

Abstract
Trehalose is a non-reducing sugar whose ability to stabilize biomolecules has brought about its widespread use in biological preservation applications. Trehalose is also an essential metabolite in a number of pathogens, most significantly the global pathogen Mycobacterium tuberculosis, though it is absent in humans and other mammals. Recently, there has been a surge of interest in modifying the structure of trehalose to generate analogues that have applications in biomedical research and biotechnology. Non-degradable trehalose analogues could have a number of advantages as bioprotectants and food additives. Trehalose-based imaging probes and inhibitors are already useful as research tools and may have future value in the diagnosis and treatment of tuberculosis, among other uses. Underlying the advancements made in these areas are novel synthetic methods that facilitate access to and evaluation of trehalose analogues. In this review, we focus on both aspects of the development of this class of molecules. First, we consider the chemical and chemoenzymatic methods that have been used to prepare trehalose analogues and discuss their prospects for synthesis on commercially relevant scales. Second, we describe ongoing efforts to develop and deploy detectable trehalose analogues, trehalose-based inhibitors, and non-digestible trehalose analogues. The current and potential future uses of these compounds are discussed, with an emphasis on their roles in understanding and combatting mycobacterial infection.
Keywords: Trehalose, analogue, chemical synthesis, chemoenzymatic synthesis, biocatalysis, imaging, inhibitors, mycobacteria, biopreservation
INTRODUCTION
Trehalose (1) is a C2-symmetric, non-reducing disaccharide consisting of two glucose residues connected by a 1,1-α,α-glycosidic linkage (Figure 1). It is abundant and widely distributed in nature, as it occurs in bacteria, yeast, fungi, plants, and invertebrates.1 The distinctive structure and exceptional stability of trehalose give rise to its main biological function, which is stress protection.1 The ability of trehalose to stabilize a wide range of biomaterials—including proteins, DNA, cells, and tissues—has been exploited for myriad preservation applications in the pharmaceutical, food, and cosmetic industries.2 Beyond bioprotection, trehalose and its naturally occurring derivatives have various other functions, depending on the organism. For example, insects store high levels of blood trehalose, which can be rapidly utilized to permit flight.3 In plants, trehalose’s biosynthetic precursor, trehalose-6-phosphate (2), has been implicated in plant growth regulation and development.4 Some actinobacteria express lentztrehalose A (3), which has a 2,3-dihydroxy-3-methylbutoxy group that renders the molecule resistant to degradation.5 Trehalose is also essential for growth and virulence of globally significant pathogens such as Mycobacterium tuberculosis, which not only uses trehalose for energy storage and stress resistance, but also incorporates trehalose into virulence-associated cell envelope glycolipids (e.g., 4 and 5).6
Figure 1.

Structures of trehalose and a few naturally occurring trehalose derivatives.
Synthetic trehalose analogues, which contain structural modifications designed to impart particular properties or functions to the native disaccharide, are of interest for numerous applications. In relation to the expansive use of trehalose as a bioprotectant and food additive, there is considerable interest in developing metabolically stable versions of trehalose that, upon ingestion by a human, would not be broken down into glucose by the intestinal trehalase enzyme.7 Indeed, trehalase-resistant trehalose analogues, including lentztrehalose A (3), may retain desirable properties (e.g., biomolecular stabilization, sweetness) while increasing residence time in the body and reducing caloric intake.5,7 Due to the importance of trehalose to various pathogens (e.g., M. tuberculosis) and insect vectors of pathogens (e.g., mosquitoes), as well as the lack of trehalose in mammals, recent research has focused on targeting trehalose metabolism in these organisms with analogues designed to facilitate disease diagnosis, treatment, or prevention of transmission.8,9 Unfortunately, the difficulties associated with trehalose analogue synthesis have impeded progress in the development and applications of these compounds. Despite steady advances in the stereoselective construction of carbohydrates and carbohydrate analogues over the past few decades, the unique 1,1-α,α-glycosidic linkage of trehalose has rendered analogue synthesis very challenging. In turn, it has been challenging to access trehalose analogues on a laboratory scale for research purposes, let alone on an industrial scale for commercial applications.
In this review, we discuss progress in the synthesis and applications of trehalose analogues. After a primer on trehalose metabolism to orient readers to this review’s content, we provide an overview of the synthetic approaches used to produce trehalose analogues. Although chemical synthesis methods are briefly discussed in this review, we point readers to two excellent reviews by the Kulkarni group focusing on chemical methods for 1,1-α,α-glycosylation and trehalose desymmetrization.10,11 It is noted that since 2013, a number of new reports describing chemical syntheses of various mycobacterial glycolipids have been published as well.12–18 Here, we emphasize recent advances in trehalose analogue synthesis using chemoenzymatic methods, which have been inspired by naturally occurring trehalose biosynthetic pathways. The bulk of this review is devoted to the various emerging applications of trehalose analogues, which range from the detection and treatment of pathogenic organisms to the development of non-digestible food additives. For readers interested in the latter topic, we highly recommend Desmet and co-workers’ recent review, which mainly focuses on the biocatalytic production of hydrolytically stable trehalose analogues and their potential for use in the food industry.7 We attempt to touch on most applications currently being pursued for trehalose analogues, paying particular attention to their current use and future potential in tuberculosis research and related areas. Overall, we seek to illustrate how rationally manipulating the structure of trehalose can engender this unique disaccharide with properties or functions that can benefit a wide range of fields.
TREHALOSE METABOLISM IN NATURE
We begin this review with an overview of trehalose biosynthesis and utilization pathways that exist in nature. The purpose of this section is two-fold. First, it illustrates the biosynthetic pathways that have inspired chemoenzymatic methods to synthesize trehalose and trehalose analogues on laboratory and industrial scales. Second, it provides background for understanding many of the present and possible future biomedical and biotechnological applications of trehalose analogues.
Five pathways for de novo biosynthesis of trehalose have been reported (Figure 2). In some organisms, multiple pathways may coexist. The most common route of trehalose biosynthesis in nature is the trehalose-6-phosphate synthase/trehalose phosphatase (TPS/TPP) pathway, which was first identified in yeast19 and subsequently identified in many other eukaryotic and prokaryotic organisms, including bacteria, archaea, insects, and plants.1 Commonly referred to as the OtsAB pathway in Escherichia coli,20 the two-step TPS/TPP pathway converts glucose-6-phosphate and uridine diphosphate (UDP)-glucose into trehalose-6-phosphate, which is subsequently dephosphorylated to yield trehalose (Figure 2B). In some organisms, including species in the Sulfolobus and Mycobacterium genera, trehalose can be biosynthesized via the two-step TreY/TreZ pathway, which converts maltooligosaccharides into trehalose.21,22 In this pathway, TreY isomerizes the reducing end of the maltooligosaccharide substrate to generate maltooligosyltrehalose, which is then hydrolyzed by TreZ to give trehalose (Figure 2B). Another trehalose biosynthesis pathway relies on trehalose synthase (TreS), which isomerizes the disaccharide maltose to trehalose (Figure 2C).23 While the TreS pathway is involved in trehalose biosynthesis in vivo in some organisms, it appears to work in the opposite direction in Mycobacterium,24 instead converting trehalose to maltose, which is ultimately used to produce α-glucans.25,26 A fourth pathway, found in archaea, utilizes the trehalose-synthesizing glycosyltransferase TreT, which converts glucose and UDP-glucose into trehalose in one step (Figure 2D).27 Finally, trehalose phosphorylase (TreP) is thought to break down trehalose to glucose and either α- or β-glucose-1-phosphate, depending on whether the TreP is retaining or inverting, respectively.7 However, it is possible that TreP might also catalyze the reverse reaction, trehalose biosynthesis, in some organisms in vivo (Figure 2E). The exploitation of some of these pathways for the laboratory synthesis of trehalose and trehalose analogues is covered later in this review.
Figure 2.

Known trehalose biosynthesis (A–E) and degradation (F) pathways existing in nature. Note: TreP may catalyze both trehalose biosynthesis and trehalose degradation.
The degradation of trehalose to glucose, which is catalyzed by the enzyme trehalase, is an important process in a multitude of organisms. In most scenarios, trehalose degradation occurs for the purposes of either (i) restoring cellular homeostasis after stress-induced trehalose overproduction; or (ii) producing glucose to meet energy and/or carbon demands.1 Interestingly, although mammals do not have pathways for trehalose biosynthesis, they do possess intestinal and renal trehalase,28,29 the former of which is known to be involved in the hydrolysis of ingested trehalose to glucose.30 The presence of trehalase in humans has possible ramifications for many applications of trehalose and trehalose analogues, as discussed later.
In addition to biosynthetic and degradative pathways for trehalose, some organisms possess other pathways involving trehalose. Most notably, trehalose is central to the construction of the complex cell envelope of the Corynebacterineae, which include mycobacterial pathogens such as M. tuberculosis. Here, we summarize trehalose metabolic pathways that are conserved in mycobacteria (Figure 3) because they are valuable targets for the development of probes and inhibitors, as covered in detail later. For trehalose biosynthesis, mycobacteria use either the TPS/TPP or TreY/TreZ pathways.6,22,24,31 In an important pathway, trehalose is also lipidated, undergoing 6-O-mycoloylation by Pks13 to generate trehalose monomycolate [TMM (4), structure shown in Figure 1].32 After crossing the plasma membrane via MmpL3,33,34 TMM donates its 6-O-mycoloyl group to two major acceptor molecules in service of constructing the unique mycobacterial outer membrane, or “mycomembrane.” These processes, which are catalyzed by the Ag85 complex, involve mycoloyl group transfer to either (i) terminal residues of the arabinogalactan polymer, generating arabinogalactan-linked mycolates (AGM); or (ii) a second molecule of TMM, generating trehalose dimycolate [TDM (5), shown in Figure 1].35–37 AGM is the foundational inner leaflet component of the mycomembrane, while TDM resides in the mycomembrane’s outer leaflet and is an important contributor to mycobacterial pathogenesis.38–40 When AGM and TDM are formed, free trehalose is released and reclaimed by the cell through the trehalose-specific transporter SugABC-LpqY.41 This trehalose recycling step is essential for M. tuberculosis virulence and underscores the significance of trehalose uptake pathways, which appear to exist in many organisms. In addition to serving as the essential mediator of mycomembrane biosynthesis, trehalose is also incorporated into numerous other mycobacterial glycolipids, such as sulfolipids,42 polyacyltrehaloses,43 lipooligosaccharides,44 and the recently identified trehalose polyphleates.45 For a more extensive discussion of trehalose metabolism in mycobacteria, we refer readers to a recent review by Kalscheuer and Koliwer-Brandl.6
Figure 3.

Mycobacterial trehalose metabolism.
The pathways described above, which can be categorized into trehalose biosynthesis, degradation, uptake, or elaboration to glycolipids, have important implications for the development and applications of trehalose analogues. Even the limited overview of trehalose metabolism provided above hints at various possible uses of trehalose analogues. Trehalose-based probes are critical tools for unraveling the complex functional and regulatory aspects of trehalose metabolism across all domains of life. Trehalose analogues containing detectable moieties—the archetypal example being 14C-labeled trehalose—have applications ranging from basic biological inquiry to the detection of pathogens. Likewise, trehalose-based inhibitors can be useful as probes of trehalose-processing enzymes, and perhaps as therapeutic compounds in the future. The presence of trehalose uptake pathways in pathogens suggests the possibility of delivering trehalose-linked diagnostic or therapeutic cargo using Trojan horse-type strategies, which are of particular interest because trehalose metabolism is largely absent from mammals. On the other hand, mammals do have the ability to degrade trehalose into glucose. This must be considered in the contexts of pathogen targeting and consumption of native trehalose from food and pharmaceutical products. In this respect, non-degradable trehalose analogues may be of value for numerous applications. Lastly, trehalose biosynthetic pathways have the potential to be adapted for the chemoenzymatic synthesis of analogues, which is of high interest due to the drawbacks of chemical synthesis. Recent efforts in this area are highlighted in the next section of this review.
APPROACHES TO TREHALOSE ANALOGUE SYNTHESIS
The development of synthetic methods to access trehalose derivatives has primarily been motivated by interest in the bioactive glycolipids of M. tuberculosis, such as TMM (4), TDM (5), and sulfolipid-1. Because these molecules have important immunomodulatory properties, it is necessary to have access to pure and structurally well-defined samples to elucidate their structure–activity relationships (SARs). Earlier reviews by Asselineau in 197846 and Timmer and Stocker in 201247 provided excellent accounts of the structures, activities, and chemical syntheses of naturally occurring trehalose-containing glycolipids. The decades-long work in this field has led to the development of numerous useful synthetic methods, while simultaneously calling attention to the significant challenges associated with synthesizing trehalose derivatives. Here, we discuss these challenges and provide an overview of the different approaches that have been used to synthesize modified trehaloses, including traditional chemical approaches and more recent chemoenzymatic approaches.
Chemical synthesis of trehalose analogues
The chemical synthesis of carbohydrates and their analogues has been driven forward by the development of reliable and general methods for the regioselective functionalization of hydroxyl groups and the stereoselective construction of glycosidic bonds. However, trehalose has an atypical 1,1-α,α-glycosidic linkage that joins two glucose units at their anomeric positions, making the molecule C2-symmetric. Many established carbohydrate synthesis methods are not well-suited to these structural features, so trehalose-specific synthetic methods have been developed. Generally, there are two chemical synthesis strategies that can be used to access trehalose analogues, including (i) stereoselective 1,1-α,α-glycosylation reaction between two appropriately functionalized glucoses (Figure 4A); (ii) desymmetrization of natural trehalose using regioselective protection/deprotection steps (Figure 4B).
Figure 4.

Chemical synthesis of trehalose analogues via (A) 1,1-α,α-glycosylation between monosaccharide derivatives and (B) regioselective functionalization of trehalose. LG, leaving group; PG, protecting group; X, structural modification.
With respect to the chemical glycosylation approach (Figure 4A), 1,1-α,α-glycosidic bonds remain among the most difficult to forge with good stereoselectivity. First, the 1,1-α,α-glycosidic linkage is of the 1,2-cis variety, which is incompatible with neighboring-group participation strategies that are employed to stereoselectively form 1,2-trans linkages. Second, this type of glycosylation reaction involves the simultaneous generation of two glycosidic bonds, so three stereoisomers (α,α; α,β; β,β) are possible for symmetrical products and four stereoisomers (α,α; α,β; β,α; β,β) are possible for unsymmetrical products. Not only are these reactions generally inefficient, but the isomeric products are very difficult to separate. Reflecting these challenges, early efforts to chemically synthesize native 1,1-α,α-trehalose through reactions between protected glucose hemiacetals and various protected glucosyl donors (e.g., 1,2-epoxy sugars, glycosyl halides, glycosyl trichloroacetimidates) usually resulted in complex reaction mixtures and low to moderate yields of the desired 1,1-α,α-product.48–50 In fact, trehalose is now produced on an industrial scale by an enzymatic process due to low yields from chemical synthesis methods (discussed in further detail in the following section). Not surprisingly, classical chemical glycosylation methods have only been sparingly applied to the preparation of trehalose analogues.
A few custom chemical 1,1-α,α-glycosylation methods have been developed to overcome the problems discussed above. Two particularly inventive approaches that afforded excellent 1,1-α,α-stereoselectivity are discussed here. Ikegami and co-workers developed a method for the synthesis of ketoside trehalose analogues, which were obtained by reacting an α-configured trimethylsilyl glycoside acceptor with either an exo-glycal or a ketose donor in the presence of a Lewis acid catalyst (Figure 5A).51 Although custom sugar donors are required for this reaction, its complete 1,1-α,α-stereoselectivity and excellent yields (88–96%) make it a powerful method. As discussed later, Ikegami’s method was later applied to the synthesis of a fluorescent-tagged ketoside trehalose analogue used for the detection of M. tuberculosis.52 The Bertozzi group developed another method for chemical 1,1-α,α-glycosylation,53 which beautifully exploited intramolecular aglycon delivery54 (Figure 5B). In this case, the glycosyl acceptor and thioglycoside donor were first tethered to each other via a mixed acetal linkage, placing them in the proper orientation to undergo exclusive 1,1-α,α-glycosylation upon subsequent activation of the thioglycoside. This method, which also required specialized sugar building blocks, was used to synthesize complex sulfolipid-1 analogues from M. tuberculosis for immunological SAR studies.55,56 For a comprehensive review of chemical 1,1-α,α-glycosylation methods, readers are referred to a recent review article by Chaube and Kulkarni.10
Figure 5.

1,1-α,α-Glycosylation strategies using (A) ketoside formation and (B) intramolecular aglycon delivery to control stereochemical outcome. DMB, dimethoxybenzyl; TMS, trimethylsilyl.
A more common approach to the chemical synthesis of trehalose analogues involves regioselective functionalization of native trehalose, which is abundant, inexpensive, and of course it already contains a 1,1-α,α-glycosidic bond (Figure 4B). In this case, the main challenges include differentiation of similarly reactive hydroxyl groups (six secondary and two primary), as well as desymmetrization if unsymmetrical analogues are ultimately desired. Generally speaking, regioselective hydroxyl functionalization methods that work for simpler α-D-glucosides can also be applied to trehalose, with the obvious caveat that trehalose is a symmetrical disaccharide of glucose. A detailed review of these various methods is outside the scope of this review, but a few routinely used strategies are mentioned here. Differentiation of the primary hydroxyls at the 6-O-position(s) from the remaining hydroxyls via selective protection with bulky protecting groups [e.g., trityl,57 tosylate,58 tert-butyldimethylsilyl59 (early examples referenced)] is commonly done, often en route to bioactive glycolipids bearing 6-O-acyl groups, such as TMM and TDM analogues. Differentiation of the secondary hydroxyl groups is more challenging, but a variety of chemistries—many invoking benzylidene60 and cyclohexylidene61,62 acetal protecting groups for the 1,2- and 1,3-diols present in trehalose (early examples referenced)—have been developed for this purpose. The complete repertoire of regioselective functionalization methods for trehalose has enabled the preparation of numerous trehalose analogues, ranging from complex glycolipids to analogues bearing deoxy, fluoro, azido, and phosphate modifications. The synthesis and applications of select examples of such analogues are covered later in this review. For an in-depth discussion of regioselective trehalose functionalization methods, readers are pointed to a recent review article by Sarpe and Kulkarni.11
The two chemical synthesis approaches described above offer the flexibility to access virtually any type of trehalose analogue. However, it is clear that the unique structure of trehalose presents formidable challenges to synthetic chemists. While numerous methods have been developed to address these challenges, the chemical synthesis of trehalose analogues remains a daunting task due to lengthy multi-step syntheses, low overall yields, the need for synthetic expertise, and the generation of hazardous waste streams.
Chemoenzymatic synthesis of trehalose analogues
The existence of at least five de novo trehalose biosynthetic pathways in nature (Figure 2A–E) provides ample opportunity for the development of enzymatic methods to produce trehalose and trehalose analogues. Trehalose-synthesizing enzymes are capable of forming 1,1-α,α-glycosidic bonds with exquisite stereoselectivity from simple, unprotected substrates in aqueous conditions. Together, these features address many of the drawbacks to chemical synthesis mentioned above. Therefore, enzymatic methods have gained increasing attention due to their potential to accelerate the development and applications of trehalose and trehalose analogues.
A breakthrough in the worldwide usage of native trehalose was the invention of an industrial-scale enzymatic synthesis of trehalose beginning from starch, which is inexpensive and abundant. Developed by Hayashibara Biochemical Laboratories, this process harnesses the TreY/TreZ pathway from the soil bacterium Arthrobacter ramosus.63 While a number of steps and components are used to maximize the yield of this process, the two main steps are (i) TreY-catalyzed isomerization of starch maltooligosaccharides to maltooligosyltrehaloses; and (ii) TreZ-catalyzed hydrolysis to release free trehalose from maltooligosyltrehaloses (Figure 2B).63 According to a 2011 review on the properties and applications of native trehalose, the Hayashibara process generates at least 30,000 tons/yr of trehalose, which is then used in over 8,000 commercial products in various industries.2 This is a striking example of how biocatalysis can improve the overall efficiency and commercial viability of a synthetic process.
Despite the importance of the Hayashibara process for generating trehalose on an industrial scale, it is not easily adaptable to the production of trehalose analogues. Indeed, this would require a homogeneously modified starch substrate as well as sufficient tolerance of all enzymatic components (e.g., TreY and TreZ) for the unnatural substrate. Fortunately, other de novo pathways for trehalose biosynthesis have better characteristics for analogue synthesis, namely the utilization of simple monosaccharide-based substrates.
The TPS/TPP pathway converts glucose-6-phosphate and UDP-glucose to trehalose over two steps. In principle, this pathway could be exploited for the chemoenzymatic synthesis of trehalose analogues by using a modified glucose-6-phosphate or UDP-glucose substrate. To our knowledge, the only example of the TPS/TPP pathway being used for in-flask analogue synthesis was reported by the Barry and Davis groups in 2011 (Figure 6).52 With the target molecule being 2-deoxy-2-fluoro-D-trehalose (19F-2-FDTre, 9), 2-deoxy-2-fluoro-D-glucose (19F-2-FDG, 6) was first phosphorylated at the 6-O-position using yeast hexokinase to generate intermediate 7, which was then was reacted with UDP-glucose in the presence of E. coli TPS to form 2-deoxy-2-fluoro-D-trehalose-6-phosphate (8). Removal of the phosphate group of 8 by alkaline phosphatase, rather than TPP, afforded the desired product 9. This synthesis was quite efficient (62% yield over the final two steps), indicating that the TPS/TPP pathway has potential for trehalose analogue synthesis. However, it requires the use of three separate enzymes to generate trehalose analogues from glucose analogues, which limits the overall efficiency and increases the probability that some substrate analogues will not work. Further studies are necessary to establish the scope of this method. Additionally, the requirement for 6-O-phosphorylation of the TPS substrate means that 6-position modifications are not possible using this strategy. Finally, UDP-glucose is a moderately expensive sugar donor ($250/g), which could limit scale-up efforts.
Figure 6.

Chemoenzymatic synthesis of fluorine-modified trehalose analogue 19F-2-FDTre (9) from the corresponding glucose analogue 19F-2-FDG (6) using a three-step method based on the TPS/TPP pathway.
Both the TreP (Figure 2E) and TreT (Figure 2D) pathways are attractive for trehalose analogue synthesis from the standpoint that they directly convert glucose into trehalose in one step. A plethora of modified glucoses are commercially available, including those containing radiolabels, stable-isotope labels, click chemistry labels, and other structural modifications. In addition, the chemical synthesis of custom glucose building blocks is typically much more facile than the chemical synthesis of custom trehalose analogues. Thus, in principle, the TreP and TreT pathways are capable of converting these readily available glucose analogues into the corresponding trehalose analogues.
TreP is thought to mainly catalyze trehalose degradation to glucose and glucose-1-phosphate in vivo, but the reaction is reversible (Figure 2E).64 Both inverting and retaining TreP enzymes exist in nature, and both have been evaluated for their tolerance for unnatural monosaccharide substrates.65–71 In a promising example, the Desmet group showed that the the thermostable inverting TreP enzyme from Caldanaerobacter subterraneus exhibited good activity on a variety of naturally occurring monosaccharides (D-galactose, D-xylose, L-arabinose, D-mannose, D-fucose, and L-fucose all had >10% relative activity compared to the natural substrate D-glucose).70 Despite exhibiting promising substrate promiscuity in wild type and engineered variants, inverting TrePs suffer from the use of sugar donor β-D-glucose-1-phosphate, which is extremely expensive ($60/mg) and difficult to obtain in sufficient quantities for synthetic applications. Thus, the reaction scale is currently quite limited, which may explain the absence of product yields and characterization data in this report. Interestingly, the Desmet group is also developing enzymatic approaches to synthesize glycosyl phosphates such as β-D-glucose-1-phosphate, which, if efficient, could alleviate this problem.72,73 On the other hand, retaining TreP enzymes, which can produce trehalose from glucose and much cheaper α-D-glucose-1-phosphate ($25/g), may provide a more efficient route to trehalose analogues. Unfortunately, to date all retaining TrePs that have been evaluated have shown poor stability and expression yields, and furthermore exhibited low substrate promiscuity.74–77 Therefore, improvement of retaining TrePs through enzyme engineering will be needed for this approach to be fruitful. Additional details on TreP-catalyzed trehalose analogue synthesis can be found in Desmet’s 2015 review article.7
Like TreP, the TreT pathway is attractive due to its one-step conversion of glucose to trehalose (Figure 2D).27 In this case, however, the donor used is UDP-glucose, which as noted above is moderately expensive but much less so than β-D-glucose-1-phosphate. Early studies by the Lee group on the thermostable TreTs from Thermotoga maritima and Pyrococcus horikoshii indicated moderate to good activity on a few naturally occurring monosaccharides, such as galactose and mannose.78–80 Furthermore, the trehalose stereoisomer galactotrehalose—discussed later due to its trehalase-resistance property—was synthesized on a semi-preparative scale in 10% isolated yield (based on the acceptor galactose) and characterized by 13C NMR.80 These preliminary studies encouraged further research on the TreT pathway for trehalose analogue synthesis.
Recently, our group became interested in exploiting the thermostable TreT from Thermoproteus tenax for trehalose analogue synthesis. Intriguingly, T. tenax TreT, which was first reported by Zaparty and co-workers in 2008, is unique among TreTs because it is unidirectional, i.e. it is incapable of degrading trehalose.81 In combination with excellent thermostability and simple substrates, we predicted that the unidirectional nature of T. tenax TreT would make it ideal for synthetic applications. In our initial report in 2014, we showed that this enzyme could indeed convert a wide variety of glucose analogues to trehalose analogues in high yield (up to 99% yield as determined by HPLC, Figure 7).82 The method has now been applied to the semi-preparative synthesis of numerous analogues bearing a variety of modifications (e.g., fluoro-, deoxy-, azido-, thio-, stereochemical, and isotopic modifications), with typical isolated yields of 60–80%.82–84 The rapid and facile procedures we have developed for TreT-catalyzed synthesis and product purification have facilitated access to novel trehalose analogues for mycobacteria research, as discussed in more detail later. As mentioned above, the cost of UDP-glucose is a limitation, plus T. tenax TreT showed poor activity on glucose analogues bearing 4-position modifications. Additional research will be necessary to address these issues.
Figure 7.

One-step TreT-catalyzed synthesis of trehalose analogues (9–24). Yields were determined by HPLC. X and Y represent structural modifications. N.D., not detected.
In sum, chemoenzymatic synthesis using de novo trehalose synthesis enzymes is emerging as a powerful approach to the production of trehalose analogues. In particular, the TreP and TreT pathways, which allow one-step synthesis of trehalose analogues from glucose analogues, are of interest for further development and scale-up. The Hayashibara process for trehalose synthesis provides a model for how thermostable trehalose-synthesizing enzymes might be adapted to large-scale analogue production in the future. Compared to chemical synthesis, chemoenzymatic methods are straightforward, convenient, environmentally friendly, and very high-yielding for relatively simple analogues (e.g., monofunctionalized or isotope-labeled analogues). However, biocatalytic approaches are dependent on enzyme substrate promiscuity, which cannot be easily controlled. Thus, for more complex trehalose analogues, such as glycolipids, chemical synthesis remains the best option due to its versatility.
APPLICATIONS OF TREHALOSE ANALOGUES
With an understanding of how trehalose analogues are produced, we now shift our focus to their applications in biomedical research and biotechnology. Given the increasing focus on using trehalose analogues in the study and targeting of pathogens, we first discuss compounds that have possible applications relating to the investigation, diagnosis, or treatment of diseases, namely those caused by mycobacteria. Then, we consider non-digestible trehalose analogues, which have several possible applications, including biopreservation. Due to the recent comprehensive review on the chemical synthesis and biological activities of mycobacterial trehalose glycolipids,47 we do not discuss this topic. For each type of trehalose analogue covered herein, we discuss current use(s), method(s) of synthesis, and future directions for development and use.
Detectable trehalose analogues
14C-labeled trehalose
Trehalose analogues modified with isotopic labels and other types of detectable chemical handles are invaluable research tools that may also have applications in medical imaging and diagnostics. The characterization of many trehalose metabolic pathways (e.g., those shown in Figures 2 and 3) has been facilitated by the classical reagent in this category, 14C-trehalose (25, Figure 8), whose stable 12C atoms have been exchanged for radioactive 14C atoms. This modification enables sensitive tracking of trehalose in biological samples using techniques such as liquid scintillation counting and chromatography–autoradiography. Thus, 14C-trehalose serves as a metabolic radiotracer whose abundance in a sample can be quantified and any apparent metabolic alterations to the molecule (e.g., elaboration to glycolipids or degradation to glucose) can be observed.
Figure 8.
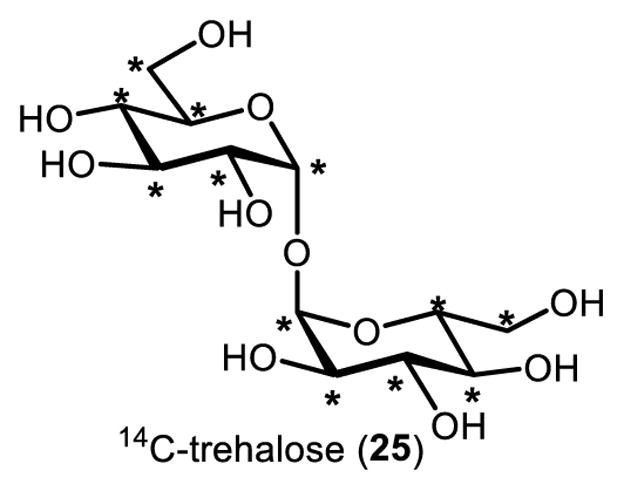
Uniformly labeled 14C-trehalose. Asterisk (*) indicates a 14C-labeled atom.
Mapping trehalose metabolism using 14C-trehalose has been reported in a variety of organisms, including Escherichia coli,85 Thermococcus litoralis,86 and Saccharomyces cerevisiae,87 to highlight a few examples that encompass all three domains of life. Of particular interest to the biomedical research community, 14C-trehalose has been used to elucidate trehalose metabolic pathways in pathogenic organisms, such as M. tuberculosis, thus presenting new opportunities for drug and diagnostic development. For instance, 14C-trehalose has been used to investigate the role of the Ag85 complex in M. tuberculosis cell envelope biosynthesis,36 a pathway that is essential for mycobacterial growth and virulence and is therefore an attractive drug target. In another example, Kalscheuer and colleagues used 14C-trehalose to characterize a novel virulence-associated trehalose transporter in M. tuberculosis, SugABC-LpqY.41 In addition to M. tuberculosis, 14C-trehalose has been used to study trehalose metabolism in related species, including the non-pathogenic model organism M. smegmatis and the industrial fermentation species Corynebacterium glutamicum.35,88 Although studying metabolism is the most common application of 14C-trehalose, there have been other pursuits using the molecule as well. For instance, Hayek and co-workers used 14C-trehalose to investigate the disaccharide for the long-term preservation of human pancreatic endocrine cells, which are used in tissue transplantation in diabetic patients.89 In this case, 14C-trehalose was used to track the intracellular accumulation of trehalose in pancreatic islet cells, which confirmed the successful translocation of exogenous trehalose into the cells.89
14C-trehalose can be acquired through either commercial purchase or in-house synthesis, both having advantages and drawbacks. Purchasing 14C-trehalose is labor-free but presents a tremendous monetary cost ($2,500–5,000/50 μCi). Of the reported methods for synthesizing 14C-trehalose,90–92 the most effective method to date employs a mutant strain of E. coli that is unable to grow on glucose and is capable of overexpressing the trehalose-synthesizing TPS/TPP enzymes from an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible plasmid.92 Thus, this method calls for the incubation of the E. coli strain with exogenous 14C-glucose, IPTG-induced gene expression, and synthesis and purification of 14C-trehalose from the cells. This method is low-cost and generates pure 14C-trehalose product with a reported overall yield of approximately 80%.92 However, 14C-trehalose must be synthesized and purified from cells, which can be a time consuming process and may be inaccessible to some labs. Furthermore, the 14C-glucose starting material or 14C-trehalose product within cells can be funneled to other pathways, decreasing yield and increasing radioactive waste. Additional research aimed at improving the efficiency of 14C-trehalose synthesis could bring down reagent costs and simplify in-house synthesis. Certainly, the chemoenzymatic approaches to trehalose analogue synthesis discussed above could be applied to the production of 14C-trehalose. For example, the unidirectional TreT enzyme from T. tenax81,82 could provide a rapid and high-yielding synthesis of 14C-trehalose from relatively inexpensive 14C-glucose, without the need for cell culture or laborious purification steps.
Stable isotope-labeled trehalose
Related to 14C-trehalose, stable isotope-labeled versions of trehalose can also be beneficial for research purposes. For example, symmetrically 13C- and 2H-labeled trehalose are commercially available and can be used in combination with spectroscopic and spectrometric methods, e.g. NMR and mass spectrometry, to study the structure and metabolism of trehalose and its derivatives. In addition, custom stable isotope-labeled trehaloses have also been chemically synthesized for specific applications. For example, asymmetrical [1-13C]-trehalose was synthesized using a 1,1-α,α-stereoselective glycosylation method and used to elucidate the conformation and dynamics of trehalose in aqueous solution by NMR.93 Recently, 2H-labeled trehalose was chemically synthesized through a one-step ruthenium-catalyzed deuteration of trehalose, and the product was used in conjunction with nuclear reaction analysis to evaluate trehalose uptake by M. smegmatis.94
Fluorescent-labeled trehalose
14C-trehalose is a powerful tool for studying trehalose metabolism, but radiotracers have a number of drawbacks. Radioactive facilities must be available and users must go through extensive training to work with radioisotopes. Such work also has safety concerns and generates radioactive waste streams that are difficult and expensive to expose of. Stable isotope-labeled trehalose analogues are safer and easier to use, but their detection in samples can be difficult and of low sensitivity. From an experimental standpoint, both 14C-containing trehalose radiotracers and stable isotope-labeled trehalose analogues cannot be adapted to the study of intact, single cells. Therefore, alternative means to track trehalose metabolism are of interest for many experiments.
In 2011, the Barry and Davis groups reported the first trehalose-based fluorescent probe, named FITC-Tre (26, Figure 9A).52 FITC-Tre is a ketoside trehalose analogue containing a thiourea-linked fluorescein group at its 2’-position. The probe is conveniently deployed in a onestep metabolic labeling approach and it is capable of reporting on trehalose metabolism in living, single cells through fluorescence microscopy. The initial publication on FITC-Tre demonstrated its ability to label live M. tuberculosis cells through Ag85-mediated incorporation into surface glycolipids such as TDM (refer to Figure 3 for a depiction of this metabolic pathway). This work had two important implications. First, it revealed that extracellular Ag85 had extraordinarily high substrate tolerance, since it accepted a substrate analogue bearing a large fluorophore. Second, because the Ag85 pathway is absent from humans and other mammals, probes such as FITC-Tre could potentially be used to track mycobacterial trehalose metabolism in vivo. As a demonstration of this capability, FITC-Tre was used for the specific imaging of intracellular M. tuberculosis within macrophages (Figure 9B). These findings suggest that trehalose-based probes could be useful for the detection and investigation of M. tuberculosis in complex environments, perhaps within a human patient or patient sputum sample. As discussed below, this concept is now being furthered through the development of trehalose-based tuberculosis diagnostic tools.
Figure 9.

(A) Structure of FITC-Tre (26). (B) Metabolic labeling of M. tuberculosis within host macrophages using FITC-Tre (image reprinted from ref. 52 with permission). Scale bar, 5μm. (C) Final steps in the chemical synthesis of FITC-Tre. Cbz, carboxybenzyl; FITC, fluorescein isothiocyanate; TMSOTf, trimethylsilyl trifluoromethanesulfonate.
The synthesis of FITC-Tre was accomplished via chemical synthesis (Figure 9C). The key step in the synthesis employed Ikegami’s 1,1-α,α-glycosylation method51 to couple two building blocks, ketose donor 27 and protected glucosamine hemiacetal acceptor 28. Upon treatment with Lewis acid catalyst TMSOTf, monosaccharide derivatives 27 and 28 combined with good stereoselectivity to form α,α-ketoside 29 in 45% yield, while the undesired α,β-isomer was generated in less than 10% yield. Disaccharide 29, which contained a Cbz-protected amine at the 2’-position, then underwent deacetylation and hydrogenolysis to remove the hydroxyl and amine protecting groups. The free amine of the intermediate was then chemoselectively conjugated to FITC, generating FITC-Tre (26) in 67% yield over the final three steps. While this synthesis benefited from a glycosylation method with good stereoselectivity (6:1 α,α:α,β), it did require the use of custom monosaccharide building blocks, each of which required several steps to prepare from commercially available materials. Nonetheless, FITC-Tre represented a major advance in the development of trehalose analogues for studying mycobacteria, as exemplified by the additional work in this area discussed below. Furthermore, FITC-Tre has inspired next-generation fluorogenic trehalose analogues that produce fluorescence only upon binding to mycobacteria, which are currently under development.
Azide-labeled trehalose analogues
Fluorescent-labeled trehalose analogues such as FITC-Tre operate through one-step metabolic labeling, which is advantageous for some applications but has limitations for others. First, the fluorophore is a large modification that can impact the route or efficiency of metabolic incorporation and potentially affect the function of labeled biomolecules. Second, because in this approach trehalose is pre-labeled with a fluorophore, the probe has limited versatility. If a different functionality (e.g., a different fluorophore or an affinity tag) is desired, then a new probe must be synthesized and evaluated.
The bioorthogonal chemical reporter strategy, pioneered by the Bertozzi group, is a two-step labeling approach that is complementary to one-step approaches.95,96 The chemical reporter is an unnatural metabolite containing a small, abiotic functional group that does not interact or react with endogenous biomolecules. After metabolic incorporation of the chemical reporter into the cell, it can be detected via selective bioorthogonal reaction with a secondary reagent containing a fluorophore. The most commonly used reporter group is the azide, which is small, inert, and can selectively react with either (i) terminal alkyne-containing reagents through Cu-catalyzed azide–alkyne cycloaddition (CuAAC; “click” reaction)97,98; or (ii) cyclooctyne-containing reagents through strain-promoted azide–alkyne cycloaddition (SPAAC; “Cu-free click” reaction).99 Compared to one-step labeling, the chemical reporter strategy allows for more efficient metabolic incorporation and lower structural/functional perturbation due to the small size and inertness of the azide. In addition, the modular nature of the bioorthogonal reaction confers high experimental flexibility, allowing virtually any type of cargo to be delivered to the labeled metabolite in its native environment.
In 2012, the Bertozzi group developed a series of detectable trehalose analogues bearing azides at all positions of the disaccharide (17–20, Figure 10A).100 Referred to as TreAz analogues, these chemical reporters were shown to be metabolically incorporated into surface-bound trehalose glycolipids in various mycobacterial species, including M. smegmatis, M. bovis, and M. tuberculosis. Both CuAAC and SPAAC were used to deliver fluorophores to TreAz-labeled cells and permit subsequent detection by flow cytometry and fluorescence microscopy (Figure 10B). Interestingly, it was shown through biochemical analysis that different TreAz analogues were incorporated by distinctive metabolic pathways. 3-TreAz (18) behaved similarly to FITC-Tre, undergoing extracellular incorporation into trehalose glycolipids via the Ag85 complex. However, 2-, 4-, and 6-TreAz (17, 19, and 20, respectively) were first imported into the cell via the trehalose transporter SugABC-LpqY, then incorporated into trehalose glycolipids from the inside-out. This finding, which indicates that TreAz analogues accurately mimic native trehalose in the cellular environment, reflects the subtleness of the azide modification to the disaccharide’s structure and function. In addition to facilitating basic biological inquiries into trehalose metabolism, the TreAz two-step labeling strategy allows any type of chemical payload to be delivered to specifically to mycobacteria—potentially within a host—opening up new possibilities for tuberculosis diagnostic and therapeutic applications.
Figure 10.

(A) Structures of 2-, 3-, 4-, and 6-TreAz (17–20). (B) Metabolic labeling of M. smegmatis with TreAz followed by CuAAC with an alkyne-modified fluorophore (image reprinted with permission from ref. 100). Scale bar, 5 μm. (C) An example of the approach used to synthesize TreAz analogues. Differentiation of the 2- and 3-O-positions of trehalose (1) was accomplished in two steps, giving an intermediate (30), which was elaborated to 2- and 3-TreAz (17 and 18). (D) Rapid TreT-catalyzed synthesis, purification, and administration of 6-TreAz (20) to M. smegmatis for a metabolic labeling experiment. CSA, camphor sulfonic acid; DCC, N,N’-dicyclohexylcarbodiimide; DMAP, 4-dimethylaminopyridine.
In Bertozzi’s work, the synthesis of TreAz analogues 17–20 was accomplished using previously established methods for regioselective functionalization of trehalose. The most difficult of these syntheses were 2- and 3-TreAz. These target compounds both originated from a common synthetic intermediate (30), which was obtained from trehalose via Wallace and Minnikin’s desymmetrizing cyclohexylidenation reaction101 followed by regioselective 2-O-benzoylation59 (Figure 10C). Intermediate 30 could be elaborated to either 2- or 3-TreAz by carrying out multistep double displacement sequences to replace the hydroxyl group with an azido group, followed by deprotection. The syntheses of 2- and 3-TreAz required 7–9 steps and proceeded in very low overall yield from trehalose. 4- and 6-TreAz were also accessed via reported trehalose desymmetrization methods, with the former taking 6 steps100,102 from trehalose and the latter taking 4 steps.103 Although these syntheses were successful and provided enough material for evaluation, they were inefficient in terms of step count, time, and yield.
Our group’s chemoenzymatic method for synthesizing trehalose analogues, reported in 2014, proved to be beneficial for accessing some TreAz analogues.82 The TreT enzyme was capable of generating 3- and 6-TreAz from the corresponding commercially available azido glucoses in a single step in 95 and 99% yield (HPLC), respectively. In addition, a purification protocol was developed that utilizes spin dialysis and ion exchange to deliver pure products quickly and in aqueous solution. Using this method, 6-TreAz was synthesized and purified in 60 min starting from 6-azido-6-deoxy-D-glucose (31), and it could be rapidly deployed in metabolic labeling experiments (Figure 10D).84 Thus, chemoenzymatic synthesis employing TreT is preferable to chemical synthesis for accessing 3- and 6- TreAz on a semi-preparative scale. However, the wild-type TreT enzyme has not been able to generate 2- or 4-TreAz from the corresponding azido glucoses. At present, efforts are being made to address these limitations in reaction scope.
Alkyne-labeled trehalose monomycolate (TMM) analogues
FITC-Tre and TreAz analogues are valuable probes for tracking trehalose metabolism that have complementary attributes. Both probes operate by metabolic labeling of trehalose glycolipids. Another major envelope component of mycobacteria is AGM, which is generated through the Ag85-catalyzed transfer of mycoloyl lipid chains from TMM to terminal residues of the arabinogalactan polymer (refer to Figure 3).35–37 This step in trehalose metabolism is essential for mycobacterial growth and virulence. Our group posited that a TMM-based chemical reporter would serve as a cellular probe of this important biochemical process and—because only mycobacteria are known to possess TMM-processing enzymes—potentially have enhanced specificity for detecting/delivering cargo to the cell surface of mycobacteria as compared to FITC-Tre or TreAz.
In 2016, we reported the development of a TMM-mimicking reporter called O-AlkTMM (32), which contained a truncated, linear acyl chain with a terminal alkyne connected to the 6-O-position of trehalose (Figure 11A).104 As hypothesized, metabolic labeling experiments confirmed that O-AlkTMM predominantly incorporated into the AGM layer via the action of Ag85. O-AlkTMM also exhibited high specificity for mycobacteria, as it robustly labeled M. smegmatis and the related species C. glutamicum, but it did not label Gram-negative E. coli or Gram-positive Bacillus subtilis.
Figure 11.

(A) Structure of O-mycoloylation probe O-AlkTMM (32). (B) Simultaneous metabolic labeling of M. smegmatis with 6-TreAz and O-AlkTMM followed by sequential SPAAC and CuAAC reactions to deliver different fluorophores to trehalose glycolipids and AGM (image reprinted with permission from ref. 104). Scale bar, 5 μm. (C) Chemical synthesis of O-AlkTMM using a 4-step trehalose desymmetrization approach.
Because O-AlkTMM has a chemical reporter group (terminal alkyne) that is orthogonal to the chemical reporter of 6-TreAz (azide), these two probes were used to simultaneously detect trehalose glycolipids and AGM in whole cells of M. smegmatis using two-color fluorescence microscopy (Figure 10B).104 Such experimental flexibility will facilitate the study of cell envelope dynamics in intact mycobacterial cells. Recently, we also observed that O-AlkTMM can label and facilitate the identification of proteins modified by mycoloyl groups in C. glutamicum.105 This form of protein post-translational modification (PTM)—termed protein O-mycoloylation—was recently discovered in Corynebacterineae106,107 but its scope and functional consequences remain unknown. O-AlkTMM will be a useful tool for identifying and characterizing proteins that contain this PTM and for probing the link between protein lipidation and trehalose metabolism in C. glutamicum and related organisms.
O-AlkTMM, along with several related TMM analogues with varying chain lengths and chemical reporter groups, were synthesized chemically by employing a trehalose desymmetrization approach originally developed by the Kulkarni group (Figure 11C).108 After per-trimethylsilylation of trehalose, the intermediate was selectively desilylated at the 6-O-positions under basic conditions to give diol 33. Mono-6-O-acylation of 33 was carried out with 6-heptynoic acid (or other azido or alkynyl fatty acids) in the presence of DCC and DMAP, followed by acid-mediated desilylation to give O-AlkTMM (or related chemical reporters). This procedure is straightforward, relatively fast, and provides high purity product. Other methods have been developed to directly mono-6-O-acylate unprotected trehalose either chemically or enzymatically, although removing all side products from the target compound is challenging.109,110 If these purification issues can be addressed, such one-step methods offer a powerful approach to the synthesis of TMM-mimicking probes.
18F-labeled trehalose analogues
Tracking trehalose metabolism in large animals, including humans, could be desirable for numerous reasons. For example, trehalose is used in thousands of food products and is often taken as a dietary supplement.2 In addition, it is being explored as a potential therapeutic compound for neurodegenerative diseases due to its ability to prevent protein aggregation.111 Elucidating the biodistribution and pharmacokinetics of orally or intravenously administered trehalose could be highly informative in these pursuits. In addition, microbes that populate humans—whether it be commensal gut bacteria or pathogenic organisms such as M. tuberculosis—could potentially be imaged by monitoring trehalose uptake processes that are absent from the host.112,113 Unfortunately, fluorophore- and bioorthogonal tag-labeled trehalose analogues (e.g., FITC-Tre, TreAz, and O-AlkTMM) depend on optical imaging modalities and are therefore not well-suited to in vivo imaging in animals. Furthermore, bioorthogonal chemical reporters require two steps to permit detection, which is a challenge in complex organisms.
In their 2011 paper on FITC-Tre, Barry and Davis proposed the idea of 18F-modified trehalose as a radiotracer to image M. tuberculosis infection in vivo.52 18F-modified metabolites can be visualized by positron emission tomography (PET) imaging, which enables rapid, real-time monitoring of metabolic processes in living organisms in three dimensions. Indeed, 2-deoxy-2-[18F]fluoro-D-glucose (18F-2-FDG) in conjunction with PET imaging has been used for decades to facilitate the detection and treatment of cancer.114 Currently, bacteria-specific PET radiotracers that can image wild-type M. tuberculosis are in high demand but are not yet available.112 An 18F-modified trehalose analogue capable of specifically detecting M. tuberculosis infection in animal models and humans would be an important tool in tuberculosis drug development and clinical diagnostics.
The synthesis of 18F-modified compounds is very challenging due to the short half-life of 18F (t1/2 = 110 min). To be viable for PET imaging applications, synthetic methods involving 18F must be rapid and convenient. Toward this goal, in 2011 Barry and Davis developed a chemoenzymatic synthesis of the non-radioactive molecule 19F-2-FDTre (9) modeled after the TPS/TPP pathway, as described earlier in this review (Figure 6). Since this initial report, the method has been adapted by the same groups to synthesize radioactive 18F-2-FDTre, which in preliminary studies has shown promise for imaging tuberculosis lesions in animal models.115 A very attractive feature of this synthesis is that it utilizes commercially available 18F-FDG as a starting material. A drawback of the method is that it requires three enzymes to convert 18F-2-FDG into 18F-2-FDTre, which limits the overall efficiency of the process.
Our group has also made progress toward the development of 18F-modified trehalose analogues. In 2016, we reported the use of TreT catalysis to quantitatively convert non-radioactive 19F-2-FDG (6) into 19F-2-FDTre (9) in a single step in only 15 minutes (Figure 12).83 The purification steps took 45 minutes, resulting in the production of a concentrated aqueous solution of pure 19F-2-FDTre in a total of 60 minutes. This timeframe is compatible with the short half-life of 18F, so the method should be adaptable to the synthesis of 18F-2-FDTre. In addition to 19F-2-FDTre, we synthesized 19F-modified trehalose analogues containing the fluorine atom at the 3’-, 4’-, and 6’-positions (19F-3-, 19F-4-, and 19F-6-FDTre, compounds 14–16, shown in Figure 7), mainly through chemoenzymatic synthesis. This panel of analogues was subjected to conformational analysis by NMR and molecular modeling, which showed that there was negligible difference in structure between native trehalose and the fluorine-modified versions. In addition, three of the four analogues, including 19F-2-FDTre, were shown to be taken up through the trehalose-specific transporter SugABC-LpqY in whole cells of M. smegmatis. Thus, this work established a robust method for the rapid and convenient synthesis of fluorine-modified trehalose analogues, confirmed that the analogues mimic the structure of native trehalose, and demonstrated that they could bind to mycobacteria through a non-mammalian trehalose recycling pathway. Together, the pioneering work of the Barry and Davis groups,52,115 combined with our contributions,82,83 should help to pave the way for continued development of trehalose-based PET imaging probes for tuberculosis diagnostics and other applications.
Figure 12.

Rapid TreT-catalyzed synthesis of 19F-2-FDTre (9).
Trehalose-based inhibitors
The essentiality of trehalose metabolism in various pathogens, coupled with the lack of trehalose metabolism in humans (excepting trehalose degradation), makes it an attractive target for drug development. For example, M. tuberculosis has several trehalose-processing proteins that are required for virulence and are under intensive investigation as drug targets, including Pks13, MmpL3, Ag85, and SugABC-LpqY. Much of the recent progress in this area was covered in a 2016 review by Thanna and Sucheck.116 Other prokaryotic pathogens also have trehalose metabolic pathways involved in physiology and pathogenesis.8,9 The importance of trehalose metabolism to insects3 may provide opportunities for the development of new insecticides. In a similar vein, insects that act as disease vectors, such as mosquitoes for malaria, could also be targeted with trehalose metabolism inhibitors to prevent disease transmission. Common drug discovery approaches, such as natural products and high-throughput screening (HTS), will continue to be the main drivers of identifying inhibitors of trehalose metabolism. Indeed, HTS-based searches for anti-mycobacterial compounds have yielded numerous drug-like small molecules that act on trehalose metabolism, some of which are presently being pursued as possible treatments for tuberculosis.116 Another approach that has shown promise is the rational design of inhibitors based on the structure of trehalose or trehalose derivatives. In keeping with the theme of this review, here we discuss the synthesis and evaluation of trehalose-based inhibitors in mycobacteria and other systems.
6-TreAz (20, Figure 10) was the first trehalose analogue shown to have anti-mycobacterial activity. In 1997, Belisle and co-workers found that 6-TreAz inhibited growth of M. aurum (MIC 200 μg/mL).36 6-TreAz-treated bacteria showed marked reductions in TMM, TDM, and AGM, suggesting that the compound affected trehalose-mediated mycomembrane biosynthesis, perhaps via inhibition of Ag85.36 Following on Belisle’s discovery of 6-TreAz activity, Barry and Davis chemically synthesized a panel of over 20 trehalose and ketoside trehalose analogues and assayed them against purified Ag85 enzyme and whole cells of M. tuberculosis (compounds 2, 9, 16, 20, 34–49, Figure 13).52 The chemical synthesis of these compounds involved a combination of stereoselective 1,1-α,α-glycosylation (Figure 4A) and regioselective functionalization of trehalose (Figure 4B). These analogues, which had a variety of modifications on the sugar rings (e.g., stereochemical inversion, deoxygenation, halogenation), were largely ineffective at inhibiting growth of M. tuberculosis. However, consistent with the earlier report from Belisle, it was found that analogues containing modifications at the 6-position, including 6-TreAz (20) and 19F-6-FDTre (16), exhibited 50% growth inhibition (MIC50) of M. tuberculosis at concentrations in the 100–200 μg/mL range. In a subsequent report, the same groups developed a trehalose analogue bearing an electrophilic fluorophosphonate at the trehalose 6-position (50, Figure 13) and demonstrated its ability to selectively and covalently inhibit Ag85 in vitro, although bacterial growth inhibition was not evaluated.117
Figure 13.

Panel of trehalose analogues evaluated for inhibition of M. tuberculosis growth in ref. 52. The MIC value for each compound is given in parentheses.
Modified versions of TDM have also been explored as anti-mycobacterial agents due to their potential ability to inhibit Ag85. Reynolds and co-workers synthesized a panel of symmetrical 6,6’-diamino-6,6’-dideoxytrehalose derivatives bearing N,N’-dialkylamino groups and 6,6’-bis(sulfonamido) groups and evaluated growth inhibition in an attenuated strain of M. tuberculosis and clinical isolates of M. avium.118 While several compounds exhibited activity, N,N’-dioctylamino- and N,N’-didodecylamino-substituted TDM analogues (52 and 53) were the most potent (MIC 2–8 μg/mL) and acted on both M. tuberculosis and M. avium (Figure 14A). These structures were chemically synthesized through reaction of benzoyl-protected 6,6’-dibromo-6,6’-dideoxytrehalose derivative 51 with excess alkylamine to effect simultaneous N-substitution and debenzoylation, although the two-step yields were quite low (Figure 14A). The Chang group also chemically synthesized a series of TDM analogues and screened them against non-pathogenic M. smegmatis.119 The most active of these compounds contained “reversed” amide linkages stemming from each 6-position of trehalose. For example, compound 56 was obtained in four steps from diol 54—first sequential Swern oxidation and Horner–Wadsworth–Emmons reaction to give 55, then aminolysis and reduction—but the MICs of these derivatives were quite high (Figure 14B).119 The Ronning and Sucheck groups chemically synthesized and evaluated a few related TDM-inspired analogues (57–59), also obtained from intermediate 55, but they lacked activity against purified Ag85 and whole cells of M. smegmatis (Figure 14C).120
Figure 14.

TDM analogues developed as potential inhibitiors of mycobacterial growth. (A) N,N’-dialkylated 6,6’-diamino-6,6’-dideoxytrehalose derivatives 52 and 53 from ref. 118 (accessed through N-substitution of dibromide 51) exhibited good activity against an attenuated strain of M. tuberculosis. (B) TDM analogue 56 bearing a “reversed” acyl linkage from ref. 119 showed moderate activity against M. smegmatis. (C) TDM analogues 57–59 from ref. 120 displayed no activity against M. smegmatis.
Overall, trehalose analogues have had mixed success as inhibitors of mycobacteria, with some of the trehalose- and TDM-based structures discussed above showing activity against whole cells. Additional research is needed to determine whether these compounds can be optimized to attain higher potency in growth inhibition assays. All of the trehalose-based inhibitors discussed were synthesized chemically. Chemoenzymatic approaches could increase synthetic efficiency for some simple inhibitors, e.g. compounds 16 and 20, which can be made in high yield in one step using TreT catalysis.82 Trehalose analogues may also have anti-biofilm activity, because trehalose-mediated export of mycolic acids is essential for biofilm formation in mycobacteria.121,122 Biofilm-like clusters of mycobacteria observed in TB animal models have been shown to correlate with bacterial persistence and drug tolerance, so anti-biofilm agents are of interest.123 Preliminary work from our lab indicates that some trehalose analogues inhibit biofilm formation at lower concentrations than they inhibit growth. For both growth and biofilm inhibitors, efforts will be required to elucidate the mechanism(s) of action of trehalose analogues in cells, which may involve Ag85 inhibition as previously suspected, but could conceivably involve a number of other essential steps in cell envelope biosynthesis (Figure 3).
Beyond anti-mycobacterial activity, trehalose-based inhibitors are of possible value in several additional areas. Other pathogens (or vectors that harbor them) have pathways for trehalose biosynthesis, degradation, and uptake that could be targeted for inhibition with trehalose analogues. Trehalose metabolism is critical to a number of bacterial pathogens, including E. coli, Pseudomonas aeruginosa, and Burkholderia pseudomallei, to name a few.8,9 Recently, the trehalose transporter AgTreT1 in the mosquito Anopheles gambiae was shown to be important to malaria infection caused by Plasmodium falciparum, suggesting that AgTreT1-mediated trehalose uptake may be a target for preventing malaria transmission.124 Trehalose breakdown into glucose by trehalase is critical for insect flight, making it a possible target for the development of novel insecticides. For example, trehazolin (60) and validoxylamine A (61) are naturally occurring insecticidal trehalose-like trehalase inhibitors (Figure 15A).125–127 Trehalase-resistant trehalose analogues are of interest for other biotechnological applications too, as discussed in the next section of the review. Naturally occurring trehalose derivatives (and their synthetic analogues) have also shown interesting bioactivity in mammalian systems. For instance, brartemicin (62), a trehalose 6,6’-bisbenzoyl derivative, was isolated from Nonomuraea and shown to prevent tumor cell invasion (Figure 15B).128 Based on this discovery, the Liu group chemically synthesized brartemicin analogues bearing modified aryl rings that were either ester- or amide-linked to the trehalose core.129,130 Of the these compounds, 63 and 64 matched or slightly improved upon the inhibitory activity of the parent compound, while exhibiting no cytotoxicity.
Figure 15.

(A) Naturally occurring trehalase inhibitors trehazolin (60) and validoxylamine A (61). (B) Brartemicin (62) and synthetic analogues (63 and 64) are being investigated as anti-invasive compounds with possible cancer chemotherapy applications.
Non-digestible trehalose analogues
Native trehalose belongs to a class of molecules called compatible solutes, which function to protect biomolecules from chemical stresses.131 Trehalose can protect biomolecules from osmotic stress,132 thermal stress,133,134 oxidative damage,135 and even radioactive stress.136 These abilities have made trehalose a sought-after additive for applications that require long-term stabilization of reagents and medicines. For instance, trehalose is frequently an additive to vaccine formulations.137 While trehalose is a powerful protein stabilizer, sweetening agent, and vaccine preservative, its use is somewhat limited by the fact that it can be consumed as a carbon source by most microorganisms. As noted earlier, trehalase splits trehalose into two glucose monomers for facile entry into traditional metabolic pathways.138 While mammals do not synthesize trehalose themselves, they are able to break down trehalose present in the food they ingest. Trehalase is found across broad taxa, but in mammals the enzyme is only known to be expressed in the brush border of the intestine and the kidneys.139
Several trehalose analogues are trehalase-resistant, and thus have limited ability to contribute to caloric intake by humans or be used as a carbon source by potential microbial contaminants. Thus, trehalase-resistant trehalose analogues have potential as low calorie food additives and stabilization agents. The analogues have varying degrees of resistance to trehalase, depending on the structural modification (Figure 16). Galactotrehalose (α-D-glucopyranosyl-α-D galactopyranoside, 23), a trehalose analogue where one glucose is replaced by galactose, acts as a competitive inhibitor of trehalase.80 It inhibits trehalase in the high micromolar/low millimolar range. More importantly, mammalian trehalase is unable to degrade galactotrehalose, making it a prime candidate for applications that benefit from resistance to degradation. For an excellent discussion of galactotrehalose, see Desmet’s review.7
Figure 16.

Trehalose analogues that are resistant to degradation by trehalase, including trehalose epimers 21 and 23 and naturally occurring lentztrehaloses A–C (3, 65, 66).
Similarly, mannotrehalose (α-D-glucopyranosyl-α-D-mannopyranoside, 21), has one of the glucose monomers replaced by the 2-position epimer, mannose. Mannotrehalose had no detectable activity against rat intestinal trehalase, indicating it could be used in comparable applications as galactotrehalose.80 Additionally, mannotrehalose has been shown to inhibit other intestinal disaccharidases such as sucrase, maltase, and isomaltase.140 Ryu et al have suggested that by inhibiting intestinal disaccharidases, non-digestible trehalose analogues such as mannotrehalose (and by extension, galactotrehalose) would limit caloric intake, ultimately leading to a reduction in obesity and diabetes.140 Additionally, non-digestible trehalose analogues could act as calorie-free food substitutes in low-calorie diet foods, simply by their inability to enter glycolysis. However, calorie-free food substitutes have a checkered history in both implementation and public acceptance, and the relative sweetness of these analogues has not been studied in great detail. Both galacto- and mannotrehalose have been synthesized on small scales enzymatically,71,78,80,82 and are excellent candidates for production via TreT or TreP catalysis assuming these methods can be adapted to permit large-scale synthesis.
Recently, a new class of trehalose analogues was discovered in the actinomycete genus Lentzea. Unlike the unnatural galactotrehalose and mannotrehalose analogues discussed above, lentztrehalose analogues are found naturally in Lentzea sp. The lentztrehalose class is modified at the 4-position with a 2,3-dihydroxy-3-methylbutoxy moiety (lentztrehalose A, 3), which can be di-dehydroxylated (lentztrehalose B, 65) or cyclized (lentztrehalose C, 66).5 Lentztrehaloses do not inhibit trehalase like galactotrehalose or mannotrehalose. However, they are resistant to trehalase degradation, up to a point. Unlike galactotrehalose and mannotrehalose, trehalase has limited activity on the lentztrehalose class, and can break them apart given sufficient concentration and time.141 Lentztrehalose A was tested against several mammalian cell lines and had low cytotoxicity, and was nontoxic when administered both orally and intraveneously in a mouse model.141 It has also been shown to induce autophagy, which has implications for the treatment of neurodegenerative diseases, as discussed below.142 A recent report disclosed an efficient 6-step chemical synthesis of lentztrehalose A starting from trehalose, which allowed determination of the natural compound’s absolute configuration and should facilitate future SAR studies.143 Overproduction of lentztrehaloses in culture may provide an efficient route for large-scale production, although further optimization will be needed.142
Aside from dietary applications, non-digestible trehalose analogues may be useful in protein stabilization applications. There is a great deal of precedent for using trehalose to stabilize biomolecules from osmotic, thermal, and oxidative stress, as mentioned above. These properties have allowed trehalose to be employed as a food preservative, vaccine preservative, and general protein stabilizer.2 However, the problem with adding any carbon source to food packaging or pharmaceutical is that it makes the item more prone to colonization by unwanted microorganisms. The non-digestible analogues galactotrehalose, mannotrehalose, and the lentztrehaloses should provide similar levels of protection as trehalose. However, these analogues’ reduced reactivity with microbial trehalases may have the added benefit of limiting unwanted microbial contamination.
Finally, there may be applications for non-digestible trehalose analogues in diseases of protein aggregation. Trehalose moderates symptoms in disease models of Huntington’s and Alzheimer’s diseases by reducing protein aggregation.144–146 However, trehalose has also recently been shown to activate autophagy in both Huntington’s and Alzheimer’s models. Trehalose easily crosses the blood brain barrier, and inhibits solute carrier 2A (SLC2A) class transmembrane saccharide transporters. This inhibition leads to cyclic AMP activation and ultimately autophagy.147 Much like the caloric examples cited above, it is likely this process could be increased by using a trehalose analogue that cannot be degraded by trehalase before it reaches its target. In this regard, there may be value in molecules such as lentztrehalose, which is resistant to degradation and exhibits autophagic induction activity. However, whether non-digestible trehalose analogues cross the blood brain barrier and inhibit SLC2A transporters has yet to be demonstrated.
Other applications
Applications of trehalose analogues are by no means limited to the areas that we focused on in this review. For example, immunostimulatory TDM from mycobacteria is a key component of Freund’s adjuvant, which has been used for decades in vaccines. Simplified synthetic analogues of TDM, such as trehalose dibehenate, exhibit beneficial immunological characteristics and possess better toxicity profiles.148 Recently, there have been reports of acetylated versions of trehalose with increased membrane permeability, which allow for enhanced uptake by mammalian cells and subsequent deacetylation by intracellular esterases.149 This strategy for intracellular delivery of trehalose could prove invaluable for stabilizing cell lines and other biological products. The importance of trehalose metabolism in plant growth and development4 may provide opportunities for the development of trehalose analogues that manipulate plant signaling for agricultural applications. Excitingly, during the proof stage of preparing this manuscript, the Davis group reported photo-activatable, plant-permeable analogues of trehalose-6-phosphate.150 When administered to plants, these compounds underwent sunlight-mediated release of trehalose-6-phosphate, providing increased grain yield and enhanced recovery from stress.150 In addition to biomedical and biotechnological uses, trehalose analogues will continue to play an important role in various areas of fundamental research, including SAR studies on bioactive trehalose derivatives, characterization of trehalose-processing enzymes, and determination of the in vivo functions of trehalose metabolic pathways in numerous living systems.
CONCLUSIONS AND FUTURE DIRECTIONS
Trehalose analogues have a breadth of emerging applications in biomedicine and biotechnology, as well as basic research. The difficulty of trehalose analogue synthesis has slowed the development and utilization of these compounds. Encouragingly, though, just like the applications of native trehalose escalated dramatically after the Hayashibara trehalose synthesis process was introduced, progress in the development of trehalose analogues has also correlated with the introduction of improved methods for their synthesis. Trehalose-specific chemical synthesis strategies involving stereoselective α,α-glycosylation or regioselective functionalization of trehalose offer high flexibility with respect to the target molecule structure, but they are generally lengthy, tedious, and inefficient. On the other hand, chemoenzymatic approaches to analogue synthesis can be exceptionally fast, simple, and efficient, but they have limitations with respect to structural flexibility and scale-up. Thus, although it is now possible to generate and evaluate trehalose analogues in a research setting, their commercial adoption will require further synthetic improvements in most cases. In our view, the most impactful improvement will involve scaling up chemoenzymatic syntheses of trehalose analogues.
In the meantime, there are several key areas where trehalose analogues have shown promise, warranting further research. Detectable trehalose analogues bearing stable isotopes, radionuclides, fluorophores, and click chemistry labels enable the study of trehalose metabolism in various contexts and may prove useful as diagnostic tools in the future. Toward this end, a better understanding of the specificity of trehalose-based probes for various microbial species is needed, as is a clearer picture of how these compounds are (or are not) metabolized by humans. Of particularly interest in this area are 18F-modified trehalose analogues, which could provide an unprecedented ability to visualize trehalose metabolism in vivo in animal models and humans. Fortunately, the past few years have witnessed the creation of novel synthetic methods that allow the rapid synthesis and evaluation of trehalose analogues containing fluorine and other detectable tags. The development of trehalose-based inhibitors has yielded some compounds with anti-mycobacterial activity, but their generally low potency suggests limited potential as drugs for treating tuberculosis. More likely, trehalose-based inhibitors will be useful as probes of trehalose-processing enzymes in vitro or in cells. Future efforts in this area should seek to better understand inhibitors’ mechanism of action, improve their potency, and explore their uses outside of mycobacteria. Finally, significant progress has been made in identifying trehalose analogues that maintain native trehalose’s beneficial features for food and pharmaceutical applications, but are resistant to enzymatic degradation. In these regards, molecules such as galacto- and mannotrehalose exhibit attractive properties, but they remain challenging to synthesize on a large scale.
Trehalose analogues are an exciting class of molecules with important applications on the horizon. Chemists’ ability to tailor the structure of trehalose for specific applications will continue to be of paramount importance in these pursuits.
Acknowledgments
This work was funded by a grant from the National Institutes of Health (R15 AI117670) to B. M. S. and P. J. W., as well as Cottrell College Scholar Awards from the Research Corporation to B. M. S. (22525) and P. J. W. (20185).
References
- 1.Elbein AD, Pan YT, Pastuszak I, Carroll D. Glycobiology. 2003;13:17R. doi: 10.1093/glycob/cwg047. [DOI] [PubMed] [Google Scholar]
- 2.Ohtake S, Wang YJ. J Pharm Sci. 2011;100:2020. doi: 10.1002/jps.22458. [DOI] [PubMed] [Google Scholar]
- 3.Thompson SN. Adv Insect Physiol. 2003;31:205. [Google Scholar]
- 4.Van Dijck P, Thevelein JM, Avonce N, Leyman B, Iturriaga G. Plant Physiol. 2004;136:3649. doi: 10.1104/pp.104.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wada S, Ohba S, Someno T, Hatano M, Nomoto A. J Antibiot. 2014;67:319. doi: 10.1038/ja.2013.143. [DOI] [PubMed] [Google Scholar]
- 6.Kalscheuer R, Koliwer-Brandl H. Microbiol Spectr. 2014;2 doi: 10.1128/microbiolspec.MGM2-0002-2013. [DOI] [PubMed] [Google Scholar]
- 7.Walmagh M, Zhao R, Desmet T. Int J Mol Sci. 2015;16:13729. doi: 10.3390/ijms160613729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tournu H, Fiori A, Van Dijck P. PLoS Pathog. 2013;9:e1003447. doi: 10.1371/journal.ppat.1003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz S, Van Dijck P. Virulence. 2016 doi: 10.1080/21505594.2016.1216295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaube MA, Kulkarni SS. Trends Carbohydr Res. 2013;4:1. [Google Scholar]
- 11.Sarpe VA, Kulkarni SS. Trends Carbohydr Res. 2013;5:8. [Google Scholar]
- 12.Lemétais A, Bourdreux Y, Lesot P, Farjon J, Beau JM. J Org Chem. 2013;78:7648. doi: 10.1021/jo4012255. [DOI] [PubMed] [Google Scholar]
- 13.Gau B, Lemetais A, Lepore M, Garcia-Alles LF, Bourdreux Y, Mori L, Gilleron M, De Libero G, Puzo G, Beau JM, Prandi J. ChemBioChem. 2013;14:2413. doi: 10.1002/cbic.201300482. [DOI] [PubMed] [Google Scholar]
- 14.Geerdink D, Minnaard AJ. Chem Commun. 2014;50:2286. doi: 10.1039/c3cc48087a. [DOI] [PubMed] [Google Scholar]
- 15.Sarpe VA, Kulkarni SS. Org Lett. 2014;16:5732. doi: 10.1021/ol5027987. [DOI] [PubMed] [Google Scholar]
- 16.Chaube MA, Kulkarni SS. Chem Eur J. 2015;21:13544. doi: 10.1002/chem.201502521. [DOI] [PubMed] [Google Scholar]
- 17.Sarpe VA, Jana S, Kulkarni SS. Org Lett. 2016;18:76. doi: 10.1021/acs.orglett.5b03300. [DOI] [PubMed] [Google Scholar]
- 18.van der Peet PL, Gunawan C, Torigoe S, Yamasaki S, Williams SJ. Chem Comm. 2015;51:5100. doi: 10.1039/c5cc00085h. [DOI] [PubMed] [Google Scholar]
- 19.Cabib E, Leloir LF. J Biol Chem. 1958;231:259. [PubMed] [Google Scholar]
- 20.Strøm AR, Kaasen I. Mol Microbiol. 1993;8:205. doi: 10.1111/j.1365-2958.1993.tb01564.x. [DOI] [PubMed] [Google Scholar]
- 21.Maruta K, Mitsuzumi H, Nakada T, Kubota M, Chaen H, Fukuda S, Sugimoto T, Kurimoto M. Biochim Biophys Acta. 1996;1291:177. doi: 10.1016/s0304-4165(96)00082-7. [DOI] [PubMed] [Google Scholar]
- 22.De Smet KAL, Weston A, Brown IN, Young DB, Robertson BD. Microbiology. 2000;146:199. doi: 10.1099/00221287-146-1-199. [DOI] [PubMed] [Google Scholar]
- 23.Nishimoto T, Nakano M, Nakada T, Chaen H, Fukuda S, Sugimoto T, Kurimoto M, Tsujisaka Y. Biosci Biotechnol Biochem. 1996;60:640. doi: 10.1271/bbb.60.640. [DOI] [PubMed] [Google Scholar]
- 24.Miah F, Koliwer-Brandl H, Rejzek M, Field RA, Kalscheuer R, Bornemann S. Chem Biol. 2013;20:487. doi: 10.1016/j.chembiol.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalscheuer R, Syson K, Veeraraghavan U, Weinrick B, Biermann KE, Liu Z, Sacchettini JC, Besra G, Bornemann S, Jacobs WR. Nat Chem Biol. 2010;6:376. doi: 10.1038/nchembio.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandra G, Chater KF, Bornemann S. Microbiology. 2011;157:1565. doi: 10.1099/mic.0.044263-0. [DOI] [PubMed] [Google Scholar]
- 27.Qu Q, Lee SJ, Boos W. J Biol Chem. 2004;279:47890. doi: 10.1074/jbc.M404955200. [DOI] [PubMed] [Google Scholar]
- 28.Yoneyama Y, Lever JE. J Cell Physiol. 1987;131:330. doi: 10.1002/jcp.1041310305. [DOI] [PubMed] [Google Scholar]
- 29.Dahlqvist A. Anal Biochem. 1968;22:99. doi: 10.1016/0003-2697(68)90263-7. [DOI] [PubMed] [Google Scholar]
- 30.Ruf J, Wacker H, Kieckebusch V, James P, Mantei N. J Biol Chem. 1990;265:15034. [PubMed] [Google Scholar]
- 31.Woodruff PJ, Carlson BL, Siridechadilok B, Pratt MR, Senaratne RH, Mougous JD, Riley LW, Williams SJ, Bertozzi CR. J Biol Chem. 2004;279:28835. doi: 10.1074/jbc.M313103200. [DOI] [PubMed] [Google Scholar]
- 32.Gavalda S, Bardou F, Laval F, Bon C, Malaga W, Chalut C, Guilhot C, Mourey L, Daffé M, Quémard A. Chem Biol. 2014;21:1660. doi: 10.1016/j.chembiol.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Grzegorzewicz AE, Pham H, Gundi VAKB, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SEM, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M, Korduláková J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M. Nat Chem Biol. 2012;8:334. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, 3rd, Boshoff HI. Antimicrob Agents Chemother. 2012;56:1797. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sathyamoorthy N, Takayama K. J Biol Chem. 1987;262:13417. [PubMed] [Google Scholar]
- 36.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Science. 1997;276:1420. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 37.Backus KM, Dolan MA, Barry CS, Joe M, McPhie P, Boshoff HI, Lowary TL, Davis BG, Barry CE., 3rd J Biol Chem. 2014;289:25041. doi: 10.1074/jbc.M114.581579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. Proc Natl Acad Sci USA. 2008;105:3963. doi: 10.1073/pnas.0709530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marchand CH, Salmeron C, Bou Raad R, Méniche X, Chami M, Masi M, Blanot D, Daffé M, Tropis M, Huc E, Le Maréchal P, Decottignies P, Bayan N. J Bacteriol. 2011;194:587. doi: 10.1128/JB.06138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welsh KJ, Hunter RL, Actor JK. Tuberculosis. 2013;93(Suppl 0):S3. doi: 10.1016/S1472-9792(13)70003-9. [DOI] [PubMed] [Google Scholar]
- 41.Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR. Proc Natl Acad Sci U S A. 2010;107:21761. doi: 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Middlebrook G, Coleman CM, Schaefer WB. Proc Nat Acad Sci U S A. 1959;45:1801. doi: 10.1073/pnas.45.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rousseau C, Neyrolles O, Bordat Y, Giroux S, Sirakova TD, Prevost MC, Kolattukudy PE, Gicquel B, Jackson M. Cell Microbiol. 2003;5:405. doi: 10.1046/j.1462-5822.2003.00289.x. [DOI] [PubMed] [Google Scholar]
- 44.Burguière A, Hitchen PG, Dover LG, Kremer L, Ridell M, Alexander DC, Liu J, Morris HR, Minnikin DE, Dell A, Besra GS. J Biol Chem. 2005;280:42124. doi: 10.1074/jbc.M507500200. [DOI] [PubMed] [Google Scholar]
- 45.Burbaud S, Laval F, Lemassu A, Daffé M, Guilhot C, Chalut C. Cell Chem Biol. 2016;23:278. doi: 10.1016/j.chembiol.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 46.Asselineau C, Asselineau J. Prog Chem Fats Other Lipids. 1978;16:59. doi: 10.1016/0079-6832(78)90037-x. [DOI] [PubMed] [Google Scholar]
- 47.Khan AA, Stocker BL, Timmer MS. Carbohydr Res. 2012;356:25. doi: 10.1016/j.carres.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Lemieux RU, Hendriks KB, Stick RV, James K. J Am Chem Soc. 1975;97:4056. [Google Scholar]
- 49.Chittenden GJF. Carbohydr Res. 1969;9:323. [Google Scholar]
- 50.Ronnow TECL, Meldal M, Bock K. Tetrahedron: Asymmetry. 1994;5:2109. [Google Scholar]
- 51.Namme R, Mitsugi T, Takahashi H, Ikegami S. Eur J Org Chem. 2007;(22):3758. [Google Scholar]
- 52.Backus KM, Boshoff HI, Barry CS, Boutureira O, Patel MK, D’Hooge F, Lee SS, Via LE, Tahlan K, Barry CE, 3rd, Davis BG. Nat Chem Biol. 2011;7:228. doi: 10.1038/nchembio.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pratt MR, Leigh CD, Bertozzi CR. Org Lett. 2003;5:3185. doi: 10.1021/ol034836t. [DOI] [PubMed] [Google Scholar]
- 54.Cumpstey I. Carbohydr Res. 2008;343:1553. doi: 10.1016/j.carres.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 55.Leigh CD, Bertozzi CR. J Org Chem. 2008;73:1008. doi: 10.1021/jo702032c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilmore SA, Schelle MW, Holsclaw CM, Leigh CD, Jain M, Cox JS, Leary JA, Bertozzi CR. ACS Chem Biol. 2012;7:863. doi: 10.1021/cb200311s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bredereck H. Ber. 1930;63:959. [Google Scholar]
- 58.Helferich B, von Stryk F. Ber. 1941;74:1794. [Google Scholar]
- 59.Lin FL, van Halbeek H, Bertozzi CR. Carbohydr Res. 2007;342:2014. doi: 10.1016/j.carres.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ali Y, Hough L, Richardson AC. Carbohydr Res. 1970;14:181. [Google Scholar]
- 61.Wallace PA, Minnikin DE, Besra GS, Bolton RC, McNeil MR, Ridell M, Simpson KE, Glushka J, Halbeek H, van Brennan PJ, Minnikin DE, Gautier N, Marín LML, Lanéelle M-A, Daffé M, Hamid ME, Fraser JL, Wallace PA, Besra GS, Goodfellow M, Minnikin DE, Ridell M, Ridell M, Wallerström G, Minnikin DE, Bolton RC, Magnusson M, Baer HH, Wu X, Bisset FH, Evans ME, Parrish FW, Neises B, Steglich W. J Chem Soc, Chem Commun. 1993;31:1292. [Google Scholar]
- 62.Wallace PA, Minnikin DE. Carbohydr Res. 1994;263:43. doi: 10.1016/0008-6215(94)00164-2. [DOI] [PubMed] [Google Scholar]
- 63.Maruta K, Nakada T, Kubota M, Chaen H, Sugimoto T, Kurimoto M, Tsujisaka Y. Biosci, Biotechnol, Biochem. 1995;59:1829. doi: 10.1271/bbb.59.1829. [DOI] [PubMed] [Google Scholar]
- 64.Luley-Goedl C, Nidetzky B. Biotechnol J. 2010;5:1324. doi: 10.1002/biot.201000217. [DOI] [PubMed] [Google Scholar]
- 65.Belocopitow E, Marechal LR, Gros EO. Carbohydr Res. 1971;19:268. doi: 10.1016/s0008-6215(00)81631-6. [DOI] [PubMed] [Google Scholar]
- 66.Wannet WJ, Op den Camp HJ, Wisselink HW, van der Drift C, Van Griensven LJ, Vogels GD. Biochim Biophys Acta. 1998;1425:177. doi: 10.1016/s0304-4165(98)00066-x. [DOI] [PubMed] [Google Scholar]
- 67.Maruta K, Watanabe H, Nishimoto T, Kubota M, Chaen H, Fukuda S, Kurimoto M, Tsujisaka Y. J Biosci Bioeng. 2006;101:385. doi: 10.1263/jbb.101.385. [DOI] [PubMed] [Google Scholar]
- 68.Inoue Y, Ishii K, Tomita T, Yatake T, Fukui F. Biosci Biotechnol Biochem. 2002;66:1835. doi: 10.1271/bbb.66.1835. [DOI] [PubMed] [Google Scholar]
- 69.Chaen H, Nakada T, Mukai N, Nishimoto T, Fukuda S, Sugimoto T, Kurimoto M, Tsujisaka Y. J Appl Glycosci. 2001;48:135. [Google Scholar]
- 70.Van der Borght J, Chen C, Hoflack L, Van Renterghem L, Desmet T, Soetaert W. Appl Environ Microbiol. 2011;77:6939. doi: 10.1128/AEM.05190-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van der Borght J, Soetaert W, Desmet T. Biotechnol Progr. 2012;28:1257. doi: 10.1002/btpr.1609. [DOI] [PubMed] [Google Scholar]
- 72.Van der Borght J, Desmet T, Soetaert W. Biotechnol J. 2010;5:986. doi: 10.1002/biot.201000203. [DOI] [PubMed] [Google Scholar]
- 73.Chen C, Van der Borght J, De Vreese R, D’hooghe M, Soetaert W, Desmet T. Chem Commun. 2014;50:7834. doi: 10.1039/c4cc02202e. [DOI] [PubMed] [Google Scholar]
- 74.Saito K, Kase T, Takahashi E, Horinouchi S. Appl Microbiol Biotechnol. 1998;64:4340–5. doi: 10.1128/aem.64.11.4340-4345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saito K, Yamazaki H, Ohnishi Y, Fujimoto S, Takahashi E, Horinouchi S. Appl Microbiol Biotechnol. 1998;50:193. doi: 10.1007/s002530051276. [DOI] [PubMed] [Google Scholar]
- 76.Eis C, Nidetzky B. Biochem J. 1999;341:385–393. [PMC free article] [PubMed] [Google Scholar]
- 77.Goedl C, Griessler R, Schwarz A, Nidetzky B. Biochem J. 2006;397:491. doi: 10.1042/BJ20060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ryu SI, Kim JE, Huong NT, Woo EJ, Moon SK, Lee SB. Enzym Microb Technol. 2010;47:249. [Google Scholar]
- 79.Woo EJ, Ryu SI, Song HN, Jung TY, Yeon SM, Lee HA, Park BC, Park KH, Lee SB. J Mol Biol. 2010;404:247. doi: 10.1016/j.jmb.2010.09.056. [DOI] [PubMed] [Google Scholar]
- 80.Kim HM, Chang YK, Ryu SI, Moon SG, Lee SB. J Mol Catal B Enzym. 2007;49:98. [Google Scholar]
- 81.Kouril T, Zaparty M, Marrero J, Brinkmann H, Siebers B. Arch Microbiol. 2008;190:355. doi: 10.1007/s00203-008-0377-3. [DOI] [PubMed] [Google Scholar]
- 82.Urbanek BL, Wing DC, Haislop KS, Hamel CJ, Kalscheuer R, Woodruff PJ, Swarts BM. ChemBioChem. 2014;15:2066. doi: 10.1002/cbic.201402288. [DOI] [PubMed] [Google Scholar]
- 83.Rundell SR, Wagar ZL, Meints LM, Olson CD, O’Neill MK, Piligian BF, Poston AW, Hood RJ, Woodruff PJ, Swarts BM. Org Biomol Chem. 2016;14:8598. doi: 10.1039/c6ob01734g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meints LM, Poston AW, Piligian BF, Olson CD, Badger KS, Woodruff PJ, Swarts BM. J Vis Exp. doi: 10.3791/54485. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ehmann U, Forkl H, Klein W, Rimmele M, Postma P. J Bacteriol. 1990;172:3450. doi: 10.1128/jb.172.6.3450-3461.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xavier KB, Martins LO, Peist R, Kossmann M, Boos W, Martins GIaO, Peist R, Kossmann M, Xavier KB. J Bacteriol. 1996;178:4773. doi: 10.1128/jb.178.16.4773-4777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kotyk A, Michaljanicová D. J Gen Microbiol. 1979;110:323. doi: 10.1099/00221287-110-2-323. [DOI] [PubMed] [Google Scholar]
- 88.Tropis M, Meniche X, Wolf A, Gebhardt H, Strelkov S, Chami M, Schomburg D, Krämer R, Morbach S, Daffé M. J Biol Chem. 2005;280:26573. doi: 10.1074/jbc.M502104200. [DOI] [PubMed] [Google Scholar]
- 89.Beattie GM, Crowe JH, Lopez AD, Cirulli V, Ricordi C, Hayek A. Diabetes. 1997;46:519. doi: 10.2337/diab.46.3.519. [DOI] [PubMed] [Google Scholar]
- 90.Brand B, Boos W. Appl Environ Microbiol. 1989;55:2414. doi: 10.1128/aem.55.9.2414-2415.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stambuk BU, Crowe JH, Crowe LM, Panek AD, de Araujo PS. Anal Biochem. 1993;212:150. doi: 10.1006/abio.1993.1305. [DOI] [PubMed] [Google Scholar]
- 92.Horlacher R, Peist R, Boos W. Appl Env Microbiol. 1996;62:3861. doi: 10.1128/aem.62.10.3861-3863.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Batta G, Kövér KE, Gervay J, Hornyák M, Roberts GM. J Am Chem Soc. 1997;119:1336. [Google Scholar]
- 94.Lowery R, Gibson MI, Thompson RL, Fullam E. Chem Commun. 2015;51:4838. doi: 10.1039/c4cc09588j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sletten EM, Bertozzi CR. Angew Chem Int Ed. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Grammel M, Hang HC. Nat Chem Biol. 2013;9:475. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 98.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 99.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Swarts BM, Holsclaw CM, Jewett JC, Alber M, Fox DM, Siegrist MS, Leary JA, Kalscheuer R, Bertozzi CR. J Am Chem Soc. 2012;134:16123. doi: 10.1021/ja3062419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wallace PA, Minnikin DE. J Chem Soc Chem Commun. 1993:1292. [Google Scholar]
- 102.Bassily RW, El-Sokkary RI, Silwanis BA, Nematalla AS, Nashed MA. Carbohydr Res. 1993;239:197. doi: 10.1016/0008-6215(93)84215-r. [DOI] [PubMed] [Google Scholar]
- 103.Hanessian S, Lavallee P. J Antibiot. 1972;25:683. doi: 10.7164/antibiotics.25.683. [DOI] [PubMed] [Google Scholar]
- 104.Foley HN, Stewart JA, Kavunja HW, Rundell SR, Swarts BM. Angew Chem Int Ed. 2016;55:2053. doi: 10.1002/anie.201509216. [DOI] [PubMed] [Google Scholar]
- 105.Kavunja HW, Piligian BF, Fiolek TJ, Foley HN, Nathan TO, Swarts BM. Chem Commun. 2016;52:13795. doi: 10.1039/c6cc07143k. [DOI] [PubMed] [Google Scholar]
- 106.Huc E, Meniche X, Benz R, Bayan N, Ghazi A, Tropis M, Daffé M. J Biol Chem. 2010;285:21908. doi: 10.1074/jbc.C110.133033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huc E, de Sousa-D’Auria C, de la Sierra-Gallay IL, Salmeron C, van Tilbeurgh H, Bayan N, Houssin C, Daffé M, Tropis M. J Bacteriol. 2013;195:4121. doi: 10.1128/JB.00285-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sarpe VA, Kulkarni SS. J Org Chem. 2011;76:6866. doi: 10.1021/jo200979n. [DOI] [PubMed] [Google Scholar]
- 109.Paul NK, Twibanire JD, Grindley TB. J Org Chem. 2012;78:363. doi: 10.1021/jo302231v. [DOI] [PubMed] [Google Scholar]
- 110.Raku T, Kitagawa M, Shimakawa H, Tokiwa Y. J Biot. 2003;100:203. doi: 10.1016/s0168-1656(02)00224-9. [DOI] [PubMed] [Google Scholar]
- 111.Emanuele E. Curr Drug Targets. 2014;15:551. doi: 10.2174/1389450115666140225104705. [DOI] [PubMed] [Google Scholar]
- 112.Johnson DH, Via LE, Kim P, Laddy D, Lau CY, Weinstein EA, Jain S. Nucl Med Biol. 2014;41:777. doi: 10.1016/j.nucmedbio.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ankrah AO, van der Werf TS, de Vries EFJ, Dierckx RAJO, Sathekge MM, Glaudemans AWJM. Clin Transl Imaging. 2016;4:131. doi: 10.1007/s40336-016-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, Oster ZH, Sacker DF, Shiue CY, Turner H, Wan CN, Wolf AP, Zabinski SV. J Nucl Med. 1980;21:670. [PubMed] [Google Scholar]
- 115.Conference Proceedings abstract. J Label Compd Radiopharm. 2015;58:S75–S411. [Google Scholar]
- 116.Thanna S, Sucheck SJ. Med Chem Commun. 2015;7:69. doi: 10.1039/C5MD00376H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barry CS, Backus KM, Barry CE, Davis BG. J Am Chem Soc. 2011;133:13232. doi: 10.1021/ja204249p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rose JD, Maddry JA, Comber RN, Suling WJ, Wilson LN, Reynolds RC. Carbohydr Res. 2002;337:105. doi: 10.1016/s0008-6215(01)00288-9. [DOI] [PubMed] [Google Scholar]
- 119.Wang J, Elchert B, Hui Y, Takemoto JY, Bensaci M, Wennergren J, Chang H, Rai R, Chang CW. Bioorg Med Chem. 2004;12:6397. doi: 10.1016/j.bmc.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 120.Sucheck SJ. Mol BioSyst. 2009;5:945. doi: 10.1039/b902284h. [DOI] [PubMed] [Google Scholar]
- 121.Ojha AK, Trivelli X, Guerardel Y, Kremer L, Hatfull GF. J Biol Chem. 2010;285:17380. doi: 10.1074/jbc.M110.112813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kulka K, Hatfull G, Ojha AK. J Vis Exp. 2012:e3820. doi: 10.3791/3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Orme IM. Tuberculosis. 2014;94:8. doi: 10.1016/j.tube.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu K, Dong Y, Huang Y, Rasgon JL, Agre P. Proc Natl Acad Sci U S A. 2013;110:17504. doi: 10.1073/pnas.1316709110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wegener G, Tschiedel V, Schlöder P, Ando O. J Exp Biol. 2003;206:1233. doi: 10.1242/jeb.00217. [DOI] [PubMed] [Google Scholar]
- 126.Asano N, Takeuchi M, Kameda Y, Matsui K, Kono Y. J Antibiot. 1990;43:722. doi: 10.7164/antibiotics.43.722. [DOI] [PubMed] [Google Scholar]
- 127.Gibson RP, Gloster TM, Roberts S, Warren RAJ, De Gracia IS, Chiara JL, Davies GJ. Angew Chem Int Ed. 2007;46:4115. doi: 10.1002/anie.200604825. [DOI] [PubMed] [Google Scholar]
- 128.Igarashi Y, Mogi T, Yanase S, Miyanaga S, Fujita T, Sakurai H, Saiki I. J Nat Prod. 2009;72:980. doi: 10.1021/np9000575. [DOI] [PubMed] [Google Scholar]
- 129.Jiang Y, Tang L, Miyanaga S, Igarashi Y, Saiki I, Liu Z. Bioorg Med Chem Lett. 2011;21:1089. doi: 10.1016/j.bmcl.2010.12.133. [DOI] [PubMed] [Google Scholar]
- 130.Jiang Y, Li S, Liu Y. Chem Biol Drug Des. 2015;86:1017. doi: 10.1111/cbdd.12569. [DOI] [PubMed] [Google Scholar]
- 131.Empadinhas N, da Costa MS. Int Microbiol. 2006;9:199. [PubMed] [Google Scholar]
- 132.Purvis JE, Yomano LP, Ingram LO. Appl Environ Microbiol. 2005;71:3761. doi: 10.1128/AEM.71.7.3761-3769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kandror O, DeLeon A, Goldberg AL. Proc Natl Acad Sci U S A. 2002;99:9727. doi: 10.1073/pnas.142314099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Avanti C, Saluja V, van Streun EL, Frijlink HW, Hinrichs WL. PLoS One. 2014;9:e86244. doi: 10.1371/journal.pone.0086244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.da Costa Morato Nery D, da Silva CG, Mariani D, Fernandes PN, Pereira MD, Panek AD, Eleutherio EC. Biochim Biophys Acta. 2008;1780:1408. doi: 10.1016/j.bbagen.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 136.Yoshinaga K, Yoshioka H, Kurosaki H, Hirasawa M, Uritani M, Hasegawa K. Biosci Biotechnol Biochem. 1997;61:160. doi: 10.1271/bbb.61.160. [DOI] [PubMed] [Google Scholar]
- 137.Alcock R, Cottingham MG, Rollier CS, Furze J, De Costa SD, Hanlon M, Spencer AJ, Honeycutt JD, Wyllie DH, Gilbert SC, Bregu M, Hill AV. Sci Transl Med. 2010;2:19ra12. doi: 10.1126/scitranslmed.3000490. [DOI] [PubMed] [Google Scholar]
- 138.Divate NR, Chen GH, Wang PM, Ou BR, Chung YC. Bioengineered. 2016 doi: 10.1080/21655979.2016.1207019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arguelles JC. J Mol Evol. 2014;79:111. doi: 10.1007/s00239-014-9645-9. [DOI] [PubMed] [Google Scholar]
- 140.Ryu Kim JE, Nguyen TH, Woo EJ, Moon SG, Lee SBSI. Enzym Microb Technol. 2010;47:249. [Google Scholar]
- 141.Wada SI, Sawa R, Ohba SI, Hayashi C, Umekita M, Shibuya Y, Iijima K, Iwanami F, Igarashi M. J Agric Food Chem. 2016;64:7121. doi: 10.1021/acs.jafc.6b02782. [DOI] [PubMed] [Google Scholar]
- 142.Wada S, Kubota Y, Sawa R, Umekita M, Hatano M, Ohba S, Hayashi C, Igarashi M, Nomoto A. J Antibiot. 2015;68:521. doi: 10.1038/ja.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang M, Wada S-I, Amemiya F, Watanabe T, Shibasaki M. Chem Pharm Bull. 2015;63:961. doi: 10.1248/cpb.c15-00600. [DOI] [PubMed] [Google Scholar]
- 144.Fernandez-Estevez MA, Casarejos MJ, Lopez Sendon J, Garcia Caldentey J, Ruiz C, Gomez A, Perucho J, de Yebenes JG, Mena MA. PLoS One. 2014;9:e90202. doi: 10.1371/journal.pone.0090202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N. Nat Med. 2004;10:148. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 146.Du J, Liang Y, Xu F, Sun B, Wang Z. J Pharm Pharmacol. 2013;65:1753. doi: 10.1111/jphp.12108. [DOI] [PubMed] [Google Scholar]
- 147.DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, Chi M, Newberry EP, Chen Z, Finck BN, Davidson NO, Yarasheski KE, Hruz PW, Moley KH. Sci Signal. 2016;9:ra21. doi: 10.1126/scisignal.aac5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Holten-Andersen L, Doherty TM, Korsholm KS, Andersen P. Infect Immun. 2004;72:1608. doi: 10.1128/IAI.72.3.1608-1617.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Abazari A, Meimetis LG, Budin G, Bale SS, Weissleder R, Toner M. PLoS One. 2015;10:e0130323. doi: 10.1371/journal.pone.0130323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Griffiths CA, Sagar R, Geng Y, Primavesi LF, Patel MK, Passarelli MK, Gilmore IS, Steven RT, Bunch J, Paul MJ, Davis BG. Nature. 2016 doi: 10.1038/nature20591. [DOI] [PubMed] [Google Scholar]