

Abstract
Current efforts in precision oncology largely focus on the benefit of genomics-guided therapy. Yet, advances in sequencing techniques provide an unprecedented view of the complex genetic and non-genetic heterogeneity within individual tumors. Herein, we outline the benefits of integrating genomic and transcriptomic analyses for advanced precision oncology. We summarize relevant computational approaches to detect novel drivers and genetic vulnerabilities, suitable for therapeutic exploration. Clinically relevant platforms to functionally test predicted drugs/drug combinations for individual patients are reviewed. Finally, we highlight the technological advances in single-cell analysis of tumor specimens. These may ultimately lead to the development of next generation cancer drugs, capable of tackling the hurdles imposed by genetic and phenotypic heterogeneity on current anticancer therapies.
Precision Medicine Aims to Address Inter- and Intratumor Heterogeneity
Precision medicine aims to use multiple types of data to classify patients into groups that will most likely respond to a given treatment. The identification of biomarkers that correlate with response to therapy or function in disease initiation/progression -- therefore representing therapeutic targets themselves -- is fundamental in this process [1]. Determination of molecular biomarkers is not limited to a specific methodology, and DNA, RNA, proteins, metabolites or microorganisms can individually, or in combination, serve as biomarkers. With cancer primarily being a genetic disease, precision oncology has largely focused on the determination of genetic biomarkers and multiple clinical trials to test whether targeting these genetic alterations in cancer can prolong survival. Remarkable success in applying genomics-driven cancer therapy has been noted [2], yet, serious criticism remains regarding this genomics-focused precision oncology concept, including scientific, social, ethical and economical aspects [2–5]. In this review, we focus on the biological rationale for precision oncology and outline current efforts and achievements of implementing precision oncology in the clinic, while highlighting promising routes to overcome the limitations of genomic-focused approaches. The current availability of screening platforms and the armamentarium of anticancer drugs now allows us to recognize and address inter-tumor heterogeneity (the different molecular characteristics observed between patients). We outline how the simultaneous assessment of genomic and transcriptomic data, combined with functional testing, can serve to overcome hurdles imposed by inter-tumor heterogeneity. In addition, we discuss the major limitations of prolonged response to current anticancer therapies, including intra-tumor heterogeneity (ITH); namely, differences in the molecular makeup of tumor cells within individual patients. We have only begun to decipher and address therapeutically such challenges.
The Technical and Molecular Basis for Precision Oncology
The ability to detect mutations in a tumor sample was one of the first milestones in recognizing the genetic events that underlie the cellular transformation process, denoting an early phase of genetic-based evidence for cancer occurrence and development. Improved technologies enabling the detection of such mutations in non-neoplastic tissues (including bodily fluids), has allowed the early detection of somatic oncogenic mutations such as Ras mutations and hotspot p53 tumor suppressor mutations [6–8]. While these developments reflect advances made already in the 80’s, it has taken another generation to better establish the importance of mutation frequency, its variability in the transformed tissue, and its causative role. This growing understanding has been a prerequisite for the introduction of mechanism-based therapies into clinical practice. Commonly known as targeted therapies, these therapeutic approaches are based on small molecules or monoclonal antibodies that inhibit oncogenic drivers [9–14], or target genetic vulnerabilities (e.g. Poly (ADP-ribose) polymerase PARP inhibitors in tumors with homologous recombination deficiency [15]). Several years of clinical experience with targeted agents -- and especially of the resistance to drugs – has led to the recognition of the central role of genetic heterogeneity and plasticity of growth-promoting signaling pathways in determining a patient’s individual response. A notable example is the targeting of BRAF mutations that are present in more than 40% of melanomas [16]. Although targeting recurrent BRAF-mutation(s) by mutant-specific BRAF inhibitors demonstrated great clinical success [9, 17], understanding the complex feedback and cross-talk between key players of the altered RAS/RAF/MEK/ERK signaling axis became necessary for optimizing therapy. Accordingly, in terms of clinical outcomes, combined BRAF and MEK inhibition proved superior over single agent use[18]. Furthermore, new generations of specific BRAF inhibitors are currently in the pipeline, finely-tuned to overcome mutation-driven altered signaling events in the RAS/RAF/MEK/ERK pathway [18]; these might be expected to outperform previous inhibitors of this pathway. Similar undertakings may be required to target deregulated signaling pathways arising from other mutations in different tumors, where a driver mutation is known, and where drugs targeting a given driver may exist.
Beyond direct targeting of genomic alterations, the impact of differentiation hierarchies, epigenetic alterations and the role of the microenvironment in driving tumor pathogenesis has become increasingly recognized. Accordingly, therapeutic approaches that aim to restore normal differentiation programs such as all-trans retinoic acid in acute promyelocytic leukemia and neuroblastoma have been developed [19]. Along these lines, drugs are/have been developed to reprogram epigenetic marks and restore normal gene expression programs, such as various HDAC inhibitors [20], in addition to drugs that interfere with tumor-microenvironment crosstalk, including angiogenesis inhibitors [21] and immunotherapeutic agents [22]
The search for cancer vulnerabilities in specific cancer types has been facilitated by numerous technological advances yielding large-scale molecular profiling of major cancer types [23, 24]. This system-based analysis of tumor samples, together with massive hypothesis-based research, has significantly changed our understanding of cancer biology (Key Figure, Figure 1): Carcinogenesis is generally considered to be driven by the natural selection of continuously acquired genetic and epigenetic variation in individual cells [25]. These converge on common phenotypic characteristics for cancer cells, including sustained proliferation, migration, invasion, and/or resistance to apoptosis [26]. Tissue microenvironments provide the fitness selection defining spatial and temporal changes in environmental pressures. These influence the evolutionary path of any given cancer cell, resulting in (epi-)genetically heterogeneous subpopulations. Diversity within cancer populations is not limited to the genome, and dynamic variations in differentiation hierarchies, transcriptional signals and the proteomic landscape add to the phenotypic heterogeneity observed within tumors [27]. Indeed, cancer cells do not exist as isolated entities, but rather, engage in heterotypic interactions with stromal cells and cooperate with adjacent tumor subclones; this is important, as it can result in increased robustness of a tumor [28].
Figure 1, Key Figure.

Determinants of Tumor Pathogenesis and Measures to Inform Precision Therapy
The genetic and phenotypic characteristics of a patient’s tumor are influenced by tissue/cell type specific factors, germline genetic background, lifestyle factors, as well as the number and type of previous anticancer drugs received [25, 27, 28]. Each individual cell is further influenced by, first, the proximity to and the integrity of the tumor vasculature; second, the biochemical and biophysical properties of the surrounding extracellular matrix (ECM); third, competing/cooperating interactions between individual tumor cells or tumor and stromal cells (among which are cancer associated fibroblaststs (CAFs), endothelial cells (ECs), and bone marrow-derived cells (BM-DCs)); and fourth, anti-tumor immunity. These factors further shape the geno- and phenotypic properties of a tumor in a spatial and temporal manner. While the genomic analysis of a tumor biopsy at the time of diagnosis identifies genetic vulnerabilities, the inclusion of transcriptomic data holds the additional potential of identifying non-genetic vulnerabilities by considering pathway activity and the composition of the tumor microenvironment. The integrated analysis of genomic and transcriptomic data is therefore a valuable tool to inform precision therapy.
Moreover, large-scale sequencing of human cancer genomes and transcriptomes have identified nearly 200 “consensus” driver genes (of which ~15% were identified primarily using DNA sequencing of cancer genomes [29]) and an additional 300 putative driver genes have been suggested [30, 31]. The pathways in which these genes function are also emerging [32–36]. Coupled with the success seen using targeted therapies in certain cancer subtypes [9–14], these efforts have contributed to laying the basis for precision molecular oncology: patients are treated according to the molecular makeup of their tumors rather than solely based on tumor histology, type, grade and stage (Figure 1).
Clinically Relevant “Omics“ Approaches
Genomics-driven Cancer Therapy in Clinical Testing
While at present only a small proportion of cancer patients benefit from targeted therapies, great efforts are ongoing to extend the scope of precision oncology to a broader spectrum of patients (reviewed in [2]). Massive inter-tumor heterogeneity that has been rigorously documented through large-scale DNA and RNA sequencing, as well as DNA copy number and DNA methylation profiling (e.g. TCGA, ICGC, and others) [23, 24] (see supplementary Table S1 for relevant data sources). However, unexpected similarities between tumors of different tissues-of-origin have been uncovered, while certain tumors have been found to be more similar at the molecular level to tumors from a different tissue-of-origin [37]. These similarities, together with the detection of rare variants within well-characterized driver genes suggest that approved targeted therapies might be effective in diverse tumor types with distinct molecular alterations [32, 36]. This has resulted in the initiation of clinical programs that evaluate whether molecular profiling of patients is clinically feasible and importantly, whether treating patients based on their genomic profiles might be beneficial relative to a given standard of care, or a physician’s treatment choice (see Table 1 for examples of programs/studies). In addition to the identification of new putative driver genes found in a low percentage of patients with less common cancer types or subtypes, several novel clinical hypotheses have been generated but await verification. Recently, Foundation Medicine reported that in a targeted sequencing study of 63,220 tumors, more than 75% of patients presented a mutation in at least one of 10 cancer driver genes, and more than 25% of patients presented a known driver mutation within these genes [38]. Accordingly, in silico computational studies predict that up to 90% of patients may benefit from molecularly-guided therapy when biomarkers of uncertain clinical significance, as well as off-label and investigational drugs are considered to inform therapy [39, 40]. To test this multitude of novel clinical hypotheses, new adaptive trial designs, including basket and umbrella trials have been employed [41, 42] (Table 2). Basket trials are designed to test the effects of a single (or a few) drug(s) in a variety of cancer types (or possibly subtypes) using specific mutation(s) as biomarker(s). By contrast, umbrella trials are designed to test the impact of specific drugs on different mutations within the same cancer type.
Table 1.
Studies Evaluating Feasibility and Clinical Benefit of Molecular Profiling
| Study | Tumor Types | Screening platform | #Pts sequenced | # Pts on matched therapy | Type of therapy | Endpoint/Results |
|---|---|---|---|---|---|---|
| IMPACT MD Anderson [226] NCT00851032 Retrospective, non-randomized; | All; advanced, refractory | Hotspot sequencing 11 genes; FISH (ALK) | 1144 | 175 | Mono- and combination; Phase I therapies; N.S. | Higher ORR and longer PFS compared to un-matched therapy |
| IMPACT/COMP ACT [48] NCT01505400 Retrospective, observational, non-randomized | Advanced cancers and phase I candidate s | MALDI-TOF MS hotspot panel (23 genes) or targeted NGS panel (48 or 50 genes) | 1640 | 245 | N.S.; investigation al agents of 277 trials including 89 genotype matched trials | Genotyped matched therapy improved response |
| PREDICT [227] NCT02478931 Retrospective, correlative, non-randomized | All; | NGS; (182 or 236 gene panel and 14 or 19 rearrangements | 347 | 87 | N.S. | More patients with SD ≥ 6 month; 45% with extended PFS of 30% compared to previous therapy |
| Bisgrove[228] NCT00530192 Prospective, single-arm Phase I | All; advanced, refractory | IHC, FISH, gene expression | 86 | 68 | Mono- and combination; N.S. | 27% with extended PFS of 30% compared to previous therapy |
| Genomic Profiling in Phase I[46] NCT02437617 Prospective, non-randomized; | All; advanced, refractory | Panel NGS (236 genes); standard biomarker | 339 | 122 | Mono- and combination; Phase I/II therapies; N.S. | High matching score associated with higher SD ≥6 months/PR/CR, longer PFS and survival |
| MOSCATO 01 [44] NCT01566019 Prospective, non-randomized | All; advanced, refractory | CGH array; panel NGS (WES and RNAseq in 2014 included) | 843 | 199 | Phase I drugs and off-label drugs; N.S. | 33% with extended PFS of 30% compared to previous therapy |
| WinTher NCT01856296 Prospective, non-randomized | All; advanced, refractory | DNA (236 genes) and RNA in tumor and normal matched tissue; | To be 200 | N. A. | N.S. Chosen based on DNA analysis, if no actionable mutation, then based on RNA analysis | Estimated completion in 2018 |
| SHIVA [45] NCT01771458 Prospective, randomized | All; advanced, refractory | Panel hotspot NGS (46 genes); CNV | 741 | 99 | erlotinib, lapatinib + trastuzumab, sorafenib, imatinib, dasatinib, vemurafenib, everolimus | Median PFS in experimental group (matched therapy) not significantly longer than in the control group |
| NCI-MPACT NCT01827384 Prospective, Phase II randomized feasibility study | All, advanced solid tumors | NGS (4000 variants across 143 genes); Activation of RAS/RAF or PI3K pathway; Inactivation of DNA repair pathway; | To be 700 | N.A. | Carboplatin, Everolimus, Temozolomi de, Trametinib, Veliparib, AZD1775 | Estimated completion in 2019; Outcome measures: ORR; compare genotype matched vs physicians choice |
AZD1775, WEE1 inhibitor; IHC, Immunohisochemistry; NGS, next generation sequencing; FISH, fluorescent in situ hybridization; PFS, progression free survival; ORR, overall response rate; SD, stable disease; PR, partial response; CR, complete response; CGH, comparative genomic hybridization; WES, whole exome sequencing; N.A., not applicable; N.S., not specified; Pts, patients;
Table 2.
Representative Basket and Umbrella Trials
| Study | Tumor types | Screening platform | Biomarkers tested | Drugs | Endpoint/results |
|---|---|---|---|---|---|
| BATTLE NCT00409968, NCT00411671, NCT00411632, NCT00410059, NCT00410189 Prospective, adaptively randomized, umbrella trial | Chemorefr actory NSCLC | non-NGS, mutation analysis, FISH, IHC; | mutation/ amplification: EGFR, KRAS/BRAF, CCND1; protein expression: VEGF, RXR, cyclinD1 | erlotinib, sorafenib, vandetanib, erlotinib + bexarotene | Better DCR for EGFR+erlotini b and KRAS/BRAF +sorafenib; |
| BATTLE-2 NCT01248247 2-stage Phase II umbrella design | Advanced NSCLC | NGS; DNA, mRNA, RPPA, IHC | IHC: pAKT, PTEN, HIF1a, LKB1; Mutation: P13KCA, BRAF, AKT1, HRAS, NRAS, MEK1, MET, CTNNB1, LKB1; | Erlotinib (ctrl) sorafenib, MK-2206+erlotinib; MK-2206+ selumetinib | 8-week disease control rate |
| Lung-MAP NCT02154490 Randomized Phase II/III umbrella design | Advanced, recurrent squamous cell lung carcinoma | FMI Foundatio nOne platform; IHC; | PI3KCA, CDK4/6, CCND1/2/3, FGFR1/2/3, HGF/c-MET | taselisib, palbociclib, AZD4547, erlotinib, erlotinib+rilotumu mab, ipilimumab, nivolumab, durvalumab | PFS, ORR, OS |
| I-SPY-2 NCT01042379 Randomized open label Phase II umbrella design (adjuvant setting) | Stage III breast cancer | Conventio nal, MRI; | HER2, hormone receptor, and Mammaprint; Bayesian marker-adaptive trial designs | HER2+: Neratinib, MK2206+Trastuzu mab, T-DM1+Pertuzumab, Trebananib +Trastuzumab, Pertuzumab+ Trastuzumab HER2−: veliparib, MK2206, Ganitumab+metfor min, Trebananib | Pathological complete response; Several drugs graduated to Phase III trials [229–231] |
| ALCHEMIST NCT02194738; NCT02193282; NCT02201992; NCT02595944 Screening study and accrual to Phase III randomized treatment studies (adjuvant setting) | Resectable non-squamous NSCLC | NGS, tissue and blood; germline + somatic alteration s | EGFR mutation; ALK rearrangements (FISH); PD-L1 expression (IHC) | Erlotinib, Critozinib, Nivolumab | OS |
| SAFIR02 Breast NCT02299999 Phase II randomized umbrella design | HER2 negative recurrent and/or metastatic breast cancer | CGH array, hotspot sequencin g | To be determined during the study | AZD2014, AZD4547, AZD5363, Sapitinib Selumetinib, Vandetanib, Bicalutamide, Olaparib, durvalumab | PFS as compared to standard maintenance |
| SAFIR02 Lung NCT02117167 Phase II randomized umbrella design | EGFR and ALK WT recurrent and/or metastatic NSCLC | CGH array, hotspot sequencin g | To be determined during the study | AZD2014, AZD4547, AZD5363, Sapitinib Selumetinib, Vandetanib, durvalumab | PFS as compared to standard maintenance |
| Lung MATRIX NCT02664935 Phase II non-randomized umbrella design | Advanced, pretreated NSCLC | 28-gene NGS platform | Mutation: FGFR2/3, TSC1/2, LKB1, KRAS+RbWT, NF1, NRAS, PIK3CA, AKT1, EGFR+ EGFRT790 LoF: p16+RbWT, PTEN Amp: CDK4+RbWT, CCDN1+RbWT, MET, PIK3CA; Rearranged: ROS1 | AZD4547, AZD2014,Palbocicli b, Crizotinib, Selumetinib, AZD5363, Osimertinib, durvalumab | ORR, PFS |
| FOCUS 4 [232] PhaseII/III randomized umbrella design | Advanced/ metastatic, untreated colorectal cancer | Hotspot-seq biomarker only; IHC, | BRAF, PIK3CA, KRAS, NRAS; PTEN, MMR, | BRAFi+panitumum ab+/−MEKi, Aspirin, AKTi+MEKi, HER1/2/3i, | ORR, PFS, OS |
| V-BASKET [49] NCT01524978 flexible, early phase 2, basket study | Solid tumors, multiple myeloma | Mutation analysis with local method | BRAFV600 | Vemurafenib monotherapy; Vemurafenib + Cetuximab in CRC | Efficacy in NSCLC, ECD, and LCH |
| CUSTOM [47] NCT01306045 Biomarker-derived, Multiarm, Multihistology Phase II, basket trial | NSCLC, SCLC, thymic malignancy | NGS | Mutation: AKT1, BRAF, EGFR, ERBB2, HRAS, KIT, KRAS, NRAS, PDGFRA, PIK3CA, PTEN; Amplification: ERBB2, PIK3CA, PDGFRA; Fusion: ALK; | erlotinib, selumetinib, MK2206, lapatinib, sunitinib | Targeting EGFR and ALK offers benefit; design not feasible for most arms |
| NCI-MATCH NCT02465060 Non-randomized Phase II; basket trial | Advanced, recurrent, refractory solid tumors, lymphoma, myeloma | NGS (4000 variants across 143 genes) | Mutations: AKT1, BRAF V600; BRAF non V600, BRCA1/2, cKIT, DDR2, dMMR, EGFR, EGFRT790M, FGFR1/2/3, GNAQ/GNA11, HER2, MET ex14 sk, mTOR, NF1, NRAS, PI3KCA, PTEN, SMO/PTCH1, TSC1/2;Transloc ations: ROS1, ALK; Amplification: CCDN1/2/3, CDK4/6, HER2, MET; Loss: NF2, PTEN; Fusion: NFRK | Ado-trastuzumab emtansine, Afatinib, AZD4547, AZD5363, AZD1775, Osimertinib, Binimetinib, Crizotinib, Dabrafenib+tramet inib, Dasatinib, Defactinib, GSK2636771, Larotectinib, Nivolumab, Palbociclib, Sunitinib, TAK-228, Taselisib, Trametinib, Trastuzumab, Vismodegib | ORR |
AZD4547, FGFR inhibitor; AZD5363, AKT1/2/3 inhibitor; AZD1775, WEE1 inhibitor; AZD2014, inhibitor of mTORC1/2; CGH, comparative genomic hybridization; ECD, Erdheim-Chester disease; GSK2636771, inhibitor of PI3K beta; LCH, Langerhans’ cell histiocytosis; NGS, next generation sequencing; MK2206, inhibitor of AKT1/2/3; NSCLC, non small cell lung cancer; OS, ocerall survival; ORR, overall response rate; PFS, progression free survival; RPPA, reverse phase protein array; SCLC, Small cell lung cancer; TAK-228, dual mTORC1/2 inhibitor;
The majority of these studies profile the mutation status of a few dozen or hundreds of selected genes [2]. This is based on the fact that although whole genome sequencing (WGS) can detect DNA sequence variants as well as focal and large chromosomal rearrangements, deletions or amplifications, it is difficult to identify driver events within large chromosomal abnormalities. Therefore, clinically valuable sequencing approaches can be reduced to either the whole exome (WES) or targeted exomes (panel sequencing) of cancer-related genes. These approaches are often combined with the analysis of some well-characterized intronic regions (e.g. ALK, RET1, ROS, BCR) that are frequently rearranged in cancer genomes [2] (Figure 1). Furthermore, targeted sequencing has the advantage of yielding a high sequencing depth, which is important to be able to infer clonality of a detected driver event. Determining the clonal distributation of identified alterations should be a priority in precision oncology trials given that targeting trunk mutations appears to be crucial to maximizing the efficacy of targeted therapies [43].
Most studies have demonstrated that genomics-guided therapy improves patient outcomes when well-characterized biomarker-drug pairs -- with strong clinical or preclinical evidence-- are used (Table 1). For example, the MOSCATO 01 trial [44] that used only last-generation drugs with high affinity to a specific target, achieved positive results, whereas the SHIVA study [45], which heavily relied on everolismus -- a drug weakly affecting the PI3K/AKT/mTOR pathway, indicated that genomic profiling did not result in patient benefit. Furthermore, emerging evidence suggests that targeting multiple drivers by combination therapy is superior over single-agent use [46] as patients with advanced tumors frequently exhibit multiple aberrations detected by genomic profiling. Despite these advances, several challenges remain: First, trial recruitment of patients with rare mutations is difficult [47] and only a small percentage of patients (2–5%) undergoing genomic profiling have been subsequently treated with off-label drugs or been enrolled in genotype-matched trials [46, 48]. Noteworthy, the recent report on the positive outcomes of the MOSCATO 01 trial indicates that match-rate can be improved (19%) when performed within a big cancer center offering access to a variety of clinical trials [44]. Second, the presence of validated genetic biomarkers does not strictly predict a response to targeted therapies to different tumor types [49], given that the effect of therapies is known to be context-dependent (as seen, e.g., by the lack of response of BRAFV600-positive colorectal cancer to BRAF inhibitors which show good clinical responses to melanomas carrying the same mutation [50]). On a positive note, identifying such genomic-context effects has already expanded the use of inhibitors, such as against BRAF [49] or PARP [51], and may result in expedited approval of investigational drugs. Finally, identifying high-confidence biomarkers to guide specific drug treatments remains a challenge. One approach to expanding the biomarker landscape may be to determine the differential molecular profile of patients showing a dramatic response to targeted therapy versus non-responsive ones. An increasing number of publications report such “exceptional responders”, which has led to the identification of rare genomic events likely to predict the response or resistance to targeted therapies [52–62]. Taken together, sequencing efforts of cancer genomes within clinical trials or by research initiatives such as the TCGA and ICGC initiatives (see supplementary Table S1) are expected to improve the identification of driver mutations, as well as patient stratification strategies associated with these. This in turn may expand the scope of genomics-based precision oncology to a broader spectrum of patients.
Limitations of Using Genomics as a Single Approach for Biomarker Identification
While genomic profiling provides valuable information regarding genetic mutation / amplification / deletion and certain epigenetic modifications (e.g. methylation), there are certain inherent limitations of using an approach that simply tests the presence or absence of genetic driver events to inform therapeutic decision-making. This includes limitations of using genomics as a single platform for biomarker identification; indeed, some cancer types, such as prostate cancer or some pediatric malignancies have very few or even no recurrent mutations detected, indicating that other types of somatic variation, may be potent drivers of oncogenesis [36]. Furthermore, no genetic alterations have been found to correlate with well-characterized predictive biomarkers such as the expression of estrogen receptor or androgen receptor, in breast or prostate cancer, respectively. In addition, genomic profiling does not provide sufficient information regarding the activity of actual protein products mediating oncogenic or tumor suppressor gene functions. In other words, variations in oncogenes/tumor suppressor genes do not necessarily predict activation of the corresponding biological pathway, and vice versa: cancer driver pathways can be active without the presence of a mutation(s) [63]. Finally, novel biomarkers linked to non-genetic vulnerabilities, such as those involving cancer cell reliance on stress response or metabolic pathways, may be able to predict responses to autophagy inhibitors or drugs inhibiting antioxidant enzymes, and these need to be defined [64].
The most comprehensive approach to overcome these challenges and to elucidate cancer vulnerabilities is the simultaneous characterization of the genome, epigenome, transcriptome, proteome, and metabolome of tumors and their surrounding stroma; indeed, these are all crucial parameters to defining cellular phenotypes involved in cancer pathogenesis, as well as in characterizing responsiveness to therapy [65] (Box 1). As these parameters are dynamic entities (e.g. changes in responses to external stimuli), they are expected to show spatial heterogeneity (geno- or phenotypic distinct clones may show different growth kinetics or survival rates dependent on their location). Coonsequently, an analysis of multiple biopsies and longitudinal follow-up of patients would ideally be performed to predict the initial responses to therapy and to identify putative mechanisms of drug resistance. Although such comprehensive approaches are not yet feasible for routine clinical practice, current state-of-the-art technologies are already enabling the combination of at least two different omics platforms for cancer analysis, genomics with epigenomics, and/or transcriptomics. As discussed below, combined genomic and transcriptomic analysis, together with functional testing of omics-derived treatment predictions, are expected to overcome many of the challenges that current precision oncology-based trials are facing.
Box 1. Relevant “Omics” for Precision Oncology.
Epigenetic profiling holds great promise in deciphering the cellular states and characterizing phenotypic heterogeneity. The importance of epigenetic reprogramming in cancer is evidenced by the fact that chromatin regulators are often mutated [32, 36] and widespread epigenetic changes throughout cancer genomes can be identified, intricately linked to the activity of known tumor promoters/suppressors such as EGFR [193] or TP53 [194]. There are two general classes of drugs targeting the epigenome: (i) broad reprogrammers, which include inhibitors of DNA methyltransferases, histone deacetylases or bromodomain and extra-terminal motif proteins, and (ii) targeted therapies that pin specific activating mutations in DNA-modifying enzymes such as EZH2, or in enzymes whose mutations have a profound effect on epigenetic pathways, e.g. IDH1/2 [20]. Currently, there are no epigenetic drug-sensitivity biomarkers that would predict the response to these approved or investigational drugs. Therefore, the addition of epigenetics in clinical practice awaits the identification of epigenetic marks that mediate distinct tumor phenotypes of clinical relevance (such as mesenchymal differentiation, stemness, dormancy or therapy resistance) [65].
Proteomics combined with genomic data likely reveal the most accurate information on the activity state of individual genes. The proteome represents the ideal readout to define a cell’s functional state in response to internal or external perturbations, and proteogenomic analysis is being integrated in large-scale characterization efforts of the TCGA [195–197]. This integration has the power to nominate driver genes from large chromosomal deletions or amplifications and can identify new driver clusters that are not easily found in transcriptomics signatures [195, 196, 198]. Although TCGA analysis has long included antibody-based phosphoprotein analyses, the comprehensive proteomic characterization based on mass spectrometry increases the breadth of phosphoproteomics data and importantly, allows for the identification of post-translational modifications beyond phosphorylation [199]. The latter may represent important biomarkers for drugs that do not target kinases such as the identification of “acetylation-signatures” in serous ovarian cancer, and which may predict responses to HDAC inhibitors [197]. While it is expected that future technologies will provide the platform for large-scale proteomic assessment of tumor samples, current proteomic analysis requires a large amount of tissue, is costly, labor-intensive and lacks the analytical validity and sensitivity that genomics provides.
Emerging metabolome and microbiome data, are expected to provide important additions to genomics: Rewired metabolic pathways in tumors provide alternate fuel sources that can be targeted, and the mutations and/or deregulated expression of metabolic genes have been linked to tumor propensity for metastasis or therapeutic resistance [200]. Translating this knowledge in the clinic will require further preclinical analysis, especially given the differences between cancer cell metabolism in vitro and in vivo [201, 202]. Microbiome-based data is a likely addition in the more distant future, which might provide novel putative biomarkers and means to monitor predicted therapeutic responses, and possibly, improvements.
Transcriptomics as a Valuable Measure to Improve Biomarker Identification
At present, the most common way to enhance genomic information available to us is by the inclusion of transcriptomic analyses (Figure 1). RNA sequencing (RNA-seq) technologies allow the mapping of the entire transcriptome or select gene expression networks, and are readily available, becoming economically feasible. The ability to decipher the landscape of gene expression offers important steps over acquiring genomic data alone. First, aside from the ability of RNA-seq to detect splice variants [66], RNA-seq can also detect novel or known gene fusions, which have been identified as drivers of disease in a variety of rare and common tumors [66–69]. The latter are promising therapeutic targets as the inhibition of fusion-genes is often associated with striking efficacy, as exemplified by targeting the BCR-ABL fusion in chronic myeloid leukemia, or targeting RET, ALK, ROS, FGFR or BRAF fusions in various malignancies (reviewed in [2]). Second, transcriptomics can provide indirect information about protein expression status; knowing that a candidate gene harboring certain mutation(s) is also expressed (and to what level) is valuable in establishing the importance and contribution of this gene to the tumor phenotype. Third, beyond providing information about the expression of tumor driver genomic variations, the inclusion of transcriptomics allows the mapping of non-oncogene vulnerabilities, and provides information about oncogenic pathway activities, even in the absence of mutated driver genes [63, 70]. One such example is the BRAF-mutation signature in colon cancer that can be found in BRAF-mutated but also BRAF-wildtype tumors, and characterizes (in addition to the KRAS and PI3K signatures) patients resistant to EGFR inhibition [71]. The BRAF-signature can not only serve as a resistance biomarker but has been recently suggested to serve as a sensitivity biomarker for mitotic poison drugs such as vinorelbine [72]. Another notable example is that of BRCA-associated signatures [73], where tumors (such as breast, ovarian or prostate tumors among others [66, 73]) sharing similar molecular signatures to BRCA-mutant tumors may also respond to similar therapeutic approaches, even when lacking specific BRCA mutations [73–75]. Fourth, transcriptomes, by contrast to DNA, are tissue- and cell-type specific [65]; this is often considered a disadvantage for RNA analysis of bulk tumor samples because in samples with a high proportion of stromal cells, massive computational deconvolution is necessary to extract the transcriptional profile of interest, as all cells within the biopsy contribute to the RNA pool [65]. However, tissue specificity can provide important clinical information about tumor histology and tumor origin, which is of high relevance in patients with cancer of unknown primary tumors [37]. Additionally, cell-type specific transcriptomes can reveal certain aspects of the immune status of tumor samples that may be of therapeutic relevance [76, 77]. High overall mutational load within tumors (e.g. highly mutated human tumors such as melanoma, lung cancer or mismatch-repair deficient colon cancer) has been reported to correlate with therapeutic responses to immune-checkpoint inhibitors (e.g. drugs targeting CTLA-4 or the PD1/PDL1 axis) [76, 77]; however, these factors have not been strictly linked, and long-term responses to checkpoint inhibition have been observed for a broad mutational spectrum of cancers [76, 77]. Notably, integrating genomic and transcriptomic data holds promise for the identification of patients that can benefit from immune-checkpoint inhibitors. For example, the transcriptomic analysis of responders to CTLA-4-blockade (ipilimumab) has revealed that the expression of cytolytic effector genes (e.g. granzyme A and perforin) positively correlate with patient response (complete or partial response to ipilimumab, or stable disease with overall survival > 1 year by RECIST criteria) [76]. Furthermore, the expression of immune checkpoint regulators have correlated with increased patient survival [76]. In addition, transcriptomic signatures that significantly correlate with resistance to anti-PD1 therapy in melanoma have also been identified [77].
Although the practical utility of RNA-seq in the clinic has been challenging, technological advances allowing the application of RNA-seq to clinically-relevant specimens (including formalin-fixed, paraffin-embedded tissues), along with efforts to benchmark data analysis pipelines (ICGC-TCGA DREAM Somatic Mutation Calling Challenge – RNA) [78], have set the basis to move RNA-seq into routine clinical practice. Valuable transcriptomic information can thus be combined with genomic data to establish new blueprints that provide multidimensional insight into the characteristics of a given tumor biopsy. Such combinations can benefit from innovative computational approaches which may identify novel master regulators, not seen in either analysis alone.
Analysis Approaches to Determine Molecular Subtypes and Cancer Vulnerabilities
To overcome the challenges of inter-tumor heterogeneity in determining molecular-guided therapy, the identification and characterization of molecular subtypes of cancer and the mutations that drive cancer have been an urgent priority. The promise of characterization of tumors with molecular subtypes or biomarkers is two-fold. The first major goal is to find molecular biomarkers of patient prognosis or of effective drug treatments. The second major goal is to develop a better mechanistic model for understanding the role of the tumor’s genome, transcriptome, methylome, epigenome and environmental alterations in driving its initiation and evolution. Extensive clinical efforts now provide us with an unprecedented view on the genomic (and transcriptomic) landscape of all advanced cancer types [79], in addition to the datasets provided by the TCGA and ICGC, which have focused on the sequencing of common cancer types early in disease progression. The following sections describe related computational approaches, and for an expanded summary of references on these and additional topics (intra-tumor heterogeneity and single cell analysis approaches) the reader is referred to supplementary tables S1–S3.
Approaches for Tumor Subclassification
Methods for identifying molecular subtypes generally fall into two categories, based on whether data from a single platform or multiple platforms is being used. For single platform data (e.g. gene expression), any off-the-shelf clustering algorithms can be used, though choosing the method depends on the type of data being clustered. The more challenging case is clustering patients with data from multiple platforms, especially because there is often a data type that is missing, as not all measurements are performed in every patient. Researchers have taken multiple approaches (see references in Table S2A). Some methods search for a “consensus” after clustering patients by each platform separately [37], or cluster with protein-protein interactions [29], or patient similarity networks [80, 81]. Other methods formulate the problem as a “multi-view” matrix factorization and dimension reduction, or as a probabilistic model (reviewed in [82]). In all cases, a key challenge is the selection of features from each platform as inputs to the clustering algorithms; for example, it is possible to summarize mutations, gene expression, and DNA methylation events as binary alterations [80], and then treat any missing data as a non-alteration event. We anticipate that recent advances in methods for learning low-dimensional representations of multiple data types such as deep neural nets [83] will soon be applied in molecular classification of tumors, given the amount of molecular cancer data being produced and the successful application of deep neural nets in areas of computer vision, natural language processing, and biology [84]. Initial molecular subtype studies have often focused on clustering samples into subtypes based on gene expression in a single cancer type, which have provided robust biomarkers and subgroups, coherent with patient survival profiles (e.g, in breast cancer [85] or colorectal cancer (CRC) [86]). These studies typically reveal a more refined set of clusters than those defined by known histopathological markers, with more coherent survival profiles of the samples/patients composing them. While some of the molecular clusters strongly overlap with known histologically-based clusters, others are surprisingly composed of samples with distinct histopathological markers, but with similar transcriptomic profiles and survival rates. More recent analyses have clustered multiple platforms across multiple cancer types, and, as outlined before, identified molecular similarities between tumors of different tissue-of-origin. For example, one study analyzed TCGA data from >3000 samples across 12 cancer types, and found that while most cancers could be classified based on their histology, ~10% could be classified as belonging to an “integrated” subtype, i.e. including cancers from multiple tissues-of-origin in the same subtype [37]. Furthermore, grouping samples from different tissue types yielded improved predictive power for patient prognosis, potentially reflecting the value of molecular features (such as common mutations) for predicting survival [37]. The FDA recently approved the drug pembrolizumab (immune-checkpoint PD-1 blockade), used across many cancer types – with demonstrated effectiveness in colorectal, endometrial, pancreas, thyroid, and eight other cancer types-- based on the presence of a specific (mismatch repair deficiency) signature [87]. These studies demonstrate the promise of classifying tumors using molecular features, which can give additional insights into prognosis and treatment beyond tissue-of-origin.
Approaches to Identifying Genetic Drivers
While as few as 3–8 somatic mutations are required to drive cancer [36], identifying the entire set of driver mutations in any given tumor is a difficult biological and computational problem. The observation that relatively few mutations occur in a significantly recurrent manner across tumors, holds, despite the development of sophisticated statistical tools for evaluating the significance of mutations. Researchers have developed multiple different classes of tools that consider different information about somatic mutations, including the predicted functional impact [88] or conservation across populations [89]. Other methods attempt to classify driver mutations by identifying hotspots in the protein sequence or structure [90–92], or targets of recurrent copy number aberrations [93]. Some methods also consider side information such as a gene’s replication timing and expression [94] [95], or per patient, and/or per gene mutation rates [96] (see references summarized in Table S2B).
Despite these advances, in most cancer datasets there is a “long tail” of genes with infrequent mutations, where the drivers are statistically indistinguishable from passenger mutations [32]. One report illustrated the depth of this problem by estimating the number of samples required to detect driver mutations with a given frequency in a given cancer type through saturation analysis [34]. The cancer type in question was critical because of the high variance in background mutation rates in different cancers (e.g. breast cancer, prostate cancer, etc.). For example, they showed that up to 5300 samples would need to be sequenced to detect drivers occurring at 2% above the high background mutation rate in melanoma [34]. This presents a particular challenge for rare cancer types, especially since cancer types continue to be divided into different subtypes [34].
The observed inter-tumor mutational heterogeneity is widely believed to be due, in part, to mutations targeting pathways or “cancer hallmarks” [26], where each pathway includes multiple genes such that many different combinations of aberrations can affect hallmark pathways and drive cancer. Thus, by uncovering the genes in these pathways, it may be possible to identify “hidden” driver mutations in the “long tail”, i.e. the set of mutations that are indistinguishable from passengers without considering prior knowledge such as pathways. To date, researchers have developed multiple classes of methods that use different side information to identify the pathways/hallmarks targeted in cancer (see Table S2B). One group of methods searches for significantly mutated groups of genes in known pathway databases [97] and protein interaction networks [35, 93, 98–100]. Other methods search for functional mutations that co-occur with sample-level events [101, 102], which can be viewed as a supervised learning task. These approaches have been used on cancer cell line drug sensitivity and gene dependency/addiction (i.e. conditional essentiality) data to generate testable hypotheses, but are less well-suited for predicting coarse measurements with many factors such as overall survival. Another promising approach has been to search for groups of genes with mutually exclusive mutations [103–107]. However, these approaches also require large sample sizes through saturation analysis [108], and, depending on the relative rate of driver/passenger mutations, such sample sizes can be even larger than those required by the recurrent mutation detection methods described above. Finally, going beyond coding region mutations, researchers are beginning to uncover recurrent and functional mutations in non-coding regions of the genome that might play a role in dysregulated gene expression, as is the case of the TERT promoter, shown to be mutated for the first time in melanoma [109], and more recently, in 43 tumor types [79], with significant association to poor survival in cutaneous melanoma, bladder urothelial carcinoma, and papillary thyroid cancer [79]. Larger whole-genome sequencing efforts such as those from the ICGC are likely to uncover more of these non-coding mutations due to increased statistical power.
Integrating Genomic and Clinical Data
Current efforts linking genomic mutation data with clinical data to assist in therapeutic decisions build and use knowledge banks (see Table S3). These include web tools that provide data and text summaries of the frequency, mechanisms, and druggable targets of known driver mutations [110]. Multiple tools now include “interpretations” or summaries of the driver mutations written by clinicians – including the Precision Medicine Knowledgebase (at Weill Cornell) and the Personalized Cancer Therapy knowledge base (at MD Anderson) – or by the “crowd” [111, 112] (see list of references in Table S2C). A related approach recently explored leveraging existing ‘omics datasets for the interpretation of variants in newly sequenced samples, in acute myeloid leukemia[113]. For example, one study recently demonstrated the use of this approach by building survival models that linked genomic and clinical data, and then using these models to choose treatment(s) and predict survival for new acute myeloid leukemia patients [113]. Regularized regression on both genomic and clinical features was performed on these models to predict overall survival; the authors used these to identify additional interventions that could potentially increase overall survival, and extraneous interventions that could be removed for some patients without decreasing overall survival [113]. However, effectively integrating annotations and clinical knowledge of known variants with ‘omic databases in an automated manner for the interpretation of patient molecular data, and creating features from molecular data for input into survival models, remains a key challenge. This appears to be largely due to the fact that most clinical data still need to be extracted from free text, and the pertaining electronic medical record (EMR) systems are mostly not standardized.
Identifying Cancer Vulnerabilities on the Basis of Genetic Interactions
Another way to guide precision therapy is based on identifying and utilizing genetic interactions, in particular, by harnessing the concept of Synthetic Lethal interactions (SLi). SLi describe the relationship between two genes where an individual inactivation of either gene results in a viable phenotype, while their combined inactivation is lethal for the cancer cell [114, 115]. SLi have long been considered a foundation for the development of selective anticancer therapies [64, 114, 115], which aim to inhibit the Synthetic Lethal (SL) partner of a gene that is inactivated de novo in cancer cells. As this SLi partner gene is most likely to be inhibited only in the tumor, this treatment will thus primarily kill these cancer cells but not healthy ones. Thus, this offers a complementary approach for predicting patient drug responses to sequence-based cancer precision medicine strategies. This might be achieced by (i) going beyond existing precision oncology approaches based on actionable mutations (i.e. mutation that can be targeted by specific small molecule inhibitors) in a few hundred cancer driver genes, and examining the whole genome, thereby covering all possible changes that might have occurred in a tumor. These might uncover many more treatment options for patients whose tumors do not bear actionable mutations. (ii) SLi are well poised to offer effective options for potentially treating heterogeneous tumors, presumably impacting differenting subclones, and overall resulting in more effective tumor eradication with reduced likelihood of drug resistance.
Given this promising potential, extensive experimental efforts have aimed to tease out the wiring of genetic interactions in cancer cells based on single (isogenic) cell lines [116–121] or on large-scale genetic knockout-based screens [122–126]. However, due to the large combinatorial space of pairwise interactions that need to be surveyed, these screens have probed only a small fraction of the coverage offered by SLi: each screen typically scans a few thousand candidate SL partners of just one “anchor” cancer driver gene of interest (e.g., KRAS or VHL), altogether covering a mere fraction of the 500 million gene pairings in the human genome. Yet, with these screens, several SL interactions have been successfully uncovered to date; apart from examining the effect of PARP inhibitors in patients with BRCA-mutated breast and pancreatic tumors, a growing number of other treatments targeting SL-based cancer specific vulnerabilities are currently being clinically investigated [127].
Aiming to bypass the limitations of current experimental techniques in probing the vast space of potential SLi, various computational approaches have been developed to identify such candidate SLi (see references in Table S2D). These include applying various machine learning methodologies to predict genetic interactions in different species [128–131], and in cancer (employing yeast SLi) [119, 132], utilizing metabolic modeling [133, 134], evolutionary characteristics [119, 129], transcriptomic profiles [101, 135], and more recently, by mining cancer patient data [136–138] (Table S2D). One recent study evaluated the TCGA copy number and transcriptomics data to identify, as candidate SLis, gene pairs that are almost never found inactivated in the same tumors [136]. The study demonstrated that gene pair interactions (a subset of which was validated in experimental screens) could be successfully used to predict the survival of breast cancer patients in an independent dataset [136]. The pair was also used to predict in vitro drug responses in order to identify novel drug repurposing indications for potentially treating renal cancer [136]. Unlike the approach of using expression and copy number data ([136]), an algorithm was recently developed to mine pan-cancer human tumor data and define mutation-specific SL interactions for specific cancers [139]. Its SL predictions were validated against published SL screens and one specific SL gene pair interaction between mutated IDH1 and acetyl-CoA carboxylase 1 (ACACA) in leukemia was experimentally validated; this interaction attenuating tumor growth in patient-derived xenografts (PDX) [139]. Finally, certain predicted SL interactions where shown to successfully predict drug sensitivity, thus serving as biologically interpretable biomarkers of the latter [139]. Overall, while these studies have laid a solid basis for some of these genome-wide approaches, extensive research is warranted to further elucidate the potential of SLi based approaches in precision oncology. Moroever, for clinical trials, there is an important unmet need to specifically design and test SLi-based approaches that may uncover a wide range of tumor-specific vulnerabilities.
The Use of In Vitro and In Vivo Models for Guiding Precision Therapy
The increased functional annotation of genetic variants of unknown significance [140], along with systematic high throughput drug screens in 1000 cancer cell lines [141] or 1000 PDX models [142] has significantly increased our understanding of the relationship between genotype and drug sensitivity. We are beginning to understand the molecular profiles of patients responding to conventional chemotherapy, such as to temozolomide [143] and other DNA-damaging agents [144]. Due to their well-characterized clinical benefit in unstratified patient cohorts, these remain equally valuable therapy choices in addition to targeted therapies. With this ever-increasing number of validated biomarkers and available drugs, it is expected that molecular profiling will reveal multiple potentially actionable alterations, which may be treated with a multitude of drugs/drug combinations. Prioritizing predicted treatments requires functional testing, especially in cases where the drug-biomarker association has not been clinically validated. Undoubtedly, there will still be patients whose molecular analysis is either not feasible or does not reveal targetable alterations, for which alternate routes to inform therapy are necessary. For this purpose, several in vitro or in vivo patient-derived functional platforms (e.g. PDX or organoid models) have been developed that mimic the native features of tumors more closely than conventional cell culture drug screening platforms [145].
PDX models offer one attractive approach, as tumor heterogeneity is maintained in these models at least in early passages (for comprehensive review see [146]). In addition, clinical studies have demonstrated remarkable correlations between drug activity in the PDX model and a patient’s clinical outcome [146–149]. However, not all human tumor samples grow in mice following subcutaneous or orthotopic implantation , and the long time span needed for tumor development and expansion to test multiple drugs/drug regimens restricts this approach to patients with a less aggressive disease course [146]. Serial passaging is not only accompanied by the substitution of human stroma with murine components, eventually affecting clonal evolution [146], but also results in extensive mouse colonies and hence, logistical difficulties and rapidly expanding costs. Finally, although humanized mouse models with a (partially) competent “human” immune system have been developed, the remaining technical and biological difficulties of generating these mice, restrict the use of PDX models in studying immunotherapeutic approaches as well as the effects of immunity on the efficacy of other drugs in pre-clinical models [146]. Nonetheless, alternate, in vitro or ex vivo models may substitute the extensive use of PDX.
Patient-derived 3D organoids provide a practical alternative (see other models in Box 2). Organoids are established by dissociating and embedding tissue in an cell-free extracellular matrix (matrigel or collagen), which can be expanded in a growth factor-enriched medium [150]. Organoids from pancreatic [151], colon [152–154], gastric [155], prostate cancer [156] and brain tumors/metastasis [157] have been established, and have the advantage of 3D growth of normal and cancer tissue, recapitulating copy number and mutation spectra, as well as other physiologically relevant aspects of disease progression in vitro [150–158]. Organoids can be established in culture from needle biopsies within a relative short time period, and have also been generated from circulating tumor cells [156]. Organoids can serve as a model system to perform high-throughput screens within a clinically relevant time frame: in a larger precision oncology study, organoids were established from fresh tissue available from 38% of 145 patients [158]. In addition, PDX models were successfully established from these organoids in 19 of 22 attempts [158]. High-throughput drug screens were performed (160 drugs, including chemotherapy and targeted therapy) in 2D cultures from 4 patients, and the best “hits” (drugs that most effectively decreased cell viability in vitro) were verified in 3D organoid cultures. Selected treatments were then tested in combination, to identify effective combination therapies. The best hits of single and combination therapies from 2 patients were further tested in 3D organoids and in PDX models, validating tumor responses in vivo, and compared to the efficacy of current patient treatments [158]. In addition, potential drug toxicities were evaluated (e.g. trametinib and afatinib led to significant weight loss in mice) [158]. For both cases, the combination of targeted therapies was superior over standard chemotherapy [158]. This study underscores the potential use of functional screens in patients where no targeted therapies are available, and the possibility of identifying effective drugs/ drug combinations [158]. The study further demonstrated that therapy recommendations could be retrieved within a clinically relevant time frame (between 7 and 13 weeks) [158], highlighting the importance of defining regulatory routes that might simplify off-label drug access for late-stage patients, often not eligible for clinical trial enrollment (see Outstanding Questions). Therefore, the combination of molecular profiling (genomics and transcriptomics) and functional testing holds promise for determining effective combination therapies for individual cancer patients.
Box 2. Valuable Models for Guiding Precision Therapy.
In addition to PDX and organoids, conditional reprogramming (CR) of patient-derived primary epithelial tumor cells or organotypic cultures are among the possibilities to test selected treatments. Patient-derived cell lines via CR can be rapidly established [203] and are suitable to screen large drug libraries [204, 205], or to test drug combinations to overcome acquired resistance to targeted therapy [206]. While phenotypic features and the genetic heterogeneity of the original tumor are retained in short term CR cultures, the enrichment of specific cell populations, including non-transformed epithelial cells in this model, requires cross-verification of pheno- and genotypic features of donor tissues and CR cells. The lack of a 3D environment may be partially overcome by culturing CR cells in sophisticated 3D artificial organotypic cultures [207], of which fully automated 1,536-well high throughput screening platforms have recently been described [208]. Although these artificial microenvironments lack the heterogeneity observed in patient tumors, they may allow testing tumor cell behaviors in the context of different organ microenvironments shown to influence drug responses [208].
In organotypic slice cultures [209] or organ explants [210], either thin slices of the tumor sample or minced tumor tissues are maintained in culture. The biggest advantage of these culture types is that they retain cancer associated stromal cells, preserve tumor–stroma interactions, signaling pathways and gene expression profiles [211]. Improvements include the use of autologous serum and patient-specific stromal-matrix proteins to more closely resemble individual microenvironmental conditions [212], aiming to accurately predict responses to anticancer drugs.
However, not all tissues are suitable to generating thin slices (e.g. soft, mucinous or fatty tissue), where firm tissue consistency is required [213, 214]. Another drawback of slice cultures is the loss of viability within 5 to 7 days [211]. As the median time frame for molecular profiling and data processing in precision oncology trials is 2–4 weeks, the method is not suitable for testing genomics-guided therapies derived from the same biopsy, unless combined with other models. Recently, organotypic slice cultures established from pancreatic ductal adenocarcinoma PDX models were used to screen against clinically relevant drug regimens in a 96-well format, demonstrating consistency between sensitivity of organotypic cultures and the clinical responses of donor patients [215].
Outstanding Questions Box.
Can the integration of transcriptomic information in clinical decision-making improve patient outcomes? There is a strong biological rationale to support this hypothesis. However, the necessary steps to enable integration of distinct data platforms and to rigorously test the clinical value of transcriptomics remain to be assessed.
How many platforms within the tumor microenvironment (immune, microbiome, metabolome) need to be integrated to provide a multi-dimensional map of the complex tumor landscape? Will it allow a more accurate prediction of regulatory nodes and possible therapeutic modalities?
Preclinical patient-derived models (PDX and/or organoids) demonstrate a strong correlation with patient outcomes. Can regulatory guidelines be defined to render an easier use of off-label drugs in the clinic when based on positive outcomes taken from such models?
How many single cells (and from how many tumors) will we need to sequence to accurately depict the complex heterogeneity observed within patients? Can machine-learning approaches be used to predict a tumor’s behavior?
Are CTCs a reliable source to comprehensively map tumor heterogeneity and do they accurately define phenotypic heterogeneity of advanced disease? How can we improve the isolation of these valuable cells?
How can intra-tumor heterogeneity be exploited therapeutically?
Clinical Management of Therapy Resistance in the Precision Oncology Era
Although genomics-guided therapy is associated with prolonged progression-free survival, cancer patients can generally develop resistance to drugs within 6–12 months, even when trunk mutations are targeted (Figure 2). Such acquired resistance to targeted drugs may be explained by the selection of resistant cancer cells that are present prior to therapy or that are generated de novo as a result of genomic instability. Sequential therapy of 2nd, 3rd and even 4th generation inhibitors that specifically address emerging mutations within the original target (i.e. EGFR inhibitors [159–163]) or drugs targeting newly established driver mutations (e.g. MET amplification in EGFR inhibitor -resistant colorectal cancers [164]) have been used to overcome resistance. Alternatively, combined inhibition of multiple pathways [165] or vertical pathway inhibition (targeting multiple proteins within one pathway) have been suggested to prolong progression-free survival by pre-empting resistance in a pro-active manner and inhibiting the selection of resistant clones [166]. As ana example, targeted therapy has been used against BRAF and MEK in melanoma to counteract feedback regulatory loops and achieve efficient pathway inhibition [18]. Although these approaches are suitable to prolong progression-free survival, management of resistant disease remains dismal/short-lived, partly because multiple resistance mutations (in addition to other mechanisms) can occur simultaneously. This heterogeneity was recently demonstrated in a colorectal cancer patient, where a MEK1 mutation was detected in a liver metastasis biopsy, conferring resistance to cetuximab [167]. Treatment of this patient with the combination of panitumumab and trametinib resulted in regression of the biopsied liver metastasis; however, other liver metastases progressed while on treatment. Analysis of circulating tumor DNA (ctDNA) revealed a previously unrecognized KRAS mutation, which was later found in a biopsy from a progressing liver metastasis, highlighting the challenges of combating polyclonal resistance [167]. To address these issues, more general approaches have been suggested, such as interfering with tumor evolutionary programs, for instance, by increasing genomic instability to lethal levels (e.g. PARP inhibitors in tumors with homologous recombination deficit) [25]. These have been promising strategies to exploit genome instability - as one driving force of heterogeneity -- therapeutically [25]. However, even this approach is accompanied by resistance [25]. Finally, there is growing interest in applying intermittent treatment doses, or adjusting drug doses to limit the evolutionary pressure imposed by a given drug [25]. Such adaptive therapy may serve to maintain a drug-sensitive population, with the goal of stabilizing the tumor size rather than eliminating the tumor. Preliminary evidence for the putative benefit of such drug “holidays” comes from colorectal cancer patients receiving EGFR therapy [168], also suggested for melanoma [169, 170] and recently, breast cancer models [171]. Advances in the characterization of ctDNA now make it possible to carefully evaluate these different methods to combat genetic resistance to targeted drugs in the clinical setting [167, 168, 172–175]. Furthermore, the reappearance of the driver mutation or the appearance of previously undetected mutations associated with resistance to targeted therapy in the blood can enable early detection of therapy failure (before tumor imaging indicates relapse [173, 174]), and might also identify new potential drivers suitable for guiding second-line therapy [167, 168, 172–175], a promising approach for the management of resistant disease.
Figure 2. Targeted Therapy and Mechanisms of Acquired Resistance.

Major classes of current FDA-approved targeted therapies include a. drugs targeting oncogenic drivers or drugs targeting other genetic vulnerabilities, e.g. PARP inhibitors in tumors with HR deficiency; b. drugs that aim to increase the anti-tumor immune-response or c. inhibit neo-angiogenesis. Numerous genetic as well as non-genetic mechanisms (green boxes) of acquired resistance to targeted therapeutics are known, which likely act in concert to mediate the largely short-lived response to these drugs. d. Routes to monitor emerging resistance as well as the suitability of liquid biopsies as compared to tumor biopsies to inform second-line therapy are displayed. Abbreviations: APC, antigen presenting cell; CAFs, cancer associated fibroblasts; ctDNA, circulating tumor DNA; CTC, circulating tumor cell; EMT, epithelial-mesenchymal transition; HR, homologous recombination; NSCLC, non small cell lung cancer; RTK, receptor tyrosin kinase; SCLC, small cell lung cancer;
In addition to the Darwinian-like evolution of genomic alterations in resistant clones under therapeutic pressure, tumors evolve resistance to therapy by adaptively rewiring transduction networks to support the signaling processes required for survival/tumor maintenance in a post-genomic, transient and dynamic manner [176] (Figure 2; Table S2F; S3). One such example is the ability of BRAF-inhibitor sensitive MITFhigh/AXLlow melanoma cell populations to readily switch into a MITFlow/AXLhigh drug resistant population [177, 178]; these cells have been shown to pre-exist in treatment-naïve samples by single-cell RNA-seq [179]. Another example of such phenotypic plasticity has been recently reported in ER+HER2− breast cancers [180]. Following multiple courses of therapy, HER2+ cells lacking gene amplification have been found to emerge in addition to HER2− cells in patient tumors, and among circulating tumor cells (CTCs); this may be indicative of non-genetic mechanisms involved in HER2 upregulation [180]. Furthermore, characterization of patient-derived CTCs revealed that although both, HER2+ and HER2− CTCs maintained tumor-initiating potential in ortothopic xenograft experiments, HER2+ cells were highly proliferative and sensitive to chemotherapy, whereas HER2− CTCs exhibited a slow proliferation rate, upregulated NOTCH signaling, and were chemoresistant [180]. Of note, cells could interconvert between a HER2+ chemosensitive, and a HER2− chemoresistant state, and with chemotherapy a HER2+ population could shift towards a HER2− phenotype [180]. Accordingly, targeting NOTCH in combination with chemotherapy (paclitaxel) suppressed tumor growth in mice, whereas either treatment alone was inefficient in limiting tumor growth [180]. Therefore, rapid interconversion of CTCs between distinct functional states may contribute to acquired resistance to therapy; consequently, it is possible that combination therapy might improve therapeutic responses and delay the onset of resistance. Other non-genetic routes to escape targeted therapy can include the epithelial to mesenchymal transition (EMT), a developmental program that is often hijacked by cancer cells [181]. Preclinical models have associated the EMT program with chemoresistance [182, 183] and a subset of non small cell lung cancer (NSCLC) patients resistant to EGFR-targeted therapy were shown to display an increase in “mesenchymal” cancer cells [184]. Additionally, tumors can also undergo “histological transformations” – as shown for EGFR inhibitor-resistant NSCLC patients whose tumors converted to a small cell lung cancer (SCLC) phenotype, escape therapy [184]. Finally, vascular mimicry, a phenomenon where tumor cells transdifferentiate into endothelial-like cells that can form matrix-rich, vascular-like, perfused channels, have also been proposed to contribute to resistance to anti-angiogenic therapy, but further testing of this mechanism will be required to better understand its role in resistance [185].
Multiple resistance mechanisms (genetic and non-genetic) can act in concert to confer resistance to targeted therapy (Figure 2). Indeed, in a recent melanoma study [186], analysis of patient-matched melanoma tumors biopsied before therapy and during disease progression demonstrated that, in contrast to heterogeneous genetic mechanisms that could result in acquired resistance, transcriptomic signatures were highly recurrent in serial biopsies; these indicated that a multitude of genetic and epigenetic events within the tumor compartment converged on specific genes (c-MET,LEF1,YAP1) and pathways to mediate resistance, consistent with a canalization evolutionary process [186]. Additionally, this acquired resistance signature correlated with changes in the tumor immune microenvironment, including depletion of intratumoral CD8+ T cells, exhaustion of tumor-reactive CD8+ T cells, and loss of antigen presentation; these have been previously linked to resistance to anti-PD-1 salvage therapy in melanoma patient biopsies, in support of the presumed role of CD8+ T cell exhaustion in the development of resistance [77, 186]. Furthermore, these findings suggest that first-line therapy with immune-checkpoint inhibitors followed by BRAF targeted therapy upon relapse in BRAF-mutant melanomas, may be superior over BRAF-inhbitor frontline therapy, a hypothesis that is currently being tested in a clinical trial (NCT02224781).
The mere follow-up of genomic alterations in ctDNA will unlikely result in satisfactory management of resistant disease as adaptive mechanisms on transcriptional and signaling levels can be missed by genomic analysis alone (Figure 2). The use of CTCs may be preferred to monitor resistant disease, as they have a better overall prognostic value, provide an additional opportunity to characterizing genetic and non-genetic intratumor heterogeneity (ITH), and are suitable for functional studies [187]. The previously described in vitro/ex vivo models may serve to detect relevant signaling nodes and counteract adaptive signaling by combinatorial therapy to delay onset of resistance. As suggested for the selection of first line therapies, a comprehensive genomic, transcriptomic and functional analysis of resistant disease is required to overcome these challenges. In that case, it may be possible to mine the increasingly available ‘omics data from large cohorts of patients and identify genetic interactions that mediate network-wide signaling alterations conferring resistance. Such an approach could exploit the much less studied type of genetic interactions, termed synthetic rescues (SRs), also known as suppression interactions [188–192]. Such SRs denote a functional interaction between two genes where the targeting of one gene is compensated by the altered activity of another gene (termed the rescuer gene); this can restore and rescue cell fitness, leading to drug resistance. Like SLs, SR interactions could in principle be identified by mining omics data from large cohorts of pretreated tumor samples, taking into consideration that functional alterations might already be occurring during the natural evolution of tumorigenic populations.
Concluding Remarks
The Road Ahead
Despite the limitations of genomics-driven cancer therapy, current efforts have demonstrated that this approach has the potential to improve clinical outcomes – although at this time, only for a minority of patients. In addition, it has laid the basis for the necessary infrastructure to expand on the concept. Future precision oncology treatment will need to evolve to consider the broader landscape of genetic and epigenetic changes that take place in a tumor, as in its microenvironment, which comprises metabolic as well as immunologic changes, in addition to the influence exerted by the microbiome. Consequently, we must understand that targeting a single pathway in a tumor is in most cases, not sufficient to achieve a sustained response, and we must enforce this principle. We have outlined how the inclusion of transcriptomic data could serve to stratify patients, suggest combination therapies, or define novel vulnerabilities to improve upon current precision oncology trials (Figure 3 and Box 3). Patient-derived ex vivo/ in vivo models can serve to identify the toxicity and efficacy of combination therapies, link genomics to drug responses, and when such information is included in a mineable database, can potentially serve to inform therapeutic decision-making. The limiting factor in performing multiple omics approaches and functional testing, aside from cost, is tissue availability, and one important future step will be to improve the methods to retrieve sufficient tumor material. As such, the use of CTCs is especially appealing, as theses can be non-invasively isolated and may better reflect the prevailing ITH (see Outstanding Questions).
Figure 3. Precision Oncology Workflow.
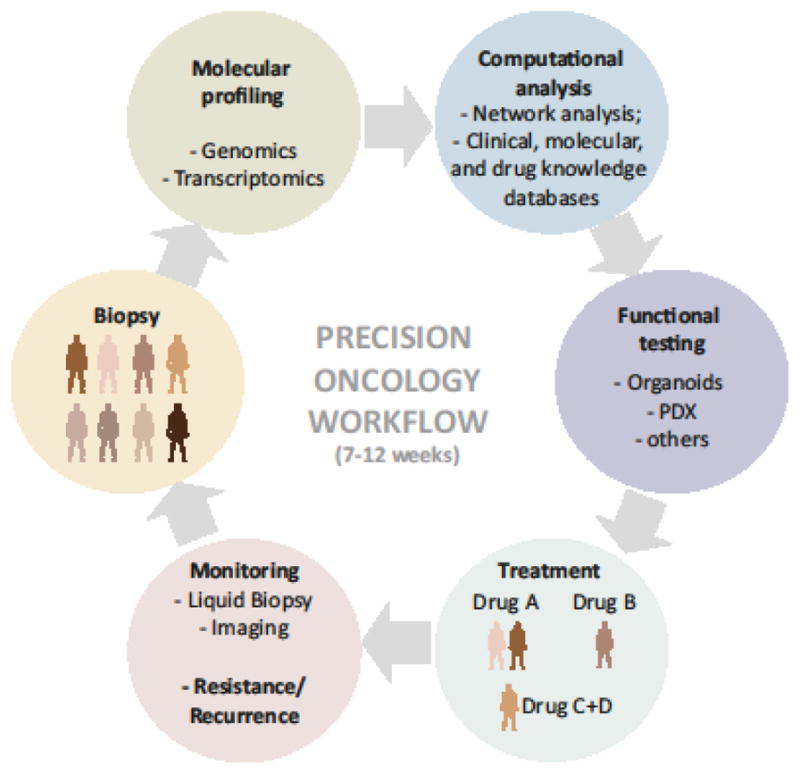
In this proposed precision oncology workflow, patient molecular profiles are used to suggest first-line (combination) therapies. Functional models serve to test safety and efficacy of selected drugs or screen for drugs in cases when molecular profiling is not informative. Patients are closely monitored during the course of therapy to detect resistance. Molecular profiling, computational analysis and functional testing of resistant tumors are repeated upon onset of resistance to determine second-line therapy options.
Box 3. Clinicians’ Corner.
Targeted drugs are largely based on defined drugs (small molecules or biologic antibodies) designed to inhibit specific oncogenic mutations or target key regulatory nodes that drive tumorignesis or underlie cancer vulnerabilities. Usually, the presence of drug-specific biomarkers enables stratification of patients for therapy and monitoring drug effectiveness. Given the success of targeted therapies, together with the recognition that different tumor types share driver / master regulators, the use of drugs that target common regulatory nodes in a histology-agnostic manner is being evaluated in clinical trials.
Clinical experience with genomics-guided cancer therapy supports the notion that genomic profiling can improve patient outcomes. The degree of success can be associated with the ability to verify the role of a targeted mutation/alteration in tumor development, or vice versa, that is, whether the drug can efficiently attenuate the tumorigenic effects orchestrated by the genetic alteration.
Precision oncology cannot be limited to genetics to predict responses to therapy, nor can it be limited to a single “omics” based approach. Multiple drivers can underlie tumor heterogeneity, which in turn can confer resistance, metastasis and dormancy. It also requires the targeting of master regulators that are influenced by epigenetic and microenvironmental-based pathways. The inclusion of additional platforms, of which at this time, the most suitable appears to be transcriptomics, (with future inclusion of metabolome and microbiome analysis) is highly desirable to identifying such master regulators and designing more precise putative therapeutic modalities.
Advances in computational approaches to mine and integrate the multitude of datasets that become available to us are expected to allow better sub-classification of patient cohorts into subpopulations able to respond to a given therapy. In addition, integration of multiple data platforms is expected to drive the identification of novel vulnerabilities, which will further add to the armamentarium of current anticancer therapies. This may likely result in newer stratification methods to identify patients that might benefit from a given precision oncology approach.
The implementation of powerful ex vivo or improved in vivo PDX models in the planning of clinical practice is encouraged. By using these models, a multitude of available drugs and predictive biomarkers might be assessed to evaluate therapeutic options, as well as their combinations and delivery sequence / approaches.
ITH is clearly the biggest obstacle we need to overcome in order to achieve a sustained therapeutic response. With rapid technological advances, we are acquiring the toolbox to comprehensively characterize the complex heterogeneity of tumors at the single cell level (Box 4). However, computing the data from different platforms of 1000 cells remains a challenge. Among the questions that emerge is whether it will be possible to develop multiplex-based approaches, or use machine-learning techniques to compute tumor trends in order to predict sub-cluster and clonal behaviors, and this may be potentially addressed in the near future (see Outstanding Questions).
Box 4. Available tools to decipher Intra-Tumor-Heterogeneity.
ITH manifests as differences in genetic, epigenetic and signaling networks of individual tumor cells coupled with heterogeneity within the stromal compartment [25, 27]. While ITH is influenced by the inherent tumor genetic makeup, epigenetic states (influenced by the location of tumor cells), as well as microenvironmental factors have been recognized as being equally important in dictating the diverse cellular states that drive the primary tumor or its metastatic lesions. Those include the proximity to endothelial cells, cancer associated fibroblasts, immune cells, as well as the nutrient and oxygen availability and biophysical properties of the extracellular matrix [28, 200]. The development of single cell separation and analysis methods, has provided critical insights into the complex heterogeneity of tumors, where multiple clusters of genetically [216] and phenotypically (more so) distinct populations exist. The resulting cell-to-cell variability in stemness and differentiation programs, proliferation and quiescence markers, as well as in the expression of predictive biomarkers [179, 180, 217, 218] define a tumor’s propensity for therapeutic resistance, metastasis and dormancy.
Active research is being undertaken to overcome limitations in single cell methods, including single-cell DNA [219–221] and single cell RNA-seq [222–224] throughput methods, as well as sampling bias. These limitations might be partially resolved by using liquid biopsies [187], and by performing tissue dissociation and dissection of spatial information [225], methods which are currently underway.
Among the first series of steps needed to provide important insights into the complex nature of malignanices, the inclusion of technological and computational advances in clinical trials stands out. These should aim to map and analyze the impact of complex genetic and non-genetic heterogeneity during cancer progression and therapy regimens to help pave the way towards the next generation of cancer therapeutics, capable of overcoming the challenges imposed by the defying intratumor heterogeneity of cancers.
Supplementary Material
TRENDS BOX.
Genomics-driven cancer therapy benefits a subset of patients, although, there are clear shortcomings to this approach
Using genomics as a single “biomarker” to inform therapy is insufficient to comprehensively predict efficient therapeutic approaches. By providing information about active pathways, the inclusion of transcriptomic data reveals a more comprehensive and thus accurate, molecular profile, which likely improves the choice therapy.
Available patient-derived functional models (e.g. organoids or PDX) are promising for testing multiple drugs/drug combinations in a clinically-relevant time frame.
Mining available datasets can allow to comprehensively map the processes that drive cancer and reveal novel vulnerabilities.
Intra-tumor heterogeneity remains one of the biggest challenges in reaching sustained therapeutic responses to cancer treatment. Inclusion of additional factors (immune, metabolome, microbiome) could pinpoint novel putative therapeutic approaches and combinational drug therapies, in an effort to overcome tumor heterogeneity.
Acknowledgments
Support by NCI grant (R35CA197465-01), and a Hervey Family Fund (to ZR) are gratefully acknowledged.
Glossary Box
- Actionable mutations
gene alterations that can be specifically targeted with an approved or investigational drug. The term does not provide information about drug efficacy.
- Acquired resistance
(or secondary resistance), indicates that a tumor which initially responded to therapy becomes resistant to this treatment during the course of therapy. In contrast, in intrinsic (primary) resistance, no responses agiansta tumor are noted upon initiation of therapy.
- Afatinib
tyrosine kinase inhibitor of EGFR (ErbB1), HER2 (ErbB2) and HER4 (ErbB4).
- Angiogenesis
blood vessel formation.
- Basket trial
histology-agnostic trial design allowing to test the efficacy of specific drug(s) in molecularly-stratified patients. It evaluates whether a biomarker (signature) is predictive for drug response irrespective of tumor histology.
- Binary alterations
binary classification of a molecular event, such as somatic mutations (present or absent), gene expression (upregulated or downregulated), or DNA methylation (hypo- or hyper-methylated).
- Biomarker
a molecular characteristic with a correlative or functional association with disease risk, prognosis (prognostic biomarker) or response to treatment (predictive biomarker).
- Canalization evolutionary process
describes the stability of a phenotype despite variation in the genotype.
- Cancer hallmarks
cellular and molecular functions required for cancer development and progression. Hallmarks are sometimes described by a set of genes that perform a specific function.
- Clone
one or more cells derived from and genetically identical to a single ancestor cell. Accordingly, subclones share many of the genetic features of the initial ancestor cell, but contain additional genetic alterations.
- Conditional reprogramming
technique used to establish patient-derived cell cultures from healthy or diseased (e.g. tumor) tissue.
- Copy number profiling
the genome-wide screening for gene copy number variations (gains or losses).
- Circulating tumor cells (CTCs)
tumor cells that can be found in and isolated from the circulation of blood and/or lymphatic system of cancer patients.
- Circulating tumor DNA (ctDNA)
circulating, cell-free tumor DNA that can be isolated from whole blood of cancer patients.
- Cetuximab
anti-EGFR monoclonal antibody that binds to the extracellular domain of EGFR and prevents its dimerization.
- Differentiation hierarchies
relates to differences in the impact of phenotype seen in an isogenic population of cancer cells; e.g. in their ability to metastasize or form tumors upon serial transplantation on immunodeficient mice (i.e. cancer stem cells).
- Deep neural nets
neural network of multiple layers often used for supervised learning; at each layer, a function is applied to the input from the previous layer.
- Dimension reduction
selecting a subset of features, or combining features, from a dataset (e.g. principal component analysis).
- DNA methylation profiling
genome-wide screening for variations in DNA methylation status (hyper- or hypomethylation).
- Driver
usually refers to a genetic event that is shown to elicit phenotypic changes associated with tumor initiation and progression (see oncogenic drivers). In a broader definition, the term can also be used to describe non-genetic and/or non-cell autonomous alterations that can alter certain aspects of disease progression.
- Epigenetic alterations
heritable trait not explained by changes in DNA sequence but by changes in gene expression. Common examples often observed in cancer cells include promoter hypermethylation or aberrant histone modifications (e.g. acetylation).
- Evolutionary pressure
(or selection pressure); any change in the microenvironment (e.g. cancer therapies) that leads to a selection of clones that have a growth advantage under these new conditions.
- Functional mutations
somatic mutations that change the phenotype of a cancer cell or tumor.
- Gene fusions
hybrid genes that combine parts of two or more original genes. Fusion genes originate from chromosomal rearrangements (i.e. deletions, translocations, tandem duplications, inversions).
- Genetic interaction
functional interplay between multiple genes and their corresponding gene products that has an impact on the cellular phenotype.
- Genotype-matched trials
when a clinical trial is selected for a patient based on his/her genotype. This can be a study that only accepts patients with a given mutation, but also any study that either analyses a drug that is likely to be effective in the context of a given genotype, or inhibits a pathway that is directly linked to a mutation identified in the patient considered to be genotype-matched.
- HDAC (histone deacetylases) inhibitors
HDACs remove acetyl groups from histone lysine residues of but also from non-histone substrates. HDAC inhibitors can have transcription-dependent effects (e.g. relief of transcriptional repression of tumor suppressor genes) but also transcription-independent effects due to altered acetylation (and activity) of non-histone substrates, e.g. involved in cell proliferation or cell death.
- Homologous recombination deficiency
defect in double-strand break repair by homologous recombination repair associated with high levels of genomic instability. These defects were originally identified in BRCA1/2-deficient tumors, but can be observed in the absence of BRCA-mutations, a phenomenon generally referred to as ‘BRCA-ness’.
- ICGC
International Cancer Genome Consortium (ICGC) has the goal to generate comprehensive catalogues of genomic abnormalities (somatic mutations, abnormal expression of genes, epigenetic modifications) in tumors from 50 different cancer types and/or subtypes and make the data available to the entire research community to accelerate research into the causes and control of cancer.
- ICGC-TCGA DREAM Somatic Mutation Calling Challenge – RNA
The DREAM Challenges are crowdsourcing (open science efforts) challenges examining questions in biology and medicine. In the RNA-Challenge“, leaders from ICGC and The Cancer Genome Atlas (TCGA) have come together to develop a Challenge to assess the accuracy of methods to work with cancer RNA Sequencing data.
- Immune-checkpoint inhibitors
drugs that block immune-inhibitory signals (such as PD1, PD-L1 or CTLA4) expressed on/by tumor or immune cells. Inhibition of these factors can unleash, in some cases, a robust and durable antitumor immune response.
- “Integrated” subtype
a subtype of cancer that includes patients with tumors from multiple tissues-of-origin.
- Inter-tumor heterogeneity
differences in the molecular makeup of tumor cells between patients.
- Intra-tumor heterogeneity
differences in the molecular makeup of tumor cells within individual patients.
- Match-rate
frequency by which genomic alterations detected in a patient cohort can be matched to targeted therapies.
- Methylome
collection of all DNA methylation markers in a single cell or a population of cells.
- MOSCATO 01
prospective clinical trial evaluating whether high-throughput genomic analyses can improve patient outcomes.
- “multi-view” matrix factorization
factorization of multiple data matrices into a lower-dimensional space, where each data matrix gives a different “view” of the data (e.g. gene expression and DNA methylation views).
- Off-label drug use
a drug is used for a purpose not specified in the marketing authorization determined by a licensing body such as the FDA (e.g. drug used for a different type of cancer than the one it is approved to treat).
- Oncogenic drivers
genetic events associated with tumor initiation and progression, including metastasis and therapy resistance.
- Orthotopic implantation
a form of xenograft experiment where (patient-derived) tumor cells are implanted into the organ of origin to maintain the tissue-specific environment.
- Paclitaxel
chemotherapeutic drug that binds tubulin and inhibits the disassembly of microtubules, thereby resulting in the inhibition of cell division.
- Panitumumab
monoclonal antibody that binds to and inhibits (autocrine) stimulation of the epidermal growth factor receptor (EGFR)
- Passenger mutations
somatic mutations with no obvious role (owing to their inability to cause phenotypic changes) in cancer.
- Patient-derived xenograft (PDX)
xenograft model wheretumor cells (derived from a biopsy or bodily fluids) are subcutaneously or orthotopically engrafted into immune-incompetent mice, without any intermediate growth or modification in culture.
- Phenotypic heterogeneity
differences in phenotypic characteristics (e.g. potential to form metastasis or resist therapy) of genetically diverse but also isogenic tumor cells.
- Rare variants
genetic variations (i.e. mutations) within a particular gene that occur in less than 1% of patients.
- Recurrent mutation
somatic mutation that occurs in a statistically significant number of times in a cohort of sequenced tumors.
- Sample-level events
measurements of individual tumors, patients, or cancer cell lines (e.g. drug sensitivity).
- Saturation analysis
modeling of sample complexity to determine the number of samples required to detect some event as statistically significant.
- Sequencing depth
reflects the average number of times a given region has been sequenced by independent reads.
- Serial passaging
serial transplantation of tumors (usually in the flank of nude mice) for tumor expansion.
- SHIVA study; the first randomized
open-label, controlled phase II study that evaluated whether targeted treatment based on molecular testing improved patient outcomes as compared to conventional treatments across all tumor types.
- Somatic variation
genetic mutations occuring in a somatic (body) cell.
- Splice variants
transcripts (mRNAs) resulting from the alternative splicing of different exons in genes.
- Synthetic rescue
type of genetic interaction where the mutation/deletion of one gene rescues the lethality or growth defect of a cell mutated/deleted for another gene.
- Suppression interactions
phenotypic defects caused by a mutation in a particular gene are rescued by a mutation/deregulation in a second gene
- Targeted therapies
drugs that either target molecular alterations specific to cancer cells (e.g. mutated, amplified or epigenetically up/downregulated signaling proteins), or target immune cells to increase anti-cancer immunity.
- TCGA
The Cancer Genome Atlas (TCGA) collaboration between the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI); it has generated comprehensive, multi-dimensional maps of key genomic changes in 33 types of cancer.
- Temozolomide
chemotherapeutic drug in the class of alkylating agents exerting cytotoxicity by inhibiting DNA replication.
- Trametinib
targeted therapy that specifically binds and inhibits MEK1/2.
- Trunk mutations
genetic variations occurring in early tumor evolution and common to most clones.
- Umbrella trial
study design evaluating the efficacy of multiple drugs within a biomarker-stratified single cancer type. They allow the evaluate of multiple biomarker-drug combinations in a histology-dependent manner.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer. 2016;16(8):525–37. doi: 10.1038/nrc.2016.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman DM, et al. Implementing Genome-Driven Oncology. Cell. 2017;168(4):584–599. doi: 10.1016/j.cell.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prasad V. Perspective: The precision-oncology illusion. Nature. 2016;537(7619):S63. doi: 10.1038/537S63a. [DOI] [PubMed] [Google Scholar]
- 4.Tannock IF, Hickman JA. Limits to Personalized Cancer Medicine. N Engl J Med. 2016;375(13):1289–94. doi: 10.1056/NEJMsb1607705. [DOI] [PubMed] [Google Scholar]
- 5.Voest EE, Bernards R. DNA-Guided Precision Medicine for Cancer: A Case of Irrational Exuberance? Cancer Discov. 2016;6(2):130–2. doi: 10.1158/2159-8290.CD-15-1321. [DOI] [PubMed] [Google Scholar]
- 6.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–31. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sidransky D. Nucleic acid-based methods for the detection of cancer. Science. 1997;278(5340):1054–9. doi: 10.1126/science.278.5340.1054. [DOI] [PubMed] [Google Scholar]
- 8.Soussi T. The history of p53. A perfect example of the drawbacks of scientific paradigms. EMBO Rep. 2010;11(11):822–6. doi: 10.1038/embor.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobleigh MA, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 11.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 12.Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 14.Saltz LB, et al. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 15.Ledermann J, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 18.Johnson DB, et al. Emerging targeted therapies for melanoma. Expert Opin Emerg Drugs. 2016;21(2):195–207. doi: 10.1080/14728214.2016.1184644. [DOI] [PubMed] [Google Scholar]
- 19.Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol. 2011;6:345–64. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- 20.Jones PA, et al. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–41. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- 21.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438(7070):967–74. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 22.Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015;27(4):439–49. doi: 10.1016/j.ccell.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research N et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International Cancer Genome C et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168(4):613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Marusyk A, et al. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 28.Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15(8):473–83. doi: 10.1038/nrc3971. [DOI] [PubMed] [Google Scholar]
- 29.Hofree M, et al. Challenges in identifying cancer genes by analysis of exome sequencing data. Nat Commun. 2016;7:12096. doi: 10.1038/ncomms12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forbes SA, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Hofree M, et al. Network-based stratification of tumor mutations. Nat Methods. 2013;10(11):1108–15. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence MS, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leiserson MD, et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015;47(2):106–14. doi: 10.1038/ng.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoadley KA, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–44. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartmaier RJ, et al. Genomic analysis of 63,220 tumors reveals insights into tumor uniqueness and targeted cancer immunotherapy strategies. Genome Med. 2017;9(1):16. doi: 10.1186/s13073-017-0408-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dienstmann R, et al. Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov. 2015;5(2):118–23. doi: 10.1158/2159-8290.CD-14-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubio-Perez C, et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015;27(3):382–96. doi: 10.1016/j.ccell.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Biankin AV, et al. Patient-centric trials for therapeutic development in precision oncology. Nature. 2015;526(7573):361–70. doi: 10.1038/nature15819. [DOI] [PubMed] [Google Scholar]
- 42.Rashdan S, Gerber DE. Going into BATTLE: umbrella and basket clinical trials to accelerate the study of biomarker-based therapies. Ann Transl Med. 2016;4(24):529. doi: 10.21037/atm.2016.12.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Tourneau C, et al. Treatment Algorithms Based on Tumor Molecular Profiling: The Essence of Precision Medicine Trials. J Natl Cancer Inst. 2016;108(4) doi: 10.1093/jnci/djv362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Massard C, et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017;7(6):586–595. doi: 10.1158/2159-8290.CD-16-1396. [DOI] [PubMed] [Google Scholar]
- 45.Le Tourneau C, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–34. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 46.Wheler JJ, et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016;76(13):3690–701. doi: 10.1158/0008-5472.CAN-15-3043. [DOI] [PubMed] [Google Scholar]
- 47.Lopez-Chavez A, et al. Molecular profiling and targeted therapy for advanced thoracic malignancies: a biomarker-derived, multiarm, multihistology phase II basket trial. J Clin Oncol. 2015;33(9):1000–7. doi: 10.1200/JCO.2014.58.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stockley TL, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016;8(1):109. doi: 10.1186/s13073-016-0364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hyman DM, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015;373(8):726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kopetz S, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. Journal of Clinical Oncology. 2010;28(15_suppl):3534–3534. [Google Scholar]
- 51.Kaufman B, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–50. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Ahmadie H, et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 2014;4(9):1014–21. doi: 10.1158/2159-8290.CD-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banerji U, et al. Abstract LB-66: Results of two phase I multicenter trials of AZD5363, an inhibitor of AKT1, 2 and 3: Biomarker and early clinical evaluation in Western and Japanese patients with advanced solid tumors. Cancer Research. 2014;73(8 Supplement):LB-66. [Google Scholar]
- 54.Cappuzzo F, et al. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N Engl J Med. 2006;354(24):2619–21. doi: 10.1056/NEJMc060020. [DOI] [PubMed] [Google Scholar]
- 55.Hammerman PS, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 2011;1(1):78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iyer G, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;338(6104):221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao RG, et al. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013;73(16):5195–205. doi: 10.1158/0008-5472.CAN-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Serra V, et al. Clinical response to a lapatinib-based therapy for a Li-Fraumeni syndrome patient with a novel HER2V659E mutation. Cancer Discov. 2013;3(11):1238–44. doi: 10.1158/2159-8290.CD-13-0132. [DOI] [PubMed] [Google Scholar]
- 59.Subbiah V, et al. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J Hematol Oncol. 2014;7:8. doi: 10.1186/1756-8722-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Allen EM, et al. Genomic Correlate of Exceptional Erlotinib Response in Head and Neck Squamous Cell Carcinoma. JAMA Oncol. 2015;1(2):238–44. doi: 10.1001/jamaoncol.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagle N, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371(15):1426–33. doi: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagle N, et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014;4(5):546–53. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen JC, et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell. 2014;159(2):402–14. doi: 10.1016/j.cell.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo J, et al. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alizadeh AA, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(8):846–53. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fishbein L, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell. 2017;31(2):181–193. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kato S, et al. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin Cancer Res. 2017;23(8):1988–1997. doi: 10.1158/1078-0432.CCR-16-1679. [DOI] [PubMed] [Google Scholar]
- 69.Stransky N, et al. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alvarez MJ, et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48(8):838–47. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tian S, et al. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut. 2013;62(4):540–9. doi: 10.1136/gutjnl-2012-302423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vecchione L, et al. A Vulnerability of a Subset of Colon Cancers with Potential Clinical Utility. Cell. 2016;165(2):317–30. doi: 10.1016/j.cell.2016.02.059. [DOI] [PubMed] [Google Scholar]
- 73.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 74.Konstantinopoulos PA, et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol. 2010;28(22):3555–61. doi: 10.1200/JCO.2009.27.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Larsen MJ, et al. Classifications within molecular subtypes enables identification of BRCA1/BRCA2 mutation carriers by RNA tumor profiling. PLoS One. 2013;8(5):e64268. doi: 10.1371/journal.pone.0064268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Allen EM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hugo W, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ewing AD, et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat Methods. 2015;12(7):623–30. doi: 10.1038/nmeth.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zehir A, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ciriello G, et al. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang B, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11(3):333–7. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 82.Meng C, et al. Dimension reduction techniques for the integrative analysis of multi-omics data. Brief Bioinform. 2016;17(4):628–41. doi: 10.1093/bib/bbv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fakoor R, et al. Using deep learning to enhance cancer diagnosis and classification. Proceedings of the ICML Workshop on the Role of Machine Learning in Transforming Healthcare; 2013. p. 28. [Google Scholar]
- 84.Ching T, et al. Opportunities And Obstacles For Deep Learning. Biology And Medicine. 2017 doi: 10.1098/rsif.2017.0387. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dienstmann R, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17(2):79–92. doi: 10.1038/nrc.2016.126. [DOI] [PubMed] [Google Scholar]
- 87.Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reva B, et al. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang MT, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34(2):155–63. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao J, et al. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Med. 2017;9(1):4. doi: 10.1186/s13073-016-0393-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miller ML, et al. Pan-Cancer Analysis of Mutation Hotspots in Protein Domains. Cell Syst. 2015;1(3):197–209. doi: 10.1016/j.cels.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jia P, Zhao Z. VarWalker: personalized mutation network analysis of putative cancer genes from next-generation sequencing data. PLoS Comput Biol. 2014;10(2):e1003460. doi: 10.1371/journal.pcbi.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Polak P, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518(7539):360–364. doi: 10.1038/nature14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mermel CH, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wendl MC, et al. PathScan: a tool for discerning mutational significance in groups of putative cancer genes. Bioinformatics. 2011;27(12):1595–602. doi: 10.1093/bioinformatics/btr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Glaab E, et al. EnrichNet: network-based gene set enrichment analysis. Bioinformatics. 2012;28(18):i451–i457. doi: 10.1093/bioinformatics/bts389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vandin F, et al. Algorithms for detecting significantly mutated pathways in cancer. J Comput Biol. 2011;18(3):507–22. doi: 10.1089/cmb.2010.0265. [DOI] [PubMed] [Google Scholar]
- 100.Vanunu O, et al. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010;6(1):e1000641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim JW, et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat Biotechnol. 2016;34(5):539–46. doi: 10.1038/nbt.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Knijnenburg TA, et al. Logic models to predict continuous outputs based on binary inputs with an application to personalized cancer therapy. Sci Rep. 2016;6:36812. doi: 10.1038/srep36812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ciriello G, et al. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22(2):398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim YA, et al. WeSME: uncovering mutual exclusivity of cancer drivers and beyond. Bioinformatics. 2017;33(6):814–821. doi: 10.1093/bioinformatics/btw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leiserson MD, et al. A weighted exact test for mutually exclusive mutations in cancer. Bioinformatics. 2016;32(17):i736–i745. doi: 10.1093/bioinformatics/btw462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miller CA, et al. Discovering functional modules by identifying recurrent and mutually exclusive mutational patterns in tumors. BMC Med Genomics. 2011;4:34. doi: 10.1186/1755-8794-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vandin F, et al. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012;22(2):375–85. doi: 10.1101/gr.120477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vandin F, et al. On the Sample Complexity of Cancer Pathways Identification. J Comput Biol. 2016;23(1):30–41. doi: 10.1089/cmb.2015.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang FW, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–9. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ainscough BJ, et al. DoCM: a database of curated mutations in cancer. Nat Methods. 2016;13(10):806–7. doi: 10.1038/nmeth.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Griffith M, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49(2):170–174. doi: 10.1038/ng.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leiserson MD, et al. MAGI: visualization and collaborative annotation of genomic aberrations. Nat Methods. 2015;12(6):483–4. doi: 10.1038/nmeth.3412. [DOI] [PubMed] [Google Scholar]
- 113.Gerstung M, et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat Genet. 2017;49(3):332–340. doi: 10.1038/ng.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brough R, et al. Searching for synthetic lethality in cancer. Curr Opin Genet Dev. 2011;21(1):34–41. doi: 10.1016/j.gde.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 115.Hartwell LH, et al. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278(5340):1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 116.Lord CJ, et al. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 2008;7(12):2010–9. doi: 10.1016/j.dnarep.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 117.Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin SA, et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med. 2009;1(6–7):323–37. doi: 10.1002/emmm.200900040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Srivas R, et al. A Network of Conserved Synthetic Lethal Interactions for Exploration of Precision Cancer Therapy. Mol Cell. 2016;63(3):514–25. doi: 10.1016/j.molcel.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang T, et al. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168(5):890–903. e15. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang X, et al. Widespread genetic epistasis among cancer genes. Nat Commun. 2014;5:4828. doi: 10.1038/ncomms5828. [DOI] [PubMed] [Google Scholar]
- 122.Aguirre AJ, et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016;6(8):914–29. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cheung HW, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108(30):12372–7. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cowley GS, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marcotte R, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2(2):172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marcotte R, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell. 2016;164(1–2):293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beijersbergen RL, et al. Synthetic Lethality in Cancer Therapeutics. Annual Review of Cancer Biology. 2017;1(1):141–161. [Google Scholar]
- 128.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327(5964):425–31. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu X, et al. Genome evolution predicts genetic interactions in protein complexes and reveals cancer drug targets. Nat Commun. 2013;4:2124. doi: 10.1038/ncomms3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Madhukar NS, et al. Prediction of Genetic Interactions Using Machine Learning and Network Properties. Front Bioeng Biotechnol. 2015;3:172. doi: 10.3389/fbioe.2015.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhong W, Sternberg PW. Genome-wide prediction of C. elegans genetic interactions. Science. 2006;311(5766):1481–4. doi: 10.1126/science.1123287. [DOI] [PubMed] [Google Scholar]
- 132.Conde-Pueyo N, et al. Human synthetic lethal inference as potential anti-cancer target gene detection. BMC Syst Biol. 2009;3:116. doi: 10.1186/1752-0509-3-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Folger O, et al. Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol. 2011;7:501. doi: 10.1038/msb.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Frezza C, et al. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 2011;89(3):213–20. doi: 10.1007/s00109-011-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang K, et al. Genome-wide identification of post-translational modulators of transcription factor activity in human B cells. Nat Biotechnol. 2009;27(9):829–39. doi: 10.1038/nbt.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jerby-Arnon L, et al. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell. 2014;158(5):1199–209. doi: 10.1016/j.cell.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 137.Park S, Lehner B. Cancer type-dependent genetic interactions between cancer driver alterations indicate plasticity of epistasis across cell types. Mol Syst Biol. 2015;11(7):824. doi: 10.15252/msb.20156102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Szczurek E, et al. Synthetic sickness or lethality points at candidate combination therapy targets in glioblastoma. Int J Cancer. 2013;133(9):2123–32. doi: 10.1002/ijc.28235. [DOI] [PubMed] [Google Scholar]
- 139.Sinha S, et al. Systematic discovery of mutation-specific synthetic lethals by mining pan-cancer human primary tumor data. Nat Commun. 2017;8:15580. doi: 10.1038/ncomms15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Berger AH, et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. 2016;30(2):214–228. doi: 10.1016/j.ccell.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Iorio F, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016;166(3):740–54. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gao H, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–25. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 143.Nagel ZD, et al. DNA Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res. 2017;77(1):198–206. doi: 10.1158/0008-5472.CAN-16-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Reinhold WC, et al. DNA-Targeted Precision Medicine; Have we Been Caught Sleeping? Trends Cancer. 2017;3(1):2–6. doi: 10.1016/j.trecan.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Friedman AA, et al. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15(12):747–56. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Byrne AT, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17(4):254–268. doi: 10.1038/nrc.2016.140. [DOI] [PubMed] [Google Scholar]
- 147.Garralda E, et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin Cancer Res. 2014;20(9):2476–84. doi: 10.1158/1078-0432.CCR-13-3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hidalgo M, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10(8):1311–6. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stebbing J, et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer. 2014;120(13):2006–15. doi: 10.1002/cncr.28696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fatehullah A, et al. Organoids as an in vitro model of human development and disease. Nat Cell Biol. 2016;18(3):246–54. doi: 10.1038/ncb3312. [DOI] [PubMed] [Google Scholar]
- 151.Boj SF, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–38. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141(5):1762–72. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 153.van de Wetering M, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933–45. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weeber F, et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc Natl Acad Sci U S A. 2015;112(43):13308–11. doi: 10.1073/pnas.1516689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nadauld LD, et al. Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol. 2014;15(8):428. doi: 10.1186/s13059-014-0428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gao D, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–87. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hubert CG, et al. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016;76(8):2465–77. doi: 10.1158/0008-5472.CAN-15-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pauli C, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017;7(5):462–477. doi: 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ercan D, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. 2015;21(17):3913–23. doi: 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Janne PA, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–99. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 161.Jia Y, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–32. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sequist LV, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–9. doi: 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]
- 163.Zhou W, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–4. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bardelli A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–73. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bozic I, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shahbazian D, et al. Vertical pathway targeting in cancer therapy. Adv Pharmacol. 2012;65:1–26. doi: 10.1016/B978-0-12-397927-8.00001-4. [DOI] [PubMed] [Google Scholar]
- 167.Russo M, et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016;6(2):147–153. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Siravegna G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Das Thakur M, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494(7436):251–5. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sun C, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508(7494):118–22. doi: 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 171.Enriquez-Navas PM, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24. doi: 10.1126/scitranslmed.aad7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Diaz LA, Jr, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Girotti MR, et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016;6(3):286–99. doi: 10.1158/2159-8290.CD-15-1336. [DOI] [PubMed] [Google Scholar]
- 174.Misale S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Murtaza M, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–12. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 176.Kolch W, et al. The dynamic control of signal transduction networks in cancer cells. Nat Rev Cancer. 2015;15(9):515–27. doi: 10.1038/nrc3983. [DOI] [PubMed] [Google Scholar]
- 177.Konieczkowski DJ, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4(7):816–27. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Muller J, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. 2014;5:5712. doi: 10.1038/ncomms6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–96. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jordan NV, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537(7618):102–106. doi: 10.1038/nature19328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nieto MA, et al. Emt: 2016. Cell. 2016;166(1):21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 182.Fischer KR, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–6. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zheng X, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sequist LV, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dey N, et al. Evading anti-angiogenic therapy: resistance to anti-angiogenic therapy in solid tumors. Am J Transl Res. 2015;7(10):1675–98. [PMC free article] [PubMed] [Google Scholar]
- 186.Hugo W, et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015;162(6):1271–85. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Alix-Panabieres C, Pantel K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016;6(5):479–91. doi: 10.1158/2159-8290.CD-15-1483. [DOI] [PubMed] [Google Scholar]
- 188.Fong CY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538–42. doi: 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Miyamoto DT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351–6. doi: 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Motter AE, et al. Predicting synthetic rescues in metabolic networks. Mol Syst Biol. 2008;4:168. doi: 10.1038/msb.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rathert P, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525(7570):543–547. doi: 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.van Leeuwen J, et al. Exploring genetic suppression interactions on a global scale. Science. 2016;354(6312) doi: 10.1126/science.aag0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu F, et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Mol Cell. 2015;60(2):307–18. doi: 10.1016/j.molcel.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhu J, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525(7568):206–11. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mertins P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang B, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513(7518):382–7. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhang H, et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell. 2016;166(3):755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Drake JM, et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell. 2016;166(4):1041–1054. doi: 10.1016/j.cell.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–55. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 200.Senft D, Ronai ZA. Adaptive Stress Responses During Tumor Metastasis and Dormancy. Trends Cancer. 2016;2(8):429–442. doi: 10.1016/j.trecan.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Davidson SM, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23(3):517–28. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hensley CT, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164(4):681–94. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liu X, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12(2):439–451. doi: 10.1038/nprot.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Beglyarova N, et al. Screening of Conditionally Reprogrammed Patient-Derived Carcinoma Cells Identifies ERCC3-MYC Interactions as a Target in Pancreatic Cancer. Clin Cancer Res. 2016;22(24):6153–6163. doi: 10.1158/1078-0432.CCR-16-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Saeed K, et al. Comprehensive Drug Testing of Patient-derived Conditionally Reprogrammed Cells from Castration-resistant Prostate Cancer. Eur Urol. 2017;71(3):319–327. doi: 10.1016/j.eururo.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 206.Crystal AS, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–6. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ridky TW, et al. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat Med. 2010;16(12):1450–5. doi: 10.1038/nm.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kenny HA, et al. Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat Commun. 2015;6:6220. doi: 10.1038/ncomms7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Vaira V, et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc Natl Acad Sci U S A. 2010;107(18):8352–6. doi: 10.1073/pnas.0907676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Freeman AE, Hoffman RM. In vivo-like growth of human tumors in vitro. Proc Natl Acad Sci U S A. 1986;83(8):2694–8. doi: 10.1073/pnas.83.8.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Meijer TG, et al. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci OA. 2017;3(2):FSO190. doi: 10.4155/fsoa-2017-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Majumder B, et al. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat Commun. 2015;6:6169. doi: 10.1038/ncomms7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Holliday DL, et al. The practicalities of using tissue slices as preclinical organotypic breast cancer models. J Clin Pathol. 2013;66(3):253–5. doi: 10.1136/jclinpath-2012-201147. [DOI] [PubMed] [Google Scholar]
- 214.Naipal KA, et al. Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer. 2016;16:78. doi: 10.1186/s12885-016-2119-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Roife D, et al. Ex Vivo Testing of Patient-Derived Xenografts Mirrors the Clinical Outcome of Patients with Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2016;22(24):6021–6030. doi: 10.1158/1078-0432.CCR-15-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–60. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tirosh I, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539(7628):309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Gawad C, et al. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175–88. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 220.Vitak SA, et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat Methods. 2017;14(3):302–308. doi: 10.1038/nmeth.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zahn H, et al. Scalable whole-genome single-cell library preparation without preamplification. Nat Methods. 2017;14(2):167–173. doi: 10.1038/nmeth.4140. [DOI] [PubMed] [Google Scholar]
- 222.Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161(5):1187–201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Macosko EZ, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161(5):1202–14. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zheng GX, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tsoucas D, Yuan GC. Recent progress in single-cell cancer genomics. Curr Opin Genet Dev. 2017;42:22–32. doi: 10.1016/j.gde.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tsimberidou AM, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18(22):6373–83. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Schwaederle M, et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol Cancer Ther. 2016;15(4):743–52. doi: 10.1158/1535-7163.MCT-15-0795. [DOI] [PubMed] [Google Scholar]
- 228.Von Hoff DD, et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28(33):4877–83. doi: 10.1200/JCO.2009.26.5983. [DOI] [PubMed] [Google Scholar]
- 229.Park JW, et al. Adaptive Randomization of Neratinib in Early Breast Cancer. N Engl J Med. 2016;375(1):11–22. doi: 10.1056/NEJMoa1513750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rugo HS, et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N Engl J Med. 2016;375(1):23–34. doi: 10.1056/NEJMoa1513749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tripathy D, et al. Adaptively randomized trial of neoadjuvant chemotherapy with or without the Akt inhibitor MK-2206: Graduation results from the I-SPY 2 Trial. Journal of Clinical Oncology. 2015;33(15) [Google Scholar]
- 232.Kaplan R, et al. Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol. 2013;31(36):4562–8. doi: 10.1200/JCO.2013.50.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.