

Summary
Mitochondrial network remodeling between fused and fragmented states facilitates mitophagy, interaction with other organelles and metabolic flexibility. Aging is associated with a loss of mitochondrial network homeostasis, but cellular processes causally linking these changes to organismal senescence remain unclear. Here, we show that AMP-activated protein kinase (AMPK) and dietary restriction (DR) promote longevity in C. elegans via maintaining mitochondrial network homeostasis and functional coordination with peroxisomes to increase fatty acid oxidation (FAO). Inhibiting fusion or fission specifically blocks AMPK- and DR-mediated longevity. Strikingly however, preserving mitochondrial network homeostasis during aging by co-inhibition of fusion and fission is sufficient itself to increase lifespan, while dynamic network remodeling is required for intermittent fasting-mediated longevity. Finally, we show that increasing lifespan via maintaining mitochondrial network homeostasis requires FAO and peroxisomal function. Together these data demonstrate that mechanisms that promote mitochondrial homeostasis and plasticity can be targeted to promote healthy aging.
eTOC Blurb
Dysfunctional mitochondrial dynamics are a hallmark of aging, but the physiological consequences are unknown. Weir et al. show that mitochondrial network remodeling is required for AMPK- and dietary restriction-mediated longevity, and identify a downstream role for fatty acid oxidation and peroxisome function in C. elegans.
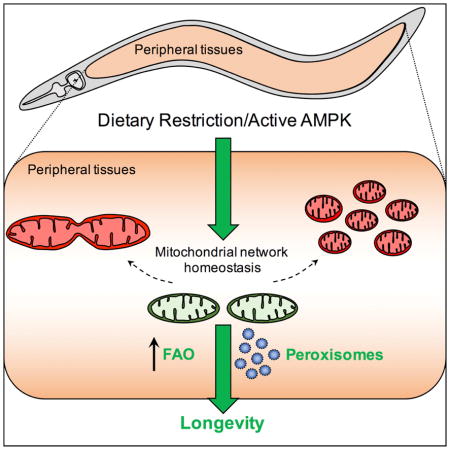
Introduction
Dynamic remodeling of mitochondrial networks by fusion and fission promotes maintenance of cellular homeostasis. Dysregulation of mitochondrial dynamics and aberrant mitochondrial morphology are hallmarks of aging and are thought to contribute to the pathology of numerous age-related pathologies including Alzheimer’s and Parkinson’s disease (Bonda et al., 2011; Leduc-Gaudet et al., 2015; Sebastián et al., 2016). Mitochondrial dynamics regulate several key cellular functions, including metabolic plasticity, mitochondrial turnover, and inter-organelle communication (Wai and Langer, 2016). However, how these functions are affected by deregulation of mitochondrial dynamics during aging, and whether alternate pro-longevity interventions require different mitochondrial network states and outputs remains unclear. Age-onset appearance of swollen, fragmented mitochondria has been reported across species (Jiang et al., 2015; Leduc-Gaudet et al., 2015; Sebastián et al., 2016), suggesting mitochondrial fragmentation (driven by fission) is pro-aging. Supporting this hypothesis, studies in yeast show inhibition of fission increases longevity, while unopposed fission shortens lifespan (Scheckhuber et al., 2007, 2014). In addition, fragmentation of mitochondria has been associated with age in multiple studies in C. elegans (Jiang et al., 2015; Regmi et al., 2014), while maintenance of fused networks often correlates with lifespan extension (Chaudhari and Kipreos, 2017; Houtkooper et al., 2013). However, mitophagy, the process by which damaged mitochondria are degraded and recycled, requires fission to remove damaged mitochondria and has been shown to be pro-longevity in Drosophila (Rana et al., 2013; Ulgherait et al., 2014). It therefore remains unclear under which contexts and in which cell types specific states of the mitochondrial network are pro-aging or pro-longevity.
AMP-activated protein kinase (AMPK), a highly conserved regulator of energy homeostasis, links energetics to the rate of aging and is implicated as a modulator of lifespan extension by dietary restriction (DR) (Fontana and Partridge, 2015). Activating AMPK increases lifespan in C. elegans and Drosophila, while pharmacological activation via metformin treatment promotes healthy aging in mice (Apfeld et al., 2004; Mair et al., 2011; Martin-Montalvo et al., 2013; Stenesen et al., 2013). Considerable interplay exists between AMPK and mitochondrial function; AMPK induces mitochondrial biogenesis (Reznick et al., 2007; Zong et al., 2002), and has emerging roles in the regulation of both mitochondrial metabolism and dynamics (Burkewitz et al., 2015; Kang et al., 2016; Toyama et al., 2016). We previously reported that AMPK increases lifespan in C. elegans via remodeling of systemic mitochondrial metabolism, which correlates with altered mitochondrial morphology in peripheral tissues (Burkewitz et al., 2015). However, the spatial requirement and physiological role of mitochondrial network remodeling that causally link AMPK and longevity is unknown.
Here, we show that AMPK and DR maintain youthful mitochondrial network morphology with age. We demonstrate that inhibiting fusion blocks AMPK- and DR-mediated lifespan extension, and that fusion is specifically required in peripheral tissues for AMPK-mediated longevity. However, promoting fusion directly is not sufficient to increase lifespan, and inhibiting fission also specifically blocks AMPK- and DR-mediated longevity. Strikingly, preserving mitochondrial network homeostasis with age by dual inhibition of fusion and fission is sufficient to increase lifespan. Finally, using targeted metabolomics and genetic analyses we identify a downstream role for peroxisomal function and fatty acid metabolism in AMPK- and DR-mediated longevity. Collectively, our data suggest that in the context of AMPK activation and DR, functional coordination between mitochondrial networks and peroxisomes causally links mitochondrial dynamics to longevity. Further, maintaining mitochondrial network homeostasis and plasticity may represent a novel therapeutic strategy to promote healthy aging.
Results
AMPK maintains youthful mitochondrial morphology and requires fusion to increase lifespan
To assess age-related changes to mitochondrial network morphology in vivo, we generated a C. elegans strain expressing GFP targeted to the outer mitochondrial membrane in body-wall muscle cells. In young wild-type animals, mitochondria appear tubular and exist in highly interconnected, well-organized networks (Figure 1A). There is a progressive loss of network connectivity from day 1 to day 15, indicated by a decrease in the area:perimeter ratio, as well as a reduction in mitochondrial content (Figure 1A and 1B). Muscle fiber organization does not overtly change between day 1 and day 8, confirming that decreased mitochondrial network connectivity is not due to age-related degeneration of muscle structure (Figure 1C). We hypothesized that if the observed age-related changes to mitochondrial networks correlate with lifespan, these would be delayed in long-lived models. To test this, we used a transgenic strain expressing a truncated AMPKα2 catalytic subunit (AAK-2). This truncation results in constitutively active (CA)-AAK-2 and extends C. elegans lifespan (Burkewitz et al., 2015; Mair et al., 2011). Mitochondrial networks of CA-AAK-2 animals resemble wild-type networks on day 1, but remain unchanged from day 1 to day 5, with no significant difference in either network connectivity or mitochondrial content (Figures 1D and 1E). At day 7, CA-AAK-2 animals still have increased network connectivity and mitochondrial content compared to wild-type, but by day 9 there are no significant differences in either measurement (Figures 1D and 1E). Together, these data demonstrate a correlation between mitochondrial morphology and rate of aging, and show that AMPK activation delays age-dependent changes to mitochondrial networks.
Figure 1. AMPK maintains youthful mitochondrial morphology with age and requires fusion to increase lifespan.

(A) Fluorescence images of mitochondrial networks in worm body wall muscle cells on day 1, 5, 8 and 15 of adulthood, visualized with outer mitochondrial membrane-targeted GFP (myo-3p::tomm20(1-49aa)::gfp). Scale bar = 20μm. (B) Quantification of (A): Mean ± SEM of n=21–26 muscle cells from different worms, pooled from 2 independent experiments. **p<0.01, ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (C) Body wall muscle fibers on day 1, 5 and 8, visualized with MYO-3::GFP. (D) Mitochondrial networks in muscle cells of wild-type worms and worms expressing CA-AAK-2 on day 1, 5, 7 and 9, visualized with outer mitochondrial membrane-targeted RFP (myo-3p::tomm20::mrfp). (E) Quantification of (D): Mitochondrial network area:perimeter ratio (left) and mitochondrial content (right, % mitochondrial coverage of cell area). Mean ± SEM of n=36–56 muscle cells from 3 independent experiments. *p<0.05, **p<0.01,***p<0.001, ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (F) Mitochondrial networks in muscle cells of wild-type (top) and fzo-1 null (bottom) worms on day 1. (G) and (H) Survival curves demonstrating that CA-AAK-2 extends lifespan in wild-type (G), but not fzo-1 mutant worms (H). (I) Survival curves of fzo-1 mutant and wild-type worms are not significantly different. (J) Mitochondrial networks from day 1 worms fed empty vector (EV) control or eat-3 RNAi. (K) and (L) Survival curves demonstrating that CA-AAK-2 extends lifespan on EV (K), but not eat-3 RNAi (L). See Table S1 for lifespan statistics.
Given that fragmented networks are associated with loss of AMPK-mediated longevity (Burkewitz et al., 2015), and that AMPK maintains fused mitochondrial morphology (Figure 1D and 1E), we asked if mitochondrial fusion is required for AMPK-mediated lifespan extension. To test this we used an fzo-1(tm1133) mutant strain, which has a null mutation in the single C. elegans homologue of the mammalian mitofusins (Mfn) (Kanazawa et al., 2008). FZO-1 mediates outer mitochondrial membrane fusion, and fzo-1(tm1133) mutants therefore have highly fragmented mitochondrial networks (Figure 1F). To determine if fusion is required for AMPK-mediated longevity, we tested if activating AMPK increases lifespan in this fusion-deficient genetic background. In a wild-type background, CA-AAK-2 robustly increases lifespan (Figure 1G). However, in fzo-1 mutants CA-AAK-2 completely fails to extend lifespan (Figure 1H), indicating that fusion is indeed required for AMPK-mediated longevity. Importantly, the lifespan of fzo-1 mutants is not significantly different from wild-type animals, confirming that suppression of AMPK-mediated longevity is not due to sickness or basal shortening of lifespan in fzo-1 mutants (Figure 1I). To further interrogate the role of mitochondrial fusion in AMPK-mediated longevity, we tested if inhibiting inner membrane fusion affects lifespan. The C. elegans gene eat-3 encodes a protein homologous to mammalian OPA1, which is required for inner membrane fusion (Kanazawa et al., 2008). RNAi of eat-3 results in fragmented mitochondria, detected using a transgenic strain expressing mitochondrial matrix targeted-GFP (Figure 1J). CA-AAK-2 does not significantly increase lifespan in the presence of eat-3 RNAi (Figure 1K and L), indicating that both inner and outer mitochondrial membrane fusion are required for AMPK-mediated longevity. Together, these data demonstrate that AMPK-dependent maintenance of mitochondrial network homeostasis is required for lifespan extension.
Fusion is required for DR-mediated longevity
Having determined that fusion is required for AMPK-mediated longevity, we tested whether mitochondrial network remodeling is a global modulator of other longevity pathways. Lifespan can be increased by mutation or knockdown of electron transport chain (ETC) components (Dillin et al., 2002). To determine if lifespan extension via reduced ETC function requires fused mitochondrial networks we performed RNAi of cco-1, which encodes a subunit of cytochrome c oxidase-1, in wild-type and fzo-1 mutants. cco-1 RNAi significantly increases lifespan in both genetic backgrounds, showing that outer membrane fusion is not required for reduced ETC function-mediated longevity via cco-1 RNAi. The percent lifespan extension is reduced in fzo-1 mutants however, suggesting that fusion may contribute to reduced ETC function longevity (Figure 2A and 2B, Table S1). We next tested whether fusion is required for reduced insulin/insulin-like growth factor signalling (rIIS) longevity (Kenyon et al., 1993). Suggesting outer membrane fusion is not required, rIIS via RNAi of the insulin/IGF receptor ortholog daf-2 significantly increases lifespan of both wild-type and fzo-1 mutants (Figure 2C and 2D). Finally, we examined whether network remodeling occurs in response to DR, which robustly extends lifespan across the evolutionary spectrum (Fontana and Partridge, 2015). Examination of mitochondrial networks with age in eat-2(ad1116) mutants, a genetic model of DR, revealed a similar phenotype to that seen in CA-AAK-2 animals: age-related changes are delayed by DR, with eat-2 mutants displaying increased network connectivity and content at day 8 and day 15 compared to wild-type animals (Figure 2E and 2F). Diet-restricting wild-type worms by feeding diluted bacteria for the first 3 days of adulthood also results in striking changes to mitochondrial network morphology (Figure 2G). Suggesting that the morphological changes are driven by fusion, fzo-1 mutant networks appear unchanged in response to DR (Figure 2G). To determine if fusion is required for DR-mediated longevity, we assayed the lifespan of wild-type and fzo-1 mutants that were fed either ad libitum (AL) or DR. DR significantly increases the lifespan of wild-type animals (Figure 2H), but completely fails to do so in an fzo-1 null background (Figure 2I), demonstrating that fusion is necessary for DR-mediated longevity. Together our data show that, rather than being required for all longevity pathways, fusion is specifically required for AMPK- and DR-mediated longevity.
Figure 2. Fusion is required for DR-mediated longevity.

(A) and (B) cco-1 RNAi extends lifespan in wild-type (A) and fzo-1 mutants (B). (C) and (D) daf-2 RNAi extends lifespan in wild-type (C) and fzo-1 mutants (D). (E) Mitochondrial networks in wild-type and eat-2 mutant worms on day 1, 5, 8 and 15. (F) Quantification of (E): Mitochondrial content (left) and area:perimeter ratio (right). Mean ± SEM of n=8–13 muscle cells from different worms. *p<0.05, **p<0.01,***p<0.001 by t test. (G) Mitochondrial networks in day 4 worms fed ad libitum (AL, top panels) or diet-restricted (DR, bottom panels). DR for the first 4 days of adulthood induces mitochondrial network remodeling in wild-type worms, while networks remain fragmented in fzo-1 mutants. (H) and (I) Survival curves demonstrating that DR extends lifespan compared to AL controls in wild-type (H), but not in fzo-1 mutants (I). See Table S1 for lifespan statistics.
Fusion is required in peripheral tissues for AMPK-mediated longevity
Mitofusins are expressed ubiquitously, but tissue-specific roles are emerging for their functions, including their role in metabolic plasticity (Dietrich et al., 2013; Schneeberger et al., 2013; Schrepfer and Scorrano, 2016). Having established that fusion is required for AMPK longevity, we examined if fusion is required ubiquitously or only in specific tissues to increase lifespan. Expressing fzo-1 cDNA under the control of the ubiquitous promoter sur-5p rescues the fragmented mitochondrial morphology in fzo-1 mutants, confirming that the transgene is functional (Figure 3A). Furthermore, ubiquitous rescue of fzo-1 completely restores lifespan extension by CA-AAK-2 (Figure 3B). Given the importance of fusion in the central nervous system for regulation of whole-body energy homeostasis (Dietrich et al., 2013), we examined whether fusion in neurons is also required for AMPK-mediated longevity. However, neuronal-specific expression of fzo-1 using the pan-neuronal rab-3 promoter fails to rescue lifespan extension by AAK-2 activation (Figure 3C). Ruling out neurons as the key site of action, we next asked if fusion in peripheral tissues underlies AMPK-mediated longevity. Supporting this hypothesis, rescuing fzo-1 only in muscle cells (myo-3 promoter) or only in the intestine (ges-1 promoter) partially restores CA-AAK-2 lifespan extension (Figure 3D, 3E and S1). Indicating that rescue of AMPK-mediated longevity is not due to fzo-1 expression level alone, global or tissue-specific rescue of fzo-1 in fzo-1 mutants alone marginally increase lifespan, but to a lesser extent than in the presence of CA-AAK-2 (Figure 3F, Table S1). Given the requirement for fusion in the intestine as well as muscle for AMPK-mediated longevity, we investigated whether mitochondrial morphology also undergoes age-related changes in the intestine, as it does in muscle cells (Figure 1). To do this, we generated a transgenic strain expressing outer mitochondrial membrane targeted-GFP in the intestine. Similar to our observations in muscle cells, mitochondrial content and size decrease significantly between day 1 and day 8, although the area:perimeter ratio does not significantly change (Figure 3G and 3I). CA-AAK-2 worms express GFP ubiquitously, precluding visualization of the mitoGFP reporter. Therefore, to address whether AMPK delays age-related changes to mitochondrial networks, we pharmacologically activated AMPK by treating worms with the biguanide phenformin, an indirect agonist which increases AMPK phosphorylation at Thr172, a site in the activation loop associated with increased activity (Figure 3H) (Cabreiro et al., 2013). Strikingly, phenformin treatment maintains mitochondrial size with age, and delays the reduction in mitochondrial content (Figure 3G and 3I). We previously reported that mitochondrial fragmentation in peripheral tissues correlates with suppression of AMPK longevity (Burkewitz et al., 2015); these data now show that fusion in peripheral tissues is required for lifespan extension via AMPK activation.
Figure 3. Fusion is required in peripheral tissues for AMPK-mediated longevity.

(A) Mitochondrial networks in wild-type and fzo-1 mutant muscle cells on day 1. Ubiquitous rescue of fzo-1 (sur-5p::fzo-1) rescues the fragmented mitochondrial morphology in fzo-1 mutants (bottom panel). (B) Ubiquitous rescue of fzo-1 fully restores lifespan extension by CA-AAK-2 in fzo-1 mutants. (C)–(E) Survival curves with tissue-specific rescue of fzo-1. Neuronal rescue of fzo-1 does not restore CA-AAK-2 lifespan extension (rab-3p::fzo-1, C), whereas muscle (myo-3p::fzo-1, D) or intestinal rescue (ges-1p::fzo-1, E) result in partial rescue. (F) Survival curves of fzo-1 mutants with ubiquitous or tissue-specific rescue of fzo-1. See Table S1 for lifespan statistics. (G) Mitochondrial networks in intestinal cells on day 1 and 8 with or without phenformin (4.5mM) treatment from L4 stage. Scale bar = 20μm. (H) p-AMPK levels are increased in wild-type worms treated with phenformin for 24h from L4. 3 biological replicates of lysates from 300–500 worms. (I) Quantification of (G): Mitochondrial content (left), size (middle) and area/perimeter ratio (right). Mean ± SEM of n=25–26 intestinal cells from 2 independent experiments. *p<0.05, **p<0.01,****p<0.0001 by t test.
Promoting fusion does not increase lifespan
Since fusion is required for both AMPK and DR-mediated longevity, we hypothesized that driving fusion directly might be sufficient to increase lifespan. To test this we used a drp-1(tm1108) mutant strain; DRP-1 is a conserved GTPase that catalyzes mitochondrial fission, and loss of function results in unopposed fusion of the network. This is evident in drp-1 mutants, which have increased mitochondrial network connectivity compared to wild-type controls (Figure 4A–C). Contrary to our hypothesis however, drp-1 mutants are not long-lived, and have identical lifespan to that of wild-type animals (Figure 4D). Similarly, driving fusion by ubiquitous overexpression of fzo-1 has no effect on lifespan (Figures 4A and 4E). Since promoting fusion is not sufficient to promote longevity, we next hypothesized that AMPK increases lifespan not by increasing fusion, but by preserving the balance between fusion and fission to maintain mitochondrial network homeostasis. Supporting this hypothesis, CA-AAK-2 fails to extend lifespan in drp-1 mutants (Figure 4F), showing that inhibiting fission also blocks AMPK-mediated longevity. Furthermore, lifespan extension via DR is suppressed in drp-1 mutants (Figure 4G; Table S1). Confirming the specificity of this pathway to AMPK and DR, knockdown of daf-2 (Figure 4H and (Yang et al., 2011)) and cco-1 (Figure 4I) both increase lifespan in drp-1 mutants demonstrating that, like fusion, fission is also dispensable for these longevity pathways. These data also highlight that genetic manipulation of mitochondrial morphology is not universally disruptive to lifespan. Taken together, our results suggest that AMPK and DR increase lifespan by maintaining a balance between fusion and fission, thereby preserving mitochondrial network homeostasis.
Figure 4. Driving fusion does not increase lifespan.

(A) Mitochondrial networks in muscle cells of wild-type, drp-1 mutant, and wild-type worms overexpressing fzo-1 (sur-5p::fzo-1) on day 1. (B) Mitochondrial networks in intestinal cells from wild-type and drp-1 mutants on day 1. (C) Mitochondrial area:perimeter ratio showing that mitochondrial networks are more connected in drp-1 mutants. Mean ± SEM of 8 muscle cells and 10 intestinal cells from different worms. *p<0.05, ****p<0.0001 by t test. Driving fusion by deletion of drp-1 (D) or overexpressing fzo-1 (E) has no effect on lifespan. Lifespan extension by CA-AAK-2 (F) and DR (G) is suppressed in drp-1 mutants. RNAi knockdown of daf-2 (H) and cco-1 (I) extend lifespan in drp-1 mutants. See Table S1 for lifespan statistics.
Maintaining mitochondrial network homeostasis is sufficient to extend lifespan
To determine if preserving balance between fission and fusion is sufficient to promote longevity, we crossed drp-1 mutants with fzo-1 mutants to generate animals deficient in both fission and fusion. Co-deletion of fission and fusion machinery components results in morphologically balanced mitochondrial networks: mitochondrial morphology in drp-1;fzo-1 double mutant animals is comparable to that of wild-type animals on day 1 of adulthood, with no significant differences in mitochondrial size or network connectivity (Figures 5A, 5B and S1A). Wild-type, drp-1 mutants and fzo-1 mutants all exhibit age-dependent changes to mitochondrial morphology, including a significant reduction in mitochondrial connectivity and content in all 3 genotypes (Figures 5A, 5B and S1B). Conversely however, drp-1;fzo-1 double mutants maintain both mitochondrial morphology and content with age, showing that mitochondrial network homeostasis is preserved as these animals age (Figures 5A, 5B, S2A and S2B). To determine whether preserving network homeostasis affects mitochondrial function with age we measured oxygen consumption in live, old (day 11) animals. Basal respiration is increased in drp-1;fzo-1 double mutants compared to wild-type worms (Figure 5C). Maximal respiration is not different however, suggesting that increased basal respiration in drp-1;fzo-1 double mutants is not solely due to increased mitochondrial content (Figure S2C). Furthermore, the effects of co-inhibiting fusion and fission on respiration are conserved in mammalian cells. We knocked down Drp1 via shRNA in Mfn2 null mouse embryonic fibroblasts (MEFs) to generate cells deficient in both fusion and fission, and measured oxygen consumption in the absence of glucose to force dependence on mitochondrial oxidative phosphorylation for respiration. Similar to drp-1;fzo-1 worms, Mfn2;Drp1-deficient MEFs have increased basal respiration compared to wild-type, while maximal respiration and spare respiratory capacity are not significantly different (Figures 5D and S2E–G). Next, we asked whether preserving network homeostasis affects lifespan. Remarkably, while individual deletion of either drp-1 or fzo-1 does not alter lifespan, co-deletion of drp-1 and fzo-1 increases lifespan (Figure 5E), demonstrating that maintaining mitochondrial network homeostasis with age is sufficient to promote longevity.
Figure 5. Maintaining mitochondrial network homeostasis is sufficient to increase lifespan under ad libitum fed but not intermittent fasting conditions.

(A) Mitochondrial networks in muscle cells of wild-type, drp-1 mutant, fzo-1 mutant, and drp-1;fzo-1 double mutant worms on day 1 and 8. (B) Quantification of (A): drp-1;fzo-1 double mutants maintain connected mitochondrial networks with age. Mean ± SEM of n=11–18 muscle cells from different worms. **p<0.01, ****p<0.0001 by t test. See also Figure S2. (C) Basal oxygen consumption rate (OCR) is increased in old (day 11) drp-1;fzo-1 double mutant worms vs wild-type (WT). Mean ± SEM of 10 replicates, *p<0.05 by t test. (D) Raw averaged traces of oxygen consumption from WT and Mfn2;Drp1-deficient MEFs in the presence of 20mM galactose, 4mM pyruvate and 2mM glutamine. Mean ± SEM of n=3–5 independent experiments. See also Figure S2. (E) Survival curves demonstrating that drp-1;fzo-1 double mutant worms are long-lived. (F) Representative images showing that mitochondrial networks undergo remodeling in response intermittent fasting (IF) in wild-type but not drp-1;fzo-1 mutant worms. (G) Mitochondrial network area:perimeter ratio on day 4, 6, 8, and 10, from wild-type and drp-1;fzo-1 mutant worms fed ad libitum (AL) or with IF. Worms in the IF group were fasted from day 2–4 and day 6–8. Mean ± SEM of n=12–26 muscle cells from 2 independent experiments. *p<0.05, ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. See also Figure S3. (H) and (I) Survival curves showing that IF extends lifespan of wild-type but not drp-1;fzo-1 mutant worms. See Table S1 for lifespan statistics.
Mitochondrial network plasticity is required for intermittent fasting (IF)-mediated longevity
Fusion and fission are integral processes with many important roles for cellular and metabolic health. It was therefore surprising to find that drp-1;fzo-1 double mutants, which lack the ability to undergo these processes, are long-lived. Intrigued by this result, we hypothesized that morphologically balanced yet static networks may be beneficial under normal laboratory conditions, but that upon exposure to environmental conditions requiring metabolic adaptation, a lack of network plasticity would be detrimental. To test this, we exposed wild-type and drp-1;fzo-1 double mutant animals to a dietary regimen of IF, previously reported to increase lifespan in C. elegans, and confer numerous health benefits in mammals (Honjoh et al., 2009; Mattson et al., 2016). Animals in the IF group underwent cycles of AL feeding for 2 days, followed by 2 days without food. Mitochondrial networks in wild-type animals undergo striking remodeling in response to IF; networks cycle between being less connected after fasting, and more fused upon refeeding, with corresponding fluctuations in mitochondrial size (Figures 5F, 5G, S3A and S3B). Furthermore, whereas mitochondrial network connectivity, size and content decrease with age in worms fed AL, these parameters are maintained between day 1 and day 10 by IF. While drp-1;fzo-1 double mutant networks are protected from age-related changes, they are unable to adapt to IF, with no change in connectivity between fasted and fed states (Figures 5F and 5G). To determine if network remodeling in response to IF is required for IF-mediated longevity, we assayed lifespan in wild-type and drp-1;fzo-1 double mutants under AL and IF conditions. IF increases lifespan in wild-type animals, but fails to do so in drp-1;fzo-1 double mutants (Figures 5H and 5I). In addition, IF-mediated longevity is blocked or partially suppressed by inhibition of fission or fusion individually (Figures S3C-F; Table S1). These data show that while maintaining morphologically balanced mitochondrial networks with age is sufficient to increase lifespan under basal AL conditions, network plasticity is required to increase lifespan in response to changing nutrient availability.
Maintaining mitochondrial network homeostasis increases lifespan via FAO and coordination with peroxisomes
Different mitochondrial network states have been associated with longevity, but it is unclear what functional outputs specifically couple network state to longevity. Mitophagy, facilitated by network fragmentation, was shown to be partially required for lifespan extension in several longevity pathways (Palikaras et al., 2015). Knockdown of mitophagy genes pink-1 and pdr-1 (homologous to mammalian parkin) does not shorten the lifespan of drp-1;fzo-1 animals however (Figure S4A). Similarly, pink-1 and pdr-1 RNAi do not increase wild-type lifespan (Figure S4B), together suggesting that altered mitochondrial turnover (either increased or decreased) does not underlie the longevity of drp-1;fzo-1 animals. Given recent studies demonstrating that mitochondrial morphology directly modulates cellular metabolism (Wai and Langer, 2016), we next hypothesized that maintaining mitochondrial network homeostasis might increase lifespan via metabolic reprogramming. To examine how manipulating mitochondrial network architecture affects metabolic state we performed targeted metabolite profiling in young adult drp-1 mutants, fzo-1 mutants and drp-1;fzo-1 double mutants. Consistent with fused networks promoting oxidative metabolism and altering protein biosynthesis (Wang et al., 2015), drp-1 mutants have decreased lactate, increased succinate, and increased levels of most amino acids (Figures 6A and S4C). fzo-1 mutant amino acid and acylcarnitine levels are broadly decreased and increased, respectively, suggesting protein and fatty acid metabolism may be perturbed by fragmented networks (Figures 6B and S4C–E). Interestingly, rather than directly mirroring wild-type animals or either of the single mutants, the metabolic profile of drp-1;fzo-1 double mutants appears more complex; several TCA intermediates are uniquely decreased in drp-1;fzo-1 double mutants (Figure 6A), while amino acid levels are similar to fzo-1 mutant levels, with a general trend towards decreased levels compared to wild-type animals (Figure S4C). Suggesting that fatty acid oxidation (FAO) is altered, very long- and lo ng-chain acylcarnitines are markedly elevated, with a similar trend in medium- and short-chain acylcarnitines (Figures 6B, S4D and S4E). Increased steady state acylcarnitines could be indicative of either increased flux of FAs through the FAO pathway, or decreased flux leading to an accumulation of FAs. To explore this further, we assayed FAO capacity in wild-type and Mfn2;Drp1-deficient MEFs by measuring palmitate oxidation. FAO capacity is increased in Mfn2;Drp1-deficient cells, shown by elevated basal and maximal respiration (Figure 6C). Next, to test if increased FAO is required for drp-1;fzo-1 longevity, we inhibited FAO by treating worms with the carnitine palmitoyltransferase (CPT) inhibitor perhexiline (PHX) (Kennedy et al., 1996). PHX suppresses drp-1;fzo-1 longevity (Figures 6D and 6E), suggesting that maintaining network homeostasis increases lifespan via increased FAO.
Figure 6. Fatty acid oxidation and peroxisome function are required for drp-1;fzo-1-mediated longevity.

(A) and (B) Metabolomic analyses of wild-type, drp-1 mutant, fzo-1 mutant, and drp-1;fzo-1 double mutant worms on day 1. Levels of organic acids (A) and very long-/long-chain acylcarnitines (B) are expressed as log2 fold change relative to wild-type levels. Mean ± SEM of 5–6 biological replicates. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by two-way ANOVA with Tukey’s multiple comparisons test. Statistics shown are relative to wild-type levels. See also Figure S4 for amino acids, short- and medium-chain acylcarnitines. (C) Basal and maximal oxygen consumption during palmitate oxidation showing total fatty acid oxidation (FAO) capacity is increased in Mfn2;Drp1-deficient MEFs. Palmitate-specific oxidation was obtained by taking the rate of palmitate oxidation in the absence of etomoxir and subtracting the rate in the presence of etomoxir (40μM), corrected for non-mitochondrial respiration rates. Mean ± SEM of n=3 replicates, *p<0.05 by t test. (D) and (E) Inhibiting FAO with perhexiline (PHX) suppresses drp-1;fzo-1-mediated longevity. (F) Fluorescence images of peroxisomes in L4 wild-type and drp-1;fzo-1 mutant worms, visualized with vha-6p::mRFP-PTS1. (G) Quantification of (H): Peroxisomes are larger in drp-1;fzo-1 mutant worms. Median and interquartile range of n=1261–4762 peroxisomes from 2 independent experiments. ****p<0.0001 by t test. (I) and (J) prx-5 RNAi suppresses drp-1;fzo-1-mediated longevity. See Table S1 for lifespan statistics.
Functional interplay exists between mitochondria and peroxisomes in the regulation of FAO, and oxidation of very long- and long-chain fatty acids takes place in peroxisomes (Wanders et al., 2015). In addition, peroxisome import and protein expression decrease with age (Narayan et al., 2016). We therefore asked if peroxisomes have a role in drp-1;fzo-1 longevity. Peroxisomal dynamics and size are remodeled according to cellular metabolic requirements, and increased peroxisome size has been linked to elevated expression of β-oxidation enzymes in yeast (Oeljeklaus et al., 2016; Smith et al., 2000). To determine if peroxisome size and morphology are altered in drp-1;fzo-1 double mutant animals we used a transgenic strain expressing RFP fused to a peroxisome targeting sequence driven by an intestinal promoter (vha-6p::mRFP-PTS1). Peroxisome morphology is more fused in drp-1;fzo-1 double mutant animals, and peroxisomes are significantly larger compared to those in wild-type animals (Figures 6F and 6G). To determine if peroxisomal function is required for drp-1;fzo-1 longevity we performed RNAi of prx-5 (ortholog of mammalian peroxisome biogenesis factor 5 (PEX5)). PRX-5/PEX5 is involved in the import of peroxisomal matrix enzymes (Narayan et al., 2016). Consistent with this and as previously reported (Narayan et al., 2016), prx-5 RNAi blocks import into peroxisomes, resulting in the mRFP-PTS1 fluorescence signal appearing diffuse, compared to punctate signal characteristic of peroxisomes in the control (Figure S5). Demonstrating that peroxisome function is required for drp-1;fzo-1 longevity, prx-5 RNAi blocks the increased lifespan of drp-1;fzo-1 double mutant animals (Figures 6H and 6I). Moreover, RNAi of the peroxisomal-specific enzyme dhs-28, which catalyzes the final step in peroxisomal β-oxidation, also suppresses drp-1;fzo-1 longevity (Table S1). Together, this suggests preserving mitochondrial network homeostasis increases lifespan via mitochondria-peroxisome coordination to increase FAO.
Peroxisome function is involved in AMPK and DR-mediated longevity
The requirement for both fusion and fission suggests that AMPK increases lifespan by maintaining mitochondrial network homeostasis. In support of this, activation of AMPK has no additive effect on drp-1;fzo-1 longevity (Figures 7A and 7B; Table S1), suggesting these longevity interventions act in the same pathway. Therefore, we hypothesized that AMPK also requires FAO and peroxisome function to increase lifespan, and thus may share a common metabolic profile with drp-1;fzo-1 animals. We performed metabolite profiling in young adult drp-1, fzo-1 and drp-1;fzo-1 mutants, with and without CA-AAK-2. Hierarchical clustering analysis revealed that CA-AAK-2 in conjunction with either drp-1 or fzo-1 deletion clusters with each single mutant alone (Figure S6). Conversely, whereas drp-1;fzo-1 double mutants cluster with fzo-1 mutants, drp-1;fzo-1;CA-AAK-2 worms cluster more closely with wild-type animals (Figure S6). Despite this interaction, we did not detect a common metabolic signature between CA-AAK-2 worms and drp-1;fzo-1 double mutants. Interestingly, a recent study in C. elegans that examined the effect of age on metabolite profiles reported that FA levels in wild-type and aak-2 null mutants are very similar at day 1. However, there is an increase in wild-type FA levels at day 7 that is blunted in aak-2 mutants (Gao et al., 2017), which led us to hypothesize that activating AMPK might increase lifespan via a shift to FAO as animals age. In support of a causal role for FAO, treatment with PHX has no effect on wild-type animals but reduces lifespan extension by 50% in CA-AAK-2 animals (Figures 7C and 7D; Table S1).
Figure 7. AMPK- and DR-mediated longevity involves peroxisome function.

(A) and (B) Survival curves showing that CA-AAK-2 has no additive effect on the lifespan of drp-1;fzo-1 double mutant worms. (C) and (D) Inhibiting fatty acid oxidation with PHX partially suppresses CA-AAK-2-mediated longevity. (E) Peroxisome size in YA wild-type and eat-2(ad1116) mutants. Median and interquartile range of n=2174–3187 peroxisomes, *p<0.05 by t test. (F) DR by bacterial dilution maintains peroxisome density (% cell coverage) with age. Mean ± SEM of n=18–25 worms. **p<0.01, ****p<0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (G) and (H) prx-5 RNAi reduces DR-mediated longevity. (I) CA-AAK-2 does not alter peroxisome size in young adult (YA) stage worms. Median and interquartile range of n=879–918 peroxisomes. (J) Phenformin treatment from YA stage maintains peroxisome density between YA and day 8. Mean ± SEM of n=11–35 worms. ***p<0.001 by one-way ANOVA with Tukey’s multiple comparisons test. (K) and (L) prx-5 RNAi suppresses CA-AAK-2-mediated longevity. See Table S1 for lifespan statistics.
Next, we examined whether FAO and peroxisome function are involved in DR-mediated longevity. Suggesting a role for both, we recently identified KEGG pathways upregulated by DR (Heintz et al., 2017), and found a significant enrichment of peroxisomal genes by DR in young animals, and in both peroxisomal and FA metabolism genes in old animals (Figure S7, (Heintz et al., 2017)). DR slightly decreases peroxisome size in young adults (Figure 7E), however whereas wild-type peroxisome content decreases with age, this is maintained between young adulthood and day 8 in diet-restricted worms (Figure 7F). Moreover, prx-5 and dhs-28 RNAi reduce lifespan extension by DR (Figures 7G and 7H; Table S1), showing that peroxisomal function is causally involved in DR-mediated longevity. Finally, we asked if peroxisome function is required for AMPK-mediated longevity. Supporting a specific role for peroxisomes with age, CA-AAK-2 does not change peroxisome size in young adults (Figure 7I), but as with DR, activating AMPK prevents the age-related decrease in peroxisome content (Figure 7J). Crucially, prx-5 RNAi suppresses CA-AAK-2 lifespan extension (Figures 7K and 7L; Table S1), demonstrating that peroxisome function is required for AMPK-mediated longevity. Collectively, these data suggest that AMPK and DR increase lifespan via maintaining mitochondrial network homeostasis and plasticity with age, involving altered FAO and crosstalk with peroxisomes.
Discussion
Previous studies in yeast and C. elegans implicated fused mitochondrial networks as being pro-longevity (Chaudhari and Kipreos, 2017; Houtkooper et al., 2013; Jiang et al., 2015; Scheckhuber et al., 2007). However, recent reports using C. elegans and Drosophila have also linked increased fission to longevity (Han et al., 2017; Rana et al., 2013), suggesting that specific network states are not universally pro- or anti-aging, and the relationship between network state and longevity is more complex. Our study demonstrates that AMPK and DR require both fusion and fission to increase lifespan, implying that maintaining the balance or capacity to remodel networks between fused and fragmented/divided states is vital for these longevity pathways. Further, our data shed light on the physiological significance of network remodeling, revealing a novel role for metabolic reprogramming and functional coordination between mitochondria and peroxisomes.
Supporting a model in which the requirement for different network states is context-specific, we find that fusion and fission are not universally required to increase lifespan, as both daf-2 and cco-1 knockdown increase lifespan with or without fzo-1 or drp-1. Our study also shows that fusion is required specifically in peripheral tissues for AMPK-mediated longevity, suggesting that both pathway-specific and tissue-specific requirements exist for particular network states. In addition, DR not only fails to increase lifespan in fusion-deficient animals, but results in shortened lifespan compared to control animals fed AL, underscoring the critical importance of mitochondrial fusion for the beneficial effects of DR. Nutrient deprivation in cells induces fusion, likely to maintain ATP production since mitochondria in fused networks are thought to be more respiration-efficient and are protected from degradation by mitophagy (Liesa and Shirihai, 2013). It will be interesting to determine if these outputs of fusion are also required for DR-mediated longevity. Of note, a parallel study showed that eat-3 and fzo-1 RNAi suppressed or reduced daf-2(e1370) mutant longevity, respectively. This suggests the requirement for fusion may be dependent on precise levels of IIS and/or fusion, and points to a specific role for inner mitochondrial membrane fusion worthy of further exploration. (Chaudhari and Kipreos, 2017). Together, this highlights that understanding the functional consequences of mitochondrial network remodeling, as well as the physiological contexts in which they are required, is crucial in order to harness potential therapeutic benefits.
Unraveling the relative importance of mitochondrial network morphology versus network plasticity (i.e the capacity to remodel networks) for longevity will aid advances in the field. We show that maintaining mitochondrial network homeostasis by co-inhibition of fusion and fission is sufficient to increase lifespan, suggesting that, under basal conditions, maintaining youthful morphology is advantageous over a plastic network that becomes vulnerable to age-related fragmentation. However, active mitochondrial network remodeling between more and less fused states is required for IF-mediated longevity, implying that maintaining the capacity to remodel networks is necessary for adaptation to different conditions. Contrary to our C. elegans data, a study performed in yeast showed shortening of replicative lifespan upon co-inhibition of fusion and fission (Bernhardt et al., 2015). This apparent discrepancy, together with our observations that inhibiting fusion or fission individually does not affect lifespan in C. elegans, may indicate an evolutionary shift in the effects of altered mitochondrial morphology on lifespan. Furthermore, recent work performed in mice demonstrated that concomitant deletion of Mfn1 in Mff mutant (fission-deficient) mice restores mitochondrial network balance and leads to rescue of mitochondrial function and tissue integrity (Chen et al., 2015). Our data further show that co-inhibiting fusion and fission increases FAO capacity in mammalian cells, which is required for drp-1;fzo-1 longevity. A critical next step will be to determine if the potential to promote healthy aging by restoring network homeostasis is conserved in mammals.
AMPK and DR delay age-related changes to mitochondrial networks, suggesting that age-related loss of mitochondrial homeostasis can be targeted to promote longevity. Recent studies have demonstrated that AMPK promotes both fusion and fission under different conditions, via direct and indirect mechanisms (Kang et al., 2016; Toyama et al., 2016; Wang et al., 2017). Our data show that chronic activation of AMPK maintains mitochondrial network homeostasis, however under normal physiological conditions the capacity to activate AMPK diminishes with age (Reznick et al., 2007). Taken together, this could point to dysfunctional AMPK activity as an underlying cause of age-related loss of network homeostasis. In addition we show phenformin, which indirectly activates AMPK, also delays age-related changes to both mitochondria and peroxisomes. For future studies it will be important to determine if the impact of phenformin on mitochondria results exclusively from AMPK action, and whether a direct AMPK agonist such as A-769662 elicits the same effects (Cool et al., 2006).
Moving forward, identifying the specific mechanisms that couple mitochondrial network remodeling to longevity will be informative, and our data implicate FAO and mitochondria-peroxisome crosstalk for AMPK and DR-mediated longevity. Mitochondria and peroxisomes function coordinately in several pathways, and share several components of the fission machinery including DRP1 (Rambold et al., 2015; Schrader, 2006; Sugiura et al., 2017). Peroxisome protein expression and import were recently shown to decrease with age in C. elegans (Narayan et al., 2016), however until now, it was unknown whether a longevity-associated role existed for interplay between these organelles. Our data reveal that AMPK and DR specifically modulate peroxisomes with age, suggesting the functional requirement - perhaps involving a metabolic shift towards increased FAO - may be of particular importance in later life. Determining if the metabolic flexibility required for contexts such as IF involves coordinated regulation of mitochondria and peroxisomes will be an exciting area for future studies.
Collectively, our study identifies mitochondrial network remodeling involving communication with peroxisomes as a downstream mechanism by which DR and AMPK increase lifespan. Targeting mechanisms that maintain mitochondrial homeostasis and plasticity in the elderly may therefore represent a novel strategy to restore metabolic homeostasis and promote healthy aging.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William Mair (wmair@hsph.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C. elegans strains CU5991 (fzo-1(tm1133) II), CU6372 (drp-1(tm1108) IV), RW1596 (stEx30[myo-3p::GFP + rol-6(su1006)]), VS10 (hjIs37[vha-6p::mRFP-PTS1 + Cbr-unc-119(+)]), VS15 (hjIs8 [ges-1p::GFP-PTS1]), DA1116 (eat-2(ad1116) II) and N2 wild-type C. elegans strains were obtained from the Caenorhabditis Genetic Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). The eat-3(tm1107) strain was a gift from Alexander van der Bliek. The mRFP mitochondrial reporter strain was supplied by Sean Curran/Paul Sternberg. The GFP muscle mitochondrial reporter strain (myo-3p::tomm-20(1-49aa)::GFP) was generated in the lab by PCR amplification from genomic DNA encoding amino acids 1-49 of TOMM-20. GFP was cloned directly downstream and the construct driven by a muscle-specific (myo-3) promoter. The GFP intestinal mitochondrial reporter was generated by subcloning the intestine-specific ges-1 promoter in place of the myo-3 promoter. Worms were grown and maintained on standard nematode growth media (NGM) seeded with Escherichia coli (OP50-1), and maintained at 20°C.
Microbe strains
E. coli (OP50-1) bacteria was cultured overnight in LB at 37°C, after which 100 μl of liquid culture was seeded on plates to grow for 2 days at room temperature. RNAi experiments were performed on E. coli (HT115) expressing dsRNA against the gene noted or an empty vector control. HT115 was cultured overnight in LB containing carbenicillin (100μg ml−1) and tetracycline (12.5μg ml−1). daf-2, cco-1 and pdr-1 RNAi constructs were obtained from the Vidal library. All other RNAi constructs were obtained from the Ahringer library.
Cell lines
Stable Mfn2;Drp1-deficient MEFs were generated from Mfn2-null MEFs (ATCC CRL-2993; sex unknown) transfected with DNM1L shRNA lentiviral transduction particles (Sigma Mission SHCLNV-NM_152816) after puromycin (5μg/mL) selection. Control MEFs were transfected with scrambled shRNA lentiviral transduction particles (Sigma Mission SHC202) and subjected to the same puromycin selection process. A multiplicity of infection of two was used, and five different shRNAs were tested. Cells generated using clone TRCN0000321169 were used as this clone gave the most efficient silencing.
METHOD DETAILS
Rescue constructs
fzo-1 rescue strains were generated by subcloning fzo-1 cDNA downstream of a 3xFLAG tag in a standard expression vector based on the Fire Lab C. elegans Vector Kit. Expression was driven by the following promoters: sur-5p (ubiquitous expression), rab-3p (neuron-specific), myo-3p (muscle-specific), and ges-1p (intestine-specific).
Lifespans
Lifespan experiments were performed as described previously (Burkewitz et al., 2015). All experiments were performed on standard NGM at 20°C. All lifespans were performed on OP50-1, except for RNAi experiments which were performed on HT115. RNAi experiments were performed from hatch in all cases, on standard NGM containing carbenicillin (100μg ml−1). Expression of dsRNA was induced by pipetting 100μl IPTG solution (100mM, containing carbenicillin (100μg ml−1) and tetracycline (12.5μg ml−1)) onto HT115 lawns prior to introduction of worms. Worms were synchronized by timed egg lays using gravid adults. When the progeny reached adulthood (~72 hr), 100–150 worms were transferred to fresh plates with 20 worms per plate. Worms were transferred to fresh plates every other day until reproduction had ceased (day 10–12). Survival was scored every 1–2 days and a worm was deemed dead when unresponsive to 3 taps on the head and tail. Worms were censored due to contamination on the plate, leaving the NGM, eggs hatching inside the adult or loss of vulval integrity during reproduction. Lifespan analysis was performed using GraphPad Prism.
Plate dietary restriction assays
Solid plate-based DR assays were performed as described previously (Heintz et al., 2017). Plates were prepared in advance and stored at 4°C. Ad libitum (AL) plates were prepared with a bacterial concentration of 1011 cfu/ml and DR plates with 108 cfu/ml bacterial concentration. 5-Fluoro-2′-deoxyuridine (FUDR) was added on top of the bacterial lawn (100μl; 1mg ml−1) 24 hours before worms were introduced to the plates on the first day of adulthood. FUDR was added on top of the bacterial lawn (100μl; 1mg ml−1) 24 hours before worms were introduced to the plates on the first day of adulthood. Worms were transferred off FUDR-containing plates once reproduction had ceased (7–10 days). FUDR was also used for eat-2(ad1116) lifespans.
Intermittent fasting
Synchronized populations of late L4 stage worms (100–180) were transferred to fresh NGM plates seeded with OP50-1 and FUDR. On day 2 of adulthood, worms in the IF group were transferred to NGM plates containing carbenicillin (100μg ml−1) and FUDR, without any bacteria. Worms in the control group were transferred to NGM plates seeded with OP50-1 and FUDR. On day 4 of adulthood, worms in both the IF and control groups were transferred to NGM plates seeded with OP50-1. Worms underwent 3 cycles of IF, where 1 cycle = 2 days in the absence of bacteria followed by 2 days in the presence of bacteria. Use of FUDR was ceased at day 10 of adulthood. On day 12, all worms were transferred to NGM plates seeded with OP50-1 for the remainder of their lifespan.
Phenformin treatment
L4 stage worms were transferred to NGM plates containing 4.5mM phenformin and seeded with OP50-1. FUDR was used for lifespan experiments to prevent excessive censoring due to internal hatching.
Immunoblotting
After 24h exposure to 0mM or 4.5mM phenformin from L4 stage, day 1 adult worms (300–500 worms/replicate) were harvested and washed 3 times in M9, pelleted, and snap frozen in liquid nitrogen. Samples were resuspended in 75μL RIPA buffer supplemented with protease and phosphatase inhibitors before lysis via sonication (Qsonica Q700). Lysates were centrifuged at 14000g, 5 min, 4°C to remove debris before measuring protein concentration using the BCA assay (Pierce PI23227). Lysates were adjusted to 1μg/ml and Laemmli buffer + β-mecaptoethanol was added before boiling at 95°C, 5 min. 20μg protein/sample was loaded on a 7.5% acrylamide gel for separation by SDS-PAGE. Proteins were transferred to PVDF membrane and blocked with 5% BSA before incubation with primary antibodies overnight at 4°C. Primary a ntibodies and dilutions: phospho-AMPKα Thr172 (Cell signaling #2535, 1:1000); β-actin (Abcam #8226, 1:1000). After incubation, membranes were washed 3 times in TBS-T before incubation with HRP-conjugated secondary antibody (anti-mouse or anti-rabbit, Cell Signaling, 1:2000) for 1h at RT. Bands were visualized on a Gel Doc system (Bio Rad) using ECL Western Blotting Detection Reagent (GE Healthcare).
Perhexiline (PHX) treatment
A stock concentration of 100mM was made by dissolving PHX in DMSO, which was diluted to a working concentration of 2.5mM with water. 100μl of 2.5mM PHX was pipetted onto OP50-1 bacterial lawns prior to transferring worms to plates. Worms were exposed to PHX from hatch.
Microscopy, mitochondrial and peroxisomal image analysis
Worms were anaesthetized for 20 min in 0.2 mg ml−1 tetramisole/M9, mounted on 2% agarose pads on glass slides under coverslips, and subsequently imaged on Zeiss Discovery V8 and ApoTome.2-equipped Imager M2 microscopes with Axiocam cameras. Mitochondrial network morphology was detected using strains expressing either mRFP or GFP targeted to the outer mitochondrial membrane. Images were taken of >10 muscle or intestine cells from at least 10 worms per condition, with a minimum of 2 independent experiments performed. Muscle mitochondria were analyzed in cells midway between the pharynx and vulva, or vulva and tail. Intestinal mitochondria were analysed in the posterior intestinal cell adjacent to the tail. Quantification was performed with ImageJ, using a macro to measure mitochondrial content, size and network connectivity (Dagda et al., 2009). Peroxisomes were detected in worms expressing intestinal mRFP-PTS1 or GFP-PTS1. Optical sections of intestinal mid-sections were captured at 63x magnification. To quantify peroxisomes, images were thresholded using ImageJ and the “moments” method. Peroxisome size was quantified using the entire image and a minimum pixel area of 10 to define particles. To measure peroxisome density, an area of the intestine was defined between two visible intestinal nuclei. Peroxisome density was calculated as a percentage of pixels in the image that were defined as peroxisomes (minimum pixel area of 10). Peroxisomes were quantified in at least 10 worms per condition from 2 independent experiments.
Oxygen consumption assays
Oxygen consumption was measured using a Seahorse XF96 analyzer (Agilent Technologies).
Worms
Worms were washed from NGM plates, washed 3 times with M9 buffer and transferred to a 96 well plate (10 worms/well). Basal respiration was measured 10 times followed by the addition of FCCP (10μM) to measure maximal respiration, which was measured 6 times. Oxygen consumption rates were normalized to the number of worms in each well. Values were calculated from the mean of 10 wells per condition.
Cells
24h before the assay, 20000 cells were cultured in 100μL DMEM supplemented with 10% cosmic calf serum in a seahorse 96 well plate. 3h before the assay, cells were carefully washed three times in PBS and incubated in 100μL of Seahorse DMEM without glucose supplemented with 20mM galactose, 2mM glutamine, 4mM pyruvate and 0.3% w/v fatty-acid free bovine serum albumin, pH 7.4, 37°C. The respiratory control and oxidative phosphorylation capacity was assayed by addition of 4μM oligomycin in port A, 9μM FCCP in port B, and 4μM rotenone plus 4μM antimycin A in port C. FCCP titration was performed to determine the optimal concentration to measure maximal respiration. Fatty acid oxidation (FAO) was measured using the XF Palmitate-BSA FAO Substrate with the XF Cell Mito Stress Test (Agilent Seahorse), according to manufacturer’s instructions. 3h before the assay, cells were incubated in FAO buffer (111mM NaCl, 4.7mM KCl, 1.25mM CaCl2, 2mM MgSO4, 1.2mM NaH2PO4, 5mM HEPES) supplemented with 2.5mM Glucose and 0.5mM carnitine, pH7.4, 37°C. 100μL FAO buffer was added to the cells 45 min before the assay. 40μM etomoxir was added to control wells 15 mins before the assay. BSA control or palmitate conjugated with BSA was added to the appropriate wells immediately before the run. The respiratory control and the oxidative phosphorylation capacity was assayed as described above. FCCP was titrated under FAO assay conditions and 12μM was used to measure maximum respiratory capacity.
Metabolomics
Synchronized worm populations grown on NGM seeded with OP50-1 were collected on day 1 of adulthood. Worms were washed 3 times with M9 buffer before resuspending pellets 1:1 with 0.6% formic acid. Pellets were lysed by sonication, diluted 1:1 with acetonitrile and snap frozen. Metabolite profiling was performed as described previously (Burkewitz et al., 2015). Metabolite levels were normalized to protein concentration measured from an aliquot taken prior to acetonitrile addition. 5–6 biological replicates were used.
QUANTIFICATION AND STATISTICAL ANALYSIS
ImageJ, including the ‘Mitochondrial Morphology’ macro (Dagda et al., 2009) was used for quantification of images. GraphPad Prism was used for statistical analyses. Statistical details of experiments can be found in the figure legends. The Log-rank (Mantel-Cox) method was used to compare survival curves. Two-way ANOVA with Tukey’s multiple comparisons test was used to analyze the metabolomics data. Student’s t test or one-way ANOVA with Tukey’s multiple comparisons test were used to analyze all other data sets. For all experiments p values <0.05 were considered significant.
Supplementary Material
Survival curve p values calculated with Log-rank (Mantel-Cox) test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Table S2. AL and DR gene expression in ‘Peroxisome’ KEGG pathway on day 3. Related to Figures 7 and S7.
Table S3. AL and DR gene expression in ‘Peroxisome’ KEGG pathway on day 15. Related to Figures 7 and S7.
Table S4. AL and DR gene expression in ‘Fatty acid metabolism’ KEGG pathway on day 15. Related to Figures 7 and S7.
Highlights.
AMPK and DR maintain mitochondrial network homeostasis with age
Mitochondrial fusion and fission are required for DR- and AMPK-mediated longevity
Preserving mitochondrial homeostasis increases lifespan via fatty acid oxidation
These longevity pathways require coordination between mitochondria and peroxisomes
Acknowledgments
This work was supported by funding from The Lawrence Ellison Medical Foundation (U54CA155626), The Glenn Foundation for Medical Research, the National Institutes of Health (NIH, 1R01AG044346 (W.B.M) and 1R01AG045351 (M.D.H)), and the American Diabetes Association/Canadian Diabetes Association PF-3-13-4342 (F.K.H). We thank the Caenorhabditis Genetic Center for providing strains. We are grateful to Gökhan Hotamisligil for support and access to resources. We also thank the Mair laboratory members for comments and discussion on the project and manuscript.
Footnotes
Author Contributions
H.J.W and W.B.M designed the study and wrote the manuscript. H.J.W performed most experiments and analyzed results. P.Y and K.B performed peroxisome imaging experiments. C.E performed the immunoblotting. R.L.G generated the MEF cell line and performed oxygen consumption assays. P.Y, C.E and R.L helped with lifespan repeats. F.K.H and M.D.H performed the metabolite profiling and analyzed the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apfeld JG, O’connor TM. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt D, Muller M, Reichert A, Osiewacz H. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. 2015;5:7885. doi: 10.1038/srep07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonda DJ, Smith MA, Perry G, Lee HG, Wang X, Zhu X. The mitochondrial dynamics of Alzheimer’s disease and Parkinson’s disease offer important opportunities for therapeutic intervention. Curr Pharm Des. 2011;17:3374–3380. doi: 10.2174/138161211798072562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Morantte I, Weir H, Yeo R, Zhang Y, Huynh F, Ilkayeva O, Hirschey M, Grant A, Mair W. Neuronal CRTC-1 Governs Systemic Mitochondrial Metabolism and Lifespan via a Catecholamine Signal. Cell. 2015;160:842–855. doi: 10.1016/j.cell.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari SN, Kipreos ET. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun. 2017;8:182. doi: 10.1038/s41467-017-00274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ren S, Clish C, Jain M, Mootha V, Jm M, Chan DC. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol. 2015;211:795–805. doi: 10.1083/jcb.201507035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Dietrich M, Liu Z, Horvath T. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 2013;155:188–199. doi: 10.1016/j.cell.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106–118. doi: 10.1016/j.cell.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao AW, Chatzispyrou IA, Kamble R, Liu YJ, Herzog K, Smith RL, van Lenthe H, Vervaart MAT, van Cruchten A, Luyf AC, et al. A sensitive mass spectrometry platform identifies metabolic changes of life history traits in C. elegans. Sci Rep. 2017;7:2408. doi: 10.1038/s41598-017-02539-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Sivaramakrishnan P, Lin CCJ, Neve IAA, He J, Tay LWR, Sowa JN, Sizovs A, Du G, Wang J, et al. Microbial Genetic Composition Tunes Host Longevity. Cell. 2017;169:1249–1262. e13. doi: 10.1016/j.cell.2017.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz C, Doktor TK, Lanjuin A, Escoubas C, Zhang Y, Weir HJ, Dutta S, Silva-García CG, Bruun GH, Morantte I, et al. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature. 2017;541:102–106. doi: 10.1038/nature20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2009;457:726–730. doi: 10.1038/nature07583. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HCC, Hsu JMM, Yen CPP, Chao CCC, Chen RHH, Pan CLL. Neural activity and CaMKII protect mitochondria from fragmentation in aging Caenorhabditis elegans neurons. Proc Natl Acad Sci U S A. 2015;112:8768–8773. doi: 10.1073/pnas.1501831112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa T, Zappaterra M, Hasegawa A, Wright A, ENS, Buttle K, KM, Mannella C, Bliek A. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008;4:e1000022. doi: 10.1371/journal.pgen.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SWS, Haydar G, Taniane C, Farrell G, Arias IM, Lippincott-Schwartz J, Fu D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS One. 2016;11:e0165638. doi: 10.1371/journal.pone.0165638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52:273–280. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Leduc-Gaudet JP, Picard M, St-Jean Pelletier F, Sgarioto N, Auger MJ, Vallée J, Robitaille R, St-Pierre DH, Gouspillou G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget. 2015;6:17923–17937. doi: 10.18632/oncotarget.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W, Morantte I, Rodrigues A, Manning G, Montminy M, Shaw R, Dillin A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature. 2011;470:404–408. doi: 10.1038/nature09706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev. 2016;S1568–1637:30251–30253. doi: 10.1016/j.arr.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan V, Ly T, Pourkarimi E, Murillo AB, Gartner A, Lamond AI, Kenyon C. Deep Proteome Analysis Identifies Age-Related Processes in C. elegans. Cell Syst. 2016;3:144–159. doi: 10.1016/j.cels.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeljeklaus S, Schummer A, Mastalski T, Platta HW, Warscheid B. Regulation of peroxisome dynamics by phosphorylation. Biochim Biophys Acta. 2016;1863:1027–1037. doi: 10.1016/j.bbamcr.2015.12.022. [DOI] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–528. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. doi: 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A. 2013;110:8638–8643. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regmi SG, Rolland SG, Conradt B. Age-dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging. 2014;6:118–130. doi: 10.18632/aging.100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznick RM, Zong H, Li J, Morino K, Moore IK. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckhuber C, Erjavec N, Tinazli A, Hamann A, Nystrom T, Osiewacz H. Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol. 2007;9:99–105. doi: 10.1038/ncb1524. [DOI] [PubMed] [Google Scholar]
- Scheckhuber C, Wanger R, Mignat C, Osiewacz H. Unopposed mitochondrial fission leads to severe lifespan shortening. Cell Cycle. 2014;10:3105–3110. doi: 10.4161/cc.10.18.17196. [DOI] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich M, Sebastian D, Imbernon M, Castano C, Garcia A, Esteban Y, AGF, Rodriguez I, Bortolozzi A, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M. Shared components of mitochondrial and peroxisomal division. Biochim Biophys Acta. 2006;1763:531–541. doi: 10.1016/j.bbamcr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to Metabolism. Mol Cell. 2016;61:683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Sebastián D, Sorianello E, Segalés J, Irazoki A, Vanessa RB, Sala D, Planet E, Antoni BL, Muñoz JP, Manuela SF, et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016;35:1677–1693. doi: 10.15252/embj.201593084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Brown TW, Eitzen GA, Rachubinski RA. Regulation of Peroxisome Size and Number by Fatty Acid β-Oxidation in the Yeast Yarrowia lipolytica. J Biol Chem. 2000;275:20168–20178. doi: 10.1074/jbc.M909285199. [DOI] [PubMed] [Google Scholar]
- Stenesen D, Suh JM, Seo J, Yu K, Lee KSS, Kim JSS, Min KJJ, Graff JM. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 2013;17:101–112. doi: 10.1016/j.cmet.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, Mattie S, Prudent J, McBride HM. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature. 2017;542:251–254. doi: 10.1038/nature21375. [DOI] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulgherait M, Rana A, Rera M, Graniel J, Walker DW. AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 2014;8:1767–1780. doi: 10.1016/j.celrep.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Langer T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wanders RJA, Waterham HR, Ferdinandusse S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front Cell Dev Biol. 2015;3:83. doi: 10.3389/fcell.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ishihara T, Ibayashi Y, Tatsushima K, Setoyama D, Hanada Y, Takeichi Y, Sakamoto S, Yokota S, Mihara K, et al. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia. 2015;58:2371–2380. doi: 10.1007/s00125-015-3704-7. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, Zou MH. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes. 2017;66:193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Chen D, Lee SS, Walter L. The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell. 2011;10:724–728. doi: 10.1111/j.1474-9726.2011.00711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Survival curve p values calculated with Log-rank (Mantel-Cox) test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Table S2. AL and DR gene expression in ‘Peroxisome’ KEGG pathway on day 3. Related to Figures 7 and S7.
Table S3. AL and DR gene expression in ‘Peroxisome’ KEGG pathway on day 15. Related to Figures 7 and S7.
Table S4. AL and DR gene expression in ‘Fatty acid metabolism’ KEGG pathway on day 15. Related to Figures 7 and S7.