

Abstract
Lymphodepletion chemotherapy followed by infusion of CD19-targeted chimeric antigen receptor (CAR)-modified T (CAR-T) cells can be complicated by neurologic adverse events (AEs) in patients with refractory B cell malignancies. In 133 adults treated with CD19 CAR-T cells we found that acute lymphoblastic leukemia, high CD19+ cells in bone marrow, high CAR-T cell dose, cytokine release syndrome, and preexisting neurologic comorbidities were associated with increased risk of neurologic AEs. Patients with severe neurotoxicity demonstrated evidence of endothelial activation, including disseminated intravascular coagulation, capillary leak, and increased blood-brain barrier (BBB) permeability. The permeable BBB failed to protect the CSF from high concentrations of systemic cytokines including IFN-γ, which induced brain vascular pericyte stress and their secretion of endothelium-activating cytokines. Endothelial activation and multifocal vascular disruption were found in the brain of a patient with fatal neurotoxicity. Biomarkers of endothelial activation were higher before treatment in patients who subsequently developed grade ≥4 neurotoxicity.
Keywords: CAR, chimeric antigen receptor, CD19, neurotoxicity, cytokine release syndrome
INTRODUCTION
Lymphodepletion chemotherapy followed by infusion of autologous T cells that are genetically modified to express a CD19-specific CAR (CD19 CAR-T cells) has produced high response rates in phase 1 studies in refractory CD19+ B-cell acute lymphoblastic leukemia (B-ALL), chronic lymphocytic leukemia (CLL), and non-Hodgkin’s lymphoma (NHL) (1–6). When CD19-specific CAR-T cells encounter CD19+ target cells in vivo, signaling through the CAR induces CAR-T cell proliferation, cytokine secretion, and target cell lysis (7). Thus, most patients develop cytokine release syndrome (CRS), a systemic inflammatory response initiated by T cell activation and characterized by fever and hypotension within the first 2 weeks after CAR-T cell infusion (8, 9). Neurologic adverse events (AEs) are frequently observed in association with or following CRS occurring with CD19 CAR-T cell therapy, and in rare instances can be fatal (1–6); however, the pathogenesis of neurotoxicity remains obscure.
Here, we provide a detailed clinical, radiologic and pathologic characterization of neurotoxicity resulting from CD19 CAR-T cell therapy. The data provides strong evidence for cytokine-mediated endothelial activation causing coagulopathy, capillary leak, and blood-brain barrier (BBB) disruption, which allows transit of high concentrations of systemic cytokines into the CSF. In autopsy studies of 2 patients who had fatal toxicity, we identified evidence of endothelial activation that in one patient resulted in loss of cerebral vascular integrity manifesting as multifocal hemorrhage. We developed a predictive classification tree algorithm based on the presence of fever and high serum IL-6 and MCP-1 concentrations to identify patients within the first 36 hours after CAR-T cell infusion who are at high risk of subsequent severe neurotoxicity and might be candidates for early intervention. Patients with evidence of endothelial activation before treatment were at increased risk of neurotoxicity after CAR-T cell infusion.
RESULTS
Neurologic adverse events after CD19 CAR-T cell immunotherapy
We studied neurologic AEs in 133 adults with refractory B-ALL, NHL or CLL who received lymphodepletion chemotherapy (Supplementary Table 1) followed by infusion of CD19 CAR-T cells engineered to express a CD19-specific CAR containing a 4-1BB costimulatory domain (Supplementary data) (5, 6). Within 28 days of CD19 CAR-T cell infusion 53/133 patients (40%) had one or more grade ≥1 neurologic AEs (Figure 1A–B), presenting a median of 4 days after CAR-T cell infusion. The median time from onset of neurotoxicity to the highest neurotoxicity grade was 1 day (range 0–19) and the median duration of reversible neurologic AEs was 5 days (range 1 – 70 days). Forty-eight of 53 patients with any neurologic AE (91%) also had CRS (Figure 1C). Five patients with neurologic AEs (9%) did not develop CRS; however, all neurologic AEs in patients without CRS were mild (grade 1), subjective, and transient. CRS with fever (≥38°C) preceded the onset of neurotoxicity in all patients who developed grade ≥3 neurotoxicity (n=28). In patients with CRS, neurotoxicity presented a median of 4.5 days (range 2–17 days) after the first fever. Fever occurred earlier after CAR-T cell infusion in patients who subsequently developed grade ≥3 neurotoxicity compared to those who developed grade 1–2 neurotoxicity (p=0.0007). However, the time from CAR-T cell infusion to the onset of neurotoxicity (p=0.17) as well as the time to the maximum grade of neurotoxicity (p=0.78) were similar (Figure 1D). These data show that an early onset of CRS after CAR-T cell infusion is associated with a higher risk of subsequent developing severe neurotoxicity.
Figure 1. Frequency, kinetics, and treatment of neurotoxicity.

A: The numbers of patients with each overall neurotoxicity grade are shown for the entire cohort and each disease. The diameters of each pie chart indicate the relative size of each subgroup. B: The swimmer plot (bottom) shows the kinetics of the severity of neurotoxicity in each patient who developed neurotoxicity through 28 days after CAR-T cell infusion (n=53). Each row represents one patient and the colors indicate the highest grade of neurotoxicity recorded on each day. Six patients died within 28 days of CAR-T cell infusion (neurotoxicity, n=3; disease progression, n=1; CRS, n=2). The graph (top) shows the mean of the highest grade of neurotoxicity occurring in all patients on each day after CAR T cell infusion. C: Numbers of patients with each grade of neurotoxicity and CRS. D: Cumulative incidences of fever, any grade of neurotoxicity, and the peak grade of neurotoxicity are shown for patients with grade 1–2 and grade ≥3 neurotoxicity.
Among the 53 patients with neurotoxicity the most common finding was delirium with preserved alertness (35 of 53, 66%; Supplementary Table 2), which was grade ≤2 in 13 of 35 patients (37%), and present for a median of 4 days (range 1–24 days). Headache was observed in 29 of 53 patients (55%), was grade ≤2 in 26 of 29 patients (90%), and persisted for a median of 3 days (range 1–12 days). Grade 1–2 headache was the only neurologic AE in 9 patients. A decreased level of consciousness occurred in 13 of 53 patients (25%), and in 6 cases was associated with coma requiring ventilatory support. In those who recovered, the median duration of the decreased level of consciousness was 2 days (range 1 – 12 days). Language disturbance was observed in 18 of 53 patients (34%) for a median of 4 days (range 1–9) and was accompanied in 15 of 18 patients (83%) by decreased level of consciousness and/or delirium, which complicated the distinction between impaired attention and aphasia as the etiology of language disturbance. Focal neurologic deficits, ataxia, and other abnormal movements were infrequent. Seizures occurred in 4 of 53 patients (8%). Seizures in 2 patients with grade 5 neurotoxicity occurred in the absence of a prior seizure history. The other 2 patients were among 6 in the study with an antecedent seizure history. Macroscopic intracranial hemorrhage (ICH) was rare (1 of 53; 2%) and focal large vessel ischemic stroke was not observed by imaging.
Of the entire cohort of 133 patients treated with lymphodepletion chemotherapy and CAR-T cells, 7 (5%) developed grade ≥4 neurotoxicity. Six of these 7 patients were treated during the dose-escalation phase of the protocol and received CAR-T cell doses that were subsequently determined to be above the maximum tolerated dose (MTD) for each disease and tumor burden. Only 1 of 79 patients (1.3%) treated after completion of the CAR-T cell dose-escalation phase developed grade ≥4 neurotoxicity. Four of 133 patients (3%) died due to neurotoxicity: one from multifocal brainstem hemorrhage and edema associated with DIC, 2 from acute cerebral edema, and one developed cortical laminar necrosis with a persistent minimally conscious state until death 4 months after CAR-T cell infusion. With the exception of those with fatal neurotoxicity and one patient in whom a grade 1 neurologic AE resolved 2 months after CAR-T cell infusion, neurotoxicity completely resolved in all patients by day 28 after CAR-T cell infusion (Figure 1B).
Of 53 patients with neurotoxicity, 23 underwent brain magnetic resonance imaging (MRI) within 28 days of CAR-T cell infusion. Acute abnormalities on MRI were evident in 7 of 23 patients (30%), 4 of whom had fatal neurotoxicity, indicating that an abnormal MRI scan during acute neurotoxicity is associated with a high risk of a poor outcome. T2/FLAIR changes indicative of vasogenic edema, leptomeningeal enhancement, and/or multifocal microhemorrhages were present in a majority of patients with severe neurotoxicity and abnormal MRI scans. Contrast enhancement, consistent with breakdown of the BBB, was also seen in some T2/FLAIR lesions (Figure 2A–F). One patient developed extensive cortical diffusion restriction indicative of cytotoxic edema (Figure 2G–I), which appeared distinct from vasogenic edema observed in other patients and evolved into cortical laminar necrosis. Electroencephalography (EEG) was performed in 17 of 53 patients during acute neurotoxicity. Diffuse slowing was present in 13 of 17 patients (76%). Focal slowing was noted in one patient (6%) with known epilepsy, and clinical and subclinical seizures were observed in one patient. EEG was normal in 2 of 17 patients (12%).
Figure 2. Brain magnetic resonance imaging (MRI) findings in patients with severe neurotoxicity after CD19 CAR-T cell immunotherapy.

A–B: Symmetric edema of deep structures in a patient with grade 5 neurotoxicity. FLAIR hyperintensities were seen in the bilateral thalami (A, arrowheads) and the pons (B, arrowheads), consistent with vasogenic edema. Punctate hemorrhages in the most affected areas are seen as T2 dark lesions (B, arrow). C: Global edema with blurring of the gray-white junction (stars) and slit-like ventricles (arrowhead) on FLAIR imaging in a patient with grade 5 neurotoxicity. D: Diffuse leptomeningeal enhancement (arrowheads) in a patient with grade 5 neurotoxicity. E–F: White matter FLAIR hyperintensities (E, arrowheads) that in some cases were contrast enhancing (F, arrowheads; T1 + gadolinium) in a patient with grade 3 neurotoxicity without focal neurologic deficits on exam. G–I: Cytotoxic edema of the cortical ribbon is seen on diffusion weighted imaging (G, arrowheads) and concomitant cortical swelling on FLAIR (H, arrowheads). In the same patient, injury progressed to irreversible cortical laminar necrosis indicated by T1 hyperintensities within the cortical ribbon 10 days later (I, arrowheads).
Treatment of neurotoxicity
Tocilizumab, an antagonistic IL-6R monoclonal antibody, effectively ameliorates fever and hypotension in most patients that develop severe CRS after CD19 CAR-T cells, and is frequently administered with or without corticosteroids to patients with neurotoxicity (5, 6). Twenty of 53 patients diagnosed with neurotoxicity (38%) received tocilizumab (4–8 mg/kg IV) and dexamethasone (10 mg I.V. bid), one (2%) received tocilizumab alone, and 6 (11%) received dexamethasone alone either before or after the onset of neurotoxicity (Supplementary Figure 1). Fourteen patients received 1 dose of tocilizumab, 5 received 2 doses, and two received 3 doses. A median of 2 doses (range 1–31) of dexamethasone 10 mg I.V. b.i.d. were administered and one patient received methylprednisolone (1000mg I.V. × 9). In 14 of the 21 patients (67%) who received tocilizumab the peak grade of neurotoxicity occurred after the first dose of tocilizumab, and in 8 of those patients the first presentation of neurotoxicity occurred after tocilizumab had been administered for CRS. In patients in whom neurotoxicity resolved, the time from the first tocilizumab and/or dexamethasone dose to resolution of neurotoxicity (median 4 days, range 1–64 days) was longer than the time to resolution of fever (temperature <38°C for at least 48 hours, median 0.4 days, range 0–3.8 days, p<0.0001), suggesting that once established, neurotoxicity is less responsive than CRS to interventions that suppress IL-6 activity or CAR-T cell function.
Baseline characteristics associated with subsequent neurotoxicity
We analyzed the baseline patient characteristics to identify factors associated with an increased risk of subsequent neurotoxicity. In univariate analyses (Table 1), neurotoxicity of any grade was more frequent in younger patients (p=0.094), those with B-ALL (p=0.084), a high burden of tumor (p=0.072) and CD19+ cells in bone marrow (p=0.062), and a high CAR-T cell dose (p<0.0001). The presence of any pre-existing neurologic comorbidity was also associated with neurotoxicity (p=0.0059). The infused CAR-T cell dose was the only factor associated with the occurrence of more severe neurotoxicity (grade ≥3 versus grade 1–2, p=0.014). The selection of CD8+ T cell subset in CAR-T cell manufacturing, the patient’s sex and race, the number of prior chemotherapy regimens, previous hematopoietic stem cell transplantation, and pretreatment performance score were not associated with neurotoxicity in univariate analyses. Multivariable analysis showed that preexisting neurologic comorbidities (p=0.0023), and factors associated with increased in vivo CAR-T cell numbers, including cyclophosphamide (Cy) and fludarabine (Flu) lymphodepletion (p=0.0259), higher infused CAR-T cell dose (p=0.0009), and higher burden of malignant CD19+ B cells in marrow (p=0.0165) were associated with an increased risk of neurotoxicity (Table 1).
Table 1.
Factors associated with neurotoxicity
| NEUROTOXICITY CTCAE GRADE | GRADE 0a | GRADE 1–2a | GRADE 3–5a | TOTAL | UNIVARIATEb | MULTIVARIABLEc | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| OVERALL, n (%) | 80 (60) | 25 (19) | 28 (21) | 133 (100) | |||
| AGE, n (%) | <40 years | 11 (41) | 10 (37) | 6 (22) | 27 | 0.094 | |
| 40–60 years | 42 (66) | 8 (13) | 14 (22) | 64 | |||
| >60 years | 27 (64) | 7 (17) | 8 (19) | 42 | |||
| SEX, n (%) | Male | 59 (63) | 17 (18) | 17 (18) | 93 | 0.4 | |
| Female | 21 (53) | 8 (20) | 11 (28) | 40 | |||
| DIAGNOSIS, n (%) | ALL | 22 (47) | 11 (23) | 14 (30) | 47 | 0.084 | |
| CLL | 16 (67) | 2 (8) | 6 (25) | 24 | |||
| NHL | 42 (68) | 12 (19) | 8 (13) | 62 | |||
| RACE, n (%) | White | 62 (56) | 22 (20) | 26 (24) | 110 | 0.17d | |
| Not white | 18 (78) | 3 (13) | 2 (9) | 23 | |||
| PRIOR THERAPIES | Median (range) | 4 (1, 11) | 4 (1, 10) | 4 (1, 11) | 4 (1, 11) | 0.5 | |
| TRANSPLANT | Auto | 17 (68) | 5 (20) | 3 (12) | 25 | 0.5 | |
| HISTORY, n (%) | Allo | 14 (50) | 8 (29) | 6 (21) | 28 | ||
| KARNOFSKY SCOREe, n (%) | 60 – 70 | 7 (50) | 3 (21) | 4 (29) | 14 | 0.5 | |
| 80 – 90 | 65 (61) | 18 (17) | 23 (22) | 106 | |||
| 100 | 8 (62) | 4 (31) | 1 (8) | 13 | |||
| PRE-EXISTING NEUROLOGIC COMORBIDITIES, n (%) | Any | 26 (45) | 16 (28) | 16 (28) | 58 | 0.0059g | 0.0023g |
| PNf | 14 (47) | 7 (23) | 9 (30) | 30 | 0.2 | ||
| CNS involvement | 6 (43) | 5 (36) | 3 (21) | 14 | 0.2 | ||
| Headache disorder | 6 (43) | 5 (36) | 3 (21) | 14 | 0.2 | ||
| Other | 5 (50) | 2 (20) | 3 (30) | 10 | 0.7 | ||
| ICHh | 4 (67) | 1 (17) | 1 (17) | 6 | 1 | ||
| Seizures | 2 (33) | 2 (33) | 2 (33) | 6 | 0.3 | ||
| Cog impairmenti | 1 (25) | 2 (50) | 1 (25) | 4 | 0.1 | ||
| MTX CNS toxicityj | 1 (50) | 1 (50) | 0 | 2 | 0.4 | ||
| MARROW DISEASE, % | Median (range) | 0.6 (0, 97) | 0.4 (0, 93) | 25.8 (0, 97) | 1.3 (0, 97) | 0.072 | 0.0165 |
| TOTAL CD19+ CELLS IN MARROW, % | Median (range) | 5.3 (0, 99) | 12.4 (0, 93) | 29.1 (0, 97) | 8.8 (0, 99) | 0.062 | |
| CD8+ CENTRAL MEMORY ENRICHED CAR-T CELLSk, n (%) | Selected | 48 (67) | 11 (15) | 13 (18) | 72 (54) | 0.242 | |
| LYMPHODEPLETION | Cy/Flu | 58 (56) | 23 (22) | 23 (22) | 104 | 0.11 | 0.0259 |
| REGIMENl, n (%) | Non-Cy/Flu | 22 (76) | 2 (7) | 5 (17) | 29 | ||
| CAR-T CELL DOSE, n (%) | 2×105 cells/kg | 20 (57) | 10 (29) | 5 (14) | 35 | <.0001 | 0.0009 |
| 2×106 cells/kg | 55 (64) | 15 (17) | 16 (19) | 86 | |||
| 2×107 cells/kg | 5 (42) | 0 | 7 (58) | 12 | |||
|
| |||||||
| CYTOKINE RELEASE | None (G 0) | 35 (88) | 5 (13) | 0 | 40 | <0.0001 | n/a |
| SYNDROME, n (%) | Mild (G 1–2) | 44 (57) | 19 (25) | 14 (18) | 77 | ||
| Severe (G 3–5) | 1 (6) | 1 (6) | 14 (88) | 16 | |||
Percentages are shown in parentheses.
Two-sided p-values calculated based on Kruskal-Wallis test for continuous variables, and based on Fisher’s Exact test for categorical variables.
Stepwise multivariable proportional odds models were performed to assess the impact of baseline factors on the occurrence of neurotoxicity (grade 0 vs 1–2 vs 3–5), where log10 values were used to transform data as appropriate, with 0.001 substituting for marrow disease values of 0. Although the lymphodepletion regimen did not significantly impact the risk of neurotoxicity in the univariate model, it was included in the multivariable model because of its association with increased in vivo CAR-T cell proliferation. After controlling for CAR-T cell dose and pre-treatment tumor burden, the lymphodepletion regimen was found to have a significant impact on the risk of neurotoxicity. CRS was not included in the stepwise multivariable model, because it is not a pre-treatment variable. The percentage of all CD19+ cells in bone marrow was not included in the stepwise multivariable model since it strongly correlates with the percentage of marrow CD19+ abnormal B cells (r = 0.99, p<0.0001). Only variables with a p-value <0.05 were retained in the final model.
White versus non-white
Karnofsky performance score prior to lymphodepletion
Peripheral neuropathy
None versus any
Intracranial hemorrhage
Cognitive impairment
CNS toxicity from prior intra-thecal methotrexate use
CAR-T cells manufactured from CD4+ T cells and central memory enriched CD8+ T cells
Cy/Flu regimens include both cyclophosphamide and fludarabine
Severe neurotoxicity is more frequent in patients with severe CRS and is associated with systemic vascular dysfunction
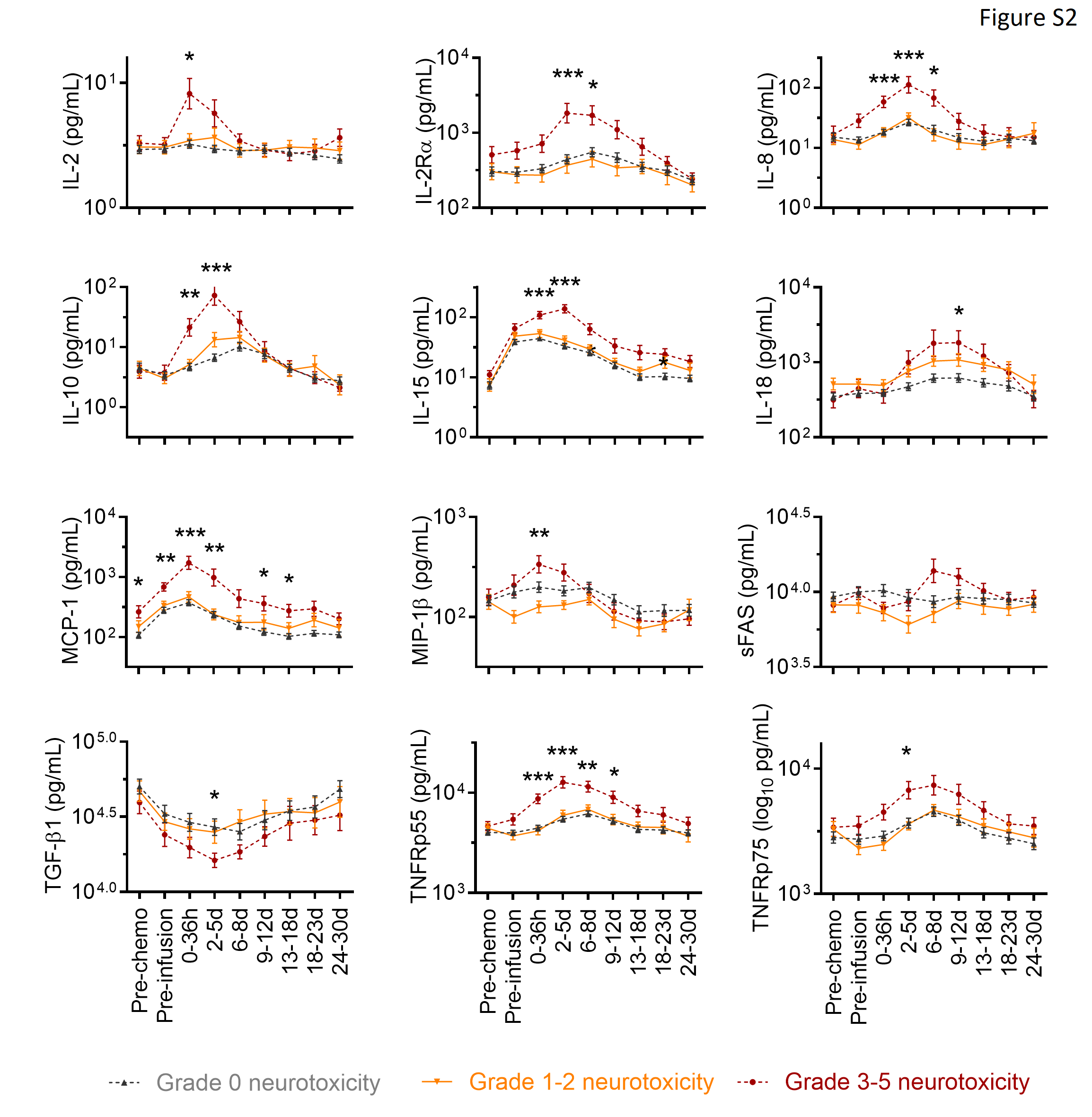
Consistent with the baseline factors that were associated with more severe neurotoxicity, we found that patients who developed grade ≥3 neurotoxicity had more severe CRS (p<0.0001; Figure 1C) and earlier and higher CD4+ and CD8+ CAR-T cell peak counts in blood compared to those with grade ≤2 neurotoxicity (Figure 3A). Patients with grade ≥3 neurotoxicity also had earlier and higher fever, more severe hemodynamic instability and tachypnea, and more severe hypoproteinemia, hypoalbuminemia and weight gain, consistent with loss of vascular integrity and systemic capillary leak (Figure 3B). Severe neurotoxicity was accompanied by disseminated intravascular coagulation (DIC), with elevated PT, aPTT and d-dimer beginning 2–5 days after CAR-T cell infusion, prolonged thrombocytopenia, and a late reduction in fibrinogen to a nadir approximately 1–2 weeks after CAR-T cell infusion (Figure 3C). The severity of neurotoxicity correlated with higher peak concentrations of CRP, ferritin, and multiple cytokines, including those that activate endothelial cells (ECs), such as IL-6, IFN-γ, and TNF-α (Figure 3D, Supplementary Figure 2). In line with the association between early onset CRS and subsequent development of severe neurotoxicity (Figure 1D), an earlier peak of the IL-6 serum concentration was associated with a higher risk of grade ≥4 neurotoxicity (Supplementary Figure 3). Within the first 6 days after CAR-T cell infusion 5 of 5 patients (100%) with an IL-6 concentration ≥ 501 pg/mL developed grade ≥4 neurotoxicity, whereas only 2 of 11 patients (18%) who reached the same serum IL-6 concentration more than 6 days after CAR-T cell infusion developed grade ≥4 neurotoxicity. These findings indicate that neurotoxicity is associated with vascular dysfunction and early onset of high concentrations of serum cytokines.
Figure 3. Severe neurotoxicity is associated with vascular dysfunction.

A: Absolute counts of CD4+/EGFRt+ and CD8+/EGFRt+ CAR-T cells in blood, and the percentages of CD4+/EGFRt+ cells within CD4+ T cells and of CD8+/EGFRt+ cells within CD8+ T cells in the indicated time windows after CAR-T cell infusion are shown in patients without neurotoxicity (grey) or with grade 1–2 (orange) or ≥3 (red) neurotoxicity. B: Minimum (min) or maximum (max) values of vital signs, serum protein and albumin concentration, and body weight are shown within the indicated time periods. Pre-chemo, before lymphodepletion chemotherapy. Pre-infusion, before CAR-T cell infusion. SBP, systolic blood pressure. DBP, diastolic blood pressure. HR, heart rate. RR, respiratory rate. C: Minimum (min) or maximum (max) values of coagulation parameters are shown within the indicated time periods. PT, prothrombin time. APTT, activated partial thromboplastin time. D: Maximum serum CRP, ferritin, IFN-γ, IL-6, and TNF-α concentrations within indicated time periods are shown, according to severity of neurotoxicity. Within each time window in all figures, the y-axis shows the mean +/− standard error of the mean (SEM) of the values for all patients. *0.001<p<0.005, **0.0001<p<0.001, ***p<0.0001 for the indicated time points for the comparison of grade 0 vs 1–2 vs 3–5 neurotoxicity. P values for TNF-α at 0–36 hours and 2–5 days after CAR-T cell infusion were 0.038 and 0.022, respectively.
Endothelial activation in patients with severe neurotoxicity
The clinical evidence of vascular leak and DIC in patients with severe neurotoxicity was consistent with widespread endothelial activation. The angiopoietin (Ang)-Tie2 axis regulates the balance between endothelial quiescence and activation (10). Ang-1 is produced constitutively, primarily by vascular pericytes and platelets, and when bound to the endothelial Tie2 receptor Ang-1 favors EC quiescence and stabilization. Ang-2 is stored in endothelial Weibel-Palade bodies and released upon EC activation by stimuli including inflammatory cytokines, displacing Ang-1 and causing increased activation and microvascular permeability. We evaluated the concentrations of Ang-2 and Ang-1 in serum from patients 1 week after CAR-T cell infusion and found that the serum Ang-2 concentration (p=0.0003) and the Ang-2:Ang-1 ratio (p=0.0014) were higher in patients with grade ≥4 neurotoxicity compared to those with grade ≤3 neurotoxicity (Figure 4A). To confirm the presence of in vivo endothelial activation in patients with severe neurotoxicity, we evaluated the concentration of von Willebrand Factor (VWF), a glycoprotein involved in hemostasis that like Ang-2 is stored in endothelial Weibel-Palade bodies and released on EC activation (11). Compared to patients with grade 0–3 neurotoxicity after CAR-T cell infusion, those with grade ≥4 neurotoxicity had higher concentrations of VWF in serum (p=0.004), which in some patients was 4–5 fold higher than those observed in pooled serum from healthy donors (Figure 4B). IL-8 is sequestered with vWF in Weibel-Palade bodies and was also elevated during severe neurotoxicity (Supplementary Figure 2) (12). These findings indicated endothelial activation and Weibel-Palade body release during severe neurotoxicity.
Figure 4. Endothelial activation in neurotoxicity associated with CD19 CAR-T cell immunotherapy.

A: Ang-1 (left) and Ang-2 (center) concentrations and the Ang-2:Ang-1 ratio (right) in serum collected approximately 7 days after CAR-T cell infusion from a subset of patients with grade 0–3 (n=52) or ≥4 (n=7) neurotoxicity. The median (bar) and interquartile range are shown. Each point represents data from one patient. B: VWF concentration in serum from patients with grade 0–3 (n=45) or grade ≥4 (n=7) neurotoxicity. Serum was collected approximately one week after CAR-T cell infusion. Data represent the fold change from the VWF concentration in normal reference plasma (CRYOcheck, Precision Biologic, Dartmouth, NS, Canada; VWF 12.2 μg/mL). C: Ang-2:Ang-1 ratios in serum collected before lymphodepletion chemotherapy from patients who subsequently developed grade 0–3 (n=49) or ≥4 (n=6) neurotoxicity. The median and interquartile range are shown. D: VWF string unit formation in HUVECs incubated with serum collected from day 3–5 from patients who received CD19 CAR-T cells and developed grade ≥4 neurotoxicity (n=4) or from healthy donors (n=4). E: VWF string unit formation in HUVECs incubated with serum collected from patients with grade ≥4 (n=3) or grade 0–3 (n=6) neurotoxicity between day 7 and 14 after CAR-T cell infusion. The mean value of 2 samples collected on days 7 and 10 was used for one patient without neurotoxicity. F: HMW and LMW VWF multimers in serum from patients with grade ≥4 (n=5) compared to grade 0–3 (n=6) neurotoxicity.
We considered that patients with evidence of endothelial activation before embarking on CAR-T cell immunotherapy might be at higher risk of subsequent cytokine-mediated vascular injury and neurotoxicity. We evaluated Ang-2 and Ang-1 concentrations in serum from patients prior to commencing lymphodepletion chemotherapy and found that the Ang-2:Ang-1 ratio was higher in patients who subsequently developed grade ≥4 neurotoxicity compared to those with grade ≤3 neurotoxicity (Figure 4C), suggesting that before lymphodepletion biomarkers of endothelial activation might identify patients at high risk of subsequent neurotoxicity. Furthermore in paired samples between day 0 and day 1 after CAR-T cell infusion, the magnitude of the change in Ang-2 concentration correlated with increasing severity of subsequent neurotoxicity (grade 0–2, median 80 ng/mL; grade 3, median 394 ng/mL; grade ≥4, median 6392 ng/mL; p=0.0039), indicating that endothelial activation occurs early after CAR-T cell infusion and precedes the onset of neurotoxicity (Figure 1D).
We hypothesized that cytokines or other factors in serum of patients treated with CAR-T cell infusion induced EC activation. We found that serum collected from patients with evidence of early neurotoxicity 3–5 days after CAR-T cell infusion induced the formation of a greater number of VWF-platelet strings on human umbilical vein ECs (HUVECs) compared to serum from normal donors (Figure 4D, Supplementary Figure 4). Formation of these strings requires release of endothelial VWF stores and subsequent platelet attachment. When we examined VWF-platelet string formation in HUVECs incubated with serum collected 7–14 days after CAR-T cell infusion, we found lower VWF-platelet string formation using serum from patients with grade ≥4 neurotoxicity compared to serum from patients with grade 0–3 neurotoxicity (Figure 4E). Lower VWF-platelet string formation occurred despite the presence of higher serum concentrations of IL-6, IFN-γ, Ang-2, and VWF in patients with grade ≥4 neurotoxicity. We considered that the low string unit formation during severe neurotoxicity might be due to relative deficiency of high molecular weight (HMW) VWF multimers, which bind activated endothelium more efficiently than low molecular weight (LMW) VWF multimers and can be consumed during acute presentations of thrombotic thrombocytopenic purpura (TTP) (13, 14). Indeed, we found that patients with grade ≥4 neurotoxicity had a lower fraction of HMW VWF multimers in serum and a higher fraction of LMW VWF multimers compared to those with grade ≤3 neurotoxicity (Figure 4F). We also examined the levels and activity of ADAMTS13, a protease that cleaves HMW VWF from activated endothelium (11). When the ADAMTS13 activity was normalized to the VWF serum level (ADAMTS13:VWF ratio), we found that during grade ≥4 neurotoxicity the ADAMTS13:VWF ratio was lower than observed with grade 0–3 neurotoxicity (grade ≥4 versus 0–3; 26.4% vs 35.2%; p=0.007), suggesting that patients with grade ≥4 neurotoxicity inefficiently remove bound HMW VWF multimers from activated endothelium. Together, these data suggest that serum from patients with neurotoxicity induces activation of ECs, which release and bind VWF, and in severe cases causes sequestration of HMW VWF multimers and contributes to the consumptive coagulopathy.
The blood-brain barrier does not shield the CSF from high serum cytokine concentrations during acute neurotoxicity
The presence of endothelial activation and systemic capillary leak raised the possibility that severe neurotoxicity after CD19 CAR-T cells might be associated with disruption of the BBB. No evidence of infectious etiology was found in CSF from patients with neurotoxicity, and only one patient had concurrent detection of leukemia blasts in CSF. CSF during acute neurotoxicity had a high protein concentration and leukocyte count compared to CSF collected before lymphodepletion, consistent with increased permeability of the BBB (Figure 5A). We did not detect a correlation between the CSF protein concentration and leukocytosis and the severity of neurotoxicity, in part because CSF sampling from patients with the most severe toxicity was frequently unable to be performed due to concurrent coagulopathy. Both CD4+ and CD8+ CAR-T cells were detected in the CSF by flow cytometry (CD4+/EGFRt+, median 2.6 cells/μL; CD8+/EGFRt+, median 2.1 cells/μL). CAR-T cells comprised a higher fraction of the CD4+ T cell subset in CSF compared to blood (Figure 5B), indicating that distinct mechanisms might regulate the migration of CD4+ and CD8+ CAR-T cells across the BBB and their retention or proliferation in CSF or brain parenchyma. CD4+ and CD8+ CAR-T cells persisted in CSF at high frequency in a subset of patients after recovery from and/or stabilization of neurotoxicity, but were infrequent in CSF from patients who had not previously developed neurotoxicity (Figure 5C). These CSF analyses show that protein and cells, including CAR-T cells, transit into the CSF in patients with neurotoxicity, consistent with increased permeability of the BBB.
Figure 5. Increased permeability of the BBB during neurotoxicity.

CSF was collected from patients before CAR-T cell infusion (Pre), during acute neurotoxicity (Acute), and after recovery from acute neurotoxicity or ≥21 days after CAR-T cell infusion in those without neurotoxicity (Recovery). A: Protein concentration and WBC counts in CSF in patients who did (red) or did not (grey) develop neurotoxicity. Each point represents data from a single patient. Box and whisker plots show the interquartile range. B: Paired CSF and blood samples collected on the same day from individual patients with neurotoxicity, showing CD4+ and CD8+ CAR-T cells as a percentage of total CD4+ and CD8+ cells, respectively. Each line represents data from a single patient. NS, not significant. C: CD4+ and CD8+ CAR-T cells as percentages of total CD4+ and CD8+ cells, respectively, in CSF. Each point represents data from a single patient. Box and whisker plots show the interquartile range. D: Concentrations of cytokines in paired serum and CSF samples obtained from patients who developed neurotoxicity. Box and whisker plots show the median (bar) and interquartile range (box). Each point represents data from one patient. *p<0.05, **p<0.01, ***p<0.001. Paired tests were used to compare serum and CSF cytokines at a single timepoint. Unpaired tests were used for comparisons between Pre and Acute timepoints. E: IL-6 and VEGF concentrations in supernatant from pericytes cultured with medium alone, IFN-γ or TNF-α. Data are representative of 6 experiments and are expressed as the fold change (mean +/− SEM) compared to culture in medium alone. F: PDGFRβ and activated caspase-3 expression by human brain vascular pericytes incubated with IFN-γ. Data are expressed as the fold change (mean +/− SEM) compared to culture in medium alone.
Serum cytokines can access the CSF through saturable transporters, circumventricular organs, and during BBB breakdown (15). To determine if increased BBB permeability during severe CRS would permit transit of plasma cytokines into the CSF, we evaluated cytokine concentrations in paired blood and CSF samples obtained before lymphodepletion and during acute neurotoxicity. Prior to lymphodepletion there was a cytokine concentration gradient between serum and CSF, with IFN-γ, TNF-α, and the TNF-α stabilizing soluble receptors, TNFR p55 and TNFR p75, being higher in blood (Figure 5D). During acute neurotoxicity, the concentrations of IFN-γ, TNF-α, IL-6, and TNFR p55 had increased markedly, and were comparable between serum and CSF, suggesting that either the BBB did not prevent high plasma cytokine concentrations from transitioning into the CSF, or that there was local cytokine production in the CSF (Figure 5D).
We considered that high concentrations of cytokines crossing the BBB might activate brain vascular pericytes, which together with ECs play an important role in maintenance of the BBB (16, 17). We evaluated the effects of IFN-γ and TNF-α by exposing primary human brain vascular pericytes in vitro to concentrations of these cytokines that are observed in patients with severe neurotoxicity. Pericytes exposed to IFN-γ secreted increased amounts of IL-6 and vascular endothelial growth factor (VEGF), each of which activates endothelial cells and increases BBB permeability (Figure 5E) (10). Incubation of pericytes with TNF-α induced an even more marked increase in IL-6 secretion (Figure 5E). IFN-γ also induced downregulation of PDGFRβ and upregulation of cleaved caspase-3 expression, consistent with induction of pericyte stress (Figure 5F) (17). Together, these findings show that increased permeability of the BBB allows transit into CSF of high concentrations of serum cytokines, including IFN-γ and TNF-α, which induce pericyte stress and secretion of cytokines that promote a further increase in BBB permeability.
Endothelial activation and vascular disruption in fatal neurotoxicity
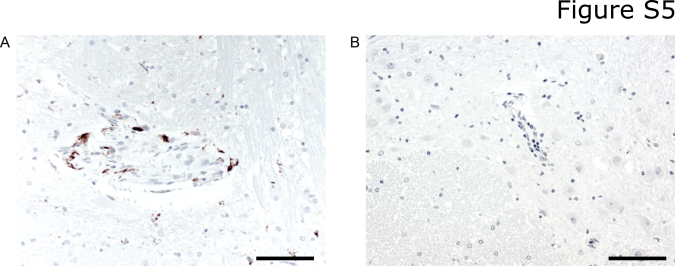
We examined autopsy tissue from the brains of 2 patients who developed fatal CRS and neurotoxicity after CD19 CAR-T cell therapy to evaluate endothelial activation and vascular injury in severe neurotoxicity. One patient died 13 days after CAR-T cell infusion with CRS and neurotoxicity characterized by brainstem hemorrhage and edema. Neuropathologic examination showed multifocal microhemorrhages and patchy parenchymal necrosis in the pons, medulla and spinal cord. Red blood cell (RBC) extravasation was observed from multiple vessels in both morphologically affected and non-affected areas of the brain parenchyma (Figure 6A). Associated with small areas of infarction were more severe vascular lesions with karyorrhexis, fibrinoid vessel wall necrosis (Figure 6B), and perivascular CD8+ T cell infiltration (Figure 6C), representing the vast majority of the brain T cells by IHC. Flow cytometry on fresh pons tissue obtained at autopsy showed T cells constituted 56.9% of CD45+ cells and that most of the T cells in the brain were CAR-T cells, with 93% of the T cells in the pons expressing the EGFRt transduction marker (CD4−/CD8+ CAR-T cells, 51.9%: CD4+/CD8− CAR-T cells, 48.1%). This was consistent with the data from CSF collected at autopsy in which 82% of the CD45+ cells were CD3+ T cells and of these 94.9% were CAR-T cells (CD4−/CD8+ CAR-T cells, 48%: CD4+/CD8− CAR-T cells, 42.5%). We observed intravascular VWF binding and CD61+ platelet microthrombi (Figure 6D, E), consistent with endothelial activation, and CD31 immunohistochemistry (IHC) demonstrated disrupted endothelium in some vessels (Figure 6F). Reactive microglia were noted in a perivascular distribution, but marked and diffuse microglial activation was not observed (Supplementary Figure 5). No CD79a+ tumor cells were detected (Supplementary Figure 5). The brain of a patient who died due to severe CRS with multiorgan failure and grade 4 neurotoxicity (on day 3 after CAR-T cell infusion) also showed intravascular CD61+ platelet microthrombi.
Figure 6. Endothelial activation and vascular disruption in CAR-T cell neurotoxicity.
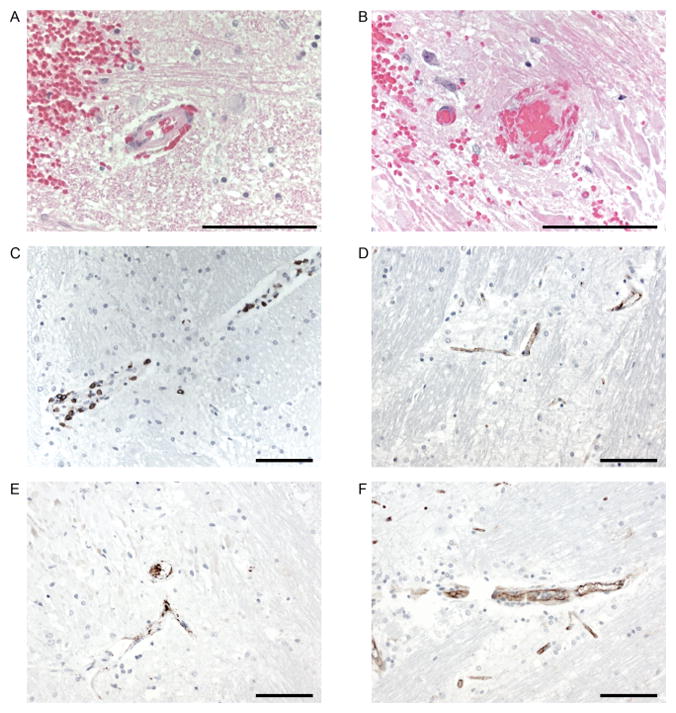
A: Hematoxylin and eosin staining of medulla showing red blood cell extravasation into the surrounding parenchyma and Virchow-Robin space in the setting of minimal arteriolar wall disruption. B: Hematoxylin and eosin staining showing fibrinoid vessel wall necrosis and vascular occlusion. C: Perivascular CD8+ T cell infiltration. D: IHC for VWF showing VWF binding to capillaries. E: IHC for CD61 demonstrates intravascular microthrombi. F: IHC for CD31 showing reduplicated and disrupted endothelium. Size bars (100μm) are shown.
Identification of patients at high risk of subsequent neurotoxicity
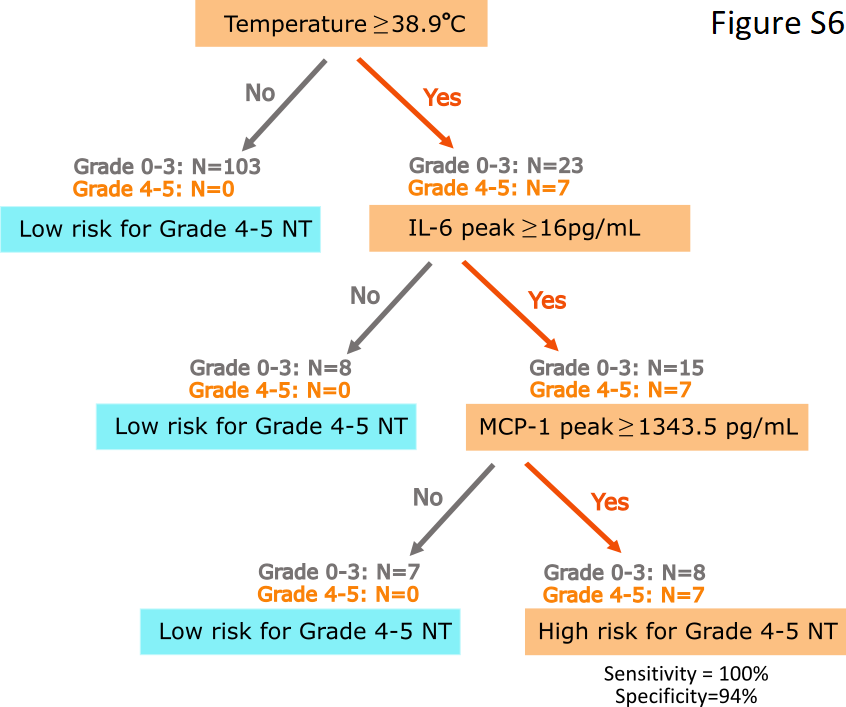
Early identification of patients at risk of developing the most severe neurotoxicity might allow intervention with tocilizumab and/or corticosteroids, enabling reduction in serum cytokine concentrations that could mitigate or prevent subsequent toxicity. Fever ≥38.9°C occurring within 36 hours of CAR-T cell infusion had a 100% sensitivity for subsequent grade ≥4 neurotoxicity; however, the specificity was only 82%, in part due to other causes of fever in these patients with chemotherapy-induced neutropenia. Because IL-6, IFN-γ, MCP-1, IL-15, IL-10, and IL-2 were higher (p<0.001) within the first 36 hours after CAR-T cell infusion in those who subsequently developed grade ≥4 neurotoxicity, we investigated whether testing of serum cytokines could better identify patients at risk of severe neurotoxicity compared to evaluation of the temperature alone within 36 hours of CAR-T cell infusion. Classification tree modeling demonstrated that patients with fever ≥38.9°C and serum IL-6 ≥16 pg/mL and MCP-1 ≥1343.5 pg/mL in the first 36 hours after CAR-T cell infusion were at high risk of subsequent grade ≥4 neurotoxicity (sensitivity 100%; specificity 94%; Supplementary Figure 6). The model misclassified 8 of 133 patients, of whom only one (0.75%) did not subsequently develop moderate or severe grade 2–3 neurotoxicity and/or grade ≥2 CRS, indicating that unnecessary early intervention guided by the classification tree model is rare.
DISCUSSION
Neurologic AEs occur frequently after lymphodepletion chemotherapy and CD19 CAR-T cell immunotherapy, and the incidence of grade ≥3 neurotoxicity in our study was similar to that described in previous reports (1–4). The presentation of neurotoxicity was heterogeneous, with most events being consistent with nonspecific brain dysfunction without focal signs. The diagnosis of CAR-T cell-associated neurotoxicity required the appearance of new neurologic signs or symptoms within the first 1–3 weeks after CAR-T cell infusion without attribution to other etiologies. Imaging by MRI or CT was normal in most cases and was mainly used to exclude other possible causes of neurologic AEs; however, acute abnormalities on MRI were observed in patients with severe neurotoxicity. EEG showed diffuse slowing in most cases and was primarily used to exclude seizure activity. CSF analysis did not reveal findings specific for neurotoxicity, but did demonstrate evidence of increased BBB permeability, with transit of protein, leukocytes and elevated concentrations of serum cytokines into the CSF. Although most neurologic AEs were mild and transient, a subset of patients developed severe neurotoxicity with presentations that included focal neurologic signs, seizures, and/or cerebral edema. Six of the 7 patients with grade ≥4 neurologic AEs were treated during the dose-escalation stage of the study with CAR-T cell doses that were subsequently established to be above the MTD for the patient’s disease and tumor burden. After establishment of the CAR-T cell MTDs, the incidence of grade ≥4 neurotoxicity was only 1.3%.
Neurotoxicity was almost invariably associated with CRS and preceded by fever, allowing for hospital admission of febrile patients who received outpatient CAR-T cell immunotherapy. The association between CRS and neurotoxicity was supported by the high cytokine concentrations in serum and high CAR-T cell counts in blood in patients with severe neurotoxicity. Baseline factors, including higher disease burden, infused CAR-T cell dose and addition of Flu to Cy-based lymphodepletion that are associated with robust CAR-T cell proliferation in blood, were also associated with an increased risk of subsequent neurotoxicity. It is probable that the increase in neurotoxicity in patients who received Cy/Flu lymphodepletion in our study is due to greater in vivo CAR-T cell expansion; however, a contribution from a direct toxic effect of Flu on the brain or endothelium cannot be excluded. Neurotoxicity has been reported after immunotherapy with T cells that are engineered with CD19 CARs that incorporate either 4-1BB or CD28 costimulation, but no studies have examined whether the selection of costimulatory molecule affects risk (1–6). Neurotoxicity also occurs after immunotherapy with blinatumomab, a bi-specific anti-CD3/CD19 T cell engager (BiTE), supporting a role for T cell activation in the pathogenesis (18). It is intriguing that neurotoxicity is observed more frequently after T cell-mediated immunotherapies targeting CD19 compared to other antigens. It seems likely that the higher risk of neurotoxicity after CD19-targeted therapies reflects their capacity for robust T cell activation compared to T cell therapies targeting other antigens expressed on different tumors. Factors that could contribute to the differences in T cell activation after targeting CD19 compared to other antigens include accessibility of the tumor to T cells, the level of antigen expression, the presence of regulatory molecules, and the affinity of the antigen-targeting moiety (CAR vs TCR). A direct effect of targeting CD19 cannot be excluded.
In addition to the high serum and CSF cytokine levels in patients during acute neurotoxicity, we observed that the severity of neurotoxicity was greater in patients who reached a peak IL-6 concentration early after CAR-T cell infusion, suggesting that the rate of rise in serum cytokine concentration, as well as the peak concentration, may be a determinant of the severity of neurotoxicity. Consistent with this, we noted that patients who subsequently developed severe neurotoxicity had a shorter time to first fever and had higher serum cytokine concentrations early after CAR-T cell infusion compared to those with mild neurotoxicity.
In patients with severe neurotoxicity we noted clinical evidence of endothelial dysfunction, including vascular instability, capillary leak, DIC, and BBB disruption, associated with elevated serum biomarkers of endothelial activation (Ang-2 and VWF). The concentrations of VWF observed in a subset of patients with severe neurotoxicity were extremely high, which along with thrombocytopenia and consumption of HMW VWF multimers in these patients indicated profound endothelial activation. Similar findings have been identified in children with cerebral edema due to P. falciparum malaria and in patients with TTP (11). While endothelial activation and loss of HMW VWF multimers can be seen during acute presentations of TTP, microangiopathic RBC morphology and renal failure were not characteristic of neurotoxicity associated with CAR-T cell therapy, suggesting a distinct pathogenesis from TTP (11). Due to the small numbers of patients in the study who developed grade ≥4 neurotoxicity, the relationships between the clinical factors and these biomarkers will need validation in subsequent cohorts.
Among the cytokines that were elevated in serum from patients with severe neurotoxicity were those that induce endothelial activation (IL-6, IFN-γ, and TNF-α). IL-6, IFN-γ, and TNF-α were increased in both blood and CSF during acute neurotoxicity, consistent with either the failure of the BBB to exclude transit of the high concentrations of serum cytokines or local production of these cytokines within the CSF. Due to the risks of obtaining CSF early after CAR-T cell infusion from patients without neurotoxicity, we cannot definitively exclude the presence of a BBB leak in asymptomatic patients. However, the presence of CAR-T cells in CSF from patients after resolution of active neurotoxicity, but not from patients without prior neurotoxicity, suggests a BBB leak in asymptomatic patients is unlikely. We identified CAR-T cells in CSF and in a patient who died of severe neurotoxicity that appeared to be the major component of the T cell population in the brain, suggesting that the egress of CAR-T cells into the CNS might contribute to high CSF cytokine levels and neurotoxicity. Other cells in the CNS might contribute to high cytokine levels in CSF. We show that incubation of primary human brain pericytes with TNF-α resulted in increased secretion of IL-6, and that incubation with IFN-γ increased secretion of IL-6 and VEGF and induced pericyte stress. These findings are consistent with the combination of high serum cytokines, endothelial activation, and BBB leak leading to a cascade that progressively amplifies endothelial activation and BBB injury (Supplementary Figure 7). In support of this, autopsy studies identified evidence of endothelial activation manifest by platelet aggregation and VWF binding in small capillaries, which was accompanied by RBC extravasation, a perivascular CD8+ T cell infiltrate, vascular wall destruction, and multifocal hemorrhage. Microhemorrhages were observed on MRI scans in other patients with severe neurotoxicity. A recent study in a murine tumor model found that IFN-γ and TNF-α contributed to tumor vessel regression and occlusion, and vascular burst, respectively (19); however, the relative contributions of distinct cytokines to BBB injury were not determined in these studies.
We did not observe profound morphologic changes in microglial cells at autopsy. However, the finding of hyperferritinemia in patients with neurotoxicity raises the possibility that activation of macrophage-lineage cells may contribute to the pathogenesis of neurotoxicity, akin to the neurologic manifestations observed in hemophagocytic lymphohistiocytosis (20). Although there are as yet no animal models of neurotoxicity after CD19 CAR-T cell immunotherapy that reflect the human presentation, future improvements of these models may be of value in further exploring the pathogenesis of human CAR-T cell associated neurotoxicity.
Treatment of established neurotoxicity has largely been directed towards management of CRS by suppression of T cell activation with corticosteroids, and control of IL-6-mediated inflammatory pathways using tocilizumab. Other mainstays of management include supportive care to maintain organ function and prevent seizures. Tocilizumab and dexamethasone were highly effective at controlling fever and hypotension during CRS, but it is unclear whether the near uniform resolution of neurotoxicity is a result of these interventions or the natural history of CAR-T cell proliferation, contraction, and quiescence. Furthermore, the observation that neurotoxicity still occurred in some patients despite prior administration of tocilizumab for CRS suggests that targeting IL-6R in isolation during established CRS is insufficient to prevent subsequent neurotoxicity. Because tocilizumab may not penetrate the CNS (21, 22), we and others have considered that its use might increase CSF IL-6 levels and worsen neurotoxicity, and have suggested that siltuximab, an IL-6 antagonist, may be preferred. Strategies to target other cytokines and/or their receptors warrant further investigation. In the absence of data from randomized studies, the optimal approach to management remains unknown. In addition to regulating CAR-T cell activation and reducing production of cytokines that might activate ECs, strategies to stabilize ECs and minimize BBB disruption might be suitable to treat or prevent neurotoxicity. For example, recombinant Bow-Ang-1 was used to effectively prevent cerebral edema associated with malaria in mice (23), and modification of Tie2 signaling minimized injury associated with BBB disruption in stroke models (24). Other strategies such as hypertransfusion of platelets to augment Ang-1 levels, or plasma exchange to deplete cytokines and HMW VWF multimers could be studied in animal models or suitably designed clinical trials.
An alternative approach to the management of neurotoxicity is to prospectively identify, and either exclude or pre-emptively treat those at highest risk. Patients with preexisting neurologic comorbidities were more likely to develop neurotoxicity, although the significance is unclear because the study was not powered to determine the risk associated with individual neurologic comorbidities. Given the potential benefits of CAR-T cell immunotherapy and the almost universally poor outcomes without this approach, exclusion of patients with previous comorbidities does not seem justified; however, reduction of the CAR-T cell dose may be appropriate in a subset of high-risk patients, for example those with biomarkers that might indicate pre-existing endothelial activation, such as a high Ang-2:Ang-1 ratio in serum before lymphodepletion or CAR-T cell infusion. Our finding that the serum concentrations of IL-6 and MCP-1 within 24 hours of CAR-T cell infusion identified patients who subsequently developed severe neurotoxicity provides an opportunity to monitor concentrations of these cytokines to identify those at highest risk. Additional studies will be required to determine if pre-emptive therapy with dexamethasone, IL-6 or IL-6R blockade, or endothelial stabilizing agents will prevent severe neurotoxicity in high-risk patients.
CD19 CAR-T cell therapy is a promising and novel therapy for patients with refractory B-cell malignancies that can be complicated by neurotoxicity in a subset of patients. Identification of the pathologic characteristics of neurotoxicity, related biomarkers, and risk factors will facilitate further studies of the mechanisms of neurotoxicity, and will enhance efforts to safely deliver CAR-T cell immunotherapy.
METHODS
Patient characteristics, lymphodepletion chemotherapy and CAR-T cell infusion
We conducted a single center study of neurologic AEs in 133 patients with relapsed and/or refractory CD19+ B-ALL, NHL and CLL who received lymphodepletion chemotherapy and CD19 CAR-T cells in a phase 1/2 CAR-T cell dose escalation/de-escalation clinical trial (5, 6). The study is available at https://clinicaltrials.gov/ct2/show/NCT01865617, and was conducted with approval of the Fred Hutchinson Cancer Research Center (FHCRC) institutional review board. Informed consent was obtained from all patients. Patients received lymphodepletion chemotherapy (Supplementary Table 1) 36–96 hours before CAR-T cell infusion. CD19 CAR-T cells were manufactured in a 1:1 ratio of CD4+:CD8+ CAR-T cells as previously described (Supplementary data), and infused at one of 3 dose levels (2×105/kg, 2×106/kg or 2×107/kg) 2–4 days after completion of chemotherapy (5, 6). A non-signaling truncated human epidermal growth factor receptor (EGFRt) encoded in the transgene allowed precise enumeration of transduced CD4+ and CD8+ CAR-T cells by flow cytometry. This paper reports neurologic AEs presenting within 28 days after the first CAR-T cell infusion.
Assessment of neurologic adverse events
Neurologic symptoms and signs were prospectively assigned an AE term and maximal severity score according to the NCI Common Terminology Criteria for Adverse Events (CTCAE; version 4.03). The prospective assignment was made by the Principal Investigator and co-investigator who managed the patient. An independent retrospective review of the electronic medical record was performed by a member of the neurology team, who assigned AE terms and daily severity scores for each term according to the CTCAE version 4.03. Where a discrepancy between prospective severity scoring by the PI and co-investigator and retrospective scoring by the neurology team was identified, the final score was allocated by consensus review. Neurologic assessment was performed daily on inpatients by the managing oncology team and as clinically indicated by the neurology team. Due to the heterogeneous presentation of neurotoxicity, we retrospectively grouped AEs into term subsets (Supplementary Table 2). “Delirium” encompassed acute cognitive impairment manifesting as confusion, agitation or difficulty with attention or short-term memory, and was distinguished by preserved alertness from “decreased level of consciousness”. Additional AE subsets included “ataxia”, “focal weakness”, “generalized weakness”, “hallucinations”, “headache”, “ICH”, “language disturbance”, “oculomotor disorder”, “seizure”, “stroke”, “tremor”, “other abnormal movements”, and “visual changes”. Neurologic AEs not captured in the preceding list were designated “other”. The overall neurotoxicity grade assigned for a given patient was the highest grade of all neurologic AEs identified in that patient.
Neuroimaging
Computed tomography (CT) and MRI scans of the head were performed when clinically appropriate using standard clinical sequences. All available imaging was reviewed for this study and retrospectively classified as normal, acutely abnormal or chronically abnormal. Abnormalities on MRI scans performed during clinical neurotoxicity within 28 days after CAR-T cell infusion were designated acute when findings were consistent with an acute event (edema, blood, enhancement, or diffusion restriction) and were either new compared to prior imaging or evolved on follow-up. Abnormalities were designated chronic when there were nonspecific white matter changes that are typical sequelae of chemotherapy or age-related microvascular disease, when abnormalities were due to an unrelated prior process, and when findings were stable in baseline or follow-up scans.
CSF and blood samples
CSF was collected from patients when appropriate for clinical care: before lymphodepletion (Pre), during the presence of acute neurotoxicity (Acute), or when patients had recovered from the acute toxicities associated with CAR-T cell immunotherapy (Recovery, approximately 3 weeks or more after CAR-T cell infusion). CD4+ and CD8+ CAR-T cell counts in blood and CSF were evaluated by flow cytometry, as previously described (5, 6). The absolute CAR-T cell count was determined by multiplying the percentage of CAR-T cells identified by flow cytometry in a lymphocyte FS-SS gate by the absolute lymphocyte count established by automated hemocytometer. Patients with progressive CNS malignancy detected by flow cytometry analysis of CSF were not included in the CSF analyses. Concentrations of all cytokines except Ang-1 and Ang-2 in serum and CSF were evaluated by Luminex assay (Riverside, CA), as previously described (5, 6). Serum Ang-1 and Ang-2 concentrations were evaluated using an immunoassay-based method on the Meso Scale Discovery Quickplex SQ 120 instrument (Meso Scale Diagnostics, Rockville, MD), according to the manufacturer’s instructions.
VWF and ADAMTS13 assays
The VWF concentration in patient sera was measured by sandwich ELISA as described previously (13), using polyclonal rabbit anti-human VWF as a capture antibody and HRP-conjugated polyclonal rabbit anti-human VWF as a detection antibody (Dako, Troy, MI). ADAMTS13 activity in patient sera was measured using an enzyme-linked assay to evaluate cleavage of a horseradish peroxidase (HRP)-conjugated peptide from the VWF A2 domain as previously described (25).
Serum-induced activation of ECs
HUVECs (Lonza, Portsmouth, NH) were cultured for 7 days in parallel-plate flow chambers coated with rat tail type I collagen. Serum, either from patients or healthy donors, was incubated with HUVECs at 37°C for 30 min under static conditions. The chambers were perfused with PBS to remove serum then perfused with a suspension of fixed platelets (Dade Behring, Siemens Medical Solutions, Deerfield, IL) to decorate VWF strings attached to the surface of the HUVECs. The number and length of the VWF-platelet strings were quantified as string units on 16 random non-overlapping bright field images per chamber, as described (13). The values for string units obtained from HUVECs incubated with serum were normalized to those from HUVECs stimulated with phorbol myristate acetate (PMA), which was designated as 100%.
Cytokine stimulation of primary human brain pericytes
Primary human brain vascular pericytes were cultured in Specialty Medium (ScienCell Research Laboratories, Carlsbad, CA) alone or supplemented with IFN-γ 30ng/mL (Peprotech, Rocky Hill, NJ) after 24 and 72 hours. After 96 hours, IL-6 and VEGF concentrations were analyzed in the culture supernatant by Luminex, and PDGFRβ (Biolegend, San Diego, CA) and cleaved caspase-3 (Cell Signaling, Danvers, MA) expression on pericytes were determined by flow cytometry.
Histology and immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) brain tissue blocks were sectioned at 4 μm and mounted on positively charged slides. A hematopathologist, an anatomic transplant pathologist, and a neuropathologist examined hematoxylin and eosin-stained slides of available autopsy tissues. Histologic features were identified and graded by consensus. Immunohistochemistry was performed on brainstem sections of pons at the level of the locus coeruleus using a standard automated immunodetection system with the following antibodies: anti-CD3 (Ventana, Tucson, AZ), anti-CD8 (Ventana), CD31 (Dako), CD61 (Ventana), CD68 (Dako), CD79a (Ventana), and VWF (Dako). Appropriate positive and negative controls were included with each antibody run.
Statistical methods
Statistical methods are available in Supplementary data.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STATEMENT OF SIGNIFICANCE.
We provide a detailed clinical, radiological and pathological characterization of neurotoxicity after CD19 CAR-T cells, and identify risk factors for neurotoxicity. We show endothelial dysfunction and increased blood-brain barrier permeability in neurotoxicity, and find that patients with evidence of endothelial activation before lymphodepletion may be at increased risk of neurotoxicity.
Acknowledgments
Funding was provided by: NCI R01 CA136551; Juno Therapeutics, Inc; NIDDK P30 DK56465; NCI P30 CA15704; Life Science Discovery Fund; Bezos Family Foundation; University of British Columbia Clinical Investigator Program; R56 HL131946-01; R01 HL117639-01; R01 HL112633; R21 HL129526-01; and institutional funds from Bloodworks Northwest.
The authors acknowledge the FHCRC Cell Processing Facility and Program in Immunology; Seattle Cancer Care Alliance (SCCA) Cell Therapy Laboratory, Immunotherapy Clinic, and Pathology Laboratory; the University of Washington (UW) HIC Comparative Pathology Laboratory and UW Medical Center Immunohistochemistry Laboratory; and the UW Autopsy and Decedent Affairs staff and the Pathology and Neuropathology trainees for their help acquiring neuropathological samples.
Footnotes
Conflict of interest disclosure:
CJT, DGM and SRR receive research funding from Juno Therapeutics and hold patents and/or provisional patent applications. SRR is a co-founder of Juno Therapeutics, and holds patents. DL is an employee of and has equity in Juno Therapeutics. CY received research funding from Gilead for an unrelated project. FHCRC receives research funding from Juno Therapeutics.
References
- 1.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–9. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–38. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle CJ, Riddell SR, Maloney DG. CD19-targeted chimeric antigen receptor-modified T cell immunotherapy for B cell malignancies. Clin Pharmacol Ther. 2016 doi: 10.1002/cpt.392. [DOI] [PubMed] [Google Scholar]
- 8.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–30. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence. 2013;4:507–16. doi: 10.4161/viru.24530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwameis M, Schörgenhofer C, Assinger A, Steiner MM, Jilma B. VWF excess and ADAMTS13 deficiency: a unifying pathomechanism linking inflammation to thrombosis in DIC, malaria, and TTP. Thromb Haemost. 2015;113:708–18. doi: 10.1160/TH14-09-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romani de Wit T, de Leeuw HP, Rondaij MG, et al. Von Willebrand factor targets IL-8 to Weibel-Palade bodies in an endothelial cell line. Exp Cell Res. 2003;286:67–74. doi: 10.1016/s0014-4827(03)00094-6. [DOI] [PubMed] [Google Scholar]
- 13.Chung DW, Chen J, Ling M, et al. High-density lipoprotein modulates thrombosis by preventing von Willebrand factor self-association and subsequent platelet adhesion. Blood. 2016;127:637–45. doi: 10.1182/blood-2014-09-599530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wada H, Kaneko T, Ohiwa M, et al. Plasma cytokine levels in thrombotic thrombocytopenic purpura. Am J Hematol. 1992;40:167–70. doi: 10.1002/ajh.2830400303. [DOI] [PubMed] [Google Scholar]
- 15.Yarlagadda A, Alfson E, Clayton AH. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry (Edgmont) 2009;6:18–22. [PMC free article] [PubMed] [Google Scholar]
- 16.Armulik A, Genové G, Mäe M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 17.Rustenhoven J, Jansson D, Smyth LC, Dragunow M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol Sci. 2017;38:291–304. doi: 10.1016/j.tips.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 19.Kammertoens T, Friese C, Arina A, et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature. 2017 doi: 10.1038/nature22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trottestam H, Horne A, Aricò M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118:4577–84. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen F, Teachey DT, Pequignot E, et al. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J Immunol Methods. 2016;434:1–8. doi: 10.1016/j.jim.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112:3959–64. doi: 10.1182/blood-2008-05-155846. [DOI] [PubMed] [Google Scholar]
- 23.Higgins SJ, Purcell LA, Silver KL, et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med. 2016;8:358ra128. doi: 10.1126/scitranslmed.aaf6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurnik S, Devraj K, Macas J, et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol. 2016;131:753–73. doi: 10.1007/s00401-016-1551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu JJ, Fujikawa K, Lian EC, McMullen BA, Kulman JD, Chung DW. A rapid enzyme-linked assay for ADAMTS-13. J Thromb Haemost. 2006;4:129–36. doi: 10.1111/j.1538-7836.2005.01677.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.