

Abstract
Objective
Human adenoviruses are small double stranded DNA viruses that provoke vast array of human diseases. Next generation sequencing techniques increase genomic data of HAdV rapidly, which increase their serotypes. The complete genome sequence of human adenovirus shows that it contains large amount of proteins with unknown cellular or biochemical function, known as hypothetical proteins. Hence, it is indispensable to functionally and structurally annotate these proteins to get better understanding of the novel drug targets. The purpose was the characterization of 38 randomly retrieved hypothetical proteins through determination of their physiochemical properties, subcellular localization, function, structure and ligand binding sites using various sequence and structure based bioinformatics tools.
Results
Function of six hypothetical proteins P03269, P03261, P03263, Q83127, Q1L4D7 and I6LEV1 were predicted confidently and then used further for structure analysis. We found that these proteins may act as DNA terminal protein, DNA polymerase, DNA binding protein, adenovirus E3 region protein CR1 and adenoviral protein L1. Functional and structural annotation leading to detection of binding sites by means of docking analysis can indicate potential target for therapeutics to defeat adenoviral infection.
Electronic supplementary material
The online version of this article (10.1186/s13104-017-2992-z) contains supplementary material, which is available to authorized users.
Keywords: Human adenovirus, Hypothetical proteins, Function annotation, DNA terminal protein, DNA polymerase, DNA binding protein
Introduction
Human adenoviruses are non-enveloped dsDNA viruses of almost 35 kb in size [1]. HAdV can infect a variety of tissues and cause a wide range of complications like gastroenteritis, hepatitis, myocarditis, keratoconjunctivitis and pneumonia [2, 3]. It is contagion in nature which occurs through direct contact or fomites and virus is also resistant to various physical and chemical agents. Children younger than the age of 5 years and immune compromised persons especially the pediatric patients are most susceptible to these viruses. Worldwide 5–7% respiratory tract infections are ascribed by HAdV in pediatric patients [4] and persons of all ages are susceptible to infections caused by these viruses [5].
Seven known Human adenoviruses species from HAdV-A to HAdV-G are constitute of the genus Mastadenovirus in which all the human adenoviruses are categorized and further divided into different strains [6]. Now 67 types of HAdV have been reported [7]. Their number is rapidly increasing due to bioinformatics and genomic advances and availability of whole genome sequences [8, 9].
After an immense effort 50–60% genes have a known function in most of completely sequenced genomes. Number of genes having unknown functions called as hypothetical protein are present in each organism’s genome [10]. To understand the biology and genome of the organisms, it is important to discover the function of hypothetical proteins, despite HAdV has a small size genome but still it has a several hypothetical proteins. So, in order to treat infectious diseases such as those caused by HAdV, functional annotation of these HPs might open avenues for prioritizing novel drug targets [4].
In-silico strategies to annotate the hypothetical proteins are cost effective and fast enough to explore their function. In this study, multiple algorithm based software’s have been used for the prediction of hypothetical protein function that may lead to the identification of novel pharmacological targets for screening, drug discovery and designing for the treatment of HAdV infections [11].
Main text
Methods
Sequence retrieval
Proteins having unknown function of Human adenovirus were taken from UniProt [12, 13]. Random selection of 38 hypothetical proteins belonging to eight different types of HAdV was carried out (Additional file 1: Table S1). The sequence analysis was done by taking FASTA sequence of these proteins along with their UniProt ID. For characterization purposes, number of software based on different algorithms were used as shown in Fig. 1.
Fig. 1.
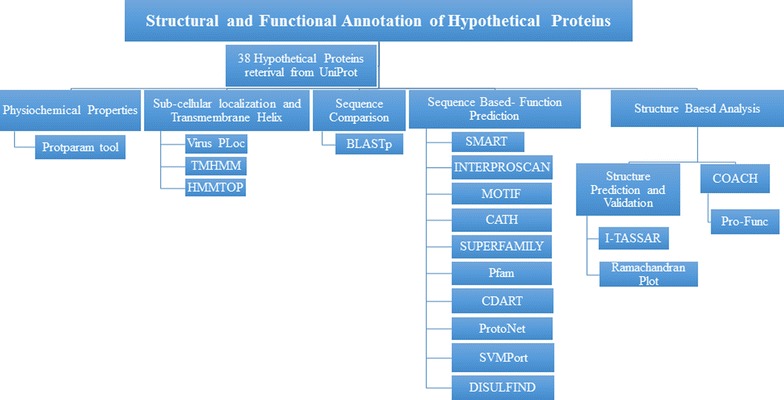
Flowchart that shows all of the tools used for functional and structural annotation of hypothetical proteins
Physicochemical characterization
Analysis of physiochemical properties of all HPs was done by online server ExPASy’s Protparam tool [14]. This server executes theoretical evaluation of physiochemical properties like isoelectric point, molecular weight, aliphatic index, grand average of hydropathicity (GRAVY) and instability index [15].
Sub-cellular localization
To predict the cellular function of a protein it is important to get information about its sub-cellular localization i.e. a protein can be present in outer membrane, inner membrane, periplasm, extracellular space or in cytoplasm [16]. Sub-cellular localization of viral proteins were predicted using Virus-PLoc [17] online server tool [18], TMHMM [19, 20] and HMMTOP [21, 22].
Sequence analogy
Most basic step in the function prediction of a protein is looking for its structural homologs in different available genomics and proteomics based databases. Popular bioinformatics tool BLASTp was used for this purposes [23, 24].
Function and disulfide bridges prediction
For precise function annotation, various tools like SVMport, ProtNet [25, 26], Pfam, Motif [27, 28], CDART [24, 29], CATH [30, 31], SMART [32, 33], Superfamily [34, 35] and InterProscan [27, 36] were used that classified all 38 proteins of HAdV into families and subfamilies on the basis of their sequence, structure and function [16, 37]. DISULFIND [38] server was used to evaluate occurrence of disulfide bonds between cysteine residues [39].
Structure prediction and validation
For prediction and validation of three dimensional I-TASSAR (Iterative Threading Assembly Refinement) [40, 41] Ramachandran Plot were used [42, 43].
Structure analysis
Functions of proteins based on structural analysis are considered more acceptable as compared to sequence based function annotation, because homologous proteins show more conserved structures in evolution than sequences [44]. For this purpose, we have used ProFunc [27] and COACH [40, 45].
Results
Random selection of 38 hypothetical proteins belonging to eight different types of HAdV was carried out from UniProt (Additional file 1: Table S1). The amino acid length of 38 randomly selected proteins of eight different types of Human Adenovirus ranges from 1198 amino acids for longest protein to 81 amino acids for shortest protein (Additional file 1: Table S1). Protparam tool has been used for the prediction of physiochemical properties of all hypothetical proteins (Additional file 2: Table S2). Subcellular localization and transmembrane helix prediction software predicated most of the HPs to be localized in the host cytoplasm and a few in-host cell membrane and nucleus (Additional file 3: Table S3). Multiple softwares were used for the function prediction of 38 hypothetical proteins (Additional file 4: Table S4, Additional file 5: Table S5). Out of 38 proteins, 6 HP’s whose function was confidently predicted by ≥ 6 software’s were confidently selected (Table 1). Confidently function predicted HPs were further used for structure prediction, structure analysis and disulphide bridges prediction. The detailed results of structure prediction and analysis are shown in Additional file 6: Table S6 and Additional file 7: Table S7. DISULFIND was unable to find disulphide bonds in any of the HP’s and characterized them as thermally unstable proteins.
Table 1.
Proteins whose function is predicted confidently along with their corresponding genomes and subcellular location
| Sr no | Uniprot ID | HAdV | Function | Confidence level | Virus-PLoc |
|---|---|---|---|---|---|
| 01 | P03269 | HAdV-2 | DNA terminal protein | 7/9 | Host nucleus |
| 02 | P03261 | HAdV-2 | DNA polymerase | 8/9 | Host nucleus |
| 03 | P03263 | HAdV-2 | DNA binding protein | 6/9 | Host cytoplasm |
| 04 | Q83127 | HAdV-7 | Adenovirus E3 region protein CR1 | 6/9 | Host cell membrane |
| 05 | Q1L4D7 | Human mastadenovirus B | Adenoviral protein L1 | 7/9 | Host cytoplasm and nucleus |
| 06 | I6LEV1 | Human mastadenovirus B | Adenoviral protein L1 | 6/9 | Host cytoplasm and nucleus |
Discussion
In this study, we carried out structural and functional annotation of 38 HPs of human adenovirus that is responsible for variety of clinical diseases. Physiochemical properties prediction showed that Isoelectric point [46] of HPs ranges from 4.1 to 12.43. Isoelectric point is pH at which the net charge on the protein is zero and at this pH the protein become less soluble, compact and stable that leads to crystallization of protein. So, the purification and crystallization of protein can be carried out by developing a buffer system with the help of computed pI [47, 48] (Additional file 2: Table S2).
The extinction coefficient of the HPs computed by Protparam tool ranges from 1490.0 to 179,580.0 M−1 cm−1 at 280 nm. This computed extinction coefficient can be helpful for quantitatively studying protein–ligand and protein–protein interaction. It is forecasted that if the instability index is less than 40 then a protein will be stable and if greater than 40 then it will be unstable. The instability index of 38 hypothetical proteins ranges from 20.1 to 106.56 and due to this only nine proteins are stable and rest is unstable. The GRAVY index of all proteins ranges from − 0.908 to 0.166 and out of 38 HPs, 32 HPs have negative GRAVY index which indicate that these proteins are non-polar in nature [49].
The detailed information about the functional and structural annotation for six hypothetical proteins is as follow:
P03269
P03269 is predicted as an adenoviral DNA terminal protein that performs function in the initiation of the viral DNA replication [50]. This protein is covalently bound to the viral DNA and acts as a primer for viral genomic replication by DNA strand displacement [51]. Seven software confidently predicted the function of this protein and Virus-PLoc server also confirmed its function by predicting its location in host nucleus. Predicted three-dimensional structure highest C-score − 2.25 (Additional file 8: Figure S1) was selected and structure verification through RAMACHANDRAN PLOT showed 76.9% residues are in most favored region and 18.8% residue are in additional allowed region (Additional file 9: Figure S2). For pharmaceutical and docking analysis, COACH has been used, out of many ligand binding sites, best ligand binding sites with maximum C-score were selected that can be used for further molecular docking analysis (Additional file 6: Table S6). Further structure based function analysis predicted adenoviral DNA terminal protein motif in HP P03269 and Ala159-Arg161, Gly558-Gly560, Leu406-Glu408, Gln241-Ala243 and Pro275-Arg277 structure motifs are also predicted to be conserved in this HP that may have a similar function (Additional file 7: Table S7). Gene Ontology analysis shows that HP P03269 may have role in the biological process of DNA replication, cellular process, cellular metabolic process, cellular biosynthetic process and biochemical function as DNA binding and nucleic acid binding.
P03261
P03261 belongs to Human adenovirus C serotype 2 and predicted to contain DNA polymerase type-B family catalytic domain and sub-cellularly localized in host nucleus. DNA-directed DNA polymerases has both exonucleases and polymerase activity and play role in the process of recombination, repair and DNA replication [52]. Out of five 3D models predicted by I-TASSAR, structure with highest C-score (− 0.20) was selected (Additional file 10: Figure S3) and structure verification shows that 67.1% residues are in favored region and 25.7% residues are in additional allowed regions of RC-plot (Additional file 11: Figure S4). ProFunc server has predicted DNA polymerase family B signature.
Gene ontology analysis showed that this HP may play its role in DNA replication and cellular process and biochemically function in nucleotide binding, nucleic acid binding and DNA-directed DNA-polymerase activity. DNA polymerase type B, organellar and viral and DNA-directed DNA-polymerase family B signature motifs in the HP these results have further validated the results of sequence based function prediction. Five other structure motifs were also identified as Leu323-Asp326, His955-Leu957, Ser926-Pro928, Leu645-Pro647 and Lys850-Asn853 (Additional file 7: Table S7).
P03263
P03263 is predicted as an adenoviral protein L1 52/55-kDa and that perform multiple functions in DNA packaging by facilitating stable interactions between empty capsid and viral DNA through its expression both in the early and late stages of infection cycle [46] (Additional file 12: Figure S5).
Model with highest C-score − 3.74 was selected and structure validation shows that 69.6% residues are in favored regions and 23.2% resides are present in the additional allowed regions of RC-plot (Additional file 13: Figure S6). Functional analysis server has verified the results of sequence based function prediction by predicting adenoviral protein L1 52/55-kDa motif in HP P03263 along with three conserved structure motifs Ala105-Ala107, Glu7-Asp9 and Asp4-Glu6 (Additional file 7: Table S7). According to gene ontology results HP P03263, HPQ83127, HP I6LEV1 are involved in the biological process of virion assembly, anatomical structure formation, anatomical structure formation involved in morphogenesis and cellular component assembly involved in morphogenesis.
Q83127
Q83127 is annotated as Adeno E3 region protein CR1 that is responsible for controlling the viral interactions with host [53]. The virus-PLoc also confirmed that this protein is a transmembrane and HMMTOP predicts 2 helices in a membrane. Three-dimensional structure with highest C-score − 4.78 (Additional file 14: Figure S7) was selected and structure verification using SAVES shows that 42.9% residues are in favored region and 44.1% residues are in additional allowed regions (Additional file 15: Figure S8). HP contains Adenovirus E3 region protein CR2 and Adenovirus E3 region protein CR1 motifs along with one conserved structural motif Gln171-Pro173 (Additional file 7: Table S7).
Q1L4D7
Q1L4D7 is predicted as adenoviral protein L1 and confidence level for this HP is seven out of nine respectively. This protein expresses in both early and late stage of viral life cycle and plays multiple roles in DNA packaging [46]. We have modeled its three-dimensional structure and out of five models with C-score − 4.53 (Additional file 16: Figure S9) was selected. RC-plot shows that 34.7% residues are in favored region and 46.0% are present in the additional allowed regions (Additional file 17: Figure S10) and contains two structure motifs Glu65-Ala67 and Val115-Gly117 (Additional file 7: Table S7).
I6LEV1
I6LEV1 is also predicted as adenoviral protein L1 like HP Q1L4D7. Structure verification of model with C-score − 4.56 (Additional file 18: Figure S11) shows that 36.0% residues are in favored regions and 43.2% residues are in additional allowed regions of RC-plot (Additional file 19: Figure S12). Sequence based function prediction verified by structural analysis and predicted three structural motifs Leu22-Leu24, Val98-Glu100 and Arg126-His128 (Additional file 7: Table S7).
To summarize, this study helped to search functionality in the hypothetical proteins of human adenovirus whose exact role in the infectious cycle was still unknown. Finally, we may emphasize that quantitative computational analysis that is carried out in the present study, may help us in better understanding of the biology of adenovirus as a whole and identify potential therapeutic leads to molecular level and may facilitate better understanding of the human biology.
Limitations
As our study is based on less sample size, increase sample size can provide more information about the function of HPs proteins and for identifying novel drug targets and this study is totally based on in silico analysis but through side by side wet lab analysis these proteins can be used for drug targeting analysis on experimental basis.
Additional files
Additional file 1: Table S1. This table reports list of 38 hypothetical proteins of human adenovirus along with their UniProt ID, corresponding genome and protein length.
Additional file 2: Table S2. This table presents list of predicted physiochemical properties of 38 human adenovirus hypothetical proteins.
Additional file 3: Table S3. This table reports list of predicted sub-cellular localization of the 38 HPs from human adeno viruses (HADVs).
Additional file 4: Table S4. This table details list of annotated function of 38 human adenovirus using BLASTp, SMART, INTERPROSCAN and MOTIF.
Additional file 5: Table S5. This table presents list of functionally annotated domain and motifs of HPs 38 s from human adenovirus by CATH, SUPERFAMILY, Pfam, CDART, ProtNet and SVMprot.
Additional file 6: Table S6. This table presents ligand binding sites prediction of hypothetical proteins of human adenovirus.
Additional file 7: Table S7. Sequence and structure motif prediction by pro-func.
Additional file 8: Figure S1. 3D structure of hypothetical protein P03269 predicted from I-TASSER.
Additional file 9: Figure S2. Evaluation of 3D structure of hypothetical protein P03269 through Ramachandran Plot.
Additional file 10: Figure S3. 3D structure of hypothetical protein P03261 predicted from I-TASSER.
Additional file 11: Figure S4. Evaluation of 3D structure of Hypothetical Protein P03261 through Ramachandran Plot
Additional file 12: Figure S5. 3D structure of hypothetical protein P03263 predicted from I-TASSER.
Additional file 13: Figure S6. Evaluation of 3D structure of hypothetical protein P03263 through Ramachandran Plot.
Additional file 14: Figure S7. 3D structure of hypothetical protein Q83127 predicted from I-TASSER.
Additional file 15: Figure S8. Evaluation of 3D structure of hypothetical protein Q83127 through Ramachandran Plot.
Additional file 16: Figure S9. 3D structure of hypothetical protein Q1L4D7 predicted from I-TASSER.
Additional file 17: Figure S10. Evaluation of 3D structure of hypothetical protein Q1L4D7 through Ramachandran Plot.
Additional file 18: Figure S11. 3D structure of hypothetical protein 16LEV1 predicted from I-TASSER.
Additional file 19: Figure S12. Evaluation of 3D structure of hypothetical protein I6LEV1 through Ramachandran Plot.
Authors’ contributions
MN, ST, MU, ZC and GA carried out characterization of hypothetical proteins following methodology designed by ST. ST and MU wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article as additional information files.
Consent to publish
Not applicable.
Ethics approval and consent to participate
The data used in this study was retrieved from online publicly available database so no ethical approval was required as not use any living organism.
Funding
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- HAdV
human adenoviruses
- HP
hypothetical protein
- GRAVY
grand average of hydropathicity
- I-TASSAR
iterative threading assembly refinement
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13104-017-2992-z) contains supplementary material, which is available to authorized users.
Contributor Information
Muhammad Naveed, Phone: 0092-3015524624, Email: naveed.quaidian@gmail.com, Email: dr.naveed@ucp.edu.pk.
Sana Tehreem, Email: sanatehreem69@yahoo.com.
Muhammad Usman, Email: meusman45@gmail.com.
Zoma Chaudhry, Email: zoomachaudhry94@gmail.com.
Ghulam Abbas, Email: imabbas2@gmail.com.
References
- 1.Davison AJ, Benko M, Harrach B. Genetic content and evolution of adenoviruses. J Gen Virol. 2003;84(11):2895–2908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 2.Robinson CM, Singh G, Lee JY, et al. Molecular evolution of human adenoviruses. Sci Rep. 2013;3:1812. doi: 10.1038/srep01812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramke M, et al. The 5′ UTR in human adenoviruses: leader diversity in late gene expression. Sci Rep. 2017;7(1):618. doi: 10.1038/s41598-017-00747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghebremedhin B. Human adenovirus: viral pathogen with increasing importance. Eur J Microbiol Immunol. 2014;4(1):26–33. doi: 10.1556/EuJMI.4.2014.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott MK, et al. Human adenovirus associated with severe respiratory infection, Oregon, USA, 2013–2014. Emerg Infect Dis. 2016;22(6):1044. doi: 10.3201/eid2206.151898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang G, Xu W. Recent advance in new types of human adenovirus. Chin J Virol. 2013;29(3):342–348. [PubMed] [Google Scholar]
- 7.Li X, et al. An outbreak of acute respiratory disease in China caused by human adenovirus type B55 in a physical training facility. Int J Infect Dis. 2014;28:117–122. doi: 10.1016/j.ijid.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lion T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev. 2014;27(3):441–462. doi: 10.1128/CMR.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sayers EW, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2011;39(suppl 1):D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivashankari S, Shanmughavel P. Functional annotation of hypothetical proteins—a review. Bioinformation. 2006;1(8):335–338. doi: 10.6026/97320630001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barragán-Osorio L, et al. Computational analysis and functional prediction of ubiquitin hypothetical protein: a possible target in Parkinson disease. Cent Nerv Syst Agents Med Chem. 2016;16(1):4–11. doi: 10.2174/1871524915666150722120605. [DOI] [PubMed] [Google Scholar]
- 12.v L, Poux S, Estreicher A, et al. The UniProtKB guide to the human proteome. Database J Biol Databases Curation. 2016;2016:bav120. doi: 10.1093/database/bav120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Consortium, U The universal protein resource (UniProt) Nucleic Acids Res. 2008;36(suppl 1):D190–D195. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook, Humana Press; 2005. pp. 571–607.
- 15.Gasteiger E, et al. Protein identification and analysis tools on the ExPASy server. Berlin: Springer; 2005. [DOI] [PubMed] [Google Scholar]
- 16.Gazi MA, et al. Functional, structural and epitopic prediction of hypothetical proteins of Mycobacterium tuberculosis H37Rv: an in silico approach for prioritizing the targets. Gene. 2016;591(2):442–455. doi: 10.1016/j.gene.2016.06.057. [DOI] [PubMed] [Google Scholar]
- 17.Shen H-B, Chou K-C. Virus-PLoc: a fusion classifier for predicting the subcellular localization of viral proteins within host and virus-infected cells. Biopolymers. 2007;85:233–40. doi: 10.1002/bip.20640. [DOI] [PubMed] [Google Scholar]
- 18.Shen HB, Chou KC. Virus-PLoc: a fusion classifier for predicting the subcellular localization of viral proteins within host and virus-infected cells. Biopolymers. 2007;85(3):233–240. doi: 10.1002/bip.20640. [DOI] [PubMed] [Google Scholar]
- 19.Krogh A, et al. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 20.Zhou H, Zhou Y. Predicting the topology of transmembrane helical proteins using mean burial propensity and a hidden-Markov-model-based method. Protein Sci Publ Protein Soc. 2003;12(7):1547–1555. doi: 10.1110/ps.0305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welner S, Nielsen M, Rasmussen M, Buus S, Jungersen G, Larsen LE. Prediction and in vitro verification of potential CTL epitopes conserved among PRRSV-2 strains. Immunogenetics. 2017;69(10):689–702. doi: 10.1007/s00251-017-1004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17(9):849–850. doi: 10.1093/bioinformatics/17.9.849. [DOI] [PubMed] [Google Scholar]
- 23.Mahram A, Herbordt MC. Fast and accurate NCBI BLASTP: acceleration with multiphase FPGA-based prefiltering. In: Proceedings of the 24th ACM international conference on supercomputing. New York: ACM; 2010.
- 24.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Li YH, Xu JY, Tao L, et al. SVM-Prot 2016: a web-server for machine learning prediction of protein functional families from sequence irrespective of similarity. PLoS ONE. 2016;11(8):e0155290. doi: 10.1371/journal.pone.0155290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasson O, et al. ProtoNet: hierarchical classification of the protein space. Nucleic Acids Res. 2003;31(1):348–352. doi: 10.1093/nar/gkg096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkataraman A, Chew TH, Hussein ZAM, Shamsir MS. A protein short motif search tool using amino acid sequence and their secondary structure assignment. Bioinformation. 2011;7(6):304–306. doi: 10.6026/007/97320630007304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32(suppl 1):D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geer LY, et al. CDART: protein homology by domain architecture. Genome Res. 2002;12(10):1619–1623. doi: 10.1101/gr.278202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knudsen M, Wiuf C. The CATH database. Hum Genom. 2010;4(3):207–212. doi: 10.1186/1479-7364-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearl FM, et al. The CATH extended protein-family database: providing structural annotations for genome sequences. Protein Sci. 2002;11(2):233–244. doi: 10.1110/ps.16802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40(D1):D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acid Res. 2000;28(1):231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson D, et al. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009;37(suppl 1):D380–D386. doi: 10.1093/nar/gkn762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson D, Madera M, Vogel C, Chothia C, Gough J. The SUPERFAMILY database in 2007: families and functions. Nucleic Acid Res. 2007;35(Database issue):D308–D313. doi: 10.1093/nar/gkl910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zdobnov EM, Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 37.Rentzsch R, Orengo CA. Protein function prediction using domain families. BMC Bioinform. 2013;14(Suppl 3):S5. doi: 10.1186/1471-2105-14-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ceroni A, Passerini A, Vullo A, Frasconi P. DISULFIND: a disulfide bonding state and cysteine connectivity prediction server. Nucleic Acid Res. 2006;34(Web Server issue):W177–W181. doi: 10.1093/nar/gkl266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ceroni A, et al. DISULFIND: a disulfide bonding state and cysteine connectivity prediction server. Nucleic Acids Res. 2006;34(suppl 2):W177–W181. doi: 10.1093/nar/gkl266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naveed M, et al. Bioinformatics based structural characterization of glucose dehydrogenase (gdh) gene and growth promoting activity of Leclercia sp. QAU-66. Braz J Microbiol. 2014;45(2):603–611. doi: 10.1590/S1517-83822014000200031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang W, Xia M, Chen J, et al. Data set for phylogenetic tree and RAMPAGE Ramachandran plot analysis of SODs in Gossypium raimondii and G. arboreum. Data Br. 2016;9:345–348. doi: 10.1016/j.dib.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naveed M, et al. In-silico analysis of non-synonymous-SNPs of STEAP2: to provoke the progression of prostate cancer. Open Life Sci. 2016;11(1):402–416. [Google Scholar]
- 44.Kumar K, et al. Structure-based functional annotation of hypothetical proteins from Candida dubliniensis: a quest for potential drug targets. 3 Biotech. 2015;5(4):561–576. doi: 10.1007/s13205-014-0256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33(suppl 2):W89–W93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gustin KE, Lutz P, Imperiale MJ. Interaction of the adenovirus L1 52/55-kilodalton protein with the IVa2 gene product during infection. J Virol. 1996;70(9):6463–6467. doi: 10.1128/jvi.70.9.6463-6467.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kantardjieff KA, Rupp B. Protein isoelectric point as a predictor for increased crystallization screening efficiency. Bioinformatics. 2004;20(14):2162–2168. doi: 10.1093/bioinformatics/bth066. [DOI] [PubMed] [Google Scholar]
- 48.School K, et al. Predictive characterization of hypothetical proteins in Staphylococcus aureus NCTC 8325. Bioinformation. 2016;12(3):209. doi: 10.6026/97320630012209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Islam M, et al. In silico structural and functional annotation of hypothetical proteins of Vibrio cholerae O139. Genom Inform. 2015;13(2):53–59. doi: 10.5808/GI.2015.13.2.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamanoi F, Stillman BW. Function of adenovirus terminal protein in the initiation of DNA replication. Proc Natl Acad Sci. 1982;79(7):2221–2225. doi: 10.1073/pnas.79.7.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lieber A, He C-Y, Kay MA. Adenoviral preterminal protein stabilizes mini-adenoviral genomes in vitro and in vivo. Nat Biotechnol. 1997;15(13):1383–1387. doi: 10.1038/nbt1297-1383. [DOI] [PubMed] [Google Scholar]
- 52.Garg P, Burgers PM. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol. 2005;40(2):115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- 53.Deryckere F, Burgert H-G. Early region 3 of adenovirus type 19 (subgroup D) encodes an HLA-binding protein distinct from that of subgroups B and C. J Virol. 1996;70(5):2832–2841. doi: 10.1128/jvi.70.5.2832-2841.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. This table reports list of 38 hypothetical proteins of human adenovirus along with their UniProt ID, corresponding genome and protein length.
Additional file 2: Table S2. This table presents list of predicted physiochemical properties of 38 human adenovirus hypothetical proteins.
Additional file 3: Table S3. This table reports list of predicted sub-cellular localization of the 38 HPs from human adeno viruses (HADVs).
Additional file 4: Table S4. This table details list of annotated function of 38 human adenovirus using BLASTp, SMART, INTERPROSCAN and MOTIF.
Additional file 5: Table S5. This table presents list of functionally annotated domain and motifs of HPs 38 s from human adenovirus by CATH, SUPERFAMILY, Pfam, CDART, ProtNet and SVMprot.
Additional file 6: Table S6. This table presents ligand binding sites prediction of hypothetical proteins of human adenovirus.
Additional file 7: Table S7. Sequence and structure motif prediction by pro-func.
Additional file 8: Figure S1. 3D structure of hypothetical protein P03269 predicted from I-TASSER.
Additional file 9: Figure S2. Evaluation of 3D structure of hypothetical protein P03269 through Ramachandran Plot.
Additional file 10: Figure S3. 3D structure of hypothetical protein P03261 predicted from I-TASSER.
Additional file 11: Figure S4. Evaluation of 3D structure of Hypothetical Protein P03261 through Ramachandran Plot
Additional file 12: Figure S5. 3D structure of hypothetical protein P03263 predicted from I-TASSER.
Additional file 13: Figure S6. Evaluation of 3D structure of hypothetical protein P03263 through Ramachandran Plot.
Additional file 14: Figure S7. 3D structure of hypothetical protein Q83127 predicted from I-TASSER.
Additional file 15: Figure S8. Evaluation of 3D structure of hypothetical protein Q83127 through Ramachandran Plot.
Additional file 16: Figure S9. 3D structure of hypothetical protein Q1L4D7 predicted from I-TASSER.
Additional file 17: Figure S10. Evaluation of 3D structure of hypothetical protein Q1L4D7 through Ramachandran Plot.
Additional file 18: Figure S11. 3D structure of hypothetical protein 16LEV1 predicted from I-TASSER.
Additional file 19: Figure S12. Evaluation of 3D structure of hypothetical protein I6LEV1 through Ramachandran Plot.
Data Availability Statement
All data generated or analyzed during this study are included in this published article as additional information files.