

Abstract
Herein, the structure resulting from in situ turnover in a chemically challenging quaternary ammonium oxidative demethylation reaction was captured via crystallographic analysis and analyzed via single-crystal spectroscopy. Crystal structures were determined for the Rieske-type monooxygenase, stachydrine demethylase, in the unliganded state (at 1.6 Å resolution) and in the product complex (at 2.2 Å resolution). The ligand complex was obtained from enzyme aerobically cocrystallized with the substrate stachydrine (N,N-dimethylproline). The ligand electron density in the complex was interpreted as proline, generated within the active site at 100 K by the absorption of X-ray photon energy and two consecutive demethylation cycles. The oxidation state of the Rieske iron–sulfur cluster was characterized by UV–visible spectroscopy throughout X-ray data collection in conjunction with resonance Raman spectra collected before and after diffraction data. Shifts in the absorption band wavelength and intensity as a function of absorbed X-ray dose demonstrated that the Rieske center was reduced by solvated electrons generated by X-ray photons; the kinetics of the reduction process differed dramatically for the liganded complex compared to unliganded demethylase, which may correspond to the observed turnover in the crystal.
Graphical Abstract
INTRODUCTION
Biological methylation has been a subject of intense study since the 1970s.1 Earlier studies were focused on the role of methylation in metabolism, e.g., mechanistic studies of enzymes in the one-carbon cycle.2,3 In the post genomic era, there is renewed interest in chemical transformations with emphasis on chemical modifications of DNA, RNA, and post-translational protein modifications (e.g., acetylation, ADP-ribosylation, lipidation, methylation, phosphorylation, proteolytic protein processing, sumoylation, sulfonation, and ubiquitination), which can further expand the information encoded by the genome by at least 1–2 orders of magnitude.4,5 Among these modifications, methylation of macromolecules (e.g., protein and DNA) has been known for decades.1
Unlike most other modifications, which are known to be reversible processes, protein N-methylation (e.g., histone N-methylations) was originally proposed to be an irreversible process because of the stability of the C–N bond.6,7 For example, the biological half-life of a histone methyl group approximately equals that of the histone itself.8,9 With the discovery of two types of histone demethylases in the past few years,10 the dynamic nature of N-methylation was recognized and mechanistic studies in this area have grown exponentially. The first histone demethylase type, lysine-specific demethylase (LSD1), is an FAD-dependent oxidase that catalyzes the demethylation of mono- or dimethylated lysine 4 on histone 3 (Scheme 1A) with formation of formaldehyde.11 After the discovery of LSD1, a second type of histone-demethylase, the α-ketoglutarate-dependent mononuclear iron oxygenases, was discovered;1,12–14 these enzymes can demethylate both di- and trimethylated lysine (Scheme 1B). DNA N-dealkylation from the N1-position of adenine and the N3-position of cytosine using a similar type of mechanism has also been reported.15,16 These proteins normally have multiple domains, e.g., the catalytic domain, substrate recognition domain, and other domains used for DNA binding or protein complex formation. Structural studies have revealed that the identity of the cofactor dictates the reaction mechanism, whereas the catalytic domain in combination with other domains controls substrate specificity.17–22 Thus, the discovery of new N-demethylases through bioinformatics may uncover other types of novel chemical demethylation mechanisms.10
Scheme 1.

Oxidative Demethylation Reactions (R = H or CH3): (A) LSD1 (a Flavoprotein)-Catalyzed Demethylation of Mono-or Dimethylated Lysine Residues on Histone Tails; (B) α-Ketoglutarate-Dependent Demethylation of Di- or Trimethylated Lysine Residues on Histone Tails; and (C) Stc2 (Rieske-Type Mononuclear Non-Heme Enzyme)-Catalyzed Oxidative Demethylation of Quaternary Ammonium
The study of small-molecule quaternary ammonium demethylases can lend insight into the potential folds and chemistries capable of supporting demethylation reactions. For this reason, we undertook the analysis of the enzyme stachydrine demethylase (Stc2), from Sinorhizobium meliloti 1021. Nitrogen-fixing soil bacteria such as S. meliloti can live in symbiosis within root nodules of legumes. Quaternary ammonium solutes such as glycine betaine and proline betaine (also known as N,N-dimethylproline or stachydrine, 1, Scheme 1C) are secreted from the roots and taken up by S. meliloti, where they can serve as carbon and nitrogen sources in conditions of low osmotic strength, and additionally as osmoprotective compounds in conditions of high osmotic stress.23–27 Under catabolic conditions, stachydrine is converted to proline in two demethylation steps with the first step dependent on stc2, stc3, and stc4 genes and the second requiring a distinct stcD2 gene, which is positioned on a separate locus.28–30 Stc2 is a putative Rieske-type oxygenase, and stc4 is proposed to serve as the reductase in the system.31 On the basis of these findings, it is proposed that the Stc2 complex oxidatively demethylates stachydrine to monomethylproline (2), which is then demethylated by StcD2 to proline (3, Scheme 1C), a flavin-dependent oxygenase. Herein, we present the chemical, structural, and spectroscopic characterization of stachydrine demethylase (Stc2) from S. meliloti 1021.
EXPERIMENTAL SECTION
Cloning
DNA for the Rieske dioxygenase subunit (Stc2) of stachydrine demethylase was amplified by PCR reaction from S. meliloti 1021 genomic DNA using the following two primers: forward GGGAATTCGACAGCTAACCCGACC and reverse CTCGAGT-CACTCCGCAGCCAC. The amplified Stc2 DNA fragment was subcloned into the EcoRI and XhoI sites of the pASK-IBA5+ vector from IBA Biotechnology Inc., which contains an N-terminal strep-tag. The identity of the resulting construct was confirmed by DNA sequencing (at GeneWiz, Inc.) and transformed into BL21(DE3)-pDB1281 competent cells for Stc2 protein overexpression.
Expression and Purification
A single colony of Stc2-pASK-IBA5+/BL21(DE3)/pDB1281 (pDB1281 is a cassette containing E. coli isc iron–sulfur cluster synthetic operon to ensure proper iron–sulfur cluster maturation (kindly provided by Dr. Dennis R. Dean at Virginia Polytechnic Institute)) was selected to inoculate 50 mL of LB medium supplemented with ampicillin (100 μg/L) and kanamycin (50 μg/L) and grown overnight at 37 °C with shaking at 170 rpm. The overnight culture was used to inoculate (in 1:200 dilution) 2 L of LB-broth supplemented with ampicillin, kanamycin, and 0.1 mM ferric chloride. The culture was incubated at 37 °C with shaking at 170 rpm and monitored by absorbance at 600 nm. Once the OD600 reached 0.1, 0.2 g of L-arabinose was added to induce isc protein production. When OD600 reached 0.5–0.6, the temperature of the cell culture was reduced to 25 °C, and Stc2 protein production was induced by anhydrotetracycline at a final concentration of 200 ng/mL. The induced cells were cultured for an additional 20–24 h with shaking at 170 rpm. The cells were harvested and stored at −80 °C.
Protein was purified in a Coy-anaerobic chamber. Cells (15 g) were resuspended in 50 mL of buffer (100 mM Tris-HCl, 150 mM NaCl, pH 7.5). Lysozyme (50 mg) and 10 μL of 100 Units/μL DNase I were added and the mixture was incubated at room temperature with gentle agitation for 15 min. The mixture was then cooled by incubating on ice for 30 min. The cells were disrupted by sonication (20 cycles; 30 s on and 1–2 min off). The supernatant and the cell debris were separated by centrifugation (38000g) for 10 min at 4 °C. Streptomycin sulfate was added to the supernatant to a final concentration of 2% (w/v). After the mixture was incubated on ice for 30 min, the DNA precipitate was removed by centrifugation at 38000g for 40 min. The supernatant was mixed with the Strep-Tactin resin (5 mL, pre-equilibrated in 100 mM Tris-HCl, 150 mM NaCl, pH 7.5 buffer) and incubated on ice for 30 min. The protein–resin mixture was loaded into a column, and the column was washed with the same buffer until OD260 was lower than 0.01. The desired Stc2 holo-enzyme was eluted with eluting buffer (10 mL of 2.5 mM desthiobiotin in 5 mM Tris-HCl buffer, pH 7.5). The elution process was repeated once following the same procedure. The purification was monitored by SDS-PAGE, and Stc2-containing fractions were combined and concentrated by ultrafiltration to ~2 mL (30–40 psi, 30 kDa/44.5 mm membrane). The protein product was aliquoted into 200–400 μL fractions, flash frozen with liquid nitrogen, and stored at −80 °C. The concentration of the protein was determined by Bradford reagent. The yield of pure protein was 10–15 mg/g wet cell paste.
Activity Assays
The substrates N,N-dimethylproline, also known as stachydrine (1), and N-methylproline (2) were synthesized as described in Supporting Information. A Stc2 protein anaerobic solution (0.4 mM in 50 mM Tris, pH 7.5) was mixed with 1.0 mM Fe(II). The mixture was reduced with 1.0 mM dithionite in an anaerobic Coy-chamber. The protein mixture was mixed with an aerobic 0.4 mM stachydrine solution and incubated at room temperature aerobically for 20 h. The formation of monomethylated proline was monitored by 1H NMR. The 1H NMR chemical shifts of the two methyl groups of stachydrine are 3.16 and 2.97 ppm, respectively. The 1H NMR chemical shift for the methyl group of monomethylated proline is at 2.80 ppm.
Stc2 Crystallization
Stc2 crystals were grown under anaerobic conditions in a Coy-chamber using the hanging drop method. Crystallization conditions were obtained from sparse matrix screening using the Index Screen (Hampton Research) and optimized using the Additive Screen (Hampton Research). Drops were equilibrated against a reservoir solution containing 15% poly(ethylene glycol) (PEG) 3350, 20% glycerol, 100 mM succinic acid buffer, pH 7.0, and 10 mM hexamine cobalt at 22 °C. The hanging drops contained equal volumes (1 μL) of the reservoir solution and protein solution (35 mg/mL in 10 mM Tris buffer, pH 7.5, 2.5 mM biotin). Small crystals (0.1 mm × 0.1 mm × 0.05 mm) appeared in 2 days. The crystals belonged to the space group P6322 with cell dimensions a = b = 97.8 Å, c = 179.4 Å. There was one protomer in the asymmetric unit, and the solvent comprised 54% of the cell.
For data collection, crystals were flash-cooled directly in liquid nitrogen and transferred to a stream of N2 gas cooled to 100 K. A single-wavelength anomalous dispersion (SAD) data set was collected at the X12C beamline of the National Synchrotron Light Source (NSLS, Brookhaven National Laboratory, Upton, NY) equipped with an ADSC Q210 detector. To measure the maximum f ″ contribution from the Fe atoms, a wavelength of 1.738 Å was chosen on the basis of the X-ray fluorescence spectrum at the iron K absorption edge. A “native” data set was also collected on the same crystal at a wavelength of 1.00 Å. Data collected to 1.9 (SAD) and 1.6 Å (native) were processed with the program DENZO.32
For phase determination, HKL2MAP33 was used to navigate the SHELX programs.34 SHELXD was used to identify heavy-atom sites using Fe-S-SAD, utilizing inherent Fe and S atoms. SHELXE was used to calculate phases based on three Fe and eight S heavy-atom sites. The program ARP/wARP was used for model building from initial phases, resulting in placement of 7–403 out of 412 residues.35 Iterative rounds of refinement and rebuilding using Phenix36 consisted of a single energy minimization molecular dynamics cycle followed by a simulated annealing molecular dynamics cycle at 5000 K, which was then followed by alternating cycles of positional and individual temperature factor refinement with manual inspection/rebuilding in COOT.37 The structure of Stc2 was determined at 1.6 Å resolution by isomorphous phasing using the protomer from the 1.9 Å structure of Stc2 (vide supra). In the later stages of refinement, water molecules were added to the model using an electron-density difference map calculated with the coefficients |Fo| – |Fc| with electron-density acceptance criteria of F ≥ 3σ(F).
Crystals of Stc2 and its proline complex were obtained by cocrystallizing 30 mg/mL Stc2 protein with 10 mM stachydrine (1) in a 1:1:0.2 μL drop ratio (protein:well:stachydrine) aerobically. Hanging drops were equilibrated against 100 mM Hepes buffer, pH 7.0, 10% PEG 3350, 10% glycerol, and 25 mM hexamine cobalt. Small trapezoidal crystals appeared within 10 min and grew to a final size of 0.2 mm × 0.1 mm × 0.02 mm within 1 day. After 24 h, crystals were moved to a drop of mother liquor containing 20% glycerol for 5 min and then either flash-cooled directly or moved into paratone to remove surface solvent before flash-cooling by plunging the loop into liquid nitrogen. X-ray data collection was performed at the NSLS, Brookhaven National Laboratory, on the X26C beamline equipped with a ADSC Q210 X-ray detector, single-crystal visible optical absorption, and resonance Raman spectroscopies. X-ray diffraction (XRD) data were collected at a wavelength of 1.0 Å. Because paratone-N gives a large background absorbance in the UV–vis region of the optical spectrum, it was omitted from all the spectroscopy studies, and a cryoprotectant of 20% glycerol in mother liquor was used. The microspectrophotometer allowed for direct characterization of the Rieske [2Fe-2S] cluster.38 Upon reduction, there is a characteristic shift of absorption from 550 nm in the oxidized form to an absorption feature at 525 nm in the reduced form. To test if reduction occurred due to X-ray exposure, the data set was parsed into a separate oxidized and reduced data set, using the first 100 images (50°) for structure determination. Data were processed to 2.2 Å resolution using HKL2000.32
The structure of the Stc2–ligand complex was solved by isomorphous phasing using the Stc2 protomer (vide supra). The ligand was modeled as proline in the final stages of refinement, after placement of water. Positioning and refinement with N,N-dimethylproline or N-monomethylproline resulted in strong (<3σ) peaks in the |Fo| – |Fc| difference map when contoured at negative values centered on the N-methyl group(s), suggesting the density does not support either mono- or dimethylated product. Proline was therefore unambiguously modeled and refined into the density corresponding to ligand.
UV–Vis Spectroscopy
The beamline at X26C is equipped with a microspectrophotometer (350–800 nm) with an approximately 25 μm diameter optical focal point coincident with that of the X-ray beam. Absorption spectra are collected from protein crystals in conjunction with XRD data as previously described.39,40 The source objective that focuses the light from a 75 W Xe arc lamp (Newport) onto the sample was mounted above the sample. The collection lens, located below the sample, was attached to a motorized x,y,z translation stage (under EPICS software control) adjacent to the optical components for Raman spectroscopy as previously described.41,42
For concurrent spectroscopy collection, we typically oriented the flat face of the crystal normal to the spectroscopy photons. Spectra were measured at 5° intervals over a 360° rotation, and the characteristic spectral peaks determined in solution for Stc2 (460 and 550 nm) were analyzed with respect to the crystal orientation. Electronic absorption and XRD data collection were integrated into the XRD data collection software (CBASS). For example, during the X-ray detector readout, the crystal was rotated to the optimum crystal orientation angle, a spectrum was collected, and the crystal was returned to the correct orientation for X-ray data collection. Thus, all spectra collected for a particular crystal were from the same orientation of the crystal. We estimated the absorbed X-ray dose via a Java tool that uses the program RADDOSE.41,43
Resonance Raman Spectroscopy
Native, oxidized, and reduced crystals were used for resonance Raman spectroscopy. Oxidized crystals were obtained by adding 5% hydrogen peroxide directly to the crystallization drop and incubating at room temperature for 5 min before freezing in liquid nitrogen. Native crystals were grown in atmospheric oxygen. Reduced crystals were obtained using the X-ray source at X26-C of NSLS to reduce the iron–sulfur cluster. Crystals were monitored for reduction by UV–vis spectroscopy by following the disappearance of the 460 nm peak and the peak shift from 550 to 525 nm.
Raman spectra were collected from the identical region of the crystal as was probed by absorption spectroscopy. Alignment of the Raman probe head (Horiba Jobin-Yvon) utilized the same motorized x,y,z translation stage as controlled the positioning of the lower absorption objective. The experimental setup has been fully described elsewhere.42 For the experiments reported herein, a 300 mW, 532 nm diode laser (Laser Quantum) was used as the excitation source. Power levels at the sample were kept below 7 mW, although levels at or below 3 mW were determined to be preferable. The spectrometer used a 1 μm entrance slit and a 600 lines/mm grating. Resolution is estimated at 6 cm−1. The system was calibrated using the laser line and the Hg line from ambient light. Total collection times ranged from 5 to 10 min. LabSpec (provided by Horiba Jobin-Yvon) is the collection software; data analysis utilizes GramsAI (Thermo Scientific) and Origin 6.0 (Microcal Software). Pre- versus post-X-ray-exposure spectra, as well as spectra from exposed versus unexposed regions of a single crystal, were compared.
Accession Codes
The coordinates, structure factors, and single-crystal spectroscopy of the oxidized Stc2 holoenzyme, and Stc2 cocrystallized aerobically with stachydrine but interpreted as proline bound, have been deposited in the PDB under accession codes 3VCA and 3VCP, respectively.
RESULTS AND DISCUSSION
Activity of Stc2
In order to enable the identification of novel demethylases, it is essential to have examples of each structural/chemical class that can carry out this reaction. Toward this goal, we undertook the structural and functional characterization of a predicted Rieske-type mononuclear non-heme iron demethylase, stachydrine demethylase.30
The activity of Stc2 as a quaternary ammonium oxidative demethylase with dithionite as the reductant was measured directly using a 1H NMR assay (see Experimental Section and Supporting Information). The 1H NMR chemical shifts of the two stachydrine methyl groups are at 3.16 and 2.97 ppm, respectively (Figure S1). In the presence of dithionite-reduced Stc2 and molecular oxygen, stachydrine was oxidatively demethylated to monomethylated proline as suggested by the 1H NMR spectrum (Figure S1), in which a new peak with a chemical shift of 2.80 ppm was assigned as the methyl group of monomethylated proline. This conclusion was substantiated by comparison to a chemically synthesized monomethylproline standard (Figure S1).
The FAD-dependent demethylase LSD1 can demethylate tertiary and secondary amines but cannot utilize quaternary ammonium compounds (e.g., trimethylated lysine in histone tails) as substrates.11 In contrast, α-ketoglutarate-dependent mononuclear iron oxygenases can demethylate either quaternary ammonium or tertiary amines, but some secondary amines do not act as substrates (e.g., monomethylated lysine in histone tails).11–15 In solution, Stc2 can oxidatively demethylate quaternary ammonium to tertiary amines (Scheme 1C), but it does not catalyze the demethylation of either tertiary amine or secondary amine substrates based on the results from 1H NMR assays using N,N-dimethylglycine, N-methylglycine, and N-methylproline as substrates (with a detection limit of 10 μM, Figure S1). Consistent with the conclusions reached in previous histone demethylase studies, demethylases have differing substrate methylation state preferences, and their active-site structures and cofactor chemistries play key roles in such selectivity.10
X-ray Crystal Structure of Stc2 as Isolated
The initial structure of the Stc2 holoenzyme was solved to 1.9 Å resolution with phases determined via Fe-S-SAD. These coordinates were then used for isomorphous phasing of a higher resolution data set collected to 1.6 Å resolution (at λ = 1.0 Å) (Table 1). The overall protomeric structure is similar to that of other Rieske-type oxygenases consisting of all α-subunits, such as 2-oxoquinoline 8-monooxygenase44 and phthalate dioxygenase,45 in contrast to 2,4-dinitrotoluene dioxygenase,46 naphthalene 1,2-dioxygenase (NDO),47 benzoate 1,2-dioxygenase,48 and others that are α-β trimers. The Stc2 protomer comprises a Rieske [2Fe-2S] domain (residues 45–153) and a catalytic domain (residues 1–44 and 154–412) (Figure 1; Figure S2 and Table S1). The Rieske domain contains 11 β-strands which position the iron–sulfur liganding residues. In the Rieske [2Fe-2S] cluster, one iron is coordinated by His88 and His109, and the second iron is coordinated by Cys86 and Cys106 (Figure 2A). The catalytic domain consists of a seven-stranded antiparallel β-sheet, with loops and α-helices connecting the β-strands. The mononuclear non-heme iron is coordinated by a 2-His-1-carboxylate facial triad49 (His204, His209, and Asp360). A well-ordered loop (residues 200–210) provides His204 and His209 (2.1 Å Fe–N distance for both His residues). An α-helix spanning the length of the catalytic domain contributes the iron-coordinating residues Asp360 (2.0 Å Fe–O distance). In addition, the mononuclear iron coordinates two solvent ligands (W1 and W2 with 2.1 and 2.4 Å Fe–O distances, respectively); a third water (W3) is nearby (2.7 Å Fe–O distance), although it is a long metal–ligand distance for a water forming a coordinate bond to Fe (Figure 3A). Although the density corresponding to W3 is elongated, the center of highest density is at the center of the ellipsoid and is inconsistent with refinement with two solvent molecules at half occupancy. Thus, the coordination geometry is five-coordinate in a square pyramidal shape or distorted octahedral by including the solvent with the long, 2.7 Å bond as a ligand (Figure 3A).
Table 1.
X-ray Data Collection and Refinement Statistics for Holoenzyme Stc2 and Proline-Bound Stc2
| Stc2 SAD | Stc2 | Stc2/proline | |
|---|---|---|---|
| space group | P6322 | P6322 | P6322 |
| cell dimens (Å) | a = b = 97.859, | c = 179.372 | a = b = 97.140, c = 180.08 |
| protomer/ASU | 1 | 1 | 1 |
| wavelength (Å) | 1.738 | 1.0 | 1.0 |
| resolution (Å) | 50–1.9 (1.97– 1.9) | 28.2–1.6 (1.66–1.6) | 39.7–2.2 (2.32–2.2) |
| obsd reflns | 1 589 393 | 2 886 811 | 143 238 |
| unique reflns | 40 656 | 68 467 | 25 766 |
| completeness (%) | 99.2 (93.8) | 99.9 (100.0) | 98.7 (99.56) |
| Rmerge (%) | 9.8 (55.7) | 8.0 (53.7) | 17.1 (60) |
| 〈I/σ〉 | 44.5 (1.89) | 40.78 (6.2) | 6.9 (2.9) |
| redundancy | 39.4 (5.6) | 16.3 (18.0) | 5.6 (5.8) |
| R/Rfree | 17.8/20.9 | 17.8/20.9 | 18.74 (24.22) |
| average temperature factors (Å2) | |||
| protein/solvent | 27.6/37.8 | 18.7/32.1 | 23.12/24.5 |
| cluster/mono Fe | 24.4/32.8 | 11.9/17.3 | 15.8/23.2 |
| ligand | na | na | 34.4 |
| rmsd bonds/angles | 0.007/1.091 | 0.005/1.042 | 0.008/1.157 |
Rmerge = Σhkl Σi|Ihkl,i – 〈Ihkl〉I|/ΣhklΣi|Ihkl,i|, where 〈Ihkl〉 is the mean intensity of the multiple Ihkl,i observations for symmetry-related reflections. Numbers in parentheses indicate highest resolution shell.
Figure 1.

Structures of the Stc2 holoenzyme protomer and the α3 trimer. (A) Protomer in the asymmetric unit depicted as a ribbon diagram colored from blue (N-terminal) to red (C-terminal). (B) Ribbon diagram of the holoenzyme with each protomer colored by subunit. The Rieske cluster (Fe, orange; S, yellow) and the mononuclear iron site are rendered as CPK spheres. All structural figures were rendered with Molscript91 and POVRAY (www.povray.org).
Figure 2.

Rieske mononuclear iron cluster. (A) Depicted as ball-and stick with electron density (gray cages) calculated with coefficients 2Fo – Fc; contoured at 1σ. (B) Interface between two Stc2 subunits (carbons of neighboring subunits in gray and brown) showing the hydrogen bond bridge between the mononuclear non-heme iron site and the Rieske [2Fe-2S] cluster. Hydrogen bonds are shown as dashed lines.
Figure 3.

Mononuclear Fe active site of Stc2 with electron density (gray cages) calculated with coefficients 2Fo – Fc and contoured at 1σ. (A) Active site of the unliganded holoenzyme. (B) Holoenzyme cocrystallized with substrate N,N-dimethylproline (proline, gray ball-and-stick, observed after in situ turnover). Coordination bonds to Fe (orange sphere) are shown.
Within one protomer, the distance between the Rieske [2Fe-2S] cluster and the mononuclear iron site is ~46 Å (Figure 1), which is too long for the efficient electron transfer necessary for catalysis. The holoenzyme exists as a homotrimer, similar to that observed in other Rieske-type oxygenases50 with a buried surface area ~570 Å2 at the subunit–subunit interface51 of the catalytic and Rieske domains with 17 hydrogen bonds and 9 salt bridges.50 Rieske-type oxygenases structurally similar to Stc2 were identified via the Dali server (see Figure S2 for sequence alignment).52 These included the putative aromatic ring hydroxylating dioxygenase (PDB 3N0Q), toluene 2,3-dioxygenase (PDB 3EN1),53 naphthalene 1,2-dioxyggenase (PDB 1NDO),47 biphenyl dioxygenase (PDB 1ULI),54 cumene dioxygenase (PDB 1WQL),55 nitrobenzene dioxygenase (PDB 2BMO),56 PAH-hydroxylating dioxygnease (PDB 2CKF),57 carbazole 1,9α-dioxygenase (PDB 3GKQ), 2-oxoquinoline 8-monooxygenase (PDB 1ZO1),58 and dicamba demethylase (PDB 3GOB).59 Overall, analysis of the rmsd values (Table S1, Supporting Information) shows higher structural similarity between the Rieske domains than between the catalytic domains, perhaps reflecting changes in specificity determinants necessary to support the differing chemistry catalyzed by these enzymes. Notably, dicamba demethylase is an O-demethylase that shares less sequence identity (14%) with Stc2 than many of the other Rieske oxygenases (Figure S2, Table S1).59–61 Thus, though both these enzymes catalyze demethylation reactions, they are not closely related by sequence and probably represent distinct evolutionary pathways from a common progenitor. The distance between the Rieske domain of one subunit and the mononuclear iron site in an adjacent subunit catalytic domain is ~12 Å (Figure 1B). Glu201 bridges the two sites from two adjacent subunits, directly forming a hydrogen bond to His109 of the Rieske [2Fe-2S] cluster (2.8 Å) on one subunit and His204 of the mononuclear iron (2.8 Å) from another subunit (Figure 2B). Studies from other Rieske-type oxygenases reveal that the homologous bridging Asp/Glu residue is essential, and ablation via mutagenesis completely abolishes activity (as seen with NDO for the Asp to Gln mutation),62 leading to the proposal that it acts as a conduit of electrons between the two cofactors. It is very likely that Glu201 plays a similar role in Stc2.
We note several unique features within the structure of resting Stc2. First, in the loop after Glu201 and spanning two of the mononuclear iron ligands (His204 and His209), a disulfide bond between Cys202 and Cys205 is present (Figure 3A). This disulfide bond is only ~4.6 Å from the mononuclear iron. It also forms one side of the putative substrate binding cavity, which in the resting state contains several solvent molecules and is adjacent to the iron-bound solvent ligands (Figure S3A). The occurrence of a disulfide bond adjacent to Rieske [2Fe-2S] clusters has been shown to structurally stabilize the cluster in cytochrome bc1 with minimal effect on its reduction potential, 63 however, no such cysteine moieties have previously been documented to be adjacent to a mononuclear non-heme iron center.
Assignment of Orthologues via Bioinformatic Analysis
The enzyme most similar to Stc2, ABF62193 from Silicibacter sp. TM1040 (PDB 3N0Q), is annotated as a putative aromatic ring dioxygenase. Little variation is seen in the catalytic domain structure between Stc2 and ABF62193 (rmsd 0.7 Å), and the distinctive disulfide bond (between Cys202 and Cys205) proximal to the active site in Stc2 is also present. Bioinformatic analysis shows the Silicibacter sp. TM1040 oxygenase gene sequence is adjacent to that of a ferredoxin reductase domain (ABF62192). Therefore, we propose that 3N0Q is an orthologue to Stc2 and probably performs a similar metabolic function. A simple BLAST search64 using Stc2 as the query sequence retrieves 148 sequences (with overall sequence identity (24–98%) which conserve the CxHC signature for the disulfide bond as well as mononuclear active-site residues (Figure S4). Sequences were found mainly within the Proteobacteria phylum (alphaproteo-bacteria, betaproteobacteria, and gammaproteobacteria) as well as the Chloroflexi, Cyanobacteria, Firmicutes, and Plantomy-cetes phyla. Based on this bioinformatic analysis, numerous orthologues and homologues of Stc2 exist that may be capable of oxidative demethylation.
X-ray Crystal Structure of Stc2 Cocrystallized with Stachydrine
In order to observe the Michaelis complex, Stc2 was aerobically cocrystallized with the substrate stachydrine and the X-ray structure was determined via isomorphous phasing with the unliganded holoenzyme model. In the 2.2 Å liganded Stc2 structure obtained, rather than the expected Stc2–substrate complex, the electron density at the mononuclear iron site was consistent with bound proline (Figure 3B). This was confirmed by examination of a Fo – Fc omit electron density map with stachydrine modeled into the density (Figure S5), which showed density with negative contours at greater than 3σ at both N-methyl positions. Notably, the proline-binding mode observed for Stc2 differs from substrate complexes previously characterized for other Rieske-type mononuclear non-heme iron oxygenases (e.g., NDO,65,66 Figure S6A). To our knowledge, there is no documented instance of iron forming a direct coordination bond to substrate in a Rieske-type oxygenase. However, in the homologues delineated above, most of the substrates are hydrophobic and devoid of chemical moieties capable of iron coordination. In contrast, stachydrine and the products of these homologues frequently include groups that can coordinate to iron (e.g., NDO in complex with product; PDB 1O7P, Figure S6B).66 In the Stc2-product structure, proline is observed to bind to the mononuclear iron as a monodentate carboxylate ligand. One of the proline carboxylate oxygens (Fe–O distance 2.4 Å) binds in the coordination site occupied by the weakly interacting solvent (with the long coordination bond), W3, in the holoenzyme structure. The second proline carboxylate oxygen (3.0 Å from Fe) is located at the position occupied by W2 in the holoenzyme structure. The electron density is consistent with one solvent molecule as a ligand to the mononuclear iron, W1 (Fe–O distance 2.1 Å), which also occurs in this position in the unliganded holoenzyme. In Stc2, the bound proline appears to form two hydrogen bond interactions between its amine moiety and the carboxylate oxygen of Glu201 and the iron-bound solvent ligand. Thus, the mononuclear iron in the proline complex exhibits a distorted square pyramidal coordination geometry. To the extent that the observed ligand complex reflects features of a catalytically competent substrate complex, such five-coordinate geometry leaves one site open for oxygen binding and activation. Alternatively, the single solvent ligand could be displaced by oxygen binding. Thus, molecular oxygen could bind in either an end-on mode or a side-on bidentate fashion.
Stc2 retains the hydrophobic pocket used as the substrate binding site in the “classic” Rieske dioxygenases.65 For example, the active sites of NDO and Stc2 are compared in Figure S7. Residues comprising the hydrophobic pocket are Leu212, Phe216, Ala223, Tyr273, Leu297, Trp305 Phe316, and Trp353. Close to the mononuclear iron site, only two polar residues are found, Asn199 and His307, and neither forms a hydrogen bond to proline in the Stc2–product complex. We note that Arg200 and Arg260 residues form the outer border of the substrate binding cavity (Figure S3A), close to the channel that provides access to bulk solvent, and that these residues are conserved in two-thirds of the proposed Rieske demethylases (vide infra, Figure S4).
Single-Crystal UV–Vis Spectroscopy Correlated with X-ray Diffraction
To assess the origin of the reducing electrons necessary to support two consecutive oxidative demethylation reactions as observed in the Stc2-proline complex, correlated single-crystal spectroscopy was undertaken. More than 90% of the X-ray photons that do interact with the sample do not yield a diffraction pattern, but rather deposit energy into the crystal.68,69 For example, X-ray photons absorbed by the sample liberate two electrons from biological elements (photoelectric effect). They have sufficient energy to induce several hundred further ionization events that can propagate for up to 4 μm70 and, consequently, may reduce samples in the X-ray beam.41,65 We have measured electronic absorption spectra as a function of absorbed X-ray dose (Figure 4) using crystals grown under the same conditions as the ligand complex with 20% glycerol as the cryo-protectant to test for reduction of the samples.
Figure 4.

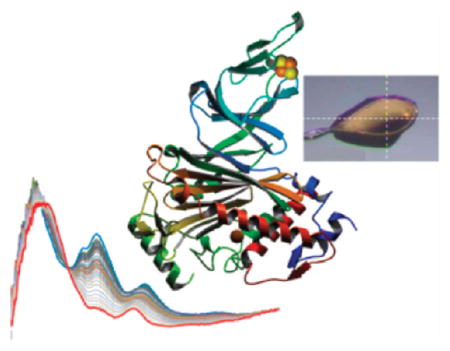
Optical absorption spectra of oxidized Stc2 collected between XRD images demonstrate that the Rieske cluster is reduced by solvated electrons. (A) After every XRD image (1–15) or after every 10th image (21–291), the crystal was rotated back to the perpendicular orientation (inset image) from which the single-crystal absorption spectra were collected. Each spectrum in the family has been normalized to the initial absorbance value at 750 nm. The crystal rotation axis (horizontal, y) as well as the spectroscopy (vertical, z) and the X-ray (horizontal, x) photon axes intersect at the white dashed cross-hairs. The crystal was held at 100 K; it measured ~300 μm × 325 μm × 130 μm; the X-ray beam was 100 μm × 100 μm; and the spectroscopy beam was focused to ~25 μm diameter. (B) Change in absorption at either 460 or 550 nm as a function of X-ray image or absorbed X-ray dose (solid lines) fitted to a power-law series (dashed lines).
Prior to X-ray exposure, in crystals oxidized with 5% H2O2, two absorption peaks were observed at 460 and 550 nm (Figure 4), which correlate well with solution UV–vis spectroscopy for a [2Fe-2S]2+ state in Stc2. This spectroscopic analysis does not provide information related to the redox state of the mononuclear iron center. After the X-ray exposure required for each diffraction image, the intensity of the peak at 460 nm decreased, and the peak at 550 nm was reduced in intensity and shifted to 525 nm. This observation was further supported by plotting the change in absorption at 460 and 550 nm as a function of absorbed X-ray dose (Figure 4B). The observed changes in absorbance can be fit reasonably well either to a power-law series or to several exponential processes. The majority of the observed spectral change occurred with exposure to only 0.5 MGray (roughly 25° or 50 frames of XRD data (~50% of a data set)). The absorption spectrum collected after the diffraction data set was complete was very similar to that obtained by chemical reduction of the Stc2 Rieske cluster to the [2Fe-2S]+ state in solution (Figure S8). Although the crystal started in the oxidized state, it was rapidly reduced in the X-ray beam; therefore, we conclude that a better electronic description for the unliganded and liganded holoenzyme structures described (vide supra) is that the majority of the Rieske center is reduced.
Single-Crystal Resonance Raman Spectroscopy before and after X-ray Diffraction
To further address the redox state of the Rieske cluster within Stc2, we used a 532 nm excitation laser to collect resonance Raman spectra from the same single crystals at 100 K.42 We used enzyme crystals poised in several redox states using 5% H2O2 or dithionite, before and after XRD data collection, and correlated these results with optical absorption spectroscopy (Figure 5, Table 2). Comparison of the results with published solution Raman spectra of other Rieske-type [2Fe-2S] proteins allowed us to assign the majority of the observed features.71–73 The hydrogen peroxide-treated, oxidized crystals have several strong Raman features in the region from 250 to 500 cm−1 (Table 2). Peaks at 321, 335, and 360 cm−1 were assigned to Fe–S(Cys) stretching with weak contributions from Fe–N(His) stretching. Peaks observed at 376, 405, and 429 cm−1 are characteristic of Fe–S(bridge) stretching. The band observed at 268 cm−1 in the oxidized spectra is consistent with Fe(III)–N(His) stretching.71
Figure 5.

Resonance Raman spectra collected from single crystals of Stc2 at 100 K using a 532 nm excitation laser, 600 lines/mm monochromator grating, and 1 mm entrance slit to the spectrometer. (A) Raman spectrum of an H2O2-treated Stc2 crystal prior to exposure to X-rays. The spectrum is the average of 240 spectra, each having a 5 s acquisition time. Laser power at the sample was set at 3 mW. (B) An expanded view of the low-frequency region of the spectrum of a H2O2- treated Stc2 crystal. (C) The low-frequency region of the Raman spectrum of Stc2-stachydrine complex following collection of XRD data. The spectrum is the average of 10 spectra, each having a 40 s acquisition time. Laser power at the sample was set at 4.5 mW.
Table 2.
Observed Raman Peaks for Stc2 Crystals and Selected Rieske Proteins (cm−1)
| Stc2 crystal, oxidizeda | T4MOC ferredoxin, oxidizedb | putiredoxin, oxidizedc | Stc2 assignments by comparison with T4MOC and Putiredoxin |
|---|---|---|---|
| 268 | 265 | 291 | Fe–N(His); contributions from Fe–S(Cys); His protonation state undetermined |
| 321 | 321 | 320 | Fe–S(Cys) stretch; contributions from Fe–N(His), Cys ligand deformation, and Fe-bridge S |
| 335 | 332 | 338 | |
| 358 | 350 | 350 | |
| 375 | 362 | 370 | Fe-bridge S stretch |
| 406 | 410 | 400 | |
| 430 | 433 | 426 |
| Stc2 crystal, reducedd | T4MOC ferredoxin, reducedb | putiredoxin, reducedc | |
|---|---|---|---|
| 292 | 297 | 307 | undetermined |
| 314 | 315 | 319 | Fe-bridge S stretch |
| 335 | |||
| 377 | 377 | 381 | |
| 411 | 408 | 406 |
H2O2-treated Stc2 crystals with 532 nm excitation; sample held at 100 K.
Data from ref 78 with 514 nm excitation; samples held at 15 K.
Data from ref 92 with 406.7, 457.9, or 530.9 nm excitation; samples held at 17 K.
Stc2 crystals after XRD with 532 nm excitation; sample held at 100 K. Some weak peaks from the oxidized form are also present.
We also collected resonance Raman spectra from Stc2 crystals after X-ray data collection or an X-ray dose equivalent to that used to obtain the high-resolution structures. In every case, the optical absorption spectrum for the crystals was bleached, and the resonance Raman peaks from this region of the crystal showed significant changes in the 250–500 cm−1 low-frequency region (Table 2, Figure 5C). In particular, the changes included a reduction in number and a shift of the Fe–S(bridge) stretching modes from 376, 405, and 429 cm−1 to 377 and 411 cm−1. The peak at 314 cm−1 was assigned to the Fe–S(Cys) stretch mode. A new peak at 292 cm−1 was also observed when the crystal was reduced by X-ray exposure.
Comparison of the results to those reported for other reduced Rieske iron–sulfur cluster-containing proteins in solution was performed.50,71,73 Overall our data are consistent with the reduction of Rieske [2Fe-2S]2+ to [2Fe-2S]+ upon X-ray exposure. Thus, by comparison to literature values, the Raman data presented here are consistent with the histidine-liganded iron in the ferrous state, whereas the cysteine-liganded iron remains in the ferric state. The electrophilic nature of the imidazole makes the histidine-liganded iron more susceptible to reduction compared to that coordinated by the electron-donating thiolate.71,74–77 Thus, the two irons form a spin-localized Fe(II)/Fe(III) cluster in the reduced state. Many of the characteristic Raman spectral changes which have been noted in the solution studies of Rieske-containing proteins are also observed with nearly identical spectra in our X-ray exposed Stc2 crystals. In other cases, the matches are not as straightforward. For example, our feature at 292 cm−1 may correspond to the Raman peak at 297 cm−1 noted in the spectrum of the dithionite-reduced Rieske protein toluene-4-monooxygenase from Pseudomonas mendocina.78 The 297 cm−1 peak was assigned to a Fe–S(bridge) stretch mode, based on comparisons with reduced adrenodoxin and “red paramagnetic protein” from Clostridium pasteurianum.73
X-ray Crystallographic and Spectroscopic Analysis of the Stc2-Stachydrine Complex
Catalysis by Stc2 is an O2-dependent process and also requires two electrons (see Scheme 1C). Moreover, in most members of the multicomponent Rieske oxygenase family, it is likely that O2 reactivity at the mononuclear iron site is gated by both organic substrate binding and by reduction of the Rieske cluster as dictated in part, by other components in the system.79–82 This ensures that a reactive oxygen species is not generated unless all of the reagents needed for catalysis are present. By extension, to the catalytic cycle of similar enzymes, stachydrine binding to oxidized Stc2 should not result in catalysis until electrons are provided. Therefore, we crystallized oxidized Stc2 in the presence of 1 mM substrate stachydrine under aerobic conditions to trap the Michaelis complex. As described above, the electron density observed in the structure (vide supra) was consistent with proline (3) bound and may derive from two demethylation steps.
The single-crystal optical absorption spectrum of Stc2 cocrystallized with stachydrine (identical to crystals used for data collection of the complex) is shown in Figure 6A. Resonance Raman spectra collected from these crystals also indicate that the majority of the Rieske clusters before data collection are in the oxidized state. However, upon exposure to X-ray photons at 100 K, as is necessary for crystallographic analysis, the Rieske cluster is reduced by the X-ray beam (vide supra). The kinetics of the reduction process for the enzyme–substrate crystals as a function of X-ray dose reveals a dramatically different reduction profile compared to unliganded Stc2 (Figure 6B). The absorption peak at 460 nm is quickly partially reduced then exhibits a more stable phase between ~0.33 and 1.25 MGr. The latter phase is followed by a second rapid phase that subsequently tapers off. The plateau between ~0.33 and 1.25 MGr may be due to the consumption of electrons in the demethylation process. This curve shape is very different from that observed for unliganded or fully oxidized Stc2 (Figure 4B), which fits to a normal power-law series. The kinetic trace and the final optical spectrum suggest that full reduction of the Rieske cluster was achieved by the end of the diffraction data set. Resonance Raman spectra collected from this crystal after XRD were consistent with those of the fully reduced cluster (Figure 5B).
Figure 6.

(A) Optical absorption spectra of the aerobic Stc2–stachydrine complex (Stc2-Subst) collected between XRD frames (see legend to Figure 4 for conditions). The crystal was held at 100 K; the size of the crystal was ~623 μm × 500 μm × 150 μm; the X-ray beam was 100 μm × 100 μm; and the spectroscopy beam was focused to ~25 μm diameter. (B) Change in absorption at 460 nm as a function of X-ray image or absorbed X-ray dose (blue solid lines) fitted to a polynomial series (green dashed lines). For comparison, the change in absorbance as a function of dose for oxidized Stc2 in the absence of substrate (Stc2-ox, from Figure 4) is shown with the solid red line and power-law fitted (the dashed black line).
The results above suggest turnover in the crystal that may be initiated by solvated electrons resulting from the absorption of X-ray photons. Consequently, the XRD data set was reexamined with respect to absorbed dose. When the first 100 images (50°) of diffraction data were used for the structure factors, the negative features associated with stachydine N-methyl groups remained. The amount of X-ray dose after 100 images of data collection is approximately 0.9 MGray, which falls within the plateau region observed (Figure 6B) between 0.3 and 1.2 MGray. Thus, the reactivity indicated by the plateau region is consistent with the observation of the proline-Stc2 complex. The solvated electrons created by the X-ray photons circumvent the need for the reducing protein components in the natural Stc2 system or for dithionite in the in vitro assay. Control experiments in which solutions of stachydrine in the absence of enzyme were exposed to a 10 times greater X-ray dose did not show any loss of methyl groups as analyzed by mass spectrometry (Figure S9), which suggests that the formation of proline in the proline-Stc2 complex is enzyme-dependent.
Our results from the 1H NMR assays indicate that Stc2 in solution can use quaternary ammonium compounds (e.g., stachydrine, 1) as a substrate but does not appear to catalyze turnover of tertiary amines (e.g., N-monomethylproline, 2). These results are also consistent with genetic studies of S. meliloti, discussed above and illustrated in Scheme 1C. The results from the X-ray structure determination of Stc2 cocrystallized with stachydrine suggest that Stc2 is capable of demethylating both N,N-dimethylstachydrine and N-methyl-proline. One possible explanation for the discrepancy between the solution studies and crystallographic results is that, in the crystalline state, N-monomethylproline (2) cannot be released from the active site and proceeds through a second round of demethylation. Indeed, in the structure of the Stc2–proline complex, the active site is closed from bulk solvent by a loop (residues 215–233; residues 225–229 within the loop are disordered; see Figure S3A,B). B-factors in the loop are considerably higher than the average for the protein atoms in the structure (43.9 Å2 for loop vs 16.1 Å2 for protein), consistent with the hypothesis that it is more flexible. Movement of the homologous gating loop has been demonstrated in dicamba demethylase, in which open and closed gate conformations are observed.59 It appears that the loop plays a role in controlling access to or dissociation from the active site by solvent, substrates, and product molecules. Thus, the effective concentration of the N-methylproline in the crystal is much higher than that achieved in the single-turnover assays performed in solution. Notably, the bound conformation of substrate need not change for the second demethylation of N-monomethylproline to occur since enantiomers of amines interconvert rapidly by inversion of the nitrogen unshared electron pair.83
Another possible explanation for the second turnover observed in the crystallographic results could derive from the X-ray-dependent photoelectric effect. Our results suggest a substrate–iron complex that also includes a solvent molecule in the coordination sphere. The ejection of an electron from the iron complex will likely occur with an energy of approximately 7100 eV.84,85 The XRD data were collected with 1 Å X-ray photons (12 398 eV). The incident X-ray photons have sufficient energy to eject a core electron from the iron to yield oxidized Fe–O(H)x species directly. Thus, it is possible that conversion of stachydrine to proline within the active site may also be driven, in part, by core electron ejection from the iron–ligand complex.
Proposed Stc2 Reaction Mechanism
For α-ketogluta-rate-dependent histone demethylases,1,11–14 N-demethylation is achieved by the oxidative hydroxylation of the N-methyl group. The resulting N-methoxyl group is unstable and decomposes spontaneously to form formaldehyde as the oxidative demethylation byproduct. On the basis of these N-demethylation precedents and general properties of Rieske-type oxygenases,86 we propose a mechanistic model for Stc2-catalyzed oxidative demethylation of stachydrine (Scheme 2). The reaction cycle starts with oxidized Rieske cluster and substrate binding to the mononuclear Fe(II) site. In the structure of the Stc2–proline complex, proline binds to the mononuclear iron site through its carboxylate moiety by displacing one iron solvent ligand observed in the substrate free Stc2 structure (Figures 3A and 4). The proline observed in this structure is proposed to be generated in situ through X-ray-derived photoelectric effects and two consecutive demethylation cycles (Scheme 2). Therefore, the structure observed might also reflect a potential binding mode for stachydrine to Stc2. The binding order of O2 to the Fe(II) and the injection of an electron through the Rieske cluster is not well established in the Stc2 reaction cycle as it is in NDO.87 Based on known mononuclear iron enzyme models in the literature, the reaction likely goes through the oxidative hydroxylation of the stachydrine methyl group by high-valent iron–oxo species.88,89 The hydroxylated product spontaneously decomposes to produce formaldehyde.1,11–14 In solution studies via NMR, only a single demethylation cycle occurs, but in the crystal a second demethylation cycle occurs, resulting in proline production. In the crystalline state, the dissociation of the N-methylproline may be blocked by crystal packing and the loop covering the substrate binding/product release path. Therefore, in the aerobic crystal we propose a second round of hydroxylation and formaldehyde release to yield proline.
Scheme 2.

Proposed Stc2 Oxidative Demethylation Mechanism
CONCLUSIONS
In this work, we demonstrated that Stc2 is a demethylase, which is capable of oxidatively demethylating a quaternary ammonium. Notably, the correlation of X-ray data collection with UV–vis and resonance Raman spectroscopy was key to defining the redox state and ligand identity observed in the crystal structure. The formation of product in the crystal was consistent with the dose-dependence of the reduction of the Rieske center by the X-ray beam. The product-bound complex showed significant differences at the catalytic site compared to classic Rieske-type oxygenases, in which substrates do not directly interact with the mononuclear iron site. Instead, we propose on the basis of the structure of the Stc2 product complex that the substrate of Stc2, stachydrine, directly coordinates to the mononuclear non-heme iron center. How such a coordination mode modulates the activities of the non-heme iron center to meet the challenges of such a unique transformation, demethylation of a quaternary ammonium, awaits further elucidation.
In addition to prokaryotic examples of Rieske demethylases such as Stc2, phylogenetic analysis of Rieske and Rieske-type proteins suggests that they are conserved in both prokaryotes (e.g., cytochrome bc1 and b6 f complexes) and eukaryotes (e.g., cytochrome bc1, choline monooxygenase, CMP-Neu5Ac hydroxylase).90 Although the homotrimeric, α3 type of structure found in Stc2 is common, the presence of many Rieske-type oxygenases in the form α3β3, which suggests that recruiting other domains to achieve more complex functions, might be possible.90 Due to the importance of N-methylations in epigenetic and post-translational control, in the future it is essential to uncover the possible scaffolds and chemistries that can support the N-demethylation reactions.
Supplementary Material
Acknowledgments
The authors thank Dr. Alexei S. Soares for assistance in initial phase determination of the apo structure. This work was supported by NSF Grant CHE 0748504 (to P.L.). E.C. was supported by the Boston University Undergraduate Research Opportunities Program. Data for this study were measured at Beamline X12C and X26C of the National Synchrotron Light Source (NSLS). Support for A.M.O. and D.S.-M. was from the US Department of Energy (DOE) Office of Biological and Environmental Research (FWP BO-70) and from the National Center for Research Resources of the National Institutes of Health (2-P41-RR012408). The NSLS was supported by the DOE Office of Basic Energy Sciences (DE-AC02-98CH10886).
Footnotes
Synthesis of stachydrine and accompanying NMR spectra, structural homology analysis, sequence alignments of homologues and orthologues, table of rmsd values of structural homologues, figures showing active site, electron density maps, and overlay with NDO, solution spectra of Stc2, and mass spectrometry data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cantoni GL. Annu Rev Biochem. 1975;44:435. doi: 10.1146/annurev.bi.44.070175.002251. [DOI] [PubMed] [Google Scholar]
- 2.Benkovic SJ. Annu Rev Biochem. 1980;49:227. doi: 10.1146/annurev.bi.49.070180.001303. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee R, Ragsdale SW. Annu Rev Biochem. 2003;72:209. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 4.Biel M, Wascholowski V, Giannis A. Angew Chem, Int Ed. 2005;44:3186. doi: 10.1002/anie.200461346. [DOI] [PubMed] [Google Scholar]
- 5.Walsh CT. Posttranslational modification of proteins: Expanding Nature’s inventory. Roberts and Co. Publishers; Greenwood Village: 2006. [Google Scholar]
- 6.Chinenov Y. Trends Biochem Sci. 2002;27:115. doi: 10.1016/s0968-0004(02)02058-3. [DOI] [PubMed] [Google Scholar]
- 7.Bannister AJ, Schneider R, Kouzarides T. Cell. 2002;109:801. doi: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- 8.Byvoet P, Shepherd GR, Noland BJ, Hardin JM. Arch Biochem Biophys. 1972;148:558. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- 9.Thomas G, Hempel K, Lange HW. Hoppe-Seylers Z Physiol Chem. 1972;353:1423. [PubMed] [Google Scholar]
- 10.Mosammaparast N, Shi Y. Annu Rev Biochem. 2010;79:155. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Cell. 2004;119:941. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 12.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Cell. 2006;125:467. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 13.Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Genes Dev. 2006;20:1557. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. Nature. 2006;442:307. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 15.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Nature. 2002;419:174. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 16.Falnes PO, Johansen RF, Seeberg E. Nature. 2002;419:178. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 17.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Nature. 2007;448:87. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 18.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Nat Struct Mol Biol. 2007;14:689. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, Shi Y, Zhang G. Cell. 2006;125:691. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, Hansen K, Shi Y, Zhang G. Proc Natl Acad Sci USA. 2007;104:10818. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stavropoulos P, Blobel G, Hoelz A. Nat Struct Mol Biol. 2006;13:626. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 22.Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Mol Cell. 2006;23:377. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 23.Phillips DA, Joseph CM, Maxwell CA. Plant Physiol. 1992;99:1526. doi: 10.1104/pp.99.4.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fougere F, Le Rudulier D. J Gen Microbiol. 1990;136:157. doi: 10.1099/00221287-136-1-157. [DOI] [PubMed] [Google Scholar]
- 25.Amin US, Lash TD, Wilkinson BJ. Arch Microbiol. 1995;163:138. doi: 10.1007/BF00381788. [DOI] [PubMed] [Google Scholar]
- 26.Trinchant JC, Boscari A, Spennato G, Van de Sype G, Le Rudulier D. Plant Physiol. 2004;135:1583. doi: 10.1104/pp.103.037556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zahran HH. J Biotechnol. 2001;91:143. doi: 10.1016/s0168-1656(01)00342-x. [DOI] [PubMed] [Google Scholar]
- 28.Goldmann A, Boivin C, Fleury V, Message B, Lecoeur L, Maille M, Tepfer D. Mol Plant-Microbe Interact. 1991;4:571. doi: 10.1094/mpmi-4-571. [DOI] [PubMed] [Google Scholar]
- 29.Phillips DA, Sande ES, Vriezen JAC, de Bruijn FJ, Le Rudulier D, Joseph CM. Appl Environ Microbiol. 1998;64:3954. doi: 10.1128/aem.64.10.3954-3960.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burnet MW, Goldmann A, Message B, Drong R, El Amrani A, Loreau O, Slightom J, Tepfer D. Gene. 2000;244:151. doi: 10.1016/s0378-1119(99)00554-5. [DOI] [PubMed] [Google Scholar]
- 31.Mason JR, Cammack R. Annu Rev Microbiol. 1992;46:277. doi: 10.1146/annurev.mi.46.100192.001425. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Pape T, Schneider TR. J Appl Crystallogr. 2004;37:2. [Google Scholar]
- 34.Sheldrick GM. Acta Crystallogr A. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 35.Morris RJ, Perrakis A, Lamzin VS. Methods Enzymol. 2003;374:229. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 36.Adams PD, et al. Acta Crystallogr D: Biol Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr D: Biol Crystallogr. 2010;66:486. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elsen NL, Moe LA, McMartin LA, Fox BG. Biochemistry. 2007;46:976. doi: 10.1021/bi0616145. [DOI] [PubMed] [Google Scholar]
- 39.Yi J, Orville AM, Skinner JM, Skinner MJ, Richter-Addo GB. Biochemistry. 2010;49:5969. doi: 10.1021/bi100801g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orville AM, Lountos GT, Finnegan S, Gadda G, Prabhakar R. Biochemistry. 2009;48:720. doi: 10.1021/bi801918u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orville AM, Buono R, Cowan M, Heroux A, Shea-McCarthy G, Schneider DK, Skinner JM, Skinner MJ, Stoner-Ma D, Sweet RM. J Synchrotron Radiat. 2011;18:358. doi: 10.1107/S0909049511006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoner-Ma D, Skinner JM, Schneider DK, Cowan M, Sweet RM, Orville AM. J Synchrotron Radiat. 2011;18:37. doi: 10.1107/S0909049510033601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paithankar KS, Garman EF. Acta Crystallogr D: Biol Crystallogr. 2010;66:381. doi: 10.1107/S0907444910006724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martins BM, Svetlitchnaia T, Dobbek H. Structure. 2005;13:817. doi: 10.1016/j.str.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 45.Tarasev M, Kaddis CS, Yin S, Loo JA, Burgner J, Ballou DP. Arch Biochem Biophys. 2007;466:31. doi: 10.1016/j.abb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parales RE, Emig MD, Lynch NA, Gibson DT. J Bacteriol. 1998;180:2337. doi: 10.1128/jb.180.9.2337-2344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Structure. 1998;6:571. doi: 10.1016/s0969-2126(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 48.Yamaguchi M, Fujisawa H. J Biol Chem. 1982;257:12497. [PubMed] [Google Scholar]
- 49.Hegg EL, Que L., Jr Eur J Biochem. 1997;250:625. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 50.Ferraro DJ, Gakhar L, Ramaswamy S. Biochem Biophys Res Commun. 2005;338:175. doi: 10.1016/j.bbrc.2005.08.222. [DOI] [PubMed] [Google Scholar]
- 51.Krissinel E, Henrick K. J Mol Biol. 2007;372:774. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 52.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Bioinformatics. 2008;24:2780. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friemann R, Lee K, Brown EN, Gibson DT, Eklund H, Ramaswamy S. Acta Crystallogr D. 2009;65:24. doi: 10.1107/S0907444908036524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T. J Mol Biol. 2004;342:1041. doi: 10.1016/j.jmb.2004.07.062. [DOI] [PubMed] [Google Scholar]
- 55.Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, Wakagi T. J Bacteriol. 2005;187:2483. doi: 10.1128/JB.187.7.2483-2490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friemann R, Ivkovic-Jensen MM, Lessner DJ, Yu CL, Gibson DT, Parales RE, Eklund H, Ramaswamy S. J Mol Biol. 2005;348:1139. doi: 10.1016/j.jmb.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 57.Jakoncic J, Jouanneau Y, Meyer C, Stojanoff V. Biochem Biophys Res Commun. 2007;352:861. doi: 10.1016/j.bbrc.2006.11.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen GS, Zavialov A, Gursky R, Ehrenberg M, Frank J. Cell. 2005;121:703. doi: 10.1016/j.cell.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 59.D’Ordine RL, Rydel TJ, Storek MJ, Sturman EJ, Moshiri F, Bartlett RK, Brown GR, Eilers RJ, Dart C, Qi Y, Flasinski S, Franklin SJ. J Mol Biol. 2009;392:481. doi: 10.1016/j.jmb.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 60.Wang X, Li B, Herman PL, Weeks DP. Appl Environ Microbiol. 1997;63:1623. doi: 10.1128/aem.63.4.1623-1626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dumitru R, Jiang WZ, Weeks DP, Wilson MA. J Mol Biol. 2009;392:498. doi: 10.1016/j.jmb.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parales RE, Parales JV, Gibson DT. J Bacteriol. 1999;181:1831. doi: 10.1128/jb.181.6.1831-1837.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leggate EJ, Hirst J. Biochemistry. 2005;44:7048. doi: 10.1021/bi050189x. [DOI] [PubMed] [Google Scholar]
- 64.McGinnis S, Madden TL. Nucleic Acids Res. 2004;32:W20. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karlsson A, Parales J, Parales R, Gibson D, Eklund H, Ramaswamy S. J Biol Inorg Chem. 2005;10:483. doi: 10.1007/s00775-005-0657-1. [DOI] [PubMed] [Google Scholar]
- 66.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Science. 2003;299:1039. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 67.Quaye O, Lountos GT, Fan F, Orville AM, Gadda G. Biochemistry. 2008;47:243. doi: 10.1021/bi7017943. [DOI] [PubMed] [Google Scholar]
- 68.Garman EF. Acta Crystallogr D: Biol Crystallogr. 2010;66:339. doi: 10.1107/S0907444910008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J. Nature. 2000;406:752. doi: 10.1038/35021099. [DOI] [PubMed] [Google Scholar]
- 70.Sanishvili R, Yoder DW, Pothineni SB, Rosenbaum G, Xu S, Vogt S, Stepanov S, Makarov OA, Corcoran S, Benn R, Nagarajan V, Smith JL, Fischetti RF. Proc Natl Acad Sci USA. 2011;108:6127. doi: 10.1073/pnas.1017701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuila D, Schoonover JR, Dyer RB, Batie CJ, Ballou DP, Fee JA, Woodruff WH. Biochim Biophys Acta. 1992;1140:175. doi: 10.1016/0005-2728(92)90007-o. [DOI] [PubMed] [Google Scholar]
- 72.Kuila D, Fee JA, Schoonover JR, Woodruff WH, Batie CJ, Ballou DP. J Am Chem Soc. 1987;109:1559. [Google Scholar]
- 73.Rotsaert FJ, Pikus JD, Fox BG, Markley JL, Sanders-Loehr J. J Biol Inorg Chem. 2003;8:318. doi: 10.1007/s00775-002-0417-4. [DOI] [PubMed] [Google Scholar]
- 74.Cline JF, Hoffman BM, Mims WB, LaHaie E, Ballou DP, Fee JA. J Biol Chem. 1985;260:3251. [PubMed] [Google Scholar]
- 75.Fee JA, Findling KL, Yoshida T, Hille R, Tarr GE, Hearshen DO, Dunham WR, Day EP, Kent TA, Munck E. J Biol Chem. 1984;259:124. [PubMed] [Google Scholar]
- 76.Kuila D, Fee JA. J Biol Chem. 1986;261:2768. [PubMed] [Google Scholar]
- 77.Zu Y, Couture MM, Kolling DR, Crofts AR, Eltis LD, Fee JA, Hirst J. Biochemistry. 2003;42:12400. doi: 10.1021/bi0350957. [DOI] [PubMed] [Google Scholar]
- 78.Rotsaert FJ, Pikus JD, Fox BG, Markley JL, Sanders-Loehr J. J Biol Inorg Chem. 2003;8:318. doi: 10.1007/s00775-002-0417-4. [DOI] [PubMed] [Google Scholar]
- 79.Gassner GT, Ballou DP, Landrum GA, Whittaker JW. Biochemistry. 1993;32:4820. doi: 10.1021/bi00069a017. [DOI] [PubMed] [Google Scholar]
- 80.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. J Am Chem Soc. 2003;125:2034. doi: 10.1021/ja028781m. [DOI] [PubMed] [Google Scholar]
- 81.Wolfe MD, Altier DJ, Stubna A, Popescu CV, Munck E, Lipscomb JD. Biochemistry. 2002;41:9611. doi: 10.1021/bi025912n. [DOI] [PubMed] [Google Scholar]
- 82.Tarasev M, Rhames F, Ballou DP. Biochemistry. 2004;43:12799. doi: 10.1021/bi0490587. [DOI] [PubMed] [Google Scholar]
- 83.Wade LG. Organic chemistry. 6. Pearson Prentice Hall; Upper Saddle River, Nj: 2006. [Google Scholar]
- 84.de Groot F. Chem Rev. 2001;101:1779. doi: 10.1021/cr9900681. [DOI] [PubMed] [Google Scholar]
- 85.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297. [Google Scholar]
- 86.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Science. 2003;299:1039. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 88.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM. Biochemistry. 2005;44:8138. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 89.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Krebs C. Nat Chem Biol. 2007;3:113. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 90.Schmidt CL, Shaw L. J Bioenerg Biomembr. 2001;33:9. doi: 10.1023/a:1005616505962. [DOI] [PubMed] [Google Scholar]
- 91.Kraulis PJ. J Appl Crystallogr. 1991;24:946. [Google Scholar]
- 92.Fu W, Drozdzewski PM, Davies MD, Sligar SG, Johnson MK. J Biol Chem. 1992;267:15502. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.