

Abstract
Background
CALGB 100104 (Alliance) studied lenalidomide vs. placebo following autologous stem cell transplant (ASCT) for newly diagnosed myeloma patients, demonstrating improved time to progression (TTP) and overall survival (OS), and an increase in second primary malignancies (SPM) for lenalidomide at 34-months median follow-up. Here we report an updated intent-to-treat analysis at 91-months median follow-up.
Methods
Patients were eligible if they had active myeloma, had received at most two induction regimens and had achieved stable disease or better in the first 100 days after ASCT. In this phase 3 study, 460 patients were randomised in a double-blind manner to either lenalidomide (n=231) or placebo (n=229) utilizing a permutated-block randomisation with fixed block size. Randomisation was stratified by three factors: normal or elevated β2-microglobulin level at registration (≤2·5 mg/L vs > 2·5 mg/L), prior use or nonuse of thalidomide during induction therapy, and prior use or nonuse of lenalidomide during induction therapy. The starting dose was 10 mg daily, escalated to 15 mg daily after three months. The primary endpoint was TTP (time of progressive disease or death from any cause) using intent-to-treat analysis. After three interim analyses, the study was unblinded at median follow-up of 18 months and 86/128 placebo patients without progressive disease chose to cross over to lenalidomide. This study is registered with ClinicalTrials.gov identifier NCT00114101; new patients are no longer being recruited, but some patients remain on treatment and in follow-up.
Findings
The median TTP for lenalidomide is 57·3 months (95% CI 44·2–73·3) and 28·9 months (95% CI 23·0–36·3) for placebo (hazard ratio (HR): 0·57, 95% CI 0·46–0·71, p<0·0001). The TTP benefit with lenalidomide was observed regardless of whether patients were in a complete response at time of randomisation or whether they had received thalidomide or lenalidomide induction therapy. The most common grade 3–4 adverse events were neutropenia (116 (50%) of 231 patients in the lenalidomide arm and 37 (16%) of 229 patients in the placebo arm) and thrombocytopenia (34 patients (15%) in the lenalidomide arm and 11 patients (4·8%) in the placebo arm. Eighteen haematological (7·8%) and 14 solid tumour (6·1%) SPMs have been diagnosed following randomisation and prior to disease progression in the lenalidomide arm vs. three haematological (1·3%) and nine solid tumour (3·9%) SPMs in the placebo arm. Of the placebo SPMs, three haematological and five of nine solid tumour SPMs were in the crossover subgroup.
Interpretation
Despite an increase in haematological adverse events and SPMs, lenalidomide maintenance therapy following ASCT significantly improves TTP and can be considered a standard of care.
Introduction
Despite improvements in survival of newly diagnosed myeloma patients as a consequence of induction therapy with novel agents followed by consolidation with high dose melphalan and autologous stem cell transplant (ASCT), the majority of patients will suffer disease relapse/progression. There has been significant interest in the role of maintenance therapy following ASCT in order to delay disease relapse/progression and prolong survival. A number of randomised studies evaluated maintenance therapy with thalidomide, the first generation immunomodulatory agent (IMiD), following ASCT. In aggregate, while a benefit for progression-free survival (PFS) was observed, a consistent overall survival benefit has not been demonstrated and prolonged therapy with thalidomide has been limited by this agent’s side effect profile.1 Lenalidomide, the second generation IMiD, has a more favourable side-effect profile and therefore has been studied in the context of post-ASCT maintenance therapy.
Cancer and Leukemia Group B (CALGB) 100104, in collaboration with the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) and the Eastern Cooperative Oncology Group (ECOG) randomised 460 patients to lenalidomide or placebo maintenance following ASCT. CALGB is now part of the Alliance for Clinical Trials in Oncology. Initial publication demonstrated that lenalidomide maintenance was associated with significantly longer time to disease progression (TTP) (median TTP of 46 months for the lenalidomide group and 27 months for the placebo group, p<0·0001) as well as a significant improvement in overall survival at median follow-up of 34 months.2 Eight percent of patients in the lenalidomide group developed second primary malignancies (SPMs) before disease progression compared with three percent in the placebo group. Three other large randomised studies (IFM 2005-02, GIMEMA RV-209, and Myeloma XI study) evaluated the role of lenalidomide maintenance following ASCT.3–5 While both the IFM 2005-02 and GIMEMA RV-209 studies reported significant improvements in PFS in the lenalidomide groups, there was no significant improvement in overall survival. The survival data for the Myeloma XI trial are not yet mature. SPM rates (excluding noninvasive skin cancers) of 7·5%3 and 2·8%4 were observed in the lenalidomide treatment groups of the IFM 2005-02 and GIMEMA RV-209 studies, respectively. A recent meta-analysis that included CALGB 100104, IFM 2005-02, and GIMEMA RV-209 found that lenalidomide maintenance significantly improves overall survival.6 Here we present an updated analysis of CALGB 100104 which provides long-term follow-up data with respect to TTP, overall survival and SPMs.
Methods
Study design and patient selection
Adults aged 18 to 70 years were enrolled across 47 centers in the United States. Patients were eligible if they had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had symptomatic disease requiring treatment, had received no more than 12 months of any prior therapy, were within 12 months of initiation of induction therapy and had received at most two induction regimens. Patients were excluded if they had disease progression during induction therapy or if they had previously undergone a prior peripheral blood, bone marrow, or solid organ transplant. Patients with stable disease or better (marginal, partial, or complete response) in the first 100 days after ASCT were eligible. Additional details regarding inclusion and exclusion criteria can be found in the supplemental material. Each participant signed an IRB-approved, protocol-specific informed consent in accordance with federal and institutional guidelines.
Randomisation and masking
Patients were randomly assigned (1:1) in a double-blinded manner to initiate either lenalidomide or placebo between day 90 and day 100 after ASCT. Randomisation was stratified by three factors: normal or elevated β2-microglobulin level at registration (≤ 2·5 mg/L vs. > 2·5 mg/L), prior use or nonuse of thalidomide during induction therapy, and prior use or nonuse of lenalidomide during induction therapy. A permutated-block randomisation with a block size of six and equal allocation between two arms was utilized for each stratum. The study was unblinded on December 17, 2009 after the third interim analysis. A detailed description of the randomisation, unblinding, and interim analyses is included in the supplemental material. Of the 128 eligible patients without progressive disease in the placebo group, 86 chose to cross over and receive lenalidomide therapy.
Procedures
All patients with stable disease or better were scheduled to start therapy between day 100–110 post-ASCT. All patients started on two capsules (10 mg of lenalidomide, or placebo) per oral daily. After three months, the dose could be escalated to three capsules (15 mg) daily. Response and progression were initially defined using the European Blood and Marrow Transplant Group7 and were subsequently changed to the International Myeloma Working Group (IMWG) criteria.8 Criteria for continued treatment were decided by the local centers. Dose modifications are detailed in the supplemental material. Disease progressions (see supplemental material for definition), deaths, responses at day 100, one-year, two-year, and three-year post-ASCT were determined at the treating center and centrally reviewed according to the IMWG criteria, with the exception that stringent CR (sCR) was not utilized as a response category as the majority of patients did not undergo free light chain testing. The central review was done in a blinded manner by four of the authors (SAH, PLM, SG, EAS).
Outcomes
The primary endpoint was time to progression (TTP), defined as time to progressive disease or death from any cause after transplantation. Secondary objectives included overall survival (defined as the length of time from ASCT to death from any cause), assessment of complete response rate, and determination of feasibility of long-term administration of lenalidomide. The assessment of second primary malignancies was an exploratory objective not specified in the protocol.
Statistical analysis
For sample size calculations, a total of n=462 patients were planned to be randomised over a period of 33 months. Accounting for a drop-out rate of 15%, this would require registering n=554 patients over this period. Under an equal allocation scheme (i.e., 231 patients per arm), a planned accrual period of 33 months and a follow-up period of 30 months, this design would have a power of at least 0·9 for the log-rank test to compare a median TTP of 2 years for the control arm and 2·8 years for the experimental arm (HR=1·4) with one-sided alpha = 0·05. The primary statistical analysis was conducted using the log-rank test and the secondary analysis was conducted using the Cox regression method. To assess the occurrence of second primary malignancies (SPM) reported post-randomisation, the non-protocol endpoint of event-free survival (EFS), defined as time of first event (SPM, progressive disease, or death) is considered. Interim analyses were planned on a semi-annual basis to coincide with the semi-annual meetings of the CALGB Data and Safety Monitoring Board (DSMB) starting from about 21 months. Superiority tests were conducted using a group sequential test design by Emerson and Fleming9 and futility tests were conducted to test if the HR of TTP between the two arms was <1·4 with one-sided alpha=0·05. The survival functions are estimated using the Kaplan-Meier estimator.10 The Cox score statistic is used to test discrepancy between survival distributions adjusting for baseline patient characteristics.11 Hazard ratios are estimated using a Cox model under the implicit assumption of proportional hazards. To assess cause-specific (progression, death, and SPM) risk, the cumulative incidence curves are estimated using Kaplan-Meier estimators12 and compared using the log-rank test proposed by Gray.13 All analyses are right censored and per protocol used ASCT as the reference date except the EFS analysis, which uses the randomisation date. The complete response rates at one year after transplant for the lenalidomide and placebo arms were compared by Chi-square testing. The difference in the most frequent grade 3 and higher at least possibly treatment-related adverse events proportion between independent patient groups is tested using Fisher’s test and estimated using a conditional maximum likelihood estimator of the odds ratio.14 The analyses were conducted by the Alliance Statistics and Data Center using the R Statistical Environment [R Development Core Team 2011] along with the survival and cmprsk extension packages and SAS © 9·4 TS Level 1M3 for Windows [SAS Institute, Cary, NC, USA]. A detailed description of the statistical considerations, including the design and analysis methods, has been provided in the supplemental material. All analyses were based on the study database frozen on October 19, 2016.
Role of the funding source
The National Cancer Institute (NCI) sponsored the study. Lenalidomide and placebo were provided by Celgene (Summit, New Jersey, United States) to the NCI, which in turn provided the study drugs to the investigators. Celgene was not involved in the study design, conduct of the study, or in the analysis or reporting of the data. All authors had access to the data through the Alliance Statistics and Data Center, which collected the data.
RESULTS
Four hundred sixty patients, out of 568 enrolled patients, were randomised between April 14, 2005 and July 2, 2009: 231 to the lenalidomide group and 229 to the placebo group. As has been previously reported, patients were evenly distributed by age, sex, disease stage, β2-microglobulin level at registration (supplemental material).2 The disposition of the patients, including reasons for ineligibility, is shown in the CONSORT diagram (Figure 1 and supplemental material). The reasons for treatment discontinuation are summarised in the supplemental material.
Figure 1.

CONSORT flow diagram of patient disposition at the current data cut-off.
The median follow-up for the updated survival analysis, as of October 19, 2016 was 91 months. One-hundred forty-six of the 231 patients in the lenalidomide group (63%) as compared with 176 of the 229 patients in the placebo group (77%) had progressive disease or had died (Figure 2A). The median TTP was 57·3 months (95% CI 44·2–73·3) in lenalidomide group and 28·9 months (95% CI 23·0–36·3) in the placebo group with a hazard ratio of 0·57 (95% CI 0·46 to 0·71; p<0·001). TTP analyses comparing Alliance (EMA) and FDA censoring rules are presented in the supplemental material. A total of 88 out of the 231 patients in the lenalidomide group have died (38%) compared with 120 out of 229 patients in the placebo group (52%) (Figure 2B). The median OS is 113·8 months (95% CI 100·4-not reached) in the lenalidomide group and is 84·1 months (95% CI 73·8–106·0) in the placebo group (hazard ratio 0·61; 95% CI, 0·46 to 0·80; p<0·0004). The rate of overall survival at five years is 76% (95% CI, 70% to 81%) among patients in the lenalidomide group and 64% (95% CI, 58% to 70%) among patients in the placebo group.
Figure 2.
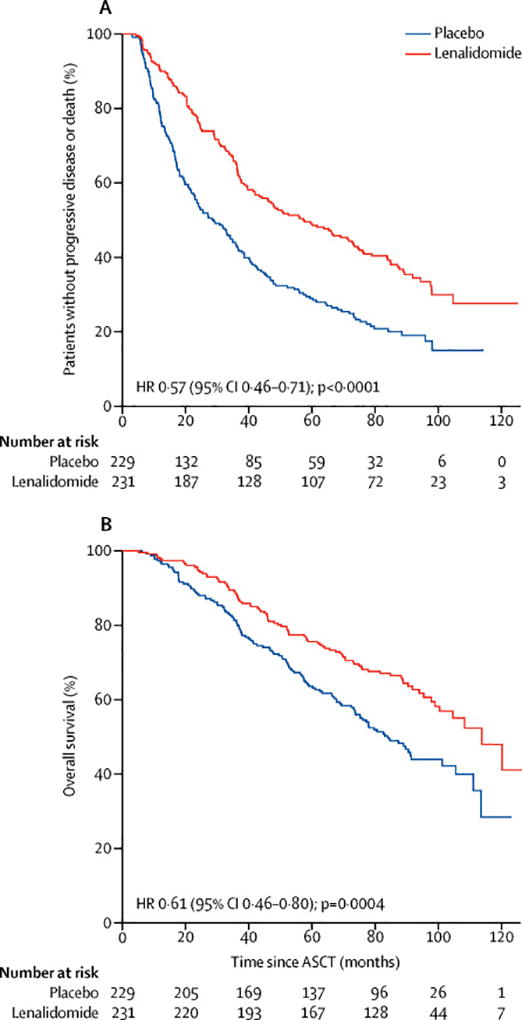
Kaplan-Meier estimates of time to progression (A) and overall survival (B). ASCT denotes autologous stem cell transplant.
The median time to crossover to lenalidomide for those placebo patients who chose to crossover at time of study unblinding was 11 months (range 2·6–49·7 months). Similar hazard ratios for TTP and overall survival were observed when placebo patients crossing over within 6 months of randomisation (n=19) (TTP hazard ratio 0·58, 95% CI 0·47-0·72, p<0·0001; overall survival hazard ratio 0·61, 95% CI 0·47-0·81, p=0·0004) or within 12 months (n=46) (TTP hazard ratio 0·53, 95% CI 0·43-0·66, p<0·0001; overall survival hazard ratio 0·58, 95% CI 0·44-0·76, p<0·0001) of randomisation were included in the lenalidomide arm (supplemental material). The median time on treatment for the lenalidomide arm was 31·0 months (95% CI 24·8–35·8 months) and was 18·1 months (95% CI 17·1–22·6 months) for the placebo arm (p<0.0001). Within the placebo group, the median time on treatment for the crossover patients was 30·7 months (95% CI 27·1–37·4) and 14·5 months (95% CI 12·5–17·3) for those patients who did not crossover. Treatment-free intervals were not determined. The median dose for the time on treatment for the lenalidomide arm was 6.8 mg daily.
For those patients who experienced disease progression, the median survival time following progression was similar amongst the two treatment arms (42·6 months (95% CI 36·4–60·5) for lenalidomide and 39·2 months (95% CI 31·2–45·0) for placebo; hazard ratio 0·83 (95% CI 0·61 to 1·13, p=0.23)) (Figure 3). Similarly, no differences in survival time after progression were observed when placebo patients crossing over within six months of randomisation (hazard ratio 0·84; 95% CI 0·62-1·13, p=0.24) or within 12 months (hazard ratio 0·85; 95% CI 0·64 to 1·15, p=0.30) of randomisation were included in the lenalidomide group (supplemental material). Time to second objective disease progression (PFS2) could not be calculated as neither the date of second progression nor the start of third-line therapy was routinely collected from the sites.
Figure 3.
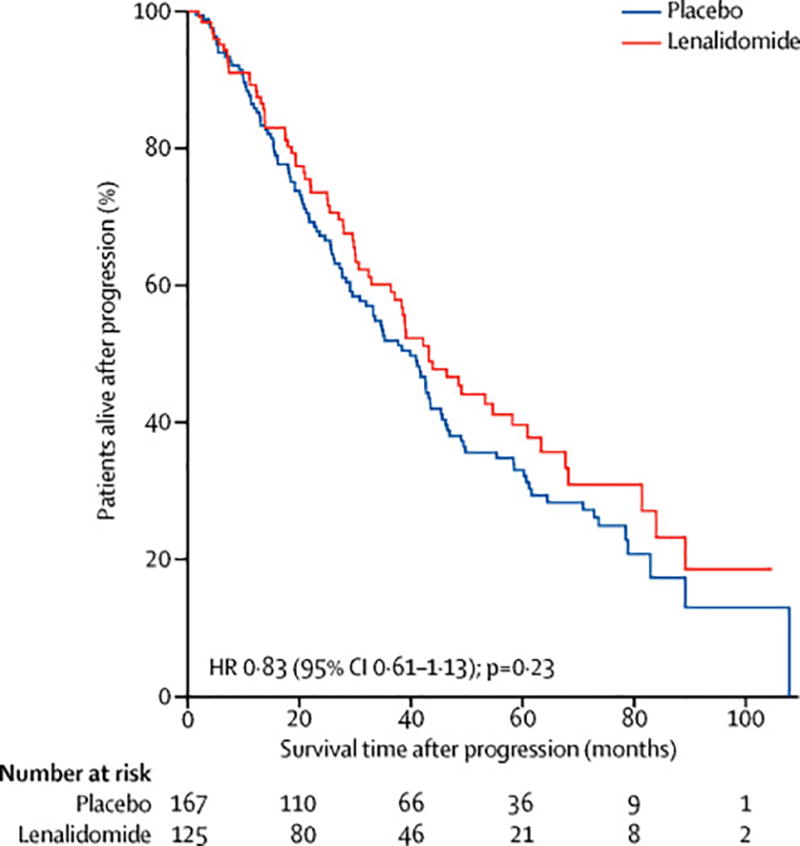
Kaplan-Meier estimates of survival time following progression.
Patients were stratified based on prior lenalidomide or thalidomide therapy, beta-2 microglobulin level (elevated vs normal), and investigator-reported complete response status (complete response or no complete response) at time of randomisation. Figure 3 shows the forest plots comparing the relative influences of these stratification factors on TTP and overall survival. There was a benefit of lenalidomide maintenance for TTP across all stratification groups (Figure 4A). Patients who received lenalidomide induction and lenalidomide maintenance (n=84) had a median TTP of 71·6 months (95% CI 57·3–97·8) vs. 46·0 months (95% CI 37·2–73·1) (p=0·21) for those who received lenalidomide maintenance in the absence of prior lenalidomide induction (n=147) (supplemental material). There was a benefit of lenalidomide maintenance on overall survival regardless of whether patients received prior lenalidomide induction or not (Figure 4B). The median overall survival did not differ significantly for patients who received lenalidomide maintenance with prior lenalidomide induction (104·7 months, 95% CI 97·8-not reached) vs those who received lenalidomide maintenance without prior lenalidomide induction (113·8 months, 95% CI 90·5-not reached) (p=0·078) (supplemental material). For patients in the placebo arm, the presence or absence of lenalidomide induction did not impact median TTP or overall survival (supplemental material). The median TTP for patients in the placebo group who were not in complete response at time of randomisation (n=60) was 19.8 months (95% CI 14·6–36·8) vs 33.0 months (95% CI 23·6–40·6) for patients in complete response (n=153) (p=0·37) (supplemental material). For the lenalidomide arm, the median TTP for patients not in complete response at time of randomisation (n=86) was 37·2 months (95% CI 28·9–60·1) vs 66·7 months (95% CI 50·6–94·3) (n=128) (p=0·0018). The median overall survival for these groups were 90·6 months (95% CI 72·5-not reached) (placebo, non-complete response), 80·6 months (95% CI 68· 9-not reached) (placebo, complete response), 97·8 months (95% CI 75· 0-not reached) (lenalidomide, non-complete response) and not reached (95% CI 100· 4-not reached) (lenalidomide, complete response) (supplemental material).
Figure 4.
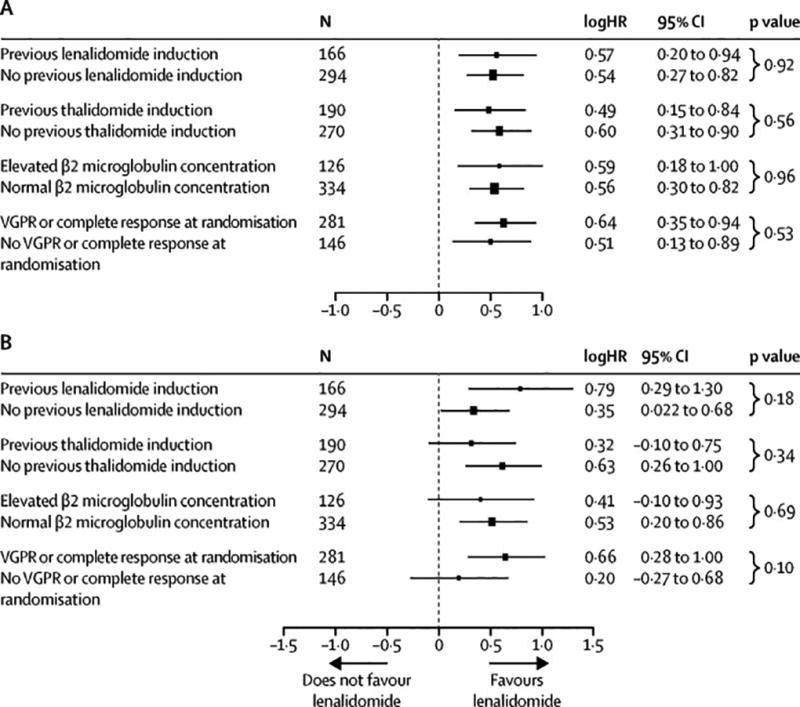
Forest plot of time to progression (A) and overall survival (B). Hazard ratios from subgroup analyses are shown on a natural-log scale. The radii of the circles are proportional to the inverse of the square of the standard error.
Centralized review was performed to assess response (per IMWG criteria) at time of randomisation as well as 1-, 2-, and 3-years post-ASCT (Table 1). Similar complete response rates were observed in the placebo and lenalidomide arms at 1 year (Chi-square testing of complete response/no-complete response vs lenalidomide/placebo, p-value pf 0·78). However, by year 3, the majority of patients who remained in complete response or VGPR were either in the lenalidomide arm or in the crossover arm. There were not reliable data regarding the numbers of patients in biochemical relapse at each time point.
Table 1.
Adjudicated response rates
| Placebo (n=229)
|
Lenalidomide (n=231) | ||||
|---|---|---|---|---|---|
| No Crossover (n=143) |
Crossover (n=86) | All Placebo (n=229) |
|||
|
| |||||
| Time of Randomisation | CR | 33 (14%) | 20 (9%) | 53 (23%) | 48 (21%) |
| VGPR | 60 (26%) | 40 (17%) | 100 (44%) | 80 (35%) | |
| PR | 32 (14%) | 20 (9%) | 52 (23%) | 78 (34%) | |
| SD | 4 (2%) | 1 (<1%) | 5 (2%) | 7 (3%) | |
| Rel/PD | 3 (1%) | 0 (0%) | 3 (1%) | 1 (<1%) | |
| NEa | 11 (5%) | 5 (2%) | 16 (7%) | 17 (7%) | |
| Off-studyb | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| VGPR/CR rate | 41% | 26% | 67% | 55% | |
|
| |||||
| One year post-ASCT | CR | 26 (11%) | 22 (10%) | 48 (21%) | 46 (20%) |
| VGPR | 32 (14%) | 36 (16%) | 68 (30%) | 65 (28%) | |
| PR | 8 (3%) | 13 (6%) | 21 (9%) | 36 (16%) | |
| SD | 0 (0%) | 0 (0%) | 0 (0%) | 2 (1%) | |
| Rel/PD | 15 (4%) | 6 (3%) | 13 (6%) | 19 (8%) | |
| NEa | 26 (11%) | 2 (1%) | 28 (12%) | 50 (22%) | |
| Off-studyb | 36 (16%) | 7 (3%) | 43 (19%) | 13 (6%) | |
| VGPR/CR rate | 25% | 25% | 51% | 48% | |
|
| |||||
| Two years post-ASCT | CR | 16 (7%) | 17 (7%) | 33 (14%) | 41 (18%) |
| VGPR | 8 (3%) | 22 (10%) | 30 (13%) | 43 (19%) | |
| PR | 2 (1%) | 13 (6%) | 15 (7%) | 19 (8%) | |
| SD | 0 (0%) | 0 (0%) | 0 (0%) | 1 (<1%) | |
| Rel/PD | 10 (4%) | 9 (4%) | 12 (5%) | 22 (10%) | |
| NEa | 29 (13%) | 13 (6%) | 42 (18%) | 61 (26%) | |
| Off-studyb | 78 (34%) | 12 (5%) | 90 (39%) | 44 (19%) | |
| VGPR/CR rate | 10% | 17% | 27% | 36% | |
|
| |||||
| Three years post-ASCT | CR | 5 (2%) | 8 (3%) | 13 (6%) | 30 (13%) |
| VGPR | 3 (1%) | 19 (8%) | 22 (10%) | 24 (10%) | |
| PR | 1 (<1%) | 4 (2%) | 5 (2%) | 8 (3%) | |
| SD | 0 (0%) | 0 (0%) | 0 (0%) | 1 (<1%) | |
| Rel/PD | 8 (3%) | 6 (3%) | 9 (4%) | 21 (9%) | |
| NEa | 31 (14%) | 27 (12%) | 58 (25%) | 84 (36%) | |
| Off-studyb | 95 (41%) | 22 (10%) | 117 (51%) | 63 (27%) | |
| VGPR/CR rate | 3% | 12% | 15% | 23% | |
Not evaluable because of missing data
Off-study because of prior disease relapse/progression
Reported salvage therapies were reviewed and the confirmed initial salvage therapies were compiled (supplemental material). Of those patients with confirmed initial salvage regimens, more patients received lenalidomide or a lenalidomide-containing regimen at time of relapse in the placebo arm (73 out 104) than in the lenalidomide (32 out of 101) or crossover arms (18 out of 42). Thirty-five patients were reported to have received a second ASCT at some point, as well as seven who underwent allogeneic transplant and one patient who received a second ASCT and an allogeneic transplant from a haploidentical donor.
Grade 3 and higher haematological and non-haematological adverse events that occurred after randomisation were previously reported for the lenalidomide and placebo arms.2 With longer follow-up, there have been a small number of additional adverse events reported (Table 2 and supplemental material). The adverse events in the placebo arm are now further categorized into the placebo crossover group and the non-crossover group. The majority of the haematologic adverse events in the placebo patients occurred in those who crossed over to receive lenalidomide. A total of three grade 5 adverse events occurred (two in the lenalidomide group and one in the placebo non-crossover group).
Table 2.
Adverse Eventsa
| Arm | Grade 1 n (%) |
Grade 2 n (%) |
Grade 3 n (%) |
Grade 4 n (%) |
Grade 5 n (%) |
|
|---|---|---|---|---|---|---|
|
| ||||||
| Haematologic | ||||||
|
| ||||||
| Hemoglobin | Len (n=231) | 15 (6%) | 6 (3%) | 9 (4%) | 2 (1%) | 0 (0%) |
| PBO (n=143) | 3 (2%) | 3 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 1 (1%) | 1 (1%) | 1 (1%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Leukocytes (total WBC) | Len (n=231) | 4 (2%) | 5 (2%) | 28 (12%) | 3 (1%) | 0 (0%) |
| PBO (n=143) | 2 (1%) | 1 (1%) | 1 (1%) | 1 (1%) | 0 (0%) | |
| CO (n=86) | 1 (1%) | 2 (2%) | 9 (10%) | 1 (1%) | 0 (0%) | |
|
| ||||||
| Lymphopenia | Len (n=231) | 2 (1%) | 2 (1%) | 20 (9%) | 1 (0%) | 0 (0%) |
| PBO (n=143) | 1 (1%) | 1 (1%) | 1 (1%) | 1 (1%) | 0 (0%) | |
| CO (n=86) | 1 (1%) | 1 (1%) | 5 (6%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Neutrophils | Len (n=231) | 14 (6%)) | 36 (16%) | 82 (35%) | 34 (15%) | 0 (0%) |
| PBO (n=143) | 12 (8%) | 10 (7%) | 7 (5%) | 4 (3%) | 0 (0%) | |
| CO (n=86) | 8 (9%) | 15 (17%) | 26 (30%) | 4 (5%) | 0 (0%) | |
|
| ||||||
| Platelets | Len (n=231) | 75 (32%) | 33 (14%) | 23 (10%) | 11 (5%) | 0 (0%) |
| PBO (n=143) | 28 (20%) | 3 (2%) | 0 (0%) | 7 (5%) | 0 (0%) | |
| CO (n=86) | 29 (34%) | 8 (9%) | 3 (3%) | 2 (2%) | 0 (0%) | |
|
| ||||||
| Non-Haematologic | ||||||
|
| ||||||
| Conduction abnormality | Len (n=231) | 0 (0%) | 0 (0%) | 1 (0%) | 0 (0%) | 0 (0%) |
| PBO (n=143) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (1%) | |
| CO (n=86) | 0 (0%) | 0 (0%) | 1 (1%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Fatigue | Len (n=231) | 10 (4%) | 9 (4%) | 0 (0%) | 0 (0%) | 0 (0%) |
| PBO (n=143) | 5 (3%) | 1 (1%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 4 (3%) | 6 (7%) | 0 (0%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Rash | Len (n=231) | 22 (10%) | 22 (10%) | 9 (4%) | 0 (0%) | 0 (0%) |
| PBO (n=143) | 10 (7%) | 7 (5%) | 1 (1%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 5 (6%) | 4 (5%) | 1 (1%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Diarrhea | Len (n=231) | 54 (23%) | 36 (16%) | 12 (5%) | 0 (0%) | 0 (0%) |
| PBO (n=143) | 15 (10%) | 3 (2%) | 2 (1%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 9 (10%) | 12 (14%) | 3 (3%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Febrile neutropenia (fever of unknown origin) | Len (n=231) | 2 (1%) | 0 (0%) | 14 (6%) | 1 (0%) | 0 (0%) |
| PBO (n=143) | 1 (1%) | 0 (0%) | 2 (1%) | 1 (1%) | 0 (0%) | |
| CO (n=86) | 1 (1%) | 0 (0%) | 1 (1%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Infection (documented clinically or microbiologically) | Len (n=231) | 1 (0%) | 4 (2%) | 13 (6%) | 2 (1%) | 0 (0%) |
| PBO (n=143) | 0 (0%) | 2 (1%) | 3 (2%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 0 (0%) | 2 (1%) | 4 (5%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Infection with normal ANC or grade 1 or 2 neutrophils | Len (n=231) | 0 (0%) | 6 (3%) | 13 (6%) | 0 (0%) | 1 (0%) |
| PBO (n=143) | 0 (0%) | 3 (2%) | 3 (2%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 1 (1%) | 9 (10%) | 1 (1%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Pain | Len (n=231) | 7 (3%) | 4 (2%) | 6 (3%) | 0 (0%) | 0 (0%) |
| PBO (n=143) | 5 (3%) | 2 (1%) | 6 (4%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 3 (3%) | 10 (12%) | 2 (2%) | 0 (0%) | 0 (0%) | |
|
| ||||||
| Vascular | Len (n=231) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (0%) |
| PBO (n=143) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
| CO (n=86) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | |
Adverse events that occurred in at least 10% for grades 1–2, at least 2% for grades 3–4, or any grade 5.
Abbreviations: ANC (absolute neutrophil count); CO (crossover); Len (lenalidomide); PBO (placebo).
The SPMs that were diagnosed after the time of randomisation and before the time of myeloma progression and receipt of salvage therapy are shown in Table 3. To date, a total of 18 haematological (7·8%), 14 solid tumour (6·1%), and 11 (4·8%) noninvasive SPMs have been diagnosed in the lenalidomide arm vs. a total of three haematological (1·3%), nine solid tumour (3·9%), and six (2·6%) noninvasive SPMs in the placebo arm. Of the placebo arm SPMs, three of three haematological, five of nine solid tumour, and five of six noninvasive SPMs were in the crossover group. Of the twenty-one patients with haematological SPMs, nine received a thalidomide-containing induction regimen, six received a lenalidomide-containing regimen, and six contained an anthracycline-containing induction regimen (supplemental material). Details of the solid tumour SPMs are shown in the supplemental material. The majority of the solid tumour SPMs were reported within the first three years after randomisation, but with longer follow-up, haematological SPMs continue to develop (supplemental material). The median times to haematological or solid tumour SPMs were similar for both treatment groups: 60·8 months (95% CI 36·1-not reached) (placebo, haematological) vs 49·8 months (95% CI 35·7–83·5) (lenalidomide, haematological) (p=0·86), 27·0 months (95% CI 18·1-not reached) (placebo, solid tumour) vs 21·7 months (95% CI 16·6-not reached) (lenalidomide, solid tumour) (p=0·45) (supplemental material). SPMs were also reported after myeloma progression and initiation of salvage therapy, including one haematological, four solid tumour, and two noninvasive SPM in the lenalidomide arm, and five haematological, two solid tumour, and two noninvasive SPMs in the placebo arm (supplemental material). Of those placebo arm SPMs, one haematological and one noninvasive SPM occurred in the crossover subgroup. Finally, one haematological, seven solid tumour, and two noninvasive SPMs were reported in enrolled patients who were never randomised (supplemental material).
Table 3.
Second primary malignancies
| SPM type | |||
|---|---|---|---|
|
| |||
| Treatment Arm | Haematologic (n) | Solid tumour (n) | Noninvasive (n) |
|
| |||
| Len (231) | MDS/AML (10) | Breast (3) | SCC (5) |
| B-cell ALL (6) | Colon (3) | BCC + SCC (3) | |
| Hodgkin lymphoma (1) | Prostate (2) | DCIS (2) | |
| Waldenstrom macroglobulinemia (1) | Endometrial (2) | BCC (1) | |
| Glioblastoma multiforme (1) | |||
| Melanoma (1) | |||
| Papillary Thyroid (1) | |||
| Salivary gland carcinoma (1) | |||
|
| |||
| Placebo (229) | |||
| Crossover to Len (86) | B-cell ALL (2) | Melanoma (2) | BCC (3) |
| MDS (1) | Endometrial (1) | BCC + SCC (2) | |
| Renal cell (1) | |||
| Invasive SCC (1) | |||
| Breast (1) | SCC (1) | ||
| No crossover (143) | Melanoma (1) | ||
| Ovarian/endometrial (1) | |||
| Lung carcinoid (1) | |||
Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; BCC, basal cell carcinoma; DCIS, ductal carcinoma in situ; MDS, myelodysplastic syndrome; SCC, squamous cell carcinoma.
Note: Two patients had MDS/AML and SCC, two patients had MDS and colon cancer, one patient had MDS and melanoma, one patient had breast and endometrial cancer, and one patient had endometrial cancer and SCC.
The cumulative incidence risks (CIR) of progressive disease, death, and SPM by treatment arm were analyzed. As shown in Figure 5, the CIR of progressive disease/death is higher for placebo compared with lenalidomide (p<0·0001) while the CIR of developing a SPM is higher for lenalidomide (p=0·0073) (Figure 5A). The CIR of death from any cause is higher with placebo than with lenalidomide (p<0.0001) (Figure 5B). The CIR of death from myeloma is higher in the placebo group (p<0·0001) while the CIR of death from SPM is higher in the lenalidomide group (p=0·031) (Figure 5C). The CIR curves for haematological vs solid tumour SPMs are shown in the supplemental material. To assess for the potential impact of crossover of placebo patients to lenalidomide on these risks, the CIR of progressive disease/death and SPM were assessed for the placebo patients who did not cross over and for the crossover patients (supplemental material). The risks of progressive disease, death, or SPM are not statistically significantly different between the lenalidomide and crossover arms. The median event-free survival (EFS; time to progressive disease, SPM, or death from any cause) for placebo was 27·0 months (95% CI 21·8–34·9) and 44·2 months (95% CI 37·3–56·1) for lenalidomide with a hazard ratio of 0·63 (95% CI 0·51-0·78) (p<0.0001) (supplemental material).
Figure 5.
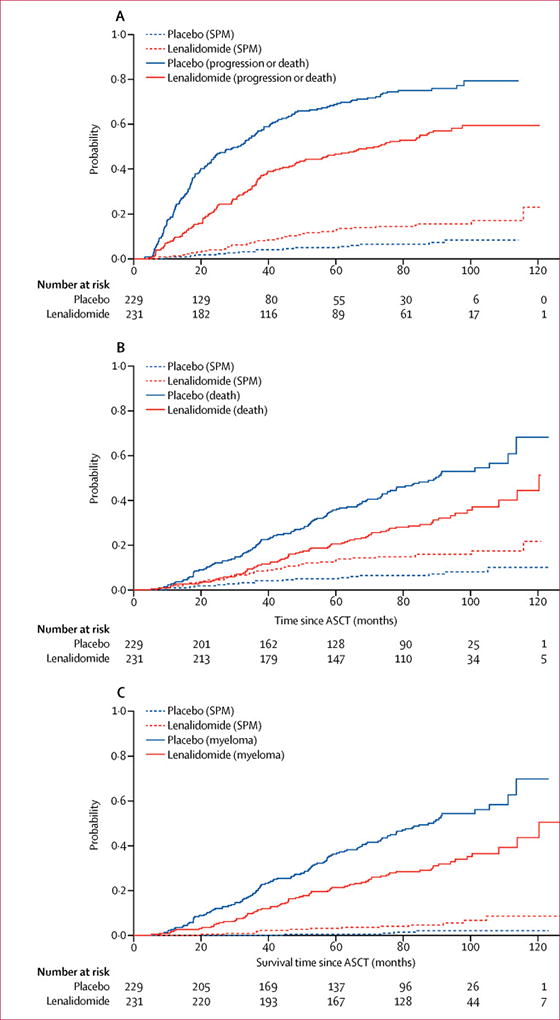
Cumulative incidence risk (CIR) of progressive disease, death, and second primary malignancies (SPMs) by treatment arm. A) The CIR of progressive disease or death from any cause is higher with placebo compared to lenalidomide (p<0·0001). The CIR of developing a SPM is higher with lenalidomide compared with placebo (p=0·0073). B) The CIR of death from any cause is higher with placebo compared with lenalidomide (p<0·0001). C) The CIR of death from myeloma is higher with placebo than with lenalidomide (p<0·0001) while the CIR of death from SPM is higher with lenalidomide than placebo (p=0·031).
DISCUSSION
This updated analysis of the phase 3 randomised trial evaluating lenalidomide vs placebo maintenance following ASCT demonstrates a persistent TTP and overall survival benefit for lenalidomide treatment. This benefit is maintained despite the crossover of a majority of eligible patients on the placebo arm to lenalidomide at the time of study unblinding. Survival after progression does not differ between the two treatment groups. The benefit derived from lenalidomide maintenance is independent of induction therapy as well as complete response status at time of randomisation. Lenalidomide maintenance is associated with an increased risk of haematological SPMs. This study, as well as a number of transplant and non-transplant randomised studies, have demonstrated benefit with prolonged lenalidomide therapy.3–5, 15, 16 A meta-analysis of the CALGB, IFM and GIMEMA studies found that lenalidomide maintenance significantly improves overall survival (median overall survival not yet reached for lenalidomide vs. 86 months for control (HR =0·74, log-rank p=0·001)), regardless of response achieved post-ASCT.6
The study most directly comparable to CALGB 100104 is the IFM 2005-02 trial, which also evaluated lenalidomide vs placebo maintenance therapy following ASCT. While a PFS benefit with lenalidomide was observed, there was no difference in overall survival between the study arms.3 There are multiple factors which may contribute to the difference in survival outcomes between the studies, including the types of induction regimens, consolidation therapy, numbers of transplants, the duration of lenalidomide maintenance and available salvage therapies. Overall, the IFM 2005-02 study population was exposed to more traditional chemotherapy while the CALGB 100104 study population was exposed to more novel agent-based therapy. The extent to which pre-transplant therapy determines response to subsequent salvage therapies remains to be determined. However, our analysis reveals that patients progressing on lenalidomide maintenance have a similar overall survival after progression as do placebo patients after progression (Figure 3), which suggests that prolonged lenalidomide maintenance does not confer disease resistance.
The optimal dose, schedule, and duration of lenalidomide maintenance continue to be topics of much discussion. The present study as well as the GIMEMA RV-209 and Myeloma XI studies involved lenalidomide maintenance therapy continued until progression while the IFM 2005-02 study discontinued lenalidomide maintenance after a median time of two years (range 1–3 years) due to concerns regarding SPM risk.3–5 The IFM 2009 study which assessed the timing of ASCT,17 incorporated one year of lenalidomide maintenance while the ongoing American study (DETERMINATION Trial) is identical in design except that maintenance is continued until progression. Future comparison of survival outcomes and toxicities, including the incidence of SPMs, between the IFM 2009 and DETERMINATION studies will provide important information regarding the impact of maintenance duration. The American and French studies (e.g., CALGB 100104, BMT CTN 0702, IFM 2005-02, and IFM 2009)2, 3, 17, 18 have utilized continuous dosing of lenalidomide while other European studies (e.g., Myeloma XI, GIMEMA RV-209, RV-MM-EMN-441, EMN02/HO95)4, 5, 19, 20 have utilized a 21/28 day schedule. In the absence of a prospective study comparing the two dosing schedules, it is difficult to conclude from the previously conducted studies whether one schedule is more optimal than the other from either a survival or toxicity perspective. However, it should be noted that the largest of the studies, CALGB 100104 and Myeloma XI, have found very similar median TTP/PFS durations for the lenalidomide arms (57·3 and 60 months respectively) despite the different dosing schedules. As ongoing and planned studies involving the addition of other agents to lenalidomide maintenance progress, it is likely that the optimal dosing, schedule and duration of lenalidomide maintenance will continue to evolve.
Central review of the response data (Table 1) revealed that, despite significant efforts by participating sites to provide follow-up data, there were missing data at later time points. These include bone marrow biopsies, 24-hour urine quantitations and incomplete serum and urine electrophoresis analyses. The independent review committee reviewed all available primary source data. Despite these limitations, this review assessment did reveal that surprisingly, there was little difference in the complete response/VGPR rate between the two arms at one year. However, the majority of patients in complete response/VGPR by year 3 were in either the lenalidomide arm or the crossover subgroup of the placebo arm.
This updated analysis of CALGB 100104 continues to show an increased risk of SPMs associated with lenalidomide maintenance therapy, although the risks of progressive disease and death due to myeloma were substantially higher than the risk of SPM in both cohorts. SPM risk is associated with multiple factors, including the underlying disease, age, and myeloma therapy. There were patients enrolled but never randomised who developed SPMs (supplemental material). This is consistent with the underlying SPM risk in this patient population. The risk of malignancy increases with advancing age and age-related clonal hematopoiesis is associated with increased risk of haematological malignancies.21, 22 Multiple studies have demonstrated that monoclonal gammopathy of undetermined significance and myeloma, even in the absence of therapy, are associated with increased risk of haematological malignancies, particularly MDS/AML.23–25 This finding implicates the presence of an intrinsic defect in the hematopoietic system in patients with plasma cell dyscrasias. An increased risk of solid tumours in myeloma patients has also been reported.26 There was not a predominant solid tumour SPM type found in the present study. With respect to lenalidomide, a meta-analysis of 3254 newly diagnosed patients treated on seven randomized phase 3 trials revealed that the cumulative 5-year incidence of all SPMs at 5 years was 6·9% in patients who received lenalidomide vs. 4·8% in those who did not (p=0·037).27 An increase in haematological malignancies (3·1% vs. 1·4%, p=0·029) but not solid tumours was observed.
Since the time of first publication of CALGB 100104, four new solid tumour SPMs and ten new haematological SPMs were reported in the lenalidomide group. It appears that the risk of solid tumours may be primarily incurred during the first several years of lenalidomide therapy post-transplant, with haematological malignancies continuing to be diagnosed with later follow-up (Supplemental Figures 6–7). However, given the overall small number of SPMs, it is difficult to draw any definite conclusions regarding the temporal association between SPM type and lenalidomide exposure. Genetic analysis, particularly of the haematological SPMs, is needed to better determine the mechanisms by which lenalidomide contributes to the pathogenesis of SPMs. While MDS/AML has previously been associated with high-dose melphalan and myeloma therapy, the appearance of B-cell ALL SPMs has been somewhat unexpected. Further studies are needed to determine whether the effects of lenalidomide on IKZF1, a transcription factor which has been associated with B-cell ALL,28–30 contribute to the development of ALL as a SPM.
In conclusion, our study demonstrates that lenalidomide maintenance therapy post-ASCT confers significant TTP and overall survival benefit. The overall survival data, which shows a median overall survival of 9·5 years (from time of ASCT), provides a new benchmark for survival, particularly noteworthy as this study was conducted in an era where triplet regimens containing IMiD/PI were not routinely used. Cytogenetic and FISH testing of diagnostic samples was not available for the majority of patients, thus the impact of lenalidomide maintenance on different cytogenetic risk groups cannot be determined. Preliminary results of the Myeloma XI trial demonstrated benefit in high and low cytogenetic risk patients.5 This report demonstrates that patients in CR post-ASCT benefit from lenalidomide maintenance, however, the CR response was determined by numbers of bone marrow plasma cells and immunofixation testing. The newly revised IMWG criteria now include minimal residual disease assessment and multiple studies have demonstrated superior outcomes for patients who achieve MRD-negativity post-ASCT.31–33 Thus the extent to which lenalidomide maintenance improves survival outcomes for MRD-negative vs MRD-positive patients remains to be determined. One limitation of the study is that quality of life data were not prospectively collected. However, this study is amongst a growing number of studies demonstrating the feasibility of long-term maintenance therapy with lenalidomide. Lenalidomide maintenance until progression post-ASCT may be considered a standard of care and should form the backbone of future maintenance studies incorporating novel agents such as monoclonal antibodies or vaccine-based approaches.
Supplementary Material
Research in context panel.
Evidence before the study
We searched PubMed up until 10/19/2016 for any clinical trial publications with the terms multiple myeloma, thalidomide, lenalidomide, maintenance, and autologous stem cell transplant (ASCT). These terms were also searched in conference abstracts from the American Society of Hematology, the American Society of Clinical Oncology, and the European Hematology Association. At the time the present study was initiated, the use of thalidomide was being studied, however no publications had yet been published. Preliminary results from two randomised studies had been presented in conference abstracts. No clinical studies had been conducted in this setting with lenalidomide. Since this study was initiated, there have been four other randomised phase 3 studies which have assessed lenalidomide maintenance following ASCT for myeloma: one placebo-controlled, two without a placebo, and one involving lenalidomide vs lenalidomide-prednisone maintenance. Data from the other lenalidomide vs placebo/no treatment studies have demonstrated a significant progression-free survival (PFS) benefit but not overall survival (OS) benefit for lenalidomide maintenance.
Added value of this study
We report the long-term follow-up of this randomised phase 3 study evaluating lenalidomide vs placebo maintenance therapy following single ASCT. This analysis confirms the TTP and overall survival benefit of lenalidomide maintenance, regardless of the response achieved post-ASCT. This analysis provides a detailed update of the SPMs that have been observed and confirms that while lenalidomide maintenance is associated with an increased risk of both haematological and solid tumour SPMs, the risk is off-set by the magnitude of the TTP and overall survival benefit.
Implications of all the available evidence
This updated analysis, in concert with the available literature, confirms that continuous lenalidomide maintenance post-ASCT until disease progression provides significant TTP and overall survival benefit, and can therefore be considered a standard of care.
Acknowledgments
The study team would like to thank the patients and families who participated in this study and the clinical teams who provided care for the patients. We wish to acknowledge the efforts of the research nurses, data coordinators, and investigators who participated in the data cleaning efforts. We would also like to acknowledge those members of the Alliance who assisted with the protocol development and amendments, including Michael Kelly, Destin Carlisle and Guadalupe Aquino. We would like to thank Michelle Maglio for administrative support. We thank John Postiglione for his efforts on this study. Finally, we wish to honor the memory of Dr. Dan Sargent who tragically died in 2016. Dr. Sargent was the head of the Alliance Statistics and Data Center and facilitated the publication of the first report of the CALGB 100104 study as well as the update. He provided sage advice throughout the analysis.
Declaration of interests:
Dr. Anderson reports personal fees from Celgene, Millennium Takeda, Gilead, Bristol Myers Squibb. Dr. Bashey has nothing to disclose. Ms Boyd has nothing to disclose. Dr. Callander has nothing to disclose. Dr. Devine has nothing to disclose. Dr. Gentile has nothing to disclose. Dr. Giralt reports research funding and personal fees from Celgene and Takeda, research funding from Sanofi, personal fees from Jazz and Amgen. Dr. Hari reports grants and personal fees from Celgene. Ms Hars has nothing to disclose. Dr. Hassoun reports grants from CALGB/Alliance during the conduct of the study and grants from Celgene. Dr. Hofmeister reports other support (local principal investigator for clinical trial using drug made by Celgene) from Celgene. Dr. Holstein reports personal fees from Celgene, Takeda and Amgen. She received travel support and honoraria for response adjudication of this study by the Alliance for Clinical Trials in Oncology. Dr. Horowitz has nothing to disclose. Dr. Hurd has nothing to disclose. Dr. Isola has nothing to disclose. Dr. Jiang has nothing to disclose. Dr. Jung has nothing to disclose. Dr. Landau reports grants from Celgene during the conduct of the study, personal fees from Spectrum Pharmaceuticals, Prothena, Janssen, and Takeda. Dr. Linker has nothing to disclose. Dr. Maziarz has nothing to disclose. Dr. McCarthy reports research support and personal fees from Celgene, and personal fees from Bristol Myers Squibb, Karyopharm, Gamida Cell, Janssen, Sanofi, and The Binding Site. He received travel support and honoraria for response adjudication of this study by the Alliance for Clinical Trials in Oncology. Dr. McClune has nothing to disclose. Dr. Moreb has nothing to disclose. Dr. Owzar has nothing to disclose. Dr. Pasquini reports personal fees from Atara Biotherapeutics, Pfizer, and Baxalta. Dr. Qazilbash has nothing to disclose. Dr. Richardson reports personal fees from Celgene. Dr. Rodriguez has nothing to disclose. Dr. Schlossman has nothing to disclose. Ms Schultz has nothing to disclose. Dr. Shea has nothing to disclose. Dr. Smith has nothing to disclose. Dr. Stadtmauer reports personal fees from Celgene and Takeda. Dr. van Besien has nothing to disclose. Dr. Vij reports personal fees from Celgene, Bristol Myers Squibb, Janssen, AbbVie, Karyopharm and research support and personal fees from Amgen and Takeda. Dr. Weisdorf has nothing to disclose. Ms Wilson has nothing to disclose.
Funding source: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA031946, U10CA033601, U10CA180821 and U10CA180882 (to the Alliance for Clinical Trials in Oncology),U01HL069294, U10CA021115, , U10CA004457, U10CA007968, U10CA016450, U10CA021060, U10CA032291, U10CA047559, U10CA059518, U10CA077298, U10CA077440, U10CA077651, U10CA077658, U10CA138561, U10CA180791, U10CA180799, U10CA180833, U10CA180838, U10CA180850, U10CA180858, U10CA180866, and U10CA180867. Support for this study was provided in part by the Blood and Marrow Transplant Clinical Trials Network through grant #U10HL069294 from the National Heart, Lung, and Blood Institute (NHLBI) and the National Cancer Institute. Support was also provided in part by the Eastern Cooperative Oncology Group (ECOG) supported by CA21115. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This study was also supported in part by Celgene Corporation.
The National Cancer Institute (NCI) sponsored the study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ClinicalTrials.gov Identifier: NCT00114101
Author contributions:
Holstein: Literature search, figures, data collection, data analysis, data interpretation, writing
Jung: data analysis, data interpretation, writing
Richardson: data interpretation, writing
Hofmeister: data interpretation, writing
Hurd: data interpretation, writing
Hassoun: data interpretation, writing
Giralt: data interpretation, writing
Stadtmauer: data interpretation, writing
Weisdorf: data interpretation, writing
Vij: data interpretation, writing
Moreb: data interpretation, writing
Callander: data interpretation, writing
van Besien: data interpretation, writing
Gentile: data interpretation, writing
Isola: data interpretation, writing
Maziarz: data interpretation, writing
Bashey: data interpretation, writing
Landau: data interpretation, writing
Martin: data interpretation, writing
Qazilbash: data interpretation, writing
Rodriguez: data interpretation, writing
McClune: data interpretation, writing
Schlossman: data interpretation, writing
Smith: data interpretation
Hars: data analysis, data interpretation, figures, writing
Owzar: study design, data interpretation, writing
Jiang: data analysis, data interpretation, figures, writing
Boyd: data collection, data analysis
Schultz: data collection, data analysis
Wilson: data collection, data analysis
Hari: data interpretation, writing
Pasquini: data interpretation, writing
Horowitz: data interpretation, writing
Shea: study design, data interpretation, writing
Devine: study design, data interpretation, writing
Linker: study design, data interpretation, writing
Anderson: study design, data interpretation, writing
McCarthy: Literature search, study design, data collection, data analysis, data interpretation, writing
Contributor Information
Sarah A. Holstein, University of Nebraska Medical Center, Omaha, NE.
Sin-Ho Jung, Alliance Statistics and Data Center, Duke University, Durham, NC.
Paul G. Richardson, Dana-Farber/Partners CancerCare, Boston, MA.
Craig C. Hofmeister, The Ohio State University Comprehensive Cancer Center, Columbus, OH.
David D. Hurd, Wake Forest Baptist Medical Center, Winston-Salem, NC.
Hani Hassoun, Memorial Sloan Kettering Cancer Center, New York, NY.
Sergio Giralt, Memorial Sloan Kettering Cancer Center, New York, NY.
Edward A. Stadtmauer, University of Pennsylvania, Philadelphia, PA.
Daniel J. Weisdorf, University of Minnesota, Minneapolis, MN.
Ravi Vij, Washington University School of Medicine, St. Louis, MO.
Jan S. Moreb, UF Health, University of Florida, Gainesville, FL.
Natalie S. Callander, University of Wisconsin, Madison, WI.
Koen van Besien, Weill Medical College of Cornell University, New York, NY.
Teresa G. Gentile, State University of New York Upstate Medical University, Syracuse, NY.
Luis Isola, Mount Sinai School of Medicine, New York, NY.
Richard T Maziarz, Oregon Health & Science University, Portland, OR.
Asad Bashey, Blood and Marrow Transplant Program at Northside Hospital, Atlanta, GA.
Heather Landau, Memorial Sloan Kettering Cancer Center, New York, NY.
Thomas Martin, University of California at San Francisco, San Francisco, CA.
Muzaffar H Qazilbash, MD Anderson Cancer Center, Houston, TX.
Cesar Rodriguez, Wake Forest Baptist Medical Center, Winston-Salem, NC.
Brian McClune, University of Minnesota, Minneapolis, MN.
Robert L. Schlossman, Dana-Farber/Partners CancerCare, Boston, MA.
Scott E. Smith, Alliance for Clinical Trials in Oncology, Chicago, IL; Loyola University, Chicago, IL.
Vera Hars, Alliance Statistics and Data Center, Duke University, Durham, NC.
Kouros Owzar, Alliance Statistics and Data Center, Duke University, Durham, NC.
Chen Jiang, Alliance Statistics and Data Center, Duke University, Durham, NC.
Molly Boyd, Alliance Statistics and Data Center, Mayo Clinic, Rochester, MN.
Chelsea Schultz, Alliance Statistics and Data Center, Mayo Clinic, Rochester, MN.
Marcia Wilson, Alliance Statistics and Data Center, Mayo Clinic, Rochester, MN.
Parameswaran Hari, Medical College of Wisconsin, Milwaukee, WI.
Marcelo C. Pasquini, Medical College of Wisconsin, Milwaukee, WI.
Mary M. Horowitz, Medical College of Wisconsin, Milwaukee, WI; Blood and Marrow Transplant Clinical Trials Network, Rockville, MD.
Thomas C. Shea, UNC Lineberger Cancer Center, University of North Carolina, Chapel Hill, NC.
Steven M. Devine, The Ohio State University Comprehensive Cancer Center, Columbus, OH.
Charles Linker, University of California at San Francisco, San Francisco, CA.
Kenneth C. Anderson, Dana-Farber/Partners CancerCare, Boston, MA.
Philip L. McCarthy, Roswell Park Cancer Institute, Buffalo, NY.
References
- 1.Morgan GJ, Gregory WM, Davies FE, et al. The role of maintenance thalidomide therapy in multiple myeloma: MRC Myeloma IX results and meta-analysis. Blood. 2012;119(1):7–15. doi: 10.1182/blood-2011-06-357038. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770–81. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attal M, Lauwers-Cances V, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782–91. doi: 10.1056/NEJMoa1114138. [DOI] [PubMed] [Google Scholar]
- 4.Palumbo A, Cavallo F, Gay F, et al. Autologous transplantation and maintenance therapy in multiple myeloma. N Engl J Med. 2014;271(10):895–905. doi: 10.1056/NEJMoa1402888. [DOI] [PubMed] [Google Scholar]
- 5.Jackson GH, Davies FE, Pawlyn C, et al. Lenalidomide is a highly effective maintenance therapy in myeloma patients of all ages; results of the phase III Myeloma XI study. Blood (ASH Abstracts) 2016;128:1143. [Google Scholar]
- 6.Attal M, Palumbo A, Holstein SA, et al. Lenalidomide (LEN) maintenance (MNTC) after high-dose melphalan and autologous stem cell transplant (ASCT) in multiple myeloma (MM): A meta-analysis (MA) of overall survival (OS) J Clin Oncol. 2016;34(15 suppl):8001. [Google Scholar]
- 7.Blade J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102(5):1115–23. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 8.Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467–73. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 9.Emerson SS, Fleming TR. Symmetric group sequential test designs. Biometrics. 1989;45(3):905–23. [PubMed] [Google Scholar]
- 10.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 11.Cox DR, Oakes D. Monographs on Statistics and Applied Probability 21. Chapman & Hall; 1984. Analysis of Survival Data. [Google Scholar]
- 12.Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. 2. Hoboken: John Wiley & Sons, Inc; 2002. [Google Scholar]
- 13.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16(3):1141–54. [Google Scholar]
- 14.Fisher RA. The logic of inductive inference. J R Stat Soc. 1935;98:39–54. [Google Scholar]
- 15.Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2012;366(19):1759–69. doi: 10.1056/NEJMoa1112704. [DOI] [PubMed] [Google Scholar]
- 16.Benboubker L, Dimopoulos MA, Dispenzieri A, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371(10):906–17. doi: 10.1056/NEJMoa1402551. [DOI] [PubMed] [Google Scholar]
- 17.Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376(14):1311–20. doi: 10.1056/NEJMoa1611750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (Len) and dexamethasone (RVD) consolidation with Len maintenance (ACM), tandem autoHCT with Len maintenance (TAM) and autoHCT with Len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702-StaMINA trial) Blood (ASH Abstracts) 2016;128:LBA-1. [Google Scholar]
- 19.Gay F, Oliva S, Petrucci MT, et al. Chemotherapy plus lenalidomide versus autologous transplantation, followed by lenalidomide plus prednisone versus lenalidomide maintenance, in patients with multiple myeloma: a randomised, multicenter, phase 3 trial. Lancet Oncol. 2015;16(16):1617–29. doi: 10.1016/S1470-2045(15)00389-7. [DOI] [PubMed] [Google Scholar]
- 20.Cavo M, Palumbo A, Zweegman S, et al. Upfront autologous stem cell transplantation (ASCT) versus novel agent-based therapy for multiple myeloma (MM): a randomized phase 3 study of the European Myeloma Network (EMN02/HO95 MM trial) J Clin Oncol. 2016;34(15 suppl):8000. [Google Scholar]
- 21.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tzeng HE, Lin CL, Tsai CH, et al. Time trend of multiple myeloma and associated secondary primary malignancies in Asian patients: a Taiwan population-based study. PLoS One. 2013;8(7):e68041. doi: 10.1371/journal.pone.0068041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Razavi P, Rand KA, Cozen W, et al. Patterns of second primary malignancy risk in multiple myeloma patients before and after the introduction of novel therapeutics. Blood Cancer J. 2013;3:e121. doi: 10.1038/bcj.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mailankody S, Pfeiffer RM, Kristinsson SY, et al. Risk of acute myeloid leukemia and myelodysplastic syndromes after multiple myeloma and its precursor disease (MGUS) Blood. 2011;118(15):4086–92. doi: 10.1182/blood-2011-05-355743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasskarl J, Ihorst G, De Pasquale D, et al. Association of multiple myeloma with different neoplasms: systematic analysis in consecutive patients with myeloma. Leuk Lymphoma. 2011;52(2):247–59. doi: 10.3109/10428194.2010.529207. [DOI] [PubMed] [Google Scholar]
- 27.Palumbo A, Bringhen S, Kumar SK, et al. Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: a meta-analysis of individual patient data. Lancet Oncol. 2014;15(3):333–42. doi: 10.1016/S1470-2045(13)70609-0. [DOI] [PubMed] [Google Scholar]
- 28.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 29.Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7101):110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 30.Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP) Blood. 2009;114(10):2159–67. doi: 10.1182/blood-2008-08-173963. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328–46. doi: 10.1016/S1470-2045(16)30206-6. [DOI] [PubMed] [Google Scholar]
- 32.Paiva B, Vidriales MB, Cervero J, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112(10):4017–23. doi: 10.1182/blood-2008-05-159624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparatmeter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540–7. doi: 10.1200/JCO.2012.46.2119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.