

Abstract
The synthesis of ethyne-linked porphyrin nanorings has been achieved by template-directed Sonogashira coupling. The cyclic hexamer and octamer are predicted by density functional theory to adopt low symmetry conformations, due to dihedral twists between neighboring porphyrin units, but their symmetries are effectively D6h and D8h, respectively, in solution by 1H NMR. The fluorescence spectra indicate that the singlet excited states of these nanorings are highly delocalized.
π-Conjugated macrocycles are useful for testing the limits of aromaticity and electronic delocalization,1,2 and they are prototype materials for achieving efficient transport of charge and electronic excitation.3 Even when these macrocycles are not aromatic, their excited states are generally more delocalized than those of the corresponding linear systems, because every site in a symmetric ring is identical whereas the sites in a linear chain are inevitably nonequivalent.4,5 The enhanced electronic delocalization in ring structures explains why many natural light-harvesting systems consist of cyclic arrays of chlorophyll units.6 Covalent cyclic oligomers also tend to have less conformational disorder than linear chains, particularly when the π-system is strained, and they can reveal how strain and curvature influence π-conjugation.7 The curved concave interiors of belt-shaped macrocycles result in high affinities for suitable guests, as illustrated by the binding of C60 inside [6]cyclo-para-phenyleneacetylene8 and porphyrin nanocages,9 and by the coordination of oligo-pyridine templates inside zinc-porphyrin nanorings.10 Alkyne bridges are often used to avoid steric clashes in the design of π-conjugated chains, as illustrated for the case of porphyrin oligomers in Figure 1a: a direct meso–meso bond causes major steric interactions, forcing the porphyrin units into perpendicular planes and preventing π-conjugation;11 the insertion of a C2 bridge alleviates this clash, although steric interactions still destabilize the coplanar conformation and the dihedral angle is about 35° allowing substantial π-overlap;12,13 the C4 bridge results in a preference for the coplanar conformation.14 Butadiyne-linked porphyrin oligomers have provided many insights into the optical, electronic and supramolecular behavior of cyclic arrays.2,4,5,10,15,16 Here we report an investigation of π-conjugated nanorings consisting of porphyrins linked by single acetylene (C2) bridges: the cyclic hexamer c-P6 and the cyclic octamer c-P8 (Figure 1, Scheme 1).
Figure 1.
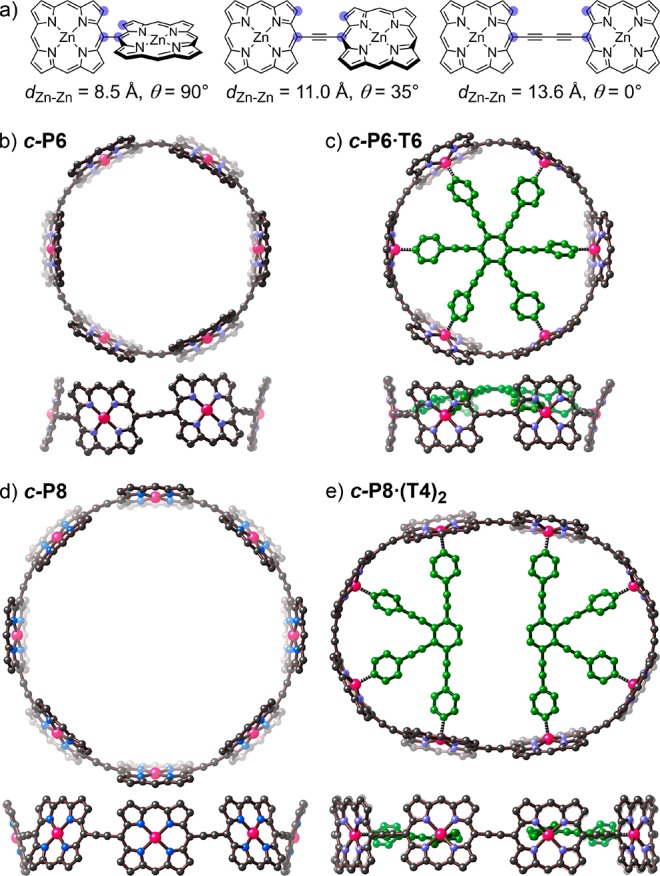
(a) Types of linear porphyrin–porphyrin connection. Zn–Zn distances and dihedral angles θ (measured from the carbon atoms indicated by blue circles from density functional theory (DFT)-optimized geometries). (b–e) Calculated geometries of c-P6, c-P6·T6, c-P8 and c-P8·(T4)2. Two orthogonal views are shown for each structure. Solubilizing aryl groups and hydrogen atoms are omitted for clarity. (B3LYP/6-31G*; see SI for details.).
Scheme 1. Synthesis of c-P8 and c-P6.

Reaction conditions: (a) 2, Pd2dba3, PPh3, CuI, PhMe/i-Pr2NH (2:1), 50 °C, 15 h, 13%; (b) TBAF, CH2Cl2, 25 °C, 10 min, 79%; (c) T4, Pd2dba3, PPh3, CuI, PhMe/i-Pr2NH (10:1), 40 °C, 2 d, 32%; (d) SEC pyridine/PhMe (1:1), 99%; (e) 1, Pd(PPh3)4, PPh3, CuI, PhMe/i-Pr2NH (2:1), 50 °C, 15 h, 30%; (f) TBAF, CH2Cl2, 25 °C, 10 min, 95%; (g) T6, Pd(PPh3)4, PPh3, CuI, PhMe/i-Pr2NH (2:1), 40 °C, 16 h, 15%; (h) SEC (pyridine/PhMe 1:1), 99%. Ar: 1,3-bis(trihexylsilyl)phenyl. Syntheses of 1, 2, 3, T4 and T6 are outlined in the SI.
Linear ethyne-linked porphyrin oligomers exhibit unique properties, including intense absorption, fluorescence and electroluminescence at near-IR wavelengths,17−19 and quasi-ohmic charge transport.20,21 No curved ethyne-linked porphyrin arrays have been reported previously. Their synthesis is more challenging than that of the C4-linked analogues because they are more strained, due to the shorter acetylenic link and the dihedral twist, discussed above. Additionally, Sonogashira cross-coupling requires asymmetric building blocks and is generally accompanied by competing Glaser homocoupling. Here we show that oligo-pyridine templates can be used to bend C2-linked zinc porphyrin oligomers into cyclic conformations and to direct the synthesis of C2-linked nanorings.
The key to the synthesis of a porphyrin nanoring lies in the design of a suitable template,22 and this requires knowledge of the geometry of the target ring. The DFT-optimized structure of c-P6 (Figure 1b) has D3d symmetry because neighboring porphyrins adopt staggered conformations due to the repulsion between β-pyrrole hydrogen atoms (dihedral angle: 25°). The larger c-P8 ring has a similar staggered conformation (dihedral angle: 30°) resulting in D4d symmetry. The decrease in dihedral angles on moving from a linear connection (35°) to the 8-ring (30°) to the 6-ring (25°) shows the balance between ring strain and dihedral twist, and it suggests that macrocyclization will enhance π-conjugation. By considering homodesmotic reactions,23 the strain energies of c-P6 and c-P8 were estimated to be 132 and 100 kJ/mol, respectively (see SI). These strain energies are comparable to that in cyclopropane (115 kJ/mol), but they are spread over many bonds, so that large changes in reactivity are not expected.
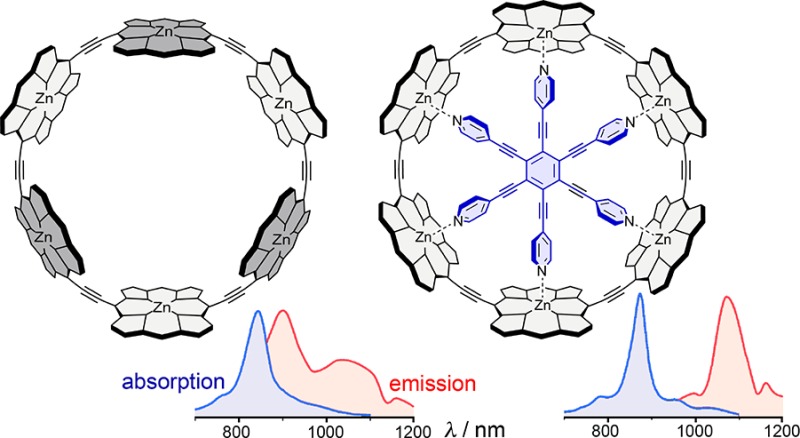
The Zn–Zn diameter of c-P6 (20.6 Å) implies that an ideal template for this nanoring would have a N–N diameter of 15.5 Å. This value is calculated from the zinc to pyridine nitrogen bond length in simple zinc porphyrin complexes (dZn-Py = 2.16 ± 0.03 Å) and the distance of the 5-coordinate zinc atom from the plane of the porphyrin (dZn-Porph = 0.37 ± 0.06 Å).24 The template T6 (Scheme 1) has a calculated N–N diameter of 16.6 Å, making it too large for the cavity of c-P6 and causing it to adopt a domed conformation in c-P6·T6 (Figure 1c). The mean porphyrin-porphyrin dihedral angle in c-P6·T6 (18°) is smaller than in the unbound c-P6 nanoring (25°) showing that both components distort when they form the 1:1 complex.
The cyclic octamer c-P8 has a calculated Zn–Zn diameter of 27.8 Å. We decided to test the synthesis of this nanoring by using two equivalents of a tailor-made four-legged T4 template to direct the formation of the c-P8·(T4)2 complex (Figure 1e and Scheme 1). This bimolecular templating strategy25 has two advantages over a classical 1:1 approach: (a) it avoids the need to synthesize a complementary eight-legged template, and (b) it is more tolerant of templates that are not exactly the right size, as illustrated by the observation that T4 is not domed in c-P8·(T4)2.
Initially, we attempted the synthesis of the 8-porphyrin nanoring, c-P8, because its lower strain made it look more accessible. The first hurdle was to prepare the linear octamer l-P8 (Scheme 1). Therien and co-workers have reported the synthesis of C2-linked linear porphyrin oligomers of this type up to the heptamer by palladium-catalyzed Sonogashira coupling, with or without a copper(I) cocatalyst.18,20 We found that standard conditions using Pd(PPh3)4 and CuI, in tandem with NBS bromination, gave efficient access to trimer 2 from monomer 1. Elongation of trimer 2 by two porphyrin units and subsequent desymmetrization by statistical deprotection yielded asymmetric pentamer 3. Statistical coupling of 3 to trimer 1 followed by deprotection gave the octamer l-P8 with the desired bromine/acetylene terminal groups. Sonogashira coupling of the linear octamer in the presence of template T4 afforded c-P8·(T4)2 in 32% yield, and addition of pyridine liberated the free nanoring c-P8.
Encouraged by the successful synthesis of c-P8, we attempted to synthesize the symmetric 6-porphyrin ring c-P6. Binding of the linear hexamer l-P6 to its six-legged template T6 and subsequent cross-coupling gave c-P6·T6 in 15% yield. Size exclusion chromatography in the presence of pyridine yielded the template-free ring c-P6. Both c-P6 and c-P8 are soluble in common organic solvents and show good stability under ambient conditions.
The 1H NMR spectra of c-P6·T6 and c-P6 reveal that these molecules have D6h symmetry on the NMR time scale (see SI for full assignments from 2D techniques). The signals for the aryl protons ortho to the porphyrin in c-P6·T6 are split due to the different environments inside and outside the nanoring. The same splitting is observed for the template-free c-P6; however, the resonances are broader indicating conformational exchange. As expected, the template signals in c-P6·T6 are substantially shielded by the ring current of the porphyrin subunit, for example the α-pyridine protons resonate at 2.56 ppm. The larger nanoring c-P8 gives a similar 1H NMR spectrum to c-P6, with two sharp β-pyrrole proton resonances indicating high symmetry (D8h). Complexation of two T4 templates leads to an elliptical distortion and all the resonances are split as expected for the effective D2h symmetry of c-P8·(T4)2.
The complexes c-P6·T6 and c-P8·(T4)2 are too stable for their formation constants to be determined from direct UV–vis-NIR titrations, but their stabilities can be determined by competition with pyridine.10 Previously, we reported that strongly coordinating ligands, such as quinuclidine, are required to displace a hexadentate template from a butadiyne-linked 6-porphyrin ring;10,24 however, the complexes presented here are dissociated by addition of pyridine. These titrations both give isosbestic spectra and denaturation of each complex was assumed to follow an all-or-nothing two-state equilibrium between the template-porphyrin complex and the pyridine-porphyrin complex.10 Independent determination of the binding constant for pyridine to a single zinc porphyrin unit then enabled calculation of the formation constant (all values in toluene at 289 K; see SI). The formation constant of c-P6·T6 is Kf ≈ 5 × 1028m–1 (logKf = 28.6 ± 0.4) which corresponds to an average statistically corrected effective molarity of EM = 9 ± 2 m. The 1:2 complex c-P8·(T4)2 has a formation constant of Kf ≈ 9 × 1040m–2 (logKf = 41.0 ± 0.2) and an average effective molarity of EM = 36 ± 5 m. Although these are high association constants, they are dramatically lower than those for the corresponding complexes of butadiyne-linked porphyrin nanorings, reflecting the poor geometrical complementarity of these templates and the dihedral twist required for template binding, as discussed above.
The absorption and fluorescence spectra, and fluorescence quantum yields, of the C2-linked linear and cyclic hexamers are compared in Figure 2 (and Figure S25). All the compounds give intense B and Q absorptions bands at around 500 and 900 nm, respectively, as reported previously for linear oligomers of this type.12,17,18 The sharper and more structured absorption spectra of the template complexes c-P6·T6 and c-P8·(T4)2 reflect their well-defined conformations.
Figure 2.

Steady-state absorption (continuous lines) and fluorescence (dashed) spectra at 295 K of l-P6 in toluene/1% pyridine, c-P6·T6 in toluene and c-P6 in toluene/1% pyridine. Quantum yields are given in %. The apparent dip in the fluorescence spectra at 1140 nm is due to absorption by the solvent.
In general, the transition to the first excited state of a symmetrical π-conjugated macrocycle is dipole-forbidden, although vibrations that break the symmetry cause a partial transfer of oscillator strength from higher-lying allowed transitions, in a process known as Herzberg–Teller coupling.5,24,26,27 Two manifestations of this effect are (a) low fluorescence quantum yield, because the slow radiative S1–S0 transition is unable to compete with nonradiative decay, and (b) red-shifted fluorescence, because the vibronic coupling shifts the emission to lower energies. Comparison of the fluorescence spectra of l-P6 and c-P6·T6 illustrates both these points: the fluorescence quantum yield is 1000-fold smaller in the ring compound and the emission band is red-shifted by 164 nm (l-P6: 909 nm; c-P6·T6: 1073 nm). This demonstrates that the first excited state is fully delocalized around the nanoring in c-P6·T6. Removing the template allows more conformational disorder, reducing the symmetry, and increasing the fluorescence quantum yield while shifting the fluorescence to higher energy in c-P6. The lower symmetry and greater disorder in c-P8 and c-P8·(T4)2 result in higher fluorescence quantum yields.
In summary, we have synthesized the first examples of ethynylene-linked porphyrin nanorings. Binding of linear porphyrin oligomers to complementary templates overcomes the energy penalty required to create these macrocycles. DFT calculations indicate that the nanorings c-P6 and c-P8 adopt low symmetry conformations (D3d and D4d, respectively) due to dihedral twists between neighboring porphyrin units; however, the symmetries are effectively D6h and D8h, respectively, at room temperature in solution on the 1H NMR time scale. The fluorescence spectrum of the c-P6·T6 complex shows all the signs of a dipole-forbidden S1–S0 transition, confirming that its singlet excited state is delocalized over the whole π-system. Many of the features of these C2-linked oligomers are similar to those of the C4-linked analogues, but the C2-linked nanorings have more red-shifted absorption and emission bands, indicating that there is stronger electronic coupling between the porphyrin units, despite the steric clash between the β-pyrrole hydrogen atoms.
Acknowledgments
We thank the Swiss National Science Foundation (P2BSP2_168919), ERC (320969), EPSRC, the Wellcome Trust and the EC H2020 SYNCHRONICS and MULTI-APP (643238 and 642793) for funding, the EPSRC UK National Mass Spectrometry Facility at Swansea University and Renée Haver for MALDI spectra and the University of Oxford Advanced Research Computing Service (ARC) for support. M.J. thanks Oxford University for a Scatcherd European Scholarship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10710.
The authors declare no competing financial interest.
Supplementary Material
References
- a Iyoda M.; Yamakawa J.; Rahman M. J. Angew. Chem., Int. Ed. 2011, 50, 10522. 10.1002/anie.201006198. [DOI] [PubMed] [Google Scholar]; b Spitler E. L.; Johnson C. A. II; Haley M. M. Chem. Rev. 2006, 106, 5344. 10.1021/cr050541c. [DOI] [PubMed] [Google Scholar]; c Grave C.; Schlüter A. D. Eur. J. Org. Chem. 2002, 2002, 3075.. [DOI] [Google Scholar]; d Kennedy R. D.; Lloyd D.; McNab H. J. Chem. Soc., Perkin Trans. 1 2002, 1601. 10.1039/b109576p. [DOI] [Google Scholar]
- Peeks M. D.; Claridge T. D. W.; Anderson H. L. Nature 2017, 541, 200. 10.1038/nature20798. [DOI] [PubMed] [Google Scholar]
- Ball M.; Zhong Y.; Fowler B.; Zhang B.; Li P.; Etkin G.; Paley D. W.; Decatur J.; Dalsania A. K.; Li H.; Xiao S.; Ng F.; Steigerwald M. L.; Nuckolls C. J. Am. Chem. Soc. 2016, 138, 12861. 10.1021/jacs.6b05474. [DOI] [PubMed] [Google Scholar]
- Tait C. E.; Neuhaus P.; Peeks M. D.; Anderson H. L.; Timmel C. R. J. Am. Chem. Soc. 2015, 137, 8284. 10.1021/jacs.5b04511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson P.; Kondratuk D. V.; Menelaou C.; Gong J. Q.; Anderson H. L.; Herz L. M. J. Phys. Chem. Lett. 2014, 5, 4356. 10.1021/jz5022153. [DOI] [PubMed] [Google Scholar]
- Cogdell R. J.; Gall A.; Köhler J. Q. Rev. Biophys. 2006, 39, 227. 10.1017/S0033583506004434. [DOI] [PubMed] [Google Scholar]
- Segawa Y.; Yagi A.; Matsui K.; Itami K. Angew. Chem., Int. Ed. 2016, 55, 5136. 10.1002/anie.201508384. [DOI] [PubMed] [Google Scholar]
- Kawase T.; Tanaka K.; Fujiwara N.; Darabi H. R.; Oda M. Angew. Chem., Int. Ed. 2003, 42, 1624. 10.1002/anie.200250728. [DOI] [PubMed] [Google Scholar]
- a Song J.; Aratani N.; Shinokubo H.; Osuka A. J. Am. Chem. Soc. 2010, 132, 16356. 10.1021/ja107814s. [DOI] [PubMed] [Google Scholar]; b Mulholland A. R.; Woodward C. P.; Langford S. J. Chem. Commun. 2011, 47, 1494. 10.1039/C0CC04474A. [DOI] [PubMed] [Google Scholar]; c Zhu B.; Chen H.; Lin W.; Ye Y.; Wu J.; Li S. J. Am. Chem. Soc. 2014, 136, 15126. 10.1021/ja507531b. [DOI] [PubMed] [Google Scholar]
- Hogben H. J.; Sprafke J. K.; Hoffmann M.; Pawlicki M.; Anderson H. L. J. Am. Chem. Soc. 2011, 133, 20962. 10.1021/ja209254r. [DOI] [PubMed] [Google Scholar]
- a Khoury R. G.; Jaquinod L.; Smith K. M. Chem. Commun. 1997, 1057. 10.1039/a701218g. [DOI] [Google Scholar]; b Yoshida N.; Ishizuka T.; Osuka A.; Jeong D. H.; Cho H. S.; Kim D.; Matsuzaki Y.; Nogami A.; Tanaka K. Chem. - Eur. J. 2003, 9, 58. 10.1002/chem.200390004. [DOI] [PubMed] [Google Scholar]
- Lin V. S.-Y.; Therien M. J. Chem. - Eur. J. 1995, 1, 645. 10.1002/chem.19950010913. [DOI] [Google Scholar]
- Rintoul L.; Harper S. R.; Arnold D. P. Phys. Chem. Chem. Phys. 2013, 15, 18951. 10.1039/c3cp53396d. [DOI] [PubMed] [Google Scholar]
- Peeks M. D.; Neuhaus P.; Anderson H. L. Phys. Chem. Chem. Phys. 2016, 18, 5264. 10.1039/C5CP06167A. [DOI] [PubMed] [Google Scholar]
- Peeks M. D.; Tait C. E.; Neuhaus P.; Fischer G. M.; Hoffmann M.; Haver R.; Cnossen A.; Harmer J. R.; Timmel C. R.; Anderson H. L. J. Am. Chem. Soc. 2017, 139, 10461. 10.1021/jacs.7b05386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-P.; Shen Y.-F.; Zhu B.-Y.; Wu J.; Li S. Chem. Commun. 2016, 52, 10205. 10.1039/C6CC04556A. [DOI] [PubMed] [Google Scholar]
- Lin V. S.-Y.; DiMagno S. G.; Therien M. J. Science 1994, 264, 1105. 10.1126/science.8178169. [DOI] [PubMed] [Google Scholar]
- Duncan T. V.; Susumu K.; Sinks L. E.; Therien M. J. J. Am. Chem. Soc. 2006, 128, 9000. 10.1021/ja061897o. [DOI] [PubMed] [Google Scholar]
- Ostrowski J. C.; Susumu K.; Robinson M. R.; Therien M. J.; Bazan G. C. Adv. Mater. 2003, 15, 1296. 10.1002/adma.200305228. [DOI] [Google Scholar]
- Susumu K.; Frail P. R.; Angiolillo P. J.; Therien M. J. J. Am. Chem. Soc. 2006, 128, 8380. 10.1021/ja0614823. [DOI] [PubMed] [Google Scholar]
- Li Z.; Park T.-H.; Rawson J.; Therien M. J.; Borguet E. Nano Lett. 2012, 12, 2722. 10.1021/nl2043216. [DOI] [PubMed] [Google Scholar]
- Anderson S.; Anderson H. L.; Sanders J. K. M. Acc. Chem. Res. 1993, 26, 469. 10.1021/ar00033a003. [DOI] [Google Scholar]
- Liu P.; Hisamune Y.; Peeks M. D.; Odell B.; Gong J. Q.; Herz L. M.; Anderson H. L. Angew. Chem., Int. Ed. 2016, 55, 8358. 10.1002/anie.201602909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprafke J. K.; Kondratuk D. V.; Wykes M.; Thompson A. L.; Hoffmann M.; Drevinskas R.; Chen W.-H.; Yong C. K.; Kärnbratt J.; Bullock J. E.; Malfois M.; Wasielewski M. R.; Albinsson B.; Herz L. M.; Zigmantas D.; Beljonne D.; Anderson H. L. J. Am. Chem. Soc. 2011, 133, 17262. 10.1021/ja2045919. [DOI] [PubMed] [Google Scholar]
- Liu S.; Kondratuk D. V.; Rousseaux S. A. L.; Gil-Ramírez G.; O’Sullivan M. C.; Cremers J.; Claridge T. D. W.; Anderson H. L. Angew. Chem., Int. Ed. 2015, 54, 5355. 10.1002/anie.201412293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kärnbratt J.; Chang M.-H.; Herz L. M.; Albinsson B.; Anderson H. L. Angew. Chem., Int. Ed. 2008, 47, 4993. 10.1002/anie.200801188. [DOI] [PubMed] [Google Scholar]
- Yong C. K.; Parkinson P.; Kondratuk D. V.; Chen W.-H.; Stannard A.; Summerfield A.; Sprafke J. K.; O’Sullivan M. C.; Beton P. H.; Anderson H. L.; Herz L. M. Chem. Sci. 2015, 6, 181. 10.1039/C4SC02424A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.