

Abstract
As the length of molecular dynamics (MD) trajectories grows with increasing computational power, so does the importance of clustering methods for partitioning trajectories into conformational bins. Of the methods available, the vast majority require users to either have some a priori knowledge about the system to be clustered or to tune clustering parameters through trial and error. Here we present non-parametric uses of two modern clustering techniques suitable for first-pass investigation of an MD trajectory. Being non-parametric, these methods require neither prior knowledge nor parameter tuning. The first method, HDBSCAN, is fast—relative to other popular clustering methods—and is able to group unstructured or intrinsically disordered systems (such as intrinsically disordered proteins, or IDPs) into bins that represent global conformational shifts. HDBSCAN is also useful for determining the overall stability of a system—as it tends to group stable systems into one or two bins—and identifying transition events between metastable states. The second method, iMWK-Means, with explicit rescaling followed by K-Means, while slower than HDBSCAN, performs well with stable, structured systems such as folded proteins and is able to identify higher resolution details such as changes in relative position of secondary structural elements. Used in conjunction, these clustering methods allow a user to discern quickly and without prior knowledge the stability of a simulated system and identify both local and global conformational changes.
Graphical Abstract
1. INTRODUCTION
Molecular dynamics (MD) is an increasingly powerful1,2 and prolific3–7 tool generating enormous data sets over ever longer time scales8–10 and larger numbers of atoms.7,11 Especially useful for predicting dynamics and structures currently inaccessible to experimental techniques for systems on desired time scales too large for ab initio methods, MD simulations simplify interactions among atoms to classical force laws and propagate equations of motion forward in time with numerical integration techniques.12 Each integration step—typically covering 1–4 fs—generates a set of coordinates for every atom in the system. Even simulations of short (nanosecond) time scales for systems with a few hundred atoms result in data sets too large for direct analysis by a human.
A typical way of dealing with such large data sets is to bin data into groups based on some similarity metric.13–16 For example, clustering methods take a set of samples (frames in the MD context) each with some number of features (atomic coordinates, dihedral angles, etc.) and assigns a label (cluster number) to each sample such that samples with the same label are similar while samples with different labels are dissimilar. This vague notion of similar is usually determined, directly or indirectly, by parameters specified by the user. Such a parameter might be the desired number of clusters (e.g., K-Means17–19), the diameter of a cluster (e.g., quality threshold20) or how sharply peaked the Gaussian distributions within the data are believed to be (e.g., mean shift21). Many such clustering methods exist,22,23 and some have been adapted to or specially created for MD.24–34 However, parameter selection for these algorithms requires some a priori knowledge or trial and error from the user—a requirement that can be prohibitive when initially exploring a trajectory. Shao et al. in 2007 offered a critical review of clustering algorithms applied to MD, discussing the bias introduced by parameter selection.34
With this sort of first-pass exploration in mind, we demonstrate the usefulness of two recently developed clustering techniques for analyzing MD trajectories. The first method, Hierarchical Density-Based Spatial Clustering of Applications with Noise (HDBSCAN),35,36 has two parameters. The first parameter, minimum cluster size, is intuitively set by answering the question, “What’s the smallest cluster I would care about?” If the user has no answer to this question, then there is the obvious choice of a minimum cluster size of 1. The second parameter, minimum samples, is less intuitive and defines what a neighborhood is in the sense of K-Nearest Neighbors.37 The algorithm’s authors recommend setting the minimum samples value equal to the minimum cluster size value,35 such that the minimum cluster size is also the minimum neighborhood size in the algorithm’s initial density estimate, reducing this algorithm to a single parameter. Therefore, the sensible default of 1 for minimum cluster size makes this method an excellent exploratory, first-pass clustering algorithm. The second method, Intelligent Minkowski-Weighted K-Means (iMWK-means) with explicit rescaling followed by K-Means38 (herein called “Amorim–Hennig” after the two authors proposing it), uses a parameter that conceptually reduces to a selection of distance metric.38,39 For MD trajectories we select the parameter analogous to a Euclidean distance metric (see Methods). Having no other parameters beyond the distance metric, this method is likewise excellent for first-pass, exploratory clustering.
As the name implies, HDBSCAN (abbreviated “HDB” in figure captions) is a density-based clustering algorithm that yields a hierarchical clustering. However, rather than cutting the resulting dendrogram at one place, it selects clusters from multiple levels of the tree. Like other density-based clustering algorithms,40 HDBSCAN searches for regions of feature space with a high density of points separated by regions with low density. Rather than having the user specify a quantitative meaning for high density, the HDBSCAN algorithm instead scans over various definitions of high density and keeps clusters from various values.35,36 That is, HDBSCAN can find clusters of varying densities and arbitrary shape, making it a flexible algorithm appropriate for initial exploration of a trajectory. Additionally, HDBSCAN can label a sample as “noise” (“−1” in all figures herein) if it does not fall into any cluster (or if it falls into a cluster smaller than the minimum cluster size). In the present study, we apply HDBSCAN exclusively to MD trajectories. However, it has been tested in a variety of contexts of which we cite a few examples.36,41–45 For example, HDBSCAN has been used to improve querying databases for location-based services41 and anomaly detection in signal processing.43 (For a more complete conceptual explanation and a toy example of HDBSCAN, see Methods.)
Amorim–Hennig (abbreviated “A-H” in figure captions) is a variant of K-Means that does not require specification of the number of clusters. Rather, it uses a clustering method iK-Means,39,46 which can be used to overestimate the number of clusters in a data set,46 to set a maximum number of clusters and then applies a rescaled variant38 of iMWK-Means47 which selects a number of clusters—less than or equal to the maximum number—by optimizing some cluster validity index48,49 (i.e., scoring metric). In our examples below, we use the silhouette index, which quantifies how similar a given member of a given cluster is to every other member of that cluster.50 In 2015, de Amorim and Hennig discussed various cluster validity indices that can be used with this clustering algorithm.38 For a more complete comparison of cluster validity indices, see the extensive comparative study by Arbelaitz et al.48 and the R package NbClust.51 Amorim–Hennig clustering is iterative, using the result of clustering round i to assign feature weights that are used in round i+1. The algorithm iterates until no change occurs between rounds. After the optimal feature weights are determined, the weighted data are then passed to the standard, likewise iterative, K-Means algorithm.38 While K-Means is biased toward spherical Gaussian clusters and the feature rescaling is similarly biased when using the euclidean distance metric as we do here, the spherical clusters in the final rescaled data are not necessarily spherical in the original data. Therefore, like HDBSCAN this method is capable of returning clusters of varying shape and density. Additionally, Amorim–Hennig addresses noise in data through its feature rescaling, i.e., a noise point would receive a low weight, whereas HDBSCAN labels the noise point as such. Like HDBSCAN, Amorim–Hennig has been tested in a variety of contexts.38,52–56 For example, Amorim–Hennig has been applied to data mining for tumor subtype discovery52 and investigating the topology of neural systems.53 (For a more complete conceptual explanation of Amorim–Hennig, see Methods.)
In the present study, we test both clustering methods on all-atom MD trajectories of both proteins and nucleic acids. Our first protein example is the MutSα complex57—a DNA mismatch repair protein complex—in the presence of cisplatinated cis-diamminedichloroplatinum(II)–DNA. This complex is of particular interest due to the chemotherapeutic nature of DNA treated with cisplatin.58–61 This complex has been the subject of MD simulations with clustering analysis to identify rare-conformations that might be involved in apoptotic pathways in order to identify possible new lead compounds for drug discovery.62–67 Next, we examine the NF-κB Essential Modulator (NEMO) zinc finger domain. It is a 28-residue zinc-binding protein with a 3CYS1HIS active site and is a known ubiquitin binder.68 NEMO is a diverse signaling protein that contributes to cellular regulation processes including apoptosis, oncogenesis, and inflammatory responses.68–70 We present clustering results of two biological configurations of NEMO: one with protonated active-site cysteines (CYS) and zinc absent, the other with deprotonated cysteines and a tetragonally coordinated zinc in the active site (CYNZN). Next, we present a folding simulation of the fast-folding villin headpiece protein.71 In the course of exploring folding processes and the formation of secondary structures, the clustering methods presented here offer a way to define the intermediate states and reveal portions of the folding pathway. Such simplification of MD trajectories may speed drug and chemotherapeutic development by identifying structural ensembles.
We conclude our protein examples with unliganded apothrombin in a solution environment with sodium ions. Thrombin has been shown to induce tumor growth, metastasis, angiogenesis,72 and even tumor invasion via interactions on cell surfaces.73 Additionally, thrombin is a central protease with allosteric regulation in the coagulation cascade.74–77 Its different activities as a procoagulant and anticoagulant are thought to highly correlate to thrombin’s conformational states.78–81 In particular, in the presence of sodium, thrombin can adopt a pro-coagulant (the so-called “fast” form) conformational state, which is structurally distinct from the anti-coagulant (the so-called “slow” form) one when sodium is absent.82–90 However, as the protein is not rigid, our clustering results help illustrate the conformational ensembles of the thrombin fast form.
We also include two nucleic acid examples: First is a therapeutic 10mer of FdUMP (5-fluoro-2′-deoxyuridine-5′-O-monophosphate, also called F10) in both stabilizing and destabilizing solvent conditions. The fluoridated oligonucleotide F10 is cytotoxic91–94 and is demonstrably more efficacious as a therapeutic and better tolerated in vivo95–98 than the widely used 5-fluorouracil (5-FU).99,100 Second, we present clustering on a trajectory of thrombin-binding aptamer, a single-stranded DNA 15mer (herein “15-TBA”).101 With a guanine-enriched sequence, 15-TBA can fold into a G-quadruplex structure and recognize fibrinogen’s binding site on thrombin.74,101
2. RESULTS AND DISCUSSION
2.1. Proteins
2.1.1. MutSα
Clustering on protein α carbon atom coordinates with HDBSCAN on two concatenated trajectories (totaling 500 ns) of MutSα in the presence of cisplatinated DNA assigned each concatenated trajectory to its own cluster (Supporting Information, Figure S1). This clustering result indicates that in both environments MutSα exhibits a stable global structure, as is expected of a folded, functional protein. As will become apparent with additional examples, this assignment to one or two clusters by HDBSCAN is typical for stable systems such as folded proteins—e.g., this MSH26 heterodimer of MutSα.
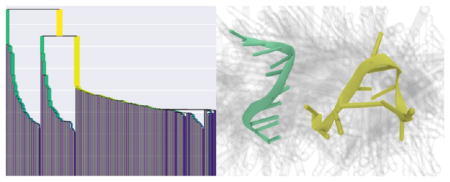
On the same concatenated trajectories of MutSα, Amorim–Hennig splits the first of the two concatenated trajectories into two bins and assigns the second trajectory to the same bin as the initial structure of both simulations (Figure 1). Visualizing these clusters (see Methods), we see that the differentiation between clusters 1 and 2 (Figure 1c,d) is the loss of secondary structure as the β sheet in the bottom right of the figures is lost, along with the α helix in the lower left of each panel. Though the nucleic acid’s atoms were not involved in clustering, we notice that the loss of β sheet is accompanied by a shift of the DNA toward the center of the protein.
Figure 1.

By plotting the MutSα cluster time series (a), we see Amorim–Hennig splits the first of the two concatenated trajectories into two bins and assigns the second trajectory to the same bin as the initial structure of both simulations. Comparing clusters 0 (b) and 1 (c), we see the overall protein close in on itself. From cluster 0 (b) to 2 (d), we see a β sheet in the upper left form and a loop near the bottom right move away from the larger structure. We see another β sheet near the bottom right of the protein that appears in cluster 1 (c) but not in 0 (b) or 2 (d). The representative (solid) structure in each panel is the frame closest to the average structure by RMSD. The protein is colored by secondary structure in VMD’s102 NewCartoon drawing method: α helices are magenta, β sheets are yellow, π helices are dark blue, and loops are cyan. Nucleic acid is colored all light blue. Shadows are 50 evenly sampled frames from the cluster.
In this first example, we see the usefulness of HDBSCAN to quickly indicate the stability of a system, which will become clearer by contrast with upcoming examples of highly unstable, disordered systems (see Figures 3, 6a, and 7, below). Additionally, we see that while HDBSCAN detects large-scale, global shifts in protein structure (Figures 3 and 7), Amorim–Hennig detects local, small-scale shifts, such as changes in relative position of nearby secondary structures (see Figures 1, 2, and 5). In this example (Figure 1) and all that follow, we visualize structures from the results of whichever clustering method we found to be most meaningful and include structures from the other technique in Supporting Information.
Figure 3.

By plotting the zinc-ubound NEMO cluster time series (a), we see HDBSCAN yielded 976 clusters with 42% of the trajectory frames labeled as noise (−1), indicating a disordered system. Visualization of the top four non-noise clusters by population shows that HDBSCAN has captured intermediate stages of secondary structure formation and destabilization. The protein is colored by secondary structure in VMD’s NewCartoon drawing method.
Figure 6.

HDBSCAN identified stable (a) vs unstable systems (b), and Amorim–Hennig provided finer resolution on structural changes (c–f). Visual comparison of the Amorim–Hennig clusters of the second of four trajectories (respectively beginning at frame 1, 1001, 6001, and 10 001) confirms that the method uncovered distinct conformations. The fluoridated DNA strand is colored by residue number drawn with VMD’s NewCartoon method.
Figure 7.

HDBSCAN clustering output suggests that the unbound thrombin aptamer is generally unstable but has some long-lived states. On this system, Amorim–Hennig binned the system into what appear to be compactness-based bins (Supporting Information, Figure S4). Thrombin aptamer DNA strand is colored by residue number drawn with VMD’s NewCartoon method. Visualized clusters are from HDBSCAN.
Figure 2.

Clustering on α carbon coordinates of a zinc-bound protein of human NEMO zinc finger with HDBSCAN yielded (a) 22 clusters, with only 2.1% of the 98 304 trajectory frames labeled as noise, (c) 75% of them placed into the most populated cluster, and (d) 11% in the second highest population cluster. Amorim–Hennig clustering yields (b) two bins with a distinct structural difference. This clustering reveals that the loop between the α helix and β sheet straightens and elongates (panels e and f).
Figure 5.

Amorim–Hennig (a) identified stable states of the system across the five concatenated trajectories. These clusters (c–f) are differentiated primarily by modes of the γ loop, shown at the bottom of each panel: the so-called “60s loop” shown in dark blue at the right of each panel and exosite II shown at the top of each panel. HDBSCAN (b) identified transient states among these stable states (Supporting Information, Figure S4).
2.1.2. NEMO Zinc Finger
Clustering on α carbon coordinates of a zinc-bound protein of human NEMO zinc finger with HDBSCAN yielded 22 clusters, with only 2.1% of the 98 304 trajectory frames labeled as noise (Figure 2a) and 75% of them placed into the most populated cluster (Figure 2c). Here we see HDBSCAN’s utility in identifying a stable system. Additionally, HDBSCAN has binned the trajectory frames into a manageable number of distinct conformations. Comparing the top two clusters by population (Figure 2c,d) reveals that HDBSCAN detected the N-terminus destabilization in the form of partial destabilization of the α helix on the right of each panel. In cluster 1 (Figure 2c) we see more turns in the α helix than in cluster 11 (Figure 2d).
By comparison, on the non-zinc-bound structure of NEMO zinc finger, HDBSCAN yielded 976 clusters, with 42.03% of the 125 000 trajectory frames labeled as noise (Figure 3a). This high-number, high-noise clustering result suggests that the zinc-less NEMO structure is intrinsically disordered, as there were no stable structural bin into which many frames could be placed. The highest population non-noise cluster contains just 2.35% of the trajectory with the next highest population cluster containing 1.25%. Visualization of the top four non-noise clusters by population (Figure 3b–f) shows that HDBSCAN has captured intermediate stages of secondary structure formation and destabilization. While 976 clusters is still excessive for analysis by a person, analyzing the highest population HDBSCAN clusters provides a quick overview of what types of structures form in simulations of this disordered, unstable protein. We again see HDBSCAN’s utility in identifying the stability of a system, and here we see HDBSCAN’s ability to identify distinct structural ensembles within an unstable system.
On the zinc-bound NEMO structure, Amorim–Hennig clustering yields two bins with a distinct structural difference. The loop between the α helix and β sheet straightens and elongates (top of Figure 2e,f). Using the shadows—representing the width of the underlying distribution—to judge uncertainty in the clusters, we see that frames within cluster 0 (Figure 2e) generally exhibit this loop straightening. Amorim–Hennig has uncovered a distinct shift in secondary structure. Here we see the utility of Amorim–Hennig to identify finer details (compared to HDBSCAN) within a relatively stable system.
On the non-zinc-bound NEMO structure, Amorim–Hennig clustering once again yields two bins. However, visualizing these two clusters (Supporting Information, Figure S2) reveals that the frames within each cluster have little structural similarity. By comparison to its performance on a relatively stable system, zinc-bound NEMO, we see that Amorim–Hennig provides meaningful clusters for a stable system but fails to do so for an unstable one. Additionally, one might run a second pass of the most populated clusters from HDBSCAN with the Amorim–Hennig algorithm for higher resolution of the conformational fluctuations.
2.1.3. Villin Headpiece
Amorim–Hennig clustering on α carbon atoms of three concatenated 6 μs trajectories of villin headpiece yielded nine clusters (Figure 4a). Calculating the average RGYR of each cluster revealed that the Amorim–Hennig clusters were differentiated primarily by the level of compactness of the clusters. Cluster 5 (Figure 4d) is the most compact with an RGYR of 12.48 Å. Cluster 7 (Figure 4e) is the least compact with an RGYR of 25.17 Å. We also see additional searching for the final folded conformation when comparing clusters 0, 5, and 8 (Figure 4c, d, and f, respectively).
Figure 4.

(a) Amorim–Hennig divided this folding simulation of villin headpiece into nine clusters (b) representing various states of foldedness (c–f). All visualized clusters are from Amorim–Hennig clustering. Cluster 7 (e) is the most compact, while cluster 5 is the least (d). Comparing clusters 5 (d), 7 (e), and 8 (f), we see additional searching for a final, folded conformation. HDBSCAN (b) also detected multiple stable structures, which upon visualization turned out to be stable folding intermediates (Supporting Information, Figure S3).
The HDBSCAN in Figure 4b also recognized several stable structures. Visualizing the top six clusters by population revealed that HDBSCAN had found stable folding intermediates (Supporting Information, Figure S3). In this example, both clustering methods identified distinct conformations. However, on this system Amorim–Hennig was most useful in identifying potential folding pathways, that is, the conformations of various stages of folding as evaluated with RGYR.
2.1.4. Thrombin
Amorim–Hennig clustering on α carbon atoms of five concatenated trajectories (10 000 frames each) of thrombin in the presence of sodium, a known binder to the fast form of thrombin, yielded six clusters—mostly splitting across trajectories (Figure 5a). These clusters primarily represent various fluctuations of several functional sites including the γ loop, 60s loop, and exosite II (Figure 5). The visualization of the additional frames from each cluster as shadows shows that each of these clusters is relatively tight, indicating that this system is highly stable. Additionally, while HDBSCAN finds additional transient states, the HDBSCAN time series (Figure 5b) is dominated by stable structures, differentiated primarily by individual simulations. Visualizations of representative structures of the most populated clusters from HDBSCAN (Supporting Information, Figure S4) show that the structural differences between the dominant clusters mainly occur at the flexible γ loop and the light-chain termini, likely due to the high variation in position from their mobility. This suggests, for the globally stable thrombin, HDBSCAN mainly captures the large scale conformational shifts, while the Amorim–Hennig clustering is able to pick out higher resolution details.
2.2. Nucleic Acids
2.2.1. F10
Clustering on heavy atom coordinates of F10 in 150 mM NaCl with HDBSCAN yields mostly noise (−1) and 126 clusters, the largest of which comprises 1.4% of the trajectory frames (Figure 6a and Supporting Information, Figure S5). This clustering output from HDBSCAN implies that F10 is highly unstable in these solvent conditions, as expected of a single strand of DNA in the presence of monovalent ions.103–107 On this same system, Amorim–Hennig places all frames into two bins with little structural similarity within each bin (Supporting Information, Figure S6). As seen with non-zinc-bound NEMO (Figure 3), we once again find that Amorim–Hennig performs poorly on unstable systems.
Clustering on heavy atom coordinates of F10 in another solvation condition, 150 mM CaCl2, with HDBSCAN yields less than 1% noise (−1) and six clusters (Figure 6b and Supporting Information, Figure S7). Both F10 trajectories are concatenated from four simulations beginning respectively at frames 1, 1001, 6001 and 10 001. For F10 in the presence of calcium, HDBSCAN primarily split up the trajectories, indicating that in each of the concatenated simulation F10 finds a different stable conformation. This high level of stability is expected of a single strand of DNA in the presence of divalent ions.103–107
Since HDBSCAN indicates F10 is stable in 150 mM CaCl2, our examples above suggest that Amorim–Hennig gives finer resolution, showing local, small-scale changes in F10’s structure. Clustering on heavy atom coordinates of F10 in 150 mM CaCl2 with Amorim–Hennig yields five clusters (Figure 6c). While this algorithm splits the first and final two trajectories crisply, suggesting highly stable structures in those trajectories (confirmed by visual inspection, Supporting Information, Figure S3), the second trajectory (frames 1001–6000) is primarily split into three clusters, indicating more structural variation in this simulation relative to the other three. Visualizing these three clusters shows three clearly distinct conformations (Figures 6d–f).
This example elucidates HDBSCAN’s utility in quickly identifying stable and unstable systems (Figure 6a compared to Figure 6b). We also see Amorim–Hennig’s utility in providing details of structural changes within a relatively stable system (Figure 6c).
2.2.2. Thrombin Aptamer
Clustering on heavy atom coordinates of the refolding 15-TBA with HDBSCAN yields 27% noise and 199 clusters (Figure 7a) the largest of which (Figure 7c) comprises 16.90% of the frames. This clustering output from HDBSCAN implies that in the refolding process, the aptamer is mostly unstable with occasional short-lived stable states (Figure 7c–f).
Interestingly, the Amorim–Hennig clustering on the same data sets of three 8.5 μs refolding simulations of the aptamer mainly outputs several long-lived clusters that are distinct from the ones corresponding to each other trajectory. Clustering on heavy-atom coordinates of 15-TBA with Amorim–Hennig yields seven clusters (Figure 7b and Supporting Information, Figure 10). Whereas the HDBSCAN clusters were fairly tight (see shadows representing uncertainty in Figures 7c–f) the Amorim–Hennig clusters are highly varied within each cluster. Here we once again see Amorim–Hennig performing poorly on an unstable system. For such systems, Amorim–Hennig favors a small number of clusters, forming highly varied clusters with little structural similarity. As seen here and in previous examples, HDBSCAN works well for deciding the stability of a system and locating distinct conformations within an unstable system. Amorim–Hennig works well for finding small structural changes within a stable system.
2.3. Summary and Additional Systems
In the above examples, we used HDBSCAN to determine the stability of systems (Figures 2a, 3a, 4b, 5b, 6a, 6b, and 7a). In the case of unstable and highly disordered systems, we used HDBSCAN to pick out distinct conformations despite a molecule’s relative disorder (Figures 3c–f and 7c–f). In the case of stable systems, we used Amorim–Hennig to find the structural details, i.e., local variations in secondary structure (Figures 1b–d, 2c,d, 4c–f, and 5c–f). Additionally, Amorim–Hennig identified distinct conformations for a nucleic acid in stabilizing salt conditions despite the fact the nucleic acid had no clear secondary structure (Figure 6d–f).
Beyond these examples, we ran clustering trials on several more systems in the course of investigating these two algorithms. We have summarized these additional examples, along with the ones presented above in Table 1. Here we present the system time and memory consumed by each clustering run along with a brief written summary of the clustering results. Across these additional examples, the trend of HDBSCAN being useful for (1) determining the stability of a system and (2) picking out distinct conformation ensembles from disordered systems continues. Similarly, Amorim–Hennig continues to be useful for finding fluctuations of various secondary structures within stable, ordered systems. Put simply, HDBSCAN is about the big picture, and Amorim–Hennig is about the details.
Table 1.
Summary of All Protein and Nucleic Acid Systems Investigated with HDBSCAN and Amorim–Hennig Methodsa
| biopolymer | atoms | frames | HBDSCAN
|
Amorim–Hennig
|
||||
|---|---|---|---|---|---|---|---|---|
| memory (kb) | time ([hh]:mm:ss) | result | memory (kb) | time ([hh]:mm:ss) | result | |||
| MutSα in presence of cisplatinated DNA | 1829 | 5 000 | 1049724 | 04:41 | 1 state per trajectory | 16307300 | 33:28 | 3 stable states |
| MutSα in presence of fluoridated DNA | 1829 | 5 000 | 3080308 | 27:40 | 1 state per trajectory | 122362552 | 03:45:46 | 1 state per trajectory |
| MutSα in presence of mismatched DNA | 1829 | 5 000 | 446560 | 03:32 | 1 state per trajectory | 16755112 | 55:12 | 3 stable, 1 transient state |
| NEMO-CYNZN | 28 | 98 304 | 1017248 | 15:46 | 1 stable state, 75% of frames | 7027188 | 34:22 | 4 stable states |
| NEMO-CYS | 28 | 98 304 | 2380156 | 05:02:34 | 42% noise, 2.4% most populated state | 64461880 | 120:00:00 | 2 frequently switching states |
| SufC108–116 | 246 | 1 000 | ** | 00:33 | 30% noise, 49% most populated state | 2597720 | 04:37 | 4 stable, 3 transient states |
| SufCD108–116 | 671 | 701 | ** | 00:12 | 18% noise, 44% most populated state | 146248 | 02:05 | 2 stable states |
| villin headpiece | 64 | 30 605 | 1570492 | 34:20 | stable segments with periods of instability | 33896452 | 04:25:57 | stages of folding |
| F10 (CaCl2) | 197 | 16 000 | 204536 | 04:15 | 1 dominant state per trajectory | 10567036 | 15:55 | 1 dominant state per trajectory |
| F10 (NaCl) | 197 | 16 000 | 1049908 | 05:38 | 74% noise, 1.4% most populated state | 8715716 | 33:48 | 2 frequently switching states |
| F10 (MgCl2) | 197 | 1 000 | ** | 00:10 | 64% noise, 6.2% most populated state | 2608468 | 09:18 | 5 frequently switching states |
| thrombin aptamer | 315 | 25 770 | 1256036 | 14:14 | 27% noise, 17% most populated state | 18838956 | 01:42:49 | various levels of compactness |
| thrombin (KCl) | 295 | 5 000 | 139136 | 01:08 | 29% noise, 14% most populated state | 7904572 | 01:13:29 | 2 stable, 1 transient state(s) |
| thrombin (NaCl) | 295 | 50 000 | 1489776 | 53:12 | 1 or 2 dominant states per trajectory | 27198820 | 23:32:15 | various loop configurations |
Atom counts are the number actually used in clustering—heavy atoms for nucleic acids and α carbons for proteins. On three systems (noted with **) HDBSCAN clustering completed too quickly for the distributed computing environment to record the amount of memory consumed. Visualization of structures from additional systems and brief analysis are given in Supporting Information, Figures S11–S20.
We also notice a correlation between a biopolymer’s average Root Mean Square Fluctuation (RMSF) and the method that seemed to give the most meaningful clusters (as discussed above and in Table 1). For systems with an average RMSF less than 2 Å, we consistently observed distinct conformational changes across Amorim–Hennig clusters and gained little information beyond deciding the stability of the system from HDBSCAN clusters (Supporting Information, Figure S22). This quantitative observation is consistent with our qualitative conclusion that Amorim–Hennig is the better choice for stable systems. Similarly, we observed that HDBSCAN provided more meaningful clusters for polymers with an average RMSF larger than 5 Å (Supporting Information, Figure S22), which is likewise consistent with our conclusion that HDBSCAN is best for more systems with higher structural variance. Systems with average RMSFs between 2 and 5 Å had no clear pattern (Supporting Information, Figure S22).
Since Amorim–Hennig is a K-Means variant that optimizes a scoring metric, one might wonder how searching over values of k with traditional K-Means clustering and selecting the k with the highest, say, silhouette score might compare to Amorim–Hennig using silhouette index as the value to optimize. We compared these two methods (Supporting Information, Figure S21) and found that Amorim–Hennig tends toward more, tighter clusters than K-Means using a k that maximizes the silhouette score.
3. METHODS
3.1. Data Generation and Feature Selection
With the exception the simulations of F10 in the presence of magnesium,117 all of the simulation data sets analyzed here are unpublished and generated in the course of ongoing research projects by the authors. They are each from all-atom MD simulations carried out by at least one of this study’s authors. These simulations calculate atomic trajectories based on Newton’s laws of motion. For a recent review of the utility of such MD trajectories, see Godwin et al.4 The exact simulation parameters used for generating each data set are detailed in the subsections below.
For all clustering performed here, we used only atomic Cartesian coordinates as features. Some set of internal coordinate(s) for each system would be preferred, as such a selection avoids the alignment issues that tend to plague analysis of MD trajectories.29,118,119 That is, it is difficult to separate out overall and internal motion of biopolymers when using Cartesian coordinates.29 However, the selection of an internal coordinate requires some prior knowledge about the system, or introduces bias based on some assumption about the system. Therefore, we assume the philosophical position of maximal ignorance and select Cartesian coordinates as features for testing HDBSCAN and Amorim–Hennig as first-pass clustering techniques for MD data.
Beyond aligning trajectory frames to the initial structure of each respective simulation, we did not standardize data to a common scale. All features are on length scale of the particular biopolymer in a given simulation. This similarity of scale is assured by structural alignment via rigid body rotations and translations to minimize the root-mean-square deviation of atomic positions among frames. Pre-processing MD trajectories in this fashion is a common and expected practice.3,4,120,121 Selection of an additional standardization method may introduce some bias. Therefore, our guiding principle of assuming maximal ignorance reinforced our decision to not further standardize the data sets.
Generation of MD data is inherently stochastic, as simulations begin with randomly seeded initial velocities. Additionally, the equations governing the Langevin heat baths that maintain the temperature of the systems are themselves stochastic. Finally, these biopolymers undergo thermal fluctuations proportional to the ambient temperature (here 300 K). By the nature of MD trajectories, we expect small random structural variations to create noise in our data—and any such data. Therefore, clustering techniques that are able to address noise—either through feature rescaling in the case of Amorim–Hennig or labeling points as noise in HDBSCAN—fit the nature of MD data well.
Another property of MD data is that they are often high dimensional, particularly when using Cartesian coordinates for features, as we have here. The only form of dimensionality reduction applied here was the typical—and, therefore, maximally ignorant—usage of only α carbon atoms for proteins and only heavy atoms for nucleic acids. The number of atoms used in each clustering trial (α carbons for proteins and heavy atoms for nucleic acids) is listed in Table 1. Each atom has three coordinates, making the number of input features for the smallest system 3 × 28 = 84 in the case of NEMO. For the largest system, the number of features was 3 × 1829 = 5487 in the case of MutSα. We do, however, see the potential for PCA and TICA122 as pre-processing steps to reduce the data to a few high variance components. Such a dimensionality reduction would improve the time and memory taken up by clustering calculations. We did not pursue these steps here, as we quickly realized that clustering with Amorim–Hennig and HDBSCAN on Cartesian coordinates was giving us relatively fast yet interesting partitions that we considered (admittedly with some arbitrariness) good enough for a first pass exploratory clustering.
3.1.1. Common Simulation Parameters
We used simulation parameters appropriate to each system and recommended for the simulation the software used, ACEMD.123 The parameters for each simulation vary due to the nature of the projects from which they come. Rather than trying to standardize the sampling rate, statistical ensemble, solvent conditions or any other parameters, we have intentionally left them varied to demonstrate the usefulness of these clustering techniques across many contexts. However, this choice means that there is not a single methodology for all simulations. Therefore, we provide this “Common” subsection that gives parameters common to all simulated systems and then one subsection for each of our four protein and two nucleic acid examples.
All protein simulations (Figures 1–5) and the thrombin aptamer simulation (Figure 7) were run under the isothermal–isobaric ensemble (NPT) in ACEMD.123 Langevin damping124 was used with a target temperature of 300 K and damping coefficient of 0.1, and a Berendsen pressure piston125 maintained approximately 1.01325 bar with a relaxation time of 400 fs. In all simulations, hydrogen mass repartitioning as implemented in ACEMD allowed us to use 4 fs time steps in our production runs. During simulation, systems were held at 300 K using a Langevin thermostat. For VdW and electrostatic forces, we applied a 9 Å cutoff and 7.5 Å switching distance, calculating long-range electrostatics with a Smooth Particle Mesh Ewald (SPME) summation method.126,127
These simulations were run on Titan GPUs in Metrocubo workstations produced by Acellera. All systems were solvated in explicit TIP3P water,128 solvated using VMD’s102 “Add Solvation Box” extension. The CHARMM27 force field used here is based on the interaction energies of small model systems determined by both quantum mechanics computations and direct experiment.129–131
3.1.2. MutSα
Initial coordinates for simulations of the MSH2/MSH6 protein come from RCSB Protein Data Bank (PDB) ID 208E.57 For cisplatinated, carboplatinated and fluoridated uracil-containing DNA, we used additional parameters based on pre-existing cisplatin, carboplatin, and FdU parameters.65,132–134 We fitted cross-linked structures of these modified DNA strands into the mismatched DNA binding pocket seen in RCSB PDB ID 208E.57 All MutSα systems were solvated in 150 mM NaCl using VMD’s “Add Ions” plugin. Each trajectory is concatenated from two simulations of 250 ns each. Each MSH2/6 simulation was minimized using conjugate gradient minimization for 1000 time steps followed by 250 ps of thermal equilibration.
3.1.3. NEMO Zinc Finger
Initial coordinates for the NEMO simulations are based on RCSB PDB ID 2JVX.68 Each NPT simulation was first minimized using conjugate gradient minimization for 5000 time steps. Subsequent equilibration took 20 ns, typically, as measured by the RMSD of each of the eight 1 μs trajectories over time, and this was removed from each trajectory for analysis. All NEMO systems were solvated in 150 mM NaCl using VMD’s “Add Ions” extension.
3.1.4. Villin Headpiece
Initial coordinates for simulations of villin headpiece are based on RCSB PDB ID 2RJY.71 Each villin headpiece folding simulation was minimized using conjugate gradient minimization for 5000 time steps. All villin systems were neutralized and solvated in 150 mM NaCl using VMD’s “Add Ions” plugin. The trajectory presented here is from three simulations of 6 μs each concatenated together.
3.1.5. Thrombin
Initial coordinates for simulations of thrombin in the presence of sodium are based on RCSB PDB ID 4DII.101 Eighteen out of 295 missing residues in the protein were added via the structural template-based atom fill-in tool Modeler in VMD, and the missing hydrogen atoms were added by VMD using standard parameters. The system was neutralized and solvated with 125 mM NaCl. Each of these simulations was minimized using conjugate gradient minimization for 1000 time steps. The trajectory analyzed here is concatenated from five simulations of 1 μs each.
3.1.6. F10
F10 structures in Figure 6 are output from MD simulations run under the canonical ensemble. Each F10 simulation was minimized using conjugate gradient minimization for 1000 time steps. For the examples above, F10 was simulated in 150 mM NaCl or 150 mM CaCl2. Prior to structural and kinetic analysis of all F10 simulations, we concatenated data from four runs of each system (one 1 μs and three 5 μs), totaling 16 μs in each trajectory used as input for the clustering algorithms. We then resampled the data at a rate of one frame every 1 ns for a final trajectory of 16 000 frames. We used a modified version of the CHARMM27 force field with additional parameters for FdU based on experimental data and quantum mechanics computations done by Ghosh et al.134 and validated in other studies.133,135
3.1.7. Thrombin Aptamer
Three NPT MD simulations were run to investigate how a fully extended 15-TBA refolds within about 8.5 μs. The initial extended aptamer structure was taken from the unfolding simulation of the same aptamer in G-quadruplex from RCSB PDB ID 4DII.101 The system in the refolding simulations was neutralized and solvated with 125 mM KCl. The trajectory analyzed in this work is concatenated from three simulations of 8.5 μs each. Each of these simulations was minimized using conjugate gradient minimization for 1000 time steps
3.2. Clustering
3.2.1. HDBSCAN
Conceptually, HDBSCAN is a method of grouping neighborhoods. It finds places of high density separated by sparse (noise) regions. To quickly estimate local densities, HDBSCAN uses distance to the kth nearest neighbor. Imagine two points a and b in a data set such as the toy data in Figure 8a. Each of these points lies in a neighborhood specified by a core point and the number of neighbors k. The distance from the core point to the edge of the neighborhood is called the core distance. These two points have core distances corek(a) and corek(b). For these two points we can calculate the mutual reachability distance,136
Figure 8.

Using a toy two-dimensional data set generated with scikit-learn140 (a), we give a conceptual explanation of HDBSCAN’s clustering algorithm. Using mutual reachability distance as the distance metric, a minimum spanning tree (b) is constructed. This tree solves an optimization problem such that removing any edge would create disconnected components–set(s) of nodes not connected to any other set(s). Based on the distances of connected points in the minimum spanning tree, single-linkage hierarchical clustering is performed (c). Splits in the dendrogram that create clusters smaller than the minimum cluster membership are rejected, and the final clusters (d) are determined using HDBSCAN’s novel cluster stability metric.35 In panel (d), λ is inverse distance. The example here is based on a tutorial by Leland McInnes, the author of the Python HDBSCAN implementation, available at github.com/lmcinnes/hdbscan. Additionally, we have made the Python code for reproducing our specific example available at figshare.com/articles/HDBSCAN_and_Amorim-Hennig_for_MD/3398266.
where d(a,b) is the distance between a and b, i.e., Euclidean distance for this example and all MD trajectories analyzed in this study.
Given the mutual reachability distance between all points, we can construct a network that has each data point as a node with edges weighted by the mutual reachability distance. We then want to begin removing edges in descending order of their weight until we have a minimal set of edges. A minimal set of edges in this case would be one in which removing any more edges would cause disconnected components, such as removing any edge in Figure 8b. The graph constructed in such a way is called a minimum spanning tree and can be constructed programmatically using Prim’s algorithm,137–139 as was done in Figure 8b.
We then construct a dendrogram from the minimum spanning tree by performing a hierarhical clustering. Each edge in the minimum spanning tree—in ascending order or distance—is used to merge the two points at its ends into a new cluster (single-linkage clustering). Each merging is represented by two joining lines in the dendrogram of Figure 8c. Traditional hierarchical clustering now calls for cutting the tree at a certain distance (usually specified by the user). However, HDBSCAN uses its minimum cluster size parameter to cut the tree at multiple points. Walking from top to bottom of the tree, any split that causes a cluster size smaller than the minimum is rejected, and the parent cluster is kept. The resulting dendrogram is a condensed tree (Figure 8d), where the width of the remaining lines indicates the number of points in the cluster. The lines change as distance (or inverse distance λ in Figure 8d) changes to indicate the number of points that remain in that cluster as a function of distance.
HDBSCAN extracts then the most significant clusters from this condensed tree. HDBSCAN uses its own, novel cluster stability metric35 to select clusters from multiple levels of the dendrogram by maximizing cluster stability. Campello et al. claimed this algorithm finds the optimal solution for any data set and demonstrate its performance across several data sets.35 The circled regions on Figure 8d indicate the final clusters chosen using this stability metric.
We used a Python141 implementation of HDBSCAN written by Leland McInnes and available in his Github repository (see Figure 8 caption). The examples in the documentation of this repository were useful and closely followed in constructing our simple example in Figure 8d. To automate the parsing of MD trajectory data into an appropriate input format for HDBSCAN and the output of HDBSCAN into plots for quick comprehension, we used an in-house script that we have made available online for free via figshare.142 This code has as a dependency the HDBSCAN code from the Github repository mentioned above. Our code sets the default minimum cluster size to 2 but allows the user to override this default. We used a minimum cluster size of 2 in all of our MD data, as our simulation sampling rates were such that we considered singleton states to be noise. Following the algorithm authors’ suggestion,35,36 the minimum samples is set equal to the minimum cluster size, meaning that the smallest neighborhood is at least as large as the minimum cluster size. We maintain that in the absence of prior knowledge setting the minimum cluster size to 1 effectively makes this clustering method non-parametric and exceptionally useful for first-pass investigation of an MD trajectory.
We have made the Python code used to produce the example in Figure 8 available online.142 Furthermore, for those wishing to reproduce the analysis described here on one of our MD data sets, we have made the trajectory and structure data for villin headpiece used to produce Figure 4 available online (figshare.com/articles/Villin_Headpiece_Simulations/3983526).
3.2.2. Amorim–Hennig
Being a K-Means variant, Amorim–Hennig is conceptually somewhat simpler than HDBSCAN. The Amorim–Hennig algorithm forms spherical Gaussian clusters in a rescaled feature space. First, the algorithm estimates a maximum number of K-means clusters using iK-Means, and then a round of K-Means clustering is performed. The distance of each data point in each cluster to that cluster’s center is calculated, and the data are rescaled on the basis of these distances for another round of clustering. The exact weights used in rescaling depend on the cluster validity index (scoring metric) the user has chosen to optimize.
Here we use the silhouette index, which is a number on the interval [−1,1] assigned to each point. Scores generally fall between the extrema, but an understanding of these special cases provides intuition into the silhouette index. A score of 1 for a given point means that point is more similar to its own cluster than any other cluster (i.e., an ideal clustering); 0 means the point falls exactly on the boundary between two clusters; −1 means the point is more similar to a neighboring cluster than its own.
Intuitively, this feature rescaling based on the scoring metric means that in the next round of clustering tightly packed points are likely to be clustered together once again but less dense clusters may be split up and/or merged into nearby clusters. This process is repeated until clusters no longer change between rounds of clustering. The reweighted data set out of the final (converged) round of clustering is then clustered once more with K-Means.
Starting by predicting a maximum number of clusters with iK-Means,39,46 this method applies a rescaled variant38 of iMWK-Means47 to the data set, selecting a number of clusters that optimizes a scoring metric.48,49 One of the algorithm’s authors has made a Matlab implementation available on his Web site (homepages.herts.ac.uk/~comqra/). He has also made available a Python implementation of the underlying algorithm, rescaled iMWK-Means,47 on which this method expands (sourceforge.net/projects/unsupervisedpy). Using the code available on Sourceforge and the algorithm described by de Amorim and Hennig in 2015, we wrote a Python implementation of Intelligent Minkowski-Weighted K-Means (iMWK-means) with explicit rescaling followed by K-Means38 (which we have called Amorim–Hennig in this work). We have made our Python implementation available for free on figshare.142 Our code has as dependencies the modules in the Sourceforge repository mentioned above.
In our examples and the Python implementation we have made available, we use the silhouette index as our scoring metric. This particular cluster validity index quantifies how similar a given member of a given cluster is to every other member of that cluster.50 The authors’ Matlab implementation, mentioned above, provides options for using additional scoring metrics.
For the reweighting of points after each clustering iteration, we set a default Minkowski metric of 2 in our Python implementation. The Minkowski metric p is defined by the distance equation for points a and b in an N-dimensional space:
where i denotes the ith coordinate for the given point. The most common values for the Minkowski metric are p = 2, making d Euclidean distance; p = 1, making d Manhattan distance; and p → ∞, making d the Chebyshev metric.47 Some studies—of which we cite a few examples47,143–149—have probed the use of various Minkowski metrics across various contexts. The choice of the default p = 2 comes from the intuitive simplicity of the Euclidean distance metric and experimental work on various data sets by the algorithm’s authors.38,39
3.2.3. Common Computational Resources
All clustering trials were performed on the Wake Forest University DEAC Cluster, a centrally managed distributed computing environment. These calculations were allotted eight 2.4 GHz cores and up to 120GB of RAM. The MD simulations were run on Titan GPUs in Metrocubo workstations produced by Acellera.
3.2.4. Analysis and Visualization
For parsing, analyzing, and plotting clustering results, we used the Python packages MDtraj,150 a library for analysis of MD trajectories; Numpy arrays151 for in-memory data storage and processing; and Scikit-Learn,140 a machine learning library for the final K-Means iteration of Amorim–Hennig and scoring clustering results with the silhouette index. These packages are dependencies for our freely available Python scripts.142
Visualizing structures from each cluster required selecting a representative conformation for each cluster. Using NumPY arrays, we averaged the (3×atoms)-dimensional position vectors (i.e., trajectory frames) of all α carbon atoms for proteins and heavy atoms for nucleic acids. We then selected the frame whose position vector has the smallest Euclidean distance from the mean vector. Next, to show the variance within a given cluster, we selected 50 evenly sampled frames from the list of all frames within that cluster.
For the layered images (those with shadows), we separately visualized representative structures and additional conformers in VMD,102 rendered them with Tachyon152 and combined the resulting graphics into the images shown here using Pillow, a fork of the Python Image Library. This visualization method is based on previous work by our research group, explained by Melvin and Salsbury.153 The choice of 50 evenly sampled frames rather than, say, standard deviation,153 comes from both algorithms’ ability to form non-Gaussian clusters of arbitrary shape. The number of frames was selected due to memory limitations when rendering the images. By trial and error, we selected the number that we could render for all systems with the computational resources available to us. Secondary structure visualization in VMD’s NewCartoon drawing method is based on STRIDE.154
4. CONCLUSIONS
As the size of MD trajectories grows with the ever increasing computational power available to researchers, automated methods for simplifying these data sets become crucial analysis tools. The clustering algorithms applied here to MD trajectories are excellent first-pass utilities for initial investigation of MD data. Additionally, using the default parameters suggested by the respective algorithm authors effectively makes these methods non-parametric. As such, having no parameters to tune through trial and error further increases their utility for beginning analysis of a new MD trajectory.
After investigating the systems detailed in the Results and Discussion and additional systems summarized in Table 1, we find that HDBSCAN is ideal for initial investigation of a trajectory. First and foremost, HDBSCAN is fast (Table 1). The time spent on clustering using HDBSCAN—a few seconds to a few hours—is negligible compared to the time to generate a trajectory—a few days to a few weeks. Furthermore, the output of HDBSCAN provides immediate insight into the stability of the system at hand. High noise percentage and/or a high number of low population (≤1%) clusters indicates an unstable system. Additionally, for unstable and/or disordered systems, HDBSCAN performs well at picking out distinct conformation ensembles, if such distinct ensembles exist. For stable systems, HDBSCAN detects large-scale, global conformational changes—if such changes occur—or puts all frames into one cluster—if such changes do not occur. These features make HDBSCAN an excellent first-pass method.
We suspect that HDBSCAN’s superior performance on disordered systems is due in part to its ability to discard noise points that might otherwise artificially cause two clusters to merge, when in bulk the clusters are significantly different. Additionally, we intuit that the algorithm’s ability to detect large-scale changes in stable systems comes from its capability to cut the hierarchical clustering dendogram at multiple locations. Its cutting algorithm optimizes the stability of a cluster, finding the clusters that would be least influenced by small variations. Translated to structure-based clusters, this optimization would mean finding the big changes and metastable conformations while ignoring small (local) structural shifts in the case of stable protein. For a disordered system, this optimization would mean finding any points of relative stability in conformation space. Given this propensity, we strongly recommend HDBSCAN clustering for IDPs.
If HDBSCAN clustering indicates a system is stable (e.g., <50 clusters or one or more cluster with population >10%), we find Amorim–Hennig clustering to be a useful next step. For stable ordered systems such as folded proteins, Amorim–Hennig tends to form clusters distinguished by small-scale changes in secondary structure. For stable but disordered systems (i.e., systems without clear secondary structure that nonetheless undergo infrequent conformational shifts), Amorim–Hennig finds distinct, stable ensembles of conformations. Amorim–Hennig’s ability to find the details for a stable system likely comes from its feature rescaling that emphasizes tight clusters. For a stable structured protein, tight structural clusters would be differentiated by small local changes.
These two complementary clustering methods provide a great deal of initial information about systems. They are also useful for grouping trajectory frames without prior knowledge to uncover conformational shifts, as seen in the examples above. In the context of MD trajectories, HDBSCAN quickly indicates stability and detects large-scale structural changes, providing big-picture information. Amorim–Hennig provides more fine-grained details for stable systems. Used in conjunction, these two clustering methods isolate diverse conformational ensembles in MD trajectories without prior knowledge.
Additionally, we see as an important line of future investigation the combination of these two clustering methods. Since Amorim–Hennig clustering seems to focus on details, while HDBSCAN captures big-picture changes, replacing the final K-Means step of Amorim–Hennig with HDBSCAN may prove fruitful. While we plan to investigate a combination of Amorim–Hennig and HDBSCAN, we intend to do so as part of a larger study exploring many pre-processing options for clustering MD trajectories for these and other clustering algorithms.
Supplementary Material
Acknowledgments
This work was partially supported by National Institutes of Health grants R01CA129373 to F.R.S. and T32-GM095440, supporting R.L.M. and R.C.G. Crystallography & Computational Biosciences Shared Resource services were supported by the Wake Forest Baptist Comprehensive Cancer Center’s NCI Cancer Center Support Grant P30CA012197. W.G.T. was partially supported by a Wake Forest Research Fellowship from Wake Forest University’s Undergraduate Research and Creative Activities Center. F.R.S. also acknowledges a Reynolds Research leave from Wake Forest University. Computations were performed on the Wake Forest University DEAC Cluster, a centrally managed resource with support provided in part by the University. The authors thank the reviewers for particularly insightful comments.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.6b00757.
Additional cluster visualizations of MutSα, villin headpiece, thrombin, F10, and thrombin aptamer; comparison of K-Means and Amorim–Hennig; correlation of RMSF to clustering results (PDF)
References
- 1.Noé F. Biophys J. 2015;108:228–229. doi: 10.1016/j.bpj.2014.11.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borhani DW, Shaw DEJ. J Comput-Aided Mol Des. 2012;26:15–26. doi: 10.1007/s10822-011-9517-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salsbury FR., Jr Curr Opin Pharmacol. 2010;10:738–744. doi: 10.1016/j.coph.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godwin RC, Melvin R, Salsbury FR. In: Computations in Drug Discovery. Zhang W, editor. Springer; New York: 2015. pp. 1–30. [Google Scholar]
- 5.Adcock SA, McCammon JA. Chem Rev. 2006;106:1589–615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Šponer J, Cang X, Cheatham TE. Methods. 2012;57:25–39. doi: 10.1016/j.ymeth.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perilla JR, Goh BC, Cassidy CK, Liu B, Bernardi RC, Rudack T, Yu H, Wu Z, Schulten K. Curr Opin Struct Biol. 2015;31:64–74. doi: 10.1016/j.sbi.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freddolino PL, Liu F, Gruebele M, Schulten K. Biophys J. 2008;94:L75–L77. doi: 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA, Young C, Deneroff MM, Batson B, Bowers KJ, Chow E, Eastwood MP, Ierardi DJ, Klepeis JL, Kuskin JS, Larson RH, Lindorff-Larsen K, Maragakis P, Moraes MA, Piana S, Shan Y, Towles B. Millisecond-scale Molecular Dynamics Simulations on Anton. Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis; New York: ACM; 2009. pp. 65: 1–65:11. [Google Scholar]
- 10.Godwin R, Gmeiner W, Salsbury FR. J Biomol Struct Dyn. 2016;34:125–134. doi: 10.1080/07391102.2015.1015168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckhardt W, Heinecke A, Bader R, Brehm M, Hammer N, Huber H, Kleinhenz H-G, Vrabec J, Hasse H, Horsch M, Bernreuther M, Glass CW, Niethammer C, Bode A, Bungartz H-J. In: Kunkel JM, Ludwig T, Meuer HW, editors. Proceedings of Supercomputing: 28th International Supercomputing Conference, ISC 2013; Leipzig, Germany. June 16–20, 2013; Berlin/Heidelberg: Springer; 2013. pp. 1–12. Chapter 591 TFLOPS. [Google Scholar]
- 12.Karplus M, McCammon JA. Nat Struct Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 13.Odell PL, Duran BS. Cluster Analysis: A Survey. Springer; Berlin/Heidelberg: 1974. [Google Scholar]
- 14.Kaufman L, Rousseeuw PJ. Finding groups in data: an introduction to cluster analysis. John Wiley & Sons; Hoboken, NJ: 2009. Wiley Series in Probability and Statistics 344. [Google Scholar]
- 15.Jain AK, Dubes RC. Algorithms for clustering data. Prentice-Hall, Inc; Upper Saddle River, NJ: 1988. [Google Scholar]
- 16.Berkhin P. Grouping Multidimensional Data. Springer-Verlag; Berlin/Heidelberg: 2006. pp. 25–71. [Google Scholar]
- 17.Lloyd S. IEEE Trans Inf Theory. 1982;28:129–137. [Google Scholar]
- 18.Steinhaus H. Bull Acad Polon Sci. 1957:801–804. Cl. III (IV) [Google Scholar]
- 19.MacQueen J. In: Le Cam LM, Neyman J, editors. Proceedings of the Fifth Berkeley Symposium on Mathematics, Statistics, and Probability; Berkeley, CA: University of California Press; 1967. pp. 281–297. Chapter 14. [Google Scholar]
- 20.Heyer LJ, Kruglyak S, Yooseph S. Genome Res. 1999;9:1106–1115. doi: 10.1101/gr.9.11.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comaniciu D, Meer P. IEEE Trans Pattern Anal Mach Intell. 2002;24:603–619. [Google Scholar]
- 22.Barbakh WA, Wu Y, Fyfe C. Review of Clustering Algorithms. In: Kacprzyk J, editor. Non-Standard Parameter Adaptation for Exploratory Data Analysis. Springer; Berlin/Heidelberg: 2009. pp. 7–28. Studies in Computational Intelligence 249. [DOI] [Google Scholar]
- 23.Aggarwal CC, Reddy CK. Data clustering: algorithms and applications. CRC Press; Boca Raton, FL: 2013. [Google Scholar]
- 24.Bowman GR, Meng L, Huang X. J Chem Phys. 2013;139:121905. doi: 10.1063/1.4812768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torda AE, van Gunsteren WF. J Comput Chem. 1994;15:1331–1340. [Google Scholar]
- 26.Kenn M, Ribarics R, Ilieva N, Cibena M, Karch R, Schreiner W. Mol BioSyst. 2016;12:1600–1614. doi: 10.1039/c5mb00879d. [DOI] [PubMed] [Google Scholar]
- 27.De Paris R, Quevedo CV, Ruiz DD, Norberto de Souza O, Barros RC. Comput Intell Neurosci. 2015;2015:1–9. doi: 10.1155/2015/916240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fraccalvieri D, Pandini A, Stella F, Bonati L. BMC Bioinf. 2011;12:158. doi: 10.1186/1471-2105-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sittel F, Stock G. J Chem Theory Comput. 2016;12:2426. doi: 10.1021/acs.jctc.5b01233. [DOI] [PubMed] [Google Scholar]
- 30.Junlin L, Hongguang F. Pattern Recognit. 2011;44:1721–1737. [Google Scholar]
- 31.Jain A, Stock G. J Chem Theory Comput. 2012;8:3810–3819. doi: 10.1021/ct300077q. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez A, Laio A. Science (Washington, DC, U S) 2014;344:1492–1496. doi: 10.1126/science.1242072. [DOI] [PubMed] [Google Scholar]
- 33.Cabria I, Gondra I. A Mean Shift-Based Initialization Method for K-means. 2012 IEEE 12th International Conference on Computer and Information Technology (CIT); Chengdu, Sichuan, China. Oct 27–29, 2012; pp. 579–586. [DOI] [Google Scholar]
- 34.Shao J, Tanner SW, Thompson N, Cheatham TE., III J Chem Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 35.Campello RJGB, Moulavi D, Sander J. Adv Knowl Discovery Data Min. 2013;7819:160–172. [Google Scholar]
- 36.Campello RJGB, Moulavi D, Zimek A, Sander J. ACM Trans Knowl Discovery Data. 2015;10:1–51. [Google Scholar]
- 37.Altman NS. Am Stat. 1992;46:175–185. [Google Scholar]
- 38.de Amorim RC, Hennig C. Inf Sci (N Y) 2015;324:126–145. [Google Scholar]
- 39.de Amorim RC, Mirkin B. Clusters, Orders, and Trees: Methods and Applications. In: Aleskerov F, Goldengorin B, Pardalos PM, editors. Honor of Boris Mirkin’s 70th Birthday. Springer; New York: 2014. pp. 103–117. [Google Scholar]
- 40.Kriegel H-P, Kröger P, Sander J, Zimek A. Wiley Interdiscip Rev Data Min Knowl Discovery. 2011;1:231–240. [Google Scholar]
- 41.Rahman MF, Liu W, Suhaim SB, Thirumuruganathan S, Zhang N, Das G. ACM. 2016:160–172. https://arxiv.org/abs/1602.03730.
- 42.Li L, Xi Y. Research on Clustering Algorithm and Its Parallelization Strategy. 2011 International Conference on Computational and Information Sciences (ICCIS); Chengdu, China. Oct 21–23, 2011; pp. 325–328. [DOI] [Google Scholar]
- 43.Wang W, Zhang B, Wang D, Jiang Y, Qin S, Xue L. Signal Processing. 2016;126:12–17. [Google Scholar]
- 44.Jaskowiak PA. PhD thesis. Universidade de Sao Paulo; 2016. On the evaluation of clustering results: measures, ensembles, and gene expression data analysis. [Google Scholar]
- 45.Moulavi D, Jaskowiak PA, Campello RJGB, Zimek A, Sander J. Density-Based Clustering Validation SDM. 2014;839–847 [Google Scholar]
- 46.Chiang MM-T, Mirkin BJ. Classif. 2010;27:3–40. [Google Scholar]
- 47.Cordeiro de Amorim R, Mirkin B. Pattern Recognit. 2012;45:1061–1075. [Google Scholar]
- 48.Arbelaitz O, Gurrutxaga I, Muguerza J, Perez JM, Perona I. Pattern Recognit. 2013;46:243–256. [Google Scholar]
- 49.Halkidi M, Vazirgiannis M. Clustering Validity Assessment: Finding the Optimal Partitioning of a Data Set. Proceedings of the 2001 IEEE International Conference on Data Mining; Washington, DC. 2001; pp. 187–194. [Google Scholar]
- 50.Rousseeuw PJ. J Comput Appl Math. 1987;20:53–65. [Google Scholar]
- 51.Charrad M, Ghazzali N, Boiteau V, Niknafs A. J Stat Softw. 2014;61:1–36. [Google Scholar]
- 52.Liu B, Shen X, Pan W. Stat Anal Data Min ASA Data Sci J. 2016;9:106–116. doi: 10.1002/sam.11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palomo EJ, López-Rubio E. Int J Neural Syst. 2016;26:1650019. doi: 10.1142/S0129065716500192. [DOI] [PubMed] [Google Scholar]
- 54.Ushizima D, Carneiro A, Souza M, Medeiros F. Advances in Visual Computing. Springer International Publishing; Cham, Switzerland: 2015. Investigating Pill Recognition Methods for a New National Library of Medicine Image Dataset; pp. 410–419. Lecture Notes in Computer Science 9475. [DOI] [Google Scholar]
- 55.Liu B, Shen X, Pan W. Stat Med. 2016;35:2235–2250. doi: 10.1002/sim.6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Amorim RC. J Classification. 2016;33:210–242. [Google Scholar]
- 57.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese L. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 58.Drotschmann K, Topping RP, Clodfelter JE, Salsbury FR. DNA Repair. 2004;3:729–742. doi: 10.1016/j.dnarep.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 59.Topping RP, Wilkinson JC, Scarpinato KD. J Biol Chem. 2009;284:14029–14039. doi: 10.1074/jbc.M809303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vasilyeva A, Clodfelter JE, Rector B, Hollis T, Scarpinato KD, Salsbury FR. DNA Repair. 2009;8:103–113. doi: 10.1016/j.dnarep.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiricny J. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 62.Negureanu L, Salsbury FR. J Biomol Struct Dyn. 2014;32:969–992. doi: 10.1080/07391102.2013.799437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Negureanu L, Salsbury FR. J Biomol Struct Dyn. 2012;30:347–361. doi: 10.1080/07391102.2012.680034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salsbury FR, Clodfelter JE, Gentry MB, Hollis T, Scarpinato KD. Nucleic Acids Res. 2006;34:2173–2185. doi: 10.1093/nar/gkl238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Negureanu L, Salsbury FR. J Mol Model. 2013;19:4969–4989. doi: 10.1007/s00894-013-1998-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Negureanu L, Salsbury FR. J Biomol Struct Dyn. 2012;29:757–776. doi: 10.1080/07391102.2012.10507412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salsbury FR., Jr Protein Pept Lett. 2010;17:744–750. doi: 10.2174/092986610791190318. [DOI] [PubMed] [Google Scholar]
- 68.Cordier F, Vinolo E, Véron M, Delepierre M, Agou F. J Mol Biol. 2008;377:1419–1432. doi: 10.1016/j.jmb.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 69.Cordier F, Grubisha O, Traincard F, Véron M, Delepierre M, Agou F. J Biol Chem. 2009;284:2902–2907. doi: 10.1074/jbc.M806655200. [DOI] [PubMed] [Google Scholar]
- 70.Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, Ploegh HL, Smith TS. Biochem J. 2004;378:727–734. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meng J, McKnight CJ. Biochemistry. 2008;47:4644–4650. doi: 10.1021/bi7022738. [DOI] [PubMed] [Google Scholar]
- 72.Nierodzik ML, Karpatkin S. Cancer Cell. 2006;10:355–362. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 73.Radjabi AR, Sawada K, Jagadeeswaran S, Eichbichler A, Kenny HA, Montag A, Bruno K, Lengyel E. J Biol Chem. 2008;283:2822–2834. doi: 10.1074/jbc.M704855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bock LC, Griffin LC, Latham Ja, Vermaas EH, Toole JJ. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 75.Morser J. J Biol Chem. 1996;271:16603–16608. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- 76.Davie EW, Fujikawa K, Kisiel W. Biochemistry. 1991;30:10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 77.Coughlin SR. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 78.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, Bode W. Nature. 2000;404:518–25. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 79.Esmon CT. Science (Washington, DC, U S) 1987;235:1348–1352. doi: 10.1126/science.3029867. [DOI] [PubMed] [Google Scholar]
- 80.Holmer E, Kurachi K, Söderström G. Biochem J. 1981;193:395–400. doi: 10.1042/bj1930395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nemerson Y, Gentry R. Biochemistry. 1986;25:4020–4033. doi: 10.1021/bi00362a006. [DOI] [PubMed] [Google Scholar]
- 82.Wells CM, Di Cera E. Biochemistry. 1992;31:11721–30. doi: 10.1021/bi00162a008. [DOI] [PubMed] [Google Scholar]
- 83.Huntington JA, Esmon CT. Structure. 2003;11:469–479. doi: 10.1016/s0969-2126(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 84.Vindigni A, Di Cera E. Biochemistry. 1996;35:4417–4426. doi: 10.1021/bi952834d. [DOI] [PubMed] [Google Scholar]
- 85.Lai MT, Di Cera E, Shafer JA. J Biol Chem. 1997;272:30275–30989. doi: 10.1074/jbc.272.48.30275. [DOI] [PubMed] [Google Scholar]
- 86.Kroh HK, Tans G, Nicolaes GAF, Rosing J, Bock PE. J Biol Chem. 2007;282:16095–16104. doi: 10.1074/jbc.M610577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ayala Y, Di Cera E. J Mol Biol. 1994;235:733–746. doi: 10.1006/jmbi.1994.1024. [DOI] [PubMed] [Google Scholar]
- 88.Mathur A, Schlapkohl Wa, Di Cera E. Biochemistry. 1993;32:7568–7573. doi: 10.1021/bi00080a031. [DOI] [PubMed] [Google Scholar]
- 89.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. J Biol Chem. 1995;270:22089–92. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 90.Johnson DJD, Adams TE, Li W, Huntington JA. Biochem J. 2005;392:21–28. doi: 10.1042/BJ20051217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liao ZY. Cancer Res. 2005;65:4844–4851. doi: 10.1158/0008-5472.CAN-04-1302. [DOI] [PubMed] [Google Scholar]
- 92.Bijnsdorp I, Comijn E, Padron J, Gmeiner W, Peters G. Oncol Rep. 2007;18:287–291. doi: 10.3892/or.18.1.287. [DOI] [PubMed] [Google Scholar]
- 93.Gmeiner WH, Reinhold WC, Pommier Y. Mol Cancer Ther. 2010;9:3105–3114. doi: 10.1158/1535-7163.MCT-10-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gmeiner WH, Skradis A, Pon RT, Liu J. Nucleosides Nucleotides. 1999;18:1729–1730. doi: 10.1080/07328319908044836. [DOI] [PubMed] [Google Scholar]
- 95.Pardee TS, Gomes E, Jennings-Gee J, Caudell D, Gmeiner WH. Blood. 2012;119:3561–3570. doi: 10.1182/blood-2011-06-362442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu J, Kolath J, Anderson J, Kolar C, Lawson TA, Talmadge J, Gmeiner WH. Antisense Nucleic Acid Drug Dev. 1999;9:481–486. doi: 10.1089/oli.1.1999.9.481. [DOI] [PubMed] [Google Scholar]
- 97.Liu J, Skradis A, Kolar C, Kolath J, Anderson J, Lawson T, Talmadge J, Gmeiner WH. Nucleosides Nucleotides. 1999;18:1789–1802. doi: 10.1080/07328319908044843. [DOI] [PubMed] [Google Scholar]
- 98.Liu C, Willingham M, Liu J, Gmeiner WH. Int J Oncol. 2002;21:303–308. [PubMed] [Google Scholar]
- 99.Longley DB, Harkin DP, Johnston PG. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 100.Heidelberger C, Chaudhuri NK, Danneberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E, Scheiner J. Nature. 1957;179:663–666. doi: 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- 101.Russo Krauss I, Merlino A, Randazzo A, Novellino E, Mazzarella L, Sica F. Nucleic Acids Res. 2012;40:8119–8128. doi: 10.1093/nar/gks512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 103.McFail-Isom L, Sines CC, Williams LD. Curr Opin Struct Biol. 1999;9:298–304. doi: 10.1016/S0959-440X(99)80040-2. [DOI] [PubMed] [Google Scholar]
- 104.Chen H, Meisburger SP, Pabit Sa, Sutton JL, Webb WW, Pollack L. Proc Natl Acad Sci U S A. 2012;109:799–804. doi: 10.1073/pnas.1119057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen H, Meisburger SP, Pabit Sa, Sutton JL, Webb WW, Pollack L. Proc Natl Acad Sci U S A. 2012;109:799–804. doi: 10.1073/pnas.1119057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qiu X, Andresen K, Kwok L, Lamb J. Phys Rev Lett. 2007;99:038104. doi: 10.1103/PhysRevLett.99.038104. [DOI] [PubMed] [Google Scholar]
- 107.Lipfert J, Doniach S, Das R, Herschlag D. Annu Rev Biochem. 2014;83:813–841. doi: 10.1146/annurev-biochem-060409-092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Selbach BP, Pradhan PK, Dos Santos PC. Biochemistry. 2013;52:4089–4096. doi: 10.1021/bi4001479. [DOI] [PubMed] [Google Scholar]
- 109.Selbach BP, Chung AH, Scott AD, George SJ, Cramer SP, Dos Santos PC. Biochemistry. 2014;53:152–160. doi: 10.1021/bi4011978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fang Z, Dos Santos PC. MicrobiologyOpen. 2015;4:616–631. doi: 10.1002/mbo3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Black KA, Dos Santos PC. J Bacteriol. 2015;197:1952–1962. doi: 10.1128/JB.02625-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Selbach B, Earles E, Dos Santos PC. Biochemistry. 2010;49:8794–8802. doi: 10.1021/bi101358k. [DOI] [PubMed] [Google Scholar]
- 113.Rajakovich LJ, Tomlinson J, Dos Santos PC. J Bacteriol. 2012;194:4933–4940. doi: 10.1128/JB.00842-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parsonage D, Newton GL, Holder RC, Wallace BD, Paige C, Hamilton CJ, Dos Santos PC, Redinbo MR, Reid SD, Claiborne A. Biochemistry. 2010;49:8398–8414. doi: 10.1021/bi100698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fang Z, Roberts AA, Weidman K, Sharma SV, Claiborne A, Hamilton CJ, Dos Santos PC. Biochem J. 2013;454:239–247. doi: 10.1042/BJ20130415. [DOI] [PubMed] [Google Scholar]
- 116.Black KA, Dos Santos PC. Biochim Biophys Acta, Mol Cell Res. 2015;1853:1470–1480. doi: 10.1016/j.bbamcr.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 117.Melvin RL, Gmeiner WH, Salsbury FR., Jr J Phys Chem B. 2016;120:10269–10279. doi: 10.1021/acs.jpcb.6b07753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sittel F, Jain A, Stock G. J Chem Phys. 2014;141:014111. doi: 10.1063/1.4885338. [DOI] [PubMed] [Google Scholar]
- 119.Pérez-Hernández G, Paul F, Giorgino T, De Fabritiis G, Noé F. J Chem Phys. 2013;139:015102. doi: 10.1063/1.4811489. [DOI] [PubMed] [Google Scholar]
- 120.Scott WRP, Straus SK. Proteins: Struct, Funct, Genet. 2015;83:820–826. doi: 10.1002/prot.24776. [DOI] [PubMed] [Google Scholar]
- 121.Damm KL, Carlson Ha. J Am Chem Soc. 2007;129:8225–8235. doi: 10.1021/ja0709728. [DOI] [PubMed] [Google Scholar]
- 122.Naritomi Y, Fuchigami S. J Chem Phys. 2011;134:065101. doi: 10.1063/1.3554380. [DOI] [PubMed] [Google Scholar]
- 123.Harvey MJ, Giupponi G, Fabritiis GD. J Chem Theory Comput. 2009;5:1632–1639. doi: 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- 124.Feller SE, Zhang Y, Pastor RW, Brooks BR. J Chem Phys. 1995;103:4613. [Google Scholar]
- 125.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J Chem Phys. 1984;81:3684. [Google Scholar]
- 126.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089. [Google Scholar]
- 127.Harvey MJ, De Fabritiis G. J Chem Theory Comput. 2009;5:2371–2377. doi: 10.1021/ct900275y. [DOI] [PubMed] [Google Scholar]
- 128.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926. [Google Scholar]
- 129.Foloppe N, MacKerell AD., Jr J Comput Chem. 2000;21:86–104. [Google Scholar]
- 130.MacKerell AD, Banavali NK. J Comput Chem. 2000;21:105–120. [Google Scholar]
- 131.MacKerell AD, Banavali N, Foloppe N. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 132.Scheeff ED, Briggs JM, Howell SB. Mol Pharmacol. 1999;56:633–643. [PubMed] [Google Scholar]
- 133.Gmeiner WH, Salsbury F, Olsen CM, Marky LA. J Nucleic Acids. 2011;2011:1–8. doi: 10.4061/2011/631372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ghosh S, Salsbury FR, Horita DA, Gmeiner WH. Nucleic Acids Res. 2011;39:4490–4498. doi: 10.1093/nar/gkr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ghosh S, Salsbury FR, Horita DA, Gmeiner WHJ. J Biomol Struct Dyn. 2013;31:1301–1310. doi: 10.1080/07391102.2012.732343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eldridge J, Belkin M, Wang Y. Beyond Hartigan Consistency: Merge Distortion Metric for Hierarchical Clustering. Proceedings of the 28th Conference on Learning Theory; 2015; pp. 588–606. [Google Scholar]
- 137.Prim RC. Bell Syst Tech J. 1957;36:1389–1401. [Google Scholar]
- 138.Dijkstra EW. Numer Math. 1959;1:269–271. [Google Scholar]
- 139.Jarník V. Práce Morav Přírodovědecké Společnost. 1930;6:57–63. [Google Scholar]
- 140.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay É. J Mach Learn Res. 2012;12:2825–2830. [Google Scholar]
- 141.van Rossum G. Python Reference Manual. CWI, Centre for Mathematics and Computer Science; Amsterdam, The Netherlands: 1995. [Google Scholar]
- 142.Melvin R, Salsbury F. HDBSCAN and Amorim–Hennig for MD. 2016 https://doi.org/10.6084/M9.FIGSHARE.3398266.V1.
- 143.Doherty K, Adams R, Davey N. Appl Soft Comput. 2007;7:203–210. [Google Scholar]
- 144.Kivinen J, Warmuth M, Hassibi B. IEEE Trans Signal Process. 2006;54:1782–1793. [Google Scholar]
- 145.Francois D, Wertz V, Verleysen M. Knowl Data Eng IEEE Trans. 2007;19:873–886. [Google Scholar]
- 146.Rudin C. J Mach Learn Res. 2009;10:2233–2271. [Google Scholar]
- 147.Bagnall A, Janacek G. Mach Learn. 2005;58:151–178. [Google Scholar]
- 148.Filippone M, Camastra F, Masulli F, Rovetta S. Pattern Recognit. 2008;41:176–190. [Google Scholar]
- 149.Strehl A, Ghosh J, Mooney R. AAAI Technical Report WS-00-01. American Association for Artificial Intelligence; 2000. Impact of similarity measures on web-page clustering; pp. 58–64. [Google Scholar]
- 150.McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang LP, Lane TJ, Pande VS. Biophys J. 2015;109:1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.van der Walt S, Colbert SC, Varoquaux G. Comput Sci Eng. 2011;13:22–30. [Google Scholar]
- 152.Stone J. MSc thesis. Computer Science Department, University of Missouri–Rolla; 1998. An Efficient Library for Parallel Ray Tracing and Animation. [Google Scholar]
- 153.Melvin RL, Salsbury FR. J Mol Graphics Modell. 2016;67:44–53. doi: 10.1016/j.jmgm.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Frishman D, Argos P. Proteins: Struct, Funct, Genet. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.