

Abstract
Aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor is involved in regulation of many essential biological processes including vascular development and angiogenesis. 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) is an AhR ligand, which regulates immune responses and cancer cell growth. However, the roles of the ITE/AhR pathway in mediating placental angiogenesis remains elusive. Here, we determined if ITE affected placental angiogenic responses via AhR in human umbilical vein (HUVECs) and artery endothelial (HUAECs) cells in vitro. We observed that ITE dose- and time-dependently inhibited proliferation and viability of HUAECs and HUVECs, whereas it inhibited migration of HUAECs, but not HUVECs. While AhR siRNA significantly suppressed AhR protein expression in HUVECs and HUAECs, it attenuated the ITE-inhibited angiogenic responses of HUAECs, but not HUVECs. Collectively, ITE suppressed angiogenic responses of HUAECs and HUVECs, dependent and independent of AhR, respectively. These data suggest that ITE may regulate placental angiogenesis.
Keywords: ITE, Aryl hydrocarbon receptor, Angiogenesis, Endothelial cells
1. Introduction
Placental angiogenesis is an essential process to support rapid placental and fetal growth under physiologic conditions during pregnancy [1,2]. In contrast, either abnormal angiogenesis or aberrant expression of angiogenic factors is associated with various human diseases, e.g., intrauterine growth retardation and preeclampsia [3–5]. Thus, better understanding of mechanisms underlying placental angiogenesis might open a new therapeutic avenue to target these angiogenesis-related diseases [3–5].
Aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor, which is expressed in various tissues and cells including in human placentas [6] and endothelial cells [7,8]. It is well-documented that AhR is activated by many environmental toxicants (i.e., 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), which could lead to increased expression of many downstream enzymes involved in metabolism of toxicants [7]. In addition, perinatal exposure to air pollution and cigarette smoke has also been shown to trigger the AhR pathway as polyaromatic hydrocarbons are AhR ligands, leading to adverse pregnancy outcomes such as intrauterine growth restriction and low birth weight [9]. However, evidence from mouse knockout studies has defined the physiological roles of AhR in numerous biological processes including cardiovascular growth and development [10–12]. Specifically, AhR knockout can lead to formation of abnormal vascular structure in multiple organs and cardiac hypertrophy [10–12].
The physiological importance of AhR is also supported by the discovery of many AhR endogenous ligands, including 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE), which is presumingly derived from tryptophan and cysteine via a condensation reaction [13]. ITE was first identified in porcine lung tissues [14]. Recently, ITE was found to be present in human tumor cells in vitro [15]. Thus far, the physiological level of ITE has not been reported in any tissue in any species. However, ITE can regulates a variety of cellular functions. For instance, ITE can induce the differentiation of stem-like cancer cells and reduce their tumorigenic potential [15]. ITE also suppresses immunologic responses by directly targeting dendritic and T cells [16,17]. We have observed that ITE attenuates the growth of human ovarian cancer cells in vitro and in vivo [18]. Together, given that ITE does not cause significant toxicity in mice [13,16–18], ITE as an endogenous AhR ligand could be used to investigate physiological roles of AhR [17,18].
Two major AhR downstream target genes are cytochrome P450, family 1, member A1 (CYP1A1) and member B1 (CYP1B1) [7]. The AhR-induced downstream signaling molecules also include mitogen activated protein kinase 3/1 (also termed ERK1/2) and v-akt murine thymoma viral oncogene homolog 1 (AKT1) [19–22]. Once the downstream signaling pathways of AhR are activated, the ligand-bound AhR is rapidly degraded by the 26S proteasome system [7,23]. Thus, the AhR ligand-induced decreases in AhR levels and increases in CYP1A1 and CYP1B1 are two hallmarks of AhR activation.
Despite the fact that the majority of AhR ligand-induced cellular responses are mediated via AhR [7, 23,24], AhR ligands can also activate other pathways via an AhR independent manner. For example, TCDD induces antiproliferative response of human breast cancer cells (MCF-7) [25] and reduction of p16ink4a (a cell cycle regulator and tumor suppressor) in human dermal fibroblasts [26] independent of AhR. Recently, we have also revealed that AhR is not involved in mediating TCDD-inhibited angiogenic responses of human umbilical vein endothelial cells (HUVECs) [8]. These data clearly indicate that some AhR ligands are capable of functioning either dependent or independent of AhR.
Little is known about the role of the ITE in mediating placental endothelial functions. Thus, in this study, we examined if ITE affected endothelial angiogenic responses via AhR and investigated potential underlying mechanisms using primary HUVECs and human umbilical artery endothelial cells (HUAECs) as in vitro endothelial models. Both vein and artery endothelial cells were used because these endothelial cells of different origins differ tremendously in their global gene expression profiles [27–29], possibly leading to their different responses to ITE as TCDD does [8].
2. Materials and methods
2.1. Cell isolation and culture
Both HUVECs and HUAECs were isolated from umbilical cord vessels of women with normal term pregnancies using a standard collagenase enzyme digestion as described [8,28,29]. The cord collection and endothelial cell isolation protocols were approved by the Institutional Review Board of Meriter Hospital, and the Health Sciences Institutional Review Boards of the University of Wisconsin-Madison. After isolation, cells were cultured in basal RPMI 1640 media (BM; Life Technology, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS, Thermo Scientific, Waltham, MA), 1% penicillin/streptomycin, 100 mg/L heparin (EMD Chemicals, San Diego, CA), and 37.5 mg/L endothelial cell growth supplement (Millipore, Billerica, MA) under 37°C, 5% CO2, and 95% air. This supplemented medium was designated as complete growth medium (CGM) and was used to stimulate all in vitro cellular responses in this study. After verification of their endothelial phenotypes (see Supplemental Methods), cells were pooled from 5 individual cell preparations at passage 1 to reduce inter-cell preparation variations and cultured [8,28,29]. These pooled cells at passages 4–5 were used for all studies described below.
2.2. Cell proliferation, viability, and migration
Cell proliferation was assayed as described [8,18,30]. Subconfluent cells were seeded in 96-well plates (5000 and 8000 cells/well for HUVECs and HUAECs, respectively). After 16 hr of culture, cells were treated with 0.1, 1, 10, 100, or 1000 nM of ITE (Tocris Bioscience, San Diego, CA) or the vehicle (dimethyl sulfoxide, DMSO, 0.01% v/v; the maximum concentration used in the final ITE solutions; Sigma-Aldrich, St. Louis, MO) in CGM up to 6 days (6 wells/treatment) with a daily change of CGM containing ITE or DMSO. At the end of treatment, the number of cells was determined using the crystal violet method.
Cell viability was determined using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole (MTT) assay kit (Cayman Chemical Company, Ann Arbor, MI) [8,18]. This method is based on reducing the yellow MTT to violet formazan, which is catalyzed by mitochondrial dehydrogenases, and is widely used to assess cell viability [31]. The confluent cells (40,000 cells/well, 6 wells/dose) were seeded in 96-well plates. After 16 hr of culture, cells were treated with ITE (1 μM) or DMSO (0.01% v/v; 6 wells) in CGM for 4 or 6 days with a daily change of CGM containing ITE or DMSO. At the end of treatment, cells were incubated with the MTT reagent for 4 hr, followed by the cell lysis. The absorbance was read at 570 nm using the microplate reader (Biotek, Winooski, VT).
Cell migration (random cell movement) was evaluated using a FluoroBlok Insert System (8.0 μm pores; BD Biosciences, San Jose, CA) as described [28,30]. Subconfluent cells grown on 60 mm culture dishes were treated with ITE (1 μM) or DMSO (0.01% v/v) in CGM for 6 days with a daily change of CGM containing ITE or DMSO. Cells were then seeded into the insert (30,000 cells/insert; 2 wells/treatment). The bottom wells were also filled with the same medium. After 6 hr of culture, cells migrated (random movement) were stained with 0.2 μg/ml of calcein AM (cat# C3100MP, Invitrogen, Carlsbad, CA) and counted using the MetaMorph image analysis software.
2.3. Western blot analysis
Western blot analysis was conducted as described [8,28–30]. To determine the ITE’s effect on AhR protein levels, cells were treated with a single dose of ITE (1 μM) in CGM for 48, 24, 8, 2, 1, or 0 hr (cells were treated with DMSO for 48 hr). Proteins were subjected to Western blotting. The membranes were probed with the rabbit anti-AhR antibody (1:2000; Enzo Life Sciences, Plymouth Meeting, PA), followed by reprobing with a mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:10,000; Novus, Littleton, CO). Proteins were visualized using the enhanced chemiluminescence reagent (Amersham, Piscataway, NJ). Signals were recorded using an Epson Perfection 4990 Photo Scanner (Long Beach, CA) and analyzed using NIH Image J software.
2.4. Reverse transcription-quantitative PCR (RT-qPCR)
To determine ITE’s effects on its downstream gene expression, RT-qPCR was performed as described [8,28–30]. Subconfluent cells were treated with a single dose of ITE (1 μM) in CGM for 48, 24 or 0 hr (cells were treated with DMSO for 48 hr) hr. Total RNA were extracted using RNeasy Plus Mini Kit (Qiagen, Valencia, CA) and quantified by using NanoDrop 1000 Spectrophotometer (Nanodrop Technologies, Wilmington, DE). The quality and integrity of total RNA were confirmed using the Agilent RNA6000 NanoChip in the Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). Only samples with RNA integrity number scores larger than 9.0 were used. Samples of total RNA (1 μg) were reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kits (AB applied Biosystems, Waltham, Mass) in a 20μL volume.
The effects of ITE on mRNA expression of CYP1A1 and CYP1B1 were first analyzed. In parallel, TBP (TATA-BOX), ACTB (β-actin), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were run, serving as endogenous controls. All primers are shown in the Supplemental Table S1. RT-qPCR was performed using the SsoAdvanced™ Universal SYBRR® Green Supermix (Bio-Rad Laboratories, Hercules, CA) and a Roche 480 Lightcycler system according to the manufacturer’s instruction, each RT-qPCR reaction was performed in triplicate. The specificity of each primer pair set was confirmed by a dissociation curve analysis. Relative expression levels of genes of interest were normalized to the geometric mean of two out of three reference genes TBP, ACTB, and GAPHD, selected by the Qbase Plus software.
RT-qPCR was also run to determine the effects of ITE on mRNA expression of vascular endothelial growth factor A (VEGFA), vascular endothelial growth factor C (VEGFC), vascular endothelial growth factor receptor 1 (VEGFR1), and vascular endothelial growth factor receptor 1 (VEGFR2), neuropilin 1(NP-1), neuropilin 2 (NP-2), and fibroblast growth factor-1 (FGFR1) (see Supplemental Methods).
2.5. siRNA transfection
To determine the AhR role in the ITE-induced cellular response, siRNA transfection was performed as described [8,18,30]. The AhR siRNA targeting human AhR was purchased (Cat # L-004990-00-0020, Dharmacon, Chicago, IL). Scrambled siRNA (ssiRNA) with 5′-Cy3 (Sense: 5-GAGAGGUCCCUCCCAUCUUTT-3; Antisense: 5-AAGAUGGGAGGGACCUCUCTT-3) was synthesized (IDT, Coralville, IA). Subconfluent cells were transfected with 20 nM of the AhR or scrambled siRNA (the siRNA control) in the Lipofectamine RNAiMAX transfection reagent (Invitrogen) or treated with the transfection reagent alone (the vehicle control) up to 6 days. After an optimal time point was identified, additional cells were transfected for determining their proliferation, viability, and migration in response to ITE.
2.6. Cell cycle and cell apoptosis
To examine the ITE’s effects on the cell cycle and apoptosis, the flow cytometry and Western blot analysis were conducted, respectively [8] (see Supplemental Methods).
2.7. Statistics
For results from the cell proliferation assay, a factorial ANOVA was conducted to compare the main effect of ITE dose and treatment day and the interaction effect between these two factors using the SigmaStat software (Jandel Co., San Rafael, CA). When an F-test was significant, pair-wise comparisons were followed using the Holm-Sidak method. For all other assays, data were analyzed using either one-way ANOVA followed by comparisons verse the respective control using the Holm-Sidak method or Student t-test when appropriate. p < 0.05 was considered statistically significant.
3. Results
3.1. ITE inhibits proliferation and viability of HUVECs and HUAECs, as well as migration of HUAECs
For HUVECs, significant effects of ITE dose, treatment day, and their interaction on cell proliferation were observed. For HUAECs, significant effects of ITE dose and treatment day on cell proliferation were detected. As compared with the vehicle control, ITE time- and dose-dependently inhibited (p < 0.05) CGM-stimulated proliferation of HUVECs and HUAECs (Fig. 1A and B). On Day 2, ITE at all doses studied did not significantly alter proliferation of HUVECs and HUAECs. On Days 4 and 6, ITE at 0.1 nM did not significantly affect proliferation of HUVECs and HUAECs. However, ITE at doses from 1 nM to 1 μM similarly inhibited (p < 0.05) proliferation of HUVECs by 13–17% on Day 4 and 23–29% on Day 6, respectively (Fig. 1A). ITE at doses from 1 nM to 1 μM also decreased (p < 0.05) proliferation of HUAECs by 23–39% on Day 4 and 26–33% on Day 6, respectively (Fig. 1B). As ITE from 1 nM to 1 μM exhibited similar inhibition on cell proliferation, 1 μM of ITE was chosen to be used in all subsequent assays.
Fig. 1.

Effects of ITE on proliferation and viability of HUVECs and HUAECs. Proliferation (A and B): Subconfluent cells were treated with ITE or DMSO (the vehicle control) in CGM up to 6 days with a daily change of CGM containing ITE or DMSO. Cell numbers were determined by the crystal violet. Cell viability (C and D): confluent cells were treated with ITE or DMSO in CGM for 4 and 6 days with a daily change of CGM containing ITE or DMSO. Cell viability was determined by the MTT assay. Data are expressed as means ± SEM% of the control (n = 3 for the cell proliferation assay; n = 5 for the cell viability assay). Two-way ANOVA was conducted for data from the cell proliferation, followed by pair-wise comparisons using the Holm-Sidak method. Student t-test was conducted for data from the cell viability assay. a,b,c Within a day, means with different superscripts differ. *Differ from the control at each corresponding day (p < 0.05).
ITE at 1 μM decreased (p < 0.05) viability of HUVECs by 17% and 22% on Days 4 and 6, respectively (Fig. 1C). Similarly, ITE at 1 μM reduced (p < 0.05) viability of HUAECs by 15% and 18% on Days 4 and 6, respectively (Fig. 1D).
Treating cells with 1 μM of ITE for 6 days suppressed (p < 0.05) migration of HUAECs by ~ 40%, but not HUVECs (Fig. 2; Supplemental Fig. S1).
Fig. 2.

Effects of ITE on migration of HUVECs and HUAECs. Cells were treated with ITE (1 μM) or DMSO (the control) for 6 days, followed by the migration assay. The migrated cells were stained and counted. Representative images for migrated cells are shown. Data are expressed as means ± SEM% of the control (n = 4 and 5 for HUVECs and HUAECs, respectively). Student t-test was conducted. *Differ from the control within each cell type (p < 0.05). Bars, 200 μm.
3.2. ITE activates the AhR/CYP1A1 and AhR/CYP1B1 pathways
To determine ITE-induced AhR activation, Western blotting was conducted (Fig. 3A and B). The AhR protein was detected at ~95kD in HUVECs and HUAECs as reported [6,8,18,32]. A single dose of ITE at 1 μM quickly decreased (p < 0.05) AhR protein levels in both HUVECs and HUAECs, indicating that ITE indeed induced AhR activation. The ITE-induced decrease in AhR began at 1 h (by 36 and 57% in HUVECs and HUAECs, respectively), and lasted up to 48 hr (by 70% and 90% in HUVECs and HUAECs, respectively) (Fig. 3A and B). To confirm the activation of the AhR downstream signaling, RT-qPCR was conducted to quantify transcripts of CYP1A1 and CYP1B1 (Fig. 3C and D). Compared with the time 0 control, ITE elevated (p < 0.05) mRNA levels of CYP1A1 by ~ 11 and 16 fold at 24 and 48 hr, respectively in HUVECs, and by ~ 16 and 17 fold at 24 and 48 hr, respectively in HUAECs. Similarly, ITE also increased (p < 0.05) mRNA levels of CYP1B1 by ~ 278 and 347 fold at 24 and 48 hr, respectively in HUVECs, and by ~ 21 and 23 fold at 24 and 48 hr, respectively in HUAECs. The increases in fold changes of CYP1B1 mRNA levels in HUVECs are much higher than those in HUAECs. These data clearly indicate that ITE activates the AhR/CYP1A1 and AhR/CYP1B1 signaling pathways in both HUVECs and HUAECs.
Fig. 3.

Effects of ITE on AhR protein levels of and CYP1A1 and CYP1B1 mRNA levels in HUVECs and HUAECs. Protein levels (A and B): Cells were treated with a single dose of ITE (1 μM) or DMSO (0 hr, the control) up to 48 hr. Proteins were subjected to Western blotting. Representative images of Western blotting are shown. Data normalized to GAPDH are expressed as means ± SEM fold of the control (n = 4–5). mRNA levels (C and D): Cells were treated with ITE (1 μM) or DMSO (0 hr, the control) for 24 or 48 hr, followed by RT-qPCR assay. Data are expressed as means ± SEM fold of the control (n = 3). One-way ANOVA was conducted followed by comparisons verse the respective control within each cell type. *Differ from the time 0 control within each cell type (p < 0.05).
3.3. Effects of AhR siRNA on ITE-inhibited cell proliferation, viability and/or migration
To determine the role of AhR in ITE-inhibited cell responses in HUVECs and HUAECs, AhR was knocked down by the AhR siRNA (Fig. 4), followed by the cell proliferation, viability and migration assays. We previously have reported that as compared with the vehicle control and the scrambled siRNA, the AhR siRNA at 20 nM significantly decreased (p < 0.05) AhR protein levels by 93%, 81%, and 55% in HUVECs after 2, 4, and 6 days of AhR siRNA transfection, respectively, while it reduced AhR protein levels by 89%, 86%, and 58% in HUAECs, respectively, at the same time frame [8]. However, the AhR siRNA did not significantly alter ITE-inhibited proliferation and viability of HUVECs (Fig. 5A and C), while it blocked (p<0.05) ITE-inhibited proliferation and viability of HUAECs (Fig. 5B and D), and ITE-suppressed migration of HUAECs (Fig. 5E and Supplement Fig. S1).
Fig. 4.
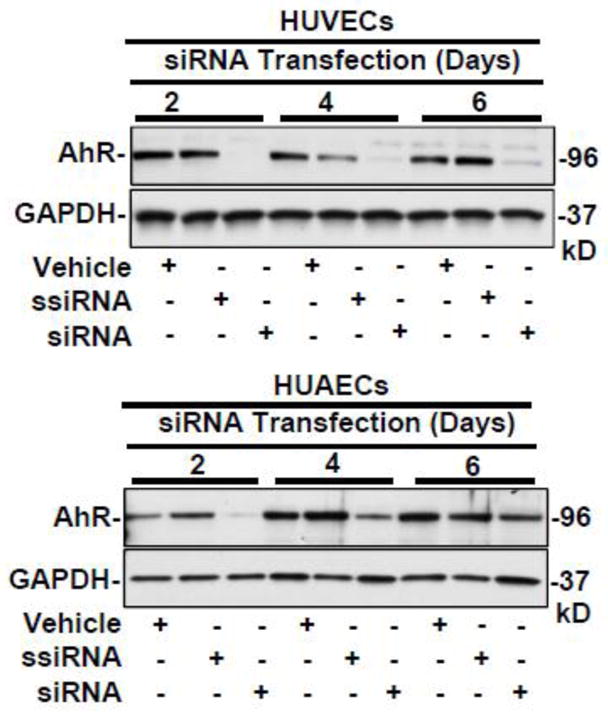
Effects of AhR siRNA on AhR protein expression in HUVECs and HUAECs. Cells were treated with the transfection reagent (vehicle) or transfected with scrambled (ssiRNA, 20 nM) or AhR siRNA (siRNA, 20 nM) for 6, 4, or 2 days. Proteins were subjected to Western blotting. Representative images were shown (n =4).
Fig. 5.

Effects of AhR siRNA on the ITE-inhibited proliferation and viability of HUVECs and HUAECs as well as migration of HUAECs. Cells were treated with the vehicle control or transfected with the scrambled (ssiRNA) or AhR siRNA (siRNA) for 2 days. After transfection, cells were treated with ITE (1 μM) for 4 days for cell proliferation (A and B) and for cell viability (C and D), or for 2 days for cell migration (E) with a daily change of ITE, followed by the cell proliferation, viability, and migration assays. Data are expressed as means ± SEM% of the vehicle (Veh; n = 5–6) in the absence of ITE. One-way ANOVA was conducted followed by comparisons verse the respective Veh in the absence of ITE. *Differ from Veh in the absence of ITE (p < 0.05).
3.4. ITE does not alter the cell cycle progression and apoptosis as well as phosphorylation of ERK1/2, AKT1, c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (p38)
As compared with the vehicle control, treatment of ITE at 1 μM up to 144 hr did not alter the percentage of cell numbers in each cell cycle phase (G0/G1, S, and G2/M) in HUVECs and HUAECs (Supplemental Fig. S2). ITE at 1 μM did not increase apoptosis and necrosis in HUVECs and HUAECs (Supplemental Fig. S3). Treatment of ITE at 1 μM up to 48 hr did not induce the formation of cleaved caspase-3 (Supplemental Fig. S4). Treatment of ITE at 1 μM also did not alter CGM-induced phosphorylation of ERK1/2, AKT1, p38, and JNK in the HUVECs and HUAECs (Supplemental Fig. S5).
3.5. ITE decreases NP-1 mRNA levels in HUVECs, but not HUAECs
RT-qPCR analysis revealed that ITE slightly but significantly decreased (p < 0.05) NP-1 mRNA levels in HUVECs, but not HUAECs (Supplemental Fig. S6). However, ITE had no significant effect on the mRNA levels of VEGFA, VEGFC, VEGFR1, and VEGFR2, NP-2, and FGFR1 in HUVECs and HUAECs (data not shown).
4. Discussion
Herein, we have demonstrated that similar to TCDD as we recently reported [8], ITE inhibits cell proliferation and viability of HUVECs and HUAECs as well as migration of HUAECs induced by the complete growth medium. Our data clearly reveal the anti-angiogenic activity of ITE in vitro. Intriguingly, these ITE-inhibited cellular responses are independent and dependent of AhR in HUVECs and HUAECs, respectively, indicating differential roles of AhR in the ITE-inhibited angiogenic responses in artery and vein endothelial cells (Table 1). Thus, although the signaling mechanisms underlying the ITE-inhibited cellular responses in HUVECs and HUAECs remain elusive, these data suggest that ITE may inhibit placental angiogenesis, which may hold a great promise of targeting angiogenesis-related diseases.
Table 1.
Summary of ITE’s and AhR siRNA’s Effects on HUVECs and HUAECs.
| HUVECs | HUAECs | |
|---|---|---|
| Cell proliferation | ↓ | ↓ |
| Cell migration | Not significant | ↓ |
| Cell viability | ↓ | ↓ |
| AhR protein levels | ↓ | ↓ |
| CYP1A1 mRNA | ↑ | ↑ |
| CYP1B1 mRNA | ↑↑ | ↑ |
| siRNA on AhR protein levels | ↓ | ↓ |
| siRNA on ITE-inhibited proliferation | Not significant | Blocked |
| siRNA on ITE-inhibited-migration | Not applied | Blocked |
| siRNA on ITE-inhibited viability | Not significant | Blocked |
The diverse functions of ITE are increasingly recognized as ITE has been shown to suppress the tumorigenic potential of stem-like cancer cells [15], autoimmunologic responses [16,17], and attenuate the growth of human ovarian cancer cells [18]. Our current data extend the role of ITE in inhibiting angiogenic response of human placental endothelial cells. To date, the physiological level of ITE and its biosynthesis process in vivo are unknown in any type of tissue of any specie. However, given the high binding affinity of ITE to AhR (the estimated Ki of ITE for AhR: ~ 3nM) [14] and its high efficacy (the estimated IC50: ~ 0.2 nM) of ITE for human ovarian cancer cell (OVCAR-3) proliferation [18], even very low levels of ITE present in placental tissues might be functional.
The current observation that ITE-suppressed cell proliferation and viability of HUVECs and HUAECs is consistent with activities of another AhR ligand, benzo[a]pyrene (B[a]P), which inhibits angiogenic factors-induced neovasculogenesis and angiogenic activity of HUVECs [33]. The ITE-suppressed cell proliferation of HUVECs and HUAECs is associated with the decreased cell viability as indicated by the decreased activity of mitochondrial dehydrogenases enzyme (intracellular NADPH-oxidoreductases) within the live cells in the MTT assay (Fig. 1C and D). Such decreases in activity of intracellular dehydrogenases enzyme might increase the accumulation of cellular reactive oxygen species, decreasing the cell viability [34]. However, even in this case, it is obvious that the ITE-decreased activity of mitochondrial dehydrogenases in HUVECs is not sufficient to suppress migration of HUVECs.
In the current study, ITE differentially regulated cellular responses in HUVECs and HUAECs. Firstly, ITE only suppressed migration of HUAECs, but not HUVECs. Secondly, ITE only slightly decreased mRNA levels of NP-1 of HUVECs, but not HUAECs. Thirdly, the decreasing effect of ITE on AhR protein levels appeared to be more potent and sustainable in HUAECs than HUVECs (Fig. 3B), whereas the promoting effect of ITE on CYP1B1 mRNA was much greater in HUVECs than HUAECs. To date, mechanisms underlying these differential regulations of ITE between HUVECs and HUAECs remain elusive. However, the failure of ITE to suppress the cell migration of HUVECs apparently is not due to the uncoupling of ITE to the AhR/CYP1A1 and AhR/CYP1B1 pathways, since ITE robustly activates both signaling pathways in HUVECs and HUAECs. Thus, other yet to be identified signaling mechanisms must be involved in such disparity. Obviously, ERK1/2, AKT1, p38, and JNK are not among the potential downstream signaling molecules because ITE does not robustly alter the activation of any of these four kinases in HUVECs and HUAECs. This notion is not surprising since ITE has also failed to inhibit the TGFβ1-induced phosphorylation of ERK1/2 or AKT1 in primary human orbital fibroblasts [35]. Other possible signaling molecules might include p21 and p27 (cell cycle regulators) as well as FAK as 3-methycholanthrene (3-MC, another AhR ligand) has been show to inhibit the proliferation and motility of HUVECs via these molecules [19,24]. In addition, since vein and artery endothelial cells are tremendously different in their global gene expression profiles [27–29], it is very likely that ITE stimulation may differentially alter gene expression profiles between artery and vein endothelial cells, leading to different angiogenic responses as we observed in the current study. Interestingly, ITE also does not slow down the cell cycle progression of either HUVECs or HUAECs, which is contrast to previous report that 3-MC arrests the cell cycle at G0/G1 [33]. In the current study, the time points (24, 36, and 144 hr) studied are within the optimal time frame to detect changes in the cell progress since the estimated doubling times are ~ 40 and 52 hr for HUVECs and HUAECs, respectively, which are similar to the previously reported ones (47 and 46 hr) for HUVECs and HUAECs, respectively [36]. Thus, it is likely that these discrepancies induced by ITE and 3-MC might be attributed to the distinct structures and characteristics of two ligands [14].
ITE-inhibited endothelial proliferation and migration are also not associated with increased apoptosis of HUVECs and HUAECs as determined using two well-established apoptosis assays. This is supported by the current observation that the numbers of HUVECs and HUAECs after ITE treatments are always higher than those initially seeded (5000 and 8000 cells/well for HUVECs and HUAECs, respectively). For example, the estimated cell numbers in the ITE (1 μM) treatment on Day 6 were 22,527 ± 2335 and 11,824 ± 3,558 for HUVECs and HUAECs, respectively.
The differential roles of AhR in mediating the ITE-inhibited endothelial responses (proliferation, migration, and viability) in HUVECs and/or HUAECs confirm the regulatory heterogeneity of HUVECs and HUAECs [27,29]. The independence of AhR in the ITE-inhibited angiogenic responses of HUVECs is not unanticipated since both AhR dependent and independent manners exist in the various cellular responses induced by AhR-ligands including TCDD [24,37,38]. We have recently reported that the TCDD-inhibited angiogenic responses of HUVECs independent of AhR [8]. This independence of AhR also happens in non-endothelial cells too. For example, ITE inhibits TGFβ1 signaling in human fibroblasts via an AhR independent manner [35]. To date, the AhR-independent signaling pathways that are involved in ITE-regulated endothelial functions in HUVECs remain unknown. Moreover, given that the AhR siRNA only partially suppresses the AhR expression in HUVECs and HUAECs, it is also possible that the decrease in AhR protein levels by AhR siRNA in HUAECs is sufficient to block ITE-inhibited cell responses in HUAECs, but not in HUVECs.
FGF2 and VEGFA are two key regulators of angiogenesis [39]. We and other researchers have shown that FGF2 and VEGFA promote placental angiogenesis [39–42]. The receptors of FGF2 include FGFR1, FGFR2, FGFR3, and FGFR4 [41], among which FGFR1 is the predominantly form present in the human umbilical endothelial cells [28,29]. The major VEGFA receptors consist of VEGFR1 and VEGFR2 as well as NP-1 and NP-2 [39]. Wu et al reported that in rats ITE increased the mRNA levels of both VEGFA and VEGFB in placental tissues [43]. However, in our current human study, ITE has only minimal effects on the mRNA expression of NP-1 in HUVECs but fails to alter mRNA expression of VEGFA, VEGFC, VEGFR1, VEGR2, FGFR1, NP-2 in both HUVECs and HUAECs. Therefore, although we do not know exactly underlying mechanisms, the ITE-inhibited angiogenesis in vitro might be not mediated via decreasing endothelial expression of these angiogenesis related genes.
In conclusion, these data indicate that AhR differentially mediates ITE-suppressed angiogenic responses of human fetoplacental vein and artery endothelial cells in vitro in association with decreased cell viability..
Highlights.
The AhR ligand, ITE suppresses growth of human umbilical endothelial cells in vitro in association with decreased cell viability.
ITE suppresses angiogenic responses of human vein and artery endothelial cells in vitro independent of and dependent on AhR, respectively.
These data suggest that ITE may regulate human placental endothelial function.
Acknowledgments
Funding information: This work was supported in part by the US National Institutes of Health Grant HD38843 (JZ), and an R & D grant from Departmental of Ob/Gyn, University of Wisconsin-Madison (JZ), the National Natural Science Foundation of China No. 81100429 (KW). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH and NNSFC.
Abbreviations
- AhR
Aryl hydrocarbon receptor
- B[a]P
benzo[a]pyrene
- CYP1A1
cytochrome P450, family 1, member A1
- CYP1B1
cytochrome P450, family 1, member B1
- BM
basal RPMI 1640 media
- CGM
complete growth medium
- FBS
fetal bovine serum
- FGF2
fibroblast growth factor-2
- FGFR1
FGF receptor 1
- FITC
fluorescein 5(6)-isothiocyanate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HPAECs
human pulmonary artery endothelial cells
- HUAECs
human umbilical cord artery cells
- HUVECs
human umbilical cord vein cells
- ITE
2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NP-1
neuropilin 1
- NP-2
neuropilin 2
- PAHs
polycyclic aromatic hydrocarbons
- RT-qPCR
Reverse Transcription-Quantitative PCR
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- VEGFA
vascular endothelial growth factor A
- VEGFC
vascular endothelial growth factor C
- VEGFR1
VEGF receptor 1
- VEGFR2
VEGF receptor 2
Footnotes
Conflict of interests: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tonini T, Rossi F, Claudio PP. Molecular basis of angiogenesis and cancer. Oncogene. 2003;22:6549–6956. doi: 10.1038/sj.onc.1206816. [DOI] [PubMed] [Google Scholar]
- 2.Wang K, Zheng J. Signaling regulation of fetoplacental angiogenesis. J Endocrinol. 2012;212:243–55. doi: 10.1530/JOE-11-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmeliet P. Manipulating angiogenesis in medicine. J Intern Med. 2004;255:538–561. doi: 10.1111/j.1365-2796.2003.01297.x. [DOI] [PubMed] [Google Scholar]
- 4.Cerdeira AS, Karumanchi SA. Angiogenic factors in preeclampsia and related disorders. Cold Spring Harb Perspect Med. 2012;1:2. doi: 10.1101/cshperspect.a006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkman J. Angiogenesis-dependent diseases. Semin Oncol. 2001;28:536–542. doi: 10.1016/s0093-7754(01)90021-1. [DOI] [PubMed] [Google Scholar]
- 6.Jiang YZ, Wang K, Fang R, Zheng J. Expression of aryl hydrocarbon receptor in human placentas and fetal tissues. J Histochem Cytochem. 2010;58:679–685. doi: 10.1369/jhc.2010.955955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Wang K, Zou QY, Magness RR, Zheng J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin differentially suppresses angiogenic responses in human placental vein and artery endothelial cells. Toxicology. 2015;336:70–78. doi: 10.1016/j.tox.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Detmar J, Rennie MY, Whiteley KJ, Qu D, Taniuchi Y, Shang X, Casper RF, Adamson SL, Sled JG, Jurisicova A. Fetal growth restriction triggered by polycyclic aromatic hydrocarbons is associated with altered placental vasculature and AhR-dependent changes in cell death. Am J Physiol Endocrinol Metab. 2008;295:E519–530. doi: 10.1152/ajpendo.90436.2008. [DOI] [PubMed] [Google Scholar]
- 10.Abbott BD, Schmid JE, Pitt JA, Buckalew AR, Wood CR, Held GA, Diliberto JJ. Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol Appl Pharmacol. 1999;155:62–70. doi: 10.1006/taap.1998.8601. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol. 1997;34:605–614. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- 12.Zhang N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J Cardiovasc Dis Res. 2011;2:91–95. doi: 10.4103/0975-3583.83033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci USA. 2002;99:14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng J, Li W, Kang B, Zhou Y, Song J, Dan S, Yang Y, Zhang X, Li J, Yin S, Cao H, Yao H, Zhu C, Yi W, Zhao Q, Xu X, Zheng M, Zheng S, Li L, Shen B, Wang YJ. Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat Commun. 2015;6:7209. doi: 10.1038/ncomms8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nugent LF, Shi G, Vistica BP, Ogbeifun O, Hinshaw SJ, Gery I. ITE, a novel endogenous nontoxic aryl hydrocarbon receptor ligand, efficiently suppresses EAU and T-cell-mediated immunity. Invest Ophthalmol Vis Sci. 2013;54:7463–7469. doi: 10.1167/iovs.12-11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li Y, Jiang YZ, Dai CF, Patankar MS, Song JS, Zheng J. An endogenous aryl hydrocarbon receptor ligand inhibits proliferation and migration of human ovarian cancer cells. Cancer Lett. 2013;340:63–71. doi: 10.1016/j.canlet.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CC, Tsai SY, Lin H, Li HF, Lee YH, Chou Y, Jen CY, Juan SH. Aryl-hydrocarbon receptor-dependent alteration of FAK/RhoA in the inhibition of HUVEC motility by 3-methylcholanthrene. Cell Mol Life Sci. 2009;66:3193–3205. doi: 10.1007/s00018-009-0102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan Z, Chang X, Puga A, Xia Y. Activation of mitogen-activated protein kinases (MAPKs) by aromatic hydrocarbons: role in the regulation of aryl hydrocarbon receptor (AHR) function. Biochem Pharmacol. 2002;64:771. doi: 10.1016/s0006-2952(02)01138-3. [DOI] [PubMed] [Google Scholar]
- 21.Tan Z, Huang M, Puga A, Xia Y. A critical role for MAP kinases in the control of Ah receptor complex activity. Toxicol Sci. 2004;82:80–87. doi: 10.1093/toxsci/kfh228. [DOI] [PubMed] [Google Scholar]
- 22.Wu R, Zhang L, Hoagland MS, Swanson HI. Lack of the aryl hydrocarbon receptor leads to impaired activation of AKT/protein kinase B and enhanced sensitivity to apoptosis induced via the intrinsic pathway. J Pharmacol Exp Ther. 2007;320:448–457. doi: 10.1124/jpet.106.111773. [DOI] [PubMed] [Google Scholar]
- 23.Fujii-Kuriyama Y, Kawajiri K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:40–53. doi: 10.2183/pjab.86.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang PH, Lin YH, Lee YH, Hou HH, Hsu SP, Juan SH. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol. 2008;215:161–171. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- 25.Yoshioka H, Hiromori Y, Aoki A, Kimura T, Fujii-Kuriyama Y, Nagase H, Nakanishi T. Possible aryl hydrocarbon receptor-independent pathway of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced antiproliferative response in human breast cancer cells. Toxicol Lett. 2012;211:257–265. doi: 10.1016/j.toxlet.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 26.Akintobi AM, Villano CM, White LA. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) exposure of normal human dermal fibroblasts results in AhR-dependent and -independent changes in gene expression. Toxicol Appl Pharmacol. 2007;220:9–17. doi: 10.1016/j.taap.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA. 2003;100:10623–10628. doi: 10.1073/pnas.1434429100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang YZ, Wang K, Li Y, Dai CF, Wang P, Kendziorski C, Chen DB, Zheng J. Transcriptional and functional adaptations of human endothelial cells to physiological chronic low oxygen. Biol Reprod. 2013;88:114. doi: 10.1095/biolreprod.113.108225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang YZ, Wang K, Li Y, Dai CF, Wang P, Kendziorski C, Chen DB, Zheng J. Enhanced cellular responses and distinct gene profiles in human fetoplacental artery endothelial cells under chronic low oxygen. Biol Reprod. 2013;89:133. doi: 10.1095/biolreprod.113.110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Wang K, Jiang YZ, Chang XW, Dai CF, Zheng J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) inhibits human ovarian cancer cell proliferation. Cell Oncol (Dordr) 2014;37:429–437. doi: 10.1007/s13402-014-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berridge MV, Tan AS, Herst PM. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127–152. doi: 10.1016/S1387-2656(05)11004-7. [DOI] [PubMed] [Google Scholar]
- 32.Juan SH, Lee JL, Ho PY, Lee YH, Lee WS. Antiproliferative and antiangiogenic effects of 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, in human umbilical vascular endothelial cells. Eur J Pharmacol. 2006;530:1–8. doi: 10.1016/j.ejphar.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 33.Li CH, Cheng YW, Hsu YT, Hsu YJ, Liao PL, Kang JJ. Benzo[a]pyrene inhibits angiogenic factors-induced alphavbeta3 integrin expression, neovasculogenesis, and angiogenesis in human umbilical vein endothelial cells. Toxicol Sci. 2010;118:544–553. doi: 10.1093/toxsci/kfq279. [DOI] [PubMed] [Google Scholar]
- 34.Wan C, Liu J, Nie X, Zhao J, Zhou S, Duan Z, Tang C, Liang L, Xu G. 2, 3, 7, 8-Tetrachlorodibenzo-P-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLoS One. 2014;9:e89811. doi: 10.1371/journal.pone.0089811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehmann GM, Xi X, Kulkarni AA, Olsen KC, Pollock SJ, Baglole CJ, Gupta S, Casey AE, Huxlin KR, Sime PJ, Feldon SE, Phipps RP. The aryl hydrocarbon receptor ligand ITE inhibits TGFβ1-induced human myofibroblast differentiation. Am J Pathol. 2011;178:1556–1567. doi: 10.1016/j.ajpath.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martín de Llano JJ, Fuertes G, Torró I, García Vicent C, Fayos JL, Lurbe E. Birth weight and characteristics of endothelial and smooth muscle cell cultures from human umbilical cord vessels. J Transl Med. 2009;7:30. doi: 10.1186/1479-5876-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 38.Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M, Fujii-Kuriyama Y. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- 39.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 40.Chung JY, Song Y, Wang Y, Magness RR, Zheng J. Differential expression of vascular endothelial growth factor (VEGF), endocrine gland derived-VEGF, and VEGF receptors in human placentas from normal and preeclamptic pregnancies. J Clin Endocrinol Metab. 2004;89:2484–2490. doi: 10.1210/jc.2003-031580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocrine-Related Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 42.Zygmunt M, Herr F, Munstedt K, Lang U, Liang OD. Angiogenesis and vasculogenesis in pregnancy. Eur J Obstet Gynecol Reprod Bio. 2003;110:S10–S18. doi: 10.1016/s0301-2115(03)00168-4. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Chen X, Zhou Q, He Q, Kang J, Zheng J, Wang K, Duan T. ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS One. 2014;9:e86549. doi: 10.1371/journal.pone.0086549. [DOI] [PMC free article] [PubMed] [Google Scholar]