

Abstract
Rationale
Ambient temperature is a risk factor for cardiovascular disease. Cold weather increases cardiovascular events, but paradoxically, cold exposure is metabolically protective due to UCP1-dependent thermogenesis.
Objective
We sought to determine the differential effects of ambient environmental temperature challenge and UCP1 activation in relation to cardiovascular disease progression.
Methods and Results
Using mouse models of atherosclerosis housed at three different ambient temperatures, we observed that cold temperature enhanced while thermoneutral housing temperature inhibited atherosclerotic plaque growth, as did deficiency in UCP1. However, while UCP1 deficiency promoted poor glucose tolerance, thermoneutral housing enhanced glucose tolerance, and this effect held even in the context of UCP1 deficiency. In conditions of thermoneutrality, but not UCP1 deficiency, circulating monocyte counts were reduced, likely accounting for fewer monocytes entering plaques. Reductions in circulating blood monocytes were also found in a large human cohort in correlation with environmental temperature. By contrast, reduced plaque growth in mice lacking UCP1 was linked to lower cholesterol. Through application of a positron emission tomography (PET) tracer to track CCR2+ cell localization and intravital 2-photon imaging of bone marrow, we associated thermoneutrality with an increased monocyte retention in bone marrow. Pharmacological activation of β3 adrenergic receptors applied to mice housed at thermoneutrality induced UCP1 in beige fat pads but failed to promote monocyte egress from the marrow.
Conclusions
Warm ambient temperature is, like UCP1 deficiency, atheroprotective, but the mechanisms of action differ. Thermoneutrality associates with reduced monocyte egress from the bone marrow in a UCP-1 dependent manner in mice and likewise may also suppress blood monocyte counts in man.
Keywords: Migration, thermogenesis, bone marrow, atherosclerosis, positron emission tomography, atherogenesis, adrenergic receptor, macrophage
Subject Terms: Animal Models of Human Disease, Inflammation, Lipids and Cholesterol, Metabolism, Vascular Biology
INTRODUCTION
Atherosclerosis remains one of the most common diseases in the United States, despite impressive gains in recent decades at reducing its prevalence. Atherosclerotic plaques are characterized by an accumulation of macrophages, lipids, smooth muscle cells and fibroblasts, and in advanced stages, necrotic debris within the arterial wall. The initial injury of the intimal monolayer of endothelial cells triggers the secretion of pro-inflammatory mediators that activate the innate and adaptive immune system and facilitate progression of the pathology1, 2. Monocyte recruitment from the blood circulation plays a crucial role in giving rise to plaque macrophages that drive plaque progression3, 4, and such recruitment along with macrophage proliferation and survival in the atherosclerotic plaque regulates overall macrophage plaque burden5–9.
Given that monocytes are recruited into plaques from blood, it is perhaps not surprising that blood monocyte counts appear to impact disease progression. That is, monocytosis and neutrophilia are well-established risk factors for disease evolution in humans10, 11, and data from preclinical mouse models demonstrated that blood monocyte levels correlate with the rate of cell recruitment to the plaque4, 12. One mechanism by which monocyte counts in the circulation are regulated is through sympathetic nerve activation in the bone marrow that regulates release of newly formed monocytes, explaining one link between stress and cardiovascular events13–15.
Sympathetic nerve activation is also well known to regulate adipose depot status, with particular impact on thermogenesis within brown and beige fat due to expression of uncoupling protein 1 (UCP1) that generates heat by uncoupling electron transport from ATP generation in the electron transport chain16. Induction of UCP1 expression and activity in brown and beige fat is often discussed as a desirable endpoint to combat obesity and insulin resistance17–20. However, for this approach to be viable, unwanted effects must be avoided. Yet, some reports indicate that cold temperature, a powerful stimulus for brown and beige fat and UCP1 activation, enhances atherosclerosis21. However, other studies suggest that thermoneutral housing at the warm temperature of 30°C—the temperature at which energy expenditure is not needed to maintain body temperature—enhances atherosclerosis22. The impact of housing temperature on blood monocyte counts has scarcely been discussed, despite reported connections of sympathetic activation to both beige fat activation and monocytosis, as discussed above.
From an epidemiological perspective, cold ambient temperature is associated with cardiovascular events23–27 and may impact blood pressure, triacylglycerides and plasma cholesterol28. However, overall, how ambient temperature affects cardiovascular disease remains unclear and whether the link is simply associative and indirect is unknown. Given the momentum behind the concept that cold exposure may be beneficial due to brown and beige fat activation and the potential health effects of such an approach, we here examined the impact of various housing temperatures on experimental atherosclerosis and addressed whether ambient temperature regulates monocyte counts in the blood. The data arising from this work indicate that cold exposure potentiates atherosclerosis. We show that thermoneutral housing protects against atherosclerosis development through, at least in part, regulation of monocyte recruitment from the bone marrow. We distinguish this mechanism from the action of UCP1 in regulating disease burden and glucose homeostasis.
METHODS
Mice
Mice were housed in specific pathogen-free animal facilities, maintained by Washington University School of Medicine. 6-week old B6 (C57Bl/6J, #000664), and Ldlr−/− (B6.129S7-Ldlrtm1Her/J, #002207) mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA). Apoe−/− (B6129P2-Apoetm1Unc/J, #002052), Cxcl12dsred (Cxcl12tm2.1sjm/J, #022458),) Cx3cr1gfp (B6.129P(Cg)-Ptprca Cx3cr1tm1litt/LittJ, #008451), and Ucp1−/− (B6.129-Ucp1tm1Kz/J, #003124) mice were purchased from Jackson Laboratories, then maintained at Washington University School of Medicine. High fat diet (Teklad TD.88137) containing 21% milk fat and 0.15% cholesterol was used for atherosclerosis studies. For cold challenge and thermoneutral housing, littermates were born at 22°C and transferred to rooms maintained continuously at 4°C or 30°C when indicated. The 30°C room was a standard room within the animal facility barrier in the same hallway as the 22°C room. By contrast, 4°C housing occurred in a cold room outside the animal facility barrier that was adapted for mouse housing and maintained with monitored standard air flow. Mice were allowed to equilibrate at new housing conditions for 7 days prior to the start of experiment or diet treatments. All facilities, inside or outside the barrier, were maintained on identical 12-hour light cycles. All experimental procedures were approved by the Washington University School of Medicine Animal Studies Committee.
Human blood analysis
Human studies were reviewed and approved by the Institutional Review Board (IRB) of the Washington University in St. Louis/Barnes-Jewish Hospital (main adult teaching hospital for Washington University School of Medicine) under protocol #201703135. These human studies were a retrospective analysis of existing data in the electronic medical record, and the IRB protocol included an approved informed consent waiver. Peripheral blood complete blood counts (CBC) with automated differentials were extracted from the electronic medical record system for outpatients tested at Barnes-Jewish Hospital (or regional affiliate clinics and hospitals) with blood draws during the second-to-third week of each month beginning March, 2016 and ending February, 2017. Monthly mean ± standard error of the mean (SEM) absolute cell counts were determined for monocytes, lymphocytes, neutrophils, eosinophils, and basophils and correlated against mean ambient temperature recorded at the Saint Louis Lambert International Airport weather station during the same time periods as the blood draws. CBC data included the age and sex of the patient, but not other factors such as ethnicity/race or disease status.
These studies were designed to have 85% power to detect a 10% difference in monocyte counts between two separate three-month periods, assuming an overall mean monocyte count of 0.5 × 103 cells/μL, equal standard deviations of 1.0 × 103 cells/μL for any given three-month period, and significance level of P<0.05. Power calculations suggested we needed to collect data for approximately 1,000 samples per month. Actual sample sizes were n=1,093 (March 2016), n=1,251 (April 2016), n=1,158 (May 2016), n=1,293 (June 2016), n=1,251 (July 2016), n=1,199 (August 2016), n=1,276 (September 2016), n=1,177 (October 2016), n=1,191 (November 2016), n=1,317 (December 2016), n=2,274 (January 2017), and n=1,036 (February 2017). Pearson linear regression was performed to test whether monthly mean cell counts correlated with monthly mean temperatures. Student’s t-tests were performed to compare average cell counts in the winter (December-February) versus summer (June–August).
Flow cytometry
Blood samples were collected from the superficial temporal vein. Then red blood cells were lysed (Pharmlyse, BD) and FACS staining was carried out. Bone marrow was extracted from the femurs and tibias. Briefly, the bones were flushed with PBS 1X and then tissue was disrupted using a 21G needle. Red blood cells were lysed and the samples were counted (Cellometer X4, Nexcelom Biosciences) and processed for staining. Samples were FcR-blocked (2.4G2, Biolegend) then stained on ice with specific antibodies; CD11b APC-Cy7 conjugated (M1/70, BD bioscience), CD115 APC conjugated (AFS98, eBioscience), Ly6C PerCP-Cy5.5 conjugated (HK1.4, Biolegend), Ly6G FITC conjugated (1A8, BD Biosciences), CD43 PE conjugated (1B11, Biolegend), CCR2 PE conjugated (475301, R&D systems), CD45 Pacific Blue conjugated (30-Fl1, Biolegend), Lin staining [CD3 PE-cy7 conjugated (145-2C11, Biolegend), CD19 PE-cy7 conjugated (eBio1D3, ebioscience), B220 PE-cy7 conjugated (RA3-6B2, eBioscience), Ter119 PE-cy7 conjugated (Ter-119 eBioscience), NK1.1 PE-cy7 conjugated (PK136, eBioscience)], CD150 PE conjugated (TC15-12Fl2.2, Biolegend), CD48 APC conjugated (HM48-1, eBioscience), c-Kit APC-efluor780 conjugated (2B8, eBioscience), Sca1 PerCP-cy5.5 conjugated (D7, eBioscience), and BrdU Fitc conjugated (Bu20a, eBioscience). Samples were run on BD LSRFortessa instrument and analysis performed using FlowJo 10 (Treestar). Monocytes were identified as CD45+ CD11b+ Ly6G− CD115+; then assayed for expression of Ly6C, CCR2, CD43, or CX3CR1. Neutrophils were identified as CD45+ CD11b+ Ly6G+ cells. Bone marrow progenitor cells were identified by expression of Lin- (CD3− B220− Gr1− Ter119−) Sca+ c-Kit+ and then subdivided according to the expression of CD150 and CD4829. Bone marrow monocytes were identified by CD45+ CD115+ Ly6G− CCR2+ Ly6C+. For proliferation assays, 1 mg BrdU (ebioscience, 00-4440-51A) was given i.p. 24 hours prior to sacrifice. For detection of BrdU, samples were labeled extracellularly, then fixed/permeabilized using BrdU staining kit (eBioscience, 14330) followed by incubation with 30 μg DNase I (eBioscience, 00-4425-10) and anti-BrdU (or isotype control) antibody staining.
Two-photon imaging
Mice were anesthetized by i.p. injection of ketamine (50 mg/kg) and xylazine (10 mg/kg), then maintained with halved doses administered every approximately 45 minutes during imaging. Animals were prepared for bone marrow imaging following a modified preparation that was previously published, without the creation of a skull-window30. Briefly, hair was shaved and skin flap cut opened. Images were collected at Washington University using a Leica SP8 2-photon microscope, equipped with a MaiTai HP DeepSee Laser (SpectraPhysics) tuned to 900 nm, and a 25x water-dipping objective. Images were collected every 30 seconds, with approximately 25 z-planes (2.5 μm step size) for each sample.
Counting of CXCL12 DsRed and CX3CR1 GFP green voxels
To count the voxels from the red and green channels, videos were initially dye separated using Leica software. In Imaris, five time frames from each video were randomly selected with the random number generator provided by www.random.org. Using Fiji, the green and red channels of each time frame were then saved separately as tiff image stacks. In Matlab, the number of voxels counted for each image stack were those above a pixel intensity threshold of 75, determined by the high background generated by the second harmonic signal. A ratio CX3CR1/CXCL12 of the voxel counts was then created to compare between conditions.
Glucose tolerance
Mice were fasted for 12 hours, then injected with 10 μL/g (20% Glucose) i.p. Blood glucose was measured (Glucocard, Vital) from the tail vein at 0, 15, 30, 45, 60, 90, and 120 minutes following glucose administration. Graphical representations were generated using GraphPad Prism 7 and the area under the curve for each animal was calculated.
Monocyte recruitment
To label classical Ly6C+ monocytes, mice were first injected i.v. with a single 200 μL dose of clodronate-loaded liposomes to deplete total circulating monocytes. Animals were then rested for 3 days and injected i.v with 200 μL of sterile fluoresbrite green (YG) 1 μm latex beads (Polysciences, Inc), diluted 1:4 from stock solution. Ly6C+ monocyte bead labeling was confirmed by flow cytometry of the blood and recruitment of Ly6C+ monocytes into plaques were assayed 2 days following bead transfer.
Body temperature
Animal body temperature was measured by rectal probe thermometer (Extech Instruments, EasyView 10).
Plaque analysis
Hearts were collected and immediately fixed in 4% paraformaldehyde containing 30% sucrose. Hearts were then embedded in OCT and cryosections (10 μm) were prepared through the aortic sinus. Samples were stained with Oil Red O to determine lipid deposition or immunostained with anti-CD68 to quantify macrophage content. Sections were blocked with 5% donkey serum in the presence of 0.1% Tx-100. Macrophages were identified in aortic sinus by CD68 (Bio-Rad) staining followed by secondary staining with anti-rat Cy3 conjugated Ab (Jackson Laboratories). CD68+, oil-red O, and plaque area was measured using image J analysis software. For en face analysis, aorta were fixed with 4% paraformaldehyde, adipose tissue was carefully removed, and the arteries were pinned onto wax dishes. Aortae were pretreated with 100% propylene glycol (sigma #P4347) for 15 minutes, then incubated with oil red o (sigma #01516) for 3 hours at room temperature. Aortae were washed with 85% propylene glycol, then washed multiple times in PBS. Aortae were imaged on a Zeiss dissecting microscope and plaque area measurements were made using Image J analysis software.
PET Imaging
For PET imaging, 0–60 min dynamic PET/CT scan was performed following injection of 64Cu-DOTA-ECL1i (3.7 MBq in 100 μL saline) via tail vein with Inveon PET/CT system (Siemens, Malvern, PA). The PET images were reconstructed with the maximum a posteriori algorithm and analyzed by Inveon Research Workplace. The organ uptake was calculated as percent injected dose per gram (%ID/g) of tissue in three-dimensional regions of interest without the correction for partial volume effect31.
Bio distribution of PET tracers
The CCR2 specific PET tracer of 64Cu-DOTA-ECL1i and CXCR4 targeted PET tracer of 64Cu-AMD3100 were synthesized following our previous reports31–34. For biodistribution studies, about 370 KBq of 64Cu-DOTA-ECL1i or 64Cu-AMD3100 in 100 μL saline (APP pharmaceuticals, Schaumburg, IL) were injected into the mice via tail vein (n= 4/group). The mice were anesthetized with inhaled isoflurane during injection and re-anesthetized before euthanasia by cervical dislocation at 1 hour post injection (n = 4/group). Organs of interest were collected, weighed, and counted in a Beckman 8000 gamma counter (Beckman, Fullerton, CA). Standards were prepared and measured along with the samples to calculate the percentage of the injected dose per gram of tissue (%ID/gram).
Beta3 agonist treatment
CL-316,243 (Sigma C5976), a β3-specific adrenoceptor agonist, or saline was administered twice-daily (1 mg/kg/day) by intraperitoneal injection for 4 weeks as previously described35, 36.
Quantitative PCR
Tissues were totally disrupted using homogenizer probe in Trizol LS (ThermoFisher Scientific, #10296010). Total RNA was extracted with chloroform followed by additional purification on QIAgen RNeasy mini columns (Product #74104) following the manufacturer’s instructions. RNA quantity was assessed on Nanodrop spectrophotometer and cDNA was prepared from 1 μg RNA using the SuperScript III reverse transcriptase kit (ThermoFisher Scientific, #18080044). The following primers (Table 1) were designed using Universal Probe Library (Roche) and used for amplification in triplicate assays with annealing temperature of 60°C. For qPCR amplification Takara SYBR mix (Takara, SYBR Premix Ex Taq II, #RR820A) was used and the assays were performed on Eppendorf cycler. For normalization between samples qPCR amplification of Hprt was performed and analyzed. ΔCt values were calculated from the raw cycle threshold (Ct values) obtained from Hprt mRNA and from the Ct values obtained for the other analyzed genes.
| Gene Target | Forward | Reverse |
| Hprt | GCTATAAATTCTTTGCTGACCTGCTG | AATTACTTTTATGTCCCCTGTTGACTGG |
| Ucp1 | GGCCTCTACGACTCAGTCCA | TAAGCCGGCTGAGATCTTGT |
Food intake protocol
For food intake studies mice were single housed for 3 days. Every day at the same hour the remaining food in the cage was weighed and recorded. Mice had unlimited access to food and water during the study, independently of the housing environmental temperature.
Plasma analysis
Mouse plasma was collected from whole blood with 5mM EDTA to limit clotting. Plasma was either used immediately or stored at −80°C until use. Total cholesterol was determined using Dako Cholesterol-E kit (product #439-17501) following manufacturer’s protocol. Plasma corticosterone was determined by ELISA following manufacturer’s protocol (Abcam #ab108821). Plasma triacylglyceride content (Infinity Triglyceride Reagent, Fisher Scientific) and non-esterified free fatty acid content (NEFA Reagents, Wako Chemicals) were analyzed by the Diabetes Model Phenotyping Core in the Diabetes Research Center at Washington University following manufacturer’s protocols. Plasma lipoprotein content was determined by mini-high-performance liquid chromatography as previously described37. In short, a LaChrom Elite HPLC system (Hitachi High Technologies) consisting of an L-2200 Autosampler with Peltier cooling, an L-2420 UV-Vis Detector, and two L-2100 SMASH pumps was used to administer ~15 mg of cholesterol from thawed plasma. Output was analyzed with ChromPerfect Spirit Chromatography Data System, version 5.5.
Statistical analysis
The statistical significance of differences in mean values was analyzed by unpaired two-tailed Student’s t test, using Prism software. P values <0.05 were considered statistically significant. Error bars show the standard error of the mean (SEM).
RESULTS
Effects of thermoneutrality on progression of atherosclerosis
To determine how environmental housing temperature influenced atherosclerosis progression, we housed male or female cohorts of Ldlr−/− mice fed a cholesterol-rich diet in rooms at different ambient temperatures: 4°C, 22°C or 30°C. In males and females, food intake greatly varied between temperature groups, with cold-challenged mice consuming ~6 g of food/day, mice housed at RT ~3 g/day, and 30°C housed animals consuming only ~2g/day (Figure 1A). Core body temperature was also remarkably distinct between mice housed at different ambient temperature, positively correlating with food consumption (Figure 1A). Mice housed at 4°C had the highest core temperature, whereas mice living at 30°C had the lowest body temperature (Figure 1A). Cold-challenge activated thermogenesis programs in brown adipose sites, and mice living at 4°C actively induced Ucp1 mRNA compared with mice housed at 22°C (Figure 1B). Ucp1 expression was below the level of detection when mice were housed at 30°C.
Figure 1. Ambient temperature regulates atherosclerosis progression.
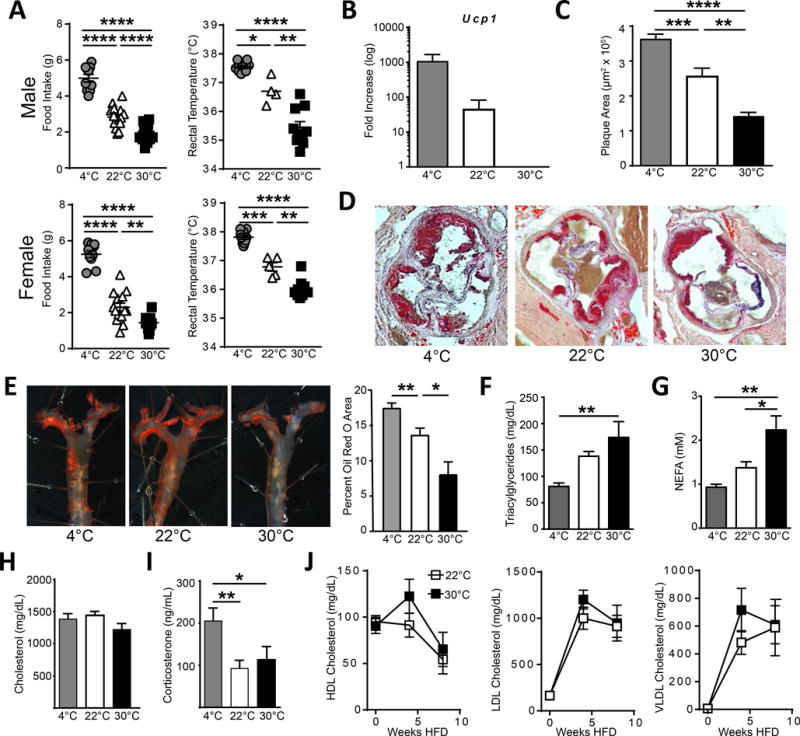
Male (top) and female (bottom) Ldlr−/− mice were housed at the indicated ambient temperature and fed HFD for four weeks, then assayed for (A) food intake (left), rectal temperature (right), and (B) UCP1 mRNA expression in brown adipose pads. C) Atherosclerotic plaque area in the aortic sinus of Ldlr−/− mice was measured following 8 weeks HFD treatment at the indicated environmental temperature. D) Representative oil red o staining to highlight regions of lipid deposition. E) Representative en face plaque staining of aortic arches and quantification following 8 weeks HFD. Triacylglycerides (F), nonesterified free fatty acids (G), and total cholesterol levels (H) were measured in the plasma from Ldlr−/− mice following 8 weeks HFD at the indicated temperatures. I) Corticosterone levels in the plasma of Ldlr−/− mice fed HFD for 8 weeks. J) Kinetic analysis of plasma lipoproteins HDL, LDL, and vLDL at zero, four, and eight weeks of HFD feeding at indicated temperatures. All experiments are representative experiments with 5–10 animals per group, repeated in two or three independent experiments. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Analysis of plasma lipoprotein composition did not uncover any significant differences in the patterns of HDL, LDL, or VLDL over time after the initiation of HFD (Figure 1H). Thus, it was unlikely that altered cholesterol metabolism accounted for the decreased plaque area in animals housed at 30°C. We conclude that thermoneutrality reduces atherosclerosis progression and does so while simultaneously reducing energy consumption and energy demand for body temperature, suggestive of a lowered whole-body metabolism.
Ambient temperature regulates monocyte recruitment to plaque
To test whether changes in plaque composition were also correlated with reduced macrophage content, we stained for CD68+ area in the aortic sinus (Figure 2A). Total macrophage (CD68+) area was consistent with total plaque measurements and reduced in Ldlr−/− mice at thermoneutral housing following 8 and 12 weeks of HFD (Figure 2A, B, and Online Figure I G). Reductions in plaque macrophages could be due to a variety of possibilities, including reduced recruitment of monocytes under thermoneutral conditions.
Figure 2. Monocyte recruitment into plaque is reduced in mice housed at thermoneutral environment.
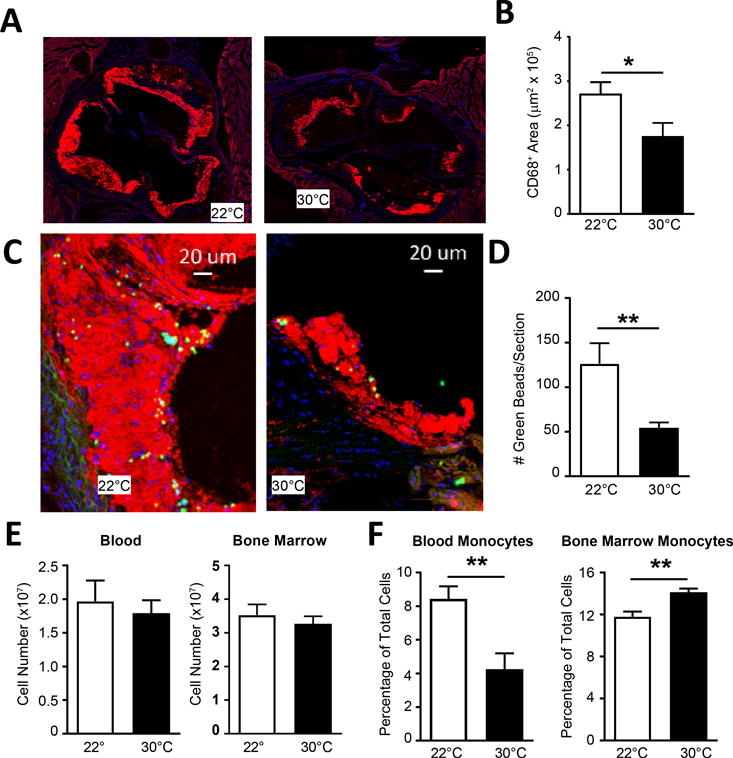
A) CD68 staining of aortic sinus of Ldlr−/− mice following 8 weeks HFD at 22°C or 30°C ambient temperature challenge, with quantification (B). C) Following clodronate liposome and bead labeling of Ly6C+ monocytes, Ldlr−/− mice on HFD were assayed for bead deposition into plaque regions of the aortic sinus, and (E) quantification of beads recruited per section. E) Total blood and bone marrow cellularity was measured following bead labeling. C) Percentage of blood and bone marrow monocytes were calculated by flow cytometry. (A-D) Data presented are combined between 3 independent experiments and include n≥10 animals per group. (E-F) Data include 4–5 animals per group and are representative of three independent experiments. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01.
We thus examined whether monocyte recruitment into plaques was reduced in animals housed at 30°C. We employed a technique that we previously developed to quantify monocyte recruitment into plaque4, 37 involving pulse-labeling of monocytes with latex particles, in which the entry of latex particle+ monocytes into plaque requires recruitment in a manner that is pertussis toxin sensitive37 and reliant on CCR24. Ldlr−/− mice housed at 22°C or 30°C and fed HFD for eight weeks were treated with clodronate-loaded liposomes to synchronize blood monocytes and deplete nonclassical Ly6C− monocytes, then 3 days later given beads i.v. to assess Ly6C+ monocyte recruitment into plaques. When we examined plaque sections for the appearance of latex bead+ monocytes (Figure 2C), bead accumulation at 30°C was only about 1/3 of that observed in sections from mice housed at 22°C (Figure 2D). Total leukocytes in blood and bone marrow were similar between the groups of mice housed at different temperatures following the labeling protocol (Figure 2E). However, Ly6C+ monocytes were dramatically reduced in the blood by approximately half and increased in the bone marrow (Figure 2F). Collectively, the data indicate that ambient temperature attenuates the recruitment of monocytes to the plaque, and further suggests the tendency of monocytes to emerge from the bone marrow in atherogenic mice is reduced when the mice are housed at 30°C. This potential retention in the bone marrow could promote a reduction in circulating monocytes and fewer monocytes being recruited to atherosclerotic plaques to promote plaque progression. We next addressed the possibility that monocytes in the circulation were reduced at higher ambient temperature, without use of monocyte ablation with clodronate-loaded liposomes.
Thermoneutral ambient temperature leads to decreased number of circulating monocytes, their accumulation in bone marrow, and reduced recruitment to atherosclerotic plaques
While total leukocytes were unchanged in the blood (Figure 3A), even in the absence of prior monocyte ablation, Ly6Chi monocytes (CD115+ CDb11+ Ly6Chi CD43− Ly6G−), gated as shown in Online Figure II, were reduced in Ldlr−/− mice on chow or following 6 weeks of HFD when housed at 30°C compared to housing at 22°C housing (Figure 3B). This drop was also observed in Apoe−/− mice housed at higher ambient temperatures and fed a HFD (Figure 3C). The counts of Ly6C− monocytes (CD115+ CD11b+ Ly6Clo CD43+ Ly6G−) also showed a shift in cell numbers in the blood (Figure 3B, C). Next, we performed BrdU labeling and chase experiments to track monocyte appearance into blood from the bone marrow. Animals housed at 22°C or 30°C and fed a HFD for 4–8 weeks received a single bolus of BrdU i.p., then were analyzed at 24 hours for blood monocyte labeling over this period of time. Representative intracellular BrdU flow cytometric staining of Ly6C+ monocytes is shown in Figure 3D. In addition to the total cell numbers of Ly6C+ monocytes being reduced (Figure 3B, C), both Ldlr−/− and Apoe−/− mouse strains on HFD showed a modest, but significant reduction in the percentage of BrdU+ monocytes that appeared in the blood following BrdU injection when housed at 30°C versus 22°C (Figure 3E).
Figure 3. Thermoneutral housing reduces circulating blood monocytes.
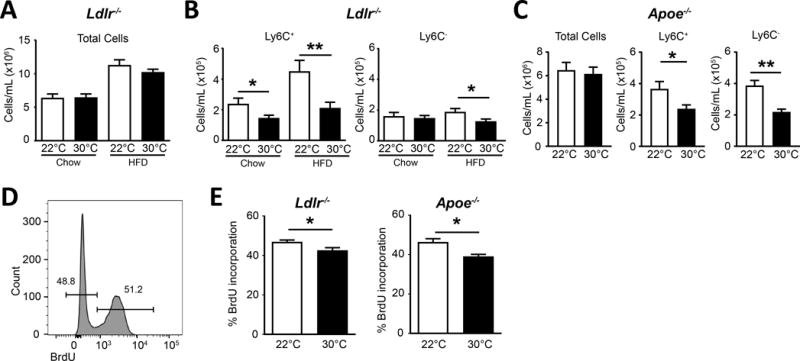
A) Ldlr−/− mice were housed at indicated temperatures and measured for total blood leukocytes on chow or HFD. B) Circulating Ly6C+ and Ly6C− monocyte number on chow or after 6 weeks of HFD treatment. C) Apoe−/− fed HFD for four weeks and assayed for total blood cell and monocyte subset numbers. D) Representative flow histogram of BrdU incorporation in Ly6C+ monocytes in the blood 24 hours following i.p. injection of 1 μg BrdU. E) BrdU incorporation in circulating Ly6C+ blood monocytes 24 hrs post BrdU injection i.p, in strains Ldlr−/− (left) and Apoe−/− (right). A-C) Data presented are combined between ≥3 independent experiments and include n≥10 animals per group. D-E) are representative experiments containing n≥4 animals per group and repeated two times each. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01.
These data prompted us to consider the possibility that monocytes accumulated in the bone marrow and were relatively less prone to enter the blood circulation. To quantify net accumulation of monocytes in the bone marrow of Ldlr−/− mice, we took advantage of a PET tracer of 64Cu-DOTA-ECL1i that specifically binds to the monocyte-selective chemokine receptor CCR232 that in turn mediates monocyte egress from bone marrow39. One advantage of the PET tracer is that most body organs can be examined simultaneously, curbing any potential selection bias with respect to the evaluation of sites where tracer might accumulate. When atherosclerotic plaque-bearing Ldlr−/− mice housed at 22°C or 30°C were imaged with 64Cu-DOTA-ECL1i, three tissues in the body showed altered patterns at the different housing temperatures. First, there was reduced tracer accumulation in the aortic arch (Figure 4A, B, Online Figure III), fitting with the reduced macrophage content in plaque previously observed (Figure 2A, B). We also observed reduced tracer uptake in brown fat (Figure 4C). Finally, bone marrow showed augmented probe accumulation (Figure 4C), indicating accumulation of CCR2+ cells, the vast majority of which are monocytes. To further investigate the concept of bone marrow retention in animals housed at 30°C we utilized another PET tracer, 64Cu-AMD3100, designed to detect CXCR440, a chemokine receptor established to promote bone marrow retention41. Biodistribution of the tracer was measured across tissues and indicated only a differential accumulation of CXCR4+ cells in the bone marrow of Ldlr−/− housed at 30°C, and no other tissue (Figure 4D), supporting the concept that 30°C housing prevents egress from the marrow.
Figure 4. PET/CT imaging identified differential CCR2+ cells in aortic arch and bone marrow of mice at thermoneutral housing.
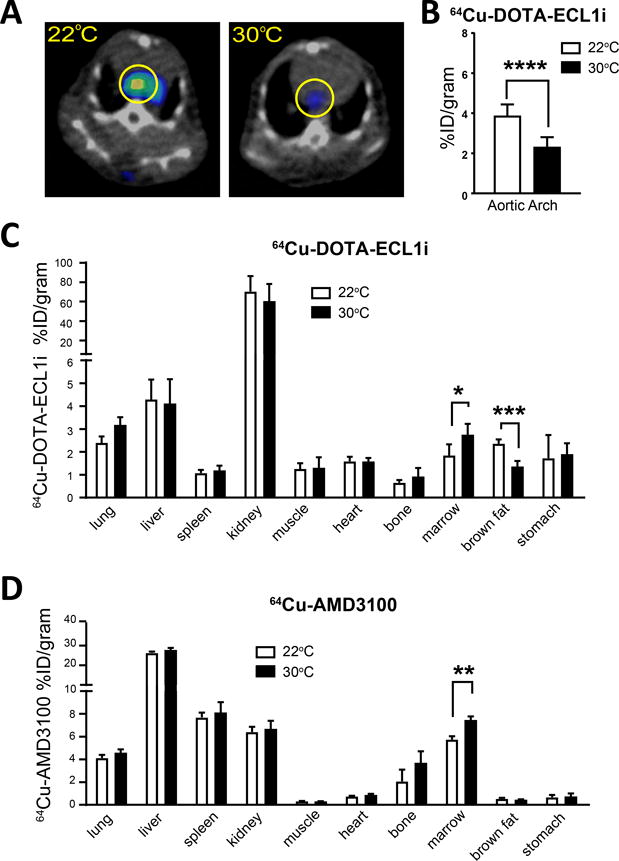
A) 64Cu-DOTA-ECL1i PET/CT images showing specific uptake at aortic arch of Ldlr−/− mice on HFD and housed at indicated ambient temperature. B) Quantification of tracer uptake in the aortic arch of Ldlr−/− mice housed in the indicated temperature. Biodistribution of C) 64Cu-DOTA-ECL1i and (D) CXCR4 tracer 64Cu-AMD3100 in Ldlr−/− mice fed HFD and housed at the indicated ambient temperature. All experiments were performed on 4–8 animals for each group and repeated in two independent experiments. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Impact of thermoneutrality in wild-type C57BL/6 mice in the absence of hypercholesterolemia
We asked next if the retention of monocytes in bone marrow was restricted to conditions of hypercholesterolemia that occured in atherogenic Apoe−/− and Ldlr−/− mouse strains. When we housed normocholesterolemic C57BL/6 wildtype mice at 22°C and 30°C, Ly6C+ monocytes were modestly but nonetheless significantly reduced in the blood of mice housed at 30°C (Figure 5A) compared to the major shifts observed in hypercholesteremic animals. Reduced monocyte egress from the marrow was also evident (Figure 5B) when using the same 24-hour BrdU labeling approach described in Figure 3.
Figure 5. Accumulation of bone marrow monocytes in mice housed at thermoneutrality.
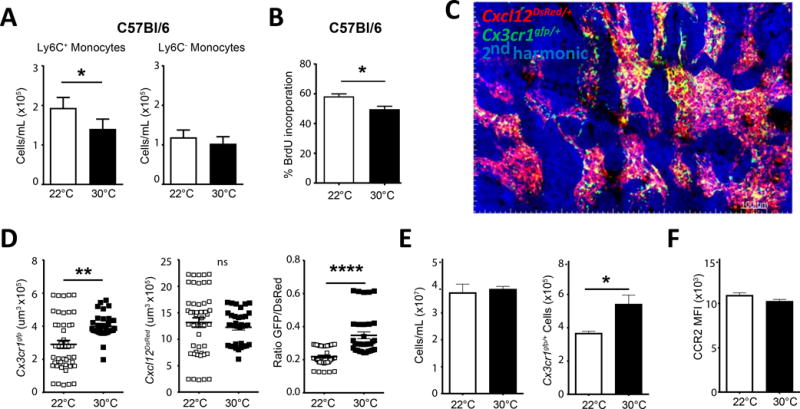
A) C57bl/6 mice were housed at 22°C or 30°C environment for seven days, then assayed for circulating monocytes (Ly6C+ and Ly6C−). B) C57bl/6 mice housed at indicated temperatures were treated with BrdU i.p. for 24hrs, then assayed for incorporation of BrdU in Ly6C+ monocytes to measure bone marrow egress C) Snapshot from 2-photon imaging approach of bone marrow in Cx3cr1gfp reporter mice crossed with Cxcl12dsred to examine monocytes within the bone marrow. D) Measurement of Cx3cr1gfp expression per frame, Cxcl12dsred expression per frame, and ratio of GFP/Dsred in bone marrow from animals housed at indicated temperature. E) Flow cytometeric analysis of Cx3cr1gfp/+ bone marrow total cellularity and GFP+ monocytes housed at indicated ambient temperature. F) CCR2 MFI for bone marrow monocytes from Cx3cr1gfp mice housed at 22°C or 30°C. (A-B, E-F) Data are representative experiments from three independent experiments with n≥5 per group. C) Image is a representative image of greater than 5 independent experiments. D) Data are combined from 5 independent experiments for each group. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Using an intravital imaging approach, we were also able to image monocytes in the bone marrow of normocholesterolemic mice in the dual reporter Cx3cr1gfp/+ Cxcl12DsRed/+ mouse (Figure 5C). Monocytes in this mouse are GFP-expressing42, and consistent with the concept that thermoneutral housing promotes monocyte retention in the bone marrow, we detected augmented GFP expression in the bone marrow compartment, defined as Cxcl12DsRed/+ area’ in mice housed at 30°C (Figure 5D and Online Movie I). By flow cytometry, there were similar total cell numbers but an increased number of CX3CR1gfp-expressing cells in the bone marrow of animals housed at 30°C (Figure 5E). However, the number of stem cells, identified as Lin− Sca1+ CD117(c-Kit)+, then separated into unique precursors by CD150 and CD48 expression cells29 (gating strategy in Online Figure IV A), was unaffected (Online Figure IV B,C). Importantly, we observed equivalent expression levels of CCR2 protein on the surface of the bone marrow monocytes, suggesting that regulation of the CCR2 receptor is not the mechanism by which monocytes are being retained in the marrow (Figure 5F, gating strategy Online Figure IV D). We conclude that monocyte retention in the bone marrow occurs when mice are housed at 30°C regardless of the presence or absence of hypercholesterolemia.
Since circulating monocyte levels are associated with vascular inflammation and reduced levels are associated with reduced risk to vascular disease, we hypothesized that a thermoneutral environment may be beneficial to overall systemic health. Mice housed at 30°C since 6 weeks of age, survived in a lifespan study a significantly longer time compared to littermate controls housed at 22°C environments (Online Figure V). The average lifespan of control animals was approximately 758 days, which is consistent with previous reports43, while almost all mice housed at thermoneutral conditions lived past 1000 days. This is a small cohort of mice, however, it suggests that modest changes in overall metabolism and inflammatory mediators may have major impact on the lifespan of the mouse.
Environmental temperature influences circulating monocytes in humans
We sought to determine whether environmental temperature correlated with circulating monocyte counts in the human patients. We obtained CBC data with automated differentials from outpatient samples over the course of a full year, with approximately 1200 samples per month, and a total of 15,516 samples derived from the Saint Louis, Missouri region (38°37′37.21″N). First we compared the average monocyte counts per month (Figure 6A), and found that monocyte counts indeed changed in a seasonal manner. These seasonal changes were independent of patient age or gender (Online Figure VI A, B). Strikingly, monocyte counts in peripheral blood were 7.4% higher in winter months compared to summer months (Online Figure VI C). Mean monthly high, average, and low environmental temperature for the region where the hospital was located confirmed that winter months had the lowest average daily temperature and summer had the highest average daily temperature (Figure 6B). Linear regression analysis indicated an inverse correlation between average human peripheral blood monocyte counts and the mean environmental temperature (Figure 6C). We were initially concerned that the elevation in monocyte counts over the winter months might link to an increase in incidence of infection. However, all other circulating cell types, including neutrophils, eosinophils, basophils, and lymphocytes were not correlated with environmental temperature (Figure 6D), and neutrophils or lymphocytes would have been expected to be most sensitive to bacterial or viral infection, respectively. Thus, a common thread for both mouse and human data is that monocyte counts are selectively altered in relation to ambient temperature. While routine examination of these outpatients did not typically involve analysis of vitamin D metabolites, which have been argued to vary seasonably in some ethnic groups above the 37th parallel44, it is unlikely that vitamin D metabolite variation accounted for these findings because in persons over 50, active vitamin D does not vary seasonably in the pattern consistent with pattern of monocyte counts in our data or consistent with vitamin D metabolite shifts in younger people45. We conclude that, remarkably, considering the immense amount of time that humans live in controlled environments (22°C indoor temperature), outdoor ambient temperature may influence monocyte counts in peripheral blood of humans.
Figure 6. Environmental temperature correlates with circulating monocyte levels in humans.
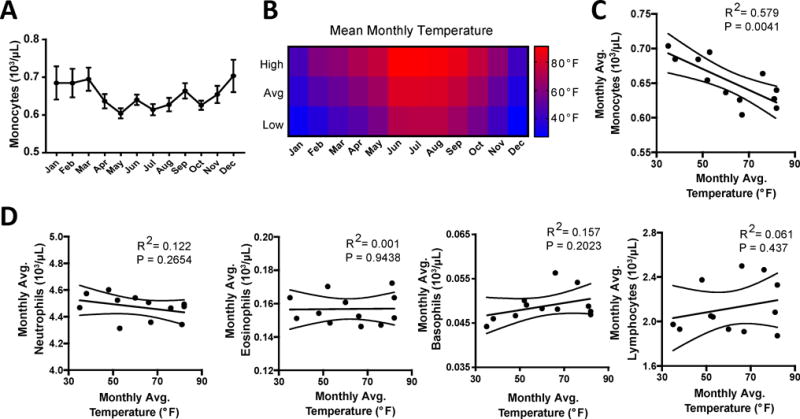
A) Peripheral blood was assayed for complete blood counts (CBC) with automated differentials for monocyte levels through a 12-month period and plotted as monthly averages. B) Monthly environmental temperature with mean high, average, and low temperatures reported for each month that peripheral blood was collected. C) Regression plot of average monthly cell numbers against average monthly temperature for monocytes. E) Regression plots for Neutrophils, Eosinophils, Basophils, and Lymphocytes numbers against average monthly temperature. Data in A and C are mean ± SEM, and represent a total cohort of n=15,516 patients, with approximately 1,200 measurements per month. Pearson linear regression analysis was performed to test correlations between monthly cell counts and environmental temperatures, p-values and R2 are reported.
Thermoneutrality and UCP1 signaling have unique downstream pathways
We next hypothesized that brown fat activation at 22°C housing might lead to augmented monocyte egress from the marrow, leading to increased atherosclerotic disease. We thus generated Ucp1−/− x Ldlr−/− mice. When housed at 22°C and fed a cholesterol-enriched diet, plaque burden in Ucp1−/− Ldlr−/− mice was reduced compared with Ldlr−/− mice and resembled that of Ldlr−/− mice housed at 30°C. These data indicate that loss of UCP1, either through genetic or environmental manipulation, was atheroprotective (Figure 7A). This was not due to major shifts in animal weight (Figure 7B). Additionally, plaque burden was not further reduced in Ucp1−/− Ldlr−/− mice housed at 30°C compared with the same strain housed at 22°C after 12 or 20 weeks HFD feeding (Figure 7C, Online Figure VII A, B), supporting the concept that UCP1 expression in Ldlr−/− mice housed at 22°C may account for the increased plaque burden observed over Ldlr−/− mice housed at 30°C.
Figure 7. Impact of UCP1 deficiency on atherosclerosis and glucose tolerance in comparison to Thermoneutrality.
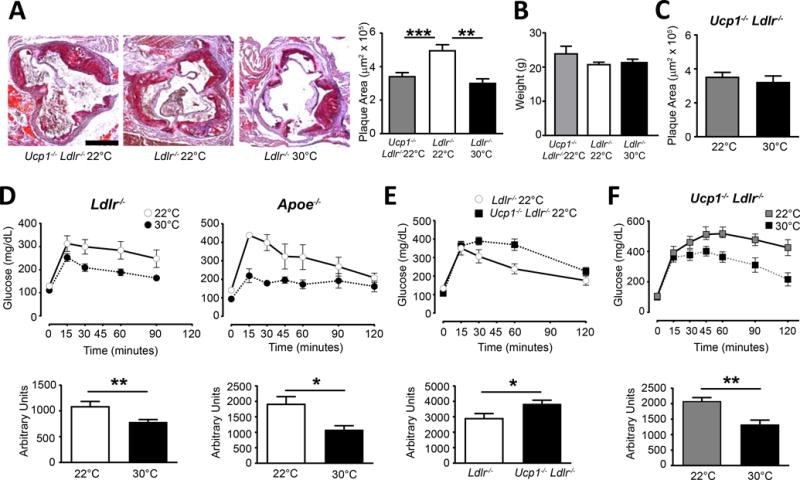
A) Oil red O lipid staining of aortic sinus from the indicated mice following HFD treatment for 8 weeks at 22°C or 30°C housing, which quantification (right). B) Total body weight in mice after HFD. C) Ucp1−/− Ldlr−/− mice fed HFD for 12 weeks and were housed at 22°C or 30°C and assayed for plaque development. D) Glucose tolerance tests (GTT) were performed on 22°C or 30°C housed Ldlr−/− and Apoe−/− mice following 8 weeks HFD. E) Ucp1−/− Ldlr−/− and Ldlr−/− mice fed HFD for 8 weeks and housed at 22°C were assayed by GTT. F) Ucp1−/− Ldlr−/− mice fed HFD were housed at 22°C or 30°C and assayed for GTT. All data are representative experiments, containing n≥5 animals per group and repeated in two or three independent times. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01, ***p≤0.001.
Changes in thermogenesis are known to affect metabolic responses in animals and humans19, 46, 47. Therefore, we next examined glucose tolerance in Ldlr−/− and Apoe−/− mice fed a cholesterol-enriched diet housed at 22°C and 30°C ambient temperatures. Mice housed at the elevated ambient temperature showed improved glucose sensitivity compared to those kept at 22°C (Figure 7D). However, consistent with previous reports with Ucp1−/− mice, Ucp1−/− Ldlr−/− mice demonstrated impaired glucose tolerance over UCP1-sufficient Ldlr−/− mice at 22°C housing (Figure 7E) despite having reduced atherosclerosis outcomes. Strikingly, when we compared glucose tolerance of Ucp1−/− Ldlr−/− mice at 22°C versus 30°C housing, the improved glucose sensitivity observed at 30°C persisted (Figure 7F). Collectively, these data implicate UCP1 in driving plaque growth in a manner independent of glucose metabolism, even as they also reveal that 30°C housing provides a means to elevated glucose sensitivity that is independent of UCP1.
Nonetheless, because our studies in Ucp1−/− Ldlr−/− mice mirrored the reduction in plaque burden observed in Ldlr−/− mice housed at 30°C, we hypothesized that UCP1 activity might be directly implicated in the regulation of monocyte output from the bone marrow. We studied C57BL/6 mice and Ucp1−/− mice in the absence of atherogenesis to address this question. In strong contrast to expectations for overlapping mechanisms UCP1 deficiency did not regulate circulating monocyte numbers (Figure 8A). Additionally, when housed at thermoneutral environment, UCP1-deficient mice did not show changes in circulating monocyte levels whereas WT mice examined side-by-side with Ucp1−/− mice did (Online Figure VII C, D) We next addressed biodistribution of 64Cu-DOTA-ECL1i tracer in Ucp1−/− mice (Figure 8B). We found a significant decrease in tracer uptake in the brown fat of Ucp1−/− mice compared to WT, which was consistent with animals housed at 30°C. White fat tracer uptake was also elevated in the absence of UCP1 (Fig. 8B). Unlike the effect of 30°C housing on accumulation of 64Cu-DOTA-ECL1i in the bone marrow, Ucp1−/− mice did not display increased tracer in the bone marrow (Figure 8B).
Figure 8. UCP1-deficiency does not drive changes in monocyte distribution following thermoneutral challenge.
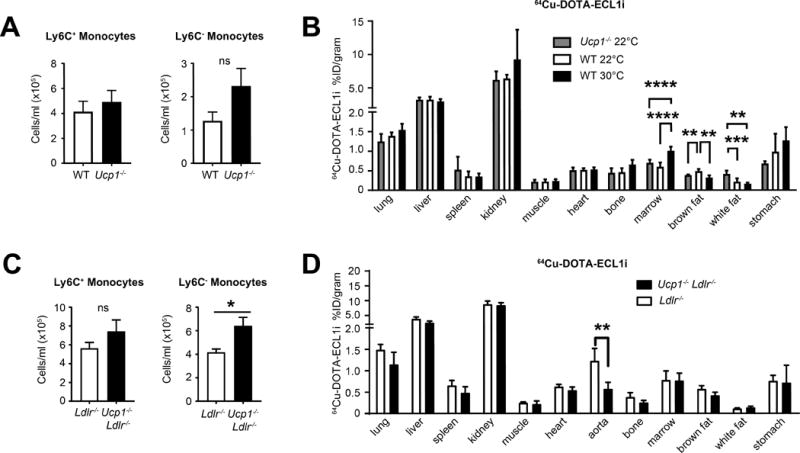
WT or UCP1−/− mice housed at 22°C ambient temperature were assayed for blood monocyte numbers (A) or biodistribution of 64Cu-DOTA-ECL1i probe (B). C) Ldlr−/− or UCP1−/− Ldlr−/− mice were housed at 22°C ambient temperature on HFD for 8 weeks, and assayed for blood leukocyte numbers. D) Ldlr−/− or Ucp1−/− Ldlr−/− mice housed at 22°C and fed HFD were assayed for 64Cu-DOTA-ECL1i biodistribution. A, C) are representative experiments n≥5 animals per group and repeated two independent times. B, D) Data are 5–8 animals combined per group. Statistical analysis was performed by unpaired student T-test, * p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
Examining Ucp1−/− Ldlr−/− mice fed HFD for 8 weeks at 22°C, we observed no change in blood monocyte numbers compared to Ldlr−/− mice, albeit a trend toward augmentation of circulating monocytes (Figure 8C), again starkly contrasting with effects of housing WT or Ldlr−/− mice at 30°C. This was accompanied by no major shifts in monocyte populations in blood or bone marrow of Ucp1−/− Ldlr−/− mice housed at 22°C and 30°C (Online Figure VII D, E). We repeated 64Cu-DOTA-ECL1i biodistribution analysis in Ucp1−/− Ldlr−/− mice on HFD to test whether hypercholesterolemia might exacerbate the monocytosis associated with thermoneutral ambient temperature. While we observed reduced tracer uptake in the aorta of the Ucp1−/− Ldlr−/− mice, consistent with reduced plaque, we did not find significant changes in other tissues, including adipose tissue compartments (Figure 8D). Finally, we asked whether Ucp1−/− Ldlr−/− mice had changes in their lipoprotein profiles that might explain the changes in atherosclerosis progression via a mechanism distinct from control of monocyte numbers in the blood. Indeed, while food intake was the same in the two groups, Ucp1−/− Ldlr−/− mice had reduced plasma cholesterol levels (Online Figure VIII A, B).In particular, Ucp1−/− Ldlr−/− mice on HFD for 8 weeks showed dramatically reduced vLDL and iLDL levels compared to controls (Online Figure VIII C, D), consistent with previous reports for loss of Ucp1 expression21, but distinct from mice housed at 30°C. Likely, this drop in plasma cholesterol largely accounts for the reduced atherosclerotic disease found in Ucp1−/− Ldlr−/− mice. Together, these studies indicate that the impact of housing temperature on monocyte egress from the bone marrow is not simply a product of UCP1 activation and possibly is unlinked to activation of beige/brown fat altogether.
β3 adrenergic receptors activation in beige fat does not promote monocyte egress from the bone marrow
Next, we considered whether retention of monocytes in the bone marrow were linked with induction of beige/brown fat. Sympathetic activation through β3 adrenergic signaling supports UCP1 expression in brown and beige fat at lower temperatures. Thus, we treated Ldlr−/− mice fed cholesterol-enriched HFD for four weeks, then began injections with β3 adrenergic receptor agonist to assess whether such stimulation would reverse the suppression in circulating monocytes observed at 30°C. Following previously described protocols35, 36, treatment with the β3 adrenergic receptor-specific agonist CL-316,243 showed little effects on circulating monocyte numbers in Ldlr−/− mice housed at 30°C on HFD (Online Figure IX A). Following 4 weeks of continuous treatment, plaque area was assessed, but activation of the β3-adrenergic receptor did not influence plaque area in male or female mice in this short period of plaque assessment (Online Figure IX B). However, indicating the efficacy of our treatment regimen to activate beige/brown fat, we observed induction of UCP1 mRNA in beige fat depots (Online Figure IX C). Additionally, body temperature was increased by the β3 adrenergic receptor agonist, returning body temperature to the equivalent of that documented at 22°C (Online Figure IX D). Furthermore, reductions in body weight were associated with animals receiving β3 adrenergic receptor agonist, consistent with what would be expected in animals with elevated activation of brown or beige fat (Online Figure IX E, F). Not only does thermoneutral temperature suppress monocyte egress from the bone marrow in a UCP1-dependent manner, but these data suggest that activation of UCP1 expression in beige adipose tissue does not reverse the atheroprotective effects of thermoneutral ambient temperature.
DISCUSSION
During the past decade, research on brown and beige adipose tissue has intensified tremendously. The discovery of thermogenic adipose tissue in adult humans, once thought to be absent after infancy, has fueled interest to promote thermogenesis in multiple metabolic disorders20, 48, 49. Evidence that thermogenically active brown fat promotes glucose disposal50 and, arguably, consumption of lipids51 are among the points of logic behind the concept. Indeed, it has evolved that cold exposure is now often viewed as a natural approach to improved metabolism. Conversely, thermoneutrality is often considered experimentally as an approach equivalent to loss of active beige or brown fat. Here we show that thermoneutrality and loss of UCP1 affect atherosclerosis similarly, by reducing disease burden. However, ultimately these manipulations diverge mechanistically: thermoneutrality governs monocyte homeostasis, whereas UCP1 expression more closely associates with cholesterol homeostasis, particularly plasma LDL accumulation.
When we started these studies, we hypothesized that the anticipated metabolic benefits of cold exposure would extend to protection against atherosclerosis. However, we were surprised to observe that thermoneutrality and genetic deficiency of UCP1 (studied in atherogenic strains of mice housed at 22°C) reduced atherosclerosis. Our findings are in general agreement with an earlier study by Dong et al., who observed, like us, that UCP1 deficiency in the context of experimental atherosclerosis is atheroprotective for reasons linked to plasma cholesterol21. They also reported that thermoneutrality was atheroprotective compared with 4°C housing. However, this study did not include a 22°C group, the most commonly analyzed in the literature, making it difficult to determine whether cold challenge increased atherosclerosis or warm temperatures were protective. We find that both conclusions are supported by our data and that experimental atherosclerosis is stepwise more severe as ambient temperature drops.
At first glance, our conclusions related to the impact of ambient temperature on atherosclerosis appear to be confounded by several studies that draw conclusions opposite to those of our study and that of Dong et al.21 First, Berbee et al. studied the impact of a pharmacological agonist to the β3 adrenergic receptor on atherosclerosis in apoE3 Leiden mice co-expressing cholesteryl ester transfer protein. In the presence of agonist, these mice showed lipid profiles consistent with reduced atherosclerosis and indeed plaque burden was lowered52. The favorable changes in lipid profiles were not recapitulated in Apoe−/− and Ldlr−/− mice. The authors argued that cold challenge would protect against atherosclerosis but did not test it directly. It is important to recognize that the work of Berbee et al. more relate to the connections between UCP1 activity and blood lipid profiles, which may respond differently between mouse and man, with the apoE3 Leiden serving as a better model for human lipoprotein pathways than the other models. The more novel aspects of our work related to thermoneutrality effects on monocytes were not examined by Berbee et al. As we show that UCP1 activation and thermoneutrality diverge with respect to regulating monocyte counts, and that human monocyte counts are reduced in warmer months of the year, it becomes difficult to argue that the study of Berbee et al. is either consistent or inconsistent with our findings.
Second, Tian et al. examined the impact of thermoneutrality on atherosclerosis. In agreement with our findings and those of others53, Tian et al. reported improved glucose clearance in mice housed at 30°C. However, they also reported worsened atherosclerosis along with modestly elevated cholesterol, albeit with minimal disease effects at the aortic arch and the most dramatic effects in the descending aorta22. Aortic sinus disease, which we quantified in the present study along with aortic disease at multiple time points, was not examined by Tian et al. Giles et al. failed to find a major shift in plasma cholesterol but nonetheless reported that enhanced atherosclerosis was worsened in Apoe−/− mice fed a high fat diet, but there was no disease change in Apoe−/− mice fed a chow diet. They then went on to report that, surprisingly, WT C57BL/6 mice developed modest, but measureable, atherosclerosis when fed a high fat diet and housed at thermoneutrality54. For unclear reasons as of yet, these two studies appear directly at odds with the conclusions from our work and that of Dong et al21. One of the critical details that deserves more attention than it has received is the conditions used for thermoneutral housing, eg., whether such housing was done within the barrier of the animal facility or in special containment units for controlling temperature, and whether air circulation and light cycles may have also varied.
To our knowledge, this is the first formal investigation of the impact of thermogenesis and thermoneutrality on leukocyte dynamics. It is interesting to note that the supplemental data included in the work of Tian et al. underscore a reduction in circulating neutrophils and the data appear to support monocyte retention in the bone marrow as well22. We documented, using 3 different techniques—flow cytometry coupled with BrdU labeling and cell counting, PET imaging, and 2-photon microscopy—that monocytes were retained in the bone marrow of mice housed at thermoneutrality, coupled with reduced circulating monocytes, regardless of diet or whether the mice were of an atherogenic strain or wildtype. However, monocyte sequestration in the bone marrow was only a feature of thermoneutrality, not UCP1 deficiency. This sequestration of monocytes in the bone marrow was associated with reduced monocyte migration into plaques, potentially explaining the reduction in aortic sinus plaque that we report. That is, the monocyte pool in the bone marrow is dependent on the dynamical equilibrium between egress (relying on the chemokine receptor CCR2) and retention through the interaction of the chemokine receptor CXCR4 expressed on hematopoietic cells and its ligand CXCL12, mainly found in the stromal compartment including CAR cells, endothelial cells and nestin+ perivascular cells55. Alteration of either of these interactions could modify the pool of monocytes in the bone marrow and peripheral blood. Blood levels of monocytes and neutrophils are strongly associated with atherosclerosis progression4, 10–12 and therefore reduced white blood cells counts provide a potential explanation accounting for the reduced plaque size at mice housed at 30°C. While it remains uncertain the extent to which this mechanism can be applied to humans, we provide data examining over 1,000 individuals per month in the St. Louis region that monocyte counts vary with outdoor temperature, in a manner that is consistent with the work in mice, including higher monocyte counts in blood during cooler weather, with effects on monocytes the most prominent among all leukocytes. We believe this retrospective analysis in humans, which should be considered with due caution in full interpretation, suggests that a prospective study on the impact of ambient temperature on monocyte dynamics may be worthwhile in future studies.
Considering that UCP1 activity promotes glucose disposal, while not linked to the regulation of monocyte dynamics in the blood and bone marrow, we propose that the field take a revised view of how to approach simultaneously optimizing metabolic and cardiovascular health. We indeed argue that our experiment using synthetic β3-agonist suggests that it is technically feasible to simultaneously activate UCP1 in the beige fat pads and retain the atheroprotective effects of thermoneutrality, including reduced monocyte mobilization into blood, whether or not β3 adrenergic receptors would be the most logical target (doubtful) to activate beige fat in a translational setting. Based on the findings here, one might envision that developing tools that recapitulate the mechanisms conferring atheroprotection in response to thermoneutral ambient temperature, without specifically ablating UCP1 activity that confers metabolic protection, may provide the most benefit to a population that encounters a high incidence of both cardiovascular and metabolic disease. In other words, we argue that it is critical in the cardiovascular research community to cease viewing cooler temperatures as a desired positive stimulus for arriving at the potential metabolic benefits of beige fat activation. By conceptually and practically unlinking UCP1 activity from the impact of ambient temperature, we propose that it might be more optimal to promote the healthful effects of warm environs—which based on our emerging survival data may extend to benefits beyond protection from atherosclerosis—while simultaneously supporting beige fat activation through mechanisms distinct from ambient temperature. Additional research is needed to test this proposed concept emerging from the present study.
Supplementary Material
Novelty and Significance.
What Is Known?
The incidence of cardiovascular events increases in colder temperatures.
Elevated levels of circulating blood monocytes are a risk factor for atherosclerosis.
What New Information Does This Article Contribute?
Warm environment protects from atherosclerosis progression in mice.
Monocyte egress from bone marrow is reduced when ambient temperature is warmer.
UCP1-deficiency protects from atherosclerosis, but does not regulate monocyte levels.
Environmental temperature has been linked to risk of cardiovascular disease, with elevated risk occurring in colder ambient temperatures. However, the effects of ambient temperature on the immune system in the context of atherosclerosis are not well known. We identify that circulating blood monocyte counts, which are also a known risk factor for atherosclerosis, are inversely correlated with environmental temperature. This association was observed in mice and in human samples collected throughout different times of the year. Animals with reduced circulating monocytes were protected from atherosclerosis. Further, deletion of UCP1, a gene associated with the activation of brown fat and the thermogenic program, was also protective to atherosclerotic disease. However, the protective effect of Ucp1 deletion was independent of monocyte levels. In addition to decreased monocyte counts and lower atherosclerosis, warmer temperatures promoted glucose tolerance and a longer lifespan. Finally, we illustrate that UCP1 activation in peripheral brown fat pads does not interfere with the protective effects of warm ambient temperature, raising the possibility that combination therapies to combat metabolic disease and atherosclerosis might be devised.
Acknowledgments
We thank the members of the Randolph Laboratory for their technical assistance and help in preparation of this manuscript.
SOURCES OF FUNDING
This work was supported by NIH R21 AG046734, RO1 HL118206, R37 AI049653, and DP1DK109668 to GJR; NIH R01 HL125655 RO1 to YL; and AHA grant 16SDGG30480008 to BHZ. JWW was supported by NIH training grant 2T32DK007120-41 and AHA grant 17POST33410473. AFE was supported by NIH training grant T32-HL07081-38. MGST was supported by NIH RO1 HL127649 and HL112276. JHC was supported by Korean Health Technology R&D Project HI15C0399, Ministry of Health, Welfare & Family Affairs, South Korea. JRB was supported by the Clinical Pathology Physician-Scientist Training Program (PSTP) at the Washington University School of Medicine. Additional technical support provided by the Washington University Diabetes Research Center, P30-DK020579.
Nonstandard Abbreviations and Acronyms
- Apoe
Apolipoprotein E
- HDL
High Density Lipoprotein
- LDLR
Low Density Lipoprotein Receptor
- PET
Positron Emission Tomography
- UCP1
Uncoupling Protein 1
Footnotes
In June 2017, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.45 days.
AUTHOR CONTRIBUTIONS
JWW, AFE, SI, and GJR wrote and edited the manuscript. JWW, AE, SI, HL, KK, BS, MJ, SK, BZ, CC, JHC, MGST, BHZ, JRB performed experiments and analyzed data. GJR and YL supervised the project and GJR, AFE, and CC conceived the start of this project.
DISCLOSURES
The authors have no disclosures or conflicts of interest to declare.
References
- 1.Gautier EL, Jakubzick C, Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1412–8. doi: 10.1161/ATVBAHA.108.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H, Horke S, Forstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014;237:208–19. doi: 10.1016/j.atherosclerosis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Swirski FK, D’Sa A, Kianpour S, Inman MD, Stampfli MR. Prolonged ovalbumin exposure attenuates airway hyperresponsiveness and T cell function in mice. Int Arch Allergy Immunol. 2006;141:130–40. doi: 10.1159/000094715. [DOI] [PubMed] [Google Scholar]
- 4.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon D, Reidy MA, Benditt EP, Schwartz SM. Cell proliferation in human coronary arteries. Proc Natl Acad Sci U S A. 1990;87:4600–4. doi: 10.1073/pnas.87.12.4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rekhter MD, Gordon D. Active proliferation of different cell types, including lymphocytes, in human atherosclerotic plaques. Am J Pathol. 1995;147:668–77. [PMC free article] [PubMed] [Google Scholar]
- 7.Katsuda S, Coltrera MD, Ross R, Gown AM. Human atherosclerosis. IV. Immunocytochemical analysis of cell activation and proliferation in lesions of young adults. Am J Pathol. 1993;142:1787–93. [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–72. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res. 2014;114:1757–71. doi: 10.1161/CIRCRESAHA.114.301174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman CM, Beilby JP, McQuillan BM, Thompson PL, Hung J. Monocyte count, but not C-reactive protein or interleukin-6, is an independent risk marker for subclinical carotid atherosclerosis. Stroke. 2004;35:1619–24. doi: 10.1161/01.STR.0000130857.19423.ad. [DOI] [PubMed] [Google Scholar]
- 11.Nasir K, Guallar E, Navas-Acien A, Criqui MH, Lima JA. Relationship of monocyte count and peripheral arterial disease: results from the National Health and Nutrition Examination Survey 1999–2002. Arterioscler Thromb Vasc Biol. 2005;25:1966–71. doi: 10.1161/01.ATV.0000175296.02550.e4. [DOI] [PubMed] [Google Scholar]
- 12.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754–8. doi: 10.1038/nm.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Courties G, Herisson F, Sager HB, Heidt T, Ye Y, Wei Y, Sun Y, Severe N, Dutta P, Scharff J, Scadden DT, Weissleder R, Swirski FK, Moskowitz MA, Nahrendorf M. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ Res. 2015;116:407–17. doi: 10.1161/CIRCRESAHA.116.305207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T, Majmudar MD, Lasitschka F, Etzrodt M, Waterman P, Waring MT, Chicoine AT, van der Laan AM, Niessen HW, Piek JJ, Rubin BB, Butany J, Stone JR, Katus HA, Murphy SA, Morrow DA, Sabatine MS, Vinegoni C, Moskowitz MA, Pittet MJ, Libby P, Lin CP, Swirski FK, Weissleder R, Nahrendorf M. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487:325–9. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golozoubova V, Cannon B, Nedergaard J. UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. Am J Physiol Endocrinol Metab. 2006;291:E350–7. doi: 10.1152/ajpendo.00387.2005. [DOI] [PubMed] [Google Scholar]
- 17.Betz MJ, Enerback S. Human Brown Adipose Tissue: What We Have Learned So Far. Diabetes. 2015;64:2352–60. doi: 10.2337/db15-0146. [DOI] [PubMed] [Google Scholar]
- 18.Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov. 2010;9:465–82. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, English J, Chatman K, Trauger SA, Doria A, Kolodny GM. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. 2015;21:33–8. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–17. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong M, Yang X, Lim S, Cao Z, Honek J, Lu H, Zhang C, Seki T, Hosaka K, Wahlberg E, Yang J, Zhang L, Lanne T, Sun B, Li X, Liu Y, Zhang Y, Cao Y. Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 2013;18:118–29. doi: 10.1016/j.cmet.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian XY, Ganeshan K, Hong C, Nguyen KD, Qiu Y, Kim J, Tangirala RK, Tonotonoz P, Chawla A. Thermoneutral Housing Accelerates Metabolic Inflammation to Potentiate Atherosclerosis but Not Insulin Resistance. Cell Metab. 2016;23:165–78. doi: 10.1016/j.cmet.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kriszbacher I, Koppan M, Bodis J. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;353:429–30. author reply 429–30. [PubMed] [Google Scholar]
- 24.Danet S, Richard F, Montaye M, Beauchant S, Lemaire B, Graux C, Cottel D, Marecaux N, Amouyel P. Unhealthy effects of atmospheric temperature and pressure on the occurrence of myocardial infarction and coronary deaths. A 10-year survey: the Lille-World Health Organization MONICA project (Monitoring trends and determinants in cardiovascular disease) Circulation. 1999;100:E1–7. doi: 10.1161/01.cir.100.1.e1. [DOI] [PubMed] [Google Scholar]
- 25.Gyllerup S, Lanke J, Lindholm LH, Schersten B. High coronary mortality in cold regions of Sweden. J Intern Med. 1991;230:479–85. doi: 10.1111/j.1365-2796.1991.tb00478.x. [DOI] [PubMed] [Google Scholar]
- 26.Donaldson GC, Keatinge WR. Early increases in ischaemic heart disease mortality dissociated from and later changes associated with respiratory mortality after cold weather in south east England. J Epidemiol Community Health. 1997;51:643–8. doi: 10.1136/jech.51.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eng H, Mercer JB. Seasonal variations in mortality caused by cardiovascular diseases in Norway and Ireland. J Cardiovasc Risk. 1998;5:89–95. [PubMed] [Google Scholar]
- 28.Luo B, Zhang S, Ma S, Zhou J, Wang B. Artificial cold air increases the cardiovascular risks in spontaneously hypertensive rats. Int J Environ Res Public Health. 2012;9:3197–208. doi: 10.3390/ijerph9093197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–30. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herz J, Zinselmeyer BH, McGavern DB. Two-photon imaging of microbial immunity in living tissues. Microsc Microanal. 2012;18:730–41. doi: 10.1017/S1431927612000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Pierce R, Luehmann HP, Sharp TL, Welch MJ. PET imaging of chemokine receptors in vascular injury-accelerated atherosclerosis. J Nucl Med. 2013;54:1135–41. doi: 10.2967/jnumed.112.114777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Li W, Luehmann HP, Zhao Y, Detering L, Sultan DH, Hsiao HM, Krupnick AS, Gelman AE, Combadiere C, Gropler RJ, Brody SL, Kreisel D. Noninvasive Imaging of CCR2+ Cells in Ischemia-Reperfusion Injury After Lung Transplantation. Am J Transplant. 2016 doi: 10.1111/ajt.13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Detering L, Sultan D, Cooper ML, You M, Cho S, Meier SL, Luehmann H, Sun G, Rettig M, Dehdashti F, Wooley KL, DiPersio JF, Liu Y. Gold Nanoclusters Doped with (64)Cu for CXCR4 Positron Emission Tomography Imaging of Breast Cancer and Metastasis. ACS Nano. 2016;10:5959–70. doi: 10.1021/acsnano.6b01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Gunsten SP, Sultan DH, Luehmann HP, Zhao Y, Blackwell TS, Bollermann-Nowlis Z, Pan JH, Byers DE, Atkinson JJ, Kreisel D, Holtzman MJ, Gropler RJ, Combadiere C, Brody SL. PET-based Imaging of Chemokine Receptor 2 in Experimental and Disease-related Lung Inflammation. Radiology. 2017;161409 doi: 10.1148/radiol.2016161409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman BM. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roth Flach RJ, Matevossian A, Akie TE, Negrin KA, Paul MT, Czech MP. beta3-Adrenergic receptor stimulation induces E-selectin-mediated adipose tissue inflammation. J Biol Chem. 2013;288:2882–92. doi: 10.1074/jbc.M112.412346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–36. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia RA, Search DJ, Lupisella JA, Ostrowski J, Guan B, Chen J, Yang WP, Truong A, He A, Zhang R, Yan M, Hellings SE, Gargalovic PS, Ryan CS, Watson LM, Langish RA, Shipkova PA, Carson NL, Taylor JR, Yang R, Psaltis GC, Harrity TW, Robl JA, Gordon DA. 11beta-hydroxysteroid dehydrogenase type 1 gene knockout attenuates atherosclerosis and in vivo foam cell formation in hyperlipidemic apoE(−)/(−) mice. PLoS One. 2013;8:e53192. doi: 10.1371/journal.pone.0053192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–7. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 40.Jacobson O, Weiss ID, Szajek L, Farber JM, Kiesewetter DO. 64Cu-AMD3100–a novel imaging agent for targeting chemokine receptor CXCR4. Bioorg Med Chem. 2009;17:1486–93. doi: 10.1016/j.bmc.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–71. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- 42.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med. 2001;194:1361–73. doi: 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kunstyr I, Leuenberger HG. Gerontological data of C57BL/6J mice. I. Sex differences in survival curves. J Gerontol. 1975;30:157–62. doi: 10.1093/geronj/30.2.157. [DOI] [PubMed] [Google Scholar]
- 44.Hagenau T, Vest R, Gissel TN, Poulsen CS, Erlandsen M, Mosekilde L, Vestergaard P. Global vitamin D levels in relation to age, gender, skin pigmentation and latitude: an ecologic meta-regression analysis. Osteoporos Int. 2009;20:133–40. doi: 10.1007/s00198-008-0626-y. [DOI] [PubMed] [Google Scholar]
- 45.Moan J, Lagunova Z, Lindberg FA, Porojnicu AC. Seasonal variation of 1,25-dihydroxyvitamin D and its association with body mass index and age. J Steroid Biochem Mol Biol. 2009;113:217–21. doi: 10.1016/j.jsbmb.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Lee P, Bova R, Schofield L, Bryant W, Dieckmann W, Slattery A, Govendir MA, Emmett L, Greenfield JR. Brown Adipose Tissue Exhibits a Glucose-Responsive Thermogenic Biorhythm in Humans. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 47.Laurila PP, Soronen J, Kooijman S, Forsstrom S, Boon MR, Surakka I, Kaiharju E, Coomans CP, Van Den Berg SA, Autio A, Sarin AP, Kettunen J, Tikkanen E, Manninen T, Metso J, Silvennoinen R, Merikanto K, Ruuth M, Perttila J, Makela A, Isomi A, Tuomainen AM, Tikka A, Ramadan UA, Seppala I, Lehtimaki T, Eriksson J, Havulinna A, Jula A, Karhunen PJ, Salomaa V, Perola M, Ehnholm C, Lee-Rueckert M, Van Eck M, Roivainen A, Taskinen MR, Peltonen L, Mervaala E, Jalanko A, Hohtola E, Olkkonen VM, Ripatti S, Kovanen PT, Rensen PC, Suomalainen A, Jauhiainen M. USF1 deficiency activates brown adipose tissue and improves cardiometabolic health. Sci Transl Med. 2016;8:323ra13. doi: 10.1126/scitranslmed.aad0015. [DOI] [PubMed] [Google Scholar]
- 48.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–25. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 49.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–8. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 50.Kwon MM, O’Dwyer SM, Baker RK, Covey SD, Kieffer TJ. FGF21-Mediated Improvements in Glucose Clearance Require Uncoupling Protein 1. Cell Rep. 2015;13:1521–7. doi: 10.1016/j.celrep.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 51.Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, Eychmuller A, Gordts PL, Rinninger F, Bruegelmann K, Freund B, Nielsen P, Merkel M, Heeren J. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–5. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 52.Berbee JF, Boon MR, Khedoe PP, Bartelt A, Schlein C, Worthmann A, Kooijman S, Hoeke G, Mol IM, John C, Jung C, Vazirpanah N, Brouwers LP, Gordts PL, Esko JD, Hiemstra PS, Havekes LM, Scheja L, Heeren J, Rensen PC. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat Commun. 2015;6:6356. doi: 10.1038/ncomms7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao C, Goldgof M, Gavrilova O, Reitman ML. Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity (Silver Spring) 2015;23:1450–9. doi: 10.1002/oby.21124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giles DA, Ramkhelawon B, Donelan EM, Stankiewicz TE, Hutchison SB, Mukherjee R, Cappelletti M, Karns R, Karp CL, Moore KJ, Divanovic S. Modulation of ambient temperature promotes inflammation and initiates atherosclerosis in wild type C57BL/6 mice. Mol Metab. 2016;5:1121–1130. doi: 10.1016/j.molmet.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–5. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.