). (F) Representative images of cartilage injury site of the lateral femoral condyle (left panel lower magnification, right panel higher magnification of white dotted line box), showing loss of Safranin-O staining and disappearance of normal nuclear staining. White dotted line box indicates the primary injury site, black dotted line box shows secondary injury site, parallel solid white lines indicate the injury border. F=femur, M=meniscus, S=synovium. Bar=100 μm. (G) Mean cartilage injury length showing no significant difference between the strains or time points. (n=4 each time point and each strain).
Statistical Analysis
All scores were evaluated by two blinded observers (XD, MFR). Data were analyzed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA) and are shown as mean ± standard error of the mean (S.E.M.). Histological scores and TUNEL counting results were analyzed by two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. The level of statistical significance was set at 0.05.
RESULTS
All the mice exhibited normal cage activity after loading and we did not observe any notable change in their movement and eating/drinking habit. We also did not observe any obvious swelling of the loaded joints. All the mice indicated in Materials and Methods were used for analysis because they had confirmed ACL rupture.
Effect of Loading on ACL
Rupture of ACL was indicated by an abrupt drop in loading wave (force) (Fig. 1C) during compression as well as by anterior dislocation of tibia (Fig. 1D and E). With regards to ACL rupture occurring solely during load-removal has not been fully determined. The numbers have not been compiled but ruptures occur before or after peak force is reached. It is possible that ruptures of the ACL during unloading may occur because the peak load needed to be reached. Therefore, if the applied load had been higher, the rupture may have occurred during the ramping (loading) portion of the cycle. Our unpublished data (MF Rai, X Duan, JD Quirk, LJ Sandell) suggest that the assessment of ACL rupture by the drop-in force is very reliable because it was found to be highly correlated with ex vivo tissue damage observed by magnetic resonance imaging.
Effect of Loading on Cartilage: Site and Magnitude of Injury
We observed multiple injuries in the non-calcified layer of cartilage of the lateral femoral condyles (Fig. 1F). The primary injury, which was localized at the posterior aspect of lateral femoral condyle, appeared to occur due to direct compression (femurtibia contact). Secondary injury, at multiple adjacent sites, was anterior to primary injury and was likely the outcome of continued compression following ACL rupture. Whether primary or secondary, all injury sites were characterized by loss of Safranin-O staining (i.e., proteoglycan loss) and chondrocyte apoptosis. No injury was observed in contralateral non-loaded control knees.
The average length of primary injury as determined by the length of Safranin-O loss remained the same over time in either strain (Fig. 1G), thus indicating that both mouse strains have similar magnitude of cartilage lesions and the lesions do not appear to progress from day 5 to 14.
Cell Apoptosis at the Site of Injury
All the enlarged images were taken from the areas indicated in Supplementary Figure S1. At day 5 post-injury, we identified apoptotic chondrocytes in the injured cartilage (Fig. 2A, day 5, arrows). The apoptotic cells in injured cartilage were characterized by TUNEL-positive (green) and DAPI-negative staining. At days 9 and 14 the calcified layer in the injured area remained the same as day 5, however the non-calcified cartilage in injured area was nearly devoid of cells, that is, neither TUNEL- nor DAPI-stained (Fig. 2A, day 9 and 14). Chondrocyte apoptosis was randomly seen in the calcified cartilage of non-loaded knees but not in the non-calcified zone. All non-loaded cells were positive for DAPI staining (Fig. 2A, Non-loaded control). Supplementary Figure S2 shows spilt channels of Figure 2A. Finally, we did not observe any significant difference for TUNEL-positive cells between LGXSM-6 (13.50 ± 1.85 cells per 1.92 million square pixels) and LGXSM-33 (13.33 ± 1.19 cells per 1.92 million square pixels) at day 5 post-injury. Quantification of TUNEL-positive cells is provided in Supplementary Figure S3.
Figure 2.
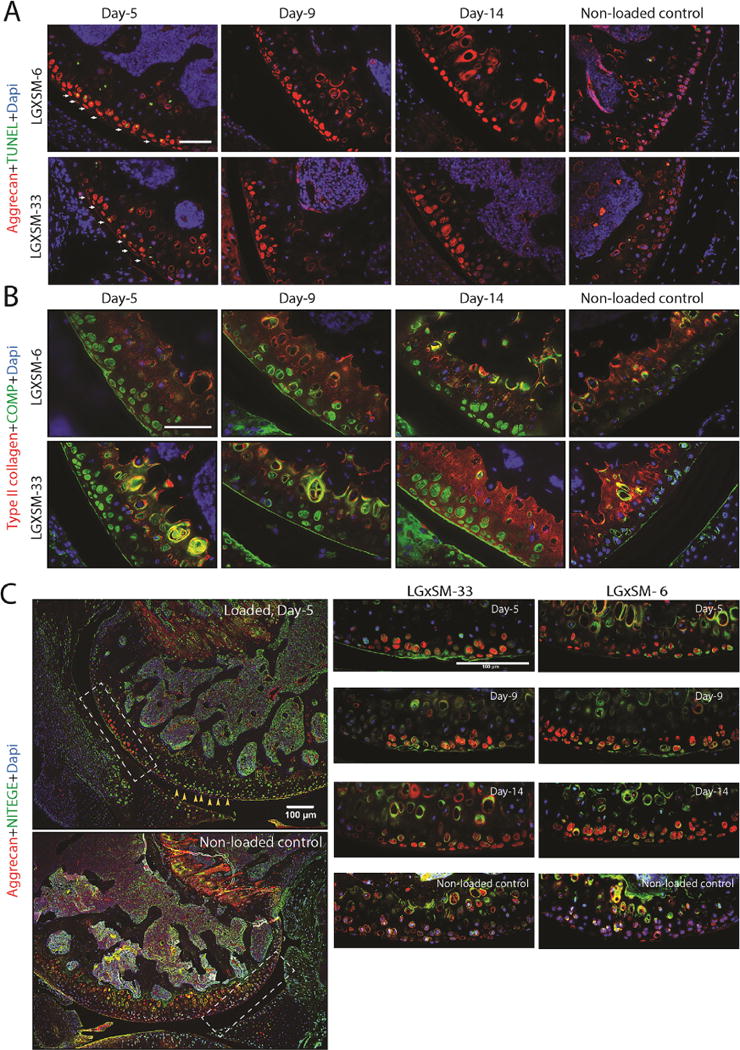
Chondrocyte apoptosis and distribution pattern of extracellular matrix proteins. (A) TUNEL assay identified apoptotic chondrocytes (green staining, white arrows) in the injured cartilage area in both strains at day 5 but not at day 9 or 14. Aggrecan (red staining) was found around the chondrocytes in the intact area and in the lacuna left by the apoptotic chondrocytes in the injured area (Bar=50 μm). (B) COMP (green staining) had the same distribution pattern as aggrecan in the injured region but could be seen less than aggrecan along the adjacent healthy cartilage surface. Type-II collagen (red staining) was uniformly found along the whole cartilage surface (Bar=50 μm). (C) The left panel shows typical images of the loaded and non-loaded control knees. NITEGE (green staining) was mainly found in the calcified layer of cartilage (hypertrophic chondrocytes) in non-loaded knees. However immunoreactivity of NITEGE was stronger in non-injured cartilage area of injured knees at all time points after injury (yellow arrows). In the injured cartilage, NITEGE was mainly in the extracellular matrix not in the dead chondrocytes (indicated by aggregated aggrecan staining without DAPI staining). Aggrecan staining (red) was undertaken to identify injury site. The area indicated by white dotted line boxes is shown at higher magnification. Bar=100 μm.
Pattern of Extracellular Matrix Proteins Distribution and CD44 Expression
The following pattern of extracellular matrix protein distribution and CD44 expression was consistent across all time points and in both mouse strains. We observed an increased presence of aggrecan (Fig. 2A, red) and COMP (Fig. 2B, green) proteins inside the empty lacunae at the site of injury. In contrast, in the non-injured area, a smooth and intense pericellular distribution pattern of these proteins was observed.
In contrast, type-II collagen protein was uniformly distributed along the entire surface of cartilage without any notable difference between injured and non-injured areas. Consistently observed by us and others, the intensity of type-II collagen protein was higher in the calcified cartilage compared to the non-calcified cartilage (Fig. 2B, red).
Previously, we reported that, in the impacted mouse knee, aggrecan was found in the injured cells, co-localizing with the TUNEL staining. In the current study, we further investigated this observation, asking whether this intracellular aggrecan was intact or fragmented, resulting in internalization, and whether it was newly synthesized. With regard to proteoglycan degradation, immunostaining for the C-terminus of the aggrecan (COOH-) cleavage product, NITEGE, revealed a higher degradation of aggrecan matrix in the intact cartilage (adjacent to the injured area) in the loaded knees compared to control knees (Fig. 2C, yellow arrows). In the injured area, NITEGE was mostly detected in extracellular matrix and not in the injured chondrocytes (Fig. 2C, green), where aggrecan and Safranin-O staining was lost. This finding indicated that injured chondrocytes did not take up the cleaved aggrecan molecules while intact chondrocytes are responding to the injury by taking up the fragmented aggrecan.
In order to determine the mechanism of aggrecan protein redistribution, we co-stained CD44 and aggrecan. CD44, a chondrocyte surface marker and a receptor for hyaluronic acid, is required to complete the process of aggrecan fragment internalization. In the intact cartilage area, we observed that CD44 protein was abundant along the plasma membrane of hypertrophic chondrocytes in the calcified layer and less so in the cells of the non-calcified zone (Fig. 3A). However, as early as day 5 after loading CD44 intensity was higher in the intact cartilage around the injured area, in both calcified and non-calcified layers, and it persisted until day 14. No CD44 expression was seen in the injured chondrocytes in any of the mouse strains or at any time point. The results from chondrocyte apoptosis, extracellular matrix proteins distribution and CD44 expression are summarized in Figure 3B.
Figure 3.
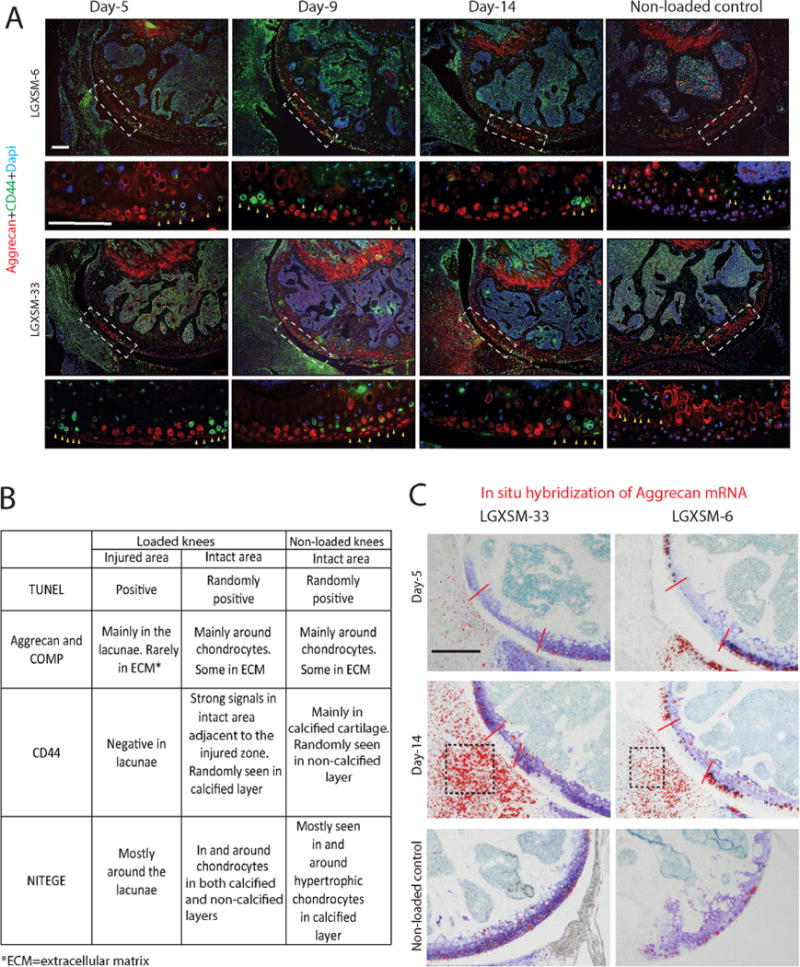
CD44 immunofluorescence and in situ hybridization of aggrecan mRNA. (A) The areas indicated by white dotted line boxes in the upper panels are shown at higher magnification in the lower panels. CD44 (green) was seen around hypertrophic chondrocytes in non-loaded control knees (yellow arrows, the right panel) and in the chondrocytes immediately around the injured zone (yellow arrows, left three panels). There was no difference in the pattern of CD44 expression between the two mouse strains. Aggrecan staining (red) was undertaken to identify injury site. Bars=100 μm. (B) Summary of changes in extracellular matrix proteins in the injured and adjacent intact areas of loaded knees and in the intact area of non-loaded knees. (C) Aggrecan mRNA, detected by in situ hybridization, was only present in the intact area of cartilage and the cartilage of non-loaded knees at all time points in both strains (red color dots show positive hybridization signals, red lines show the injured area). Aggrecan mRNA was also seen in the synovium at day 14 post-injury in both strains (black boxes). Bar = 100 μm.
In Situ Analysis of Aggrecan mRNA
In order to confirm that the aggrecan was already present in the cartilage, we probed for neo-synthesis of aggrecan mRNA. Aggrecan mRNA was detected only in the intact area of cartilage and the cartilage of non-loaded control knees at all time points in both strains (Fig. 3C). This observation indicated that up to 14 days post-injury, no cells were synthesizing aggrecan in the injured cartilage area. Chondrocytes in the intact area might be reacting to the injury and trying to produce aggrecan proteins to repair the injured cartilage. The absence of aggrecan mRNA in the injured cartilage indicated the increased presence of aggrecan protein we observed by immunostaining was not due to newly synthesized aggrecan proteins. Besides cartilage, aggrecan mRNA was also seen in the synovium at day 14 post-injury in both strains (Fig. 3C, black boxes), a finding consistent with Safranin-O staining.
Impact of Loading on Synovium: Development of Synovitis and Ectopic Chondrogenesis
Normal synovium in control knees appeared as a thin, smooth lining, with a 1–2 cell thickness intimal surface (Fig. 4, Safranin-O staining). In contrast, significant synovial cell proliferation, intimal lining cell hyperplasia and inflammatory-cell infiltration were seen as early as day 5 after loading in both strains. At day 9, loaded joints in LGXSM-33 strain exhibited proliferating synovial tissue, characterized by numerous clusters of cells with rounded, enlarged nuclei (Fig. 4, Safranin-O staining). Furthermore, there was a localized decrease in cell density and increase in proteoglycan matrix staining at the sub-intimal area. Immunofluorescence of aggrecan and COMP was intense while only weak type-II collagen staining was found (Fig. 4, day 9). At day 14, circular nodules with less cellularity but more localized type-II collagen/COMP co-staining were observed in the synovium (Fig. 4 white box). LGXSM-6 mice did not show such nodules at any time points (Fig. 4), however at day 14, increased synovial proliferation along with higher aggrecan and COMP intensities were seen. Synovitis was significantly higher in the loaded knees compared to control knees at all time points and it increased over time (Fig. 5A). Of note, there was significantly higher synovitis in LGXSM-33 than LGXSM-6 at day 9 (p=0.008) and day 14 (p=0.008) but not at day 5 (Fig. 5B). These results indicate that both mouse strains undergo similar changes in synovium initially, but LGXSM-33 surpasses LGXSM-6 later on (i.e., day 14) with the onset of gene expression characteristic of chondrogenesis.
Figure 4.
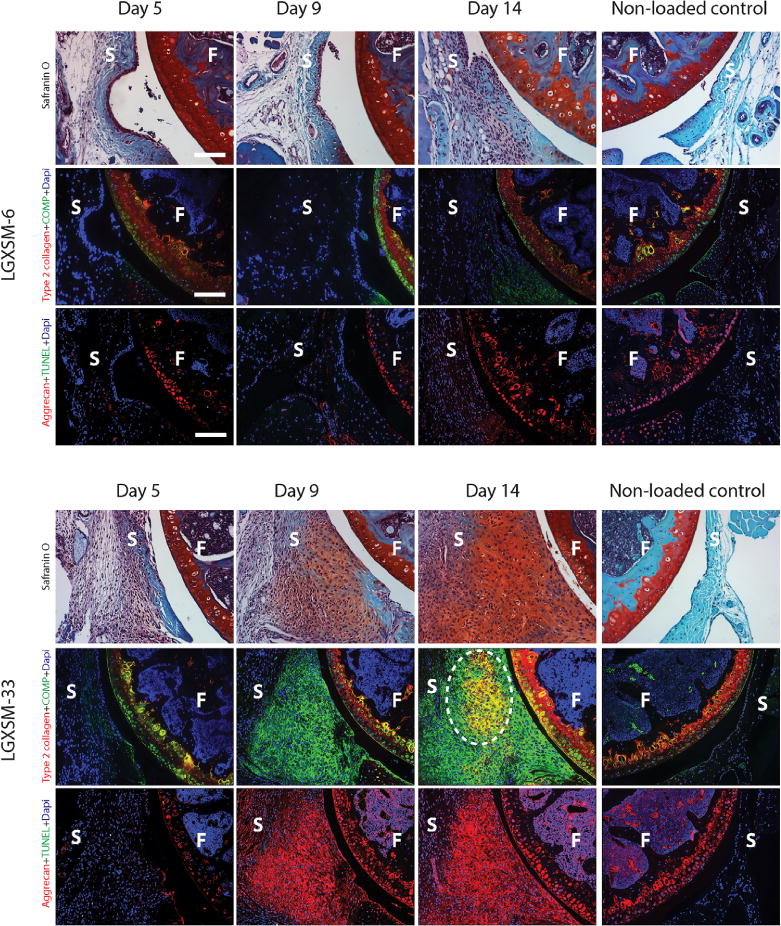
Synovial response: comparison of LGXSM-33 and LGXSM-6. Horizontal panels in each strain, from top to bottom are Safranin-O stain, immunostaining for type 2 collagen + COMP and aggrecan + TUNEL. As early as day 5 post-injury, significant synovial cell proliferation and intimal lining cell hyperplasia were observed in both strains (Safranin-O stain) compared to the non-loaded control knees (the far right column). LGXSM-33 showed higher expression of aggrecan (red) and COMP (green) in synovium than LGXSM-6 at day 9 and 14 post-injury. LGXSM-33 also developed ectopic chondrogenic nodules on day 14 after injury. These nodules were type-II collagen positive (white dotted line circle). F=femur; S=synovium; bars=50 μm.
Figure 5.
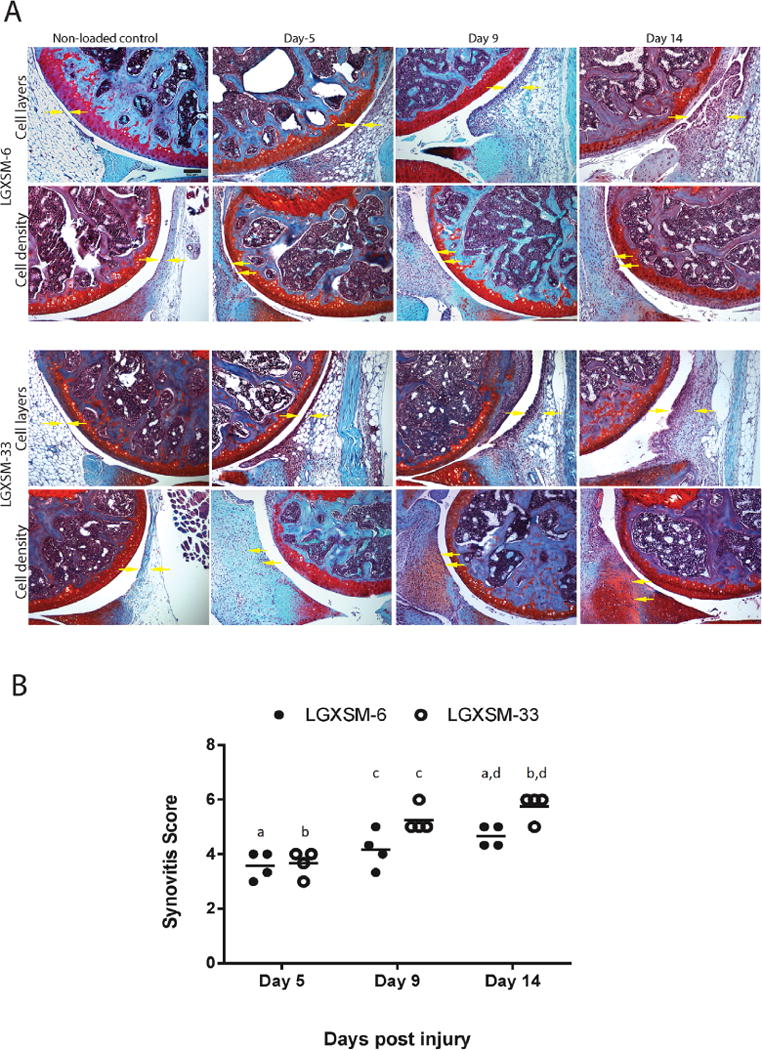
Synovitis assessment. (A) Enlargement of the synovial lining cell layer and density of the cells in the synovial stroma (yellow arrows) were individually assessed. Bar=100 μm. (B) Quantification of synovitis score at day 5, 9, and 14 post-injury indicated a trend of increased synovitis overtime following injury. There was significantly higher synovitis in LGXSM-33 compared to LGXSM-6 at day 9 and 14 post-injury. Graphs represent mean ± SEM. p-value was set at <0.05. Identical lowercase letters represent statistical difference across groups (n=4 each time point and each strain).
Source of Cells Responding to Loading
In control knees, only a small number of CIdU-positive cells (red, slow-cycling) scattered in synovium and subchondral bone marrow were observed. They likely represent normal cell proliferation in these tissues. In loaded knees, the number of CIdU-positive cells was substantially increased in the synovium and subchondral bone marrow. In chondrocytes, no CIdU-positive staining was found in either loaded or control knees. In contrast, slow-cycling, long-term retained (IdU–positive, green) cells were detected in the synovium, subchondral bone marrow and cartilage (Fig. 6).
Figure 6.
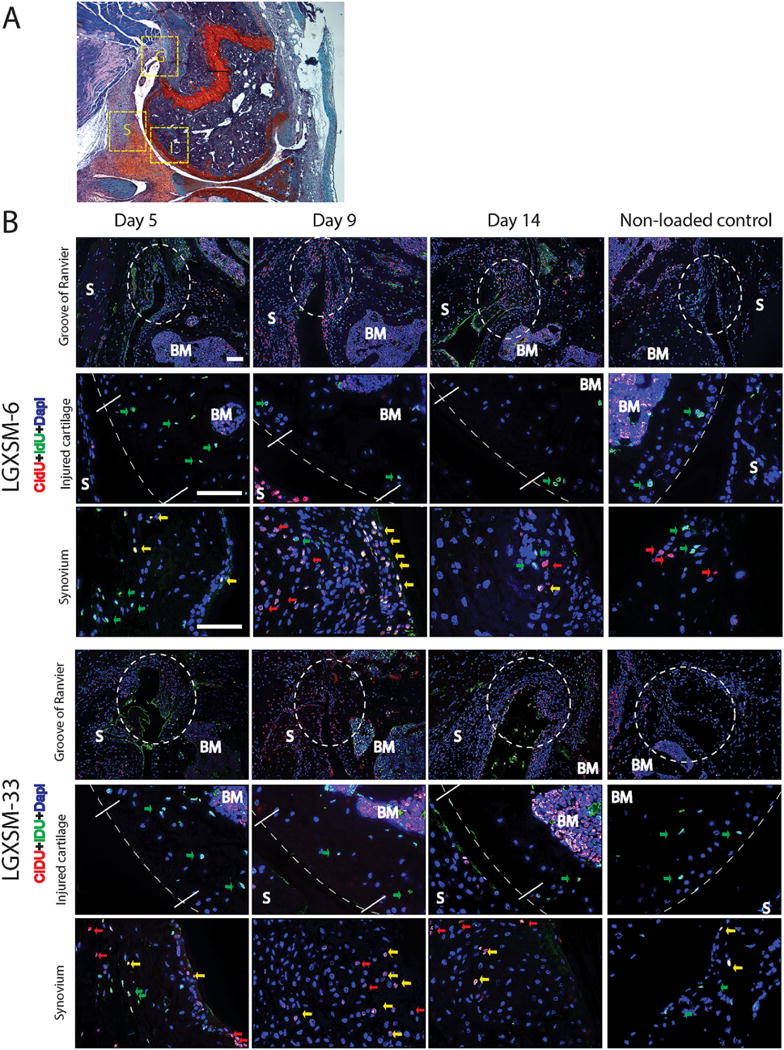
Cells responding to injury. (A) Safranin-O stained image showing areas used for the immunofluorescent images. (B) Horizontal panels from top to bottom in each strain are images taken from Groove of Ranvier (G), injured cartilage (I) and synovium (S) as indicated in Figure A. Long-term-retaining, slow-cycling IdU-positive cells (green arrows) were detected in cartilage (middle panels, dotted lines indicate the cartilage or the joint surface) and synovium (lower panels) in both strains at all time points. At 5, 9, and 14 days after injury, there was a marked proliferation of CIdU-positive cells (red arrows and yellow arrows) in the synovium (lower panels in each strain) and Groove of Ranvier (upper panels in each strain, white dotted circle), compared to non-loaded control knees in the right column. No responding cells were detected at the site of cartilage injury (middle panels). S=synovium, BM=bone marrow, G=Groove of Ranvier, I=injured cartilage. Yellow arrows indicate co-localization of IdU- and CIdU-positive cells. Bars=50 μm.
At day 5 after loading, an accumulation of IdU-and CIdU-double-positive cells were found in the synovium as well as the Groove of Ranvier (a wedge-shaped groove adjacent to the growth plate) in the injured knees of both strains (Fig. 6, day 5). This accumulation was still detectable 14 days after injury, however, with less IdU-positive cells (Fig. 6, day 14). No remarkable accumulation of CIdU-positive cells was found in control knees (Fig. 6, non-loaded control).
No fast-cycling cells were found in the injured cartilage and there were no differences in nucleoside labeling detected in the subchondral bone marrow between uninjured controls and injured mice in both strains, at all time points tested. These results indicate that the cells responding to injury arise from synovium and the Groove of Ranvier but not from cartilage or subchondral bone. There were labeled cells in subchondral bone marrow, but they did not migrate to cartilage (i.e., no CIdU-positive cells were found in cartilage) within the acute period after injury.
DISCUSSION
Here, we investigated the different responses of the two recombinant inbred mouse strains to the non-invasive mechanical injury in the early time points. We found a similar reaction in both strains in the context of cartilage phenotype and cell response at early time points (up to 14 days) after injury. However, a significant difference in synovitis was seen after day 9 post-injury suggesting strain differences in the synovial response.
Our loading model was found to be very reproducible because it resulted in an injury pattern in the genetic mouse strains in a manner similar to what we have previously reported in C57BL/6J4 and others have reported in CBA20 and FVB21 strains. The cartilage injury neither progressed to degeneration nor resulted in healing over the 14-day study period. These results were not unexpected because the injury model employed in the current study was different from what we have used previously9 in which subchondral bone was penetrated by needle to induce “full-thickness” lesions. In the prior model, healing occurred over a period of 16 weeks after injury, whereas in the current model analysis was restricted to 2 weeks post-injury. Studies have shown that partial thickness cartilage injuries do not heal or they heal poorly and over a much extended time period.22 The injury produced here did not show macroscopic damage in the cartilage however we could identify the impact zone by the loss of Safranin-O staining and lack of nuclei or, if an early time, the presence of TUNEL-positive DNA.
Studies have shown an acute chondrocyte necrosis and apoptosis in response to blunt trauma to cartilage.23 Our findings of apoptosis in the injured cartilage are in line with our previous observations4 and observations made by Aigner and colleagues.24 The programmed cell death and substantially low number of living chondrocytes in the calcified layer do not seem to impair cartilage under normal conditions.12,13 Therefore, we only compared chondrocyte apoptosis in the impacted area of non-calcified cartilage layer between the two strains, which appears to be a direct consequence of loading. We hypothesized that there would be less chondrocyte apoptosis in healers after loading and a relatively smaller area of cartilage injury, but we failed to observe differences in number of apoptotic cells and in the length of cartilage lesions. These results suggest that genetic differences do not necessarily link to greater susceptibility to mechanical damage in cartilage. Another study that used Str/ort mice25 concluded that genetic susceptibility to OA in these mice is not apparently related to greater cartilage vulnerability to trauma. It is also interesting to note that apoptosis did not progress over time (up to 14 days after loading) indicating again that chondrocytes were injured at the time of trauma and no new apoptosis occurred as a reaction to the dead cells.
Similar to C57BL/6J mice,4 the distribution pattern of several extracellular matrix proteins changed in injured knees in both strains. The mechanism of this change had not been fully studied in vivo, so we used aggrecan as an example in our study to propose a possible mechanism. Intact aggrecan is observed inside of the dead cell lacunae. However, some aggrecan in the extracellular matrix is degraded by proteoglycanases such as ADAMTS-5. Studies26,27 have found that upon aggrecan cleavage by ADAMTS-5, the C-terminus NITEGE fragments of aggrecan are internalized into the chondrocyte(s) by the hyaluronic acid cell surface receptor CD44. We found more immunostaining of NITEGE around the lacunae of dead chondrocytes in the injured zone (consistent with less Safranin-O staining) but failed to locate any CD44-positive staining at any time point. This indicates that in an acute cartilage injury, chondrocytes are not able to internalize the fragments since there is no CD44. In contrast, chondrocytes with positive CD44 signals were seen only in the area adjacent to the impacted site. The likely reason is that the mechanical loading itself injured chondrocytes and caused apoptosis. Yamamoto and colleagues28 demonstrated that dead, but not living cells, stimulated ADAMTS-5 activity. Therefore, the death of chondrocytes could stimulate aggrecan cleavage. Increased aggrecan fragments (i.e., more NITEGE staining in extracellular matrix in the injured area) called for more CD44 expression in living adjacent chondrocytes, as a receptor to fulfill the process of internalization of aggrecan fragments. The extracellular proteins were degraded (positive neoepitope NITEGE) due to mechanical impact and chondrocyte death. The apoptotic chondrocytes have lost their normal structure (no CD44 on the cell surface) and are unable to uptake or make new extracellular matrix proteins (i.e., no aggrecan mRNA was detected).
Synovial tissues are important to the overall health of the synovial joint. Previous studies have shown that changes in the synovial tissues could contribute to progression or protection of OA.4,29,30 It is increasingly appreciated that some degree of synovitis may be observed in both early and late OA, including synovial hypertrophy and hyperplasia with an increased number of lining cells, often accompanied by infiltration of the sub-lining tissue with scattered foci of inflammatory cells and eventually osteophyte formation.31–33 We observed a rapid and considerable synovial response following loading, as early as 5 days post-injury, in both strains, which extended through the entire 14-day time period. This indicates that both strains have the same initiating response of synovitis in the early days after injury. However the non-healer (LGXSM-33) strain then progressed faster. By day 9, the cells were synthesizing cartilaginous extracellular matrix molecules throughout the synovial tissue (COMP and aggrecan) and by 14 days, type-II collagen positive chondrogenic nodules were seen in the non-healer strain. It is thought that mesenchymal stem cells in the synovium are stimulated to proliferate and undergo chondrogenesis and differentiate to mature hypertrophic chondrocytes where vascular invasion occurs.34 Our findings of double-labeled cells and chondrogenesis in synovium after injury are consistent with their hypothesis. The synovial calcifications have been investigated in detail elsewhere.35
The possibility that intrinsic cells respond to injury and may participate in regeneration of cartilage has been extensively studied. Kurth and others have shown that the intra-articular tissues (e.g., synovium) contain mesenchymal stem cells in humans as well as in mice.18,36,37 From our non-invasive model, we confirmed the result of Kurth et al.18 in vivo, that IDU-positive mesenchymal stem cell (MSC)-like cells were found in synovium at all time points without any strain differences. We also found that the slow-cycling cells were in the Groove of Ranvier and became transit-amplifying cells after injury. This is consistent with Karlsson et al.38 who have demonstrated that the Groove of Ranvier is a pool of stem cells. Recent studies have shown that the bone marrow cavity underneath the subchondral bone may provide mesenchymal stromal cells to damaged cartilage to support its repair.39,40 However, we consider that the bone-marrow derived MSCs were less possible to migrate to the injured cartilage in our study, due to our partial-thickness cartilage injury model. Studies41,42 have shown the presence of stem/progenitor cells in the superficial layer in human cartilage, however it is unknown if this layer supports cartilage renewal or can provide cells for repair of cartilage. Pretzel and co-workers43 have reported that cartilage by itself may contain some mesenchymal progenitor cells, which are located in the superficial and middle zones of cartilage. But they did not show how long it took these cells to be involved in cartilage repair. Another study by Eltawil et al.44 also failed to detect proliferating cells in the cartilage adjacent to injury at 4-weeks following injury by using a joint surface injury model in DBA/1 mice. Chondrocyte proliferation has been reported to take place following injury in vitro,45 however such a model does not mimic the daily joint motions after injury, especially in the mice with ACL rupture. In agreement with Kurth et al.,18 we did not observe any CIDU-positive fast reacting cells in the injured cartilage in either strain at any time point. We propose that during the acute phase after impact (up to 14 days post-injury), no progenitor cells in cartilage responded to the injury even in the “healer” strain. However both strains demonstrated a synovial response. The independent response of cartilage and synovium has also been shown by us for mice lacking the chemokine receptor CCR5.46
CONCLUSION
Our findings indicate that both healer and non-healer mouse strains are vulnerable to 9N impact loading. Chondrocyte apoptosis occurs in the cartilage at an early time point while injury responding cells are mainly located in the synovium and the Groove of Ranvier (but not in the cartilage). We show, for the first time, the distribution of hyaluronan receptor (CD44) in healthy and injured knees in vivo in intact cartilage surrounding the impact zone. Interestingly, the cartilage response appears to be the same between the two strains, while there is a difference in the synovial response. We cannot predict what long-term outcomes this difference may lead to. Since the in vivo model described here mimics one type of blunt human knee injury resulting in an ACL tear, these results will provide a platform to compare mice with different genetic backgrounds and/or modified genes and for future treatment strategies aiming at circumventing the pathological changes following injurious loading but before the development of clinical PTOA.
Supplementary Material
Acknowledgments
We acknowledge with thanks the important technical assistance by Crystal Idleburg and Elizabeth DeLassus. These studies were supported by R01-AR063757 (Sandell), P30-AR057235 (Sandell), R01-AR063757-02S1 (Sandell, Rai), R01-AR047867 (Silva), and Pilot & Feasibility Studies grant (Rai) from P30-AR057235 (Core Center for Musculoskeletal Biology and Medicine). Dr. Rai is also supported through Pathway to Independence Award (1K99-AR064837) from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (National Institutes of Health). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Grant sponsor: National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) at National Institutes of Health; Grant numbers: R01-AR063757, P30-AR057235, R01-AR063757-02S1, R01-AR047867, 1K99-AR064837.
Footnotes
Conflict of interest: Dr. Sandell is Editor in Chief of the Journal of Orthopaedic Research. Dr. Sandell owns stock or stock options in ISTO Technologies. All other authors have nothing to disclose. None of the authors has any competing interests with regard to this manuscript.
AUTHORS’ CONTRIBUTIONS
All authors were involved in drafting and revision of the manuscript and all authors approved the final version to be published. MFR had full access to all of the data in the study and takes responsibility for the integrity and the accuracy of the data analysis. LJS, XD, and MFR: Study conception and design. XD, MFR, NH, DP, WL, and MJS: Acquisition of data. XD, MFR, NH, MJS, DP, WL, and LJS: Analysis and interpretation of data.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article.
References
- 1.Whittaker JL, Woodhouse LJ, Nettel-Aguirre A, et al. Outcomes associated with early post-traumatic osteoarthritis and other negative health consequences 3–10 years following knee joint injury in youth sport. Osteoarthritis Cartilage. 2015;23:1122–1129. doi: 10.1016/j.joca.2015.02.021. [DOI] [PubMed] [Google Scholar]
- 2.Lohmander LS, Englund PM, Dahl LL, et al. The long-term consequence of anterior cruciate ligament and meniscus injuries: osteoarthritis. Am J Sports Med. 2007;35:1756–1769. doi: 10.1177/0363546507307396. [DOI] [PubMed] [Google Scholar]
- 3.Catterall JB, Stabler TV, Flannery CR, et al. Changes in serum and synovial fluid biomarkers after acute injury ( NCT00332254) Arthritis Res Ther. 2010;12:R229. doi: 10.1186/ar3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu P, Holguin N, Silva MJ, et al. Early response of mouse joint tissue to noninvasive knee injury suggests treatment targets. Arthritis Rheumatol. 2014;66:1256–1265. doi: 10.1002/art.38375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christiansen BA, Anderson MJ, Lee CA, et al. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthritis Cartilage. 2012;20:773–782. doi: 10.1016/j.joca.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Spector TD, Cicuttini F, Baker J, et al. Genetic influences on osteoarthritis in women: a twin study. BMJ. 1996;312:940–943. doi: 10.1136/bmj.312.7036.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sandell LJ. Etiology of osteoarthritis: genetics and synovial joint development. Nat Rev Rheumatol. 2012;8:77–89. doi: 10.1038/nrrheum.2011.199. [DOI] [PubMed] [Google Scholar]
- 8.Hashimoto S, Rai MF, Janiszak KL, et al. Cartilage and bone changes during development of post-traumatic osteoarthritis in selected LGXSM recombinant inbred mice. Osteoarthritis Cartilage. 2012;20:562–571. doi: 10.1016/j.joca.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rai MF, Hashimoto S, Johnson EE, et al. Heritability of articular cartilage regeneration and its association with ear wound healing in mice. Arthritis Rheum. 2012;64:2300–2310. doi: 10.1002/art.34396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rai MF, Schmidt EJ, McAlinden A, et al. Molecular insight into the association between cartilage regeneration and ear wound healing in genetic mouse models: targeting new genes in regeneration. G3. 2013;3:1881–1891. doi: 10.1534/g3.113.007302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis JS, Hembree WC, Furman BD, et al. Acute joint pathology and synovial inflammation is associated with increased intra-articular fracture severity in the mouse knee. Osteoarthritis Cartilage. 2011;19:864–873. doi: 10.1016/j.joca.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farnum CE, Turgai J, Wilsman NJ. Visualization of living terminal hypertrophic chondrocytes of growth plate cartilage in situ by differential interference contrast microscopy and time-lapse cinematography. J Orthop Res. 1990;8:750–763. doi: 10.1002/jor.1100080517. [DOI] [PubMed] [Google Scholar]
- 13.Hunziker EB, Herrmann W, Schenk RK, et al. Cartilage ultrastructure after high pressure freezing, freeze substitution, and low temperature embedding. I. Chondrocyte ultrastructure-implications for the theories of mineralization and vascular invasion. J Cell Biol. 1984;98:267–276. doi: 10.1083/jcb.98.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Lima DD, Hashimoto S, Chen PC, et al. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis Cartilage. 2001;9:712–719. doi: 10.1053/joca.2001.0468. [DOI] [PubMed] [Google Scholar]
- 15.Patra D, Xing X, Davies S, et al. Site-1 protease is essential for endochondral bone formation in mice. J Cell Biol. 2007;179:687. doi: 10.1083/jcb.200708092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glumoff V, Savontaus M, Vehanen J, et al. Analysis of aggrecan and tenascin gene expression in mouse skeletal tissues by northern and in situ hybridization using species specific cDNA probes. Biochim Biophys Acta. 1994;1219:613–622. doi: 10.1016/0167-4781(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 17.Maslov AY, Barone TA, Plunkett RJ, et al. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurth TB, Dell’accio F, Crouch V, et al. Functional mesenchymal stem cell niches in adult mouse knee joint synovium in vivo. Arthritis Rheum. 2011;63:1289–1300. doi: 10.1002/art.30234. [DOI] [PubMed] [Google Scholar]
- 19.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mole Cell Biol. 2013;14:329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulet B, Hamilton RW, Shefelbine S, et al. Characterizing a novel and adjustable noninvasive murine joint loading model. Arthritis Rheum. 2011;63:137–147. doi: 10.1002/art.27765. [DOI] [PubMed] [Google Scholar]
- 21.Onur TS, Wu R, Chu S, et al. Joint instability and cartilage compression in a mouse model of posttraumatic osteoarthritis. J Orthop Res. 2014;32:318–323. doi: 10.1002/jor.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzgerald J, Rich C, Burkhardt D, et al. Evidence for articular cartilage regeneration in MRL/MpJ mice. Osteoarthritis Cartilage. 2008;16:1319–1326. doi: 10.1016/j.joca.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 23.Goodwin W, McCabe D, Sauter E, et al. Rotenone prevents impact-induced chondrocyte death. J Orthop Res. 2010;28:1057–1063. doi: 10.1002/jor.21091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aigner T, Hemmel M, Neureiter D, et al. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001;44:1304–1312. doi: 10.1002/1529-0131(200106)44:6<1304::AID-ART222>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 25.Poulet B, Westerhof TA, Hamilton RW, et al. Spontaneous osteoarthritis in Str/ort mice is unlikely due to greater vulnerability to mechanical trauma. Osteoarthritis Cartilage. 2013;21:756–763. doi: 10.1016/j.joca.2013.02.652. [DOI] [PubMed] [Google Scholar]
- 26.Embry JJ, Knudson W. G1 domain of aggrecan cointernalizes with hyaluronan via a CD44-mediated mechanism in bovine articular chondrocytes. Arthritis Rheum. 2003;48:3431–3441. doi: 10.1002/art.11323. [DOI] [PubMed] [Google Scholar]
- 27.Fosang AJ, Last K, Stanton H, et al. Generation and novel distribution of matrix metalloproteinase-derived aggrecan fragments in porcine cartilage explants. J Biol Chem. 2000;275:33027–33037. doi: 10.1074/jbc.M910207199. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto K, Troeberg L, Scilabra SD, et al. LRP-1-mediated endocytosis regulates extracellular activity of ADAMTS-5 in articular cartilage. FASEB J. 2013;27:511–521. doi: 10.1096/fj.12-216671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wassilew GI, Lehnigk U, Duda GN, et al. The expression of proinflammatory cytokines and matrix metalloproteinases in the synovial membranes of patients with osteoarthritis compared with traumatic knee disorders. Arthroscopy. 2010;26:1096–1104. doi: 10.1016/j.arthro.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 30.Ostalowska A, Kasperczyk S, Kasperczyk A, et al. Oxidant and anti-oxidant systems of synovial fluid from patients with knee post-traumatic arthritis. J Orthop Res. 2007;25:804–812. doi: 10.1002/jor.20357. [DOI] [PubMed] [Google Scholar]
- 31.Benito MJ, Veale DJ, FitzGerald O, et al. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64:1263–1267. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rollin R, Marco F, Jover JA, et al. Early lymphocyte activation in the synovial microenvironment in patients with osteoarthritis: comparison with rheumatoid arthritis patients and healthy controls. Rheumatol Int. 2008;28:757–764. doi: 10.1007/s00296-008-0518-7. [DOI] [PubMed] [Google Scholar]
- 33.Kawaguchi H. Mechanism underlying osteoarthritis induced by mechanical stress on joint cartilage. Clin Calcium. 2008;18:1278–1286. [PubMed] [Google Scholar]
- 34.van der Kraan PM, van den Berg WB. Osteophytes: relevance and biology. Osteoarthritis Cartilage. 2007;15:237–244. doi: 10.1016/j.joca.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Rai MF, Schmidt EJ, Hashimoto S, et al. Genetic loci that regulate ectopic calcification in response to knee trauma in LG/J by SM/J advanced intercross mice. J Orthop Res. 2015;33:1412–1423. doi: 10.1002/jor.22944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Futami I, Ishijima M, Kaneko H, et al. Isolation and characterization of multipotential mesenchymal cells from the mouse synovium. PLoS ONE. 2012;7:e45517. doi: 10.1371/journal.pone.0045517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Bari C, Dell’Accio F, Tylzanowski P, et al. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44:1928–1942. doi: 10.1002/1529-0131(200108)44:8<1928::AID-ART331>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 38.Karlsson C, Thornemo M, Henriksson HB, et al. Identification of a stem cell niche in the zone of Ranvier within the knee joint. J Anat. 2009;215:355–363. doi: 10.1111/j.1469-7580.2009.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson K, Zhu S, Tremblay MS, et al. A stem cell-based approach to cartilage repair. Science. 2012;336:717–721. doi: 10.1126/science.1215157. [DOI] [PubMed] [Google Scholar]
- 40.Koelling S, Kruegel J, Irmer M, et al. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell. 2009;4:324–335. doi: 10.1016/j.stem.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Muinos-Lopez E, Rendal-Vazquez ME, Hermida-Gomez T, et al. Cryopreservation effect on proliferative and chondrogenic potential of human chondrocytes isolated from superficial and deep cartilage. Open Orthop J. 2012;6:150–159. doi: 10.2174/1874325001206010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tallheden T, Brittberg M, Peterson L, et al. Human articular chondrocytes-plasticity and differentiation potential. Cells Tissues Organs. 2006;184:55–67. doi: 10.1159/000098947. [DOI] [PubMed] [Google Scholar]
- 43.Pretzel D, Linss S, Rochler S, et al. Relative percentage and zonal distribution of mesenchymal progenitor cells in human osteoarthritic and normal cartilage. Arthritis Res Ther. 2011;13:R64. doi: 10.1186/ar3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eltawil NM, De Bari C, Achan P, et al. A novel in vivo murine model of cartilage regeneration. Age and strain-dependent outcome after joint surface injury. Osteoarthritis Cartilage. 2009;17:695–704. doi: 10.1016/j.joca.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redman SN, Dowthwaite GP, Thomson BM, et al. The cellular responses of articular cartilage to sharp and blunt trauma. Osteoarthritis Cartilage. 2004;12:106–116. doi: 10.1016/j.joca.2002.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Takebe K, Rai MF, Schmidt EJ, et al. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthritis Cartilage. 2015;23:454–461. doi: 10.1016/j.joca.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.