

Abstract
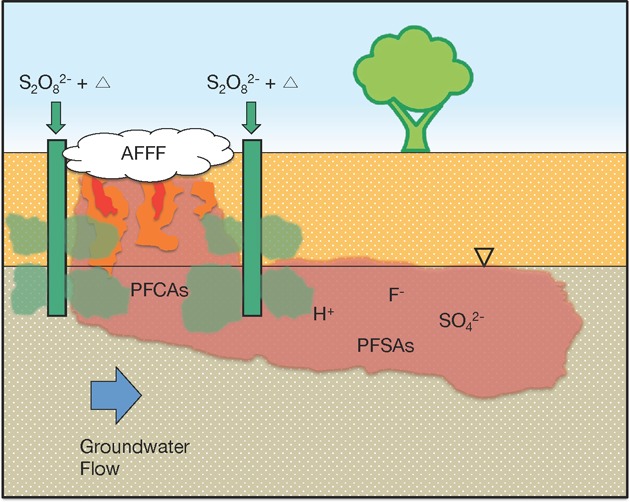
Poly- and perfluoroalkyl substances (PFASs) have been detected in an increasing number of water supplies. In many instances, the contamination is associated with the use of PFAS-containing aqueous film-forming foams (AFFF) in firefighting activities. To investigate the potential for remediating AFFF contamination in groundwater with heat-activated persulfate, PFAS oxidation and the generation of transformation products was evaluated under well-controlled conditions. Fluorotelomer- and perfluoroalkyl sulfonamide-based polyfluorinated compounds were transformed to perfluorinated carboxylic acids, which underwent further degradation under acidic conditions produced after persulfate decomposed. The presence of aquifer sediments decreased the efficiency of the remedial process but did not alter the transformation pathways. At high concentrations, the presence of organic solvents, such as those present in AFFF formulations, inhibited transformation of a representative perfluorinated compound, perfluorooctanoic acid. Heat-activated persulfate did not transform perfluorooctanesulfonic acid or perfluorohexanesulfonic acid under any conditions. Despite challenges associated with the creation of acidic conditions in the subsurface, the potential for generation of undesirable transformation products, and the release of toxic metals, heat-activated persulfate may be a useful in situ treatment for sites contaminated with polyfluoroalkyl substances and perfluorocarboxylic acids.
Introduction
The use of aqueous film-forming foams in firefighting and firefighter training exercises has led to contamination of water and soil with PFASs.1−5 AFFFs–chemical mixtures containing a combination of water, organic solvents, and fluorinated and nonfluorinated surfactants–are valued for their ability to quickly extinguish liquid fuel-based fires, and have been used for several decades by the military, operators of commercial airports, municipalities, and the chemical industry.6 PFASs are currently a required component of AFFFs under military purchasing specifications,7 but concerns over the persistence of a subclass of PFASs, perfluoroalkyl acids (PFAAs), have increased in recent years. Proximity to military firefighter training areas where AFFF was used has been linked to the presence of PFAAs in drinking water by both site investigations1,2,8 and national scale GIS analysis.9 Although PFAAs are not regulated drinking water contaminants, the U.S. EPA’s release of a combined lifetime health advisory10 of 70 ng/L for perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) has brought attention to this issue.
Numerous fluorinated surfactants other than the PFAAs have been used in AFFFs.11 The PFAS content of a particular AFFF formulation depends on the company that created the formulation, the original manufacturer of the fluorinated surfactants used, and the date of production. The main historical supplier of AFFF to the U.S. military was 3M Corporation, which manufactured PFASs through the electrochemical fluorination process.12 AFFF produced by 3M contains perfluoroalkyl sulfonates (PFSAs), perfluoroalkyl sulfonamide-based polyfluorinated compounds, and lesser amounts of perfluorinated carboxylates (PFCAs).4 The other major process used to manufacture PFASs is fluorotelomerization. Fluorotelomer-based AFFF formulations have been produced by several manufacturers and typically contain polyfluorinated compounds and lesser amounts of PFAAs.4
Unlike perfluorinated compounds, polyfluorinated compounds typically contain a perfluoroalkyl functional group and at least one nonfluorinated carbon.13 Nonfluorinated carbons are more reactive than perfluorinated carbons, which makes them more susceptible to oxidation. Some polyfluorinated compounds can produce PFAAs when the bonds linking the perfluoroalkyl moiety to nonfluorinated groups are oxidized.13 Polyfluorinated compounds that generate PFAAs upon exposure to hydroxyl radical (HO•) are referred to as PFAA-precursors. Biotransformation of sulfonamide-based PFAA-precursors typically leads to production of PFSAs,14 while biotransformation of fluorotelomer-based PFAA-precursors typically generates PFCAs and fluorotelomer (un)saturated acids.15,16 Reaction with HO• transforms Cn sulfonamide-containing precursors to equimolar quantities of the corresponding Cn PFCAs, whereas Cn fluorotelomer precursors are transformed to a mixture of PFCAs of varying carbon chain length.
Ex situ treatment using granular activated carbon is currently the main means of remediating PFAS in contaminated water sources.10 However, this approach does not treat sorbed PFAS, which may include cationic and zwitterionic PFAA-precursors that exhibit a high affinity for aquifer solids. Although PFAAs are resistant to biodegradation and hydroxyl radical-based treatment processes,17 heat-activated persulfate (S2O82–) is capable of oxidizing PFCAs.18−21 In this process, sulfate radicals (SO4•–) transform PFCAs, but only under acidic conditions22 (pH ≤ 3). The efficiency of the process decreases in the presence of sediment or high concentrations of chloride.23 With the exception of 6:2 fluorotelomer sulfonate (6:2 FtS),20 no studies to date have examined heat-activated S2O82– treatment of PFAA-precursors. Further complicating the treatment of AFFF-contaminated sites with persulfate is the potential presence of other AFFF components. For example, the solvent diethylene glycol butyl ether (DGBE) comprised 17% and 20% of two samples of AFFF manufactured by Ansul and 3M, respectively.24 DGBE is expected to reduce the efficiency of PFAS treatment by scavenging SO4•–.
To assess the feasibility of using heat-activated persulfate to remediate AFFF-contaminated groundwater and to identify terminal transformation products, batch experiments were carried out under well-controlled conditions representative of those encountered during in situ chemical oxidation (ISCO). The conversion of polyfluorinated PFAA-precursors to PFCAs, and subsequent degradation of PFCAs, was studied using samples of two representative AFFF formulations from 3M and Ansul in water and sediment slurry systems. The effect of AFFF components on degradation of PFAAs was examined using PFOA as a representative PFCA in the presence of a glycol ether and other organic solvents. Results of these experiments can be used to identify conditions under which heat-activated persulfate will be effective in the remediation of contaminated aquifers and to optimize the treatment process.
Materials and Methods
Materials
Full names and abbreviations for PFASs measured in this study are listed in Supporting Information (SI) Table S1. Analytical standards and isotopically labeled PFAS standards were purchased from Wellington Laboratories. Reagent-grade PFOA (96% purity) used in oxidation experiments was obtained from Sigma-Aldrich. Samples of AFFF manufactured by 3M and Ansul were obtained from an archive collected at U.S. military bases, as described previously.12 Prior characterization work of PFASs in contemporaneous formulations of 3M AFFF indicated that they consisted mainly of PFOS and multiple C4–C7 PFCA precursors, including, but not limited to perfluoroalkyl sulfonamide amino carboxylates (PFnSAmAs) and perfluoroalkyl sulfonamido amines (PFnSAms). Contemporaneous 3M AFFF also contained smaller amounts of C4–C7 PFSAs and C4–C8 PFCAs.4 The same study reported that the primary PFAS component of contemporaneous Ansul AFFF is 6:2 fluorotelomer thioamido sulfonate (6:2 FtTAoS). 6:2 FtTAoS, which is often referred to by the trade name Lodyne, is the main PFAS present in AFFF formulations from at least three different manufacturers. The structure of 6:2 FtTAoS is shown in Figure 1, and the structures of PFHxSAm and PFHxSAmA, two components of 3M AFFF that were present at lower concentrations, are shown in SI Figure S1.
Figure 1.

Chemical structure of 6:2 fluorotelomer thioamido sulfonate (6:2 FtTAoS).
HPLC-grade water and LC-MS-grade methanol were obtained from Fisher Scientific. All other chemicals and solvents were of the highest possible purity and were purchased from Fisher Scientific or Sigma-Aldrich. Solutions were prepared using 18 MΩ ultrapure water from a Millipore system.
Uncontaminated aquifer sediment was obtained from a surficial alluvial aquifer in Arizona, approximately 25 m below ground surface. Sediment was dried at 100 °C, homogenized, and sieved through a 600-μm sieve. The fraction that passed through the sieve was collected and used in oxidation experiments. Characterization data for the sediment are reported in SI Table S2.
Persulfate Oxidation Experiments
Batch experiments were carried out in sealed 15- or 50 mL polypropylene or polystyrene centrifuge tubes with total solution volumes of 10 or 40 mL. Concentrated AFFF stock solutions were prepared by diluting AFFF 100-fold in ultrapure water. A concentrated PFOA stock solution (0.53 mM) was also prepared in ultrapure water. Reactors were filled with ultrapure water and amended with concentrated stock solutions to obtain initial concentrations of the primary PFAS species between 0.3 and 2.5 μM. For experiments with AFFF, this corresponded to approximately a 21 000-fold dilution of the Ansul concentrate and an 83 000-fold dilution of the 3M concentrate. In some experiments, aliquots of methanol, ethanol, or DGBE were added to achieve initial concentrations ranging from 5 to 500 mM. Sediment slurry experiments were performed by adding 20 g/L of dried aquifer solids. Finally, aliquots of 500 mM Na2S2O8 were added to achieve an initial S2O82– concentration of 50 mM.
All experiments were performed in an 85 °C water bath, a temperature that is often achieved during in situ thermal treatments at hazardous waste sites.25 Reactors were placed in the hot-water bath immediately after addition of the persulfate and were removed from the bath briefly to withdraw samples. Solutions were not prewarmed, but reached 85 °C within 15 min. Most experiments employed a 50 mM aliquot of S2O82– and lasted 7.5 h. In some experiments, an additional aliquot of persulfate was added after the initial 7.5 h period to simulate the effect of additional treatment. In all cases, less than 5% of the applied S2O82– remained at the end of the experiment, as measured using the methods described below. Due to the constraints of working in a water bath, the reactors were not mixed continuously during the experiment. Instead, reactors were mixed by inverting several times at the initiation of the experiment and prior to collection of each sample. Persulfate-free control experiments were performed to assess PFAS losses unrelated to S2O82– treatment. Most experiments were performed in duplicate or triplicate, and the results presented represent averages plus or minus one standard deviation. Experiments designed to assess the effect of DGBE and ethanol were performed using a single reactor for each initial solvent concentration.
All samples for PFAS analysis were diluted with HPLC-grade water and methanol prior to measurement to bring PFAS concentrations into the instrument calibration range. To minimize losses due to salting out, experiments conducted in the absence of aquifer solids were diluted five- or 10-fold in HPLC-grade water prior to a five- to 25- fold dilution in methanol. Samples from experiments with aquifer solids were centrifuged at 10 000 rpm for 10 min. The supernatant was then subjected to the dilution procedure described above. To measure the concentration of PFASs associated with aquifer solids, the remaining aqueous supernatant was decanted and the sediments were extracted in methanol overnight on a shaker table. The extracted samples were centrifuged as described above, and the supernatant was subjected to a further 10-fold dilution in methanol. Diluted samples were stored in 1.5 mL polypropylene microcentrifuge tubes at room temperature prior to LC-MS/MS analysis, which typically occurred within 4 days.
Separate samples were collected simultaneously for other analytes. Samples for F– analysis were not diluted and were stored at 4 °C prior to analysis, which typically occurred within 2 days. Samples for total organic carbon (TOC) analysis were filtered sequentially with 0.7 and 0.2 μm syringe filters, diluted three- to 100-fold in 500 mM borate buffer to bring TOC concentrations into the instrument calibration range, and stored at 4 °C for less than 8 h before measurement. Persulfate and pH were measured immediately upon sampling, using the methods described below.
Analytical Methods
PFASs were quantified using an Agilent 6460 HPLC/MS-MS operating in negative electrospray ionization mode, as described previously.26 Briefly, samples were analyzed in 250 μL of 50:50 methanol:water mixture created by combining 125 μL of HPLC-grade water, 100 μL of diluted sample in methanol, and 25 μL of a 20 μg/L internal standard stock solution in methanol. PFAS concentrations were determined using isotope dilution with certified analytical standards. A list of ion transitions monitored and MS parameters is provided in SI Table S3. Ultrashort-chain PFAAs (C2 and C3 PFCAs and PFSAs), PFnSAmAs, and PFnSAms were not measured in this study.
Other analytes were quantified with established methods. Persulfate was measured using the KI colorimetric method27 with a PerkinElmer Lambda-14 UV spectrophotometer. Fluoride and formic acid were measured using a Dionex ICS-1100 ion chromatograph with 0.8 mM NaHCO3 and 4.5 mM Na2CO3 as the mobile phase. Fluoride was quantified by standard addition. Total organic carbon (TOC) was measured using a Shimadzu TOC-V CSH elemental analyzer.
Results and Discussion
Sediment-Free Experiments
Under the conditions studied, persulfate decomposes rapidly according to the following overall equation:
| 1 |
Sulfate radicals are produced during the initial step and in chain reactions initiated by radicals.28
In solutions of both Ansul and 3M AFFF at 85 °C, at least 95% of the added persulfate decomposed after 7.5 h, as expected from previous studies of persulfate thermolysis.29 Loss of S2O82– followed pseudo-first order kinetics (SI Figure S2a). Because the experiments were performed in the absence of a buffer, acid production from eq 1 caused the pH to drop from 4.0 to 1.5 in the Ansul experiments, and from 3.8 to 1.4 in the 3M experiments. TOC decreased from 9 mg/L to less than the method detection limit of 0.8 mg/L and from 4 mg/L to <0.8 mg/L, respectively, in the Ansul and 3M reactors (SI Figure S3). In both experiments, the initial concentration of PFAS accounted for only a small amount of the measured TOC (i.e., < 2%). Because TOC attributable to PFAS was in all cases less than the limit of detection of the TOC instrument, TOC measurements did not provide insight into PFAS transformation.
In experiments with diluted Ansul AFFF, 6:2 FtTAoS was the only PFAS detected prior to oxidation. Transformation of 6:2 FtTAoS was rapid, with 100% loss of 6:2 FtTAoS and production of a suite of short-chain PFCAs within the first hour of the experiment (Figure 2). Degradation of 6:2 FtTAoS produced mainly perfluorohexanoic acid (PFHxA), perfluoropentanoic acid (PFPeA), and perfluorobutyric acid (PFBA), as well as a small amount of perfluoroheptanoic acid (PFHpA), similar to what was observed previously4 during treatment of Ansul AFFF with HO•. After the initial conversion of 6:2 FtTAoS to PFCAs, the concentration data were consistent with the sequential −(CF2) cleavage mechanism, with PFHpA reaching its maximum concentration first, followed by PFHxA, PFPeA, and PFBA, as observed in other studies.18−20,30 In initial experiments performed with S2O82– at room temperature (data not shown), as well as in aerobic biotransformation studies,15 the primary transformation product of 6:2 FtTAoS was 6:2 FtS. The absence of detectable 6:2 FtS in the experiments performed at 85 °C indicates that 6:2 FtS is rapidly degraded by heat-activated persulfate, as has been shown previously.20 At the end of the treatment, short-chain PFCAs accounted for approximately 13% of the initial 6:2 FtTAoS on a molar basis. A 12% loss of 6:2 FtTAoS in heated controls containing AFFF without persulfate may have been due to sorption losses to the reactor walls. An attempt to obtain a mass balance on fluorine was unsuccessful due to the difficulty of accurately quantifying low F– concentrations in the presence of high concentrations of sulfate and other anions.
Figure 2.
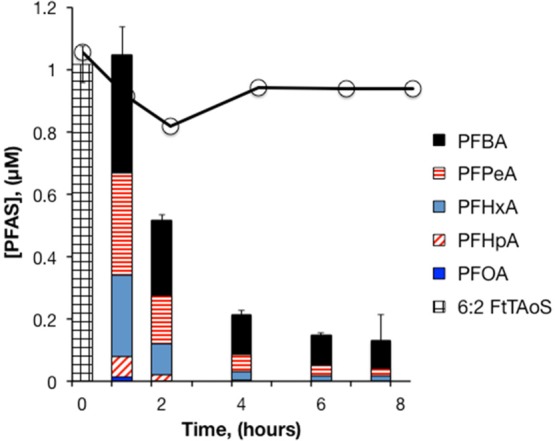
PFAS concentrations during heat-activated persulfate treatment of Ansul AFFF in water. 21 000-fold dilution of AFFF, [S2O82–]0 = 50 mM, pH0 = 3.5, T = 85 °C. Bars represent the mean of triplicate measurements. The solid line with circles represents 6:2 FtTAoS in persulfate-free controls (n = 1).
In experiments with diluted 3M AFFF, PFASs detected prior to oxidation included PFOS and smaller amounts of perfluorohexanesulfonic acid (PFHxS) and PFHxA (Figure 3a and b). No transformation of PFSAs was apparent upon treatment with heat-activated S2O82–. The PFOS concentrations decreased by approximately 40% within the first 0.5 h of the experiment in both the treatment and control reactors, and remained stable thereafter. This initial loss of PFOS may have been due to sorption to the reactor walls or some other physical phenomenon related to the AFFF components. PFCA concentrations initially increased, with a total of 150 μM of PFHxA, PFPeA, and PFBA detected after 1 h. PFCA concentrations subsequently decreased, leaving 18 μM of PFBA after 4 h of treatment. The change in PFCA concentrations with time was likely due to oxidation of sulfonamide-based polyfluorinated compounds PFHxSAm and PFHxSAmA. Although these compounds were not measured by the analytical method used in this study, they were detected in this formulation of 3M AFFF in prior work.4 Oxidation of the nonfluorinated portions of these PFAA-precursors by SO4•– yielded PFCAs as products, and these PFCAs subsequently reacted with SO4•– by the sequential chain-shortening mechanism described previously. Again, an attempt to calculate a mass balance on fluorine was unsuccessful due to the difficulty of accurately quantifying low F– concentrations.
Figure 3.
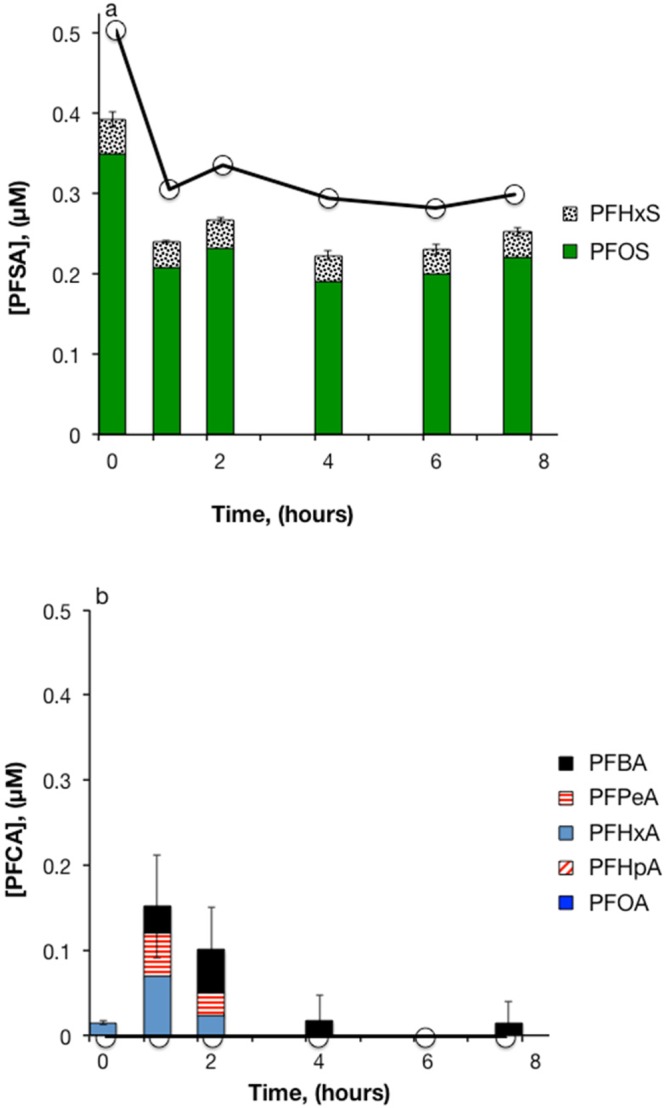
Heat-activated persulfate treatment of 3M AFFF in water. (a) PFSAs; (b) PFCAs. 83 000-fold dilution of AFFF, [S2O82–]0 = 50 mM, pH0 = 3.5, T = 85 °C. Bars represent the mean of triplicate measurements. Solid lines with circles represent the sum of PFSAs or PFCAs in persulfate-free controls (n = 1).
To verify that the decomposition of contaminants was consistent with expectations based on the rate at which persulfate undergoes thermolysis, we estimated the rates of thermolysis of persulfate under different pH conditions (SI Figure S4). Results from these experiments indicated that 87% of the persulfate should have been converted to sulfate radicals in approximately 1.7–2.5 h (i.e., at pH values between 1.5 and 3.5) and 97% of the persulfate should have been converted after 2.8–4.2 h. These predictions were consistent with observations of the transformation of PFAS in both types of AFFF (i.e., Figures 2 and 3) indicating that most of the transformation occurred through reactions with sulfate radical during the first 2 h of the experiments and that no significant changes in concentrations occurred after 4 h.
Effect of Aquifer Sediments
The rates of persulfate decomposition in sediment slurry experiments with both Ansul and 3M AFFF were similar to those observed in the sediment-free systems (SI Figure S2a, Table S4). As in the sediment-free experiments, greater than 95% of the added persulfate was decomposed after 7.5 h in the slurry reactors. In the presence of sediment slurry and AFFF, acid production from decomposition of the first 50 mM persulfate aliquot caused the pH to drop from approximately 6.5–1.6. The second 50 mM aliquot of persulfate resulted in a further pH decrease to 1.4. The initial TOC concentrations of solutions of Ansul and 3M AFFF in sediment experiments were 23 mg/L and 17 mg/L, respectively. The TOC concentrations of both AFFF solutions decreased to approximately 2 mg/L within the first hour of the oxidation treatment (SI Figure S3).
Compared to the sediment-free experiment, the presence of aquifer sediments decreased the initial aqueous concentration of 6:2 FtTAoS in solutions of Ansul AFFF by approximately 60%. This difference in initial concentration was likely due to sorption of 6:2 FtTAoS to the aquifer sediments. Treatment with 50 mM S2O82– resulted in rapid and complete removal of 6:2 FtTAoS, as in the sediment-free experiments, but degradation of short-chain PFCA intermediates was slower (Figure 4). The short-chain PFCAs accounted for 89% and 24% of the original 6:2 FtTAoS after treatment with 50 and 100 mM S2O82–, respectively. Extraction of the sediments with methanol after completion of the experiment resulted in recovery of only 1–2% of total PFAS, indicating that persulfate was able to access the initially sorbed fraction of 6:2 FtTAoS or decreased the aqueous concentration of 6:2 FtTAoS enough to drive rapid desorption.
Figure 4.
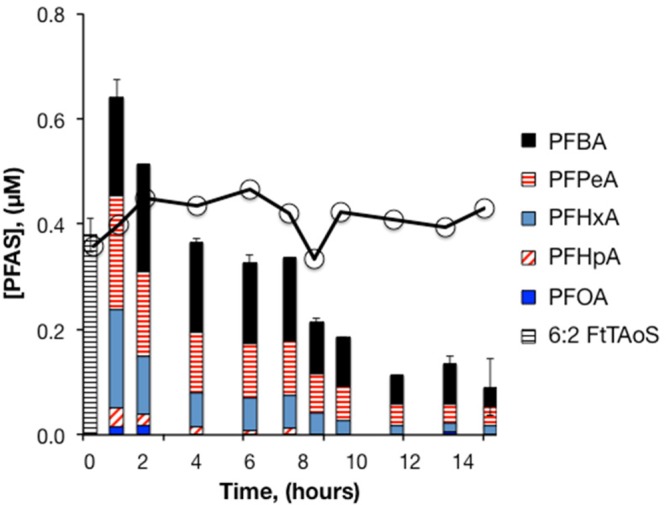
Heat-activated persulfate treatment of Ansul AFFF in aquifer sediment slurry. 21 000-fold dilution of AFFF, [S2O82–]0 = 50 mM, pH0 = 3.5, T = 85 °C. A second persulfate aliquot of 50 mM was added after 7.5 hours. Bars represent the mean of duplicate measurements. The solid line with circles represents 6:2 FtTAoS in persulfate-free controls (n = 1).
The efficiency of heat-activated S2O82– treatment of PFAS in 3M AFFF also decreased in the presence of aquifer sediments. As in the sediment-free experiments, no transformation of PFOS or PFHxS was observed (Figure 5a). Also similar to the sediment-free experiments, treatment resulted in an initial increase followed by a decrease in PFCA concentrations, as perfluoroalkyl sulfonamide-based precursors were converted to PFCAs (Figure 5b) and then subsequently oxidized. However, PFCA breakdown was slower in the sediment slurry reactors. The decrease in PFCA loss observed in slurry reactors with both 3M and Ansul AFFF was likely attributable to scavenging of SO4•– by constituents associated with the aquifer solids, such as inorganic species (e.g., chloride) or organic matter in solution or on particle surfaces. Extraction of the sediments from experiments with 3M AFFF showed that PFSAs sorbed to a greater extent than PFCAs. On average, 29% of total PFSAs were recovered by extraction, whereas 2% of total PFCAs were recovered. The initial decrease in PFOS concentration observed in both the treatment and control reactors was likely caused by sorption to the aquifer sediments and reactor walls.
Figure 5.
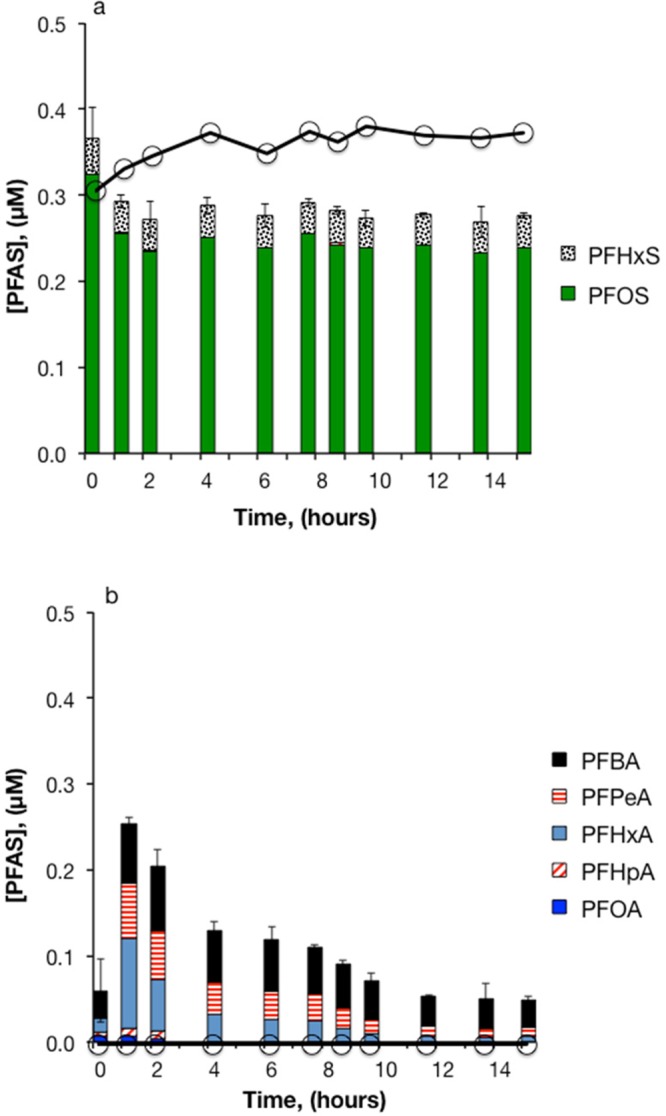
Heat-activated persulfate treatment of 3M AFFF in aquifer sediment slurry. (a) PFSAs; (b) PFCAs. 83,000-fold dilution of AFFF, [S2O82–]0 = 50 mM, pH0 = 3.5, T = 85 °C. A second persulfate aliquot of 50 mM was added after 7.5 hours. Bars represent the mean of triplicate measurements. Solid lines with circles represent the sum of PFSAs or PFCAs in persulfate-free controls (n = 1).
Effect of Solvents
The presence of DGBE decreased PFOA removal by heat-activated persulfate (Figure 6). This observation is consistent with model predictions made using the relevant reactions and second order rate constants (SI Text S1, Figures S5 and S6).
Figure 6.
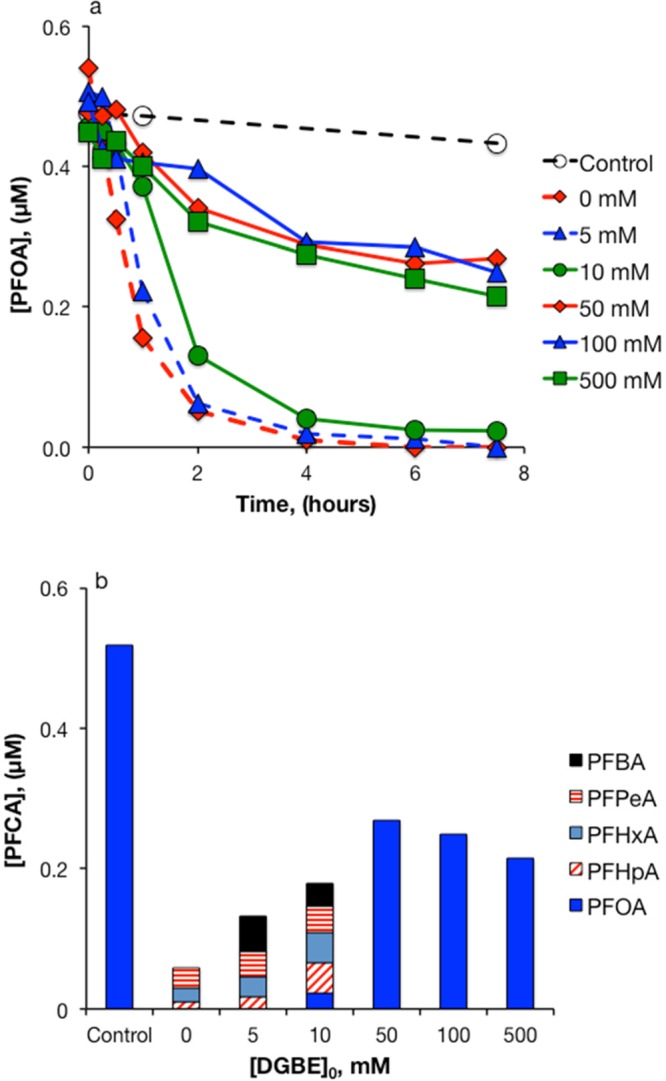
Heat-activated persulfate treatment of PFOA in the presence of varying concentrations of DGBE. (a) PFOA; (b) PFCAs after 7.5 h. [S2O82–]0 = 50 mM, T = 85 °C. All data points represent a single measurement.
In experiments with 50 mM S2O82– and varying initial concentrations of DGBE, the rate of persulfate disappearance increased at higher initial DGBE concentrations (SI Figure S2b, Table S4). Enhancement of the S2O82– disappearance rate was likely caused by radical chain reactions propagated by DGBE, as has been observed previously with benzene.28 Persulfate decay caused the pH of experiments with DGBE to drop from initial values of 3.8–4.0 to a final value of 1.3. In experiments with 5 mM and 10 mM initial concentrations of DGBE, heat-activated S2O82– resulted in greater than 95% TOC removal, but the fraction of initial TOC removed decreased with increasing initial DGBE concentration (SI Figure S7).
Results from experiments with varying concentrations of DGBE suggest that a different PFOA loss mechanism predominated at higher solvent concentrations (Figure 5). At DGBE concentrations of less than 50 mM, PFOA disappeared over the first 2 h of the experiment as shorter-chain PFCAs were produced. In the presence of 50–500 mM DGBE, about half of the PFOA disappeared gradually over the course of the 7.5 h experiment. The disappearance of PFOA at higher DGBE concentrations was not accompanied by the production of short-chain PFCAs. Similar results were obtained in experiments using methanol and ethanol instead of DGBE (SI Figures S8 and S9). Fluoride measurements from an experiment with 50 mM S2O82–, 5 μM PFOA, and 100 mM methanol showed a 44% loss of total fluorine from the system with no indication of fluoride production (Figure 7). Analysis of samples from this experiment using LC-MS/MS in full-scan mode did not indicate the formation of any transformation products.
Figure 7.
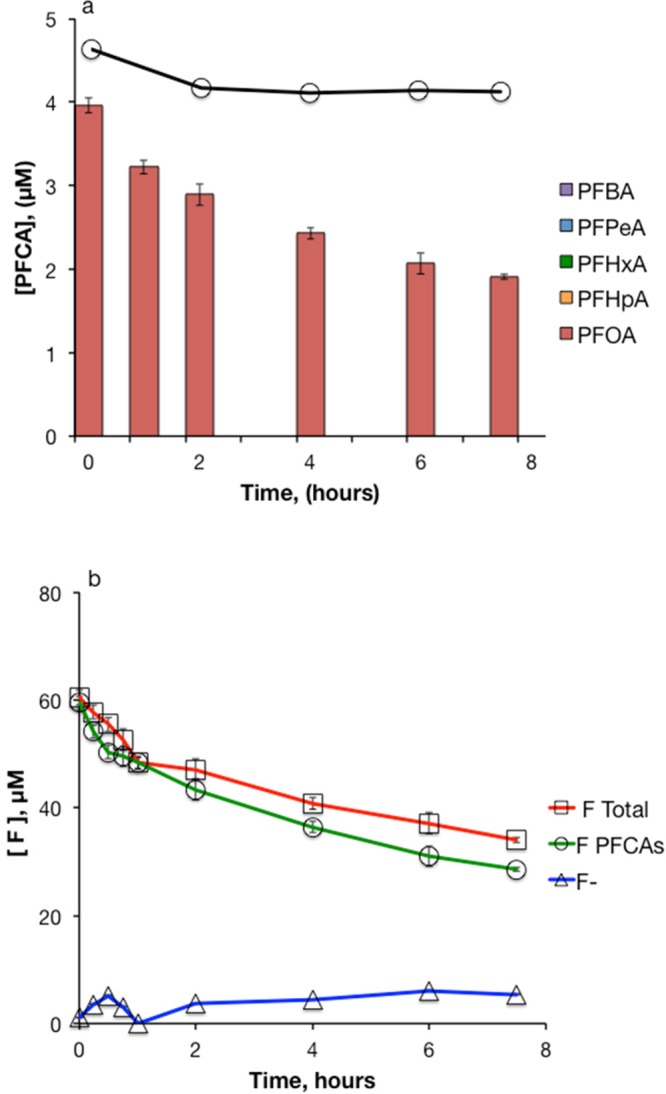
Heat-activated persulfate treatment of PFOA in the presence of methanol. (a) PFCAs. Bars represent the mean of triplicate measurements. The solid line with circles represents PFOA in persulfate-free controls (n = 1); b) Fluorine mass balance. [S2O82–]0 = 50 mM, [CH3OH]0 = 100 mM, [PFOA]0 = 5 μM, T = 85 °C. Each data point in the series “F PFCAs” represents the mean of triplicate measurements. Each data point in the series “F-“ is calculated using a linear regression of six standard addition measurements. Error bars for series “F-“, calculated as the standard error of the slope of the regression fore each standard addition curve, are too small to be visible. The series “F Total” represents the sum of “F PFCAs” and “F-”.
The lack of detectable organic transformation products or fluoride suggests that the PFOA loss observed in experiments with high solvent concentrations was due to volatilization. To test this hypothesis, S2O82–-free controls were run at varying pH values (SI Figure S10). The observed pH-dependent loss of PFOA was consistent with volatilization of the protonated form of PFOA. Although the pKa of PFOA is low (estimates range from −0.531 to 3.832), under the acidic conditions that predominated in the treatment experiments (i.e., the pH dropped below 1.8 in the persulfate-containing tests samples after 1 h) there was enough protonated PFOA for loss by volatilization to be significant over the course of the 7.5 h experiments.
Implications for Groundwater Remediation
Heat-activated persulfate under acidic conditions resulted in conversion of both fluorotelomer-based and sulfonamide-based PFAA precursors into PFCAs, which were further degraded and eventually mineralized. The efficiency of the persulfate treatment process decreased in the presence of 20 g/L of aquifer solids. In the presence of organic solvents similar to those used in AFFF, PFOA transformation was less efficient, but followed the same apparent mechanism as in ultrapure water when DGBE concentrations were below 50 mM. At higher solvent concentrations, scavenging of SO4•– by solvents prevented PFOA transformation. Some PFOA volatilized under the hot, acidic conditions typical of ISCO. Persulfate did not degrade PFOS or PFHxS.
These experiments indicate that heat-activated persulfate has the greatest potential as an in situ remedial treatment at sites where PFCAs or fluorotelomer-based precursors predominate. At sites where PFSAs are also present, heat-activated persulfate could be used as part of a treatment-train approach to reduce the contaminant mass in source zones, but groundwater extraction and ex situ treatment by physical processes would still be required. Inhibition of PFAS treatment by solvent co-contaminants will likely be less important at remediation sites than in the experiments described here, because real-world solvent concentrations are typically less than the concentrations at which PFOA transformation was inhibited in the experiments. However, treatment trains combining surfactant or cosolvent flushing with ISCO33 are likely to be ineffective for PFAS transformation due to oxidant scavenging by the surfactant or cosolvent.
Heat-activated persulfate converts PFAA-precursor compounds in AFFF, which are numerous, poorly understood, and difficult to measure, into a discrete set of PFCA transformation products. Standard analytical approaches, subsurface transport properties, toxicology data, and regulatory guidance are all better known for PFCAs than PFAA-precursors. In this way, persulfate ISCO could serve as a means of reducing the uncertainty associated with AFFF remediation and facilitate faster and more cost-effective site cleanup.
A major disadvantage of using heat-activated persulfate to treat AFFF-impacted sites is the generation of potentially harmful byproducts. Production of short-chain PFCAs is undesirable because they are mobile in the subsurface and difficult to treat using sorptive technologies. Although little is known about the mobility or treatability of PFAA-precursors, short-chain PFCAs are expected to be more mobile in groundwater34 and have been shown to be more poorly retained on granular activated carbon and ion exchange resins than the long-chain homologues.35,36 Chemical oxidation also produces ClO3– through scavenging reactions with Cl–,30 and liberation of F– from PFAS under acid conditions could generate potentially hazardous concentrations of HF (i.e., the pKa of HF is 3.2).
The potential adverse consequences of aquifer acidification resulting from S2O82– treatment include mobilization of metals through dissolution of metal oxides, and impacts to the microbial community. These impacts could be mitigated by neutralizing groundwater after PFAS treatment is complete.23 In spite of these disadvantages, heat-activated persulfate is one of the more promising options available for in situ treatment of PFAS at sites impacted by AFFF.
Acknowledgments
This study was supported by the U.S. National Institute for Environmental Health Sciences (NIEHS) Superfund Research Program (Grant P42 ES004705) and the Superfund Research Center at University of California, Berkeley. Additional support came from the Strategic Environmental Research and Development Program (SERDP ER2128). We thank our colleagues Erika Houtz, Shan Yi, and Katie Harding for insightful discussions and help in the laboratory. We also thank Dr. Christopher Higgins for helpful comments on a draft version of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.7b03969.
PFAS analytes included in PFAS analysis along with additional details of analytical methods, characteristics of sediments, experimental and modeling data on persulfate decomposition kinetics, modeling analysis of sulfate radical fate under different reaction conditions, total organic carbon concentrations measured during treatment of AFFF and solutions containing PFOA and DGBE, PFOA loss in the presence of methanol and ethanol and PFOA loss in persulfate-free controls (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Moody C. A.; Field J. A. Determination of perfluorocarboxylates in groundwater impacted by fire-fighting activity. Environ. Sci. Technol. 1999, 33 (16), 2800–2806. 10.1021/es981355+. [DOI] [Google Scholar]
- Moody C. A.; Hebert G. N.; Strauss S. H.; Field J. A. Occurrence and persistence of perfluorooctanesulfonate and other perfluorinated surfactants in groundwater at a fire-training area at Wurtsmith Air Force Base, Michigan, USA. J. Environ. Monit. 2003, 5 (2), 341–345. 10.1039/b212497a. [DOI] [PubMed] [Google Scholar]
- Moody C. A.; Martin J. W.; Kwan W. C.; Muir D. C.; Mabury S. A. Monitoring perfluorinated surfactants in biota and surface water samples following an accidental release of fire-fighting foam into Etobicoke Creek. Environ. Sci. Technol. 2002, 36 (4), 545–551. 10.1021/es011001+. [DOI] [PubMed] [Google Scholar]
- Houtz E. F.; Higgins C. P.; Field J. A.; Sedlak D. L. Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soil. Environ. Sci. Technol. 2013, 47 (15), 8187–8195. 10.1021/es4018877. [DOI] [PubMed] [Google Scholar]
- Houtz E. F.; Sutton R.; Park J.-S.; Sedlak M. Poly-and perfluoroalkyl substances in wastewater: Significance of unknown precursors, manufacturing shifts, and likely AFFF impacts. Water Res. 2016, 95, 142–149. 10.1016/j.watres.2016.02.055. [DOI] [PubMed] [Google Scholar]
- Moody C. A.; Field J. A. Perfluorinated Surfactants and the Environmental Implications of Their Use in Fire-Fighting Foams. Environ. Sci. Technol. 2000, 34 (18), 3864–3870. 10.1021/es991359u. [DOI] [Google Scholar]
- Military Specifications MIL-F-24385F: Fire Extinguishing Agents, Aqueous Film-forming Foam (AFFF) Liquid Concentrate, for Fresh and Seawater; U.S. Naval Research Laboratory: 1994. [Google Scholar]
- McGuire M. E.; Schaefer C.; Richards T.; Backe W. J.; Field J. A.; Houtz E.; Sedlak D. L.; Guelfo J. L.; Wunsch A.; Higgins C. P. Evidence of remediation-induced alteration of subsurface poly-and perfluoroalkyl substance distribution at a former firefighter training area. Environ. Sci. Technol. 2014, 48 (12), 6644–6652. 10.1021/es5006187. [DOI] [PubMed] [Google Scholar]
- Hu X. C.; Andrews D. Q.; Lindstrom A. B.; Bruton T. A.; Schaider L. A.; Grandjean P.; Lohmann R.; Carignan C. C.; Blum A.; Balan S. A. Detection of Poly-and Perfluoroalkyl Substances (PFASs) in US Drinking Water Linked to Industrial Sites, Military Fire Training Areas, and Wastewater Treatment Plants. Environ. Sci. Technol. Lett. 2016, 3 (10), 344–350. 10.1021/acs.estlett.6b00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA. Fact Sheet PFOA & PFOS Drinking Water Health Advisories. 2016.
- Barzen-Hanson K. A.; Roberts S. C.; Choyke S.; Oetjen K.; McAlees A.; Riddell N.; McCrindle R.; Ferguson P. L.; Higgins C. P.; Field J. A. Discovery of 40 Classes of Per-and Polyfluoroalkyl Substances in Historical Aqueous Film-Forming Foams (AFFFs) and AFFF-Impacted Groundwater. Environ. Sci. Technol. 2017, 51 (4), 2047–2057. 10.1021/acs.est.6b05843. [DOI] [PubMed] [Google Scholar]
- Place B. J.; Field J. A. Identification of novel fluorochemicals in aqueous film-forming foams used by the US military. Environ. Sci. Technol. 2012, 46 (13), 7120–7127. 10.1021/es301465n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck R. C.; Franklin J.; Berger U.; Conder J. M.; Cousins I. T.; de Voogt P.; Jensen A. A.; Kannan K.; Mabury S. A.; van Leeuwen S. P. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr. Environ. Assess. Manage. 2011, 7 (4), 513–541. 10.1002/ieam.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia-Avendaño S.; Vo Duy S.; Sauvé S. b.; Liu J. Generation of Perfluoroalkyl Acids from Aerobic Biotransformation of Quaternary Ammonium Polyfluoroalkyl Surfactants. Environ. Sci. Technol. 2016, 50 (18), 9923–9932. 10.1021/acs.est.6b00140. [DOI] [PubMed] [Google Scholar]
- Harding K. C.; Houtz E. F.; Yi S.; Field J. A.; Sedlak D. L.; Alvarez-Cohen L. Aerobic Biotransformation of Fluorotelomer Thioether Amido Sulfonate (Lodyne) in AFFF-Amended Microcosms. Environ. Sci. Technol. 2015, 49 (13), 7666–7674. 10.1021/acs.est.5b01219. [DOI] [PubMed] [Google Scholar]
- Wang N.; Szostek B.; Buck R. C.; Folsom P. W.; Sulecki L. M.; Gannon J. T. 8–2 Fluorotelomer alcohol aerobic soil biodegradation: Pathways, metabolites, and metabolite yields. Chemosphere 2009, 75 (8), 1089–1096. 10.1016/j.chemosphere.2009.01.033. [DOI] [PubMed] [Google Scholar]
- Vecitis C.; Park H.; Cheng J.; Mader B.; Hoffmann M. Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA). Front. Environ. Sci. Eng. China 2009, 3 (2), 129–151. 10.1007/s11783-009-0022-7. [DOI] [Google Scholar]
- Hori H.; Nagaoka Y.; Murayama M.; Kutsuna S. Efficient decomposition of perfluorocarboxylic acids and alternative fluorochemical surfactants in hot water. Environ. Sci. Technol. 2008, 42 (19), 7438–7443. 10.1021/es800832p. [DOI] [PubMed] [Google Scholar]
- Liu C. S.; Higgins C. P.; Wang F.; Shih K. Effect of temperature on oxidative transformation of perfluorooctanoic acid (PFOA) by persulfate activation in water. Sep. Purif. Technol. 2012, 91, 46–51. 10.1016/j.seppur.2011.09.047. [DOI] [Google Scholar]
- Park S.; Lee L. S.; Medina V. F.; Zull A.; Waisner S. Heat-activated persulfate oxidation of PFOA, 6:2 fluorotelomer sulfonate, and PFOS under conditions suitable for in-situ groundwater remediation. Chemosphere 2016, 145, 376–383. 10.1016/j.chemosphere.2015.11.097. [DOI] [PubMed] [Google Scholar]
- Lee Y.-C.; Lo S.-L.; Kuo J.; Lin Y.-L. Persulfate oxidation of perfluorooctanoic acid under the temperatures of 20–40 C. Chem. Eng. J. 2012, 198, 27–32. 10.1016/j.cej.2012.05.073. [DOI] [Google Scholar]
- Sun B.; Ma J.; Sedlak D. L. Chemisorption of Perfluorooctanoic Acid on Powdered Activated Carbon Initiated by Persulfate in Aqueous Solution. Environ. Sci. Technol. 2016, 50 (14), 7618–7624. 10.1021/acs.est.6b00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruton T. A.; Sedlak D. L.. Persulfate Oxidation of Perfluoroalkyl Acids Under Conditions Representative of In Situ Treatment. In preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding-Marjanovic K. C.; Yi S.; Weathers T. S.; Sharp J. O.; Sedlak D. L.; Alvarez-Cohen L. Effects of Aqueous Film-Forming Foams (AFFFs) on Trichloroethene (TCE) Dechlorination by a Dehalococcoides mccartyi-Containing Microbial Community. Environ. Sci. Technol. 2016, 50 (7), 3352–3361. 10.1021/acs.est.5b04773. [DOI] [PubMed] [Google Scholar]
- Triplett Kingston J. L.; Dahlen P. R.; Johnson P. C. State-of-the-Practice Review of In Situ Thermal Technologies. Groundwater Monit. Rem. 2010, 30 (4), 64–72. 10.1111/j.1745-6592.2010.01305.x. [DOI] [Google Scholar]
- Houtz E. F.; Sedlak D. L. Oxidative conversion as a means of detecting precursors to perfluoroalkyl acids in urban runoff. Environ. Sci. Technol. 2012, 46 (17), 9342–9349. 10.1021/es302274g. [DOI] [PubMed] [Google Scholar]
- Liang C. J.; Huang C. F.; Mohanty N.; Kurakalva R. M. A rapid spectrophotometric determination of persulfate anion in ISCO. Chemosphere 2008, 73 (9), 1540–1543. 10.1016/j.chemosphere.2008.08.043. [DOI] [PubMed] [Google Scholar]
- Liu H.; Bruton T. A.; Doyle F. M.; Sedlak D. L. In Situ Chemical Oxidation of Contaminated Groundwater by Persulfate: Decomposition by Fe(III)- and Mn(IV)-Containing Oxides and Aquifer Materials. Environ. Sci. Technol. 2014, 48 (17), 10330–10336. 10.1021/es502056d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. L.; Tratnyek P. G.; Johnson R. O. Persulfate Persistence under Thermal Activation Conditions. Environ. Sci. Technol. 2008, 42 (24), 9350–9356. 10.1021/es8019462. [DOI] [PubMed] [Google Scholar]
- Qian Y.; Guo X.; Zhang Y.; Peng Y.; Sun P.; Huang C.-H.; Niu J.; Zhou X.; Crittenden J. C. Perfluorooctanoic Acid Degradation Using UV–Persulfate Process: Modeling of the Degradation and Chlorate Formation. Environ. Sci. Technol. 2016, 50 (2), 772–781. 10.1021/acs.est.5b03715. [DOI] [PubMed] [Google Scholar]
- Goss K.-U. The pKa values of PFOA and other highly fluorinated carboxylic acids. Environ. Sci. Technol. 2007, 42 (2), 456–458. 10.1021/es702192c. [DOI] [PubMed] [Google Scholar]
- Burns D. C.; Ellis D. A.; Li H.; McMurdo C. J.; Webster E. Experimental p K a determination for perfluorooctanoic acid (PFOA) and the potential impact of p K a concentration dependence on laboratory-measured partitioning phenomena and environmental modeling. Environ. Sci. Technol. 2008, 42 (24), 9283–9288. 10.1021/es802047v. [DOI] [PubMed] [Google Scholar]
- Dugan P. J.; Siegrist R. L.; Crimi M. L. Coupling surfactants/cosolvents with oxidants for enhanced DNAPL removal: A review. Remed. J. 2010, 20 (3), 27–49. 10.1002/rem.20249. [DOI] [Google Scholar]
- Guelfo J. L.; Higgins C. P. Subsurface transport potential of perfluoroalkyl acids at aqueous film-forming foam (AFFF)-impacted sites. Environ. Sci. Technol. 2013, 47 (9), 4164–4171. 10.1021/es3048043. [DOI] [PubMed] [Google Scholar]
- Appleman T. D.; Higgins C. P.; Quinones O.; Vanderford B. J.; Kolstad C.; Zeigler-Holady J. C.; Dickenson E. R. Treatment of poly-and perfluoroalkyl substances in US full-scale water treatment systems. Water Res. 2014, 51, 246–255. 10.1016/j.watres.2013.10.067. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Ulrich B. A.; Chen B.; Higgins C. P. Sorption of Poly-and Perfluoroalkyl Substances (PFASs) Relevant to Aqueous Film-Forming Foam (AFFF)-Impacted Groundwater by Biochars and Activated Carbon. Environ. Sci. Technol. 2017, 51 (11), 6342–6351. 10.1021/acs.est.7b00970. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.