

Abstract
Cellular quiescence is a reversible mode of cell cycle exit that allows cells and organisms to withstand unfavorable stress conditions. The factors that underlie the entry, exit, and maintenance of the quiescent state are crucial for understanding normal tissue development and function as well as pathological conditions such as chronic wound healing and cancer. In vitro models of quiescence have been used to understand the factors that contribute to quiescence under well-controlled experimental conditions. Here we describe an in vitro model of quiescence that is based on neonatal human dermal fibroblasts. The fibroblasts are induced into quiescence by antiproliferative signals contact inhibition and serum-starvation (mitogen withdrawal). We describe the isolation of fibroblasts from skin, methods for inducing quiescence in isolated fibroblasts, and approaches to manipulate the fibroblasts in proliferating and quiescent states in order to determine critical regulators of quiescence.
1. Introduction
1.1 Cell quiescence and its role in human biology
Extracellular signals such as the availability of nutrients, the presence of growth factors, or the presence of cytokines can serve as a signal that cells should proliferate to generate daughter cells. When pro-proliferative signals are absent or antiproliferative signals are present, some cells have the capacity to reversibly exit the cell cycle and enter into a quiescent state [1–4]. Quiescent cells are defined by their ability to re-enter the cell cycle at a future time, when conditions are favorable for cell division. Thus, quiescence represents a reversible, non-dividing state. The reversibility of quiescent cells distinguishes them from senescent and apoptotic cells that cannot reenter the cell cycle [3], and from terminally differentiated cells that no longer divide, such as the cells that have undergone squamous maturation.
Quiescent cells in the human body include memory T cells, hepatocytes, fibroblasts and stem cells. When quiescent cells sense external stimuli to divide, they can be induced to proliferate. Often this occurs in a context in which the quiescent cells are being called on to proliferate and function as early responders in maintaining tissue homeostasis. For example, during wound healing, quiescent fibroblasts distant from the wound area become activated, resulting in their proliferation and migration to the wound site [5,6]. These activated fibroblasts synthesize extracellular matrix proteins, such as collagen, that help in closing the wound. Cancer is characterized by cells that ought to exit the proliferative cell cycle, but continue proliferating despite anti-proliferative signals. Studying the molecular processes that induce quiescence, maintain quiescence, and stimulate cells to re-enter the cell cycle may provide important insights into multiple disease states. A better understanding of the relevant factors can be gained by developing in vivo and in vitro models that mimic the biological system faithfully and in a reproducible manner.
1.2 Biological markers of quiescent cells
Quiescent cells have entered a state in which the cells have ceased dividing and no new genomic DNA is being synthesized. Recent studies have described the properties of quiescent cells [7,3,8–12], including changes in gene expression patterns [13–19], but more research is required to truly understand the molecular basis of quiescence. This leads to the question: What approaches are available to ascertain whether a cell is in a quiescent state using different cell-based techniques? What biological markers are available for this?
Table 1 summarizes some of the markers and reagents that have been used to probe for quiescent cells. The cell cycle of proliferating cells is driven by the expression of cyclins, which are proteins that activate cyclin-dependent kinases (Cdks) [20,21]. Cdks, in turn, phosphorylate key proteins at different stages of the cell cycle that allow cells to progress through the cell cycle phases [22]. The activity of these Cdks is inhibited by Cdk inhibitors such as p21 and p27 [23]. These Cdk inhibitors are often induced during quiescence [24]. Some of the markers that have been used for quiescence include reduced Cdk activity and elevated levels of cyclin-dependent kinase inhibitors.
Table 1.
Methods to identify quiescent cells
| Method |
Protein or
mRNA marker(s)/reagent |
Description | Change in levels
with quiescence (relative to dividing cells) |
Type of cells (Ref.) |
|---|---|---|---|---|
| Western blot | p27Kip1 (p27) | Cyclin-dependent kinase (Cdk) inhibitor; nuclear | Up | Fibroblasts ([24]), HUVEC ([66]) |
| p21 | Cdk inhibitor; nuclear | Up | Fibroblasts ([67])(Aster’s paper) | |
| Phosphorylated-retinoblastoma (phospo-pRB) | Phosphorylation of pRB in dividing cells activates E2F (see section 1.2). | Down | Lymphocytes ([68]) | |
| Cd82 | Cell surface protein in the tetraspanin superfamily | Up | Hematopoietic stem cells ([69]) | |
| Real time PCR | p53 | Tumor suppressor that controls proliferation, differentiation and apoptosis | Up | Hematopoietic stem cells ([70]) |
| Flow cytometry | Pyronin-Ya | Preferentially binds to RNA; emits fluorescence | Down | Fibroblasts ([32,7]), Hematopoietic stem cells (HSC)b ([33,34]) |
| Bromo-uridine (BrdU)a | Thymidine analog; incorporates into newly synthesized DNA; detected by antibody | Down | Fibroblasts ([15]) | |
| 5-ethynyl-2′ – deoxyuridine (EdU) | Thymidine analog; incorporates into newly synthesized DNA; detected by fluorescent dyes that chemically react with EdU | Down | Fibroblasts ([32] | |
| mVenus-p27K− | p27 that lacks binding to Cdk is fused to a fluorescent protein (mVenus) | Up | NIH3T3 ([48]) | |
| Reactive oxygen species (ROS) | Generated in the cell due to oxidative metabolism and stress conditions; fluorescence detection | Down | MCF7 ([71]), eMSC ([72]) | |
| Immunohistochemistry/Immunofluorescence (Tissue Staining) |
Ki-67 | Proliferation marker (nuclear) | Down | [73] |
| Fgf18 and Bmp6 | Maintenance of quiescence of hair follicle stem cells (HFSC) | Up | HFSC ([74]) | |
| Immunocytochemistry/Immunofluorescence (Cell Staining) |
E2F5 and LEK1 | Translocate to nucleus in quiescent myoblasts; fluorescence detection? | Myoblasts ([75]) | |
| Ki67 and BrdU | See above | |||
| Label retentionc (In vivo labeling) | BrDU | See above; slow-cycling cells retain BrDU | HSCb ([38,40]) | |
| Histone 2B-green fluorescent protein (H2B-GFP) | Fluorescent fusion protein that is incorporated into nucleosomes; slow-cycling cells retain H2B-GFP | Skin stem cells ([41]), HSC ([40,42])b | ||
| Live cell imaging | Cdk2 sensor | Translocates to the nucleus when active | Inactive, cytoplasmically localized | MCF10a ([10]) |
| p21 | Cdk inhibitor; nuclear | Up | MCF10a ([10]) | |
| Hypophosphorylated Rb | Rb with fewer phosphorylation events sequesters E2F | Less phosphorylated | MCF10a ([10]) | |
| Kinase assay | p33QIK, Quiescence Induced Kinase | Kinase activity goes up | NIH3T3 ([76]) |
Cells were also stained with Hoechst 33342 or DAPI. These dyes bind to A-T base pairs and therefore, measure the total DNA content
Members of retinoblastoma family of proteins (Rb/p105, p107, and Rb2/p130) bind to the activating E2F transcription factors (E2F1, E2F2 and E2F3) [25]. Binding of Rb family members results in a complex that sequesters E2F, thereby suppressing the transcription of E2F-dependent cell cycle genes [26,27]. Phosphorylation of retinoblastoma proteins by Cdks releases E2F, which allows cell cycle progression. In some models, the complex p130-E2F accumulates in quiescent cells and has been used as a marker for quiescent cells [28]. In other models, Rb itself, and not p130 or p107, is critical for maintaining quiescence [29].
Other methods to detect quiescent cells have been developed based on the lack of DNA synthesis in quiescent cells. Cells that are actively dividing incorporate nucleotides into their DNA. Addition of labeled nucleotides like bromodeoxyuridine (BrdU) can be used to detect cells that have recently replicated their DNA [15]. Quiescent cells are low or negative for this marker. Unlike BrdU, which is detected by an antibody, labeled fluorescent nucleotides have been developed with Click chemistry [30] to attach a fluorescent probe to ethinyldU (eDU) to measure DNA synthesis [31,32]. Another characteristic of many quiescent cells is low levels of total RNA. Pyronin Y stains total RNA and has been used as a quiescence marker based on FACS analysis of individual cells [33,34,7,32].
Identifying quiescent cells within an organism has mostly been based on methods that identify cells that have not divided recently. Cells that are quiescent have been identified with label-retaining assays [35–40]. With these assays, a repeated and prolonged administration of a labeled nucleotide such as tritiated thymidine ([3H]TdR) or BrdU will be taken up by the animal (pulse), followed by a chase during which labeled nucleotides are not provided. Cells that divide slowly can be identified as those that retain the label a long time after the pulse period A similar concept was developed to fluorescently label cells in living organisms that have not divided recently. An inducible histone H2B-GFP protein has been introduced into organisms [41,42]. Upon activation, the histone H2B-GFP will label every histone green. After the expression of the H2B-GFP is turned off, and endogenous, unlabeled histone H2B is the dominant form expressed, cells that retain the label will be those that have not divided, and will be enriched in quiescent cells.
The Fluorescence Ubiquitin Cell Cycle Indicator (FUCCI) cell cycle sensor can be used to monitor cell progression in living organisms, though by itself, does not specifically distinguish quiescence from the G1 phase of the cell cycle [43–47]. The Fucci system utilizes the chromatin licensing and DNA replication factor 1 (Cdt1) protein and its inhibitor geminin. With the Fucci model, Cdt1, which is expressed at high levels in G1 and declines in S phase, is labeled red. Geminin, which is expressed during S, G2 and M cell cycle phases, is labeled green. Fucci zebrafish, flies and mice have been developed and the system has been used to gain insight into the proliferative patterns of cells in vivo. Recently, the Fucci system has been combined with a method to monitor p27 levels to identify quiescent cells [48]. Levels of p27 are high in quiescent cells and two different degradation methods eliminate p27. The Skp1 (S-phase kinase-associated protein 1)-cullin-F-box (SCF) ubiquitin ligase complex SCFSkp2 binds phosphorylated p27 and mediates its degradation in S and G2 cell cycle phases [49]. KPC (Kip1 ubiquitination-promoting complex), consisting of KPC1 and −2, degrades p27 in G1 [50]. The exquisite control of p27 levels achieved by the cell have been exploited to generate a quiescent cell marker. The fluorescent protein Venus was fused to a version of p27 that does not bind CDKs (mVenus-27K-). Introducing this protein into cell allows visualization of G0 cells based on their fluorescence. Combining this method with FUCCI mice allowed sensitive detection of quiescent cells in vivo: cells that were positive for both the mVenus-27K-fluorescence and Cdt1 fluorescence were considered to be in G0, while cells positive only for Cdt1 fluorescence were considered to be in G1.
A recent exciting advance in this field was the advent of real time cell monitoring to detect quiescent cells. A CDK2 sensor has been used to monitor Cdk activity in cycling epithelial cells over several cell cycles [10]. This sensor contains four CDK consensus phosphorylation sites and the yellow fluorescent protein Venus. The sensor also contains nuclear localization and export signals so that the protein can be imported into the nucleus and exported from the nucleus based on the extent of Cdk activity in the cells. The ratio of nuclear to cytoplasmic fluorescence provides a readout for CDK2 activity. At the end of mitosis, while most cells exhibited an intermediate amount of CDK2 activity, some cells with low CDK2 activity were identified. These low CDK2 cells entered a temporary state of quiescence. Time lapse imaging followed by fixation and immunofluorescence revealed that p21 levels were an important determinant of which cells exited the cell cycle.
While these approaches have led to important discoveries about quiescence and quiescent cells, new markers, including cell surface markers, that positively identify quiescent cells and distinguish them from cells in G1 that are about to divide, and distringuish quiescent cels from cells that have irreversibly exited the cell cycle due to senescence or differentiation, would be extremey valuable for the field.
1.3 Using primary cells isolated from tissues for studying quiescence in vitro
Studying the quiescent behavior of cells using in vivo animal models is important for our understanding of quiescence because it provides information about the physiologically relevant tissue microenvironment. In addition, in vitro models of quiescence can be used to complement in vivo systems. With in vitro quiescence models, proliferating cells isolated from tissues can be induced into quiescence under different conditions (e.g. mitogen removal) in tissue culture dishes/plates. These experiments can provide a controlled environment in which cells can be subjected to specific, reproducible and well-controlled treatments and isolated for downstream analysis. Although in vitro models lack the complexity of in vivo models, they do provide a simple, readily scalable, and reproducible system for analysis of quiescence. With in vitro models, it is possible to control the specific cell types present, and the levels of extracellular matrix proteins, and signaling and growth factors.
The choice of cell type in establishing an in vitro model of quiescence is important. The cells should be amenable to easy isolation from tissues and they should be able to be cultured readily in vitro by following standard tissue culture techniques. A variety of cell types have been used for establishing in vitro models of quiescence, including pancreatic stellate cells [51], HUVEC [52], myoblasts [53], keratinocytes, astrocytes and fibroblasts described further below.
Fibroblasts are the predominant cell type in connective tissue. They secrete extracellular matrix proteins such as collagen [54]. Fibroblasts isolated from different tissues such as lung and skin have been extensively used to study quiescence in vitro [55,56,15,57,7]. Dermal fibroblasts are easy to culture and the steps to isolate them from skin have been well established [58–61]. Proliferating dermal fibroblasts can be induced into quiescence by contact inhibition or serum-starvation and these quiescent fibroblasts assume distinct morphologies and gene expression signatures compared to proliferating fibroblasts [15,19].
We present here steps to establish an in vitro model of quiescence using neonatal dermal fibroblasts. We provide protocols for the isolation of neonatal fibroblasts from foreskin, for culturing these fibroblasts in a laboratory tissue culture setting, and for inducing the fibroblasts into quiescence. We also describe methods to regulate the expression level of a specific gene via transfection of short interfering (siRNA). siRNA-based methods permit the investigation of the functional role of an individual factor in quiescence entry or exit.
2. Materials
For all of the tissue culture procedures, fibroblasts are grown in a 10-cm “tissue-culture treated” plate (10 ml culture volume) in an incubator set at 37 °C with 5% CO2 level, unless otherwise indicated. Culture volumes can be adjusted if using tissue culture plates or dishes of different sizes.
2.1 Procurement of skin tissue for fibroblast isolation
To isolate primary human dermal fibroblasts, we obtained human skin from newborns via the National Disease Research Interchange (http://ndriresource.org/) under a protocol approved by the UCLA Institutional Review Panel. Readers should follow all the safety and ethical guidelines, as mandated by their local Institutional Review Board. An authorized person who has received appropriate training, following the procedure approved by the Institutional Review Board and wearing the appropriate personal protective equipment should be responsible for skin collection.
2.2 Materials for isolating fibroblasts
Note that materials lists in 2.2.1, 2.2.6, and 2.2.9 are sufficient for one skin sample.
Antibiotic/antimycotic solution: 100 U/ml penicillin G, 100 μg/ml streptomycin, and 2.5 μg/ml amphotericin B dissolved in Dulbecco’s modified phosphate-buffered saline (DPBS) without calcium and magnesium. Filter-sterilize and store at −20°C away from light.
15 cm plate
100% ethanol
Two pairs of eye forceps
Surgical scissors
0.5% dispase solution: Add 500 mg solid dispase per 100 mL PBS. Filter-sterilize and store at −20°C in 5 or 25 mL aliquots. All the dispase may not dissolve.
Scalpel
15-ml polypropylene tube
1000 U/ml collagenase: Add 320 mg solid collagenase (318 U/mg Collagenase Type 1A) per 100 ml PBS). Filter-sterilize and store at −20°C in 5 or 25 ml aliquots.
Ice cold DMEM with 7.5% Fetal Bovine Serum (FBS)
70-mm nylon mesh strainer
100X Antibiotic/antimycotic Solution: Dissolve 10,000 units/mL Penicillin G, 10,000 μg/mL Streptomycin, and 25 μg/mL Amphotericin B in PBS. Filter sterilize and store at −20°C protected from light.
Fortified growth medium: Dulbecco’s Modified Eagle Medium (DMEM), 10% FBS, 1X antibiotic solution (#12 above), 1 mM sodium pyruvate, 10 mM HEPES buffer, 2 mM L-glutamine, 0.1 mM nonessential amino acids in Minimum Essential Medium (MEM) (1:100 of 10 mM stock)
Inverted light microscope
Number 22 disposable surgical blades
2.3 Essential tissue culture reagents for culturing fibroblasts
Vial of frozen fibroblasts
Water bath at 37°C
15-ml conical tube
Complete medium: Dulbecco’s Modified Eagle Medium (DMEM), 10% fetal bovine serum (FBS)
10-cm tissue culture-treated plate
Inverted light microscope
Dulbecco’s modified phosphate-buffered saline (DPBS) without calcium and magnesium
0.05% trypsin in DPBS
Trypan blue
Automated cell counter and disposable chamber slide or hematocytometer and hematocytometer slide
Low serum medium: DMEM and 0.1% serum
Eppendorf tubes (1.5 ml)
Cell cooling apparatus (such as Mr. Frosty™ freezing container)
2.4 Preparation of cell lysates
Cell lysis buffer (such as commercially available M-PER mammalian protein extraction reagent from Life Technologies)
Protease inhibitors
Bicinchoninic acid assay (BCA assay) kit
Soybean trypsin inhibitor
Eppendorf tubes (1.5 ml)
2.5 siRNA Transfection
Plates for transfection (depends upon the experimental design)
Transfection reagent for siRNA transfection (commercially available)
siRNAs for the genes of interest (available commercially)
3. Methods
Prewarm all the media and other reagents to 37°C using a water bath or a dry bath for at least 15 minutes. Disinfect the outside surfaces of media bottles, frozen cell vials, and other reagent tubes by spraying with 70% ethanol. All the steps are performed inside a tissue culture laminar flow hood under sterile conditions, unless otherwise indicated. The laboratory performing the fibroblast isolation procedure should have permission to work with biohazard materials and be equipped with all the instruments needed to perform mammalian cell culture in a sterile manner. All the personnel involved in performing tissue culture experiments require training in handling biohazardous materials.
3.1 Isolation of primary dermal fibroblasts
Before starting the procedure, clean all the surgical tools (number 22 disposable surgical blade, scalpel handles, eye forceps, and surgical scissors) with 70% ethanol and wrap them into a closed packet of aluminum foil before sterilizing them by autoclaving. Do not let the skin dry at any stage, as this will negatively affect fibroblast yield.
Wash the skins (5 ml of solution/skin sample) by submerging the skin in 100X antibiotic/antimycotic solution in a conical tube and then removing the solution. Repeat once.
Incubate the washed skin in 5 mL of 100% ethanol for 1 min on ice followed by washing in 5 ml 100X antibiotic/antimycotic solution for 10 min.
With two pairs of eye forceps and surgical scissors, scrape the dermal side of the tissue to recover the subcutaneous connective tissue (second layer of red tissue on the dermal side). Using either surgical scissors or a surgical scalpel and forceps, mince the tissue into small ~0.5 cm width pieces on a 15 cm plate. Incubate the pieces at 37°C for 1–4 hours in 5 ml (per skin) of 0.5% dispase solution in a 15 ml tube (see Note 1) until the epidermis detaches from the dermis. Fine mincing enhances fibroblast yield and viability, as fibroblasts will grow out from sharply cut edges.
Remove the epidermal sheet with gentle agitation or by two pairs of forceps as follows. Lay the skin sample (epidermis side up) on a 15-cm plate and with the curved flat of one pair of forceps, pin down the skin sample, while using the other pair of forceps to pinch and peel off the epidermis. Removal of the epidermis reduces keratinocyte contamination.
Using a number 22 disposable surgical blade, mince the dermal sample into 2–3 mm square pieces on a 15 cm plate. Place the pieces (~10–20) in a 15-ml polypropylene tube with 3 ml of collagenase solution. Incubate at 37° C for 1–2 hours with vigorous stirring until some of the cells are suspended in the solution. At this point, the solution should be bit turbid with some visible insoluble debris. Stop before all of the cells are dissolved in the solution.
Add 3 ml ice cold DMEM containing 7.5% FBS. Vortex the tube a few times to separate fibroblasts from collagen fibers. Filter the supernatant through a 70-μm nylon mesh strainer to remove debris.
Centrifuge the cells for 10 min at 150 × g at 4°C. Aspirate the supernatant. Resuspend the pellet in fortified growth medium and plate the cells on a 15 cm plate. This is passage 1 for this batch of fibroblasts.
Aspirate the medium after 24 hours to remove detached cells and add fresh, fortified growth medium. Confirm by light microscopy that the cells have the characteristic fibroblast morphology (spindle-shaped cells in monolayer; Figure 1).
Grow the fibroblasts until they become ~80% confluent. Passage the fibroblasts (passage # 2) to 2–3 new 15 cm plates. (See step 2–4 in section 3.2 for passaging fibroblasts. Scale the amount of each reagent for a 15 cm plate (20 ml culture volume). Use fortified growth medium instead of complete medium).
Freeze cells during passage 3 or 4 (see section 3.3 for freezing fibroblasts; scale the reagents for a 15-cm plate and use fortified growth medium instead of complete medium). Make sure the cells are in the exponential growth phase when they are collected for freezing (see Note 2).
Fig. 1.
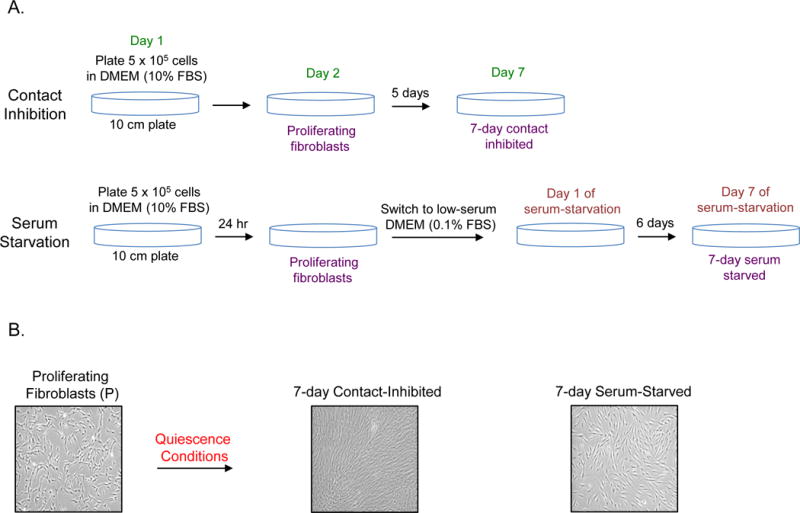
Methodology for generating proliferating, contact-inhibited, and serum-starved fibroblasts.
A. A schematic showing the protocol for generating proliferating, 7-day contact inhibited and 7-day serum starved fibroblasts is provided.
B. Images of proliferating, serum starved and contact inhibited fibroblasts are provided.
3.2 Culturing proliferating fibroblasts
Check the 10 cm plate containing fibroblasts under the microscope. Passage the cells if the confluency reaches about 80%. Use complete medium (without antibiotics) for regular culturing of cells (see Note 3)
To passage the cells, remove the medium containing dead and non-adherent cells from the plate by aspiration. Add 5–10 ml DPBS to the plate and gently swirl the plate to wash the cells. This washing step will remove any remaining complete medium that might inhibit the activity of trypsin added in the next step. Aspirate the DPBS from the plate. Add 3 ml of 0.05% trypsin solution to the plate and swirl the plate to coat the cells uniformly with trypsin. Incubate the cells in the incubator for 5 min.
After 5 min incubation, check the cells under the microscope to make sure that all the cells have been detached. If the cells are still attached, incubate the cells in the incubator for a few additional minutes. Exposure of the cells to trypsin should be minimized as trypsin affects cell viability. Tap the plate gently (if needed) to release the cells that are loosely attached to the plate. Add 10 ml of complete medium to neutralize the trypsin. Transfer the cell suspension (13 ml) to a 15-ml conical tube. Spin the tube at 500 × g for 5 min to pellet the cells.
Aspirate the supernatant (containing trypsin) from the tube. This removal of trypsin after trypsinization helps in maintaining cell viability. Resuspend the cell pellet in 1 ml of complete medium. A portion of this cell suspension is then added to a fresh tube and mixed with complete medium to achieve a final volume of 10 ml and the desired dilution of the cells. For example, to dilute the cells 4 times (4X dilution), 250 μl of cells are mixed with 9.75 ml of complete medium and then added to a 10 cm plate.
3.3 Freezing and thawing the fibroblasts
Use proliferating cells of low passage number for freezing (see Note 4). Pellet the cells as described before (see steps 2 and 3 in section 3.2). Aspirate the supernatant.
Add 3 ml of cell freezing medium to the cell pellet and resuspend the pellet by gently pipetting up and down.
Add the cell suspension to a cryovial. Using a cryo marker pen or a cryo-label suitable for liquid N2 storage, the cryovial should be labeled with following information: name of investigator, type of cells, cell source, passage number, number of cells in the vial, and date of freezing. An electronic log sheet should be maintained separately containing all the above information and other important information such as type of culture medium and freezing reagent used.
Transfer the cryovial to a cooling apparatus for slow cooling and place the apparatus inside an −80°C freezer overnight. The next day, move the cryovial to a liquid nitrogen freezer for long-term storage.
Thaw a vial of frozen fibroblasts by gently swirling the tube in the water bath set at 37°C. Transfer the cells (1 ml) to a 15 ml conical tube and add 9 ml of complete medium and mix gently by pipetting up and down using a 10 ml serological pipette. Try to avoid generation of air bubbles.
Spin the tube at 500 × g for 5 min at room temperature to harvest the cells. Remove the supernatant and add 1 ml of complete medium. Suspend the cell pellet by gently pipetting up and down a few times using a 1 ml pipet tip (see Note 5). Make sure that the cell pellet is completely suspended. Add 9 ml of complete medium and mix. Add the 10 ml of cell suspension to a 10-cm plate. Place the plate inside an incubator. Move the plate forward and backwards and sideways to distribute the cells across the surface of the plate. Swirling in a circle may result in cells piling in the middle of the plate.
3.4 Induction of fibroblast quiescence by contact inhibition
Grow fibroblasts in a 10 cm plate until the cells are about 80% confluent. Change medium after every 48 hours. Wash the cells, trypsinize, and prepare the cell pellet (see steps 2 and 3 in section 3.2). Resuspend the cell pellet in ~1 ml of complete medium (stock suspension).
Take 10 μl of cell suspension in an Eppendorf tube and add 10 μl of trypan blue. Mix well by pipetting up and down. Add 10 μl to the hematocytometer slide or into a disposable chamber slide for automatic counting. Count the cells manually using a hematocytometer or an automated cell counter by following the manufacturer’s instructions. For more detailed protocols on cell counting using trypan blue refer to [62]. Note the density (cells/ml) of live cells (see Note 6). The live cells show bright centers and dark edges upon trypan blue staining. Repeat the counting procedure. Average the two counts to get the final number for live cell density (Y cells/ml).
Calculate the volume of the stock suspension (X ml) needed for plating 5 × 105 cells per 10 cm plate using the density of live cells (Y cells/ml). Pipet the calculated amount of stock suspension (X ml) to a 15 ml conical tube and then add the appropriate amount of complete medium to make the total volume to 10 ml. Mix well and add the suspension to one or many 10 cm plate(s) (Day 1). Incubate the plate(s) in the incubator for about 24 hours.
Depending upon the strain of fibroblast, the cell confluency can be expected to reach 50–70% after 24 hours (Day 2). The cells can be harvested at this stage and the pellet can be prepared as described above (see steps 2 and 3 in section 3.2). At this stage, cell lysates from proliferating cells (P) for western blot can be prepared (see section 3.7).
Grow the cells until Day 7, change the medium every 48 hours. The cell should cover the bottom of the plate by day 3–4. By day 7, the cells will have ceased to grow due to contact inhibition (Figure 1). Prepare the cell lysates for 7-day contact inhibited cells (see section 3.7).
3.5 Induction of fibroblast quiescence by serum starvation
Plate 5 × 105 cells in a 10-cm plate (see steps 1–3 in section 3.4).
After 24 hours, wash the cells twice with 5 ml of DPBS to completely remove the medium. Add 10 ml of low serum medium (containing DMEM and 0.01% serum) to the plate. Incubate the plate in the incubator (Day 1 of serum starvation).
Change medium (low serum) every 48 hours. Continue to grow the cells until Day 7 (Figure 1). On Day 7, harvest the cells and prepare cell lysate for 7-day serum-starved cells (7dSS) (see section 3.8 for modified protocol using trypsin inhibitor).
3.6 Restimulation of quiescent fibroblasts into a proliferating state
Cells made quiescent by either contact inhibition or serum starvation can be induced to re-enter a proliferative state (restimulation). This is useful for studying the effects of specific proteins and molecular pathways on the transition of cells from a quiescent to a proliferative state.
Begin with a 10 cm plate containing fibroblasts in a contact-inhibited state (see section 3.4). Detach the cells from the plate by trypsinization and collect the cell pellet (see steps 2 and 3 in section 3.2).
Resuspend the cell pellet in 1 ml of complete medium. Take the desired amount of cell suspension (e.g. 200 μl for 5X dilution) in a 15 ml tube and add complete medium so that the final volume is 10 ml. Mix well. Add the cells to a 10 cm plate and culture them under proliferation conditions (cells should be <80% confluent). The proliferating cells can be used for downstream analysis like monitoring protein expression by western blot (see section 3.7).
For restimulating serum-starved fibroblasts, take a 10 cm plate containing serum-starved cells (see section 3.5) and aspirate the low-serum medium. Wash the cells (see step 2 in section 3.2) with 5 ml of DPBS. Detach the cells by trypsinization and neutralize the trypsin with complete medium followed by pelleting the cells by centrifugation (see steps 2 and 3 in section 3.2). Dilute the cells and plate them (see step 4 in section 3.2). This process will restimulate serum-starved cells into a proliferating state. The proliferating cells can be used for downstream analysis (see section 3.8).
The confluency of serum-starved cells is less than 100%, so depending upon the application, restimulation can be performed with (see step 3 in section 3.5) or without cell dilution (adding complete medium directly to serum-starved cells without any dilution).
3.7 Preparation of cell lysates for P, 7dCI, and restimulated cells
Detach the cells from the plate with trypsinization (see Note 7) and pellet the cells (see steps 2 and 3 in section 3.2). Resuspend the pellet in 0.5–1 ml of DPBS and transfer the cell suspension to a 1.5 ml Eppendorf tube. Keep the cell suspension on ice until the next step. Centrifuge the tube at 2500 × g for 10 min to pellet the cells again. Remove the supernatant. This step will remove any residual complete medium and trypsin. If needed, the cell pellet could be flash frozen in dry ice and stored in −80 °C freezer.
Add cell lysis buffer containing protease inhibitors to the tube and pipet up and down to suspend the cell pellet completely. Incubate the cell suspension for 10 min at room temperature with gentle shaking. Check the instructions for using the cell lysis buffer if using a commercially available one. See [63] for recipes of traditional cell lysis buffers such as NP-40 and RIPA lysis buffers.
Centrifuge the tube at 14000 × g for 10 min. Remove the supernatant (cell lysate) and transfer it to a fresh Eppendorf tube. Measure the concentration of the cell lysate using BCA assay kit by following manufacturer’s instructions. Perform western blot analysis using the collected cell lysates to probe for the proteins of interest.
3.8 Preparation of cell lysates for 7dSS cells
For a 10-cm plate containing 7dSS cells, aspirate the low-serum medium and trypsinize the cells from the plate (see steps 2 and 3 in section 3.2). Add 3 ml of soybean trypsin inhibitor (instead of complete medium) to quench the activity of trypsin. The use of soybean trypsin inhibitor ensures the cells are not stimulated by serum.
Transfer the cell suspension to a 15 ml conical tube. Spin the tube at 500 × g for 5 min to pellet the cells. Remove the supernatant. Prepare cell lysates from the cell pellets (see steps 2 and 3 in section 3.7). Perform western blot analysis using these cell lysates to probe for the proteins of interest.
3.9 Studying cell cycle exit to a quiescent state by siRNA transfection
Thaw a vial of frozen fibroblasts and grow a plate of proliferating fibroblasts (see steps 5 and 6 in section 3.3). Count the cells when the confluency reaches ~80% (steps 1 and 2 in section 3.4). Check the amount of cells to be plated for different plate formats (e.g., 6-well plate or 10-cm plate) by referring to the instruction sheet provided by the manufacturer of the transfection reagent. Plate the desired amount of cells (for the chosen plate format) depending upon the experimental goals and the amount of cells required for downstream analysis.
On the next day, check to see if the cells are at the recommended confluency for transfection. Grow the cells for a longer duration or split the cells and replate to achieve the required confluency. Perform the transfection using the desired siRNAs and transfection reagent by following the manufacturer’s instructions (see Note 8). The siRNAs for the genes of interest can be obtained commercially. Usually two or more siRNAs per gene are used. At least one control siRNA should also be used (see Note 9).
Forty-eight hours after transfection (see Note 10), switch to a low-serum medium (0.1 % serum) (see steps 1 and 2 in Section 3.5) to induce cell cycle arrest. Cells should be replated at the required confluency before adding the low serum medium.
After 24 hours, pellet the cells. Prepare cell lysates for western blot analysis, if required (see Section 3.8).
3.10 Studying cell cycle entry from a quiescent state by siRNA transfection
Take a plate of proliferating cells and switch to a low-serum medium (see steps 1 and 2 in Section 3.5).
Keep the cells in low-serum medium (0.1% serum) for 24 hours or more depending upon the experimental design. Change medium every 48 hours if serum-starving the cells for more than 48 hours.
Pellet the cells and suspend the cells in 1 ml of low-serum medium (0.1% serum) (see steps 1 and 2 of Section 3.8). Count the serum-starved cells (see step 2 of Section 3.2), and plate them according to the instruction sheet for the transfection reagent (see step 1 of Section 3.9).
On the next day, perform the transfection using the transfection reagent by following the manufacturer’s instructions.
Forty-eight hours later, switch to complete medium to induce proliferation.
Twenty-four hours after performing step 5, harvest the cells for downstream analysis, such as making cell lysates for western blot (see section 3.7).
Acknowledgments
HAC was the Milton E. Cassel scholar of the Rita Allen Foundation (http://www.ritaallenfoundation.org). This work was funded by grants to HAC from National Institute of General Medical Sciences R01 GM081686, and National Institute of General Medical Sciences R01 GM0866465. HAC is a member of the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research, the Jonsson Comprehensive Cancer Center, the UCLA Molecular Biology Institute, and the UCLA Bioinformatics Interdepartmental Program.
Footnotes
The incubation time for fibroblast release from tissue will depend upon the freshness of the dispase solution. Freshly prepared solution (same day is ideal) will decrease the incubation time. Also, the powder form of the dispase used to make the solution should be less than a year old. An alternative to dispase treatment is to incubate the skin in 0.3% trypsin solution in DPBS for 10 min. If trypsin is used, agitate the solution every 2–3 min.
Fibroblasts are generally passaged or frozen while the cells are still proliferating (i.e., exponential phase). The cells should not be allowed to reach 100% confluency for proliferating samples.
Human skin is contaminated with bacteria and other microorganisms, which need to be removed by the addition of antibiotics (penicillin and streptomycin) and antimycotics (amphotericin B). For regular culturing of fibroblasts after they are isolated from skin, the antibiotics can be omitted.
Primary cells like fibroblasts can only be passaged for a limited number of times due to a decrease in proliferative potential. The cells will ultimately become senescent upon prolonged passaging. In our laboratory, we culture the fibroblasts to ~12 passages after their isolation. For the same reason, it is also good to freeze the fibroblasts at low passage number (<7). This also allows the investigator to have reserves of fibroblasts isolated from a specific skin tissue in case an experiment needs to be repeated later. The different strains of fibroblasts isolated from the skin tissue of different infants may be heterogeneous in terms of their proliferation potential and other cellular properties.
When resuspending a cell pellet, make sure to gently pipet up and down using a pipet tip a few times (10–12 times) to obtain a uniform cell suspension and to avoid cell clumping. There should not be any visible lumps present in the suspension. A uniform cell suspension will prevent formation of cell clusters when the cells are plated.
When counting the cells, use trypan blue to count the number of live cells. Using the counts for live cells, and not total cells, for estimating the cells to be plated ensures that the same number of cells are plated in different experiments. See [62] for more information on cell counting using trypan blue.
Trypsin is used to detach the cells from the plate. If trypsin cannot be used, a cell lysis buffer can be added directly to the plate containing cells [63]. If cell lysis buffer is being used, first aspirate the medium. Wash the cells with PBS and then add appropriate amount of cell lysis buffer to the plate, maintaining a small volume to ensure proteins will be present in a high concentration. Incubate the plate for 5–10 min with gentle shaking. Collect the lysate (and debris) by tilting the plate and transfer to a 1.5 ml Eppendorf tube. Centrifuge the tube at 14000 × g for 10 min. Transfer the supernatant (cell lysate) to a fresh Eppendorf tube.
A forward transfection procedure (plating the cells a day before the transfection) is described here in detail, but transfection can also be performed by following a reverse transfection protocol, i.e., transfection and plating of the cells are performed at the same time. Check the instruction manual of the transfection reagent for more information.
siRNAs are double-stranded RNAs (21–28 bp) containing 2-nucleotide overhangs at 3′ ends. A single RNA strand of the duplex containing a region that is perfectly complementary to a region of target mRNA is incorporated into a silencing complex. This complex is then involved in recognizing and cleaving the target mRNA [64,65]. Different siRNAs targeting different regions of the same gene may have different knockdown efficiencies, so it is best to use two or more siRNAs for a gene. Follow the manufacturer’s protocol for reconstituting and storing siRNAs. Avoid repeated freeze and thaw cycles by aliquoting the reconstituted siRNAs before freezing them.
It may take about 48 hours for siRNA transfection to affect the targeted gene. Knockdown of the gene of interest should be monitored 24, 48, and 72 hours after transfection using quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) or western blot to find the time for optimal knockdown levels.
References
- 1.Coller HA. Cell biology. The essence of quiescence. Science. 2011;334(6059):1074–1075. doi: 10.1126/science.1216242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daignan-Fornier B, Sagot I. Proliferation/Quiescence: When to start? Where to stop? What to stock? Cell Div. 2011;6(1):20. doi: 10.1186/1747-1028-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14(6):329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974;71(4):1286–1290. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darby IA, Hewitson TD. Fibroblast differentiation in wound healing and fibrosis. Int Rev Cytol. 2007;257:143–179. doi: 10.1016/S0074-7696(07)57004-X. [DOI] [PubMed] [Google Scholar]
- 6.Bainbridge P. Wound healing and the role of fibroblasts. J Wound Care. 2013;22(8):407–408. 410–412. doi: 10.12968/jowc.2013.22.8.407. [DOI] [PubMed] [Google Scholar]
- 7.Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA. Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 2010;8(10):e1000514. doi: 10.1371/journal.pbio.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA. H4K20 methylation regulates quiescence and chromatin compaction. Mol Biol Cell. 2013;24(19):3025–3037. doi: 10.1091/mbc.E12-07-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polioudakis D, Bhinge AA, Killion PJ, Lee BK, Abell NS, Iyer VR. A Myc-microRNA network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res. 2013;41(4):2239–2254. doi: 10.1093/nar/gks1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, Meyer T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155(2):369–383. doi: 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014;141(24):4656–4666. doi: 10.1242/dev.106575. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, Siegenthaler JA, Dowell RD, Yi R. Foxc1 reinforces quiescence in self-renewing hair follicle stem cells. Science. 2016;351(6273):613–617. doi: 10.1126/science.aad5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. 2003;24(7):380–386. doi: 10.1016/s1471-4906(03)00141-8. [DOI] [PubMed] [Google Scholar]
- 14.Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118(5):635–648. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4(3):e83. doi: 10.1371/journal.pbio.0040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukada S, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, Yamamoto H, Miyagoe-Suzuki Y, Takeda S. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells. 2007;25(10):2448–2459. doi: 10.1634/stemcells.2007-0019. [DOI] [PubMed] [Google Scholar]
- 17.Forsberg EC, Passegue E, Prohaska SS, Wagers AJ, Koeva M, Stuart JM, Weissman IL. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS One. 2010;5(1):e8785. doi: 10.1371/journal.pone.0008785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elkon R, Drost J, van Haaften G, Jenal M, Schrier M, Oude Vrielink JA, Agami R. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol. 2012;13(7):R59. doi: 10.1186/gb-2012-13-7-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suh EJ, Remillard MY, Legesse-Miller A, Johnson EL, Lemons JM, Chapman TR, Forman JJ, Kojima M, Silberman ES, Coller HA. A microRNA network regulates proliferative timing and extracellular matrix synthesis during cellular quiescence in fibroblasts. Genome Biol. 2012;13(12):R121. doi: 10.1186/gb-2012-13-12-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol. 2008;9(11):910–916. doi: 10.1038/nrm2510. [DOI] [PubMed] [Google Scholar]
- 21.Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 22.Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & Development. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 24.Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272(5263):877–880. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- 25.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 26.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3(1):11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 27.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81(3):323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 28.Smith EJ, Leone G, DeGregori J, Jakoi L, Nevins JR. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol Cell Biol. 1996;16(12):6965–6976. doi: 10.1128/mcb.16.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo J, Longshore S, Nair R, Warner BW. Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J Biol Chem. 2009;284(1):134–140. doi: 10.1074/jbc.M806133200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Sagheer AH, Brown T. Click chemistry with DNA. Chem Soc Rev. 2010;39(4):1388–1405. doi: 10.1039/b901971p. [DOI] [PubMed] [Google Scholar]
- 31.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105(7):2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson EL, Suh EJ, Chapman TR, Coller HA. Identifying Functional miRNA Targets Using Overexpression and Knockdown Methods. In: Mallick B, Ghosh Z, editors. Regulatory RNAs: Basics, Methods and Applications. Springer Berlin Heidelberg; Berlin, Heidelberg: 2012. pp. 295–317. [DOI] [Google Scholar]
- 33.Gothot A, Pyatt R, McMahel J, Rice S, Srour EF. Functional heterogeneity of human CD34(+) cells isolated in subcompartments of the G0/G1 phase of the cell cycle. Blood. 1997;90(11):4384–4393. [PubMed] [Google Scholar]
- 34.Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bickenbach JR. Identification and behavior of label-retaining cells in oral mucosa and skin. J Dent Res. 1981;60:1611–1620. doi: 10.1177/002203458106000311011. Spec No C. [DOI] [PubMed] [Google Scholar]
- 36.Morris RJ, Fischer SM, Slaga TJ. Evidence that the centrally and peripherally located cells in the murine epidermal proliferative unit are two distinct cell populations. J Invest Dermatol. 1985;84(4):277–281. doi: 10.1111/1523-1747.ep12265358. [DOI] [PubMed] [Google Scholar]
- 37.Cotsarelis G, Sun TT, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61(7):1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 38.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci U S A. 1999;96(6):3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA, Jackson TL, Morrison SJ. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. 2007;449(7159):238–242. doi: 10.1038/nature06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 41.Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in skin. Science. 2004;303(5656):359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, Hock H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27(1):84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 44.Sakaue-Sawano A, Ohtawa K, Hama H, Kawano M, Ogawa M, Miyawaki A. Tracing the silhouette of individual cells in S/G2/M phases with fluorescence. Chem Biol. 2008;15(12):1243–1248. doi: 10.1016/j.chembiol.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 45.Sakaue-Sawano A, Hoshida T, Yo M, Takahashi R, Ohtawa K, Arai T, Takahashi E, Noda S, Miyoshi H, Miyawaki A. Visualizing developmentally programmed endoreplication in mammals using ubiquitin oscillators. Development. 2013;140(22):4624–4632. doi: 10.1242/dev.099226. [DOI] [PubMed] [Google Scholar]
- 46.Bouldin CM, Kimelman D. Dual fucci: a new transgenic line for studying the cell cycle from embryos to adults. Zebrafish. 2014;11(2):182–183. doi: 10.1089/zeb.2014.0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zielke N, Edgar BA. FUCCI sensors: powerful new tools for analysis of cell proliferation. Wiley Interdiscip Rev Dev Biol. 2015;4(5):469–487. doi: 10.1002/wdev.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, Sakaue-Sawano A, Niida A, Miyano S, Aburatani H, Kiyonari H, Miyawaki A, Kitamura T. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0-G1 transition. Sci Rep. 2014;4:4012. doi: 10.1038/srep04012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1(4):193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 50.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6(12):1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 51.Vonlaufen A, Phillips PA, Yang L, Xu Z, Fiala-Beer E, Zhang X, Pirola RC, Wilson JS, Apte MV. Isolation of quiescent human pancreatic stellate cells: a promising in vitro tool for studies of human pancreatic stellate cell biology. Pancreatology. 2010;10(4):434–443. doi: 10.1159/000260900. [DOI] [PubMed] [Google Scholar]
- 52.Vag T, Schramm T, Kaiser WA, Hilger I. Proliferating and quiescent human umbilical vein endothelial cells (HUVECs): a potential in vitro model to evaluate contrast agents for molecular imaging of angiogenesis. Contrast Media Mol Imaging. 2009;4(4):192–198. doi: 10.1002/cmmi.280. [DOI] [PubMed] [Google Scholar]
- 53.Sellathurai J, Cheedipudi S, Dhawan J, Schroder HD. A novel in vitro model for studying quiescence and activation of primary isolated human myoblasts. PLoS One. 2013;8(5):e64067. doi: 10.1371/journal.pone.0064067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alberts B, Johnson A, Lewis J. Fibroblasts and Their Transformations: The Connective-Tissue Cell Family In: Molecular Biology of the Cell 4th edn. Garland Science; New York: 2002. [Google Scholar]
- 55.Nishiyama T, Akutsu N, Horii I, Nakayama Y, Ozawa T, Hayashi T. Response to growth factors of human dermal fibroblasts in a quiescent state owing to cell-matrix contact inhibition. Matrix. 1991;11(2):71–75. doi: 10.1016/s0934-8832(11)80210-6. [DOI] [PubMed] [Google Scholar]
- 56.Bridger JM, Boyle S, Kill IR, Bickmore WA. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr Biol. 2000;10(3):149–152. doi: 10.1016/s0960-9822(00)00312-2. [DOI] [PubMed] [Google Scholar]
- 57.Boraldi F, Annovi G, Paolinelli-Devincenzi C, Tiozzo R, Quaglino D. The effect of serum withdrawal on the protein profile of quiescent human dermal fibroblasts in primary cell culture. Proteomics. 2008;8(1):66–82. doi: 10.1002/pmic.200700833. [DOI] [PubMed] [Google Scholar]
- 58.Takashima A. Establishment of fibroblast cultures. Curr Protoc Cell Biol Chapter. 2001;2:1. doi: 10.1002/0471143030.cb0201s00. Unit 2. [DOI] [PubMed] [Google Scholar]
- 59.Rittie L, Fisher GJ. Isolation and culture of skin fibroblasts. Methods Mol Med. 2005;117:83–98. doi: 10.1385/1-59259-940-0:083. [DOI] [PubMed] [Google Scholar]
- 60.Villegas J, McPhaul M. Establishment and culture of human skin fibroblasts. Curr Protoc Mol Biol Chapter. 2005;28:23. doi: 10.1002/0471142727.mb2803s71. Unit 28. [DOI] [PubMed] [Google Scholar]
- 61.Huschtscha LI, Napier CE, Noble JR, Bower K, Au AY, Campbell HG, Braithwaite AW, Reddel RR. Enhanced isolation of fibroblasts from human skin explants. Biotechniques. 2012;53(4):239–244. doi: 10.2144/0000113939. [DOI] [PubMed] [Google Scholar]
- 62.Louis KS, Siegel AC. Cell viability analysis using trypan blue: manual and automated methods. Methods Mol Biol. 2011;740:7–12. doi: 10.1007/978-1-61779-108-6_2. [DOI] [PubMed] [Google Scholar]
- 63.Ji H. Lysis of cultured cells for immunoprecipitation. Cold Spring Harb Protoc. 2010;2010(8) doi: 10.1101/pdb.prot5466. pdb prot5466. [DOI] [PubMed] [Google Scholar]
- 64.Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov. 2004;3(4):318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- 65.Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bryant P, Zheng Q, Pumiglia K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol Cell Biol. 2006;26(11):4201–4213. doi: 10.1128/MCB.01612-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perucca P, Cazzalini O, Madine M, Savio M, Laskey RA, Vannini V, Prosperi E, Stivala LA. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle. 2009;8(1):105–114. doi: 10.4161/cc.8.1.7507. [DOI] [PubMed] [Google Scholar]
- 68.Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21(14):4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hur J, Choi JI, Lee H, Nham P, Kim TW, Chae CW, Yun JY, Kang JA, Kang J, Lee SE, Yoon CH, Boo K, Ham S, Roh TY, Jun JK, Lee H, Baek SH, Kim HS. CD82/KAI1 Maintains the Dormancy of Long-Term Hematopoietic Stem Cells through Interaction with DARC-Expressing Macrophages. Cell Stem Cell. 2016;18(4):508–521. doi: 10.1016/j.stem.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 70.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, Huang G, Di Giandomenico S, Koff A, Nimer SD. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8(19):3120–3124. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dey-Guha I, Wolfer A, Yeh AC, J GA, Darp R, Leon E, Wulfkuhle J, Petricoin EF, 3rd, Wittner BS, Ramaswamy S. Asymmetric cancer cell division regulated by AKT. Proc Natl Acad Sci U S A. 2011;108(31):12845–12850. doi: 10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lyublinskaya OG, Borisov YG, Pugovkina NA, Smirnova IS, Obidina JV, Ivanova JS, Zenin VV, Shatrova AN, Borodkina AV, Aksenov ND, Zemelko VI, Burova EB, Puzanov MV, Nikolsky NN. Reactive Oxygen Species Are Required for Human Mesenchymal Stem Cells to Initiate Proliferation after the Quiescence Exit. Oxid Med Cell Longev. 2015;2015:502105. doi: 10.1155/2015/502105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Key G, Becker MH, Baron B, Duchrow M, Schluter C, Flad HD, Gerdes J. New Ki-67-equivalent murine monoclonal antibodies (MIB 1-3) generated against bacterially expressed parts of the Ki-67 cDNA containing three 62 base pair repetitive elements encoding for the Ki-67 epitope. Lab Invest. 1993;68(6):629–636. [PubMed] [Google Scholar]
- 74.Osada S, Minematsu N, Oda F, Akimoto K, Kawana S, Ohno S. Atypical Protein Kinase C Isoform, aPKClambda, Is Essential for Maintaining Hair Follicle Stem Cell Quiescence. J Invest Dermatol. 2015;135(11):2584–2592. doi: 10.1038/jid.2015.222. [DOI] [PubMed] [Google Scholar]
- 75.Reed SA, Ouellette SE, Liu X, Allen RE, Johnson SE. E2F5 and LEK1 translocation to the nucleus is an early event demarcating myoblast quiescence. J Cell Biochem. 2007;101(6):1394–1408. doi: 10.1002/jcb.21256. [DOI] [PubMed] [Google Scholar]
- 76.Wang HC, Fecteau KA. Detection of a novel quiescence-dependent protein kinase. J Biol Chem. 2000;275(33):25850–25857. doi: 10.1074/jbc.M000818200. [DOI] [PubMed] [Google Scholar]
- 77.Blanpain C, Simons BD. Unravelling stem cell dynamics by lineage tracing. Nat Rev Mol Cell Biol. 2013;14(8):489–502. doi: 10.1038/nrm3625. [DOI] [PubMed] [Google Scholar]