

SUMMARY
Suppressor mutations were isolated that obviate the requirement for essential PBP2b in peripheral elongation of peptidoglycan from the midcells of dividing Streptococcus pneumoniae D39 background cells. One suppressor was in a gene encoding a single KH-domain protein (KhpA). ΔkhpA suppresses deletions in most, but not all (mltG), genes involved in peripheral PG synthesis and in the gpsB regulatory gene. ΔkhpA mutations reduce growth rate, decrease cell size, minimally affect shape, and induce expression of the WalRK cell-wall stress regulon. Reciprocal co-immunoprecipitations show that KhpA forms a complex in cells with another KH-domain protein (KhpB/JAG/EloR). ΔkhpA and ΔkhpB mutants phenocopy each other exactly, consistent with a direct interaction. RNA-immunoprecipitation showed that KhpA/KhpB bind an overlapping set of RNAs in cells. Phosphorylation of KhpB reported previously does not affect KhpB function in the D39 progenitor background. A chromosome duplication implicated FtsA overproduction in Δpbp2b suppression. We show that cellular FtsA concentration is negatively regulated by KhpA/B at the post-transcriptional level and that FtsA overproduction is necessary and sufficient for suppression of Δpbp2b. However, increased FtsA only partially accounts for the phenotypes of ΔkhpA mutants. Together, these results suggest that multimeric KhpA/B may function as a pleiotropic RNA chaperone controlling pneumococcal cell division.
Keywords: peripheral peptidoglycan synthesis, single KH-domain protein, post-transcriptional regulation, RNA-binding protein complex, WalRK cell-wall stress response
ABBREVIATED SUMMARY
RNA binding proteins KhpA and KhpB (JAG; EloR), which are virulence factors, form a complex in cells of the Gram-positive pathogen, Streptococcus pneumoniae. KhpA/B negatively regulates ftsA expression post-transcriptionally, and FtsA overproduction in ΔkhpA/B mutants is necessary and sufficient for suppression of some, but not all, mutations in certain essential genes that mediate peptidoglycan synthesis. The combined results of this study suggest that multimeric KhpA/B may act as a pleiotropic RNA chaperone controlling cell division.
INTRODUCTION
Streptococcus pneumoniae (pneumococcus; Spn) is both a commensal bacterium of the human nasopharynx and a serious opportunistic bacterial pathogen that kills millions of people annually worldwide, despite progress in the distribution of conjugated vaccines (Ferreira & Gordon, 2015, Gratz et al., 2015, Henriques-Normark & Tuomanen, 2013, Iovino et al., 2016, Oliver & Swords, 2015, Siegel & Weiser, 2015, Vernatter & Pirofski, 2013). Of utmost concern, S. pneumoniae has emerged as a “superbug” pathogen, whose antibiotic resistance presents an imminent threat to human health (CDC, 2013, WHO, 2017). S. pneumoniae is a low-GC, Gram-positive ovoid-shaped (ovococcus) bacterium (see Fig. S1A). The ovoid shape, size, and chaining of S. pneumoniae cells are determined by the peptidoglycan (PG) cell wall, which is a mesh-like macromolecule around the cytoplasmic membrane (Egan et al., 2015, Egan et al., 2017, Turner et al., 2014, Typas et al., 2012). In Gram-positive pathogens, like S. pneumoniae (Fig. S1A), PG acts as a scaffold for attachment of other surface macromolecules involved in virulence and host interactions, such as wall teichoic acids, sortase-attached proteins, and capsule (Brown et al., 2015, Massidda et al., 2013). Steps in surface-exposed PG biosynthesis are targets for many of the antibiotics to which resistance has developed in a range of bacterial pathogens (CDC, 2013, Hakenbeck et al., 2012, Walsh & Wencewicz, 2016).
In ovococci like S. pneumoniae, the equator of newly divided cells becomes the midcell septum from which FtsZ-ring mediated division and peptidoglycan (PG) elongation occur (Fig. S1A) (Fleurie et al., 2014, Jacq et al., 2015, Massidda et al., 2013, Pinho et al., 2013, Tsui et al., 2014, Zapun et al., 2008). The septal PG synthesis machine utilizes class B PBP2x (designated here as “bPBP2x” to indicate class) and other proteins to synthesize the PG wall between daughter cells, whereas the peripheral (sidewall-like) PG synthesis machine uses class B PBP2b (designated here as “bPBP2b”) and other proteins to push PG outward from the midcell division ring to elongate cells (Berg et al., 2013, Massidda et al., 2013, Morlot et al., 2013, Tsui et al., 2014). In mid-to-late stages of cell division, bPBP2x localizes separately from bPBP2b and other proteins to complete synthesis of the septal ring (Rued et al., 2017, Tsui et al., 2014). Balance between septal and peripheral PG synthesis to produce ovoid-shaped cells is mediated by the GpsB switching protein in complexes with the StkP Ser/Thr protein kinase and other division proteins (Fleurie et al., 2014, Land et al., 2013, Rued et al., 2017). Concurrent with PG synthesis, PG hydrolases remodel the PG during closure of the septal ring and separation of daughter cells (Barendt et al., 2011, Boersma et al., 2015, Pazos et al., 2017, Sham et al., 2012, Vollmer et al., 2008).
In previous studies, we isolated suppressor mutations that allowed growth of knock-out mutants in essential PG synthesis genes, such as ΔgpsB and Δpbp2b (Rued et al., 2017, Tsui et al., 2016). Mutations that inactivate the PhpP Ser/Thr protein phosphatase suppressed ΔgpsB and led to further characterization of the effects of GpsB on protein phosphorylation catalyzed by the StkP kinase (Fleurie et al., 2014, Rued et al., 2017). Suppressor analysis of Δpbp2b led to the discovery of a new, essential protein (MltG), that likely functions as an endo-lytic transglycosylase in peripheral PG synthesis (Fig. S1B) (Tsui et al., 2016). A very recent paper independently confirmed that mltG mutations suppress Δpbp2b (Stamsås et al., 2017). A current model for the composition of the peripheral PG synthesis machine (Fig. S1B) (Massidda et al., 2013, Philippe et al., 2014) includes: bPBP2b (transpeptidase (TP) that catalyzes muropeptide crosslinks) (Berg et al., 2013, Tsui et al., 2014); class A PBP1a (designated here as “aPBP1a” that acts as a transglycosylase (TG) to catalyze glycan chain formation and as a TP) (Fenton et al., 2016, Land & Winkler, 2011, Straume et al., 2017, Tsui et al., 2016); MreC, MreD, RodZ, and CozE (regulators of aPBP1a and other proteins) (Fenton et al., 2016, Land & Winkler, 2011, Philippe et al., 2014, Straume et al., 2017, Tsui et al., 2016); RodA (likely regulator of bPBP2b (Tsui et al., 2016) with TG (Meeske et al., 2016) and/or Lipid II flippase activity (Leclercq et al., 2017, Ruiz, 2015); and MltG (likely endo-lytic transglycosylase that may release glycan chains for crosslinking) (Stamsås et al., 2017, Tsui et al., 2016). In the genetic background of the virulent serotype 2 D39 progenitor strain, these proteins are singly essential, except for aPBP1a, whose absence results in significantly smaller ovoid-shaped cells with an increased aspect ratio compared to wild-type cells (Land & Winkler, 2011, Tsui et al., 2016).
Besides mltG mutations, we identified mutations or duplications in other genes that suppresses the requirement for essential bPBP2b (Tsui et al., 2016). One of these suppressor mutations is a frameshift mutation in a small (79 amino acid) ORF that encodes a single-KH domain protein, which we designate “KhpA” (Fig. 1A). KH domains are well-characterized RNA-binding structures that contain a conserved GXXG motif between two alpha helices (Nicastro et al., 2015, Valverde et al., 2008). This conserved GXXG loop interacts with RNA backbone residues (Nicastro et al., 2015, Valverde et al., 2008). All KH domains form three-stranded β-sheets packed against three α-helices, and most prokaryotic KH domains exhibit the type II αββααβ topology (Nicastro et al., 2015, Valverde et al., 2008). Usually, KH domains are found with other domains in proteins that function by binding to RNA, such as transcription factors (e.g., NusA), ribosomal proteins, or post-transcriptional RNA modifiers. However, single-KH domain RNA-binding proteins are found in important bacterial pathogens, including Streptococcus and Enterococcus species, Bacillus anthracis, Chlamydia trachomatis, and Mycobacterium tuberculosis (Fig. S2).
Fig. 1.

Domain and modeled structures of KhpA (Spd_0675) and KhpB (Spd_1849) (not drawn to scale). A. KhpA contains only one KH (“K Homology”) RNA-binding domain (amino acids (aa) 4–75). Truncated KhpA in the original D39 Δcps Δpbp2b sup1 suppressor strain (Table S2) is indicated. The structure of KhpA was modeled as described in Experimental procedures. The location of the GXXG motif in KhpA is indicated. B. KhpB contains a Jag-N domain of unknown function, KH and R3H (“RXXXH”) RNA-binding domains. KhpB is phosphorylated by StkP at T89 and one other residue (Stamsas et al., 2017, Ulrych et al., 2016). KhpB structure (tan) was modeled on the known structure of the KhpB homologue of Clostridium symbiosum (purple), which lacks a large linker region between its JAG-N and KH domains. The large linker region of Spn KhpB, which contains phosphorylated T89, lacks predicted domains, and its structure is unknown. The locations of the GXXG (KH) and RXXXH (R3H) motifs and T89~P (Spn Linker) are marked.
In this paper, we show that the absence of KhpA drastically reduces cell size, but largely maintains cell shape. Co-immunoprecipitation (Co-IP) experiments show that KhpA forms a complex with another RNA binding protein that we designate as “KhpB,” which was previously identified by Branny, Doubravová, and coworkers as a JAG-domain protein phosphorylated by the StkP Ser/Thr protein kinase in Spn (Ulrych et al., 2016). In new work, Håvarstein and coworkers reported independently that the absence of KhpB, which they called “EloR,” suppresses the requirement for bPBP2b in Spn (Stamsås et al., 2017). Physiological experiments reported here show that the absence of the KhpA/B RNA binding protein increases the cellular concentration of the FtsA division protein through a post-transcriptional mechanism, and this increase in FtsA amount is necessary and sufficient for suppression of Δpbp2b. Moreover, patterns of suppression suggest that KhpA/B regulates expression of proteins other than FtsA, and the absence of the KhpA/B RNA binding proteins induces PG stress, resulting in a strong increase in expression of the WalRK TCS regulon. Together, these results suggest that KhpA/B is a general RNA binding protein that may act as a chaperone/regulator analogous to Hfq in Gram-negative bacteria.
RESULTS
Mutations in khpA suppress Δpbp2b
pbp2b is essential in S. pneumoniae strains (Berg et al., 2013, Stamsås et al., 2017, Tsui et al., 2014, Tsui et al., 2016). Previously, we reported the isolation of 5 independent Δpbp2b suppressor mutations in an unencapsulated (Δcps) derivative of strain D39 (Tsui et al., 2016). Strain D39 is the progenitor of numerous laboratory strains, which have accumulated additional mutations that suppress or alter cell division phenotypes (Land & Winkler, 2011, Lanie et al., 2007, Rued et al., 2017). Four of the Δpbp2b suppressors contained mutations in the mltG gene (Tsui et al., 2016). The fifth suppressor strain contained wild-type mltG+, and mutations in three different genes: spd_0268(S189C), spd_0675(frameshift), and crcB1(S72L) (Table S2). We reconstructed these mutations separately in the wild-type D39 background and found that the frameshift mutation in spd_0675 (ΔA at K71) suppressed Δpbp2b (Table 1, line 8). In addition, we constructed a Δspd_0675 markerless mutation that suppressed Δpbp2b (Table 1, line 9).
Table 1.
Suppression of mutations deficient in peripheral PG synthesis by ΔkhpA or ΔkhpB mutations or by ectopic overexpression of ftsAa.
| Amplicon | Recipient strains | Number of colonies 20 h after transformationb |
|---|---|---|
| 1. Δpbp2b<>aad9 | WT (IU1824) | 0 |
| 2. ΔmreCD<>aad9 | 0 | |
| 3. ΔrodZ<>aad9 | 0 | |
| 4. ΔrodA::Pc-erm | 0 | |
| 5. ΔmltG::Pc-erm | 0 | |
| 6. ΔcozE::Pc-erm | >500 tiny coloniesc | |
| 7. ΔgpsB<>aad9 | 0 | |
| 8. Δpbp2b<>aad9 | khpA (ΔA at K71) (IU7942) | >500 |
| 9. Δpbp2b<>aad9 | ΔkhpA (IU9036) | >500 |
| 10. ΔmreCD<>aad9 | >500 | |
| 11. ΔrodZ<>aad9 | >500 | |
| 12. ΔrodA::Pc-erm | >500 | |
| 13. ΔmltG::Pc-erm | 0d | |
| 14. cozE::Pc-erm | >500 tiny coloniesc | |
| 15. ΔgpsB<>aad9 | >500 | |
| 16. Δpbp2b<>aad9 | ΔkhpB (IU10592) | >500 |
| 17. ΔmreCD<>aad9 | >500 | |
| 18. ΔrodZ<>aad9 | >500 | |
| 19. ΔrodA::Pc-erm | >500 | |
| 20. ΔmltG::Pc-erm | 0 | |
| 21. ΔgpsB<>aad9 | >500 | |
| 22. Δpbp2b<>aad9 | ftsZ+//PZn-ftsZ+ (IU12286) | 0 |
| 23. Δpbp2b<>aad9 | ftsZ+//PZn-ftsZ+ + Zne | ~100, tiny colonies |
| 24. Δpbp2b<>aad9 | ftsA+//PZn-ftsA+ (IU12310) | 0 |
| 25. Δpbp2b<>aad9 | ftsA+//PZn-ftsA+ + Zne | >500 |
| 26. ΔmreCD<>aad9 | >500 | |
| 27. ΔrodZ<>aad9 | 0 | |
| 28. ΔrodA::Pc-erm | >500 | |
| 29. ΔgpsB<>aad9 | 0 |
Recipient strains and amplicons were obtained as described in Supplemental Table S1. Transformations with indicated amplicons were performed as described in Experimental procedures. Each transformation experiment was performed 3 times independently with similar results.
Unless indicated, transformant colonies were normal sized and generally uniform, typical of the recipient strain. See text for details about the growth properties of ΔkhpA and ΔkhpB mutants.
Direct transformation of a ΔcozE::Pc-erm amplicon into IU1824, D39, and other D39-derived stains resulted in tiny colonies that grew slowly. ΔkhpA or ΔkhpB did not rescue the growth defect of the ΔcozE::Pc-erm transformants.
Control experiments were performed showing that the ΔmltG::Pc-erm amplicon did transform a Δpbp1a mutant or a merodiploid strain expressing two copies of mltG+ (Tsui et al., 2016).
0.5 mM ZnCl2 + 0.05 mM MnSO4 were added to transformation mixes and in subsequent steps to induce expression of ftsZ or ftsA under control of the PZn zinc-inducible promoter in the ectopic bgaA site. 0.05 mM Mn2+ was added to eliminate toxicity caused by addition of 0.5 mM Zn2+ (Jacobsen et al., 2011, Martin et al., 2017a, Martin et al., 2017b). Transformants of the ftsA+//PZn-ftsA+ recipient from TSAII-BA + Zn plates re-streaked at normal densities on fresh plates containing +Zn, but did not re-streak on plates lacking +Zn. Given the relatively high frequency of transformation and normal growth, it is unlikely that these different transformants picked up additional suppressor mutations that are still dependent on +Zn and FtsA overexpression.
spd_0675 encodes a small protein of 79 amino acids with a molecular mass of 9 kDa. Spd_0675 consists of only a single KH domain (Fig. 1A) (Nicastro et al., 2015, Valverde et al., 2008) and was accordingly designated as “KhpA” (KH-domain protein A). The khpA(ΔA K71) frameshift mutation in the Δpbp2b suppressor mutant truncated the last 8 amino acids of KhpA, including 4 amino acids of the KH domain, but left the GXXG RNA-interaction loop motif intact (Fig. 1A) (Nicastro et al., 2015, Valverde et al., 2008). Modeling described in Experimental procedures predicts that the structure of KhpA has the type II αββααβ topology, characteristic of prokaryotic KH domains (Fig. 1A) (Nicastro et al., 2015, Valverde et al., 2008). KhpA is widely conserved in Gram-positive species, including several important low- and high-GC Gram-positive pathogens, including various Streptococcus species and B. anthracis, and M. tuberculosis, respectively (Fig. S2). The pneumococcal KhpA homolog is closely related to KhpA of C. trachomatis. Single-KH domain proteins like KhpA have been annotated in numerous bacterial genomes, but their functions are generally unknown. Because khpA mutations suppress the requirement for bPBP2b in peripheral PG synthesis (Table 1), we hypothesized that KhpA regulates PG synthesis in S. pneumoniae.
ΔkhpA mutations reduce growth rate, decrease cell size, minimally reduce aspect ratio, and induce expression of the WalRK TCS cell-wall stress regulon
The markerless ΔkhpA mutation removed the central 73 amino acids of khpA, leaving 4 and 2 amino acids from the N- and C-termini, respectively, fused in-frame (strain IU9036, Table S1). The ΔkhpA mutant grew ≈53% slower than the wild-type parent in BHI broth (Fig. 2A). Expression of wild-type KhpA from the ectopic bgaA site complemented growth of the ΔkhpA mutant back to the wild-type level. ΔkhpA mutant cells had shorter cell lengths (by ≈30%) and widths (by ≈23%) compared to wild-type cells; however, overall cell shape was largely maintained with a reduction in aspect ratio of the ΔkhpA mutant of only ≈10% (Fig. 2B and 2D). Notably, the relative volume of ΔkhpA cells was substantially reduced by 7ap;50% compared to parent cells (Fig. 2B and 2D). Ectopic expression of khpA+ complemented the dimension defects of the ΔkhpA mutant back almost (within 10%) to wild-type sizes. Similar growth and cell morphology defects were observed for ΔkhpA mutants of the encapsulated, virulent serotype 2 D39 and serotype 4 TIGR4 strains (Fig. S3).
Fig. 2.

ΔkhpA mutations reduce growth rate, decrease cell size, minimally reduce aspect ratio, and induce expression of the WalRK TCS stress regulon. Wide-type (WT) (IU1824), ΔkhpA (IU9036), and ΔkhpA//khpA+ (IU9621; complementation) strains were compared. A. Growth curves in BHI broth. Doubling times (DT) were calculated from 3 independent growth curves for cells growing exponentially (OD620=0.03–0.3). B. Representative phase-contrast images of cells in early exponential phase. Scale bar = 1 μm. C. Relative transcript amounts of spd_1874 and spd_0104 of the WalRK regulon in ΔkhpA and ΔkhpA//khpA+ mutants compared to WT, determined by qRT-PCR as described in Experimental procedures. Averages ± SE are shown from 3 independent experiments. D. Box-and-whisker plots (whiskers, 5 and 95 percentile) of cell length, width, aspect ratio (length/width), and relative cell volume of WT, ΔkhpA, and ΔkhpA//khpA+ cells; >100 cells from exponential cultures were measured for each strain in 2 independent experiments. For C and D, P values were obtained relative to WT by one-way ANOVA analysis (GraphPad Prism). * P<0.05, ** P<0.01, and *** P<0.001.
Proteins with KH domains generally bind RNA and function as regulators. Therefore, we performed an mRNA-Seq analysis of the ΔkhpA mutant growing exponentially in BHI broth (Table S3). Unexpectedly, the relative amounts of only 19 mRNA transcripts changed significantly (threshold 1.8-fold) in the ΔkhpA mutant compared to the parent, despite the slower growth and smaller cell size of the ΔkhpA mutant (Fig. 2A, 2B, and 2D). In particular, relative transcript amounts were greatly induced for genes controlled by the WalRK two-component system (TCS), which maintains PG homeostasis and responds to PG cell wall stress in Spn and other low-GC Gram-positive bacteria (Table S3, top) (Dubrac et al., 2008, Fritz & Mascher, 2014, Gutu et al., 2010, Ng et al., 2005, Tsui et al., 2016). Beside induction of the WalRK regulon, small changes (1.8- to 3.2-fold) in relative transcript amounts were detected for several other stress-responsive genes, possibly due to the impaired growth of the ΔkhpA mutant compared to the parent (Table S3, middle).
We confirmed induction of relative transcript amounts of two WalRK TCS regulon genes (spd_1874 and spd_0104) in the ΔkhpA mutant by qRT-PCR (Fig. 2C). Both genes, which encode extracellular proteins containing LysM domains (Barendt et al., 2011, Ng et al., 2005), were strongly induced in the ΔkhpA mutant, and the induction was complemented back to the wild-type level by ectopic expression of khpA+. Previously, we reported induction of the WalRK regulon in mutants deficient in peripheral PG synthesis, including Δpbp1a, Δpbp1b, or mltG(Y488D) (Tsui et al., 2016). Taken together, the suppression of a Δpbp2b mutation, decrease in growth rate, smaller cell size, and induction of the WalRK regulon by ΔkhpA support the hypothesis that the KhpA protein plays a fundamental role in PG homeostasis and regulating peripheral PG synthesis.
ΔkhpA suppresses ΔmreCD, ΔrodZ, ΔrodA, and ΔgpsB, but not ΔmltG and ΔcozE
Previously, we showed that the mltG(Y488D) mutation, which likely reduces MltG enzymatic activity, suppressed Δpbp2b and the requirement for all other essential genes involved in peripheral PG synthesis, including ΔmreCD, ΔrodZ, and ΔrodA (Fig. S1B) (Tsui et al., 2016). Since ΔkhpA suppressed Δpbp2b (Table 1, row 9), we tested whether ΔkhpA would suppress mutations in essential genes involved in peripheral PG synthesis. ΔkhpA suppressed ΔmreCD, ΔrodZ, and ΔrodA mutations (Table 1, lines 10–12). However, ΔkhpA did not suppress ΔmltG, nor did it improve the severely inhibited growth of a ΔcozE mutant under these conditions (Table 1, lines 13–14). Consistent with lack of suppression of ΔmltG by ΔkhpA, ΔkhpA mltG(Y488D) double mutants grew much slower than ΔkhpA or mltG(Y488D) single mutants (Fig. S4). Δpbp2b ΔkhpA, ΔmreCD ΔkhpA, ΔrodZ ΔkhpA, and ΔrodA ΔkhpA double mutants had slower growth rates and reduced cell sizes similar to those of the ΔkhpA mutant (Fig. S5), consistent with the same mechanism of peripheral PG synthesis bypass. Unexpectedly, ΔkhpA mutants also partly suppressed the requirement for the GpsB switching protein (Table 1, row 15). In contrast to the double mutants described above, the ΔgpsB ΔkhpA double mutant grew considerably slower than the ΔkhpA mutant and formed large, elongated cells, characteristic of GpsB depletion (Fig. S5B). We conclude that the absence of KhpA bypasses some, but not all of the proteins required for PG synthesis and that KhpA may regulate peripheral PG synthesis by a different pathway than GpsB function.
KhpA forms a complex with KH/R3H-domain protein KhpB (JAG/EloR) in S. pneumoniae cells
A single-KH domain protein like KhpA would recognize only about 4 nucleotides; therefore, KH domains are typically in multimeric proteins that contain additional domains (Nicastro et al., 2015, Valverde et al., 2008). Consequently, we hypothesized that KhpA may be in a complex with another RNA binding protein that would provide RNA binding strength and possibly specificity. To test this idea, we constructed a strain producing C-terminal, epitope-tagged KhpA-L-FLAG3 from its normal chromosomal locus (IU9602, Table S1). This strain, which does not exhibit any phenotypic defects of khpA mutants (Fig. 2, S5, and S6), was crosslinked with formaldehyde, and KhpA-L-FLAG3 was used as bait in co-immunoprecipitation experiments (co-IP) (see Experimental procedures) (Rued et al., 2017). After heating to reverse crosslinks, a single prominent band not seen in the control was visible on silver stained SDS-PAGE gels (Fig. 3, Lane 1). This protein was identified by mass spectrometry (MS) as Spd_1849, which is another putative RNA binding protein containing KH and R3H domains that we designated as “KhpB” (KH-domain protein B; Fig. 1B). To confirm complex formation, the reciprocal co-IP experiment with crosslinking was performed in a strain expressing functional KhpB-L-FLAG3 from its native site, which is distant from khpA (see spd gene tags, Fig. 1). A single band not present in the control was detected and confirmed to be KhpA by MS (Fig. 3, lane 3).
Fig. 3.

KhpA forms a complex with RNA binding protein KhpB in cells. Silver-stained SDS-PAGE gel from co-IP experiments, performed using KhpA-L-FLAG3 (IU9602, lane 1), WT (IU1945, lane 2 non-flagged control), or KhpB-L-FLAG3 (IU10664, lane 3) as bait (see Experimental procedures). Prey bands from lanes 1 and 3 were identified by MS as KhpB and KhpA, respectively. The experiment was performed independently twice. Indicated calculated molecular masses of bands are consistent with a standard ladder that is not shown.
Spn KhpB is a protein of 328 amino acids containing a misnamed amino-terminal “JAG-N” domain of unknown function, a long linker region, and the two tandem RNA binding domains (KH and R3H) (Fig. 1B). KhpB shows a similarly wide distribution among different bacterial species as KhpA (Fig. S2). Recently, KhpB (called “JAG”) was independently identified as a protein phosphorylated at T89 and at least one other residue, by the StkP protein kinase of Spn (Ulrych et al., 2016). Very recently, ΔkhpB (called “ΔeloR”) mutations were independently discovered as suppressors of Δpbp2b in laboratory strain R6 (Stamsås et al., 2017). We conclude that KhpA and KhpB are in a complex together in Spn cells, and given the strength and singularity of the reciprocal co-IPs (Fig. 3), it is likely that KhpA and KhpB interact directly with each other.
ΔkhpB mutants phenocopy ΔkhpA mutants exactly for growth and morphology
If KhpA and KhpB directly interact in an obligatory complex, then we would expect ΔkhpA, ΔkhpB, and ΔkhpA ΔkhpB mutants to phenocopy each other. Previously, Branny, Doubravová, and coworkers showed that ΔkhpB mutants grow slightly slower and form smaller cells with a nearly unchanged aspect ratio compared to the parent in the Rx1 laboratory strain background (Ulrych et al., 2016). This phenotype is similar to that of ΔkhpA mutants (Fig. 2). We further showed that markerless ΔkhpA, ΔkhpB, and ΔkhpA ΔkhpB mutations led to similar slower growth, decreased cell lengths and widths, minimally reduced aspect ratios, significantly decreased relative cell volumes (≈2-fold), and induction of the WalRK regulon (Fig. 4). ΔkhpA ΔkhpB Δpbp2b triple mutants showed similar slower growth and smaller cell size as ΔkhpA or ΔkhpB single mutants or ΔkhpA Δpbp2b double mutants compared to the wild-type strain (Fig. S5 and S7). ΔkhpB suppressed the same mutations in peripheral PG synthesis (Δpbp2b, ΔmreCD, ΔrodZ, ΔrodA, but not ΔmltG) and PG synthesis regulation (ΔgpsB) as ΔkhpA (Table 1, lines 9–15 and 16–21). Recently, ΔkhpB (“ΔeloR”) mutants were reported to suppress Δpbp2b and ΔrodA in laboratory strain R6, where mreCD and rodZ are not essential (Straume et al., 2017) and pbp1a is mutated (Land & Winkler, 2011, Lanie et al., 2007). After discovering the interaction between KhpA and KhpB (Fig. 3), we isolated additional spontaneous Δpbp2b suppressors, which included 3 independent knock-out mutations in khpB (sup7–9, Table S2) and a null mutation in khpA (sup10, Table S2).
Fig 4.

ΔkhpB mutants phenocopy ΔkhpA mutants exactly. WT (IU1824), ΔkhpA (IU9036), ΔkhpB (IU10592), and ΔkhpA ΔkhpB (IU10596) mutants were compared. A. Growth curves in BHI broth. Doubling times (DT) were calculated from 3 independent growth curves for cells growing exponentially (OD620=0.03–0.3). B. Representative phase-contrast images of cells in early exponential phase. Scale bar = 1 μm. C. Relative transcript amounts of spd_1874 and spd_0104 of the WalRK regulon in ΔkhpA, ΔkhpB, and ΔkhpA ΔkhpB mutants compared to WT, determined by qRT-PCR as described in Experimental procedures. Averages ± SE are shown from 3 independent experiments. D. Box-and-whisker plots (whiskers, 5 and 95 percentile) of cell length, width, aspect ratio (length/width), and relative cell volume of WT, ΔkhpA, ΔkhpB, and ΔkhpA ΔkhpB cells; >100 cells from exponential cultures were measured for each strain in 2 independent experiments. For C and D, P values were calculated as for Figure 2. ns, not significantly different.
KhpA and KhpB co-localized at all stages of the cell cycle (Fig. S8). Unlike many Spn PG synthesis and division proteins (e.g., StkP, bPBP2b, and FtsZ), KhpA and KhpB were diffuse in early divisional cells (Fig. S8B and S8C). There is some concentration of KhpA and KhpB at the midcells of dividing cells (Fig. S8B), as noted before in strain R6 (Stamsås et al., 2017); however, there is still a diffuse distribution of the two proteins throughout cells (Fig. S8B and S8C). The tendency of KhpA and KhpB to concentrate at midcells may reflect the interaction of KhpB as a substrate of the StkP protein kinase, which is confined tightly to midcells throughout division (Fig. S8B) (Beilharz et al., 2012, Fleurie et al., 2014, Morlot et al., 2013, Tsui et al., 2014). The degree of phosphorylation of MapZ/StkP and DivIVA mediated by the StkP kinase and PhpP phosphatase was reduced slightly by ≈20–40% in ΔkhpA, ΔkhpB, and ΔkhpA ΔkhpB mutants compared to the wild-type strain, perhaps with lower levels in ΔkhpB mutants (Fig. S9). These small changes in phosphorylation of cell division proteins may partly be an indirect effect of the slower growth, decreased cell size, and cell wall stress of these mutants (Fig. 2 and 4). Taken together, the exact phenocopying by ΔkhpA, ΔkhpB, and ΔkhpA ΔkhpB for growth and morphology provides strong physiological support that KhpA and KhpB interact and mediate the same regulatory pathways in pneumococcal cells.
To determine whether phosphorylation of KhpB at T89 by StkP kinase is important for KhpA/B function in the D39 genetic background, we constructed the khpB(T89A) phosphoablative mutation in the chromosome of strain IU1824 (IU12744; Table S1). khpB(T89A) did not cause the aberrant growth or morphological defects of ΔkhpA/B mutants, indicating that T89~P phosphorylation of KhpB is not obligatory for its function under these culture conditions (Fig. S10). Likewise, khpB(T89D) (IU13881) or khpB(T89E) (IU13883) phosphomimetic mutants in the D39 background were readily constructed without any indication of suppressor accumulation in transformations (few colonies; colony heterogeneity), in contrast to what was reported for laboratory strain R6 (Stamsås et al., 2017). khpB(T89D) or khp(T89E) mutants also did not exhibit ostensible growth or cell morphology phenotypes (Fig. S10). Whole-genome sequencing of khpB(T89A), khpB(T89D), and khpB(T89E) mutants confirmed that no additional suppressor mutations are present (data not shown).
KhpA and KhpB interact with a similar set of RNAs in cells
Changing the KhpA(GRKG→GDDG) and KhpA(GRKG→ARKA) nucleic acid binding motif destabilized KhpA and resulted in a ΔkhpA phenotype (Fig. S11). We performed RIP-Seq (RNA-immunoprecipitation) experiments without crosslinking using KhpA-L-FLAG3 or KhpB-L-FLAG3 as bait to demonstrate that KhpA and KhpB indeed bind to RNA in cells (see Experimental procedures) (Saadeh et al., 2015). We used the ratio of RNA reads obtained for each of the FLAG-tagged strains compared to the untagged wild-type strain as the criterion for RNA binding of RNA to KhpA or KhpB (Table S4). A separate RIP-qRT-PCR control experiment (Fig. S12) detected two transcripts enriched strongly by KhpA-L-FLAG3 and KhpB-L-FLAG3 in RIP-Seq (Table S4). In addition, a third putative RNA binding protein (Spd_1366-L-FLAG3) did not pull down the two transcripts pulled down by KhpA/B-L-FLAG3, indicating that the L-FLAG3 affinity tag was not affecting the RNAs recovered by RIP (Fig. S12). Several general conclusions emerged from the RIP-Seq experiments. With a 4-fold enrichment as the cutoff (Saadeh et al., 2015), KhpA or KhpB pulled down a remarkable overlapping set of ≈170 mRNA, tRNAs, intergenic RNAs, and putative sRNAs (Fig. 5; Table S4). The strong overlap of the RNAs pulled down by KhpA or KhpB supports the idea that KhpA and KhpB interact in cells to form a complex that mediates different RNA interactions in multiple regulatory pathways. Hence, the KhpA and KhpB proteins and ΔkhpA ΔkhpB mutants will be referred to as KhpA/B and ΔkhpA/B, respectively, in the rest of this paper. Notably, many of the mRNAs pulled down by KhpA or KhpB correspond to small reading frames annotated as hypothetical proteins (Table S4). Three mRNAs corresponding to small proteins (Spd_0023 (80 aa); Spd_0963 (44 aa); and Spd_1159 (56 aa)) and two tRNAs (tRNAThr and tRNAPro) were enriched by >10-fold by pull down with KhpA or KhpB (Table S4). The mRNAs of three cell division proteins (FtsA (2–3-fold), SepF (≈3-fold), and Spd_0703 (≈7-fold) (Barendt et al., 2011)) were preferentially pulled down by KhpA or KhpB (Table S4). An interaction of KhpA/B with a specific target near the 5′-UTR leader region of the ftsA transcript is described below.
Fig. 5.

KhpA and KhpB pull down overlapping sets of RNAs in RIP-Seq experiments. Scatter plots show the fold-changes (expressed as log2) for separate mRNAs and tRNAs (A; Table S4A) or intergenic RNAs and sRNAs (B; Table S4B) pulled down by KhpA-L-FLAG3 or KhpB-L-FLAG3 in RIP-Seq experiments, where 4-fold enrichment was used as the cutoff.
FtsA overproduction is sufficient to suppress Δpbp2b
To understand suppression of Δpbp2b by ΔkhpA/B, we selected for additional suppressor mutations and checked for mltG+, khpA+, and khpB+ alleles before whole-genome sequencing (sup6, Table S2). No mutations were detected in the sup6 suppressor other than the expected Δcps and Δpbp2b mutations. However, analysis of total genomic reads and junction points showed that sup6 contains a large tandem chromosomal duplication of the region that contains the ftsA-ftsZ operon encoding the FtsA and FtsZ division proteins (Fig. 6; Experimental procedures). This duplication in ftsA resulted in an ≈3-fold increase in FtsA cellular concentration (Fig. S13). FtsZ overproduction was previously shown to suppress Δpbp2 mutations in E. coli that lack bPBP2 required for PG side-wall elongation (Bendezu & de Boer, 2008, Vinella et al., 1993). We overproduced FtsZ by ≈1.9-fold from a Zn2+-inducible promoter in an ectopic site (Fig. S14). Overproduction of FtsZ poorly suppressed Δpbp2b, resulting in tiny colonies that acquired fast-growing suppressors upon re-streaking (Table 1, lines 22–23) (see (Tsui et al., 2016)). In contrast, overproduction of FtsA (≈2.6-fold) from an ectopic site substantially suppressed Δpbp2b and allowed colony formation and growth in BHI broth (Table 1, lines 24–25; Table 2, lines 5–6; Fig. 7A and 7B; Fig S15). A control experiment showed that ectopic overexpression of ftsA did not change the relative cellular amount of FtsZ (Fig. S15). Overproduction of FtsA resulted in slightly spherical, smaller cells than wild-type (Fig. 7C(2), S16A, and S16B), as reported previously (Mura et al., 2016). FtsA overproduction did not restore wild-type morphology to Δpbp2b cells and resulted in chains of slightly irregularly shaped, often spherical cells (Fig. 7C(4)). Depletion of excess FtsA in a ftsA+ Δpbp2b//PZn-ftsA+ mutant resulted in chains of enlarged spherical cells (Fig. 7C(4)), similar to those caused by bPBP2b depletion (Fig. 7C(3)). We conclude that ≈3-fold overproduction of FtsA alone, but not FtsZ, is sufficient to restore Δpbp2b growth, without restoring wild-type cell morphology.
Fig. 6.

The chromosomal region surrounding ftsA-ftsZ (ddl′-ylmH′) is tandemly duplicated in the Δpbp2b sup6 suppressor strain. Top, number of reads from Illumina NextSeq DNA sequencing showing that the ddl′-ylm′ region is duplicated in the Δpbp2b sup6 suppressor strain (Table S2). No other mutations were detected in this strain. The number of reads across the duplicated region is ≈twice that of the adjoining regions. As expected, there are no reads from penA (pbp2b), which is deleted in the Δpbp2b suppressor strain. Reads spanning the junction between the 3′ end of the repeat region to the 5′ end of the repeat region were identified by bioinformatic analysis described in Experimental procedures, indicating that the repeat is a single tandem duplication that occurred in place without any other obvious re-arrangements taking place. Bottom, diagram of the annotated chromosomal region of the Ω(ddl′-ylm′) region, including the ftsA-ftsZ operon, duplicated in the Δpbp2b sup6 suppressor.
Table 2.
Transformation by a Δpbp2b amplicon is dependent on overproduction of FtsAa.
| Recipient strain | Number of colonies 20 h after transformation with a Δpbp2b<>Pc-erm amplicon | Relative FtsA amountb (average ± SE) |
|---|---|---|
| 1. WT (IU1824) | 0 | ≡1.0 |
| 2. WT + Znc | 0 | 0.9 ± 0.1 |
| 3. ΔkhpA (IU9036) | >500 | 2.6 ± 1.0 |
| 4. ΔkhpA +Znc | >500 | 2.3 ±0.1 |
| 5. ftsA+//PZn-ftsA+ (IU12310) e | 0 | 1.1 ± 0.1 |
| 6. ftsA+//PZn-ftsA+ + Zn ce | >500 | 2.6 ± 0.5 |
| 7. ΔftsA//PZn-ftsA+ + Zn (IU12901) ce | 0 | 1.0 ± 0.1 |
| 8. ΔkhpA ΔftsA//PZn-ftsA+ + Zn (IU12959) ce | 0 | 1.2 ± 0.2 |
| 9. ΔftsA//PftsA-ftsA+ (IU13201) f | 0 | 0.6 ± 0.1 |
| 10. ΔkhpA ΔftsA//PftsA-ftsA+ (IU13204) f | >500 | 2.3 ± 0.2 |
| 11. ftsA(ΔUTR) (IU13641) | 0 | 0.5 ± 0.1 |
| 12. ΔkhpA ftsA(ΔUTR) (IU13672) | 0 | 0.6 ± 0.1 |
Recipient strains and amplicons were obtained as described in Supplemental Table S1. Transformations were performed as described in Experimental procedures. Each transformation experiment was performed 3 times independently with similar results.
FtsA amounts relative to WT were determined in cultures growing exponentially in BHI broth by quantitative Western blotting in the linear range of detection as described in Experimental procedures. Averages (± SE) are listed for 3 independent experiments, separate from other experiments in this paper, except for lines 11–12, which are from Figure 7.
0.5 mM ZnCl2 + 0.05 mM MnSO4 were added to induce expression of ftsA under control of the PZn zinc-inducible promoter in the ectopic bgaA site. 0.05 mM Mn2+ was added to eliminate toxicity caused by addition of 0.5 mM Zn2+ (Jacobsen et al., 2011, Martin et al., 2017a, Martin et al., 2017b).
Ectopic bgaA:: PZn-ftsA+ has the Zn2+-inducible PczcD promoter (Mu et al., 2013) fused to 22 bp from the czcD 5′-UTR leader, which is fused to the 24 bp of the ftsA 5′-UTR containing the RBS upstream of the ftsA start codon and reading frame (see Fig. 9A). bgaA:: PZn-ftsA+ does not produce most of the 74 nt ftsA 5′-UTR (ΔUTR) in its transcript.
Fig. 7.

FtsA overproduction is sufficient to suppress Δpbp2b. A. ftsA+ Δpbp2b //PZn-ftsA+ mutant (IU12707) streaked on TSAII-BA plates lacking (-Zn) or containing (+Zn) 0.5 mM ZnCl2 + 0.05 mM MnSO4. B. Representative growth curves of WT (IU1824) and Δpbp2b//PZn-pbp2b+ (IU11258) and ftsA+ Δpbp2b //PZn-ftsA+ (IU12707) mutants grown in BHI broth lacking (-Zn) or containing (+Zn) 0.5 mM ZnCl2 + 0.05 mM MnSO4 as described in Experimental procedures. Strain ftsA+//PZn-ftsA+ (IU12310) grew similarly to WT with or without Zn (Fig. S16). C. Representative phase-contrast images of cells of WT (IU1824), ftsA+//PZn-ftsA+ (IU12310), Δpbp2b//PZn-pbp2b+ (IU11258), and ftsA+ Δpbp2b//PZn-ftsA+ (IU12707) strains from growth experiments similar to Figure 5B sampled at OD620 = 0.1–0.15.
Cellular FtsA concentration is negatively regulated by KhpA/B at the post-transcriptional level
We tested whether the relative cellular amount of FtsA is increased in ΔkhpA/B mutants. Quantitative Western blotting showed that cellular FtsA concentration is induced by ≈3-fold in the ΔkhpA, ΔkhpB, or ΔkhpA/B mutants compared to the khpA+/B+ wild-type strain, where unchanging MreC amount was used as a loading control (Fig. 8A, 8B, S13, and S15). In contrast, relative FtsZ amount increased by only ≈1.5-fold in the ΔkhpA/B mutants (Fig. 8B). The linearity and reproducibility of detection in quantitative Western assays was confirmed for each protein by dilution series (Experimental procedures; Fig. S17). The increase of the relative cellular amounts of FtsA and FtsZ in ΔkhpA/B mutants was similar to that detected for ectopic overexpression of ftsA or ftsZ, respectively (Table 2, lines 3 and 6; Fig. 8B, S14, and S15); but, only overproduction of FtsA, but not FtsZ, suppressed Δpbp2b (Table 1, lines, 22–25).
Fig. 8.

FtsA amount and to a lesser extent FtsZ amount are negatively regulated by KhpA/B. WT (IU1824) and ΔkhpA (IU9036), ΔkhpB (IU10592), and ΔkhpA ΔkhpB (IU10596) mutants were compared. A. Representative Western blots of cellular amounts of FtsA, FtsZ, and MreC (control) in cells growing exponentially in BHI broth. Numbers below lanes are luminescence values relative to WT, determined in the linear range of detection as described in Experimental procedures. B. Average (± SE) cellular amounts of FtsA, FtsZ, and MreC in mutants relative to WT from 3 independent experiments. C. Average (±SE) relative transcript amounts in mutants relative to WT determined by qRT-PCR from 3 independent experiments. For B and C, P values were obtained relative to WT by one-way ANOVA analysis (GraphPad Prism).* P<0.05; ** P<0.01; *** P<0.001; ns: not significantly different.
We determined how relative ftsA and ftsZ transcript amounts tracked with protein amounts. qRT-PCR assays showed that both ftsA and ftsZ relative transcript amounts increased by ≈1.5-fold in ΔkhpA, ΔkhpB, and ΔkhpA/B mutants compared to the wild-type parent (Fig. 8C). Thus, KhpA/B exert a modest degree of negative regulation on ftsA-ftsZ co-transcription that is reflected by a corresponding change in relative FtsZ amount. In contrast, KhpA/B exert negative control that decreases the relative FtsA amount significantly more than the relative transcript amount, which is indicative of post-transcriptional regulation.
The ftsA 5′-UTR is required for FtsA overproduction, which is necessary to suppress Δpbp2b in a ΔkhpA mutant
Results presented above show that ectopic overexpression of ftsA is sufficient to suppress Δpbp2b (Table 1, line 25; Table 2, line 6). We hypothesized that post-transcriptional induction of FtsA amount is also necessary for Δpbp2b suppression in a ΔkhpA mutant. Further, we speculated that this regulation would involve the 74-nt 5′-UTR leader RNA from the start of ftsA transcription to the start codon of FtsA (Fig. 9A). Consistent with this notion, the mRNA near the start codon of ftsA is preferentially pulled down 2–3-fold by KhpA or KhpB in RIP-Seq experiments, whereas the ftsZ transcript is not pulled down (Fig. S18; Table S4). To test these hypotheses genetically, we constructed merodiploid strains in which the chromosomal copy of ftsA was wild-type or deleted, and ftsA is expressed ectopically from the native PftsA promoter (entire UTR present) or from the PZn promoter (Mu et al., 2013) fused to 22 nt from the czcD leader transcript followed by the 24 nt upstream of the FtsA start codon (ΔUTR) (Table 2 and S1; Fig. 9A). Production of FtsA solely from ectopic PZn (+Zn2+; ΔUTR) was not sufficient to suppress Δpbp2b and did not respond to ΔkhpA (Table 2, lines 7–8). By contrast, production of FtsA solely from ectopic PftsA(UTR+) was induced by ΔkhpA, thereby suppressing Δpbp2b (Table 2, lines 9–10). Last, we constructed the ΔUTR deletion (Δnt 2–50) in the ftsA chromosomal locus (Fig. 9; Table 2, lines 11–12; Table S1). The relative amount of FtsA, but not FtsZ, was reduced by about 50% in the ΔUTR mutant, possibly due to a change in segmental mRNA stability or translation of ftsA, and notably, this expression level of FtsA was now insensitive to ΔkhpA (Fig. 9B and 9C). Together, these results implicate the ftsA 5′-UTR in setting the basal amount of the FtsA protein and in the negative regulation of FtsA amount by KhpA/B. They also indicate that FtsA overproduction in ΔkhpA/B mutants is necessary for Δpbp2b suppression.
Fig. 9.

FtsA induction by the ΔkhpA mutation is lost in the absence of an intact 5′-UTR leader of the ftsA transcript. A. Nucleotide (nt) sequence of 74 nt 5′-UTR leader of the ftsA transcript. The transcription start site, indicated in red, was determined by 5′-RACE (see Experimental procedures). The 49 nt deleted in the ftsA(ΔUTR) mutant are indicated in green. The retained 24 nt containing the RBS upstream of the start codon (ATG) are marked in purple. The same 24 nt was used as the RBS to express FtsA ectopically under control of the PZn promoter (Table S1). B. Representative Western blots of cellular amounts of FtsA, FtsZ, and MreC (control) in WT (IU1824) and ΔkhpA (IU9036), ftsA(ΔUTR) (IU13641), and ΔkhpA ftsA(ΔUTR) (IU13672) mutants growing exponentially in BHI broth. Numbers below lanes are luminescence values relative to WT, determined in the linear range of detection as described in Experimental procedures. C. Average (±SE) cellular amounts of FtsA, FtsZ, and MreC in mutants relative to WT from 3 independent experiments. P values were obtained relative to WT by one-way ANOVA analysis. * P<0.05; ** P<0.01; *** P<0.001; ns, not significantly different.
FtsA overproduction only partially accounts for phenotypes of ΔkhpA mutants
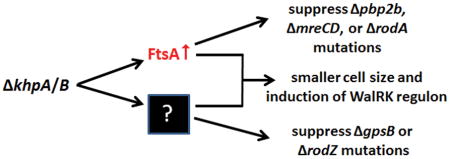
Since the relative FtsA amount is induced in ΔkhpA, ΔkhpB, and ΔkhpA/B mutants (Fig. 8B) and is necessary and sufficient for Δpbp2b suppression (Table 2), we tested whether ectopic overexpression of ftsA could account for other phenotypes of ΔkhpA or ΔkhpB mutants (Table 1). Quantitative Western blotting showed that ΔkhpA mutants and ftsA+//PZn-ftsA+ (+Zn2+) merodiploids overproduce cellular FtsA concentrations to comparable levels (Table 2, lines 3 and 6; Fig. S13 and S15). Similar to Δpbp2b, FtsA overproduction suppressed ΔmreCD and ΔrodA, but surprisingly, did not suppress ΔrodZ (Table 1, lines 25, 26, and 28), despite RodZ likely playing a role in peripheral PG synthesis (Fig. S1B) (Mura et al., 2016; Tsui et al., 2016). Likewise, increased relative FtsA amount did not suppress ΔgpsB (Table 1, line 29). As noted previously (Mura et al., 2016), overproduction of FtsA increases Spn cell sphericity and decreases relative cell volume slightly, but not nearly as drastically as ΔkhpA (Fig. S16A and S16B). In addition, overexpression of ftsA induced the cell-wall stress response mediated by the WalRK TCS (≈5-fold), but not nearly as strongly as ΔkhpA/B (≈12-fold) (Fig. S16C). We conclude that only a subset of phenotypes caused by ΔkhpA/B can be accounted for by FtsA overproduction, implying that KhpA/B regulate additional steps in PG synthesis and cell division, besides FtsA concentration (Fig. 10A).
Fig. 10.

Models for the regulation of cell division by KhpA and KhpB in S. pneumoniae D39. A. The absence of KhpA or/and KhpB increases cellular FtsA amount, which bypasses the requirement for essential bPBP2b, MreCD, and RodA. FtsA overproduction by itself partly contributes to the reduced cell size and induction of the WalRK regulon caused by ΔkhpA/B mutants. In addition, KhpA and KhpB regulate genes other than ftsA such that ΔkhpA/B mutants bypass the requirement for essential RodZ and GpsB. See text for details. B. Three models for how the KhpA/B RNA binding protein could post-transcriptionally regulate FtsA amount via the ftsA 5′-UTR leader region. Model 1 and 2 propose direct and indirect models of negative regulation of the ftsA 5′-UTR leader, whereas Model 3 is an indirect model wherein KhpA/B negatively regulates the expression or function of an unknown sRNA or protein that positively regulates FtsA production. The mutant analysis in this paper favors Model 3 (✓) as discussed in the text, although Models 1 and 2 (?) cannot be ruled out at this point.
DISCUSSION
In this paper, we report the discovery of a complex in cells between two KH-domain RNA-binding proteins, designated as KhpA and KhpB (JAG/EloR), that regulate the cellular amount of the key FtsA cell division protein in S. pneumoniae. KhpA is a small single-KH domain protein (Fig. 1A), which have been widely annotated in the genomes of Gram-positive bacteria (Fig. S2). This paper is one of the first to report a critical role of a single-KH domain protein in bacterial cell division. By contrast, the conserved KhpB(JAG/EloR) protein contains a “JAG” domain of unknown function, a large linker region of unknown structure that contains a phosphorylated Thr residue (T89), and two RNA binding domains (KH and R3H) (Fig. 1B and S2) (Ulrych et al., 2016; Stamsås et al., 2017). Several results reported here support the conclusion that KhpA and KhpB are RNA binding proteins that interact with each other in the same complex in cells. KhpA and KhpB pull down each other strongly and singularly in reciprocal co-IP experiments (Fig. 3). ΔkhpA and ΔkhpB mutants phenocopy each other by causing the same growth and cell morphology defects and cell-wall stress response (Fig. 2 and 4), and ΔkhpA and ΔkhpB mutations suppress the same set of mutations deficient in peripheral and general PG synthesis (Table 1), including Δpbp2b used in the selections that initially identified khpA and khpB(eloR) (Table 1 and S2) (Stamsås et al., 2017). Consistent with a KhpA/KhpB complex, KhpA and KhpB co-localize at all stages of cell division (Fig. S8). Finally, KhpA or KhpB separately pulled down a remarkably similar set of mRNA, sRNA, intercistronic RNA, and tRNA molecules relative to the untagged control strain in RIP-Seq experiments (Fig. 5, S12, and S18; Table S4). Together, these results support the idea that a multimeric KhpA/KhpB complex functions as a pleiotropic RNA binding protein in the control of PG synthesis and cell division in S. pneumoniae.
A Tn-Seq screen previously showed that KhpB and possibly KhpA are virulence factors required for colonization and pneumonia in a murine model of Spn TIGR4 infection (van Opijnen & Camilli, 2012). Tn-Seq data were only partially complete for KhpA, possibly because it is a small reading frame. ΔkhpA caused similar phenotypes in encapsulated TIGR4 and D39 (Fig. S3) as in unencapsulated derivatives of D39 (Fig. 2 and 4). KhpB was reported independently by three other laboratories. In a chapter of an earlier dissertation not published in a journal, Sauerbier (and Hakenbeck) found that mutant KhpB (and KhpA) imparted increased β-lactam resistance in horizontal gene transfers between Streptococcus mitis B6 and S. pneumoniae R6 (Sauerbier, 2012). In the D39 background used here, ΔkhpA/B mutants show the same sensitivity as the wild-type strain to penicillin, cefotaxime, piperacillin, and tetracycline, but are modestly resistant to gentamicin in standard disk-diffusion assays (Fig. S19). In addition, this dissertation reported induction of the WalRK TCS regulon in ΔkhpA and ΔkhpB mutants of laboratory strain R6 (Sauerbier, 2012), anticipating the similar results reported here for D39-derived strains (Fig. 4). Induction of the WalRK TCS regulon is necessary for optimal growth of a ΔkhpA mutant, since a ΔkhpA ΔwalK double mutant, lacking WalK histidine kinase, grows much slower than either ΔkhpA or ΔwalK single mutant (Fig. S20).
Recently, Branny, Doubravová and coworkers discovered KhpB, which they named “JAG,” as a substrate of the StkP Ser/Thr protein kinase, which is required for normal Spn cell division (Ulrych et al., 2016). In this study, KhpB (JAG) was shown to be phosphorylated on Thr89 and at least one other unknown site in laboratory strain Rx1. In addition, ΔkhpB(Δjag) mutations in Rx1 affected Spn cell division (Ulrych et al., 2016), and Rx1 ΔkhpB(Δjag) mutants showed slower growth rate, reduced cell size (volume), and minimally altered cell shape (aspect ratio), similar to the ΔkhpA/B mutants in the D39 progenitor background (Fig. 2 and 4). Induction of an ectopic copy of khpB+(jag+) from the PZn-inducible promoter in a Rx1 ΔkhpB(Δjag) merodiploid strain showed a marginal increase in cell length of ≈5% (Ulrych et al., 2016). Similarly, induction of ectopic khpB+(jagG+) (or separately khpA+) in a D39 Δcps khpB+(jag+) merodiploid strain did not cause substantial cell elongation (Fig. S21).
In a new report, Håvarstein and coworkers independently reported that ΔkhpB suppresses Δpbp2b or ΔrodA in the R704 derivative of laboratory strain R6 (Stamsås et al., 2017). Because of its possible regulatory involvement in peripheral PG elongation, they designated this protein as “EloR” (Stamsås et al., 2017). In our work, we have retained the name KhpB instead of EloR or JAG, because KhpB interacts with a second RNA binding protein, KhpA, and RIP-Seq and mechanistic studies described below show that KhpA/B regulation is not confined to PG elongation.
There are several striking differences between the results reported here in the D39 progenitor background and those reported in the R6 laboratory strain (Stamsås et al., 2017). We showed previously that laboratory strain R6 has accumulated ≈80 mutations that are not present in the genetic background of its progenitor D39 strain (Lanie et al., 2007, Rued et al., 2017). In particular, mreC, mreD, rodZ, and gpsB are not essential in R6 (Straume et al., 2017), whereas they are essential in D39 (Land & Winkler, 2011, Lanie et al., 2007, Rued et al., 2017). Notably, pbp1a contains two missense mutations in R6 that are not present in D39 or TIGR4, and we showed previously that swapping the wild-type allele of pbp1a from D39/TIGR4 into R6 restored essentiality of mreCD (Land & Winkler, 2011). Hence, the suppression of ΔmreCD, ΔrodZ, and ΔgpsB by ΔkhpB(ΔeloR) could not be detected in R6. Similar to R6 (Stamsås et al., 2017), we found that ΔkhpA/B reduce cell aspect ratio slightly (≈10%) (Fig. 2 and 4); but, we do not interpret this small change as indicative of a major defect in PG elongation that would cause near sphericity (Tsui et al., 2014). By comparison, we reported previously that a D39 mltG(Y488D) Δpbp2b mutant reduces aspect ratio by ≈25%, and cells appear spherical (Tsui et al., 2016). The major defect in cell morphology of ΔkhpA/khpB mutants in the progenitor D39 background is an ≈50% reduction in cell size (Fig. 2 and 4). D39 Δpbp1a cells are also significantly smaller than wild-type cells, but have a greater aspect ratio (Land & Winkler, 2011, Tsui et al., 2016). Reduced cell size was described for ΔkhpB(Δjag) mutants of strain Rx1 (Ulrych et al., 2016), but was not commented on for R6 ΔkhpB(ΔeloR) (Stamsås et al., 2017). This large reduction in size is indicative of a reduction in both septal and peripheral PG synthesis in D39 ΔkhpA/B or Δpbp1a mutants (Land & Winkler, 2011, Tsui et al., 2016).
Another major difference between the ΔkhpB phenotypes in R6 and D39 is the effect of KhpB T89 phosphorylation by StkP. In R6, a khpB/eloR(T89A) phosphoablative change phenocopied ΔkhpB, resulting in decreased growth rate, but not suppression of Δpbp2b (Stamsås et al., 2017). A khpB/eloR(T89E) phosphomimetic mutation was lethal and resulted in the accumulation of mreC truncation and rodZ null mutations, but surprisingly was not suppressed by ΔmreC (Stamsås et al., 2017). These results were incorporated into a model in which Δpbp2b or ΔrodA mutations are essential in R6 because of uncontrolled MltG lytic transglycosylase activity, which is reduced by ΔkhpB(ΔeloR) or absence of T89 phosphorylation (Stamsås et al., 2017). However, in the D39-derived strain IU1824 (Table S1), khpB(T89A) does not phenocopy ΔkhpB, and khpB(T89A), khpB(T89D) and khpB(T89E) mutants grow like wild-type in the culture conditions tested (Fig. S10). khpB(T89A), khpB(T89D), and khpB(T89E) mutants in D39 derivative IU1824 did not contain suppressor mutations based on whole-genome determinations (see Results). Thus, phosphorylation of KhpB at Thr89 does not affect KhpB function in the D39 progenitor background. A possible role of the second phosphorylation site in KhpB remains to be explored (Stamsås et al., 2017, Ulrych et al., 2016).
We reported previously that the mltG(Y488D) mutation, which likely reduces the activity of the essential MltG endo-lytic transglycosylase, suppresses the requirement for peripheral PG synthesis in D39, with some restrictions (Tsui et al., 2016). The model that peripheral PG genes are essential simply because of uncontrolled MltG activity (Stamsås et al., 2017) is not readily reconciled with the combinations of mutations that are synthetically viable in D39 or the suppression of ΔmltG by spontaneous mutations that inactivate pbp1a (Tsui et al., 2016). The differences in the phenotypes in response to KhpB phosphorylation at Thr89 in R6 versus D39 may reflect the bypass mechanism accumulated by laboratory strain R6 that is not present in the virulent D39 progenitor strain. Finally, the mechanism for the suppression of Δpbp2b by ΔkhpA/B in D39 described next only indirectly involves regulation of PG elongation.
Overproduction of FtsA as a possible mechanism of Δpbp2b suppression was first hinted at by isolation of a suppressor mutant that contained only a tandem chromosomal duplication of the ftsA-ftsZ chromosomal region (Fig. 6; Table S2). We showed that ectopic overexpression of ftsA alone is sufficient to suppress Δpbp2b, ΔmreCD, and ΔrodA (Tables 1 and 2; Fig. 7 and 10A). Overproduction of FtsA also partly can account for the smaller cell size and induction of the WalRK TCS cell-wall stress regulon in ΔkhpA/B mutants (Fig. 10 A and S16). Consistent with overproduction of FtsA as the mechanism of Δpbp2b suppression, the cellular concentration of FtsA is ≈3-fold greater in ΔkhpA/B mutants than in the wild-type parent strain (Fig.8; Table 2). There is a smaller (≈1.5-fold) increase in the relative amount of FtsZ as well. Consistent with results from Western blotting, comparable increases in the relative cellular amounts of FtsA and FtsZ were independently detected by quantitative MS-based (tandem-mass tag; TMT) proteomics of a ΔkhpA ΔkhpB mutant compared to the parent strain (data not shown). In contrast to relative protein amounts, the relative amounts of both the ftsA and ftsZ transcripts increased by the same amount (≈1.5-fold) in the ΔkhpA/B mutants compared to the parent (Fig. 8), indicating that part of the increase in relative FtsA protein amount is exerted at the post-transcriptional level. The increase in relative FtsA amount in ΔkhpA/B mutants is comparable to that attained by ectopic ftsA overexpression that suppresses Δpbp2b (Table 2). FtsA is a highly abundant protein in Spn cells (Lara et al., 2005), and there may be unknown negative feedback regulatory mechanisms that modulate FtsA amount during the cell cycle and set a limit for the level of FtsA overproduction that can be attained.
Post-transcriptional regulation of FtsA amount pointed to the 74-nt 5′-UTR leader of the ftsA-ftsZ operon (Fig. 9A) as a possible site of regulation mediated by KhpA/B. In testing this hypothesis, we were able to demonstrate that overproduction of FtsA is necessary, as well as sufficient for suppression of Δpbp2b. By using both ectopic constructs and a chromosomal deletion mutation, we found that overproduction of FtsA in cells in response to the absence of KhpA depends on an intact ftsA 5′-UTR leader region (Fig. 9; Table 2). In particular, the relative amount of FtsA, but not FtsZ, was reduced by about 50% in the chromosomal deletion of the ftsA 5′-UTR leader (ΔUTR), possibly due to a change in segmental mRNA stability or translation initiation of ftsA; but, now the expression level of FtsA was insensitive to ΔkhpA (Fig. 9). Three general models for regulation of relative FtsA amount by KhpA/B are presented in Figure 10B. In model 1, ftsA translation is negatively regulated by binding to a sRNA in the presence of KhpA/B; alternatively, KhpA/B could act as a direct negative regulator by binding to the ftsA 5′-UTR leader (not shown). Model 2 invokes indirect positive regulation by KhpA/B of a negative regulator (sRNA or protein) of ftsA translation. Model 3 is based on indirect negative regulation by KhpA/B of a positive regulator (sRNA or protein) of ftsA translation. At this point, we know that an intact ftsA 5′-UTR leader is required for increased FtsA production in the absence of KhpA/B (Fig. 9; Table 2); but, this result does not distinguish among these models. mRNA near the ftsA 5′-UTR and start codon is preferentially pulled down by KhpA or KhpB in RIP-Seq experiments (Fig. S18; Table S4), which would tend to support model 1. On the other hand, we know that the chromosomal ftsA(ΔUTR) mutation reduces relative basal amount of FtsA, without changing relative FtsZ amount, where ftsA-ftsZ are co-transcribed (Fig. 6 and 9). The reduced relative basal amount of FtsA in the ftsA(ΔUTR) mutant might suggest loss of a positive regulator of ftsA translation, as proposed for model 3. However, the ftsA(ΔUTR) mutation could change intrastrand mRNA pairing or locally destabilize the ftsA mRNA in the RBS region without affecting the stability of downstream ftsZ mRNA. Additional work is needed to determine the mechanism of ftsA regulation by KhpA/B.
The mechanism of how FtsA overproduction suppresses Δpbp2b, ΔmreCD, and ΔrodA mutations also remains to be determined (Fig. 10A). FtsA is a membrane-associated actin homologue that anchors the FtsZ ring to the cell membrane (Ortiz et al., 2016, Busiek & Margolin, 2015, Lutkenhaus et al., 2012, Juarez & Margolin, 2012). FtsA is essential in ovoid-shaped bacteria, like Spn, and has different properties than in rod-shaped bacteria (see (Mura et al., 2016). In particular, FtsA plays roles in maintaining coherent FtsZ rings and coordinating peripheral and septal PG synthesis from the midcell region of Spn cell (Fig. S1A) (Mura et al., 2016). These regulatory roles require interactions of FtsA with other septation and elongation proteins (Busiek & Margolin, 2015, Du et al., 2016, Lutkenhaus et al., 2012, Ortiz et al., 2016). It has been speculated that Spn FtsA may even assume similar functions in midcell peripheral PG synthesis as MreB, which mediates side-wall PG synthesis in rod-shaped bacteria and is absent in Spn (Fig. S1B) (Mura et al., 2016, Massidda et al., 2013). It is notable that FtsA overproduction in wild-type Spn cells stimulates septation and leads to smaller cells (Mura et al., 2016). Therefore, excess FtsA may speed up cell closure sufficiently that the requirement for full peripheral elongation diminishes. Another non-exclusive possibility is that overproduced FtsA may recruit or alter the activities of other PBPs to compensate for the absence of bPBP2b, MreCD, and RodA from the peripheral PG synthesis machine. Both of these mechanisms would result in the characteristic decrease in cell length, width, and size, but only slightly decreased ovoid shape of ΔkhpA/B mutants (Fig. 2 and 4) (Ulrych et al., 2016). Further studies of the role of FtsA in coordinating septal and peripheral PG synthesis in Spn may shed light onto the suppression mechanism (see (Mura et al., 2016)).
Finally, several pieces of evidence indicate that KhpA/B regulate genes other than ftsA in Spn. Most tellingly, excess FtsA amount did not suppress ΔgpsB or ΔrodZ (Table 1; Fig. 10A). Lack of suppression of ΔrodZ is curious, because RodZ is thought to function primarily in peripheral PG synthesis (Tsui et al., 2016, Shiomi et al., 2013, van den Ent et al., 2010, Gerdes, 2009), in contrast to GpsB whose function is not confined to this process (Rued et al., 2017, Rismondo et al., 2016, Fleurie et al., 2014, Land et al., 2013). This shared lack of suppression suggests that ΔkhpA/B changes the expression of other genes besides ftsA and that RodZ could play a role in mediating GpsB function in septal and peripheral PG synthesis (Fleurie et al., 2014, Rued et al., 2017, Tsui et al., 2016). Regulation of multiple pathways by KhpA/B is supported by results from RIP-Seq (Fig. 5; Table S4) and initial quantitative TMT-MS proteomics (data not shown), where multiple RNAs are pulled down by KhpA/B in RIP-Seq and relative amounts of numerous proteins change in ΔkhpA/B mutants compared to the parent strain. Current efforts are underway to compare and reconcile these RNA and protein data sets.
Regulation by KhpA/B likely occurs predominantly at the post-transcriptional level, since only the relative transcript amounts of genes in the WalRK TCS cell-wall stress regulon strongly change in ΔkhpA/B mutants (Fig. 2 and 4; Table S3) (Sauerbier, 2012). The hypothesis that widely distributed KhpA and KhpB (Fig. S2) form a multimeric RNA chaperone complex could address a long-standing issue in Gram-positive RNA biology. Hfq plays an indispensable regulatory role in Gram-negative bacteria (De Lay et al., 2013, Van Assche et al., 2015, Feliciano et al., 2016, Updegrove et al., 2016), whereas the roles played by Gram-positive Hfq homologues remain “elusive” (Bouloc & Repoila, 2016, Keefer et al., 2017, Rochat et al., 2012, Rochat et al., 2015). Moreover, Hfq homologues are missing from many Gram-positive species, including S. pneumoniae (Lanie et al., 2007). We speculate that a KhpA/B multimeric complex may act redundantly in Gram-positive species containing Hfq and fulfill the role of an RNA chaperone in those species that lack an Hfq homologue. The hypothesis that KhpA/B is a multimeric complex that acts as a general RNA chaperone requires future testing.
EXPERIMENTAL PROCEDURES
Bacterial strains and growth conditions
Strains used in this study are listed in Table S1. All strains were derived from strain IU1824 (D39 Δcps rpsL1), IU1945 (D39 Δcps), or encapsulated serotype 2 strain D39 strain IU1690 (Lanie et al., 2007). Strains containing antibiotic markers were constructed by transformation of competent pneumococcal cells with linear DNA amplicons synthesized by overlapping fusion PCR (Ramos-Montanez et al., 2008, Tsui et al., 2010, Tsui et al., 2016). Strains containing markerless alleles in native chromosomal loci were constructed using allele replacement via the Pc-[kan-rpsL+] (Janus cassette) (Sung et al., 2001). Primers used to synthesize different amplicons are listed in Table S1. Bacteria were grown on plates containing trypticase soy agar II (modified; Becton-Dickinson) and 5% (vol/vol) defibrinated sheep blood (TSAII-BA). Plates were incubated at 37°C in an atmosphere of 5% CO2. TSAII-BA plates for selections contained antibiotics at concentrations described previously (Tsui et al., 2010, Tsui et al., 2016). Bacteria were cultured statically in Becton-Dickinson brain heart infusion (BHI) broth at 37°C in an atmosphere of 5% CO2, and growth was monitored by OD620 as described before (Tsui et al., 2016). Mutant constructs were confirmed by PCR and DNA sequencing of chromosomal regions corresponding to the amplicon region used for transformation.
In all experiments, cells were inoculated from frozen glycerol stocks into BHI broth, serially diluted, and incubated 12–16 h statically at 37°C in an atmosphere of 5% CO2. 0.5 mM ZnCl2 + 0.05 mM MnSO4 (+Zn) was added to BHI broth cultures of merodiploid strains that require Zn2+ for growth due to ectopic expression of an essential gene (i.e., IU11258, IU12707, IU12901, and IU12959) (Tsui et al., 2016). The next day, cultures at OD620 ≈ 0.05–0.4 were diluted to OD620 ≈ 0.002 in BHI broth lacking (-Zn) or containing (+Zn) 0.5 mM ZnCl2 + 0.05 mM MnSO4 and cultured under the same conditions. Cultures were sampled in early to mid-exponential phase for different experiments as indicated. For depletion experiments, overnight cultures of Zn2+-requiring merodiploid strains were centrifuged once to remove the +Zn medium and resuspended (t = 0 in growth curves) in BHI broth lacking (-Zn) or containing (+Zn) 0.5 mM ZnCl2 + 0.05 mM MnSO4.
Transformation assays
Transformations were performed as previously described (Tsui et al., 2016, Rued et al., 2017). Δpbp2b<>aad9, Δpbp2b<>Pc-erm, ΔrodA::Pc-erm, ΔmreCD<>aad9, ΔcozE::Pc-erm, ΔmltG::Pc-erm, ΔrodZ<>aad9, and ΔgpsB<>aad amplicons, and a Δpbp1b::Pc-erm amplicon used as a positive control each contained ≈ 1 kb of flanking chromosomal DNA. Amplicons were synthesized by PCR using the primers and templates listed in Table S1.
Whole-genome DNA sequencing
Whole-genome sequencing was used to identify suppressor mutations and to verify the genomes of constructed mutants. Strains IU7476 and IU12015 containing suppressor mutations that allowed growth of a Δpbp2b mutant were isolated as described previously before (Tsui et al., 2016, Rued et al., 2017). Mutants khpB(T89A) (IU12744; IU13885), khpB(T89D), (IU13881; IU13887), or khpB(T89E) (IU13883; IU13889) were constructed and isolated from independent transformation reactions as described in Table S1. Genomic DNA preparation, DNA library construction, Illumina MiSeq or NextSeq DNA sequencing, and bioinformatics analyses were performed as described previously (Tsui et al., 2016, Rued et al., 2017). The number of reads of each base was mapped to the D39 reference genome by using the JBrowse program (Skinner et al., 2009, Westesson et al., 2013) to detect regions containing chromosomal duplications or large deletions (Rued et al., 2017).
We explicitly determined the nature of the duplication containing ftsA-ftsZ at 1,492,424 through 1,500,342 (Fig. 6) as follows. After the initial bowtie analysis we identified all the reads that were not mapped to the genome. These reads were then re-aligned using bowtie with the addition of the –local option. Reads locally mapped to the region of the deletion were then manually examined and re-aligned to the entire genome using NCBI Blastn. Reads spanning the junction between the 3′ end of the putative repeat region to the 5′ end of the repeat region were identified which is consistent with the hypothesis that the repeat was a tandem duplication that occurred in place without any other obvious re-arrangements taking place. The coverage is consistent with this being a single duplication event.
Cell length and width measurements
Cell lengths and widths of strain growing exponentially in BHI broth were measured as previously described (Tsui et al., 2016). More than 100 cells from at least 2 independent experiments were measured, and P values were obtained by one-way ANOVA analysis by using the GraphPad Prism program.
RNA preparation, mRNA-Seq, and qRT-PCR
RNA preparation, qRT-PCR and mRNA-Seq analyses of transcripts >200 nt were performed as previously described (Tsui et al., 2016, Zheng et al., 2016). Primers used for qRT-PCR are listed in Table S1. mRNA-Seq data are deposited in the GEO database (accession number GSE101583).
Co-immunoprecipitation (co-IP)
IU1945 (WT), IU9602 (khpA-L-FLAG3), and IU10664 (khpB-L-FLAG3) strains were treated with formaldehyde cross linker, and FLAG-tagged proteins were used as bait for co-IP using anti-FLAG magnetic beads as described previously (Rued et al., 2017). Eluted protein samples were mixed with same volume of 2X Laemmli sample buffer (Bio-Rad) containing 5% (vol/vol) β-mercaptoethanol (Sigma) and heated at 95°C for 1 h to break crosslinks. 20 μL of each sample were separated by SDS-PAGE on 4%–15% precast protein gels (Bio-Rad) in Tris-glycine buffer. Gels were treated with Pierce silver stain for MS (ThermoFisher) according to the manufacturer’s protocol. Prey bands were cut from gels, destained, trypsin digested, and identified by MS at the IUB core facility (Sham et al., 2011).
KhpA and KhpB structure modeling
The aa sequences of KhpA and KhpB were entered into the SWISS-Model server (Arnold et al., 2006). The PDB numbers of the templates for modeling the structures of KhpA and KhpB were 4V6W (KH domain from Drosophila melanogaster 80S ribosome) and 3GKU (KhpB homologue of Clostridium symbiosum), respectively. The resulting PDB models were visualized and aligned in PyMOL (The PyMOL Molecular Graphics System; Version 1.7.4.3; Schrödinger, LLC 2; www.pymol.org).
Quantitative Western blotting
Western blotting was performed as previously described (Tsui et al., 2014, Wayne et al., 2010, Zheng et al., 2016). Cell lysates were prepared by the FastPrep homogenizer method described before (Tsui et al., 2014, Wayne et al., 2010, Zheng et al., 2016). Protein concentrations were determined with the DCTm protein assay (Bio-Rad), and equal amounts of protein (2.5 μg) were loaded into each lane on duplicate SDS-PAGE gels. After transfer of proteins from a gel to a membrane, the membrane was cut into two sections, such that the upper section contained FtsA (50 kDa) or FtsZ (45 kDa) and the low section contained MreC (30 kDa) as a loading and blotting control. The separated membranes were labeled with anti-FtsA (1:10,000), anti-FtsZ (1:10,000), or anti-MreC (1:5,000) as primary antibodies for immunodetection (Land & Winkler, 2011, Lara et al., 2005). Chemiluminescence of protein bands in Western blots was quantitated using an IVIS imaging system as described previously (Wayne et al., 2010). To check the linearity of signal intensity versus protein amount, cell lysates from the WT strain (IU1824) were serially diluted, and different amounts of total lysate protein (1.25, 2.50, 5.00, and 10.0 μg) were subjected to Western blotting with detection using anti-FtsA, anti-FtsZ, or anti-MreC antibodies. The signal intensity obtained for each band was linear with the amount of each protein loaded (Fig. S17), confirming the linearity and reproducibility of detection of the quantitative Western blot assay. Western blotting to detect phosphorylated Spn proteins using anti-phosphothreonine (anti-pThr) antibody was performed as described previously (Rued et al. 2017).
2D immunofluorescence microscopy (IFM)
Co-localization of FLAG-tagged KhpA and HA-tagged KhpB was performed by IFM as described before (Land et al., 2013, Tsui et al., 2014)in exponentially growing cells of strain IU10602 (khpA-L-FLAG3 khpB-HA). 2D IFM images were averaged and quantitated using a previously described graphical user interface (GUI) program (Land et al., 2013). Demographs of protein localization were generated using the MicrobeJ program (Ducret et al., 2016).
5′-rapid amplification of cDNA ends (RACE)
The transcription start site of ftsA-ftsZ mRNA in wild-type cells growing exponentially in BHI broth was determined by the SMARTer-RACE 5′/3′ kit (Clontech) according to the manufacturer’s instructions.
RNA immunoprecipitation and sequencing (RIP-Seq)
Bacterial RIP-Seq was carried out by a modification of a previously published method (Saadeh et al., 2015). The method of recovery of FLAG-tagged bait proteins bound to cellular RNAs was similar to that used for co-IP described above (Rued et al., 2017), except that there was no formaldehyde crosslinking and glycine quenching steps. Briefly, strains IU1945 (WT), IU9602 (khpA-L-FLAG3), and IU10664 (khpB-L-FLAG3) were grown in 400 mL of BHI broth to OD620 ≈ 0.25–0.40. Cells were collected by centrifugation (8,000 X g for 10 min at 4°C). Cell pellets were washed once with 30 mL of cold 1XPBS (4°C) and resuspended in 2 mL of cold lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X100 (v/v) containing protease inhibitor (ThermoFisher; cat#78429)). Suspensions (2 mL) were transferred into 2 lysing matrix B tubes (1 mL each) (MB Biomedicals, Inc.). Tubes were shaken in a FastPrep homogenizer at 4°C for ten 40s cycles (4X shaking; 5 min on ice; 3X shaking; 5 min on ice; 3X shaking) at a setting of 6.0 M/s. Cell debris was removed by centrifuging at 16,000 X g for 5 min at 4°C. Protein concentration was determined by DCTM protein assay (Bio-Rad). About 1 mL of lysate with similar protein amount was added to tubes with 50 μL of anti-FLAG magnetic beads (Sigma) and rotated for 2 h at 4°C. The beads were washed 3 times with 1 mL of lysis buffer for 10 min with rotation at 4°C. FLAG3-tagged protein bound to RNA was eluted by incubation with 100 μL of FLAG elution solution (150 ng of 3X FLAG peptide per μL) (Sigma) for 30 min at 4°C.
RNA was extracted by phenol-chloroform extraction. 100 μL of phenol/chloroform/isoamyl alcohol (125:24:1, pH 4.3, ThermoFisher) was added to the eluted solution and vortexed for 10 sec before incubation at room temperature for 5 min. The mixture was centrifuged at 16,000 X g for 5 min. The upper phase was transferred to a new 1.5 mL tube. 2 volume of isopropanol and 1/10 volume of 3 M sodium acetate (pH 5.2) was added, followed by incubation for 10 min at −80°C to precipitate RNA. Precipitated material was collected by centrifugation (16,000 X g for 30 min, 4°C) and washed with 500 μL of 80% ethanol. Pellets were dried at room temperature for 10 min and resuspended in 30 μL of nuclease-free water. RNA was sequenced as described before for mRNA-Seq experiments (Tsui et al., 2016, Zheng et al., 2016). A ratio of reads for an RNA species greater than 4-fold between the FLAG-tagged KhpA or KhpB sample and the non-FLAG-tagged control sample (IU1945) was considered indicative of a possible interaction between the RNA and KhpA or KhpB (Saadeh et al., 2015). The number of reads of each base was mapped to the D39 reference genome by using the JBrowse program (Skinner et al., 2009, Westesson et al., 2013) as described above. RIP-Seq data are deposited in the GEO database (accession number GSE103735).
Disk diffusion assay for antibiotic resistance
Disk diffusion assays were performed as described before (Zheng et al. 2016). Antibiotic disks were purchased from Becton, Dickinson and Co. Disks used in assays were penicillin (10 U), cefotaxime (30 μg), piperacillin (100 μg), tetracycline (30 μg), and gentamicin (120 μg).
Supplementary Material
Acknowledgments
We thank Dhriti Sinha, Kevin Bruce, and Eric Kim for methods and constructed bacterial strains, other members of the Winkler laboratory for critical comments and discussions, Jesus Bazan, James Ford, Kurt Zimmer, Ram Podicheti, and Doug Rusch for assistance with genomic methods and bioinformatics, and Jon Trinidad for MS identifications and preliminary TMT experiments. This work was supported by NIH grant RO1GM113172 and NIH grant R01GM114315 (to M.E.W.), and by LR7/2007 grant CRP2-401 from the Autonomous Region of Sardinia (RAS) (to O.M.).
Footnotes
AUTHOR CONTRIBUTIONS
Conception or design of the study (JJZ, HCTT, MEW); acquisition, analysis, or interpretation of data (JJZ, AJP, HCTT, OM, MEW); writing the manuscript (JJZ, AJP, HCTT, OM, MEW).
References
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Barendt SM, Sham LT, Winkler ME. Characterization of mutants deficient in the L,D-carboxypeptidase (DacB) and WalRK (VicRK) regulon, involved in peptidoglycan maturation of Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol. 2011;193:2290–2300. doi: 10.1128/JB.01555-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz K, Novakova L, Fadda D, Branny P, Massidda O, Veening JW. Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proceed Nat Acad Sci USA. 2012;109:E905–913. doi: 10.1073/pnas.1119172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendezu FO, de Boer PA. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J Bacteriol. 2008;190:1792–1811. doi: 10.1128/JB.01322-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KH, Stamsås GA, Straume D, Håvarstein LS. Effects of low PBP2b levels on cell morphology and peptidoglycan composition in Streptococcus pneumoniae R6. J Bacteriol. 2013;195:4342–4354. doi: 10.1128/JB.00184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MJ, Kuru E, Rittichier JT, VanNieuwenhze MS, Brun YV, Winkler ME. Minimal Peptidoglycan (PG) Turnover in wild-type and PG hydrolase and cell division mutants of Streptococcus pneumoniae D39 growing planktonically and in host-relevant biofilms. J Bacteriol. 2015;197:3472–3485. doi: 10.1128/JB.00541-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloc P, Repoila F. Fresh layers of RNA-mediated regulation in Gram-positive bacteria. Curr Opin Microbiol. 2016;30:30–35. doi: 10.1016/j.mib.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Brown J, Hammerschmidt S, Orihuela CJ. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. Academic Press; London, UK: 2015. [Google Scholar]
- Busiek KK, Margolin W. Bacterial actin and tubulin homologs in cell growth and division. Curr Biol: CB. 2015;25:R243–254. doi: 10.1016/j.cub.2015.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. Antibiotic resistance threats in the United States, 2013. 2013 http://www.cdc.gov/drugresistance/threat-report-2013.
- De Lay N, Schu DJ, Gottesman S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Biol Chem. 2013;288:7996–8003. doi: 10.1074/jbc.R112.441386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du S, Pichoff S, Lutkenhaus J. FtsEX acts on FtsA to regulate divisome assembly and activity. Proc Nat Acad Sci USA. 2016;113:E5052–5061. doi: 10.1073/pnas.1606656113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrac S, Bisicchia P, Devine KM, Msadek T. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Molec Microbiol. 2008;70:1307–1322. doi: 10.1111/j.1365-2958.2008.06483.x. [DOI] [PubMed] [Google Scholar]
- Ducret A, Quardokus EM, Brun YV. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat Microbiol. 2016;1:16077. doi: 10.1038/nmicrobiol.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan AJ, Biboy J, van’t Veer I, Breukink E, Vollmer W. Activities and regulation of peptidoglycan synthases. Phil Trans Royal Soc Lond B, Biol Sci. 2015;370(1679) doi: 10.1098/rstb.2015.0031. pii: 20150031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan AJ, Cleverley RM, Peters K, Lewis RJ, Vollmer W. Regulation of bacterial cell wall growth. FEBS J. 2017;284:851–867. doi: 10.1111/febs.13959. [DOI] [PubMed] [Google Scholar]
- Feliciano JR, Grilo AM, Guerreiro SI, Sousa SA, Leitao JH. Hfq: a multifaceted RNA chaperone involved in virulence. Future Microbiol. 2016;11:137–151. doi: 10.2217/fmb.15.128. [DOI] [PubMed] [Google Scholar]
- Fenton AK, Mortaji LE, Lau DT, Rudner DZ, Bernhardt TG. CozE is a member of the MreCD complex that directs cell elongation in Streptococcus pneumoniae. Nat Microbiol. 2016;2:16237. doi: 10.1038/nmicrobiol.2016.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira DM, Gordon SP. Mechanisms causing inflammatory response to Streptococcus pneumoniae. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 383–400. [Google Scholar]
- Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, Grangeasse C. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet. 2014;10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz G, Mascher T. A balancing act times two: sensing and regulating cell envelope homeostasis in Bacillus subtilis. Molec Microbiol. 2014;94:1201–1207. doi: 10.1111/mmi.12848. [DOI] [PubMed] [Google Scholar]
- Gerdes K. RodZ, a new player in bacterial cell morphogenesis. EMBO J. 2009;28:171–172. doi: 10.1038/emboj.2008.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz N, Loh LN, Tuomanen EI. Pneumococcal invasion: development of bacteremia and meningitis. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 433–451. [Google Scholar]
- Gutu AD, Wayne KJ, Sham LT, Winkler ME. Kinetic characterization of the WalRKSpn (VicRK) two-component system of Streptococcus pneumoniae: dependence of WalKSpn (VicK) phosphatase activity on its PAS domain. J Bacteriol. 2010;192:2346–2358. doi: 10.1128/JB.01690-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakenbeck R, Bruckner R, Denapaite D, Maurer P. Molecular mechanisms of beta-lactam resistance in Streptococcus pneumoniae. Future Microbiol. 2012;7:395–410. doi: 10.2217/fmb.12.2. [DOI] [PubMed] [Google Scholar]
- Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harbor perspectives in medicine. 2013;3(7) doi: 10.1101/cshperspect.a010215. piia010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino F, Seinen J, Henriques-Normark B, van Dijl JM. How Does Streptococcus pneumoniae Invade the Brain? Trends Microbiol. 2016;24:307–315. doi: 10.1016/j.tim.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Jacobsen FE, Kazmierczak KM, Lisher JP, Winkler ME, Giedroc DP. Interplay between manganese and zinc homeostasis in the human pathogen Streptococcus pneumoniae. Metallomics. 2011;3:38–41. doi: 10.1039/c0mt00050g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacq M, Adam V, Bourgeois D, Moriscot C, Di Guilmi AM, Vernet T, Morlot C. Remodeling of the Z-Ring Nanostructure during the Streptococcus pneumoniae cell cycle revealed by photoactivated localization microscopy. mBio. 2015;6(4) doi: 10.1128/mBio.01108-15. pii: e01108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez JR, Margolin W. A bacterial actin unites to divide bacterial cells. EMBO J. 2012;31:2235–2236. doi: 10.1038/emboj.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefer AB, Asare EK, Pomerantsev AP, Moayeri M, Martens C, Porcella SF, Gottesman S, Leppla SH, Vrentas CE. In vivo characterization of an Hfq protein encoded by the Bacillus anthracis virulence plasmid pXO1. BMC Microbiol. 2017;17(1):63. doi: 10.1186/s12866-017-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Molec Microbiol. 2013;90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Winkler ME. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol. 2011;193:4166–4179. doi: 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol. 2007;189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Molec Microbiol. 2005;55:699–711. doi: 10.1111/j.1365-2958.2004.04432.x. [DOI] [PubMed] [Google Scholar]
- Leclercq S, Derouaux A, Olatunji S, Fraipont C, Egan AJ, Vollmer W, Breukink E, Terrak M. Interplay between penicillin-binding proteins and SEDS proteins promotes bacterial cell wall synthesis. Scientific reports. 2017;7:43306. doi: 10.1038/srep43306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkenhaus J, Pichoff S, Du S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton (Hoboken) 2012;69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JE, Edmonds KA, Bruce KE, Campanello GC, Eijkelkamp BA, Brazel EB, McDevitt CA, Winkler ME, Giedroc DP. The zinc efflux activator SczA protects Streptococcus pneumoniae serotype 2 D39 from intracellular zinc toxicity. Molec Microbiol. 2017a;104:636–651. doi: 10.1111/mmi.13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JE, Lisher JP, Winkler ME, Giedroc DP. Perturbation of manganese metabolism disrupts cell division in Streptococcus pneumoniae. Molec Microbiol. 2017b;104:334–348. doi: 10.1111/mmi.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massidda O, Novakova L, Vollmer W. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol. 2013;15:3133–3157. doi: 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature. 2016;537:634–638. doi: 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlot C, Bayle L, Jacq M, Fleurie A, Tourcier G, Galisson F, Vernet T, Grangeasse C, Di Guilmi AM. Interaction of Penicillin-Binding Protein 2x and Ser/Thr protein kinase StkP, two key players in Streptococcus pneumoniae R6 morphogenesis. Molec Microbiol. 2013;90:88–102. doi: 10.1111/mmi.12348. [DOI] [PubMed] [Google Scholar]
- Mu D, Montalban-Lopez M, Masuda Y, Kuipers OP. Zirex: a novel zinc-regulated expression system for Lactococcus lactis. App Environ Microbiol. 2013;79:4503–4508. doi: 10.1128/AEM.00866-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mura A, Fadda D, Perez AJ, Danforth ML, Musu D, Rico AI, Krupka M, Denapaite D, Tsui HT, Winkler ME, Branny P, Vicente M, Margolin W, Massidda O. Roles of the essential protein FtsA in cell growth and division in Streptococcus pneumoniae. J Bacteriol. 2017;199(3):e00608–16. doi: 10.1128/JB.00608-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng WL, Tsui HC, Winkler ME. Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphorylated VicR (YycF) response regulator in Streptococcus pneumoniae. J Bacteriol. 2005;187:7444–7459. doi: 10.1128/JB.187.21.7444-7459.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro G, I, Taylor A, Ramos A. KH-RNA interactions: back in the groove. Curr Opinion Struct Biol. 2015;30:63–70. doi: 10.1016/j.sbi.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Oliver MB, Swords WE. Pneumococcal biofilms and bacterial perisistence during otitis media. In: Brown J, Hammerschmidt S, Orihuela CJ, editors. Streptococcus pneumoniae Molecular Mechanisms of Host-Pathogen Interactions. London: Academic Press; 2015. pp. 293–308. [Google Scholar]
- Ortiz C, Natale P, Cueto L, Vicente M. The keepers of the ring: regulators of FtsZ assembly. FEMS Microbiol Rev. 2016;40:57–67. doi: 10.1093/femsre/fuv040. [DOI] [PubMed] [Google Scholar]
- Pazos M, Peters K, Vollmer W. Robust peptidoglycan growth by dynamic and variable multi-protein complexes. Curr Opin Microbiol. 2017;36:55–61. doi: 10.1016/j.mib.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Philippe J, Vernet T, Zapun A. The elongation of ovococci. Microb Drug Resis. 2014;20:215–221. doi: 10.1089/mdr.2014.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho MG, Kjos M, Veening JW. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol. 2013;11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- Ramos-Montanez S, Tsui HC, Wayne KJ, Morris JL, Peters LE, Zhang F, Kazmierczak KM, Sham LT, Winkler ME. Polymorphism and regulation of the spxB (pyruvate oxidase) virulence factor gene by a CBS-HotDog domain protein (SpxR) in serotype 2 Streptococcus pneumoniae. Molec Microbiol. 2008;67:729–746. doi: 10.1111/j.1365-2958.2007.06082.x. [DOI] [PubMed] [Google Scholar]
- Rismondo J, Cleverley RM, Lane HV, Grosshennig S, Steglich A, Moller L, Mannala GK, Hain T, Lewis RJ, Halbedel S. Structure of the bacterial cell division determinant GpsB and its interaction with penicillin-binding proteins. Molec Microbiol. 2016;99:978–998. doi: 10.1111/mmi.13279. [DOI] [PubMed] [Google Scholar]
- Rochat T, Bouloc P, Yang Q, Bossi L, Figueroa-Bossi N. Lack of interchangeability of Hfq-like proteins. Biochimie. 2012;94:1554–1559. doi: 10.1016/j.biochi.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Rochat T, Delumeau O, Figueroa-Bossi N, Noirot P, Bossi L, Dervyn E, Bouloc P. Tracking the elusive function of Bacillus subtilis Hfq. PloS One. 2015;10:e0124977. doi: 10.1371/journal.pone.0124977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rued BE, Zheng JJ, Mura A, Tsui HT, Boersma MJ, Mazny JL, Corona F, Perez AJ, Fadda D, Doubravova L, Buriankova K, Branny P, Massidda O, Winkler ME. Suppression and synthetic-lethal genetic relationships of ΔgpsB mutations indicate that GpsB mediates protein phosphorylation and penicillin-binding protein interactions in Streptococcus pneumoniae D39. Molec Microbiol. 2017;103:931–957. doi: 10.1111/mmi.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N. Lipid Flippases for bacterial peptidoglycan biosynthesis. Lipid Insights. 2015;8:21–31. doi: 10.4137/LPI.S31783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadeh B, Caswell CC, Chao Y, Berta P, Wattam AR, Roop RM, 2nd, O’Callaghan D. Transcriptome-wide identification of Hfq-associated RNAs in Brucella suis by deep sequencing. J Bacteriol. 2015;198:427–435. doi: 10.1128/JB.00711-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbier J. Fachbereich Biologie. Technische Universität Kaiserslautern; 2012. Horizontaler Gentransfer zwischen Streptococcus mitis und Streptococcus pneumoniae – Analyse der Resistenzentwicklung. [Google Scholar]
- Sham LT, Barendt SM, Kopecky KE, Winkler ME. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc Nat Acad Sci USA. 2011;108:E1061–1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham LT, Tsui HC, Land AD, Barendt SM, Winkler ME. Recent advances in pneumococcal peptidoglycan biosynthesis suggest new vaccine and antimicrobial targets. Curr Opin Microbiol. 2012;15:194–203. doi: 10.1016/j.mib.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi D, Toyoda A, Aizu T, Ejima F, Fujiyama A, Shini T, Kohara Y, Niki H. Mutations in cell elongation genes mreB, mrdA and mrdB suppress the shape defect of RodZ-deficient cells. Molec Microbiol. 2013;87:1029–1044. doi: 10.1111/mmi.12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Weiser JN. Mechanisms of Bacterial colonization of the respiratory tract. Ann Review Microbiol. 2015;69:425–444. doi: 10.1146/annurev-micro-091014-104209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamsås GA, Straume D, Ruud Winther A, Kjos M, Frantzen CA, Håvarstein LS. Identification of EloR (Spr1851) as a regulator of cell elongation in Streptococcus pneumoniae. Molec Microbiol. 2017 doi: 10.1111/mmi.13748. [DOI] [PubMed] [Google Scholar]
- Straume D, Stamsås GA, Berg KH, Salehian Z, Håvarstein LS. Identification of pneumococcal proteins that are functionally linked to penicillin-binding protein 2b (PBP2b) Molec Microbiol. 2017;103:99–116. doi: 10.1111/mmi.13543. [DOI] [PubMed] [Google Scholar]
- Sung CK, Li H, Claverys JP, Morrison DA. An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. App Environ Microbiol. 2001;67:5190–5196. doi: 10.1128/AEM.67.11.5190-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Molec Microbiol. 2014;94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Mukherjee D, Ray VA, Sham LT, Feig AL, Winkler ME. Identification and characterization of noncoding small RNAs in Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol. 2010;192:264–279. doi: 10.1128/JB.01204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, Winkler ME. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Molec Microbiol. 2016;100:1039–1065. doi: 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RD, Vollmer W, Foster SJ. Different walls for rods and balls: the diversity of peptidoglycan. Molec Microbiol. 2014;91:862–874. doi: 10.1111/mmi.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrych A, Holeckova N, Goldova J, Doubravova L, Benada O, Kofronova O, Halada P, Branny P. Characterization of pneumococcal Ser/Thr protein phosphatase phpP mutant and identification of a novel PhpP substrate, putative RNA binding protein Jag. BMC Microbiol. 2016;16:247. doi: 10.1186/s12866-016-0865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegrove TB, Zhang A, Storz G. Hfq: the flexible RNA matchmaker. Curr Opin Microbiol. 2016;30:133–138. doi: 10.1016/j.mib.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde R, Edwards L, Regan L. Structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- Van Assche E, Van Puyvelde S, Vanderleyden J, Steenackers HP. RNA-binding proteins involved in post-transcriptional regulation in bacteria. Front Microbiology. 2015;6:141. doi: 10.3389/fmicb.2015.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Ent F, Johnson CM, Persons L, de Boer P, Lowe J. Bacterial actin MreB assembles in complex with cell shape protein RodZ. EMBO J. 2010;29:1081–1090. doi: 10.1038/emboj.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Camilli A. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 2012;22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernatter J, Pirofski LA. Current concepts in host-microbe interaction leading to pneumococcal pneumonia. Curr Opin Infectious Dis. 2013;26:277–283. doi: 10.1097/QCO.0b013e3283608419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinella D, Joseleau-Petit D, Thevenet D, Bouloc P, D’Ari R. Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J Bacteriol. 1993;175:6704–6710. doi: 10.1128/jb.175.20.6704-6710.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008;32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- Walsh C, Wencewicz T. Antibiotics: Challenges, Mechanisms, Opportunities. ASM Press; Washington, DC: 2016. [Google Scholar]
- Wayne KJ, Sham LT, Tsui HC, Gutu AD, Barendt SM, Keen SK, Winkler ME. Localization and cellular amounts of the WalRKJ (VicRKX) two-component regulatory system proteins in serotype 2 Streptococcus pneumoniae. J Bacteriol. 2010;192:4388–4394. doi: 10.1128/JB.00578-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westesson O, Skinner M, Holmes I. Visualizing next-generation sequencing data with JBrowse. Brief Bioinform. 2013;14:172–177. doi: 10.1093/bib/bbr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. List of Bacteria For Which New Antibiotics Are Urgently Needed. Geneva, Switzerland: World Health Organization; 2017. http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en. [Google Scholar]
- Zapun A, Vernet T, Pinho MG. The different shapes of cocci. FEMS Microbiol Rev. 2008;32:345–360. doi: 10.1111/j.1574-6976.2007.00098.x. [DOI] [PubMed] [Google Scholar]
- Zheng JJ, Sinha D, Wayne KJ, Winkler ME. Physiological roles of the dual phosphate transporter systems in low and high Phosphate conditions and in capsule maintenance of Streptococcus pneumoniae D39. Front Cell Infect Microbiol. 2016;6:63. doi: 10.3389/fcimb.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.