

Summary
Mitochondrial fusion and fission are critical to heart health; genetically interrupting either is rapidly lethal. To understand whether it is loss of, or the imbalance between, fusion and fission that underlies observed cardiac phenotypes, we engineered mice in which Mfn-mediated fusion and Drp1-mediated fission could be concomitantly abolished. Compared to fusion-defective Mfn1/Mfn2 cardiac knockout or fission-defective Drp1 cardiac knockout mice, Mfn1/Mfn2/Drp1 cardiac triple knockout mice survived longer and manifested a unique pathological form of cardiac hypertrophy. Over time, however, combined abrogation of fission and fusion provoked massive progressive mitochondrial accumulation that severely distorted cardiomyocyte sarcomeric architecture. Mitochondrial biogenesis was not responsible for mitochondrial superabundance, whereas mitophagy was suppressed despite impaired mitochondrial proteostasis. Similar but milder defects were observed in aged hearts. Thus, cardiomyopathies linked to dynamic imbalance between fission and fusion are temporarily mitigated by forced mitochondrial adynamism at the cost of compromising mitochondrial quantity control and accelerating mitochondrial senescence.
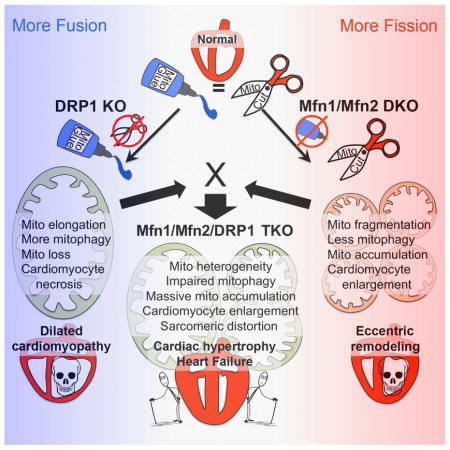
In Brief
Mitochondrial dynamism and mitophagy maintain mitochondrial fitness. The impact of fission/fusion activity versus balance is unknown. Song et al reported that mice having hearts with adynamic mitochondria live longer than fission- or fusion-defective parents, but develop mitochondrial senescence and heart failure from defective mitophagy, mitochondrial superabundance, and distorted sarcomeric architecture.
Introduction
Mitochondria confer a dual benefit to host cells by consuming toxic oxygen while generating ATP that powers cell tasks. Mitochondrial respiratory dysfunction therefore has two adverse consequences: cytotoxic oxygen radicals accumulate and ATP production declines. Both of these events contribute to senescence (Wallace, 2005).
Regulation of mitochondrial abundance and fitness are inextricably linked. Conceptually, mitochondrial abundance can be adjusted through regulated organelle production or elimination, while the fitness of mitochondrial populations is maintained by selectively purging damaged mitochondria and replacing them with newly generated healthy organelles. Thus, mitochondrial introduction through biogenesis and removal through mitophagy are homeostatically coordinated (Ploumi et al., 2016). A recent study of C. elegans identified signaling events essential to age-related engagement of mitophagy and biogenesis, without which cellular functioning was compromised and premature senescence occurred (Palikaras et al., 2015).
If mitochondrial biogenesis and mitophagy are the alpha and omega of the mitochondrial life cycle, then mitochondrial dynamics is the intervening alphabet. Because of their high minute-by-minute energy requirements, genetic deletion of mitochondrial fission and fusion factors in striated (cardiac and skeletal) muscle and neuronal tissue has provided insights into how mitochondrial dynamics factors contribute to mitochondrial quality control (Chen et al., 2007; Chen et al., 2010; Chen and Dorn, 2013; Franco et al., 2016; Lee et al., 2012). Thus, disruption in mouse hearts of mitochondrial fission induced by Dynamin-related protein1 (Drp1), which is essential for sequestering and mitophagically eliminating damaged components (Shirihai et al., 2015; Twig et al., 2008), first retards and then de-constrains mitophagy (Dorn, 2015a; Song et al., 2015a; Song et al., 2015b). Deletion in mouse hearts of Mitofusin 2 (Mfn2), which opposes Drp1 in mitochondrial dynamics and plays a central role in mitophagy as a receptor for Parkin (Gong et al., 2015), induces progressive cardiac hypertrophy with systolic dysfunction (Chen and Dorn, 2013).
Here, we interrogated a putative mitochondrial dynamism/biogenesis/mitophagy interactome by completely de-coupling mitochondrial dynamism. Using temporally-defined and cardiomyocyte-directed combined genetic ablation of Drp1 and Mfn1/Mfn2 to simultaneously abolish mitochondrial fission and fusion in adult mouse hearts we addressed three questions: 1. Is cardiomyocyte mitochondrial morphology a determinant of heart health, distinct from other factors modulated by mitochondrial dynamics? 2. Is it more important in cardiomyocytes for mitochondrial fission and fusion to be present, or to be balanced? 3. How does the complete absence of normal mitochondrial dynamism impact mitochondrial quality and quantity control? Our results point to a central role for mitochondrial fission and fusion in mitochondrial renewal, without which mitochondrial senescence is accelerated.
Results
Cardiac Drp1 overexpression provokes mitochondrial fragmentation without cardiac pathology
“Mitochondrial fragmentation” is widely regarded as harmful or pathological. Targeted cardiomyocyte deletion of fusion-promoting Mfn1 and Mfn2 in combination, or of optic atrophy 1 (Opa1), provokes mitochondrial shortening and cardiac dysfunction (Chen et al., 2011; Papanicolaou et al., 2012; Song et al., 2015b; Wai et al., 2015). However, conclusions that mitochondrial shortening causes observed cardiac phenotypes are confounded by Mfn2 functioning in mitophagy and Opa1 functioning in cristae organization (Chen and Dorn, 2013; Cogliati et al., 2013; Gong et al., 2015; Varanita et al., 2015). Likewise, cardiac dysfunction correlated with Drp1-mediated mitochondrial fission induced by myocardial injury (Disatnik et al., 2013; Ong et al., 2010) can be explained by underlying mitochondrial damage.
To determine if cardiomyocyte mitochondrial fragmentation is intrinsically damaging to hearts we overexpressed fission-promoting Drp1 in cardiomyocytes using a bi-transgenic (tet-off (Sanbe et al., 2003)) myh6 promoter system (Figure 1A). In the absence of doxycycline this system drives cardiomyocyte transgene expression from birth (Syed et al., 2004). Two independent mouse lines were established, expressing Drp1 at levels ~10- and ~25-fold normal hearts (Figure 1B; Supplemental Figure S1). Neither Drp1 transgenic line developed any cardiac phenotype through 93 weeks of age (Figures 1C–1E); detailed studies were performed in the higher expressing line.
Figure 1. Mitochondrial fragmentation induced by Drp1 is not deleterious to mouse hearts.

(A) Schematic depiction of bi-transgenic system used to target human (h) Drp1 expression to mouse cardiac myocytes.
(B) Immunoblot analysis of Drp1 (top) and GAPDH (bottom) expression in rTA transgenic control (Ctrl) and 25-fold overexpressing transgenic (TG) littermate mouse hearts. Each lane is a separate heart. Odd lane numbers are males; even lanes are females. To the right are immunoblots of other mitochondrial dynamics proteins. Mff is mitochondrial fission factor. GAPDH is loading control.
(C) Representative Masson’s trichrome stained hearts from 50 (top) and 93 (bottom) week old mice. Scale bar is 5 mm.
(D) Gravimetric heart weights indexed to body weight in aging mice (non-significant by t-test).
(E) Echocardiographic studies. (Left) Representative m-mode echos of 35 week old mice. (Right) Quantitative group data; LV EDD is left ventricular end diastolic dimension.
(F) Representative transmission electron micrographs showing mitochondrial fragmentation in Drp1 TG heart (lower panel). Quantitative data are means±SEM. Both white bars are controls, representing each of the pair of transgenes in the bi-transgenic system. NS is non-significant (ANOVA).
(G) Representative cardiac mitochondria respiration studies stimulated by glutamate/malate. Inset is quantitative group data for respiratory control ratio (state 3/state 2) (non-significant by t-test). See also Figure S1.
Forced expression of Drp1 induced mitochondrial fragmentation (Figure 1F) without affecting other mitochondrial dynamics proteins (see Figure 1B). The fragmented mitochondria were otherwise structurally unremarkable and exhibited normal mitochondrial respiration (Figure 1G). Thus, chronic Drp1-induced mitochondrial hyper-fragmentation is not intrinsically damaging either to cardiomyocyte mitochondria or to mammalian hearts. We recently reported that cardiomyocyte overexpression of Mfn2 induces mitochondrial enlargement without any cardiac pathology (Gong et al., 2015). Integrating these results we conclude that increased or decreased mitochondrial size is not necessarily the mechanism underlying cardiac phenotypes evoked by genetic deletion of mitochondrial fission of fusion factors; other functions of mitochondrial dynamics proteins require consideration.
Abolishing mitochondrial dynamism provokes mitochondrial dysfunction in cultured MEFs
Normal fibroblasts have highly dynamic mitochondria; the consequences of deleting either Drp1 or Mfn1 and Mfn2 have been described in cultured murine embryonic fibroblasts (MEFs) derived from germ-line mouse knockout lines (Chen et al., 2003; Ishihara et al., 2009). Because germ-line deletion of Drp1 or Mfn1/Mfn2 is embryonic lethal, it is not possible to abolish mitochondrial dynamism by crossing the parental lines. We worked around this problem by generating Mfn1/Mfn2/Drp1 triple floxed allele mice from which we derived MEFs. These cells express Mfn1, Mfn2 and Drp1 normally until infected with adeno-Cre, after which the targeted mitochondrial dynamics factors dissipate within ~48 hours (Figure 2A). Cell viability was not impacted (Figure 2B), but concomitant loss of Mfn1, Mfn2 and Drp1 evoked dissolution of the mitochondrial network, evoking mitochondrial fragmentation and partial depolarization (Figure 2C). Accumulation of abnormal mitochondria was associated with impaired mitochondrial translocation of the mitophagy effector Parkin (Figure 2D) and suppressed mitochondrial removal by lysosomes (Figure 2E). An adenovirus that expresses β-galatosidase did not affect these endpoints in triple floxed MEFs, nor did adeno-Cre affect these endpoints in wild-type MEFs (Supplemental Figure S2).
Figure 2. Abrogation of mitochondrial dynamism in cultured MEFs.

(A) Immunoblot analysis showing depletion of Mfn1, Mfn2 and Drp1 after adeno-Cre induced simultaneous recombination of their respective floxed genes.
(B) Live-dead studies 4 days after Cre-mediated Mfn1/Mfn2/Drp1 deletion. Controls (Pre) are Mfn1/Mfn2/Drp1 triple floxed MEFs without adeno-Cre. Means±SEM, n shown in bars.
(C) Confocal analysis of mitochondrial aspect ratio (long/short axis) and polarization states (TMRE positivity) 4 days after Cre-mediated Mfn1/Mfn2/Drp1 deletion. * is p<0.05 vs Pre (ANOVA).
(D) Mitochondrial accumulation of mcParkin at baseline and after uncoupling with FCCP 4 days after Cre-mediated Mfn1/Mfn2/Drp1 deletion. * is p<0.05 vs baseline Pre-Cre treatment; # is p<0.05 vs FCCP treated Pre (ANOVA).
(E) Lysosomal-mitochondrial overlay at baseline and upon uncoupling with FCCP 4 days after Cre-mediated Mfn1/Mfn2/Drp1 deletion. * is p<0.05 vs baseline Pre; # is p<0.05 vs FCCP-treated Pre (ANOVA).
(F) Rates of mitochondrial engulfment by lysosomes measured by red shift of adenoviral expressed mito-Keima at increasing times after Cre-mediated deletion of Drp1 alone (open circles; Drp1 floxed), Mfn1 and Mfn2 in combination (grey triangles; Mfn1/Mfn2 floxed) or Mfn1, Mfn2, and Drp1 in combination (black squares; Drp1/Mfn1/Mfn2 floxed). See also Figure S2.
Previously, we concluded that acute inhibition of mitochondrial fission by Drp1 ablation enhanced, whereas acute interruption of mitochondrial fusion by Mfn1/Mfn2 ablation suppressed, mitophagy in MEFs (Song et al., 2015b). We confirmed these findings using Mito-Keima, and further showed that acute concomitant interruption of both fusion and fission by Mfn1/Mfn2/Drp1 suppresses the rate of mitophagy, similar to interruption of fusion alone (Figure 2F). Concordance of mitochondrial parkin recruitment and static or dynamic assays of mitochondrial incorporation into lysosomes demonstrates that a dominant effect of abrogating mitochondrial dynamism in MEFs is suppression of mitophagy.
Lack of mitochondrial dynamism is less deleterious to mouse hearts than dynamic imbalance
Cardiomyocyte mitochondria appear hypodynamic compared to other cell types (Song and Dorn, 2015). Nevertheless, interrupting Drp1-mediated mitochondrial fission or Mfn1/Mfn2-mediated mitochondrial fusion in adult mouse hearts is rapidly lethal (Chen et al., 2011; Song et al., 2015b). We posited that cardiac pathology in the Mfn1/Mfn2 double deficient or Drp1-deficient hearts accrues from unopposed fission or fusion, rather than from absence of fusion or fission, respectively, ie that it is the imbalance in dynamism that is detrimental. If this were correct then simultaneously ablating fission and fusion would “rescue” the aggressive cardiomyopathies caused by interrupting fission or fusion alone. To test this notion we introduced a conditional cardiac-specific Cre transgene into Mfn1/Mfn2/Drp1 floxed mice (Figure 3A). Mice are normal until administered tamoxifen at the age of 8 weeks, which induces nuclear translocation of a modified estrogen receptor-Cre hybrid protein (MER-Cre-MER) expressed exclusively in cardiomyocytes. Thus, the Drp1, Mfn1, and Mfn2 floxed alleles are all recombined simultaneously. Mfn1 and Drp1 protein levels decreased to less than 20% of baseline 2 weeks after, and all three proteins had dissipated 8 weeks after, tamoxifen injection (Figure 3B; residual proteins are in non-myocytes).
Figure 3. Abrogation of mitochondrial dynamism in adult mouse hearts.

(A) Gene recombination design using tamoxifen-inducible cardiomyocyte-directed Cre.
(B) Immunoblot analysis of Mfn1, Mfn2 and Drp1 levels after Cre-mediated gene recombination in 8 wk old mouse hearts. * is p<0.05 vs no Cre Ctrl (ANOVA).
(C) Early lethality of cardiac Drp1 knockout (KO), Mfn1/Mfn2 double KO (DKO) delayed by Drp1/Mfn1/Mfn2 triple gene ablation. * is p<0.05 vs no Cre Ctrl; # is p<0.05 vs Mfn1/Mfn2/Drp1 TKO (Log-rank).
(D) Representative Masson’s trichrome stained TKO mouse hearts 16 weeks after combined Mfn1/Mfn2/Drp1 gene deletion. Hearts of parent lines 6 weeks after gene recombination are shown to the right. Scale is 5 mm.
(E) Gravimetric heart weight (16 wks) indexed to body weight. * is p<0.05 vs Ctrl (t-test).
(F) RT-qPCR of cardiac hypertrophy genes. * is p<0.05 vs Ctrl; # is p<0.05 vs 8wk TKO (ANOVA).
We confirmed our previous report (Song et al., 2015b) that six weeks after cardiac ablation of either Drp1 or Mfn1/Mfn2 approximately half of both mouse lines succumb (Figure 3C). Remarkably, cardiac ablation of all three genes (cardiac Mfn1/Mfn2/Drp1 TKO [triple knockout]) delayed any mortality until fourteen weeks after tamoxifen (Figure 3C).
The cardiac phenotype of cardiac Drp1/Mfn1/Mfn2 Drp1 TKO mice was distinct from both parent knockout lines. Fission-defective cardiac Drp1-deficient mice develop dilated cardiomyopathy with severe contractile depression (decreased % LV shortening) and extensive (~30%) cardiomyocyte death, whereas fusion-defective cardiac Mfn1/Mfn2-deficient mice develop eccentric cardiac remodeling (normal ventricular radius/wall thickness) and modestly impaired contractile performance with no increase in cardiomyocyte death (Song et al., 2015b). By comparison, cardiac Mfn1/Mfn2/Drp1 TKO mice developed concentric cardiac hypertrophy (Figures 3D, 3E) with increased levels of pathological “fetal” genes (Figure 3F). Thus, rebalancing mitochondrial dynamics to zero mitigated, but did not resolve, cardiomyopathies provoked by interrupting mitochondrial fission or fusion. Instead, a less aggressive phenotype of pathological cardiac hypertrophy was provoked by mitochondrial adynamism.
Mitochondrial adynamism causes unique hypertrophy-with-failure
Pathological hypertrophy is a non-specific response to mis-matched cardiac workload and myocardial functioning. Hypertrophy is the prototypical early compensatory process, but over time the heart dilates, ejection performance declines, and decompensated heart failure ensues (Diwan and Dorn, 2007). To see if Mfn1/Mfn2/Drp1 cTKO mice followed this paradigm we assessed the long-term consequences of mitochondrial adynamism. Fifty percent of cTKO mice had died 17 weeks after tamoxifen-induced target gene ablation, versus 50% lethality at only 7 weeks for the parent lines (compare Figure 4A to Figure 3C). However, rather than typical progressive cardiac dilatation, serial echocardiography revealed concomitant increases in left ventricular mass and decreased pump performance (Figure 4B) leading to pulmonary congestion that reflects overt heart failure (Figure 4C). Also in contrast to conventional decompensation of hypertrophy (Diwan and Dorn, 2007), cardiomyocyte death and drop-out were minimal, despite massive hypertrophy of individual cardiomyocytes (Figure 4D).
Figure 4. Long-term evolution of adynamic mitochondrial cardiac phenotype.

(A) Extended survival curve of cardiac Drp1/Mfn1/Mfn2 TKO mice after tamoxifen-induced gene ablation. * is p<0.05 vs no Cre Ctrl (Log-rank). Inset is representative hearts from mice that survived 24 weeks; scale is 1 mm.
(B) Echocardiography of Mfn1/Mfn2/Drp1 TKO hearts. Representative m-mode tracing are on the left. Group quantitative data for calculated left ventricular (LV) mass and % shortening of surviving mice are to the right. * is p<0.05 vs same time Ctrl (t-test).
(C) Heart and lung weights indexed to tibial length. * is p<0.05 vs same time Ctrl (t-test).
(D) Histological studies. Left, wheat germ agglutinin (WGA, green) staining and Evan’s blue (red) staining. Middle, quantitative group data for cardiomyocyte cross-sectional area (CSA). Group data for necrotic Evan’s blue stained cells, apoptotic TUNEL-positive cells, and cardiac fibrosis are to the right. Data are mean±SEM of indicated numbers; * is p<0.05 vs Ctrl; # is p<0.05 vs 8wk TKO (ANOVA).
Adynamic cardiomyocyte mitochondria exhibit abnormalities in morphometry and respiration
The above results reflect intrinsic myocardial dysfunction induced by cardiomyocyte mitochondrial adynamism. We therefore determined if cardiomyocyte mitochondria were themselves dysfunctional. By comparison to MEFs, mitochondria of normal adult cardiomyocytes appear “fragmented” at baseline; their aspect ratio is low and they are not interconnected into filamentous networks (Figure 5A). Ultrastructural examination of cardiac Mfn1/Mfn2/Drp1 TKO hearts revealed mitochondrial heterochromia, heterogeneity in size (although smaller mitochondria dominated) and “empty’ mitochondria (Figure 5A, arrows). The most striking mitochondrial abnormality in Mfn1/Mfn2/Drp1 triple deficient cardiomyocytes was their accumulation in the perinuclear region (Figure 5A, bottom panels). The proportion of each cardiomyocyte occupied by mitochondria increased from ~45% to >80% 16 weeks after combined Mfn1/Mfn2/Drp1 gene ablation (Figure 5B). Strikingly, Mfn1/Mfn2/Drp1-deficient cardiomyocytes showed evidence for peripheral displacement of sarcomeres by superabundant centrally located mitochondria (Figure 5A, bottom right; Supplemental Figure S3), suggesting that parallel development of cardiac hypertrophy and heart failure is caused by mechanical disruption of normal subcellular myofilament architecture, much like amyloid heart disease and aggresomal cardiomyopathies.
Figure 5. Structural and functional characteristics of adynamic cardiomyocyte mitochondria.

(A) Representative transmission electron micrographs of one normal Ctrl and two different Mfn1/Mfn2/Drp1 TKO mouse hearts. Note heterogeneity in mitochondrial size and occasional “ghost” mitochondrial (white arrows). Perinuclear mitochondrial accumulation peripherally displacing sarcomeres is evident in lowest magnifications of TKO hearts.
(B) Quantitative analyses of mitochondrial abundance and morphometry. Means±SEM; * is p<0.05 vs Ctrl; # is p<0.05 vs 2 wk; $ is p<0.05 vs 8 wk TKO (ANOVA).
(C) Flow cytometric analyses of isolated cardiac mitochondria. Left and middle, mitochondrial size measured by forward scatter. Right, quantitative data for mitochondrial polarization measured by DiOC6. * is p<0.05 vs Ctrl (t-test).
(D) Glutamate/malate-stimulated mitochondrial respiration; representative curves are to the left and group data are in the middle. * is p<0.05 vs Ctrl (t-test). Right, FCCP-stimulated maximal mitochondrial oxygen consumption. See also Figure S3.
Morphometric heterogeneity of cardiomyocyte Mfn1/Mfn2/Drp1 TKO mitochondria was confirmed by flow cytometric analysis. Average forward light scatter (which measures size) of cardiac mitochondria from 15–16 week old Mfn1/Mfn2/Drp1 TKO mice was modestly (~25%), but significantly, lower than normal controls (Figure 5C). Mitochondrial polarization measured by DiOC6 fluorescence was unchanged (Figure 5C).
Glutamate-stimulated respiration of Mfn1/Mfn2/Drp1 TKO cardiac mitochondria was modestly depressed, whereas FCCP-stimulated maximal (uncoupled) respiration appeared increased, suggesting modest respiratory uncoupling (Figure 5D). Together, the ultrastructural, flow cytometric and functional data reveal that concomitant interruption of mitochondrial fission and fusion in adult mouse hearts provokes accumulation of morphologically and functionally abnormal mitochondria.
Mitochondrial mass increases after mitochondrial dynamism is abrogated
We postulated that increased mitochondrial abundance directly contributed to the delayed cardiac pathology observed in TKO hearts by distorting the subcellular contractile apparatus. The proportion of mitochondrial protein in ventricular myocardium increased by ~40% after combined Mfn1/Mfn2/Drp1 gene ablation (Figure 6A). Remarkably, the ratio of mitochondrial to nuclear DNA and the transcript levels of biogenesis factors markedly decreased in the same hearts (Figures 6B and 6C), suggesting that mitochondrial accumulation was not a consequence of accelerated mitochondrial formation.
Figure 6. Mitochondrial abundance and protein content after abrogation of dynamism.

(A) Mitochondrial protein/ventricular mass. * is p<0.05 vs Ctrl (t-test).
(B) qPCR of mitochondrial DNA/nuclear DNA. * is p<0.05 vs Ctrl (ANOVA).
(C) RT-qPCR of mitochondrial biogenesis factors. * is p<0.05 vs Ctrl; # is p<0.05 vs 8wk TKO (ANOVA).
(D) Quantitative immunoblot analysis of mitochondrial outer membrane (left) and inner membrane respiratory complex (right) proteins. * is p<0.05 vs Ctrl (ANOVA).
Analyses of specific mitochondrial proteins revealed increases in outer mitochondrial membrane (OMM) proteins TOMM20, VDAC1 and Opa1 that paralleled overall mitochondrial protein levels (Figure 6D, left). By contrast, respiratory complex proteins showed little change (Figure 6C). This seeming disparity can be explained by the increase in overall organelle surface area (outer mitochondrial membrane) caused by accumulation of large numbers of smaller spheroidal mitochondria.
Cardiomyocyte mitochondrial clearance is impaired in fission/fusion defective hearts
We were intrigued that survival of mice with interrupted mitochondrial fission (Drp1 KO) or fusion (Mfn1/Mfn2 DKO) could be prolonged by their combined simultaneous disruption, but that this genetic maneuver exchanged two rapidly lethal syndromes for a different progressive one. In trying to understand the relationship between mitochondrial dynamics and age-dependent evolution of phenotypes we took note of accumulating evidence for interactions between mitochondrial elimination by mitophagy and mitochondrial formation by biogenesis (Palikaras et al., 2015). For example, genetic deletion of components that mediate mitochondrial fission and fusion in yeast produces mitophagy defects and decreased life-span (Bernhardt et al., 2015). Indeed, we observed slight increases in markers for the mitochondrial unfolded protein response (UPRmt) and mitophagy signaling associated with decreased levels of Drp1 and the inner membrane fusion protein Opa1 in aged C57BL/6 hearts obtained from the Aged Rodent Tissue bank of the National Institutes of Health, Division of Aging Biology, Biological Resources Branch (supplemental Figure S4). The link between decreased mitochondrial dynamics proteins and increased UPRmt and mitophagy markers in aged hearts prompted us to interrogate these processes in Mfn1/Mfn2/Drp1 cTKO hearts.
Levels of several UPRmt proteins, but not endoplasmic reticulum stress markers, increased in TKO hearts (Figure 7A; supplemental Figure S5). Induction of the UPRmt in response to loss of proteostasis is a hallmark of the cardiomyopathy of aging (Jensen and Jasper, 2014), but in cTKO hearts it preceded any measurable functional decline (see Figure 4B) The same stressors that induce UPRmt should simultaneously stimulate mitophagy so that mitochondrial repair and removal are coordinated (Sun et al., 2016). However, as in Mfn1/Mfn2/Drp1 deficient MEFs (vide supra), mitophagy failed to be activated in Mfn1/Mfn2/Drp1 hearts (Figure 7B). By comparison, general autophagy was induced as expected in heart failure (Nishida et al, 2009), revealing that the mitophagy defect is specific to mitochondria. The constellation of mitochondrial respiratory defects with proteotoxic stress, massive accumulation of mitochondria in the absence of increased mitochondrial biogenesis, and impaired mitophagy point to defective mitophagic mitochondrial turnover as a central underlying cause for the cardiac phenotype induced by abrogating mitochondrial dynamism.
Figure 7. Impaired proteostasis and altered mitophagy in Mfn1/Mfn2/Drp1 TKO mouse hearts.

(A) Induction of the mitochondrial unfolded protein response. Samples are myocardial homogenates; GAPDH is loading control. * is p<0.05 vs Ctrl (ANOVA).
(B) Mitophagy proteins in cardiac mitochondrial fractions. TOM20 is loading control. * is p<0.05 vs Ctrl; # is p<0.05 vs Drp1 KO (ANOVA).
(C) Macroautophagy proteins in myocardial homogenates. GAPDH is loading control. * is p<0.05 vs Ctrl (ANOVA). Bottom GAPDH is loading control for LC3 blots. See also Figures S4 and S5.
Discussion
These results dispel the notion that mitochondrial fragmentation is inherently detrimental. They further support three noteworthy conclusions: 1. An imbalance between mitochondrial fusion and mitochondrial fission (in either direction) is more deleterious than is simultaneous abrogation of both processes. 2. In hearts, suppression of mitochondrial dynamism profoundly alters the phenotypic response to, and delays lethality evoked by, disproportionate mitochondrial fusion or fission. 3. Mitochondrial dynamism is essential to replacing and revitalizing the cardiomyocyte mitochondrial collective. Our findings provide compelling evidence linking mitophagy and mitochondrial fusion/fission to mitochondrial quantity (as well as quality) control.
Abolishing mitochondrial dynamism by concomitantly ablating Drp1, Mfn1 and Mfn2 in adult mouse hearts is a completely artificial experimental model, but nevertheless provided several novel insights. Mitochondrial adynamism accelerated cardiomyocyte mitochondrial senescence, prematurely evoking a form of the “cardiomyopathy of aging” (Chaudhary et al., 2011). The dynamic nature of mammalian mitochondria is accepted as both an observational fact (Lewis and Lewis, 1914) and an important determinant of cell, organ, and organism health (Westermann, 2010). Although mitochondria in healthy adult cardiac myocytes appear almost completely static (Dorn, 2015b), perinatal and adult cardiac-directed gene ablation of the major mitochondrial dynamics proteins established the necessity for these factors to maintain cardiac health (Chen et al., 2011; Song et al., 2015b; Varanita et al., 2015; Wai et al., 2015). Cardiac ablation of the fission protein Drp1 produced, in addition to expected mitochondrial enlargement, mitochondrial loss from accelerated mitophagy. As a consequence, programmed cardiomyocyte death produced dilated cardiomyopathy (Song et al., 2015b). Remarkably, the current studies show how the seminal features of fission-defective/hyper-mitophagic hearts are corrected by also interrupting mitochondrial fusion and mitophagy through deletion of Mfn1 and Mfn2. Cardiomyocyte death decreased toward normal levels, mitochondria accumulated instead of disappeared, and the hearts responded with hypertrophy rather than dilated cardiomyopathy.
On the other hand, myocardial hypertrophy, mitochondrial accumulation, and underlying mitophagy defects in adynamic Mfn1/Mfn2/Drp1 TKO hearts echo the hallmark features of fusion-deficient Mfn1/Mfn2 DKO hearts (Song et al., 2015b). Like fission/fusion, mitophagy was moved toward a more balanced state by simultaneous ablation of Drp1 (in which mitophagy is increased) and Mfn1/Mfn2 (in which mitophagy is interrupted), which improved short-term survival, but led to mitochondrial hyper-accumulation and long-term sarcomeric distortions not observed in the parental lines.
Multiple roles of mitochondrial dynamics factors confound our understanding of mechanisms underlying how loss of mitochondrial fusion or fission compromises cell and organelle health. For example, increased mitochondrial interconnectedness promotes organelle repair and exchange of genetic information(Nakada et al., 2001; Ono et al., 2001), enhances trans-organelle signaling (Szabadkai et al., 2004; Whelan et al., 2012), and regulates mitochondrial quality during cycles of mitophagy and mitochondrial biogenesis (Martin et al., 2014; Twig et al., 2008). Conversely, mitochondrial fragmentation is associated with (although not necessarily essential for) cell division (Taguchi et al., 2007), mitochondrial triage prior to mitophagy (Twig et al., 2008), and programmed cell death (Frank et al., 2001). Moreover, in addition to promoting mitochondrial fusion, Mfn2 transduces Parkin-mediated mitophagy after its phosphorylation by PINK1 kinase (Chen and Dorn, 2013; Gong et al., 2015) and Mfn2 acts as the molecular tether connecting mitochondria to endo/sarcoplasmic reticulum (Chen et al., 2012; de Brito and Scorrano, 2008). Likewise, Opa1 regulates cristae structure and respiratory complex integrity (Cogliati et al., 2013). Although the mitochondrial dynamics proteins are best known for, and in some instances named for, their roles as fission and fusion factors as described in cultured cells, their ancillary functioning may be preeminent under in vivo conditions. The current in vitro and in vivo results emphasize orchestration of mitophagy and mitochondrial biogenesis by these mitochondrial dynamics factors.
An advantage of our studies is that we used the same genetic systems and experimental endpoints to directly compare fission-defective, fusion-defective, and dynamism-defective (lacking Mfn1, Mfn2 and Drp1) mice. Dynamism-defective mice survived much longer than mice with interruption of either the Drp1-fission or Mfn1/Mfn2-fusion pathway alone, demonstrating that equipoise between mitochondrial fusion and fission, even at very low levels, is of greater importance to the heart than is the individual integrity of either process (Bernhardt et al., 2015; Palikaras et al., 2015). The idea that balancing, rather than normalizing, mitochondrial fission and fusion could be therapeutic is supported by mechanistic studies in yeast (Bernhardt et al., 2015), worms (Palikaras et al., 2015), and a previous mouse study (Chen et al., 2015). In the latter, a lethal dilated cardiomyopathy in mice systemically lacking the Drp1 receptor Mff was rescued by concomitant deletion of the outer membrane fusion protein Mfn1. It is notable that the germ-line Mff knockout mouse, which was described as having a “partial fission defect” model, resembles adult-inducible cardiomyocyte-specific Drp1 deletion (a more complete fission defect) because increased mitophagy was associated with mitochondrial depletion. Thus, two independent approaches validate the finding that mitochondrial fission governs mitophagy (Dorn, 2015a). However, there are important differences between the cardiac Drp1-knockout and germ-line Mff knockout models. In the current and previous study (Song et al., 2015b), Drp1 was ablated exclusively in the cardiac myocytes of young adult mice, producing a phenotype that was unambiguously cell-autonomous and independent of confounding developmental effects that are observed when mitochondrial dynamism is disturbed (Kasahara et al., 2013). By contrast the Mff knockout mouse was a germ-line deletion with numerous extra-cardiac defects (including small overall size, neuromuscular problems, reduced fertility) and evidence of developmental abnormalities (such as malocclusion and kyphosis) (Chen et al., 2015). Thus, the observed decrease in life span and cardiomyopathy may have been influenced by severe non-cardiac pathologies. Moreover, whereas cardiomyocyte mitochondria of cardiac Drp1-knockout were clearly elongated (evidence of decreased fission), those of Mff-knockout mice were not (Chen et al., 2015). Indeed, the “partial fission defect” must therefore be accepted as a matter of faith. Finally re-balancing mitochondrial fission/fusion in the Mff knockout mouse in the Chen study was achieved through systemic ablation of Mfn1 or Mfn2 (but not both), which was also a germ-line/systemic genetic intervention. These confounders preclude a conclusion that balanced dynamism in cardiac myocytes is entirely responsible for observed phenotypic reversal. In the current studies, mitochondrial fission and fusion were both interrupted by ablation of the central mediators of both processes, and the consequences in the parent and combined lines are unquestionably cardiomyocyte-autonomous. Thus, while an approach of re-balancing, rather than correcting, disease-related defects in mitochondrial dynamism might be advanced based upon the previously published findings (Chen et al., 2015; Cassidy-Stone et al., 2008) in our models balanced suppression of mitochondrial dynamism was not a cure for cardiac pathology induced by ablation of either fission or fusion. Instead, dynamism-defective mice exhibited massive accumulation of mitochondria inconstant in size, density and respiratory function. Since mitochondrial biogenesis was not induced and mitophagy was impaired these features point to a defect in mitochondrial elimination. Thus, interrupted mitophagic mitochondrial turnover and renewal (quantity and quality control) are central processes driving the adynamic mitochondria cardiac phenotype.
Because mitochondrial functional impairment was mild and mitochondrial accumulation was massive in dynamism defective hearts we reason that the hypertrophy of Mfn1/Mfn2/Drp1 TKO hearts is in part a direct consequence of impaired mitochondrial quantity control; systolic dysfunction is partly attributable to mitochondrial dislocation of sarcomeric elements. It is intriguing that the ~40% increase in mitochondrial content we observed corresponds to a ~40% increase in both cardiomyocyte size and heart mass. Mitochondrial overabundance has previously been described in hearts of mice in which mitochondrial biogenesis was forcibly increased by transgenic expression of PGC1α (Russell et al., 2004) and in skeletal muscle of mice wherein mitochondrial biogenesis was disrupted though ablation of the transcription factor, mitochondrial transcription factor A (Tfam) (Wredenberg et al., 2002). In the former, it was concluded that mitochondria accumulated through increased synthesis, but in the latter as a compensatory reaction to respiratory chain defects (although ATP production was unaffected). In our mitochondrial dynamism-defective hearts mitochondria accumulated because mitophagic removal was impaired.
The current data add to evidence of mitochondrial dynamics involvement in biological events with equal or greater pathophysiological impact than simply controlling mitochondrial size. Functional interactions between mitochondrial dynamism, biogenesis and mitophagy defined through the use of genetic mouse models may have relevance to natural conditions associated with mis-expression or malfunctioning of mitochondrial fission and fusion proteins, including neurodegenerative conditions such as Charcot Marie Tooth disease and normal aging. We recently described pharmacological means of positively and negatively modulating mitochondrial fusion (Franco et al., 2016). A better understanding of the varied roles of mitochondrial dynamics together with new tools for its manipulation brings increased opportunity for therapeutic intervention.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Gerald Dorn II (gdorn@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were maintained according to the National Institutes of Health guidelines. Mice were maintained in a specific pathogen-free barrier facility maintained on a 12 hr light-dark cycle. Upon weaning, all mice were group housed, up to 5 mice of the same sex per cage. Food and water bottle were provided in a recess of the metal wire lid at the top of the cage. Cages were changed once every week.
Cardiomyocyte-specific human Drp1 overexpression (FVB/N background) was achieved using the myh6 promoter-driven doxycycline-suppressible (“tet-off”) bi-transgenic system (Sanbe et al., 2003); Because doxycycline was never administered transgene expression occurred shortly after birth (Syed et al., 2004). Experiments were undertaken with male and female mice up to 60 weeks old as indicated in the figures. No deleterious effects were detected in the hearts with human Drp1 overexpression, analyses of the influence of sex were therefore not performed.
Mfn1loxp/loxp, Mfn2 loxp/loxp (C57BL/6J background)(Chen et al., 2003; Chen et al., 2007), and Drp1loxp/loxp (C57BL/6J-129/Sv background)(Ishihara et al., 2009) mice have been described. For conditional cardiac ablation, floxed mouse lines were crossed onto the myh6-MER-Cre-Mer (C57BL/6J background) mice; gene recombination was induced by intraperitoneal administration of tamoxifen at 40 mg/kg/day for 5 continuous days at 8 weeks of age (Sohal et al., 2001). Experiments were undertaken with male and female mice at up to 24 weeks after the tamoxifen administration as indicated in the figures; analyses of the influence of sex were performed and no significant differences were detected between male and female cardiac Mfn1/Mfn2/Drp1 triple knockout mice.
M-mode echocardiography was performed on unsedated mice (Song et al., 2015); controls were age- and sex-matched tetracycline-controlled transactivator protein (tTA) transgenic littermates or Mfn1loxp/loxp+Mfn2 loxp/loxp+Drp1loxp/loxp littermates with tamoxifen administration, respectively. All experimental procedures were approved by the Washington University Institutional Animal Care and Use Committee.
Cell Lines
Mfn1/Mfn2 double floxed MEFs and Drp1 floxed MEFs were previously generated (Song et al., 2015). Mfn1/Mfn2/Drp1 triple floxed MEFs were generated from Mfn1loxp/loxp+Mfn2loxp/loxp+Drp1loxp/loxp embryos. Briefly, 6–10 mouse embryos of undetermined sex were aseptically dissected at e13.5. Heads and internal organs were discarded; bodies were rinsed twice with PBS and minced with razor blade; cells were dissociated with 10 ml 0.05% trypsin-EDTA (Thermo Fisher Scientific, 25300-054) at 37 °C for 5 min x 3 times and neutralized with fresh medium, pelleted by centrifugation at 200 g for 5 min and plated on 150-mm plastic culture dishes prior to incubation at 37 °C with 5% CO2.
MEFs were maintained in Dulbecco’s modified eagle’s medium (Thermo Fisher Scientific, 11965-084) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, 26140-079), 1x non-essential amino acids (Thermo Fisher Scientific, 11130-051), 2 mM L-glutamine (Thermo Fisher Scientific, 25030-081), 100 U/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific, 15140-122) with medium changed every other day. Cells were passaged every 4 days and used within five passages to avoid replicative senescence.
METHOD DETAILS
Western Blotting
To prepare cell lysates, MEFs were pelleted and lysed in cell extraction buffer on ice (Thermo Fisher Scientific, FNN0011) with 1 mM PMSF, protease inhibitor (Roche, 05892970001), and phosphatase inhibitor (Roche, 04906837001) according to manufacturer’s protocol.
To prepare myocardial homogenates, heart tissues were collected, snap-frozen in liquid nitrogen, and homogenized in tissue extraction reagent (Thermo Fisher Scientific, FNN0071) with protease inhibitor (Roche, 05892970001) and phosphatase inhibitor (Roche, 04906837001) as previously described (Song et al., 2015). Myocardial homogenates were collected from the supernatant after centrifugation at 3,800 g at 4 °C for 10 min.
To prepare myocardial mitochondrial fractions, heart tissues were collected, snap-frozen in liquid nitrogen, and homogenized in homogenization buffer (10 mM HEPES (pH 7.2), 320 mM sucrose, 3 mM MgCl2, 25 mM Na2P4O7, 1 mM DTT, 5 mM EGTA, 1 mM PMSF with protease inhibitor (Roche, 05892970001) and phosphatase inhibitor (Roche, 04906837001) as previously described (Gong et al., 2015). The homogenates were centrifuged at 3,800 g at 4 °C for 10 min, and the supernatants were transferred to a new tube and centrifuged at 10,000 g at 4 °C for 10 min. The resulting supernatants were removed; the pelleted mitochondrial fractions were washed once and then dissolved in 30–50 μl tissue extraction reagent (Thermo Fisher Scientific, FNN0071) with protease inhibitor (Roche, 05892970001) and phosphatase inhibitor (Roche, 04906837001).
Proteins were measured with protein assay dye reagent concentrate (Bio-Rad, 500-0006) using a spectrophotometer. Equal amount of proteins (30 μg per lane for myocardial homogenates, 7.5 μg per lane for myocardial mitochondrial fractions) for each sample were size-separated by 4–15% pre-cast gradient SDS-PAGE (Bio-Rad, 456-1084, 567-1084 and 567-1085), transferred to PVDF membranes (GE Healthcare, 10600021), and blocked with 1x phosphate-buffered saline (PBS, Thermo Fisher Scientific, 70011-069) containing 0.1% Tween-20 (Promega, H5152) plus 5% nonfat dry milk (Bio-Rad, 170-6404) and 1% bovine serum albumin (BSA, Santa Cruz Biotech, 9048-46-8) at room temperature for 1 hour, incubated with primary antibodies at room temperature for 2 hours or at 4 °C for overnight followed by incubation with horseradish peroxidase (HRP) conjugated secondary antibodies at room temperature for 1 hour, and visualized using the ECL western blotting detection reagent (Bio-Rad, 170-5060). Some immunoblot membranes were re-probed after stripping with OneMinute western blot stripping buffer (GM Biosciences, GM6001) according to the manufacturer’s protocol. The GAPDH blots shown were representative; normalized quantification for each protein was performed with the corresponding loading control.
Primary antibodies against Drp1 (1:1000, ab56788), Mff (1:500, ab129075), Mfn2 (1:300, ab56889), Opa1 (1:500, ab42364), GAPDH (1:3000, ab8245), OXPHOS (a premixed cocktail of antibodies against CI-NDUFB8, CII-SDHB, CIII-UQCRC2, CIV-MTCO1 and CV-ATP5A, 1:500, MS604), VDAC1 (1:1000, ab15895), TOMM20 (1:2000, ab186735), Hsp60 (1:2000, ab46798), LONP1 (1:500, ab103809), Catalase (1:2000, ab1877), p62 (1:1000, ab56416), LC3 I/II (1:1000, ab128025), COX IV (1:1000, ab14744), SOD1 (1:5000, ab13498), SOD2 (1:5000, ab13533) were from Abcam. Primary antibodies against Mfn1 (1:200, sc-50330), AFG3L2 (1:500, sc-84687), Ubiquitin (1:100, sc-8017), and β-Actin (1:250, sc-47778) were from Santa Cruz Biotechnology. Primary antibodies against Calreticulin (1:1000, cs12238), GRP94 (1:1000, cs2104), PERK (1:1000, cs3192), phospho-PERK (Thr980) (1:1000, cs3179), CHOP (1:1000, cs2895), LC3 II (1:1000, cs3868), and LC3 I/II (1:1000, cs4108) were from Cell Signaling Technology. Horseradish peroxidase (HRP) linked secondary antibodies anti-mouse IgG (1:3000, cs7076) and anti-rabbit IgG (1:3000, cs7074) were from Cell Signaling Technology.
Histological Studies
Mouse heart tissues were fixed with 4% formaldehyde solution in PBS with or without prior transcardial perfusion. Paraffin-embedded tissues were sectioned at a thickness of 5 μm on a Leica RM2255 rotary microtome. The sections were de-paraffinized in xylene, rehydrated with a gradient (100–50%) of ethanol, and washed in distilled water. Hematoxylin-eosin stain (Sigma, GHS116 & HT110116), Masson’s trichrome stain (Sigma, HT15, HT10132 & HT1079), and TUNEL assay (Promega, G3250) were performed as previously described (Song et al., 2015).
For immunofluorescent staining, the sections were incubated in sodium citrate buffer (Poly Scientific, S2307) at 100 °C for 20 min for antigen retrieval and then permeabilized by incubating in 1% Triton X-100 (Sigma, T9284) at room temperature for 20 min. The sections were blocked with 10% goat serum (Jackson ImmunoResearch, 005-000-121) in PBS at room temperature for 1 hr, then incubated with primary antibodies against TOMM20 (1:200, ab186735) at 4°C overnight, followed by incubation with secondary anti-rabbit (1:400, Fisher Thermo Scientific, A-11008) fluorescent antibodies at room temperature for 1 hour. The sections were then counterstained using primary antibody against Troponin T (1:400, ab10214) at room temperature for 2 hours followed by incubation with secondary anti-mouse fluorescent antibody (1:400, Fisher Thermo Scientific, A-11003) at room temperature for 1 hour. The sections were mounted with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories, H-1200) and sealed with nail polish.
For Evans blue studies, mice were intraperitoneally administered 1% Evans blue dye (Sigma, E2129) solution in PBS at 1% volume relative (ml) to body mass (g) ~24 h prior to tissue sampling as previously described (Song et al., 2015). Mouse heart tissues were fixed, embedded and sectioned as aforementioned. After de-paraffinization and rehydration, sections were stained with 20 μg/ml FITC conjugated-wheat germ agglutinin (WGA, Invitrogen, W834) at room temperature for 30 min, wash with PBS for three times and sealed with Cytoseal 60 mounting medium (Fisher Thermo Scientific, 23-244256). Evans blue positive cardiomyocytes fluoresced in red.
Transmission Electron Microscopy
Mouse heart tissues were fixed with EM fixation buffer (4% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4). A Jeol electron microscope (JEM-1400, JEOL, Tokyo, Japan) at 1,500x – 5,000x direct magnifications was used for ultrastructural examination of osmium tetroxide/uranyl acetate stained mouse heart thin sections (90 nm). Mitochondrial content (% of the areas taken by mitochondria compared to those of the cardiomyocytes), individual mitochondrial size and aspect ratio were measured using ImageJ (NIH) on transmission electron microscopic images.
Isolation of Cardiac Mitochondria
Isolation of cardiac mitochondria used a modification of a published protocol (Frezza et al., 2007). Briefly, mouse heart ventricles were collected, minced, and incubated with trypsin before homogenization with a glass/teflon Potter Elvehjem homogenizer on ice. Ventricle homogenates were centrifuged at 800 g at 4 °C for 10 min; the supernatant was collected and centrifuged at 16,000 g at 4 °C for 10 min; the resulting pellet containing normal- and small-size mitochondria were washed once and centrifuged at 16, 000 g at 4 °C for 10 min. After re-suspension, mitochondrial protein concentrations were colorimetrically measured using protein assay dye reagent concentrate (Bio-Rad, 500-0006) and subsequent mitochondrial functional assays were performed.
Mitochondrial Respiration
Respiratory activities of isolated mitochondria were assessed using a micro Clark-type electrode in a closed and magnetically stirred glass chamber as previously described (Song et al., 2015). Non-stimulated (-ADP) respiration (state 2) and ADP (Sigma, A2754)-stimulated respiration (state 3) were measured. Oligomycin (Sigma, 75351) was added to inhibit ATP synthesis. Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (Sigma, C2920) was added to stimulate maximal uncoupled respiration.
Flow Cytometric Analyses of Isolated Mitochondria
Isolated mitochondria were stained with 200 nM MitoTracker Green (Thermo Fisher Scientific, M7514) or 100 nM 3,3′-dihexyloxacarbocyanine iodide (DiOC6) (Thermo Fisher Scientific, D273) at room temperature for 20 min and washed twice with PBS. Flow cytometric analyses of mitochondrial size (forward scatter, FSC) and mitochondrial membrane potential (DiOC6 fluorescence intensity detected by FITC channel) were performed on a BD LSR II Flow Cytometer (Becton Dickinson, San Jose, CA). Data are presented as histograms for, and as bar graphs of average signal intensity of ~100,000 events.
Mitochondrial Protein Quantification
Protein concentration of the isolated cardiac mitochondria was measured with protein assay dye reagent concentrate (Bio-Rad, 500-0006) using a spectrophotometer. Data are presented as bar graph for the ratio of μg of mitochondrial protein to mg of left ventricular tissue weight.
Mitochondrial DNA Quantification
Total DNAs were extracted from cardiac tissues with DNeasy blood & tissue kit (Qiagen, 69506). The amount of mitochondrial DNA was quantified by the ratio of the relative copy numbers of mitochondrial DNA to nuclear DNA using quantitative PCR as previously reported (Chen et al. 2010).
mRNA Expression Analysis
Total RNAs were extracted from cardiac tissues with TRIzol (Thermo Fisher Scientific, 15596-026), and single strand cDNA was prepared using high-capacity cDNA reverse transcription kit with RNase inhibitor (Thermo Fisher Scientific, 4374966) following the manufacturer’s protocol. RNA expression analyses were performed using Taqman qRT-PCR master mix (Thermo Fisher Scientific, 4440038) with predesigned primer/probes sets for Tfam (Mm00447485_m1), PGC-1α (Mm01208835_m1), PGC-1β (Mm00504720_m1), PPARγ (Mm01184322_m1), ANP (Mm01255747_g1), BNP (Mm01255770_g1), SERCA (Mm01201431_m1), and GAPDH (Mm03302249_g1).
Confocal Microscopic Characterizations of Cultured MEFs
Cells were seeded on 25 mm glass round coverslips at a density of 1.0x105 cells per well in 6-well plates. Adeno-Cre (Vector Biolabs, 1794) was added after cell attaching at a multiplicity of infection (MOI) of 100 at day 0 and kept in the medium for 1, 2, or 4 days; Mfn1/Mfn2/Drp1 triple floxed MEFs without the addition of adeno-Cre, Mfn1/Mfn2/Drp1 triple floxed MEFs with the addition of adeno-β-gal (Vector Biolabs, 1080) at a MOI of 100, or wild-type MEFs with the addition of Cre (MOI 100) were used as controls (Pre), when appropriate. Adeno-mcherry-parkin (a gift from Dr. Åsa Gustafsson, University of California San Diego), adeno-mito-Keima (a gift from Dr. Lorrie Kirshenbaum, University of Manitoba) were added at MOI of 50 at 2 days prior to imaging.
Live cell imaging used a Nikon Ti confocal microscope equipped with a 60x 1.3NA oil immersion objective. Each experiment was performed for a minimum of three times (n is indicated in the bar graphs) with 15 images per treatment group.
Cell viability assay was performed using Live/Dead viability/cytotoxicity kit (Thermo Fisher Scientific, L3224) according to manufacturer’s protocol.
For visualization of mitochondria and measurement of mitochondrial membrane potential, MEFs were stained with 200 nM MitoTracker Green (Thermo Fisher Scientific, M7514), 200 nM of tetramethylrhodamine, ethyl ester (TMRE, Thermo Fisher Scientific, T-669) and 10 μg/ml Hoechst (Thermo Fisher Scientific, H3570) at 37 °C for 30 min. The coverslip was loaded onto a chamber (Warner instrument, RC-40LP) in modified Krebs-Henseleit buffer (138 mM NaCl, 3.7 mM KCl, 1.2 mM KH2PO4, 15 mM glucose, 20 mM HEPES and 1 mM CaCl2) at room temperature. Mitochondrial aspect ratio (the ratio of length/width) was quantified based on the green-stained mitochondria using ImageJ. Mitochondrial depolarization was calculated as % of the area of green mitochondria compared to that of all the mitochondria visualized on MitoTracker Green and TMRE merged images; data are presented as green ÷ (green + yellow mitochondria) × 100%.
For detection of parkin aggregation, MEFs infected with adeno-mcherry-parkin were stained with MitoTracker Green and Hoechst. Mitochondrial depolarization was induced by the addition of FCCP at 10 μm for 1 hr. Parkin aggregation was calculated as % of cells with clumping mcherry-parkin compared to all the cells.
For assessment of lysosomal-mitochondria engulfment, MEFs were stained with 50 nM LysoTracker Red (Thermo Fisher Scientific, L-7528), 200 nM MitoTracker Green and 10 μg/ml Hoechst at 37 °C for 30 min. Mitochondrial depolarization was induced by the addition of FCCP at 10 μM at 37 °C for 1 hr. Red dots represented lysosomes; yellow dots represented the co-localization of lysosomes and mitochondria. Mitochondrial depolarization was induced by the addition of FCCP (10 μM) for 1 hour. Lysosomal-mitochondria engulfment was calculated as % of lysosomes containing mitochondria (Song et al., 2015).
For quantification of mitophagic rates after mitochondrial dynamics protein deletion, MEFs were infected with adeno-mito-Keima for 2 days and stained with 200 nM MitoTracker Green for 30 min at room temperature before imaging. Mito-Keima is a fluorescent protein with mitochondrial localization signal. It emits different colored signals at neutral and acidic pHs; thus a mitochondrially localized pH-indicator protein. Every mitochondrion was visualized with green fluorescent signal from MitoTracker Green at 488 nm; lysosomal delivery of mitochondria was detected with red fluorescent signal from mito-Keima at acidic pH at 561 nm under confocal fluorescent microscope. Time-dependent mitophagy was measured as % red fluorescent signal area/cell area.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM unless otherwise indicated in figure legends. Sample number (n) indicates the number of independent biological samples (individual mice or wells of cells) in each experiment. Sample numbers and experimental repeats are indicated in the figures or methods section above. Data were analyzed using the unpaired Student’s t-test, one-way ANOVA with Tukey’s post-hoc between group comparison, as appropriate. Survival curve was analyzed using log-rank test. p < 0.05 was considered statistically significant.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Drp1 | Abcam | Cat# ab56788 |
| Rabbit monoconal anti-Mff | Abcam | Cat# ab129075 |
| Mouse monoclonal anti-Mfn2 | Abcam | Cat# ab56889 |
| Rabbit polyconal anti-Opa1 | Abcam | Cat# ab42364 |
| Mouse monoclonal anti-GAPDH | Abcam | Cat# ab8245 |
| Rabbit polyconal anti-OXPHOS | Abcam | Cat# MS604 |
| Rabbit polyconal anti-VDAC1 | Abcam | Cat# ab15895 |
| Rabbit monoconal anti-TOMM20 | Abcam | Cat# ab186735 |
| Rabbit polyconal anti-Hsp60 | Abcam | Cat# ab46798 |
| Rabbit polyconal anti-LONP1 | Abcam | Cat# ab103809 |
| Rabbit polyconal anti-Catalase | Abcam | Cat# ab1877 |
| Mouse monoclonal anti-p62 | Abcam | Cat# ab56416 |
| Rabbit polyconal anti-LC3 | Abcam | Cat# ab128025 |
| Mouse monoclonal anti-COXIV | Abcam | Cat# ab14744 |
| Rabbit polyconal anti-SOD1 | Abcam | Cat# ab13498 |
| Rabbit polyconal anti-SOD2 | Abcam | Cat# ab13533 |
| Mouse monoclonal anti-Cardiac Troponin T | Abcam | Cat# ab10214 |
| Rabbit polyconal anti-Mfn1 | Santa Cruz Biotechnology | Cat# sc-50330 |
| Rabbit polyconal anti-AFG3L2 | Santa Cruz Biotechnology | Cat# sc-84687 |
| Mouse monoclonal anti-Ubiquitin | Santa Cruz Biotechnology | Cat# sc-8017 |
| Mouse monoclonal anti-β-Actin | Santa Cruz Biotechnology | Cat# sc-47778 |
| Rabbit polyconal anti-Calreticulin | Cell Signaling Technology | Cat# 12238 |
| Rabbit polyconal anti-GRP94 | Cell Signaling Technology | Cat# 2104 |
| Rabbit polyconal anti-PERK | Cell Signaling Technology | Cat# 3192 |
| Rabbit polyconal anti phospho-PERK (Thr 980) | Cell Signaling Technology | Cat# 3179 |
| Mouse monoclonal Anti-CHOP | Cell Signaling Technology | Cat# 2895 |
| Rabbit polyconal anti-LC3 B | Cell Signaling Technology | Cat# 3868 |
| Rabbit polyconal anti-LC3 A/B | Cell Signaling Technology | Cat# 4108 |
| HRP-linked anti-mouse IgG | Cell Signaling Technology | Cat# 7076 |
| HRP-linked anti-rabbit IgG | Cell Signaling Technology | Cat# 7074 |
| Rabbit polyconal anti-Goat anti-Rabbit IgG, 488 | Fisher Thermo Scientific | Cat# A-11008 |
| Rabbit polyconal anti-Goat anti-Mouse IgG, 546 | Fisher Thermo Scientific | Cat# A-11003 |
| Bacterial and Virus Strains | ||
| Ad-CreET2 | Vector Biolabs | Cat# 1794 |
| Ad-CMV-β-Gal | Vector Biolabs | Cat# 1080 |
| Ad-mcherry-parkin | Asa Gustaffson | N/A |
| Ad-mito-Keima | Lorrie Kirshenbaum | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Evans blue | Sigma-Aldrich | Cat# E2129 |
| Adenosine 5′-diphosphate sodium salt (ADP) | Sigma-Aldrich | Cat# A2754 |
| Oligomycin A | Sigma-Aldrich | Cat# 75351 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Sigma-Aldrich | Cat# C2920 |
| Bafilomycin A1 | Sigma-Aldrich | Cat# SML1661 |
| Wheat Germ Agglutinin | Thermo Fisher Scientific | Cat# W834 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M7514 |
| MitoTracker Red CMXRos | Thermo Fisher Scientific | Cat# M7512 |
| LysoTracker Red DND-99 | Thermo Fisher Scientific | Cat# L7528 |
| Tetramethylrhodamine, Ethyl Ester (TMRE) | Thermo Fisher Scientific | Cat# T-669 |
| DiOC6(3) (3,3′-Dihexyloxacarbocyanine Iodide) | Thermo Fisher Scientific | Cat# D273 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| Critical Commercial Assays | ||
| Hematoxylin-Eosin Y solutions | Sigma-Aldrich | Cat# GHS116, HT110116 |
| Masson’s trichrome | Sigma-Aldrich | Cat# HT15, HT10132 & HT10279 |
| DeadEnd fluorometric TUNEL system | Promega | Cat# G3250 |
| DNeasy blood & tissue kit | Qiagen | Cat# 69506 |
| Live/Dead viability/cytotoxicity kit | Thermo Fisher Scientific | Cat# L3224 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Mouse: wild-type murine embryonic fibroblasts | Gerald W. Dorn II | N/A |
| Mouse: Mfn1 fl/fl x Mfn2 fl/fl murine embryonic fibroblasts | Gerald W. Dorn II | N/A |
| Mouse: Drp1 fl/fl murine embryonic fibroblasts | Gerald W. Dorn II | N/A |
| Mouse: Mfn1 fl/fl x Mfn2 fl/fl x Drp1 fl/fl murine embryonic fibroblasts | Gerald W. Dorn II | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: α-MHC promoter driven human Drp1 Tg | Gerald W. Dorn II | N/A |
| Mouse: rTA | Gossen et al., 1992 | |
| Mouse: Mfn1 fl/fl x Mfn2 fl/fl | Chen et al., 2003 | |
| Mouse: Drp1 fl/fl | Ishihara et al., 2009 | |
| Mouse: MERCreMER | Sohal et al., 2001 | |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Other | ||
Supplementary Material
Highlights.
Forced mitochondrial fragmentation in vivo did not induce cardiomyocyte apoptosis
Mitochondrial adynamism delayed cardiomyopathies induced by fission- or fusion-defect
Mitophagy defects in adynamic hearts prevented elimination of senescent mitochondria
Mitochondrial hyper-accumulation increased cardiac mass but impaired function
Footnotes
Author Contributions
Conceptualization, G.W.D.; Methodology, G.W.D., M.S., A.F.; Investigation, M.S., A.F., J.A.F. and L.Z.; Writing - Original Draft, G.W.D. and M.S.; Writing - Review & Editing, G.W.D. and M.S.; Funding Acquisition, G.W.D.; Resources, G.W.D., M.S. and L.Z.; Supervision, G.W.D..
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernhardt D, Muller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. 2015;5:7885. doi: 10.1038/srep07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary KR, El-Sikhry H, Seubert JM. Mitochondria and the aging heart. J Geriatr Cardiol. 2011;8:159–167. doi: 10.3724/SP.J.1263.2011.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen H, Ren S, Clish C, Jain M, Mootha V, McCaffery JM, Chan DC. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J Cell Biol. 2015;211:795–805. doi: 10.1083/jcb.201507035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res. 2012;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- Dorn GW., 2nd Gone fission...: diverse consequences of cardiac drp1 deficiency. Circ Res. 2015a;116:225–228. doi: 10.1161/CIRCRESAHA.114.305672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW., 2nd Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med. 2015b;7:865–877. doi: 10.15252/emmm.201404575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature. 2016 doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW., 2nd Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20:214–225. doi: 10.1016/j.cmet.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A, Cipolat S, Chen Y, Dorn GW, 2nd, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science. 2013;342:734–737. doi: 10.1126/science.1241359. [DOI] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- Lewis MR, Lewis WH. Mitochondria in Tissue Culture. Science. 1914;39:330–333. doi: 10.1126/science.39.1000.330. [DOI] [PubMed] [Google Scholar]
- Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM, Kelly DP. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res. 2014;114:626–636. doi: 10.1161/CIRCRESAHA.114.302562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–528. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012;111:1012–1026. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. Febs J. 2016 doi: 10.1111/febs.13820. [DOI] [PubMed] [Google Scholar]
- Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- Shirihai OS, Song M, Dorn GW., 2nd How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Dorn GW., 2nd Mitoconfusion: Noncanonical Functioning of Dynamism Factors in Static Mitochondria of the Heart. Cell Metab. 2015;21:195–205. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW., 2nd Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ Res. 2015a;117:346–351. doi: 10.1161/CIRCRESAHA.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015b;21:273–285. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J, Dorn GW., 2nd Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res. 2004;95:1200–1206. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P, et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–844. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:1221. doi: 10.1126/science.aad0116. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.