

ABSTRACT
Lung cancer is one of the leading causes of cancer-related mortality and is categorized into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). We present a patient with epidermal growth factor receptor (EGFR)-mutant-NSCLC who developed metastatic SCLC after initial therapy with second-generation EGFR-tyrosine kinase inhibitor, afatinib. A 65-year-old male non-smoker was diagnosed with adenocarcinoma of the right lung, stage IVA (M1a). Due to tumor positivity for EGFR-Exon 19 deletion, the patient was started on oral afatinib, which resulted in a partial response. After ten months of treatment, he presented in the office with abdominal pain, distension, weight loss and jaundice. He had diffuse skeletal and hepatic metastases on PET/CT scan with interval progression of his cancer. Although the recurrence of lung adenocarcinoma was suspected, the patient was diagnosed with SCLC on liver biopsy. He received two cycles of chemotherapy and died due to pneumonia and sepsis.
KEYWORDS: Small Cell Lung Cancer, Adenocarcinoma of Lung, Transformation, Tyrosine Kinase Inhibitors, Afatinib, Resistance, EGFR Receptors, Bronchoalveolar Lavage, Poorly differentiated Adenocarcinoma
Introduction
Lung cancer is one of the leading causes of cancer-related mortality in the United States and worldwide and is categorized into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). SCLC represents less than one-fourth of all lung cancers; the majority of patients have NSCLC with adenocarcinoma (ADC) as the most common subtype.1 The treatment approach between NSCLC and SCLC differs based upon their respective distinct clinical and molecular features. We present a patient with epidermal growth factor receptor (EGFR)-mutant-NSCLC who developed metastatic SCLC after initial therapy with a second-generation EGFR-tyrosine kinase inhibitor (TKI), afatinib.
Case history
A 65-year-old Caucasian male was diagnosed with pneumonia on x-ray in November 2015.
The patient was a lifelong non-smoker. He showed clinical improvement with oral antibiotics. After six weeks, he underwent a follow-up CT showing nodular densities in the right lung field with right hilar, right paratracheal and extensive mediastinal lymphadenopathy (LAD) (Figure 1).
Figure 1.
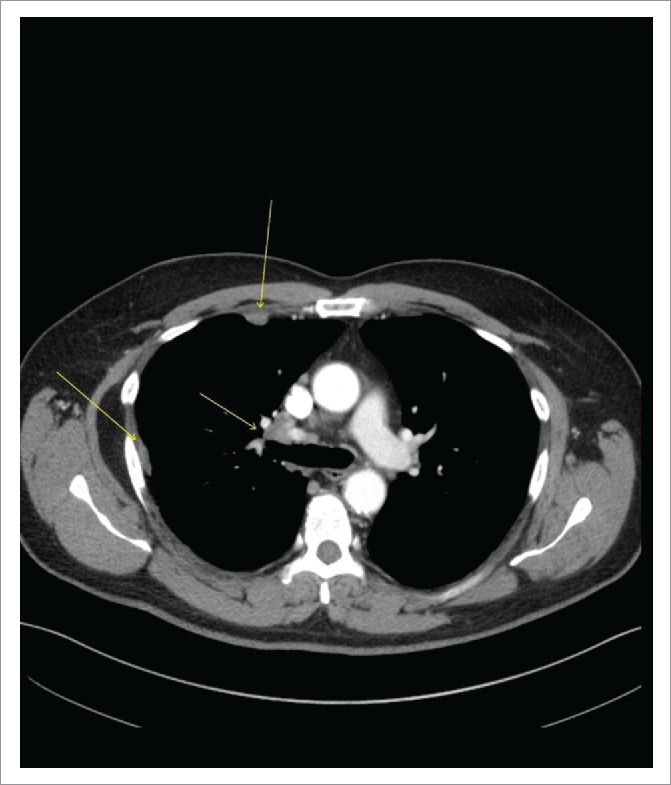
CT Chest with Contrast Demonstrating Right Hilar Disease with Right-sided Pleural Involvement (arrows).
There was a 16-mm irregular mass in the right middle lobe in addition to right-sided pleural thickening and effusion. The patient was referred to a pulmonologist for further workup which was negative for ANA, rheumatoid factor, ACE and quantiFERON-TB Gold test. He reported no appetite or weight changes. Endobronchial ultrasound (EBUS)-guided FNAC of right hilar, paratracheal and pre-carinal lymph nodes (LN) along with bronchoalveolar lavage (BAL) of the right middle lobe revealed poorly-differentiated ADC in all specimens (Figure 2A). Brain MRI ruled out metastatic lesions; however, PET/CT was positive for thoracic LAD and right pleural disease (Figure 3A, Part a). The patient was staged as IVA (M1a) of right-sided lung ADC on clinical and radiological bases and was positive for EGFR-Exon 19 deletion identified by next generation sequencing. He was started on oral afatinib 40 mg once daily as first-line therapy in January 2016 due to 1) a positive EGFR-mutant status, 2) proven superior survival with this drug compared to chemotherapy based on phase 3 studies in EGFR-mutant stage IV NSCLC patients2 and 3) per 2016 National Comprehensive Cancer Network (NCCN) guidelines.3 After four months, in May 2016 a repeat PET/CT showed significant interval improvement with only a single prominent right hilar LN as a site of disease, consistent with a partial response to afatinib (Figure 3A, Part b). In November 2016, at 10 months, he presented as an outpatient with abdominal pain, distension, weight loss and jaundice. Labs showed AST 368 U/L, ALT 243 U/L, ALP 211 U/L, LDH 1544 U/L and bilirubin 5.2 mg/dL. There was evidence of diffuse hepatic and skeletal metastases along with new cervical, thoracic and upper abdominal LAD on PET/CT scan (Figure 3B, Parts a-b). MRI brain was also positive for a solitary cerebellar lesion of six mm this time, consistent with brain metastasis. Although the recurrence of ADC was suspected, liver biopsy of the metastatic lesion revealed metastatic small cell cancer staining with synaptophysin, CD56, and P63 (Figure 2B, Parts a-d) and he was diagnosed with advanced SCLC. Based on small cell histology, T790M-mutation was not tested because it would not have affected treatment; afatinib was stopped. He started receiving chemotherapy with carboplatin and etoposide for SCLC and had radiation to his shoulder for bone pain. He completed two chemotherapy cycles with some clinical improvement. However, the patient was readmitted multiple times due to pneumonia and sepsis and died in January 2017.
Figure 2.
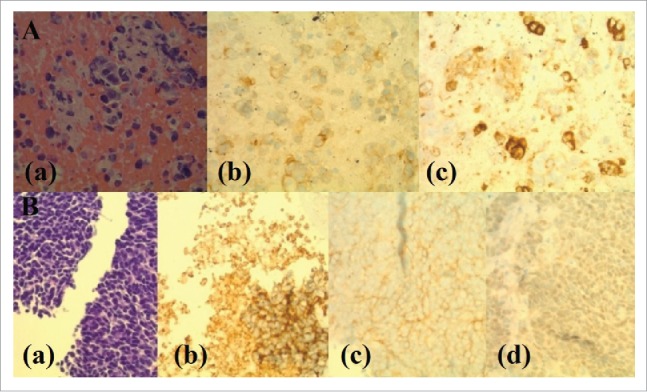
Histopathology and Immunohistochemical (IHC) Staining of Sequentially-Occurred NSCLC (ADC) and SCLC. Panel A: Histopathology and immunostaining of ADC of lung (Lymph node aspiration cytology). (a). H and E staining showing scattered enlarged cells with vesicular chromatin, visible nucleoli and moderately fine vacuolated cytoplasm. (b). IHC staining showing positive cytoplasmic stain for MOC31 (MOC31 is an immunological marker used for differentiation between metastatic ADC and lung mesothelioma where ADC is positive and mesothelioma is negative for MOC31). (c). IHC staining showing positive staining for CD15 (CD15 is a carbohydrate antigen on cells and can help during the confirmation process of lung ADC by differentiating it from mesothelioma which is negative for CD15 immunological staining). Panel B: Histopathology and immunostaining of transformed SCLC (Liver Biopsy). (a). H and E staining showing a diffuse infiltrate of polygonal neoplastic cells with a high nucleus to cytoplasm ratio, hyperchromatic nuclei, fine chromatin, inconspicuous nucleoli and focally forming vague pseudorosettes. (b). IHC staining showing positive cytoplasmic staining for synaptophysin. (c). IHC staining showing positive staining for CD56. (d). IHC staining showing very weak patchy staining for P63 (P63 is a nuclear marker used for immunostaining to differentiate squamous cell CA from ADC and is very strongly positive for SCC in contrast to ADC of the lung).
Figure 3.

PET/CT of Chest Showing Initial Response with Subsequent Progression Following TKI-Exposure (afatinib). (A). PET/CT showing interval response after 4 months treatment of afatinib. (a). FDG-avid thoracic LAD and right pleural disease before the initiation of afatinib. (b). Marked improvement of lung cancer on PET/CT showing a solitary residual right hilar LN after four months of TKI treatment. (B). PET/CT showing progression after 10 months treatment of afatinib. (a). Axial view showing a marked progression of metastatic lung disease with diffuse hepatic and skeletal involvement along with the progression of right perihilar mass. (b). Coronal view showing the same progression following post-TKI resistance.
Discussion
Among NSCLC, ADC can exhibit somatic mutations in several oncogenes, including EGFR, ALK, and ROS1, which drive the abnormal proliferation of mutant cells and can be targeted with TKIs. To date there has been substantial research in this field expanding therapeutic alternatives that have resulted in better outcomes. A large subset of advanced NSCLC with EGFR-mutations shows a promising response to TKIs making them first-line therapy. However, most of these patients develop resistance to these drugs after an average of 8–14 months that is mediated by several mutations including T790M mutation (threonine substitution with methionine at 790-amino acid position), MET and HER2 gene amplification, EGFR amplification, and PIK3CA mutation, among others.4 Additionally, T790M-mutation is the most common mutation which is present in more than 50 percent of cases5 and can be treated with the recently FDA-approved third-generation TKI, osimertinib.6
Other mechanisms of resistance, occurring less commonly, include epithelial to mesenchymal transition (EMT) and transformation from NSCLC to SCLC.4
Although these distinctive mechanisms are well-known, a heterogeneity of the resistance to TKIs in the same individual is still under study and requires more attention. Presumably, it could offer multiple alternative therapeutic solutions due to various resistant phenotypes. Although re-biopsy in the presence of a developing TKI-resistance is imperative, a single re-biopsy can often be non-representative, hence necessitating either multiple re-biopsies or additional supplemental testing. As a result, the role of liquid-based biopsy (plasma, urine) has been evolving since it could potentially detect a concomitant mechanism of resistance apart from what has already been established by a traditional re-biopsy. For example, plasma-based testing on freely-circulating tumor DNA helped in determining the concomitant resistant mechanism (T790M-mutation) in an EGFR-TKI-resistant-NSCLC in a 45-year-old man.7 On the other hand, a traditional re-biopsy revealed the emergence of small cell histology but failed to detect T790M-mutation in that specimen. Despite this discordance, the baseline EGFR-mutation (Exon 19 deletion), initially identified in pre-TKI EGFR-mutant-NSCLC, was present in both these post-TKI specimens indicating the same origin as the pre-TKI, pre-resistant NSCLC. If the plasma-based testing had not been performed, the concomitant positive T790M-mutation would have been disregarded. Hence, the authors proposed the role of heterogeneity and multiple resistance mechanisms potentially involved in the TKI-refractory cases.7
Among various resistance phenomena to TKI-exposure in EGFR-mutant-NSCLC, the transdifferentiating conversion of a NSCLC to SCLC is rare and has an unknown pathophysiology. It should also be noted that the EGFR-TKI-resistance is not limited to a single agent and has occurred in response to both first and second-generation TKIs and the same goes for SCLC transformation. Retrospectively, third-generation TKIs were developed to counteract the acquired resistance mutation of T790M. Recently, there is evidence that TKI-resistance can occur after third-generation TKIs exposure as can SCLC transformation.8
Even though the exact mechanism of this histological transition remains unclear, there are few changes evidently observed in the EGFR-mutant-transformed SCLC. Further exploration and molecular analyses reveal that this post-TKI transformed SCLC is identical to classic non-transformed SCLC in several ways. Regarding its similarity to classic SCLC, cardinal features (of transformed SCLC) acquired during post-TKI transformation include 1) universal depletion of retinoblastoma (RB) tumor suppressor, 2) decreased or absent expression of EGFR and 3) increased sensitivity to BCL-2 family inhibitors (for example, ABT-263).9
Furthermore, other related changes occurring during this transition include bi-allelic deletions or nonsense mutation of RB1 gene, heterozygosity and inactivating mutation of TP53, changes in RNA profile (mRNA and miRNA) analogous to classic SCLC and neuroendocrine differentiation.9 Given these alterations following EGFR-TKI exposure, resistant pluripotent cells are then differentiated into SCLC cells. As these cells do not require EGFR signaling, they escape the effect of EGFR-TKI and hence proliferate despite its presence.9 Although the EGFR mutation could be present to some extent in transformed SCLC, decreased or absent expression of EGFR also contributes to poor response to EGFR-TKI agents.
The nature of TKI-resistance is heterogeneous, and therefore, more than one mechanisms of resistance are possible in one individual. However, when resistance occurs in the form of a transformation, the T790M-mutation tends not to arise in the same malignancy indicating their rare concurrent pattern.10 The same reciprocal interaction was explained by Suda et al. in their scientific report. They analyzed nine EGFR-TKI-refractory metastatic lesions after a patient died with metastatic NSCLC. Out of these nine postmortem lesions, six contained SCLC, whereas two had ADC in addition to one retroperitoneal LN possessing both components. EGFR-exon 19 deletion was found in all these specimens implying one single origin. All ADC lesions harbored EGFR-T790M-mutation. Contrarily, none of the SCLC lesions harbored the T790M-mutation, indicating a reciprocal resistance pattern.11 Despite the fact that this is a predominant pattern, Fujita et al. have recently reported the concurrent existence of T790M-mutation and a SCLC transformation following TKI-resistance.10 Additionally, one author proposed the role of SCLC tumor markers (pro-gastrin-releasing peptide and neuron-specific enolase) during a TKI-resistance of SCLC as it might be helpful in early detection of transformation.12
It is also noteworthy that lung cancer is not the only malignancy involving this transformation. There are cases where transformation from ADC has occurred in patients with prostate cancer, making them resistant to the conventional treatment.13
One published study, which examined resistance to TKIs, reported five patients (14%) with transformation among 37 patients with EGFR-mutant-NSCLC.4 Other studies reported an even rarer transformation rate as a resistance mechanism, as low as 1.5 percent of cases.14 It has been hypothesized in some cases that SCLC and NSCLC co-exist from the beginning, but if the NSCLC is more extensive initially, it alone might be detected. When treated with TKIs, it could regress to the extent that the non-dominant SCLC could emerge. However, the fact that the exact EGFR-mutations of NSCLC are usually present in the transformed SCLC points towards one origin.4
Although the transformation to SCLC has usually been seen in EGFR-mutant-NSCLC, there is evidence of it taking place in cases without mutation, so called “wild-type”.14 Additionally, there are cases where transformation occurred during TKI exposure, but the patients were already exposed to chemotherapy before receiving TKIs which presents a confounding picture and demands more plausible explanations for the resistance. Similarly, in the same study, one patient who had no exposure to TKIs and was treated with surgery alone also showed transformation implying its rare occurrence in non-TKI settings as well.14 Despite these deviations, a typical patient reported in the literature is a female non-smoker who has progression after initial response to TKI and exhibits transformation from EGFR-mutant-NSCLC to SCLC.15 Our patient was a male non-smoker who had been on TKI (afatinib) for ten months for EGFR-mutant-NSCLC and showed an advanced metastatic SCLC on the second biopsy which was not expected upon initial evaluation.
Conclusion
When a rapid progression is observed in TKI settings after an initial response, potential resistance in the form of transformation should be suspected in addition to mutations; a second biopsy in these circumstances can definitively confirm histology and further guide therapy. Our patient had no evidence of an SCLC component in any tissue specimen from the first biopsy. Looking for an EGFR-exon 19 deletion in the SCLC detected in the second biopsy might have further validated transformation but it was not checked since it would not have altered his management. Further research is necessary to better define the biology and develop better treatment approaches to SCLC arising from NSCLC after EGFR-TKI exposure.
Acknowledgments
None
Conflict of interest
Y.B. has served as an advisory board member for Astra Zeneca, Bristol Meyers Squibb, Novartis and Takeda.
Disclosures funding
Y.B. laboratory has been funded by the Fox Chase Cancer Center and Temple University startup funds, DOD Career Development Award LC140074 and in part by the NCI Core Grant P30 CA006927 (to Fox Chase Cancer Center).
Funding
Y.B. laboratory has been funded by the Fox Chase Cancer Center and Temple University startup funds, DOD Career Development Award LC140074 and in part by the NCI Core Grant P30 CA006927 (to Fox Chase Cancer Center).
References
- 1.Navada S, Lai P, Schwartz A, Kalemkerian G. Temporal trends in small cell lung cancer: analysis of the national Surveillance, Epidemiology, and End-Results (SEER) database. J Clin Oncol. 2006;24(18_suppl):7082–7082. [Google Scholar]
- 2.Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O'Byrne K, Feng J, et al.. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015 Feb 28;16(2):141–51. doi: 10.1016/S1470-2045(14)71173-8. [DOI] [PubMed] [Google Scholar]
- 3.NCCN guidelines Accessed March9, 2017, at https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf.
- 4.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al.. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuiper JL, Heideman DA, Thunnissen E, Paul MA, Van Wijk AW, Postmus PE, Smit EF. Incidence of T790M mutation in (sequential) rebiopsies in EGFR-mutated NSCLC-patients. Lung cancer. 2014 Jul 31;85(1):19–24. doi: 10.1016/j.lungcan.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Goss G, Tsai CM, Shepherd FA, Bazhenova L, Lee JS, Chang GC, Crino L, Satouchi M, Chu Q, Hida T, et al.. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016 Dec 31;17(12):1643–52. doi: 10.1016/S1470-2045(16)30508-3. [DOI] [PubMed] [Google Scholar]
- 7.Alì G, Bruno R, Giordano M, Prediletto I, Marconi L, Zupo S, Fedeli F, Ribechini A, Chella A, Fontanini G. Small cell lung cancer transformation and the T790M mutation: A case report of two acquired mechanisms of TKI resistance detected in a tumor rebiopsy and plasma sample of EGFR-mutant lung adenocarcinoma. Oncol Lett. 2016;12(5):4009–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman EL, Kalsy A, et al.. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015 Jul 1;5(7):713–22. doi: 10.1158/2159-8290.CD-15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al.. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 2015 Mar 11;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujita K, Kim YH, Yoshizawa A, Mio T, Mishima M. Concomitant T790M mutation and small‐cell lung cancer transformation after acquired resistance to epidermal growth factor receptor‐tyrosine kinase inhibitor. Respirol Case Rep. 2017;5(1):e00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suda K, Murakami I, Sakai K, Mizuuchi H, Shimizu S, Sato K, Tomizawa K, Tomida S, Yatabe Y, Nishio K, et al.. Small cell lung cancer transformation and T790M mutation: complimentary roles in acquired resistance to kinase inhibitors in lung cancer. Sci Rep. 2015 Sep 24;5:14447. doi: 10.1038/srep14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furugen M, Uechi K, Hirai J, Aoyama H, Saio M, Yoshimi N, Kinjo T, Miyagi K, Haranaga S, Higa F, et al.. An autopsy case of two distinct, acquired drug resistance mechanisms in epidermal growth factor receptor-mutant lung adenocarcinoma: small cell carcinoma transformation and epidermal growth factor receptor T790M mutation. Intern Med. 2015;54(19):2491–6. doi: 10.2169/internalmedicine.54.5481. [DOI] [PubMed] [Google Scholar]
- 13.Miyoshi Y, Uemura H, Kitami K, Satomi Y, Kubota Y, Hosaka M. Neuroendocrine differentiated small cell carcinoma presenting as recurrent prostate cancer after androgen deprivation therapy. BJU Int. 2001;88(9):982–983. [DOI] [PubMed] [Google Scholar]
- 14.Ahn S, Hwang SH, Han J, Choi YL, Lee SH, Ahn JS, Park K, Ahn MJ, Park WY. Transformation to small cell lung cancer of pulmonary adenocarcinoma: clinicopathologic analysis of six cases. J Pathol Transl Med. 2016 Jul;50(4):258. doi: 10.4132/jptm.2016.04.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16(4):e165-e172. doi: 10.1016/S1470-2045(14)71180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]