

Abstract
Introduction:
Next-generation-sequencing (NGS) is increasingly used in clinical and research protocols for patients with cancer. NGS assays are routinely used in clinical laboratories to detect mutations bearing on cancer diagnosis, prognosis and personalized therapy. A typical assay may interrogate 50 or more gene targets that encompass many thousands of possible gene variants. Analysis of NGS data in cancer is a labor-intensive process that can become overwhelming to the molecular pathologist or research scientist. Although commercial tools for NGS data analysis and interpretation are available, they are often costly, lack key functionality or cannot be customized by the end user.
Methods:
To facilitate NGS data analysis in our clinical molecular diagnostics laboratory, we created a custom bioinformatics tool termed Houston Methodist Variant Viewer (HMVV). HMVV is a Java-based solution that integrates sequencing instrument output, bioinformatics analysis, storage resources and end user interface.
Results:
Compared to the predicate method used in our clinical laboratory, HMVV markedly simplifies the bioinformatics workflow for the molecular technologist and facilitates the variant review by the molecular pathologist. Importantly, HMVV reduces time spent researching the biological significance of the variants detected, standardizes the online resources used to perform the variant investigation and assists generation of the annotated report for the electronic medical record. HMVV also maintains a searchable variant database, including the variant annotations generated by the pathologist, which is useful for downstream quality improvement and research projects.
Conclusions:
HMVV is a clinical grade, low-cost, feature-rich, highly customizable platform that we have made available for continued development by the pathology informatics community.
Keywords: Bioinformatics, molecular pathology, next-generation sequencing, pathology informatics
INTRODUCTION
In the era of personalized medicine, detection of gene mutations is crucial to guiding cancer diagnosis, prognosis, and targeted treatment strategies.[1,2,3] To this end, next-generation sequencing (NGS) is routinely performed at one or more points in the cancer health-care experience, including initial diagnosis, progression after treatment, and recurrence or metastasis.[4,5] NGS tests for cancer may range in scope from targeted “hotspot” assays that detect well-characterized mutations in a small number of genes to large assays that sequence the complete coding region of many hundreds or thousands of genes.[6,7] Interpretation and maintenance of these variant datasets are challenging to clinical laboratories.[8] Documentation of regulatory compliance is also a critical but often burdensome task.[9] Importantly, low-cost, high-quality, facile bioinformatics solutions appropriate for clinical laboratories are not readily accessible. Limitations of many commercially available packages include (1) incompatibility with different instrument platforms, (2) inability to be configured for laboratory developed tests, (3) limited flexibility for customization by the end user, (4) poor integration with downstream tools and the laboratory information system, and (5) high purchase price or subscription fees.[10]
To support NGS testing in our clinical molecular diagnostics laboratory, we created the Houston Methodist Variant Viewer (HMVV). HMVV is a Java-based solution that integrates multiple sequencing instrument platforms, bioinformatics analysis, computation and storage resources, and an end-user interface. The features that we considered to be essential were (1) the user interface is visually appealing and user-friendly, (2) multiple publicly available databases can be queried, (3) a local knowledge base can be constructed and curated, (4) the query returns prompt results with low latency, and (5) the tool is easily modified to include new features as the laboratory develops new tests and scientific knowledge evolves. The critical features were determined after careful consideration of the end user pathologist's desire for functionality, IT security requirements, regulatory compliance issues, and clinical laboratory performance goals.
MATERIALS AND METHODS
Implementation of system hardware
Components of the hardware system used in our molecular diagnostics laboratory include the sequencing instruments, a Linux server, and an offsite backup storage device [Figure 1]. Our molecular diagnostics laboratory performs several different NGS assays for cancer. Depending on the NGS assay, sequencing may be performed on an Illumina MiSeq, Illumina NextSeq, Life Technologies Ion Torrent, or Life Technologies Ion Proton instrument. HMVV could also accept annotated VCF files from other sequencing platforms such as the PacBio Sequel or Oxford Nanopore MinION instruments. The Linux server (two Xeon E5-2620 v2 CPUs with 64 GB of RAM, CentOS 6.4) is configured for analysis, database hosting, and data storage. It is additionally configured with a redundant failover node if the primary system fails, and both are housed at an offsite, secure, environmentally controlled facility maintained by the hospital information technology department. Sequence data are either transferred from the instrument to the server in real-time for the Illumina instruments or as a daily rsync script for the Life Technologies instruments. Data integrity is validated nightly with an md5 checksum on a random subset of the files transferred. If the data integrity check fails, the script will email the system administrator. The local server is backed up every 24 h to a separate secure storage device, which is housed in a different physical location. The data are only accessible from the secure hospital network and are limited to specific users through hospital network credentials using the Lightweight Directory Access Protocol (LDAP).
Figure 1.
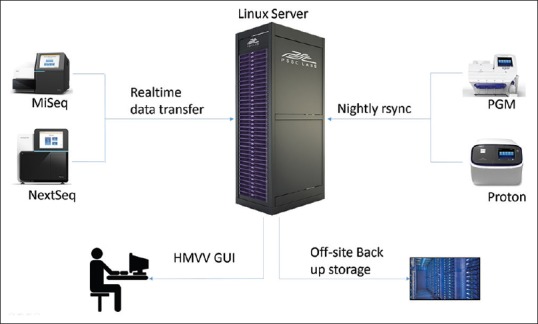
HMVV hardware configuration. HMVV is installed on a Linux server that is maintained as a clinical grade bioinformatics resource. HMVV processes the data generated by the four NGS instruments used in our clinical laboratory. Data are transferred to the server either in real time (Illumina instruments) or using a scheduled rsync command (Life Technologies instruments) and are automatically backed up daily to a separate offsite storage device. The user interfaces using HMVV. HMVV: Houston Methodist Variant Viewer
Variant analysis
The process to generate a VCF file differs for each NGS instrument [Figure 2]. For the Illumina MiSeq instrument, the onboard software generates the alignment and VCF files, which are automatically transferred to the Linux server. For the Illumina NextSeq instrument, the technologist copies the BCL files generated on the instrument to the Linux server and uses the Illumina-provided bcl2fastq script to convert the BCL files to FASTQ files. The Burrows-Wheeler Aligner (BWA-MEM version 0.7.12)[11] is used to generate an alignment file. The alignment file is sorted based on coordinate using Picard (version 1.134)[12] and indexed using SAMTools (version 1.4)[13] for fast random access and extracting alignments overlapping particular genomic regions. Next, in the variant calling process, the SAMTools mpileup and VarScan (version 2.3.9)[14] pileup2snp and pileup2indel commands call SNPs and InDels, which are then imported into a VCF file. For the Life Technologies Ion Torrent and Ion Proton instruments, the FASTQ, alignment, and VCF files are generated on the instrument server using onboard software and transferred to the Linux server daily.
Figure 2.
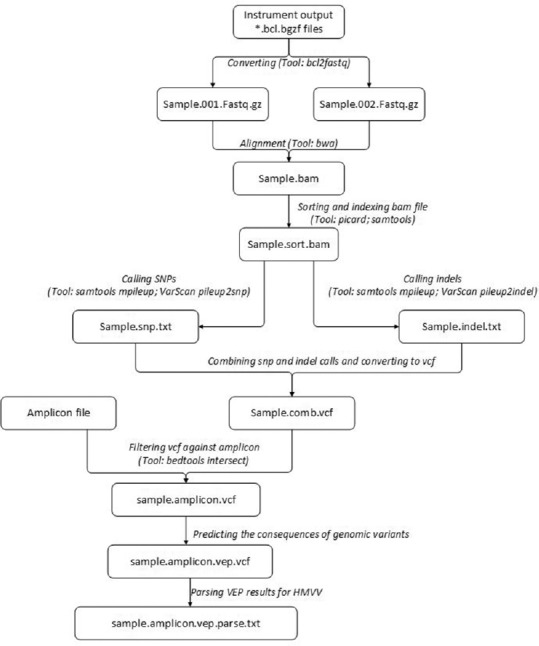
HMVV variant analysis process. Variant calls are saved in a MySQL database that is visualized by HMVV. HMVV: Houston Methodist Variant Viewer
The VCF files are then annotated with data generated by Variant Effect Predictor (VEP version 83).[15] BEDtools (version 2.17.0)[16] is used to perform genome arithmetic that predicts the affected gene, cDNA, and protein alterations. The VEP data are parsed and imported into HMVV. Once in the database, the molecular pathologist can view and annotate the variant data using HMVV.
Variant interpretation
A molecular pathologist interprets the clinical relevance of each filtered variant call and prepares the laboratory report using HMVV [Figure 3]. HMVV, built in the Java programming language, is our custom developed application, which features a graphical user interface to a MySQL database. Because it is developed in Java, HMVV can be interchangeably used on workstations running Windows, Macintosh, or Linux operating systems. HMVV queries the data stored in the MySQL database and online resources to provide rapid and user-friendly access to the biomedical information needed for the pathologist to interpret the variant calls and prepare the clinical laboratory report. When rendering variant results, HMVV cross-references local copies of G1000, ClinVar, and COSMIC databases to output the relevant details. Quick access to this information enables the molecular pathologist to quickly evaluate each variant detected.
Figure 3.

HMVV workflow. HMVV integrates all workflow from NGS data processing through variant annotation. (a) The molecular technologist enters patient metadata such as name, medical record number, and order number using the “Enter Sample” interface. (b) The “Sample List” panel shows all samples in the database for the selected assay. The sample list can be sorted by clicking any column header. (c) The “Mutation List” panel shows key information for each variant detected for a selected sample. Fields include Gene-gene name – Exon: Exon location of the detected variant, HGVSc: Human Genome Variation Society Coding DNA nomenclature, HGVSp: Human Genome Variation Society Protein nomenclature, dbSNP: Link to the variant in the dbSNP database, cosmicID: Link to the variant in the COSMIC database, Type: Variant type, including snv, deletion, insertion, indel, Genotype: Impact as predicted by Variant Effect Predictor, Life Technologies assays – altFreq: Allele frequency based on Flow Evaluator observation counts, readDP: Flow Evaluator read depth at the locus to a position and used in variant calling, altReadDP: Flow Evaluator Alternate allele observations, Illumina assays – altFreq: The percentage of reads supporting the alternate allele, readDP: Number of base calls aligned to a position and used in variant calling, altReadDP: The number of alternate calls, Occurrence: Number of previous occurrences of the detected variant in our database, Annotation: The text entered by the pathologist to generate the clinical laboratory report, (d) The “Annotation” panel allows the pathologist to designate if the variant is known to be somatic/germline/unknown and benign/likely benign/likely pathogenic/pathogenic/unknown. The pathologist can also enter text to annotate the variant, such as its likely implication to prognosis and targeted therapy, to be used in the clinical laboratory report. HMVV: Houston Methodist Variant Viewer
As recommended by multiple international expert bodies, the molecular pathologist annotates each variant based on several criteria[9,17,18] including (1) somatic or germline status of the variant, (2) cataloging of the variant in various curated public databases, (3) frequency of detection of the variant, or mutations in the gene in general, in the type of cancer tested or human malignancy overall, (4) predicted or known consequence of the variant on protein function, (5) implication of the variant, or mutations in the gene in general, on diagnosis, prognosis, or targeted therapy for the type of cancer tested or human malignancy overall, and (6) use of the variant to qualify the patient for clinical trials.
Access control
HMVV access is controlled by the Linux server credentials that are natively tied to the user's hospital credentials maintained by the department of information technology. Credentials are managed using LDAP. Therefore, the server administrator can easily grant, modify, or revoke HMVV access. Two types of HMVV users are available. Standard users have read-only access and can enter new samples with associated metadata, such as patient name and surgical pathology case number. Super users have full read and write access, including the ability to enter variant annotation information for clinical laboratory report generation and future reference. Whereas we typically grant limited access to laboratory technologists and pathology residents and fellows, full access is reserved for HMVV developers and molecular pathologists. The bioinformatics team has privileges to manage individual user accounts, which are controlled by Linux user groups.
Workflow
On completion of the bioinformatics pipeline, the molecular technologist enters each new sample into the database using the “Enter Sample” form [Figure 3a]. Users specify the assay (our laboratory offers several different NGS assays that are targeted to different cancer types or clinical indications), instrument, and instrument generated run ID. The run ID is useful for linking control samples to the patient samples performed on each assay run. HMVV will locate the sample files on the server and import them into the database.
Entered samples are viewed in the “Sample List” window [Figure 3b], which also supports sample and mutation search functions. Because it is often helpful for the molecular pathologist to visualize the sequencing alignment, HMVV can locate and load the alignment file for the selected patients into Integrated Genome Viewer (IGV)[19] by clicking the “Load IGV” button. IGV can be downloaded at https://software.broadinstitute.org/software/igv/.
The “Mutation List” window contains tabs populated with various mutation information fields for the selected sample [Figure 3c]. Users may apply filters to the data. In the current version, available filters for variants include presence in the COSMIC database, frequency compared to the wildtype sequence in the sample, minimum read depth, previous occurrences in our database, and maximum population frequency. The blue-colored text fields are clickable links to the indicated internet sites. For example, clicking on a link in the “cosmicID” column will launch the COSMIC overview for that variant in an HTML browser. Similarly, previous occurrences of the same variant can be located by clicking on the link in the “Occurrence” column.
HMVV does not natively categorize variants; rather, it provides a framework for bringing all the appropriate information into one place to facilitate the interpretation process. Considerable time and effort may be expended while researching the clinical pathological implication of each variant detected. These data, such as whether the variant is a known somatic mutation, is function altering (pathogenic), or affects patient prognosis or response to therapy, are typically included in the clinical laboratory report. HMVV allows the molecular pathologist to enter this annotation information into the database to facilitate report generation [Figure 3d] and store it for future reference.
Finally, users may select the reportable mutations and generate a human-readable text report by clicking the “Report” button. This feature is used to create the laboratory report that is entered into the laboratory information system. The mutation list can also be exported into a text file for downstream applications. At present, transfer of this text report into the LIS is a manual process (copy from HMVV and paste into LIS); however, the possibility of developing an interface in the future is under investigation.
Validation of variant calling pipeline
We use HMVV to analyze data for multiple NGS assays. Each assay was validated using nationally recognized clinical laboratory standards.[9,17,18] In brief, a combination of well-characterized control materials (Coriell Institute, Horizon DX, and Integrated DNA Technologies) and previously and prospectively tested patient samples were used. For all assays, limit of detection was determined to be 10% mutant allele frequency. Precision was determined by repeating the same samples multiple times on one sequencing run (intrarun reproducibility) and different runs performed by the same technologist (interrun reproducibility) and different technologists (intertechnologist reproducibility). Furthermore, several sequence datasets were analyzed multiple times to show reproducibility of results generated by the bioinformatics pipeline. For each assay, a trial was performed to correlate results generated by our assay to results generated by another method or a reference laboratory. Concordance >95% was considered acceptable.
RESULTS AND DISCUSSION
We have successfully developed, validated, and implemented HMVV as a bioinformatics tool for NGS data interpretation in our clinical molecular diagnostics laboratory. It is routinely used to interpret NGS data generated by multiple assays targeting gene mutations implicated in cancer. To date, we have used HMVV to evaluate more than 1100 samples. Importantly, it seamlessly accommodates data from several different NGS assays customized for different cancer types and integrates variant information from multiple public databases. Furthermore, HMVV is agnostic to the NGS sequencing instrument platform and the user's computer operating system.
Compared to the predicate method, HMVV substantially improves the user experience. Before developing HMVV, our bioinformatics workflow was cumbersome and time-consuming. First, the molecular technologist would execute a series of command line scripts to generate the list of variants detected from each sample. These variants would be imported into a spreadsheet. Then, the molecular pathologist would query a variety of online resources, one by one, to investigate the possible biological implication of each variant to determine if it should be included in the clinical laboratory report, and if so, how it should be annotated. As our clinical service grew in the number of different NGS assays offered and the number of specimens tested, we recognized the need to develop a custom bioinformatics solution. HMVV standardizes data interpretation by providing consistent centralized access to key resources such as COSMIC, dbSNP, and other online databases used by pathologists to investigate each variant detected. Technologists benefit from the user-friendly interface for entering patient metadata, ease of invoking the variant caller process without having to use the command line, and built-in storage and searchability of quality control metrics. Key to the molecular pathologist, each variant is easily queried to understand its possible biomedical implication. Furthermore, the pathologist can query the database to determine whether a particular variant has been previously detected, and if so, how it was reported. Similarly, samples can be sorted by patient name (or other text fields) to quickly compare results from previous studies. Because the annotation for each variant is saved, the historic data can guide future investigations if it is detected again in a different patient sample.
An important feature of HMVV is that it is readily customizable. Compared to the initial beta version, the current version has undergone many improvements such as inclusion of additional public databases and patient metadata fields. These enhancements have improved the user experience for technologists and pathologists, and they have facilitated quality improvement and clinical research projects. Interest in such a tool is increasing among pathologists, oncologists, and research scientists.[20]
We have made the code for HMVV publicly available at https://github.com/hmvv. We encourage HMVV users to document their experience, provide suggestions for improvement, and share self-generated modifications using the attached wiki so that others may benefit from ongoing innovation. By releasing the HMVV code, we encourage its further development. In particular, we seek opportunities to integrate HMVV into different laboratory information systems or rich-text models for reporting variant interpretations into the electronic medical record. Although commercial tools do exist, we believe that community developed, clinical grade applications such as HMVV are important to the pathology informatics community.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We thank the laboratory technologists in the Molecular Diagnostics Laboratory and the many residents and fellows in the Department of Pathology and Genomic Medicine at Houston Methodist Hospital for their contributions to HMVV.
Footnotes
Available FREE in open access from: http://www.jpathinformatics.org/text.asp?2017/8/1/44/219119
REFERENCES
- 1.Coyne GO, Takebe N, Chen AP. Defining precision: The precision medicine initiative trials NCI-MPACT and NCI-MATCH. Curr Probl Cancer. 2017;41:182–93. doi: 10.1016/j.currproblcancer.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. 2017;168:584–99. doi: 10.1016/j.cell.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Surrey LF, Luo M, Chang F, Li MM. The genomic era of clinical oncology: Integrated genomic analysis for precision cancer care. Cytogenet Genome Res. 2016;150:162–75. doi: 10.1159/000454655. [DOI] [PubMed] [Google Scholar]
- 4.Kou T, Kanai M, Yamamoto Y, Kamada M, Nakatsui M, Sakuma T, et al. Clinical sequencing using a next-generation sequencing-based multiplex gene assay in patients with advanced solid tumors. Cancer Sci. 2017;108:1440–6. doi: 10.1111/cas.13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheikine Y, Kuo FC, Lindeman NI. Clinical and technical aspects of genomic diagnostics for precision oncology. J Clin Oncol. 2017;35:929–33. doi: 10.1200/JCO.2016.70.7539. [DOI] [PubMed] [Google Scholar]
- 6.Rathi V, Wright G, Constantin D, Chang S, Pham H, Jones K, et al. Clinical validation of the 50 gene ampliSeq cancer panel V2 for use on a next generation sequencing platform using formalin fixed, paraffin embedded and fine needle aspiration tumour specimens. Pathology. 2017;49:75–82. doi: 10.1016/j.pathol.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 7.Ballester LY, Luthra R, Kanagal-Shamanna R, Singh RR. Advances in clinical next-generation sequencing: Target enrichment and sequencing technologies. Expert Rev Mol Diagn. 2016;16:357–72. doi: 10.1586/14737159.2016.1133298. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt B, Hildebrandt A. Next-generation sequencing: Big data meets high performance computing. Drug Discov Today. 2017;22:712–7. doi: 10.1016/j.drudis.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Aziz N, Zhao Q, Bry L, Driscoll DK, Funke B, Gibson JS, et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med. 2015;139:481–93. doi: 10.5858/arpa.2014-0250-CP. [DOI] [PubMed] [Google Scholar]
- 10.Loeffelholz M, Fofanov Y. The main challenges that remain in applying high-throughput sequencing to clinical diagnostics. Expert Rev Mol Diagn. 2015;15:1405–8. doi: 10.1586/14737159.2015.1088385. [DOI] [PubMed] [Google Scholar]
- 11.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. [Last accessed on 2017 Nov 5]. Available from: https://arxiv.org/abs/13033997v2 .
- 12.BroadInstitute. Picard. 2017. [Last accessed on 2017 Nov 5]. Available from: http://broadinstitute.github.io/picard/
- 13.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–5. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: A Joint consensus recommendation of the association for molecular pathology and College of American Pathologists. J Mol Diagn. 2017;19:341–65. doi: 10.1016/j.jmoldx.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A Joint consensus recommendation of the association for molecular pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foran DJ, Chen W, Chu H, Sadimin E, Loh D, Riedlinger G, et al. Roadmap to a comprehensive clinical data warehouse for precision medicine applications in oncology. Cancer Inform. 2017;16:1176935117694349. doi: 10.1177/1176935117694349. [DOI] [PMC free article] [PubMed] [Google Scholar]