

SUMMARY
Microglia play a pivotal role in maintenance of brain homeostasis, but lose homeostatic function during neurodegenerative disorders. We identified a specific apolipoprotein E (APOE)-dependent molecular signature in microglia from models of amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) and Alzheimer’s disease (AD) and in microglia surrounding neuritic β-amyloid (Aβ) -plaques in human AD brains. The APOE pathway mediated a switch from a homeostatic to neurodegenerative microglia phenotype following phagocytosis of apoptotic neurons. Triggering receptor expressed on myeloid cells 2 (TREM2) induced APOE signaling, and targeting the TREM2-APOE pathway restored the homeostatic signature of microglia in ALS and AD mouse models and prevented neuronal loss in an acute model of neurodegeneration. APOE-mediated neurodegenerative microglia led to a loss in their tolerogenic function. Taken together, our work identifies the TREM2-APOE pathway as a major regulator of microglial functional phenotype in neurodegenerative diseases and serves as a novel target to restore homeostatic microglia.
Keywords: Alzheimer’s disease, amyotrophic lateral sclerosis, APOE, microglia, multiple sclerosis, neurodegeneration, transcriptional regulation, TREM2
In Brief
Microglia change their phenotype and function during aging and neurodegeneration, but the underlying molecular mechanisms for this change remain unknown. Krasemann, Madore, et al. identify the TREM2-APOE pathway as a major regulator of microglia phenotypic change in neurodegenerative diseases, which may serve as a target to restore homeostatic microglia.
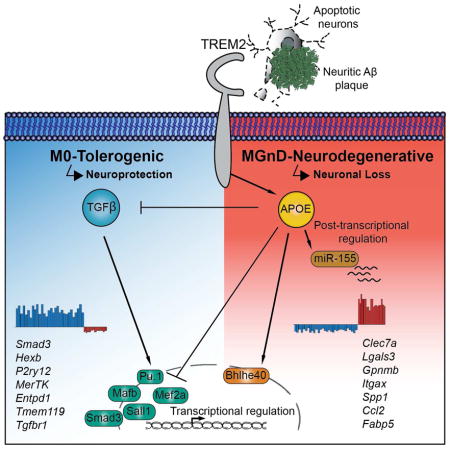
INTRODUCTION
Neuronal injury and cell death are common hallmarks of neurodegenerative processes (Mattson, 2000). It remains unclear whether neuronal cell death is a cause or result of Alzheimer’s disease (AD), especially considering that neurodegeneration is not an acute and rapid event but occurs gradually over a long period of time. However, apoptosis as well as neuritic dystrophy leading to permanent central nervous system (CNS) damage occurs in neurodegenerative diseases including AD (Masliah et al., 1998), amyotrophic lateral sclerosis (ALS) (Kostic et al., 1997) and in inflammatory autoimmune diseases such as multiple sclerosis (MS) (Ofengeim et al., 2015).
Microglia are brain resident immune cells that maintain CNS homeostasis, constantly survey their environment (Nimmerjahn et al., 2005), and react to homeostasis-perturbing elements by initiating an inflammatory reaction (Kreutzberg, 1996). They have a unique homeostatic molecular and functional signature (M0) in the healthy brain (Butovsky et al., 2014; Gautier et al., 2012; Hickman et al., 2013), which is tightly controlled by transforming growth factor β (TGFβ) signaling (Butovsky et al., 2014; Gosselin et al., 2014). However, during the course of disease, microglia lose their homeostatic molecular signature and functions (Butovsky et al., 2015; Holtman et al., 2015) and become chronically inflammatory (Perry and Holmes, 2014). Recent studies identified a common disease-associated microglia signature (Chiu et al., 2013; Hickman et al., 2013; Holtman et al., 2015; Keren-Shaul et al., 2017; Orre et al., 2014). However, the mechanisms, which regulate the microglial phenotype in disease, have not been identified.
In this study, we identified a role for apolipoprotein E (APOE) in regulating a subset of microglia, which exhibit a common disease-associated phenotype. This neurodegenerative phenotype switch is triggered by TREM2, leading to activation of APOE signaling and subsequent suppression of homeostatic microglial phenotype. A functional consequence of the activation of TREM2-APOE pathway is the loss of the ability of microglia to regulate brain homeostasis.
RESULTS
Reciprocal induction of APOE and suppression of TGFβ signaling in disease-associated microglia
To investigate underlying common molecular mechanisms that regulate microglial dysfunction, we isolated microglia and analyzed transcriptomes during aging and disease progression in mouse models of ALS (SOD1G93A, expressing human Cu,Zn superoxide dismutase mutation), AD (APP-PS1, overexpressing mutated genes for human amyloid precursor protein and presenilin 1) and MS (experimental autoimmune encephalomyelitis; EAE) (Table S1). We performed k-means clustering and identified two major gene clusters (Figure 1A). Cluster 1 was associated with loss of 68 homeostatic microglial genes, including P2ry12, Tmem119, Gpr34, Jun, Olfml3, Csf1r, Hexb, Mertk, Rhob, Cx3Cr1, Tgfbr1 and Tgfb1 and transcription factors such as Mef2a, Mafb, Jun, Sall1 and Egr1, which are enriched in adult microglia (Butovsky et al., 2014; Buttgereit et al., 2016; Matcovitch-Natan et al., 2016). Cluster 2 was associated with upregulation of 28 inflammatory molecules including Spp1, Itgax, Axl, Lilrb4, Clec7a, Ccl2, Csf1 and Apoe, of which Apoe was one of the most upregulated genes (Figure 1A and Table S1). We termed this microglial neurodegenerative phenotype as MGnD, in contrast to M0-homeostatic adult microglia phenotype (Butovsky et al., 2014; Gautier et al., 2012; Hickman et al., 2013). Linear regression analysis showed a negative correlation of Mef2a, Sall1 and Tgfbr1 with progression in EAE, SOD1 and APP-PS1 models (Figure 1B). In contrast, induction of Apoe was positively correlated with progression (Figure 1B). In EAE, we found suppression of homeostatic genes [e.g., P2ry12, Tmem119, Tgfbr1, Mafb, Mef2a, Sall1 and Egr1 (Butovsky et al., 2014; Gautier et al., 2012; Hickman et al., 2013; Matcovitch-Natan et al., 2016)] during acute phase and restoration during recovery (Figure 1C; Figures S1A–S1C and Table S1). Apoe was reciprocally upregulated (Figure 1C), which we validated by quantitative real-time PCR (qPCR) in two other MOG-induced EAE mouse models: the non-obese diabetic (NOD) (chronic-relapsing) and C57BL/6J (acute) (Figures S1D and S1E). P2ry12 immunoreactivity was lost at onset and disease peak and re-emerged during recovery (Figure S1F). APOE and TGFβ were major upstream regulators of MGnD microglia (Figure 1D and Table S1) by ingenuity pathway analysis (IPA). The top-suppresed TGFβ-dependent homeostatic genes (Butovsky et al., 2014), Olfml3, P2ry12, Tmem119, Mef2a, Jun, Sall1 and -upregulated Apoe and Axl were similarly affected during progression (Figure 1E). These results identify a common microglia molecular signature in disease that is associated with the induction of APOE and the suppression of TGFβ signaling.
Figure 1. Reciprocal induction of APOE and suppression of TGFβ signaling in disease-associated MGnD-microglia.

(A) K-means clustering of 95 significantly affected common genes in FCRLS+ microglia during aging and disease by Nanostring). Vertical lanes are biological replicates per disease stage/condition in WT-aging (n = 12), EAE (n = 18), SOD1 (n = 11) and APP-PS1 (n = 12) mice. Cluster 1: suppressed homeostatic genes; cluster 2: upregulated genes.
(B) Linear regression curve of Mef2a, Sall1, Tgfbr1 and Apoe in EAE (n = 4–6 mice/disease score) and SOD1 (n = 2–4 mice/disease score) spinal cord microglia and APP-PS1 (n = 3 mice/age) brain microglia. Thick line: 95% confidence interval of the regression line.
(C) Selected homeostatic and disease-associated microglial genes during EAE (n = 3). Data are presented as mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA followed by Dunnett’s multiple-comparison post-hoc test.
(D) IPA shows common nodes significantly affected in microglia in all three-mouse models in disease. For each molecule, the expression fold change compared to normal, homeostatic microglia is presented.
(E) Heatmap of significantly affected genes dysregulated in all 3 diseases (n = 3–4) determined by Nanostring. Vertical lanes: mean of biological replicates per disease stage/condition as indicated in (A).
The MGnD-microglia subset is associated with neuritic Aβ-plaques and diffuse neuritic dystrophy in the AD cortex
The pathology of the AD brain is characterized by a widespread neuritic dystrophy (Larner, 1995). These dystrophic axons are enlarged and show intense reactivity for phosphorylated neurofilaments (Sternberger et al., 1985). To address whether a microglial phenotype switch from M0-homeostatic to MGnD-neurodegenerative is specifically associated with neuritic dystrophy in AD, we analyzed brains from APP-PS1 mice and human AD. To distinguish M0- vs. MGnD-microglia we used P2ry12 and Clec7a mAbs and found Clec7a+P2ry12− microglia associated with Aβ-plaques in APP-PS1 mice (Figure 2A). We identified 3 microglial subsets: 1) Clec7a−P2ry12+ (not associated with Aβ-plaques); 2) Clec7aloP2ry12lo (in close proximity to Aβ-plaques); and 3) Clec7a+P2ry12− (associated with neuritic Aβ-plaques) (Figure 2A). TMEM119 protein is stable even when the messenger RNA (mRNA) is downregulated (Satoh et al., 2016) and co-localized with Aβ-plaque-associated Clec7a+ microglia (Figure S2A). Clec7a+P2ry12− microglia associated with phosphorylated neurofilaments (pNF+) in neuritic plaques in APP-PS1 mice (Figure 2B and Figure S2B). FCRLS+Clec7a+ microglia were increased in 24-month old APP-PS1 mice (Figure S2C–S2G). The phenotype of Clec7a+ microglia was similar to MGnD microglia identified in SOD1, EAE and APP-PS1 models and during aging (Figure 2C and Table S2). Among suppressed microglial homeostatic genes were P2ry12, Tmem119, Olfml3, Csf1r, Rhob, Cx3cr1, Tgfb1, Mef2a, Mafb, Sall1 and upregulated inflammatory molecules included Spp1, Itgax, Axl, Lilrb4, Clec7a, Csf1 and Apoe, of which Apoe which was among the most upregulated genes (Figures 2C and 2D and Table S2). Transition from Clec7a− to Clec7aint to Clec7ahi correlated with increased expression of Apoe and suppression of homeostatic molecules (Figure 2D). Clec7a expression increased during progression in EAE and SOD1 mice (Figures S2H–S2K). RNA sequencing (RNAseq) of the 3 microglia subsets validated the Nanostring analysis and showed only FCRLS+Clec7a+ microglia acquired an MGnD signature (Figures 2E and 2F and Table S2).
Figure 2. MGnD-microglia are associated with neuritic Aβ-plaques.

(A) Staining for P2ry12+ and Clec7a+ in Aβ-plaque microglia in 24-month-old APP-PS1 mice. MGnD around plaque (red arrows). MGnD transitioning to plaque (yellow arrows). M0-homeostatic microglia (white arrows). Scale bar, 20 μm.
(B) Staining for P2ry12+ and Clec7a+ in microglia associated with neuritic plaque (pNF) in APP-PS1 mice (24 month). Scale bar, 20 μm.
(C) Heatmap of significantly affected genes in Clec7a+ vs. Clec7aint vs. Clec7a− FCRLS+ microglia in APP-PS1 vs. WT mice (n = 5; 24 month). Selected homeostatic (blue) and inflammatory (red) genes.
(D) Selected genes shown in (C). Dot plots: mRNAs transcripts (mean ± s.e.m). *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001 by one-way ANOVA followed by Tukey’s multiple-comparison post-hoc test.
(E) Heatmap of significantly affected genes determined by RNAseq in Clec7a+ vs. Clec7a− FCRLS+ microglia in APP-PS1 vs. WT mice (n = 5; 9 month).
(F) Selected genes shown in (E). Dot plots: FPKM (mean ± s.e.m). *p < 0.05, **p < 0.01, ***p < 0.001, ****p< 0.0001 by one-way ANOVA followed by Tukey’s multiple-comparison post-hoc test.
(G) Staining for P2RY12 with Aβ or pNF in diffuse vs. neuritic plaques in human AD brain. Scale bar, 100 μm.
(H) Linear regression curve of P2RY12+ microglia with dystrophic axons in control temporal cortex and AD defined by CERAD criteria and Braak stages (n = 14) and age-matched controls (n = 10).
To investigate if a microglial phenotype was associated with neuritic plaques, we double stained AD brain for P2RY12 with Aβ or pNF. P2RY12+ microglia were preserved in Aβ diffuse plaques and P2RY12 signal was lost in pNF+ neuritic plaques (Figure 2G). TMEM119+P2RY12− microglia, and not recruited IBA1+TMEM119− peripheral myeloid cells, were associated with pNF+ neuritic plaques (Figures S2L–S2N). Loss of P2RY12+ microglia correlated with axonal dystrophy, but not with the extent of Aβ deposition (Figure 2H). We found increased APOE expression in microglia in close proximity to Aβ-plaque (Figures S2O and S2P). These data demonstrate that the microglial phenotype switch from M0 to MGnD is associated with neuritic dystrophy in APP-PS1 mice and human AD.
Phagocytosis of apoptotic neurons suppresses homeostatic microglia
To determine the mechanisms by which the MGnD are induced in neurodegeneration, apoptotic neurons (dN) were injected into the cortex and hippocampus of naïve mice. Apoptotic neurons induced the recruitment of P2ry12+ microglia towards the site of injection (Figure 3A). P2ry12+ microglia changed morphology from an M0-homeostatic non-phagocytic (MG-nΦ) phenotype to an amoeboid-phagocytic (MG-dNΦ) phenotype at the vicinity of the injection site (Figures 3A–3C and S3A). Induction of MGnD microglia was not detected in phosphate-buffered saline (PBS) injected brains (Figure S3B). Apoptotic neurons were internalized by P2ry12+ microglia (Figure 3B). Induction of Apoe was detected in MG-dNΦ microglia (Figure 3C). Increased Apoe was detected as early as 3 h post-injection and peaked at 16 h post-injection (Figure S3C). Restoration of homeostatic P2ry12+Clec7a− microglia occurred at 14 days post-injection (Figures S2D and S2E). Profiling of MG-dNΦ and MG-nΦ microglia showed that phagocytosis of dN induced a microglial molecular phenotype similar to MGnD phenotype (Figures 3D and 3E). M0-microglial homeostatic genes including Tmem119, Egr1, Hexb, Gpr34, P2ry12, Olfml3 and Tgfbr1 were suppressed in MG-dNΦ microglia, whereas Apoe was the major upregulated gene among other inflammatory molecules (Figure 3F and Table S3). This was confirmed by qPCR of microglial genes including upregulation of Apoe and microRNA miR-155 (Figures 3G and S3C and S3F). Although injured neurons express elevated Apoe (Xu et al., 2006), we observed the upregulation of Apoe in MG-dNΦ microglia phagocytosing Apoe−/− dN (Figure S3G). Injection of E.coli- or Zymosan-particles did not induce Apoe in phagocytic microglia (Figure S3H). In contrast, miR-155 was induced by dN, E.coli- and Zymosan-particles in phagocytic microglia (Figure S3I). Microglia phagocytosed injected dN, but not live neurons (Figures S3J and S3K). Upregulation of Apoe in phagocytic microglia was also induced by apoptotic monocytes (Figure S3L). We validated our paradigm of dN-induced MG-dNΦ phenotype using kainic acid as a model of acute neuronal cell death (Levesque and Avoli, 2013). We observed P2ry12−/Clec7a+ microglia 48 h post-injection (Figure S4).
Figure 3. Phagocytosis of apoptotic neurons suppresses homeostatic microglia.

(A) Staining for P2ry12+ microglia at the site of apoptotic neuron injection 16 h post-injection in WT mice (n = 6). Phagocytic (MG-dNΦ) and non-phagocytic (MG-nΦ) microglia. Scale bar, 80 μm.
(B) Orthogonal projections of confocal z-stacks show intracellular Alexa488+ apoptotic neurons co-stained with DAPI in P2ry12+ microglia (n = 6). Scale bar, 5 μm. One of two individual experiments.
(C) Staining for Apoe in P2ry12+ MG-dNΦ. White arrows: Apoe− MG-nΦ; yellow arrows: Apoe+ MG-dNΦ (n = 3). Scale bar, 15 μm.
(D) FACS-sorted CD11b+FCRLS+ MG-dNΦ containing Alexa488+ apoptotic neurons and MG-nΦ from the same injection site 16 h later.
(E) Heatmap of affected genes in MG-dNΦ (n = 4) vs. MG-nΦ (n = 4) 16 h post-injection with apoptotic neurons. One of two individual experiments.
(F) Top-20 affected genes shown in (E). Bars: mRNA Nanostring (nCounts) (mean ± s.e.m) detected in 100 ug total RNA. *p < 0.05 by Student t test, 2-tailed.
(G) qPCR validation of selected genes. Gene expression level normalized against Gapdh using ΔCt (n = 3–4/group). Results: mean normalized expression ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA followed by Tukey’s multiple-comparison post hoc test.
(H) Volcano plot of 552 TMT-mass spectrometry-identified proteins highlighting changes in MG-dNΦ vs. MG-nΦ. p < 0.05 by Student t test, 2-tailed.
(I) Circos plot: connectivity map derived from the pairwise comparison of transcriptome data from mouse AD models, ALS and Mfp2−/−, aging and irradiation shown in orange. Each line represents a pairwise data set overlap, determined using GSEA analysis and filtered by p < 0.001.
See also Figure S3, S4, S5 and Tables S3, S4.
Although in vivo LPS and IFNγ-stimulated microglia suppressed many of the homeostatic microglial genes including P2ry12, Gpr34, Fcrls, Tmem119, Olfml3, Siglech, Sall1 Hexb, Csf1r and Tgfbr1 (Figures S5A–S5C and Table S4), the expression of Apoe was suppressed in M1-microglia (Figures S5D–S5E and S5H). In contrast, Egr1, a key transcription factor in M0-homeostatic microglia (Butovsky et al., 2014; Matcovitch-Natan et al., 2016), which was most suppressed in MG-dNΦ microglia, was induced in M1-microglia (Figures S5F–S5H). Furthermore, arginase 1 (Arg1) and chitinase-3-like protein (Ym1), the ‘classical’ M2-anti-inflammatory markers (Martinez and Gordon, 2014) were induced in MG-dNΦ microglia along with inflammatory molecules including Il1b, Ptgs2 (Cox2), Ccl2, Ccl5, Tspo, Msr1 and Cebpb (Figures S5F–S5H). Thus, the MG-dNΦ molecular signature is distinct from classical M1-induced microglia.
Quantitative mass spectrometry of MG-dNΦ vs. MG-nΦ microglia showed increased Apoe and Lgals3 in MG-dNΦ microglia. In contrast, both Rgs10 and Bin1, two homeostatic microglial proteins (Butovsky et al., 2014), were suppressed (Figure 3H and Table S3). To investigate MG-dNΦ microglia in disease models, we used gene set enrichment analysis (GSEA). Circos plot network analysis presented an overlap of the phagocytic microglial phenotype with the microglia molecular signature during aging and in a model of brain irradiation. The MG-dNΦ microglia signature was enriched in microglia transcriptomes from models of AD (5XFAD), ALS (SOD1) and Mfp2 deficiency, which is associated with cerebellar degeneration (Verheijden et al., 2013). In contrast, microglial changes associated with neuropathic and chronic pain models or in Mecp2 deficiency were not connected with MG-dNΦ gene signature (Figure 3I, Table S3). Taken together these data show that MGnD microglia are induced by apoptotic neurons, which are a hallmark of neurodegeneration.
APOE regulates the transcriptional and post-transcriptional program of the MGnD phenotype and function
We reported that Apoe expression is downregulated in microglia during development, correlates with the induction of M0-homeostatic genes, and is increased in microglia progenitors from CNS Tgfb1−/− mice (Butovsky et al., 2014). To assess Apoe in the induction of MGnD microglia when phagocytosing apoptotic neurons, we analyzed Apoe−/− vs. WT transcriptomes from MG-nΦ vs MG-dNΦ microglia. We defined clusters based on differentially expressed genes between WT non-phagocytic vs. phagocytic vs. Apoe−/− phagocytic microglia (Figure 4A and Table S5). Cluster 1 showed 885 genes induced in MG-dNΦ WT microglia which were repressed in Apoe−/− MG-dNΦ microglia. 17 genes among those commonly upregulated in disease (Figure 1A) were suppressed by Apoe (Figure 4A and Table S5) including Clec7a, Gpnmb and Lgals3 (Figure 4B) and transcription factors (TFs) commonly induced in disease such as Bhlhe40 (Figure 4C). Clusters 2 and 3 showed 1,220 suppressed genes in WT MG-dNΦ which were restored in Apoe−/− MG-dNΦ microglia. Among 68 genes commonly suppressed in disease, 40 genes were restored in Apoe−/− phagocytic microglia including the major homeostatic genes Tmem119, Tgfbr2 and Csf1r (Figure 4D) and TFs such as Mef2a, Mafb and Sall1 (Figure 4E). Nanostring confirmed de-repression of major homeostatic microglial genes (Figure S6A and Table S5). Disease-associated upregulated genes including Lgals3 and Clec7a were suppressed in Apoe−/− microglia (Figure S6B). To assess an intrinsic role of APOE regulation in a cell-specific manner, we deleted Apoe in microglia using Cx3cr1CreERT2:Apoefl/fl mice. qPCR analysis confirmed Apoe deletion in Cx3cr1CreERT2:Apoefl/fl microglia as compared to Cx3cr1WT:Apoefl/fl microglia. Moreover, gene expression of Clec7a was repressed and expression of Csf1r, Tgfbr1 and Tmem119 was restored in MG-dNΦ microglia from Cx3cr1CreERT2:Apoefl/fl mice (Figure 4F). Intrinsic deletion of Apoe did not affect expression of these genes in MG-nΦ microglia (Figure 4G), demonstrating that Apoe deletion had no effect on non-phagocytosing microglia.
Figure 4. APOE regulates transcriptional and post-transcriptional program of MGnD phenotype.

(A) K-means clustering (k = 4) of 2,234 affected genes in bulk microglia (1,000 cells per animal) from WT and Apoe−/− mice. Vertical lane: mean of biological replicates of 1,000 MG-nΦ vs. MG-dNΦ cells from WT (n = 10) vs. Apoe−/− mice for MG-nΦ (n = 10) and MG-dNΦ (n = 9). Microglia isolated 16 h post-injection of apoptotic neurons.
(B–E) Selected Apoe-induced genes (B), Apoe-induced Transcription Factors (TFs) (C), Apoe-repressed genes (D) and Apoe- repressed TFs (E) from WT and Apoe−/− mice 16 h post-injection of apoptotic neurons. Data: mean ± s.e.m. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA followed by Tukey’s multiple-comparison post-hoc test.
(F) qPCR validation of selected genes in MG-dNΦ from Cx3cr1CreERT2:Apoefl/fl vs. Cx3cr1wt:Apoefl/fl controls. Expression levels normalized against Gapdh using ΔCt (n = 6–10/group). Data: mean normalized expression ± s.e.m. *p < 0.05, **p < 0.01, by Student t test, 2-tailed.
(G) qPCR validation of genes in MG-nΦ from Cx3cr1CreERT2:Apoefl/fl vs. Cx3cr1wt:Apoefl/fl controls. Expression level normalized against Gapdh using ΔCt (n = 6–10/group). Data: mean normalized expression ± s.e.m. *p < 0.05, **p < 0.01, by Student t test, 2-tailed.
(H) Staining for NeuN+ and P2ry12+ in contralateral vs. axotomized facial motor nucleus 7 d post-surgery in WT (n = 5) vs. Apoe−/− (n = 5) vs. Cx3cr1CreERT2:Apoefl/fl (n = 4) vs. Cx3cr1wt:Apoefl/fl (n = 4) control mice. Scale bar, 80 μm.
(I) Quantification of NeuN+ per facial motor nucleus in contralateral vs. axotomized nucleus 7 d post-surgery (n = 5). Data: mean ± s.e.m. *p < 0.05, **p < 0.01, ****p < 0.0001 by one-way ANOVA followed by Tukey’s multiple-comparison post-hoc test.
A recent report showed induction of Apoe among other disease-associated genes in microglia at 7 d post-facial nerve axotomy (Tay et al., 2017). We assessed the role of Apoe on neuronal survival in WT, Apoe−/−, Cx3cr1CreERT2:Apoefl/fl and control Cx3cr1WT:Apoefl/fl mice. Global and conditional deletion of Apoe in microglia reduced neuronal loss as compared to WT or Cx3cr1WT:Apoefl/fl mice in the axotomized facial motor nucleus (Figure 4H–I and Figure S6C). These data demonstrated the essential role of microglial Apoe in MGnD phenotype.
Genetic ablation of miR-155 reverses the abnormal microglial signature and ameliorates disease in SOD1 mice (Butovsky et al., 2015). In MG-dNΦ microglia from Apoe−/− mice, miR-155 expression was suppressed (Figure S6D). In contrast, expression of Apoe was unaffected in miR-155−/− MG-dNΦ microglia (Figure S6E). Thus, miR-155 expression was regulated downstream of the APOE pathway. IPA upstream-regulator analysis identified TGFβ1 as the most significantly de-repressed molecule in Apoe−/− phagocytic microglia (Figure S6F). In addition to other genes involved in inflammatory signaling, the top upstream regulator in miR-155−/− phagocytic microglia was IL-6 (Figure S6F), suggesting a different role in regulation of MGnD microglia. Egr1 transcription factor was the most de-repressed gene in the TGFβ pathway in Apoe−/− phagocytic microglia (Figure S6F). We determined the Apoe-dependent transcriptional effect in microglia by injecting recombinant Apoe intra-hippocampally in Apoe−/− mice. MGnD disease-associated genes such as Msr1, Lilrb4 and Tlr2 were induced and homeostatic transcriptional factors including Smad3, Mef2a, Mafb and Egr1 were suppressed (Figures S6G and S6H and Table S5). Thus, APOE signaling has a twofold effect on microglia regulation in neurodegeneration: 1) suppression of the microglial homeostatic transcriptional factors including Mef2a, Mafb, Smad3 and 2) induction of an inflammatory program including transcription factor Bhlhe40, Tfec, Atf3 and a both transcriptional and translational regulator miR-155.
Genetic targeting of Trem2 suppresses the APOE pathway and restores the homeostatic microglia in APP-PS1 and SOD1 mice
TREM2 is a sensor of membrane-associated lipids including phosphatidylserine (PS) focally exposed on apoptotic cells and processes or synapses of damaged neurons (Wang et al., 2015). We found blocking phosphatidylserine on apoptotic neurons with Annexin V protein (Koopman et al., 1994), reduced microglial phagocytosis by 88% (Figure S7A). Because apoptotic neurons suppressed homeostatic microglia genes (above), we asked whether they activated the APOE pathway in TREM2-dependent manner. We found that expression of homeostatic genes including P2ry12, Gpr34, Tmem119, Tgfbr1 and Csf1r, was restored in Trem2−/− MG-dNΦ as compared to WT MG-dNΦ microglia (Figure 5A), whereas Apoe and miR-155 were suppressed in Trem2−/− MG-dNΦ microglia (Figures 5B and S7B). Targeting TREM2 signaling in the facial nerve axotomy (FNA) model also ameliorated neuronal loss in Trem2−/− mice (Figure 5C). To ask whether TREM2 induced MGnD phenotype in disease, we genetically ablated Trem2 in APP-PS1 and SOD1 mice. Nanostring profiling of brain microglia from APP-PS1:Trem2−/− mice showed suppression of 7 inflammatory molecules (Trem2, Axl, Clec7a, Csf1, Itgax, Cd34 and Apoe), which were among the most upregulated genes in the common disease-associated signature (Figure 5D). Among 108 restored genes in APP-PS1:Trem2−/− mice, 54 were commonly suppressed homeostatic microglial genes in disease including P2ry12, Tmem119, Gpr34, Olfml3, Csf1r, Mertk, Rhob, Cx3Cr1, Tgfbr1 and Tgfb1 and transcriptional factors such as Mef2a, Mafb and Sall1 (Figure 5D). Moreover, we confirmed the protein level restoration of P2ry12 and suppression of Clec7a in microglia associated with Aβ-plaques in APP-PS1:Trem2−/− mice as compared to APP-PS1 mice (Figures 5E–5G). Consistent with previous findings (Jay et al., 2015), the Aβ-plaque load was decreased in APP-PS1:Trem2−/− mice (Figure 5H). In SOD1:Trem2−/− microglia, among 36 downregulated inflammatory genes, 11 genes were common to the disease-associated signature. Among 240 restored genes in SOD1:Trem2−/− mice, 66 were commonly suppressed homeostatic genes (Figure 5I). Transcription factors including Mef2a, Mafb and Sall1 and TGFβ signaling (Tgfb and Tgfbr1), which were restored in Trem2−/− microglia in disease, were restored in Apoe−/− MG-dNΦ microglia. Clec7a+P2ry12− microglia were detected in spinal cords from SOD1:Trem2+/− as compared to SOD1:Trem2−/− mice at 115 d of age (Figure 5J). Clec7a−P2ry12+ homeostatic microglia were preserved in SOD1:Trem2−/− mice (Figures 5J–5L). Expression of miR-155 was not induced in SOD1:Trem2−/− mice (Figure 5M). We previously found early induction of Apoe in SOD1 males before clinical onset (Butovsky et a., 2015). To determine the TREM2 gender-dependent regulation of microglia in SOD1 mice, we performed unbiased RNAseq of the same samples shown in Figure 5I. We found 279 commonly affected genes (cluster 2) and gender-specific genes (575 genes in females, cluster 1 and 2,639 genes in males, cluster 3) (Figure 5N and Table S6). In commonly affected genes, SOD1:Trem2+/− males show induction of Apoe as compared to female littermates (Figures 5O and 5P), which is in line with our previous report (Butovsky et al., 2015). Genetic deletion of Trem2 in males showed marked suppression of Apoe and restoration of master regulators of homeostatic microglia including Spi1 (PU.1), Smad3, Tgfbr1 and Sall1 as compared to SOD1:Trem2−/− female littermates (Figure 5P).
Figure 5. Genetic targeting of Trem2 suppresses APOE pathway and restores the homeostatic microglia in APP-PS1 and SOD1 mice.

(A) qPCR analysis of homeostatic genes in MG-dNΦ from Trem2−/− vs. WT mice (n = 4). Data: mean ± s.e.m. *p < 0.05, **p < 0.01 by one way ANOVA.
(B) qPCR analysis of Apoe and miR-155 expression in MG-dNΦ from Trem2−/− vs. WT mice (n = 3–4/group). Data: mean ± s.e.m normalized against Gapdh and U6 expression using ΔCt. *p < 0.05, by Student t test, 2-tailed.
(C) Quantification of NeuN+ per facial motor nucleus in contralateral vs. axotomized nucleus in WT vs. Trem2−/− (n = 4–5/group), 7 d post-surgery. Data: mean ± s.e.m. *p < 0.05, **p < 0.01, ****p < 0.0001 by one-way ANOVA followed by Tukey’s multiple-comparison post-hoc test.
(D) Volcano plot based on differentially expressed genes in microglia from APP-PS1 (n = 4) vs. APP-PS1:Trem2−/− (n = 5) mice at 120 d. p < 0.05 by Student t test, 2-tailed. Selected restored homeostatic (red) and suppressed inflammatory (blue) genes.
(E) Staining for P2ry12, Clec7a and Aβ plaque in APP-PS1 vs. APP-PS1:Trem2−/− mice (120 d). Scale bar, 20 μm.
(F and G) Quantification of P2ry12 and Clec7a intensity per Aβ plaque in APP-PS1 vs. APP-PS1:Trem2−/− mice (n = 5; 120 d). ***p < 0.001, by Student t test, 2-tailed.
(H) Quantification of Aβ plaque load per animal in APP-PS1 vs. APP-PS1:Trem2−/− mice (n = 5; 120 d). **p < 0.01, by Student t test, 2-tailed.
(I) Volcano plot based on differentially expressed genes in microglia from SOD1:Trem2+/− (n = 9) vs. SOD1:Trem2−/− (n = 13) mice at 115 d (neurological score 1). p < 0.05 by Student t test, 2-tailed.
(J) Staining for P2ry12, Clec7a and NeuN in SOD1:Trem2+/− vs. SOD1:Trem2−/− mice (lumbar section, 115 d). M0-homeostatic microglia (white arrows). M0 microglia transitioning to MGnD (yellow arrows). MGnD at the epicenter of the ventral horn (red arrows). Scale bar, 50 μm.
(K and L) Quantification of P2ry12 and Clec7a intensity in ventral horn of SOD1:Trem2+/− (n = 4, n = 5 females) vs. SOD1:Trem2−/− (n = 7 males, 5 females) mice (lumbar section, 115 d). **p < 0.001, ***p < 0.001 by Student t test, 2-tailed.
(M) qPCR analysis of miR-155 expression in microglia isolated from ventral horn of SOD1:Trem2+/− (n = 4) vs. SOD1:Trem2−/− (n = 5) mice (lumbar section, 115 d). Data: mean ± s.e.m normalized against U6 expression using ΔCt. *p < 0.05, by Student t test, 2-tailed.
(N) Volcano plot: differentially expressed genes in spinal cord microglia from male and female SOD1:Trem2+/− (n = 4 males, 4 females) vs. SOD1:Trem2−/− (n = 7 males, 5 females) mice at 115 d (neurological score 1). p < 0.05 by Student t test, 2-tailed. Cluster 1: female-specific differentially expressed genes; Cluster 2: differentially expressed genes in both genders; cluster 3: male-specific differentially expressed genes.
(O) Heatmap of affected genes in male and female SOD1:Trem2+/− (n = 4 males, 4 females) vs. SOD1:Trem2−/− mice (n = 7 males, 5 females) at 115 d (cluster 2). Vertical lanes: biological replicates per genotype and gender.
(P) Gender comparison of homeostatic (lower panel) and MGnD genes (upper panel) from SOD1:Trem2+/− and SOD1:Trem2−/− mice (n = 4–7/group). Data: mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by one-way ANOVA, followed by Tukey’s multiple-comparison post-hoc test.
See also Table S6.
To validate TREM2 in regulating the microglial phenotype switch in disease, we used GSEA to determine that transcripts from 5XFAD:Trem2−/− mice were enriched for the TREM2 responsive gene signature (Figures S7C–S7F and Table S6). Among the 4 disease models, TREM2-induced genes including Clec7a, Gpnmb, Fabp5, Lgals3, Csf1 and Ch25 were also induced by Apoe (Table S5).
To investigate whether findings in APP-PS1:Trem2−/− mice were found in AD subjects carrying AD-associated TREM2 variants (AD-TREM2), we investigated heterozygous carriers of R47H (AD-TREM2R47H) and R62H (AD-TREM2R62H) vs. common-variant AD-patients (AD-TREM2WT). In line with experimental data in mice (Wang et al., 2015), we found microglia were more evenly distributed within the cortex in AD-TREM2 in both types of mutations and did not cluster around Aβ plaques as in AD-TREM2WT (Figures 6A–6C). Moreover, TMEM119 intensity was significantly higher in both TREM2 mutation carriers (Figures 6D and 6E). Taken together, these results demonstrate that activation of APOE signaling by apoptotic neurons is TREM2 mediated and regulates the microglial phenotype switch in APP-PS1 and SOD1 mice.
Figure 6. TREM2 haplodeficiency in human AD lead to preservation of microglia morphology and TMEM119 immunoreactivity.

(A) Staining for Aβ (red), IBA1 (blue) in AD control subjects (AD-TREM2WT, n = 6) and TREM2 variant carriers (AD-TREM2R47H, n = 2 and AD-TREM2R62H, n = 2). Lower panels: single distribution of IBA1+ microglia in the same section areas. Scale bar, 100 μm.
(B) Higher magnification of the plaques indicated by * and + in (A). Scale bar, 50 μm.
(C) Quantification of IBA1+ microglia associated with Aβ-plaque in AD-TREM2WT and AD-TREM2Mut cortical tissue. Student’s t test, 2-tailed.
(D) Cortical tissue from AD-TREM2WT and AD-TREM2R62H cases stained with TMEM119. Panels show basis for scoring microglia staining, described in (E). Scores: − no cells stained; +/− single cells stained; + moderate number of microglia with TMEM119 reactivity; ++ most microglia are TMEM119 positive.
(E) Quantification of TMEM119 in AD-TREM2WT and AD-TREM2Mut cortical tissue. TMEM119 expression in microglia rated as in (D); the ratings are shown in numbers: − (0), +/− (1), + (2), ++ (3). Student’s t test, 2-tailed.
See also Table S7.
Activation of the APOE pathway suppresses the tolerogenic function of microglia
Genetic targeting of Apoe reduces neuroinflammation and disease in EAE (Shin et al., 2014). We tested the ability of microglia to inhibit T cell proliferation ex vivo, as it has previously been reported as a tolerogenic function (Bai et al., 2009). CSFE-labeled T cells were stimulated in vitro with anti-CD3 and CD28 mAbs (Figure 7A). M0-homeostatic microglia from spinal cord of naïve mice suppressed T cell proliferation. Stimulation of microglia with recombinant Apoe (rApoe) abrogated tolerogenic function of microglia. rApoe did not affect T cell proliferation in the absence of microglia (Figures 7A and 7B). Nonetheless, MGnD microglia from peak EAE was associated with the highest expression of Apoe (Figures 1B and 1C) and were unable to suppress T cell proliferation (Figures 7C and 7D). We then induced EAE in WT and Apoe−/− mice and found that unlike WT-EAE microglia, Apoe−/− microglia from spinal cord at disease peak suppressed T cell proliferation (Figures 7E and 7F), demonstrating the maintenance of the tolerogenic function. de novo stimulation of Apoe−/− microglia from EAE mice with rApoe inhibited their tolerogenic function (Figures 7G and 7H). Moreover, microglia isolated from SOD1 mice at disease peak, which show the highest expression of Apoe (Figure 1B), were also unable to suppress T cell proliferation (Figures 7I and 7J). In summary, we observed a loss of the tolerogenic function of microglia at peak EAE and in SOD1 mice. This loss is regulated by Apoe-dependent transcriptional effects in microglia.
Figure 7. Activation of APOE pathway suppresses tolerogenic function in microglia.

(A) CFSE+ CD3+ T cells cultured with spinal cord microglia of WT-naïve mice. Cultures treated with soluble rApoe (100 ng ml−1) for 72 h.
(B) T cell proliferation: mean ± s.e.m ***p < 0.001, Student’s t test, 2-tailed (n = 3).
(C) CFSE+ CD3+ T cells cultured for 72 h with spinal cord microglia from naive or EAE mice at disease peak.
(D) T cell proliferation: mean ± s.e.m **p < 0.01, Student’s t test, 2-tailed (n = 3).
(E) CFSE+ CD3+ T cells cultured for 72 h with spinal cord microglia from EAE WT vs. Apoe−/− mice disease peak.
(F) T cell proliferation: mean ± s.e.m ***p < 0.001, Student’s t test, 2-tailed (n = 3).
(G) CFSE+ CD3+ T cells cultured for 72 h with spinal cord microglia from EAE Apoe−/− mice at disease peak. Cultures treated with soluble rApoe (100 ng ml−1) for 72 h.
(H) T cell proliferation: as mean ± s.e.m ***p < 0.001, Student’s t test, 2-tailed (n = 3).
(I) CFSE+ CD3+ T cells cultured for 72 h with spinal cord microglia from WT vs. SOD1 mice at disease peak (neurologic score 4).
(J) T cell proliferation: mean ± s.e.m **p < 0.01, Student’s t test, 2-tailed (n = 3). Black line: isotype control. Numbers on FACS plots: representative T cell proliferation (%). Data are representative of 3 independent experiments (A–J).
DISCUSSION
We identified a molecular signature of disease-associated microglia (MGnD), which is dependent on the TREM2-APOE pathway. The MGnD phenotype was seen in several neurodegenerative models and was induced by phagocytosis of apoptotic neurons. In a mouse model and in human AD, neuritic Aβ-plaque associated microglia showed a MGnD phenotype consistent with reports of senescent microglia in aging and AD (Streit et al., 2009), which lose sensome function including TGFβ signaling (Hickman et al., 2013). Apoptotic cells are efficiently phagocytosed during nervous system development (Jacobson et al., 1997). We reported that during early stages of development microglia induce Apoe expression, which is inversely correlated with the suppression of homeostatic genes (Butovsky et al., 2014). Thus, microglia during development and in neurodegenerative processes may share common modulatory mechanisms induced by apoptotic neurons. Neuronal apoptosis is a rare event in human AD (Stadelmann et al., 1999). However, focal apoptosis (synaptosis and necroptosis) associated with phosphatidylserine exposure occurs on neuronal processes, dendrites or synapses (Mattson et al., 1998; Ofengeim et al., 2015). We found that apoptotic neurons triggered the TREM2-APOE pathway to induce MG-dNΦ and suppress the homeostatic signature, which is tightly regulated by the TGFβ pathway (Butovsky et al., 2014; Gosselin et al., 2014).
We report a role for APOE in regulating the MGnD microglial subset in a cell-autonomous manner. Senile plaques contain dystrophic neurites (Grutzendler et al., 2007) and microglia engulf amyloid fibers (Dickson et al., 1988). We suggest that MGnD microglia, amyloid plaques, and dystrophic neurites form a microenvironment highly enriched in APOE which could play a major role in AD progression. APOE both accumulates in amyloid plaques and facilitates Aβ aggregation (Kim et al., 2012). Along with APOE induction, we found increased expression of APP in EAE microglia. Chiu et al., reported increased APOE and APP expression in SOD1 microglia (Chiu et al., 2013) and Huang et al. showed that APOE stimulates APP and increases amyloid-β load in AD (Huang et al., 2017). Similarly, we found that Aβ-plaque associated microglia express a high level of APP in APP-PS1 mice. Our results are consistent with a report that increased APOE in CSF is associated with cognitive decline (Toledo et al., 2014). Thus, microglial APOE may play a detrimental role in AD.
Transcriptome profiling of disease-associated and homeostatic genes in microglia during aging and disease progression in SOD1, APP-PS1 and EAE identified APOE and TGFβ as the major upstream regulators of MGnD microglia. This phenotype was associated with neuritic plaques in APP-PS1 mice. Microglia acquire a MGnD phenotype following phagocytosis of apoptotic neurons. Interestingly, the LPS and IFNγ-induced (previously named M1) phenotype overlaps to some extent the MGnD microglia phenotype. However, microglia activated with LPS and IFNγ suppressed Apoe and induced Egr1, which are reciprocally induced and suppressed in MGnD and MG-dNΦ microglia. Macrophage lineage-determining factor PU.1 (Heinz et al., 2010) cooperates with microglia-specific enhancer regions of the Mef2 family (Butovsky et al., 2014; Matcovitch-Natan et al., 2016). Mef2a may partner with PU.1 to establish a microglia-specific signature (Lavin et al., 2014). Motif analysis of PU.1 binding revealed enrichment for Smad3, Mef2 and Mafb consensus sequences, which cooperate with PU.1 to establish microglia-specific enhancer profiles including TGFβ signaling (Butovsky et al., 2014; Gosselin et al., 2014; Matcovitch-Natan et al., 2016). We found APOE signaling suppresses PU.1, MEF2a, SMAD3 and TGFβ signaling. Deletion of Apoe in phagocytic microglia showed cell-autonomous regulation of the phenotypic switch. miR-155 is a major pro-inflammatory miRNA in ALS and the SOD1 model (Butovsky et al., 2012). Targeting miR-155 restored homeostatic microglia and attenuated disease in SOD1 mice (Butovsky et al., 2015). Here, we show that APOE signaling induces miR-155 in MGnD microglia. Targeting TREM2-APOE signaling in phagocytic microglia and in SOD1 mice suppressed miR-155 expression. Studies have shown that miR-155 directly targets Mef2a (Seok et al., 2011) and PU.1 (Lu et al., 2014). Together these data suggest that TREM2-APOE signaling via miR-155 regulates microglia enhancers, governing the core microglia-specific molecular signature.
Although TREM2 polymorphisms are associated with a risk for late onset AD (Guerreiro et al., 2013), its role in neurodegenerative diseases is controversial. Wang et al. showed that deletion of TREM2 in 5XFAD mice, another model of AD, reduced Aβ accumulation due to a dysfunctional response of microglia to cluster around Aβ plaques (Wang et al., 2015). However, Jay et al., showed that TREM2 deficiency resulted in reduced infiltration of inflammatory myeloid cells and ameliorated AD pathology (Jay et al., 2015) at early stages and exacerbated it at later stages (Jay et al., 2017). Subsequently, Wang et al., showed that amyloid-associated myeloid cells are derived from brain-resident microglia rather than from recruited peripheral monocytes (Wang et al., 2016). We found that genetic targeting of Trem2 in APP-PS1 mice at early stages restored homeostatic microglia associated with the reduction of Aβ plaques. In line with previous reports in mice and humans (Wang et al., 2015; Yuan et al., 2016), we found that microglia did not cluster around Aβ plaques in TREM2-haplodeficient AD subjects. However, TMEM119 homeostatic microglia were more preserved in AD-TREM2 variants compared to common-variant AD-subjects similar to the findings in APP-PS1:Trem2−/− mice. Consistent with our report, increased TREM2 expression is associated with the CD33 AD risk allele (Chan et al., 2015), and recent data showed that soluble TREM2 in the CSF is increased in the early symptomatic phase of AD in association with neuronal injury markers (Suarez-Calvet et al., 2016). This may reflect a dysregulation of homeostatic microglia in response to neuronal injury.
Keren-Shaul et al. reported that APOE induction is TREM2-independent in 5XFAD mice (Keren-Shaul et al., 2017), though Apoe expression in 5XFAD mice was moderately reduced in the previous study (Wang et al., 2016). We found genetic targeting of Trem2 suppresses the APOE pathway and restores homeostatic microglia in APP-PS1 and SOD1 mice. APOE is a ligand for TREM2 in microglia binds to apoptotic neurons and increases Trem2-mediated phagocytosis (Atagi et al., 2015). We found that APOE ligand suppresses major transcription factors of homeostatic microglia. Future studies will determine whether the effect is TREM2-dependent. We identified TREM2 gender-dependent regulation of microglia in SOD1 mice, which was not addressed by Wang et al., and Keren-Shaul et al.
We propose that the consequence of activating the TREM2-APOE pathway is the loss of the ability of MGnD microglia to prevent neuronal loss and provide tolerogenic signals to T cells. In the context of MS, this would amplify the pro-inflammatory properties of T cells. MGnD microglia may also be protective and represent an initial response to neuronal injury. As we showed recently, TREM2 deficiency may lock microglia in a homeostatic state (Mazaheri et al., 2017) and block essential defense functions of microglia during disease progression. In summary, we demonstrate that the TREM2-APOE pathway induces a microglia phenotype switch from a homeostatic to neurodegenerative phenotype. The modulation of the microglial neurodegenerative phenotype by targeting the TREM2-APOE pathway may serve as an approach to restore homeostatic microglia and treat neurodegenerative disorders.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Oleg Butovsky (obutovsky@rics.bwh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J, B6.Cg-Tg(SOD1*G93A)1Gur/J (SOD1G39A), B6.129P2-Apoetm1Unc/J (Apoe−/−), B6.Cg-Mir155tm1.1Rsky/J (miR-155−/−), B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT)Litt/WganJ (Cx3cr1CreERT2) mice were purchased from Jaxmice. Trem2−/− mice (Turnbull et al., 2006) were generously provided by Dr. Marco Colonna (Washington Univ.). APP-PS1 mice were kindly provided by Dr. Mathias Jucker (University of Tübingen). These mice develop amyloid plaque deposition approximately at six weeks of age in the neocortex. Deposits appear in the hippocampus at about three to four months, and in the striatum, thalamus, and brainstem at four to five months (Radde et al., 2006). These mice were analyzed at 9 and 24 months, respectively. APP-PS1:Trem2−/−, were generated by crossing APP-PS1 mice with Trem2−/− mice. These mice were analyzed at 4 months of age. Generation of Apoefl/fl mice have been described recently (Wagner et al., 2015) and these mice were used for microglia specific deletion of Apoe. If not otherwise stated, mice were female and 6–8 weeks of age at the beginning of the experiments. Mice were housed under specific pathogen free conditions. Mice from different genotypes were cohoused. Mice did not undergo any procedures prior to their stated use. All mice were housed with food and water ad libitum. Mice were euthanized by CO2 inhalation. The Institutional Animal Care and Use Committee at Harvard Medical School approved all experimental procedures involving animals.
Human specimens
Cortex samples from AD patients and respective controls from Center for Brain Research, Medical University of Vienna were examined and chosen with identical settings and were investigated to determine the degree of P2ry12 abundance associated with either diffuse or neuritic plaques. Brain tissues from AD patients with and without mutations in TREM2 were sampled and characterized at the University of Washington by David Holtzman and his team (St. Louis). Beside age, gender and disease severity (Braak stage, estimated CDR), the APOE genotype was determined for each patient (see summary in table 1). Tissues were embedded in paraffin and paraffin sections were cut at a thickness of 8μm. After deparaffination, tissue sections were stained as described in the immunohistochemistry section. All stains and subsequent analyses were performed blinded to the investigator. The cases examined in this study are summarized in Table S7. Use of post-mortem human brain tissue was approved by the Knight Alzheimer’s Disease Research Center Biospecimens Committee. The approved use of post-mortem human brain tissue obtained through the Knight ADRC was exempt from the university human studies committee review (exemption numbers 89-055, 89-056). Written informed consent was obtained from participants in studies at the Knight Alzheimer’s Disease Research center (P50 AG05681) and their collateral sources.
METHOD DETAILS
Scoring of SOD1 mice
SOD1G39A mice were assessed clinically by monitoring body weight (3 times/week) and neurological score (daily) all starting at day 60. Disease progression was documented according to established methodology provided by Prize4Life and The Jackson Laboratory. Symptomatic analysis was conducted by monitoring neurological score and weight starting at day 60 and symptomatic onset was defined as the age at which animals began to decline in weight (Boillee et al., 2006). Neurological scores for both hind legs were assessed daily for each mouse beginning at 50 days of age. The neurological score used a scale of 0 to 4 developed by ALSTDI. Criteria used to assign each score level were: 0 = Full extension of hind legs away from the lateral midline when the mouse is suspended by its tail, and mouse can hold this position for 2 seconds, suspended 2–3 times; 1 = Collapse or partial collapse of leg extension towards lateral midline (weakness) or trembling of hind legs during tail suspension; 2 = Curling of the toes and dragging of at least one limb during walking; 3 = Rigid paralysis or minimal joint movement, foot not being used for forward motion; 4 = Mouse cannot right itself within 30 seconds from either side. A score of 4 also corresponded with the humane endpoint of the study.
Conditional genetic deletion of Apoe in microglia
To induce Cre-recombinase expression, a dose of tamoxifen (75 mg/kg of body weight) in corn oil (Sigma) was injected i.p. for 5 consecutive days. For genetic depletion of APOE in microglia cells, Apoefl/fl were crossed with tamoxifen-inducible Cx3cr1CreERT2 transgenic mice. Recombination was induced in Cx3cr1CreERT2:Apoefl/fl adult mice and Cx3cr1wt:Apoefl/fl littermates were used as controls.
Generation of chimeric mice
Cx3cr1:GFP+/− chimera mice were generated as previously described (Butovsky et al., 2014). In brief, 30-day-old C57BL/6J recipient mice protected by lead head shielding were lethally irradiated (950 Rad) and transplanted with bone marrow (BM) from Cx3cr1:GFP+/− donor mice. 8 weeks post-BM transplantation, EAE was induced in these chimera mice as described below.
Immunohistochemistry
Following immersion fixation in 4% PFA, brains were dehydrated and embedded in paraffin. H&E staining was performed according to standard procedures. For immunohistochemical staining, frontal sections (2–5 μm) were collected on superfrost slides, deparaffinized in xylol and rehydrated. After heat induced antigen retrieval, the following primary antibodies were used for detection: Neurons, anti-NeuN (clone A60, Millipore, 2 μg ml−1), microglia/macrophages/monocytes, anti-Iba1 (polyclonal, #019-19741; WAKO Chemicals, 1 μg ml−1), resident microglia, anti-P2ry12 [polyclonal, 0.4 μg ml−1, validated in ref. (Butovsky et al., 2015; Butovsky et al., 2014)], anti-P2ry12 (monoclonal, 0.4 μg ml−1, validated in this study), microglia specific 4D4 (monoclonal, validated in ref. (Butovsky et al., 2012), anti-Apoe (polyclonal, AB947; Millipore, 5 μg ml−1) phosphorylated neurofilament (pNF; (clone SMI31 Biolegend; 1:2,000), and anti-human TMEM119 antibody (Sigma, HPA051870; 1:300). Detection was performed with the respective secondary antibodies and diaminobenzidine.
For immunofluorescence staining, antigen retrieval was performed for 30 min at 96°C in 10 mM citrate buffer pH 6.0. Subsequently, sections were permeabilized with 0.2% TritonX-100 (Roche) in TBS for 5 min. Tissues were blocked in Pierce Protein-Free T20 blocking buffer (Thermo Scientific) and treated with 1% Sudan Black to reduce autofluorescence. Sections were incubated with the first antibody at 4°C overnight, washed with TBS Tween (TBS-T) and incubated with secondary donkey anti-rabbit Alexa555 antibody (Life Technologies, 6 μg ml−1) or chicken anti-rabbit Alexa488, anti-mouse Alexa647 (Life Technologies, 6 μg ml−1) during 90 min. Apoe was stained with goat anti-Apoe (polyclonal, Millipore, 5 μg ml−1) and detected with the superclonal rabbit anti-goat Alexa555 (Thermo Scientific, 1 μg ml−1). Sections were washed again and slides were mounted with DAPI-Fluoromount-G (SouthernBiotech, Birmingham, USA). Staining and analyses of tissues from APP-PS1:Trem2−/−, APP-PS1, WT, Cx3cr1wt:Apoefl/fl, Apoe−/−, Trem2−/−, and Cx3cr1CreERT2:Apoefl/fl mice were performed on cryo or floating sections and the following additional antibodies were used: anti-mouse Clec7a (clone R1-8g7, Invivogene, 1:30); anti-mouse TMEM119 (clone 28-3, Abcam, 1:100); anti-Aβ (clone 6E10, Covance, 1:100 or Mob 410-05, Zytomed, 1:100); goat anti-mouse Alexa405; chicken anti-rat Alexa488 or Alexa647. Anti-Aβ antibody (clone 6E10, Covance) was applied on cryo sections (30 μm) for staining in Figure 2a, only. Sections were permeabilized with 0.2% TritonX-100 as mentioned above with subsequent antibody incubations, respectively. Data acquisition and quantification was performed using a Leica TCS SP5 confocal microscope and Leica application suite software (LAS-AF-lite). Quantification of positive staining within microglia or amyloid β (Aβ)-plaque region was performed using the LAS-AF quantification tool. Briefly, the region of interest was carefully selected and the sum of the values of the pixels in the selection was calculated. For quantification of Apoe-signal in human microglia (paraffin sections 5 μm), only cells with fully visible cell bodies, away from plaques (stained with anti-Aβ antibody Mob 410-05, Zytomed) or astrocytes were used. Selected area was 300 μm2 and 30 cells were measured each (n = 3 AD subjects vs. n = 3 controls). To investigate selected protein expression profiles in APP-PS1 versus APP-PS1:Trem2−/− mouse brains, we used floating sections (30 μm). For quantification of Clec7a signal within single plaque (stained with anti-Aβ antibody Mob 410-05, Zytomed) associated microglia in mouse brains, we measured an area of 250μm2 from 10–14 plaques each in APP-PS1 (n = 5) and APP-PS1:Trem2−/− (n = 5) mice. P2ry12 protein expression in microglia associated with Aβ plaque area was measured in 3,000 μm2 from 15 plaques per each animal in APP-PS1 (n = 5) and APP-PS1:Trem2−/− (n = 5) mice. For quantification of plaque burden, we stained for Aβ (anti-Aβ antibody Mob 410-05, Zytomed) and quantified the Aβ positive signal in the cortex of each mouse within an area of 600,000 μm2. For quantification of Clec7a and P2ry12 signal intensity in the ventral horn of SOD1: Trem2−/− (n = 13) and SOD1:Trem2+/− (n = 9) mice, 5 areas of 3,600 μm2 were measured in 3 sections per spinal cord (cryo-sections 30 μm). For quantitative evaluation of P2RY12 and dystrophic axons in human AD brains (paraffin sections 5 μm), double staining for P2RY12 with Aβ or phosphorylated neurofilament was performed on paraffin sections as described previously. After deparaffinization heat induced epitope retrieval was performed in EDTA buffer (pH: 8.5). The primary antibodies (rabbit polyclonal anti- P2RY12; mouse monoclonal anti-Aβ, Clone W02, Millipore and mouse monoclonal anti-phosporylated neurofilament, SMI31; Sternberger Monoclonals, Affiniti Res Prod.) were applied together overnight. Antibody binding was visualized with either alkaline phosphatase-conjugated anti-mouse antibodies or with biotinylated anti-rabbit secondary antibodies and peroxidase-conjugated streptavidin. Alkaline phosphatase or peroxidase reaction products were visualized by development with fast blue BB salt (blue) or amino ethyl carbazole (AEC; red), respectively.
Serial tissue sections from AD patients with and without mutations in TREM2 were stained by immunofluorescence double labeling with antibodies against IBA1, TMEM119, APOE (see above) and Aβ (Clone W02, Millipore) in the following combinations: IBA1/Aβ and TMEM119/APOE as described above. All quantifications were performed blinded to the researchers. For quantification of positive staining signal, six images through the cortical layers I–IV were taken with a 20x objective. Microglia clusters in Aβ plaques were determined in sections double stained for Aβ and Iba-1 and manually counted within 6 standardized images/case of (0.6 mm2). Images were saved as tiff-format, converted into 8-bit greyscale in ImageJ and inverted. In the resulting images, the area fraction with positive signal was calculated and plotted as percentage of cortical area covered by amyloid deposits, APOE deposits or IBA1-signal in the respective graph. The association of Iba1 positive cells associated with Aβ plaques was quantified manually by counting the number of Aβ plaques with clustered microglia. As positive plaque with microglia clusters we defined the abundance of 2–5 microglia cells surrounded by a small halo of tissue with low or without presence of microglia processes. In classical AD, such clusters or nodules are associated with neuritic or core plaques, but rarely seen in diffuse plaques.
In fluorescence staining where two primary antibodies came from the same species (rabbit), a triple staining was performed using extensive heat-induced epitope retrieval. The paraffin embedded tissue was deparaffinized and slices were steamed for 45 minutes in EDTA pH 9,0. Non-specific protein binding was blocked by incubation with diluent. The first primary Antibody (P2ry12 or Tmem119) was applied over night at 4 °C and on the following day reapplied at room temperature for 1 hour. Consequently, sections were incubated with biotinylated anti-rabbit antibody and by avidin peroxidase and catalyzed signal amplification with biotinylated tyramine. Antigen retrieval was repeated by steaming with EDTA pH 9,0 for 30 minutes, which abolishes antibody reactivity from the previous round but leaves the amplified avidin binding intact, thus preserving the localized binding of Cy2 labeled streptavidin. In a next step two additional primary antibodies were simultaneously applied overnight at 4° C (Iba-1+SMI31 or Aβ; Tmem119+SMI31 or Aβ) and reapplied the next day for one hour at room temperature. Secondary antibodies anti-rabbit Cy5 (Iba-1/Tmem119) and anti-mouse Cy3 (Smi31/ Amyloid β) used for visualizing the immunohistochemical reaction. Covering with slips was done by mounting with Gallate Geltol for 5 minutes.
For Fluoro-Jade C staining, frozen cut tissue sections were first immersed in a basic alcohol solution consisting of 1% sodium hydroxide in 80% ethanol for 5 min, then rinsed for 2 min in 70% ethanol, for 2 min in distilled water and finally, incubated in 0.06% potassium permanganate solution for 2 min. Slides were then transferred for 10 min to a 0.0001% solution of Fluoro-Jade C (Histo-ChemInc; Jefferson, AR) dissolved in 0.1% acetic acid vehicle. Slides were then rinsed three times in distilled water for 1 min, cleared in xylene and coverslipped with DPX (Sigma).
Analysis of P2RY12 and dystrophic axons in AD brains
Independent patient cohort Quantitative evaluation for P2RY12 and dystrophic axons was performed by manual counting. Plaques were identified using the anti-Amyloidβ antibody, Clone W02 (Millipore). Following immunohistochemistry on respective serial sections the previously defined regions of interest (ROI) were manually outlined. In the individual cases three different ROIs were quantified by overlaying a morphometric grid (0.27 mm2) placed within the ocular lens: two fields in areas with the highest density of P2RY12 reactive microglia, two fields in areas of intermediate density and two fields in areas of the lowest density within the respective case and lesion.
Induction of EAE
For the PLP-induced EAE, SJL/J female mice (6–8 weeks old) were immunized subcutaneously (s.c.) with 200 μg of PLP139-151 peptide in a 0.2 ml emulsion comprised of equal volumes of phosphate-buffered saline (PBS) solution. For the MOG-induced EAE, female mice (6–8 weeks) were injected s.c. in both flanks with 100 μg myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide dissolved in PBS. For both models, the solutions were emulsified in an equal volume of CFA (Difco) containing killed Mycobacterium tuberculosis strain H37RA at a final concentration of 5 mg ml−1 Mycobacterium tuberculosis H37Ra and injected twice i.p. with 200 ng pertussis toxin (List Biological Laboratories, Campbell, CA) administered on the day of immunization and 48 h later. Clinical assessment of EAE was performed daily after disease induction according to the following criteria: 0, no signs of disease; 1, loss of tone in the tail; 2, hind limb paresis; 3, hind limb paralysis; 4, tetraplegia; 5, moribund, during the duration of the acute phase (15 d), or only during the progressive or chronic phase (days 30–50). Microglia from these mice were sorted at the scores (i.e. 0, 1, 2, 3) indicated in Figures 1A and 1B and Table S1.
Isolation of primary neurons
Primary neurons were prepared from embryos at age E18.5. Cerebral hemispheres were isolated and freed from meninges. Tissues were digested with 0.25% trypsin in HBSS for 15 min at 37°C, and triturated with fire-polished glass pipettes to obtain single cell preparation. Cell suspension was filtered through a 70 and a 40 μm cell strainer and cells were collected at 1,000 g for 5 min. Cell density was then determined using a hemocytometer and cells were seeded at different densities according to the experimental design and need. We used DMEM supplemented with 10% FBS for the initial plating, and the medium was changed to Neurobasal supplemented with 1× B27 (Invitrogen) 3h later. Half medium was changed every 3 days.
Induction of apoptosis and labeling of neurons
To induce apoptosis, neurons were carefully detached from the plate surface by repeated washes with PBS. Neurons were irradiated with UV light (302 nm) with an intensity of 6 x 15 W for 15 min. From now on, neurons were kept on ice. Cells were harvested by centrifugation and the pellet processed for downstream applications. The labeling dye (1mg) was resuspended in 100 μl special anhydrous DMSO (Thermo Fisher Scientific) and immediately used. Alternatively, dissolved dye was quickly aliquoted at 5 μl and stored at −80°C until use. Neurons were carefully resuspended in 1 ml of PBS and incubated in dark for 15 min at 37°C with 2 μl of the dissolved labeling dye (Alexa488 5-SDP Ester or Alexa405 NHS Ester, Life Technologies/Thermo Fisher Scientific). To block and capture residual dye, cells were diluted with PBS, harvested by centrifugation, resuspended in 1 ml FBS and washed twice with PBS. Total apoptotic cell number was determined using Trypan Blue staining. Neurons were resuspended at a density of approximately 100,000 cells per 4 μl for stereotactic injection.
Induction of necrosis in neurons
To induce necrosis, plated neurons were irradiated with UV light for 15 min and then placed back in the incubator for 24 h. Neurons were labeled with Alexa488 5-SDP Ester (Life Technologies). To distinguish between apoptotic and necrotic cells, dying neurons were incubated with Annexin V (Biolegend) and 7-AAD (Biolegend, 5 μg ml−1) for 5 minutes before flow cytometry analysis.
Stereotactic injections
Mice were anesthetized by i.p injection of a mixture of Ketamine (100 mg kg−1), Xylazine (10 mg kg−1) and Acepromazine (3 mg kg−1). 2 μl of compound (which could be either labeled apoptotic neurons; recombinant mouse APOE (0.5 mg ml−1 My Biosource); bacterial LPS (Sigma); labeled Zymosan (Life Technologies); or labeled E.coli (Life Technologies)) or Saline solution were distributed each intrahippocampal and intracortical bilaterally (Y: ± 1.5mm; X: −2mm; Z: −2mm and −1mm) using stereotaxic equipment (Harvard Apparatus). After recovery from surgery, animals were returned to their home cages. Post-surgery (16 h or 3 days), mice were euthanized by CO2 inhalation and perfused for subsequent experiments.
Intrahippocampal injection of Kainic acid
Mice were injected into the dorsal hippocampus of both hemispheres with Kainic acid dissolved in saline (0.2 μg in 50 nl) using a microsyringe (Hamilton, Reno, Nevada) with a 26-gauge needle and the stereotaxic coordinates from bregma: anteroposterior (AP), − 1.8 mm; mediolateral (ML), 1.6 mm; dorsoventral (DV), − 1.8 mm. Control animals received 0.9% NaCl under the same surgical conditions. After 48 h, mice were euthanized by CO2 inhalation and perfused for subsequent experiments.
Facial nerve axotomy
Unilateral facial nerve transection at the stylomastoid foramen was performed in WT, ApoE−/−, Trem2−/−, and Cx3cr1CreERT2:ApoEfl/fl and Cx3cr1wt:ApoEfl/fl (control) mice (n = 4–5/group). Successful nerve injury indicated by ipsilateral whisker paresis was assessed upon recovery from mild anesthesia by isoflurane. All mice were sacrificed at 7 days post-facial nerve axotomy. Immunohistochemical staining for NeuN (clone A60, Millipore, 2 μg ml−1) and P2ry12 (polyclonal, 0.4 μg ml−1) and analyses were performed on 30 μm floating sections as described in previous section. For quantification of NeuN+ neurons, only cells within the facial motor nucleus with fully visible cell bodies and visible cytoplasm were included.
Mouse microglia isolation and sorting
Microglia isolation was performed according to our previously described protocol (Butovsky et al., 2014). Briefly, mice were transcardially perfused with ice-cold Hanks’ Balanced Salt Solution (HBSS), spinal cords and brains separately dissected. For sample post-stereotaxic injection, tissue around the site of injection was collected corresponding to the following measurements 3x4x4 mm/hemisphere and a mass in average of ~90 mg. From this tissue, ~1000 phagocytic cells and 20’000 non-phagocytic cells were sorted in average from the same site of injection for Nanostring, RNAseq and qPCR analyses. Single cell suspensions were prepared and centrifuged over a 37%/70% discontinuous Percoll gradient (GE Healthcare), mononuclear cells were isolated from the interface. In order to distinguish resident microglia from recruited myeloid cells, we used a monoclonal antibody that recognizes FCRLS, which is expressed on microglia, but not on infiltrating myeloid cells (Butovsky et al., 2014). Isolated cells were stained with anti-FCRLS [clone 4G11, 3 μg ml−1, validated in ref. ], followed by secondary detection with goat anti-rat IgG conjugated to APC [clone Poly4054, Biolegend, 0.7 μg ml−1, validated in reference (Butovsky et al., 2014)] and then CD11b-PeCy7 [clone M1/70, BD Biosciences, 2 μg ml−1, validated in reference (Butovsky et al., 2014)] antibody to specifically sort resident microglia. Phagocytic versus non-phagocytic microglia were further sorted from the FCRLS+CD11b+-population by detection of Alexa488 fluorescence. FCRLS+Clec7a+, FCRLS+Clec7aInt and FCRLS+Clec7a− cells were sorted using mClec7a (clone R1-8g7, Invivogene, 1:10) and FITC goat-anti-rat (BioLegend #405404, 1:100).
Isolation of spleenic CD11b+Ly6C+ monocytes
Spleen was dissected and homogenized using a cell strainer (70 μm). After centrifugation, the cell pellet was resuspended in ACK lysing buffer to remove red blood cells. Splenocytes were incubated with anti-CD11b magnetic beads (1:10, Miltenyi Biotec) for 15 min on ice. CD11b+ cells were collected after magnetic separation. Unspecific antigen binding sites were blocked using Fc Receptor blocking anti-CD16/32 antibodies (BD Biosciences, 5 μg ml−1) and cells were stained using anti-Ly6C-FITC and anti-CD11b-PeCy7 (BD Biosciences, 5 μg ml−1) and sorted accordingly.
Mass spectrometry analysis
Single LC-MS/MS experiment was performed on a LTQ Orbitrap Velos (Thermo Fischer) equipped with Waters (Milford, MA) NanoAcquity HPLC pump. Peptides were separated onto a 100 μm inner diameter microcapillary trapping column packed first with approximately 5 cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by ~20 cm of Reprosil resin (1.8 μm, 200 Å, Dr. Maisch GmbH, Germany). Separation was achieved through applying a gradient from 5–27% ACN in 0.1% formic acid over 180 or 400 min at 100 nl min−1. Electrospray ionization was enabled through applying a voltage of 1.8 kV using a home-made electrode junction at the end of the microcapillary column and sprayed from fused silica pico tips (New Objective, MA). The LTQ Orbitrap Velos was operated in data-dependent mode for the mass spectrometry methods. The mass spectrometry survey scan was performed in the Orbitrap in the range of 395 –1,800 m/z at a resolution of 6 × 104, followed by the selection of the twenty most intense ions (TOP20) for CID-MS2 fragmentation in the Ion trap using a precursor isolation width window of 2 m/z, AGC setting of 1,000, and a maximum ion accumulation of 200 ms. Singly charged ion species were not subjected to CID fragmentation. Normalized collision energy was set to 35 V and an activation time of 10 ms. Ions in a 10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 60 s. The same TOP20 ions were subjected to HCD MS2 event in Orbitrap part of the instrument. The fragment ion isolation width was set to 0.7 m/z, AGC was set to 10,000, the maximum ion time was 200 ms, normalized collision energy was set to 27V and an activation time of 1 ms for each HCD MS2 scan. Raw data were submitted for analysis in Proteome Discoverer 2.2.0.386 (Thermo Scientific) software. Assignment of MS/MS spectra was performed using the Sequest HT algorithm by searching the data against a protein sequence database including all entries from the Mouse Uniprot database (SwissProt 16,768 2016) and other known contaminants such as human keratins and common lab contaminants. Sequest HT searches were performed using a 20 ppm precursor ion tolerance and requiring each peptides N-/C termini to adhere with Trypsin protease specificity, while allowing up to two missed cleavages. 6-plex TMT tags on peptide N termini and lysine residues (+229.162932 Da) was set as static modifications while methionine oxidation (+15.99492 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of 1% on protein level was achieved by applying the target-decoy database search. Filtering was performed using a Percolator (64bit version). For quantification, a 0.02 m/z window centered on the theoretical m/z value of each the six reporter ions and the intensity of the signal closest to the theoretical m/z value was recorded. Reporter ion intensities were exported in result file of Proteome Discoverer 2.2 search engine as an excel tables. The total signal intensity across all peptides quantified was summed for each TMT channel, and all intensity values were normalized to account for potentially uneven TMT labeling and/or sample-handling variance for each labeled channel.
RNA isolation and Nanostring RNA counting
Total RNA was extracted using mirVanaTM miRNA isolation kit (Ambion) according to the manufacturer’s protocol. Nanostring nCounter technology (http://www.nanostring.com/) allows expression analysis of multiple genes from a single sample (Butovsky et al., 2014). We performed nCounter multiplexed target profiling of 400 to 542 microglial transcripts (MG400 and MG550, see MG custom-chip design). 100 ng of total RNA per sample were used in all described nCounter analyses according to the manufacturer’s suggested protocol (Butovsky et al., 2014).
Quantitative real-time PCR
For conventional quantitative reverse transcription polymerase chain reaction (qRT-PCR), total RNA (30 ng) with specific mRNA probes and 3 ng of RNA with specific miRNA probes (Applied Biosystems) were used after reverse transcription reaction according to the manufacturer (high-capacity cDNA Reverse Transcription Kit; Applied Biosystems). All mRNA/miRNA amplifications were performed with commercially available FAM-labeled Taqman probes (Applied Biosystems/Thermo Fisher Scientific). mRNA or miRNAs levels were normalized relative to GAPDH or U6, respectively. Real-time PCR reaction was performed using Vii7 (Applied Biosystems). All qRT-PCRs were performed in duplicate, and the data are presented as relative expression compared to GAPDH or U6 as mean ± s.e.m.
Mouse MG550 chip design
The MG550 Nanostring chip was designed using the quantitative Nanostring nCounter platform. Selection of genes is based on analyses that identified genes and proteins which are specifically or highly expressed in adult mouse microglia (Butovsky et al., 2014) plus 40 inflammation-related genes which were significantly affected in EAE, APP-PS1 and SOD1 mice. Two other versions were done after MG400 (Butovsky et al., 2014). MG468 contains additional 48 inflammation- and phagocytosis-related genes (Butovsky et al., 2015). Using this signature, we generated a new version of Nanostring-based microglia chip termed MG550 that encompasses 400 unique and enriched microglial genes we have identified previously (Butovsky et al., 2014) and additional 150 inflammation-, inflammasome- and phagocytosis-related genes.
Nanostring Data analysis
Nanostring data were normalized and analyzed using nSolver™ software. RNA ncounts were normalized using the geometric mean of 6 housekeeping genes (HKGs): Cltc, Gapdh, Gusb, Hprt, Pgk1 and Tubb5. A cutoff was introduced at the value of the highest negative control present on the chip. Fold changes were calculated using the average of each group. For each experiment, the fold changes were calculated comparing the experimental group to their appropriate controls.
RNA sequencing
1,000 FACS-sorted FCRLS+ microglia cells were lysed in TCL buffer. Smart-Seq2 libraries were prepared by the Broad Technology Labs and sequenced by the Broad Genomics Platform. cDNA libraries were generated from sorted cells using the Smart-seq2 protocol (Picelli et al., 2013). RNA sequencing was performed using Illumina NextSeq500 using a High Output v2 kit to generate 2 x 25 bp reads. RNA-Sequencing data was pooled from 3 separate experiments. Transcripts were quantified by the BTL computational pipeline using Cuffquant version 2.2.1(Trapnell et al., 2012). Raw counts were normalized using TMM normalization and then log2-transformed. Batch effects were corrected using removeBatchEffect from the R package limma (v. 3.28.21).
In vitro suppression assay
Spleens of C57BL/6J mice were gently dissociated into single-cell suspensions, and red blood cells were removed using Ammonium-Chloride-Potassium (ACK) lysis buffer (Gibco). Splenocytes were labeled with 5(6)-Carboxyfluorescein N-hydroxysuccinimidyl ester (CFSE, Molecular probes – Invitrogen, 5 μmol l−1) at 37 °C followed by staining with anti-CD3 mAb (clone 145-2C11, Biolegend, 2 μg ml−1) for 30 min on ice. Responder CFSE+ CD3+ T cells were sorted using FACS Aria II (BD Biosciences) with a purity > 98%. T cells were stimulated with mouse T cell-activator CD3 and CD28 magnetic beads (Dynabeads-Gibco). Stimulated CFSE+ CD3+ T cells were co-cultured in round bottom 96-well plates with FCRLS+ spinal cord microglia for 72 h at 37 °C in a 5% CO2 incubator. T cell-to-microglia ratio was 4:1 in a final volume of 200 μl per well in DMEM media supplemented with 10% Fetal Bovine Serum (FBS), 1% Penicillin-Streptomycin (PS, 10,000 U ml−1), L-Glutamine 2 mmol l−1, sodium pyruvate 1 mmol l−1, non-essential amino acids 0.1 mmol l−1, HEPES 5 mmol l−1 and 2-Mercaptoethanol 0.05 mmol l−1. T cell proliferation was measured by flow cytometric analysis by CFSE dilution on CD3+ T cells. Soluble recombinant ApoE (rApoe) was added into the media at 100 ng ml−1.
Heatmaps
Heatmaps and clustering were generated in R (version 3.2.0) using heatmap.2 from the gplots package and the prcomp from the stats package. For clustering, the z-scores were calculated using the mean expression of biological replicates per disease stage/condition and then subsequently clustered using K-means. A scree plot was used to assess the number of clusters.
Circos Plot
The custom-made microglia transcriptional signatures (Fig. 3I) were generated by retrieving genes upregulated at least 1.5 fold and p-value less than 0.01 (Student T-test) in disease conditions from previously identified microglia transcriptomes from mouse models of neuropathic pain (GSE60670), chronic pain (GSE71133), Rett syndrome (Mecp2−/−) (GSE66211), lipid disorder (Mfp2−/−) (GSE66420), ALS (SOD1) (GSE43366), AD (5xFAD) (GSE65067), brain irradiation (GSE55968), and aging (GSE62420). All custom signatures were derived from publicly available transcriptomes downloaded from Gene Expression Omnibus (GEO). A GSEA algorithm was applied to identify the enrichment of phagocytic microglia signaling pathways in the microglia transcriptomes in various diseases conditions. This includes Aging, Mfp2-deficiency, Alzheimer’s disease, ALS, neuropathic pain, chronic pain, and Mecp2-deficiency. GSEA assesses whether the expression of a previously defined group of related genes is enriched in one biological state. Statistical significance of GSEA results was assessed using 1,000 sample permutations. A nominal P value less than 0.001 was used to determined pairwise transcriptome connectivity. A Circos graph was generated using Circos package 0.68.12.
TREM2 pathway analysis in MGnD microglia. A GSEA algorithm was used to identify genes that are differentially expressed in microglia transcriptomes from lipid disorder (Mfp2−/−) (GSE66420), ALS (SOD1) (GSE43366), AD (5XFAD) (GSE465067), and aging (GSE62420), as compared with their control counterparts. High-scoring differentially expressed ‘leading-edge’ genes were selected on the basis of their presence in the TREM2-responsive gene signature 5xFAD:Trem2−/− and SOD1:Trem2−/− mice. The statistics were determined using Euclidean distance.
Ingenuity Pathway Analysis (IPA)
Data were analyzed using Ingenuity software (Ingenuity Systems, http://www.ingenuity.com). Differentially expressed genes (with corresponding fold changes and P values) were incorporated in canonical pathways and bio-functions and were used to generate biological networks as described previously (Butovsky et al., 2014). Briefly, uploaded data set for analysis was filtered using the following cutoff definitions: fivefold change, P < 0.01.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was done using GraphPad Prism 7 software. Data distribution was assumed to be normal, but this was not formally tested. Data are presented as mean ± s.e.m and 2-tailed Student’s t tests (unpaired) or ANOVA multiple comparison tests were used to assess statistical significance. Data collection and analysis were blindly performed. Data for each experiment were collected and processed randomly and animals were assigned to various experimental groups randomly as well. All n and P values and statistical tests are indicated in figure legends. Volcano plots were generated using Multiplot studio software. Heatmaps were generated using MEV and R-software.
DATA AND SOFTWARE AVAILABILITY
The RNAseq and Nanostring gene sequence data have been deposited in Gene Expression Omnibus (GEO) database (accession number GSE101689) and protein data has been deposited in PeptideAtlas (accession number PASS01081).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-NeuN (clone A60) | Millipore | Cat#MAB377 |
|
| ||
| Rabbit polyclonal anti-Iba1 | WAKO Chemicals | Cat#019-19741 |
|
| ||
| Rabbit polyclonal anti-P2RY12 | Butovsky et al. 2014 | N/A |
|
| ||
| Rat-monoclonal microglia-specific 4D4 | Butovsky et al. 2012 | N/A |
|
| ||
| Rat monoclonal anti-P2RY12 | This paper | N/A |
|
| ||
| Goat polyclonal anti-APOE | Millipore | Cat# AB947 |
|
| ||
| Mouse monoclonal anti-phosphorylated neurofilament (clone SMI31) | BioLegend | Cat#801601 |
|
| ||
| Rabbit polyclonal anti-human TMEM119 | Sigma | Cat# HPA051870 |
|
| ||
| Rabbit monoclonal [28-3] anti-mouse TMEM119 | Abcam | Cat#ab209064 |
|
| ||
| Rat monoclonal anti-Clec7a (clone R1-8g7) | Invivogene | Cat#mabg-mdect |
|
| ||
| Ly6C-FITC | BD Biosciences | Cat#553104 |
|
| ||
| Rat-monoclonal anti-FCRLS (clone 4G11) | Butovsky et al. 2014 | N/A |
|
| ||
| Mouse monoclonal anti-Amyloidβ (clone 6E10) | Covance | Cat#SIG-39320 |
|
| ||
| Mouse monoclonal anti-Amyloidβ (clone BAM-10) | Zytomed | Cat#Mob 410-05 |
|
| ||
| Mouse monoclonal anti-Amyloidβ (Clone W02) | Millipore | Cat#MABN10 |
|
| ||
| Rat monoclonal anti-CD11b-PeCy7 (clone M1/70) | BD Biosciences | Cat#552850 |
|
| ||
| Armenian Hamster monoclonal anti-CD3 (clone 145-2C11) | BioLegend | Cat#100302 |
|
| ||
| Super Clonal anti-goat Alexa Fluor555 | Thermo Fisher Scientific | Cat#A27017 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
|
| ||
|
| ||
| Biological Samples | ||
|
| ||
| Human cortical specimens | Massachusetts General Hospital | Massachusetts Alzheimer Disease Research Center (MADRC; NIA P50 AG005134) |
|
| ||
| Human Brain Autopsies (AD and Controls) | Center for Brain Research, Medical University of Vienna | Archival Collection; EK #: 535/2004/2017 |
|
| ||
| Human Brain specimens (AD controls and AD Trem2-polymorphisms) | Washington University | Knight Alzheimer’s Disease Research Center ( NIA P50 AG0568) |
|
| ||
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Annexin V | Biolegend | Cat#640901 |
|
| ||
| 7-AAD | Biolegend | Cat#420403 |
|
| ||
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
|
| ||
| TritonX-100 | Roche | Cat#11332481001 |
|
| ||
| Pierce Protein-Free T20 blocking buffer | Thermo Fisher Scientific | Cat#37571 |
|
| ||
| DAPI-Fluoromount-G | Southern Biotech | Cat#0100-20 |
|
| ||
| Fluoromount-G | Southern Biotech | Cat#0100-01 |
|
| ||
| Antibody diluent | DAKO | Cat#S2022 |
|
| ||
| Avidin peroxidase | Jackson | Cat#016-030-084 |
|
| ||
| Tyramine | Sigma | Cat#T-7255 |
|
| ||
| Gallate Geltol: | ||
| Glycerol | Invitrogen | Cat#15514-011 |
| Moviol | Calbiochem | Cat#475904 |
| Gallate | Sigma | Cat#P-3130 |
|
| ||
| Pertussis toxin | List Biological Laboratories | Cat#180 |
|
| ||
| CFA Complete Freund Adjuvant | Difco | Cat#231131 |
|
| ||
| myelin oligodendrocyte glycoprotein (MOG)35–55 peptide | Genemed Synthesis | Cat# MOG3555-P-5 |
|
| ||
| 5(6)-Carboxyfluorescein N-hydroxysuccinimidyl ester | Thermo Fisher Scientific | Cat#C34554 |
|
| ||
| LPS Lipopolysaccharide from Escherichia coli 0127:B8 | Thermo Fisher Scientific | Cat#L3129 |
|
| ||
| Mouse recombinant APOE (host: yeast) | My Biosource | Cat#MBS955382 |
|
| ||
| pHrodo® Green Zymosan Bioparticles® Conjugate for Phagocytosis | Life Technologies | Cat#P35365 |
|
| ||
| pHrodo® Green E. coli BioParticles® Conjugate for Phagocytosis | Life Technologies | Cat#P35366 |
|
| ||
| Kainic Acid | Sigma-Aldrich | K0250-10MG |
|
| ||
| Fluoro-Jade C | Histo-chem Inc. | #1FJC |
|
| ||
| Alexa Fluor488 5-SDP Ester | Thermo Fisher Scientific (Molecular Probes) | Cat#A30052 |
|
| ||
| Alexa Flour405 5-NHS Ester | Thermo Fisher Scientific (Molecular Probes) | Cat#A30000 |
|
| ||
| Anhydrous DMSO | Thermo Fisher Scientific | Cat#D12345 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| mirVanaTM miRNA isolation kit | Ambion | Cat#AM1561 |
|
| ||
| high-capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
|
| ||
| anti-CD11b magnetic beads | Miltenyi Biotec | Cat#130-049-601 |
|
| ||
| T cell-activator CD3/CD28 magnetic beads | Thermo Fisher Scientific | Cat#11456D |
|
| ||
| Fc Receptor blocking anti-CD16/32 antibodies | BD Biosciences | Cat#553141 |
|
| ||
| Deposited Data | ||
|
| ||
| Raw and analyzed data | This paper | GEO: GSE101689 |
|
| ||
| PeptideAtlas: PASS01081 | ||
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| NA | ||
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: C57Bl/6J | The Jackson Laboratory | JAX: 00664 |
|
| ||
| Mouse: Apoe–/– | The Jackson Laboratory | JAX: 002052 |
|
| ||
| Mouse: miR-155–/– | The Jackson Laboratory | JAX: 007745 |
|
| ||
| Mouse: Apoefl/fl | Wagner et al. 2015 | N/A |
|
| ||
| Mouse: APP-PS1 | Radde et al., 2006 | N/A |
|
| ||
| Mouse: Trem2–/– | Turnbull et al., 2006 | N/A |
|
| ||
| Mouse: Cx3cr1CreERT2 | The Jackson Laboratory | JAX: 021160 |
|
| ||
| Mouse: SOD1G93A | The Jackson Laboratory | JAX: 004435 |
|
| ||
|
| ||
| Oligonucleotides | ||
|
| ||
| Apoe (Mm01307193_g1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Clec7a (Mm01307193_g1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Csf1r (Mm01266652_m1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Egr1 (Mm00656724_m1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Gapdh (Mm99999915_g1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Gpr34 (Mm02620221_s1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| P2ry12 (Mm01950543_s1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Tgfb1 (Mm01178820_m1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Tgfbr1 (Mm00436964_m1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| Tmem119 (Mm00525305_m1) | Thermo Fisher Scientific | Cat#4331182 |
|
| ||
| miR-155 (RT002571) | Thermo Fisher Scientific | Cat#4427975 |
|
| ||
| U6 (RT001973) | Thermo Fisher Scientific | Cat#4427975 |
|
| ||
| Recombinant DNA | ||
|
| ||
| NA | ||
|
| ||
| Software and Algorithms | ||
|
| ||
| Leica application suite software (LAS-AF-lite) | Leica | http://www.leica-microsystems.com |
|
| ||
| Ingenuity software | Ingenuity Systems | http://www.ingenuity.com |
|
| ||
| Multiplot studio 1.5.20 | GParc | http://gparc.org/ |
|
| ||
| GraphPad Prism 7 | GraphPad Software | http://www.graphpad.com |
|
| ||
| FlowJo (version 10.0.8R1) | FlowJo | http://www.flowjo.com |
|
| ||
| Proteome Discoverer 2.1.0.81 software | Thermo Fisher Scientific | https://portal.thermo-brims.com/ |
|
| ||
| Sequest HT algorithm | Thermo Fisher Scientific | https://portal.thermo-brims.com/ |
|
| ||
| GSEA algorithm | http://software.broadinstitute.org/gsea/index.jsp | |
|
| ||
| oPOSSUM | oPOSSUM | http://opossum.cisreg.ca/oPOSSUM3/ |
|
| ||
| Circos package 0.68.12 | http://circos.ca/software/download/circos/ | |
|
| ||
| R software (version 3.4.0) | https://cran.r-project.org/ | |
|
| ||
| MeV software (version 10.2) | http://mev.tm4.org | |
|
| ||
| Other | ||
|
| ||
| Nanostring nCounter MG400 chip | Butovsky et al., 2014 | http://www.nanostring.com/ |
|
| ||
| Nanostring nCounter MG468 chip | Butovsky et al., 2015 | http://www.nanostring.com/ |
|
| ||
| Nanostring nCounter MG550 chip | This paper | http://www.nanostring.com/ |
|
| ||
| LTQ Orbitrap Velos | Thermo Fisher Scientific | http://planetorbitrap.com/ |
|
| ||
| C18 Reprosil resin (5 μm, 100 Å) | Dr. Maisch GmbH | http://www.dr-maisch.com/xedin.php |
|
| ||
| C18 Reprosil resin (1.8 μm, 200 Å) | Dr. Maisch GmbH | http://www.dr-maisch.com/xedin.php |
|
| ||
| Deposited Data | ||
|
| ||
| Gene expression data | GSE101689 | |
|
| ||
| Protein data | PeptideAtlas: PASS01081 | |
Supplementary Material
Nanostring MG550 chip design. Nanostring MG550 Isoform Coverage. Nanostring profile of 4D4+ microglia sorted from spinal cords of naive and EAE mice with different disease score during onset-peak stages. Nanostring profile of FCRLS+ microglia sorted from spinal cords of WT vs. EAE mice at different disease stages. Nanostring profile of FCRLS+ microglia sorted from spinal cords of WT vs. SOD1 mice at different disease scores. Nanostring chip profile of FCRLS+ microglia sorted from brains of aged WT and APP-PS1 mice. Z-score analysis of microglia transcriptomes during aging and during disease progression. Commonly affected genes in disease. Top-20 upstream regulators predicted to be affected in disease (IPA). TGFβ affected genes during disease progression.
Nanostring profile of FCRLS+ Clec7a+ microglia sorted from brains of WT C57Bl/6J and APP-PS1 mice at 24 months. SMART Seq2 profile of FCRLS+ Clec7a+ microglia sorted from brains of WT C57Bl/6J and APP-PS1 mice at 9 months.
Nanostring profile of FCRLS+ brain sorted microglia of WT mice after apoptotic AlexaFluor488+ neurons injections (Phagocytic vs. non-phagocytic microglia). Tandem TagTM mass spectrometry analysis of FCRLS+ isolated phagocytic vs. non-phagocytic microglia in WT mice after apoptotic AlexaFluor488+ neurons injections. GSEA analysis for Circos plots generation. Custom Signatures of microglia transcriptomes from mouse models of neuropathic pain, chronic pain, Rett syndrome (Mecp2−/− mice), lipid disorder (MFP2−/− mice), ALS (SOD1 mice), AD (5xFAD mice), brain irradiation and aging vs. phagocytic microglia.
Nanostring profile of FCRLS+ microglia sorted from brains of WT C57Bl/6J mice injected with PBS (n = 5 pooled mice per lane) and WT C57Bl/6J mice injected with LPS 100ng per injection site (n = 6 pooled mice per lane).
SMART Seq2 profile of WT vs. APOE-KO non-phagocytic (NP) and phagocytic (P) microglia. Z-score analysis of microglia transcriptomes of WT vs. APOE-KO non-phagocytic (NP) and phagocytic (P) microglia. Nanostring profile of FCRLS+ brain sorted microglia of WT and Apoe−/− mice after apoptotic AlexaFluor488+ neurons injections. Nanostring profile of FCRLS+ brain sorted microglia of WT and miR-155−/− mice after apoptotic AlexaFluor488+ neurons injections. Nanostring profile of FCRLS+ brain sorted microglia of Apoe−/− mice injected with PBS vs. rAPOE (500 ng).
Nanostring profile of FCRLS+ brain sorted microglia of APP-PS1 mice and APP-PS1/Trem2−/− mice. Nanostring profile of FCRLS+ brain sorted microglia of SOD1:Trem2+/− mice and SOD1:Trem2−/− mice at 115 days. SMART Seq2 profile of FCRLS+ CD11b+ in SOD1:Trem2−/− vs. SOD1:Trem2+/− microglia. TREM2 Pathway Enrichment in ALS. TREM2 Pathway Enrichment in Alzheimer’s Disease. TREM2 Pathway Enrichment in Aging. TREM2 Pathway Enrichment in Mfp2−/−.
Highlights.
Microglia associated with neuritic Aβ-plaques exhibit a neurodegenerative phenotype
Phagocytosis of apoptotic neurons suppresses homeostatic microglia
The TREM2-APOE pathway regulates neurodegenerative microglial phenotypic switch
Targeting APOE signaling restores homeostatic and tolerogenic microglia
Acknowledgments
OB: NIH-NINDS (1R01NS088137), NIH-NIA (R01AG051812), National Multiple Sclerosis Society (5092A1), Amyotrophic Lateral Sclerosis Association (2087), Nancy Davis Foundation Faculty Award; HLW: DOD (AL120029) Thome Foundation, NIH-NIA (R01AG043975) NIH-NIA (R01AG040092); CH: ERC (321366-Amyloid), DFG (EXC 1010 SyNergy), Cure Alzheimer’s Fund and MetLife Foundation Award; JH: DFG (SFB841); DMH: P50AG05681, P01-AG0399. CL: NIH-NIA (R01AG040092); TI: NIH-NIA (RF1AG054199-01); MG: DFG (GRK1459, SFB877); HL: Austrian Science Foundation Project (P27744-B27); We thank Prize4Life, a nonprofit organization dedicated to the discovery of treatments and a cure for ALS; Marco Colonna for Trem2−/− mice; microscopy Core facility/UKE; Deneen Kozoriz for FACS sorting.
Footnotes
Competing financial interests: CH: Roche advisor and collaborator. All other authors declare no conflict of interests.
Supplemental information includes seven figures and seven tables.
AUTHOR CONTRIBUTIONS
OB, SK, CM: conceived study. SK, CM: performed experiments, analyzed data, experimental design, writing. RC: Nanostring analysis, LPS-injection. CB: isolated AD microglia for RNASeq, histology, imaging. NC: SOD1:Trem2−/− mice. LB: nerve axotomy, imaging, analysis. EO, ZF, DJG, GT, KH, ZH, STS: performed experiments. RElF: neuronal culture. PCS. MG, JO: T cell assays. DH, JU: human AD-TREM2 variants. HL, TZ: histology. CL: APP-PS1 experiments. AW, JH: Apoefloxed mice. FH, FM, CH: APP-PS1:Trem2−/− mice. TI: analysis. BB: mass spec. VL, YX: network analysis. ZW, HC: circos plot. ET, AM, CM: RNAseq analyses, k-means clustering. HLW: design, editing. OB: design, analysis, wrote manuscript with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H, et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) J Biol Chem. 2015;290:26043–26050. doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Song W, Ji Y, Liu X, Tian L, Wang C, Chen D, Zhang X, Zhang M. Microglia and microglia-like cell differentiated from DC inhibit CD4 T cell proliferation. PloS one. 2009;4:e7869. doi: 10.1371/journal.pone.0007869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Annals of neurology. 2015;77:75–99. doi: 10.1002/ana.24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nature neuroscience. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, Doykan CE, Wu PM, Gali RR, Iyer LK, et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122:3063–3087. doi: 10.1172/JCI62636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M. Sall1 is a transcriptional regulator defining microglia identity and function. Nature immunology. 2016;17:1397–1406. doi: 10.1038/ni.3585. [DOI] [PubMed] [Google Scholar]
- Chan G, White CC, Winn PA, Cimpean M, Replogle JM, Glick LR, Cuerdon NE, Ryan KJ, Johnson KA, Schneider JA, et al. CD33 modulates TREM2: convergence of Alzheimer loci. Nature neuroscience. 2015;18:1556–1558. doi: 10.1038/nn.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell reports. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. The American journal of pathology. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grutzendler J, Helmin K, Tsai J, Gan WB. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Annals of the New York Academy of Sciences. 2007;1097:30–39. doi: 10.1196/annals.1379.003. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. TREM2 variants in Alzheimer’s disease. The New England journal of medicine. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nature neuroscience. 2013;16:1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W, et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta neuropathologica communications. 2015;3:31. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YA, Zhou B, Wernig M, Sudhof TC. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell. 2017;168:427–441. e421. doi: 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2017;37:637–647. doi: 10.1523/JNEUROSCI.2110-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. The Journal of experimental medicine. 2015;212:287–295. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169:1276–1290. e1217. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, Holtzman DM. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. The Journal of experimental medicine. 2012;209:2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559– 562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Larner AJ. The cortical neuritic dystrophy of Alzheimer’s disease: nature, significance, and possible pathogenesis. Dementia. 1995;6:218–224. doi: 10.1159/000106950. [DOI] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M, Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev. 2013;37:2887–2899. doi: 10.1016/j.neubiorev.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, Boiani S, Leyland R, Galloway A, Andrews S, et al. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. The Journal of experimental medicine. 2014;211:2183–2198. doi: 10.1084/jem.20140338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, Tanaka S, Hansen LA. Caspase dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer disease. Journal of neuropathology and experimental neurology. 1998;57:1041–1052. doi: 10.1097/00005072-199811000-00007. [DOI] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada Gonzalez F, Perrin P, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nature reviews Molecular cell biology. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Experimental neurology. 1998;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trumbach D, Wurst W, et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017 doi: 10.15252/embr.201743922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, et al. Activation of necroptosis in multiple sclerosis. Cell reports. 2015;10:1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I, Klooster J, Bossers K, Hol EM. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging. 2014;35:1–14. doi: 10.1016/j.neurobiolaging.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nature reviews Neurology. 2014;10:217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–946. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, Saito Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology. 2016;36:39–49. doi: 10.1111/neup.12235. [DOI] [PubMed] [Google Scholar]
- Seok HY, Tatsuguchi M, Callis TE, He A, Pu WT, Wang DZ. miR-155 inhibits expression of the MEF2A protein to repress skeletal muscle differentiation. J Biol Chem. 2011;286:35339– 35346. doi: 10.1074/jbc.M111.273276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Walz KA, Archambault AS, Sim J, Bollman BP, Koenigsknecht-Talboo J, Cross AH, Holtzman DM, Wu GF. Apolipoprotein E mediation of neuro-inflammation in a murine model of multiple sclerosis. Journal of neuroimmunology. 2014;271:8–17. doi: 10.1016/j.jneuroim.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. The American journal of pathology. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberger NH, Sternberger LA, Ulrich J. Aberrant neurofilament phosphorylation in Alzheimer disease. Proc Natl Acad Sci U S A. 1985;82:4274–4276. doi: 10.1073/pnas.82.12.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta neuropathologica. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C Dominantly Inherited Alzheimer N. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016;8:369ra178. doi: 10.1126/scitranslmed.aag1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nature neuroscience. 2017;20:793–803. doi: 10.1038/nn.4547. [DOI] [PubMed] [Google Scholar]
- Toledo JB, Da X, Weiner MW, Wolk DA, Xie SX, Arnold SE, Davatzikos C, Shaw LM, Trojanowski JQ Alzheimer’s Disease Neuroimaging I. CSF Apo-E levels associate with cognitive decline and MRI changes. Acta neuropathologica. 2014;127:621–632. doi: 10.1007/s00401-013-1236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177:3520–3524. doi: 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- Verheijden S, Bottelbergs A, Krysko O, Krysko DV, Beckers L, De Munter S, Van Veldhoven PP, Wyns S, Kulik W, Nave KA, et al. Peroxisomal multifunctional protein-2 deficiency causes neuroinflammation and degeneration of Purkinje cells independent of very long chain fatty acid accumulation. Neurobiol Dis. 2013;58:258–269. doi: 10.1016/j.nbd.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Wagner T, Bartelt A, Schlein C, Heeren J. Genetic Dissection of Tissue-Specific Apolipoprotein E Function for Hypercholesterolemia and Diet-Induced Obesity. PloS one. 2015;10:e0145102. doi: 10.1371/journal.pone.0145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. The Journal of experimental medicine. 2016;213:667–675. doi: 10.1084/jem.20151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016;92:252–264. doi: 10.1016/j.neuron.2016.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nanostring MG550 chip design. Nanostring MG550 Isoform Coverage. Nanostring profile of 4D4+ microglia sorted from spinal cords of naive and EAE mice with different disease score during onset-peak stages. Nanostring profile of FCRLS+ microglia sorted from spinal cords of WT vs. EAE mice at different disease stages. Nanostring profile of FCRLS+ microglia sorted from spinal cords of WT vs. SOD1 mice at different disease scores. Nanostring chip profile of FCRLS+ microglia sorted from brains of aged WT and APP-PS1 mice. Z-score analysis of microglia transcriptomes during aging and during disease progression. Commonly affected genes in disease. Top-20 upstream regulators predicted to be affected in disease (IPA). TGFβ affected genes during disease progression.
Nanostring profile of FCRLS+ Clec7a+ microglia sorted from brains of WT C57Bl/6J and APP-PS1 mice at 24 months. SMART Seq2 profile of FCRLS+ Clec7a+ microglia sorted from brains of WT C57Bl/6J and APP-PS1 mice at 9 months.
Nanostring profile of FCRLS+ brain sorted microglia of WT mice after apoptotic AlexaFluor488+ neurons injections (Phagocytic vs. non-phagocytic microglia). Tandem TagTM mass spectrometry analysis of FCRLS+ isolated phagocytic vs. non-phagocytic microglia in WT mice after apoptotic AlexaFluor488+ neurons injections. GSEA analysis for Circos plots generation. Custom Signatures of microglia transcriptomes from mouse models of neuropathic pain, chronic pain, Rett syndrome (Mecp2−/− mice), lipid disorder (MFP2−/− mice), ALS (SOD1 mice), AD (5xFAD mice), brain irradiation and aging vs. phagocytic microglia.
Nanostring profile of FCRLS+ microglia sorted from brains of WT C57Bl/6J mice injected with PBS (n = 5 pooled mice per lane) and WT C57Bl/6J mice injected with LPS 100ng per injection site (n = 6 pooled mice per lane).
SMART Seq2 profile of WT vs. APOE-KO non-phagocytic (NP) and phagocytic (P) microglia. Z-score analysis of microglia transcriptomes of WT vs. APOE-KO non-phagocytic (NP) and phagocytic (P) microglia. Nanostring profile of FCRLS+ brain sorted microglia of WT and Apoe−/− mice after apoptotic AlexaFluor488+ neurons injections. Nanostring profile of FCRLS+ brain sorted microglia of WT and miR-155−/− mice after apoptotic AlexaFluor488+ neurons injections. Nanostring profile of FCRLS+ brain sorted microglia of Apoe−/− mice injected with PBS vs. rAPOE (500 ng).
Nanostring profile of FCRLS+ brain sorted microglia of APP-PS1 mice and APP-PS1/Trem2−/− mice. Nanostring profile of FCRLS+ brain sorted microglia of SOD1:Trem2+/− mice and SOD1:Trem2−/− mice at 115 days. SMART Seq2 profile of FCRLS+ CD11b+ in SOD1:Trem2−/− vs. SOD1:Trem2+/− microglia. TREM2 Pathway Enrichment in ALS. TREM2 Pathway Enrichment in Alzheimer’s Disease. TREM2 Pathway Enrichment in Aging. TREM2 Pathway Enrichment in Mfp2−/−.