

Abstract
Synaptic transmission controls brain activity and behaviors, including food intake. Leptin, an adipocyte-derived hormone, acts on neurons located in the lateral hypothalamic area (LHA) to maintain energy homeostasis and regulate food intake behavior. The specific synaptic mechanisms, cell types, and neural projections mediating this effect remain unclear. In male mice, using pathway-specific retrograde tracing, whole-cell patch-clamp recordings and post hoc cell type identification, we found that leptin reduces excitatory synaptic strength onto both melanin-concentrating hormone- and orexin-expressing neurons projecting from the LHA to the ventral tegmental area (VTA), which may affect dopamine signaling and motivation for feeding. A presynaptic mechanism mediated by distinct intracellular signaling mechanisms may account for this regulation by leptin. The regulatory effects of leptin depend on intact leptin receptor signaling. Interestingly, the synaptic regulatory function of leptin in the LHA-to-VTA neuronal pathway is highly sensitive to energy states: both energy deficiency (acute fasting) and excessive energy storage (high-fat diet-induced obesity) blunt the effect of leptin. These data revealed that leptin may regulate synaptic transmission in the LHA-to-VTA neurocircuitry in an inverted “U-shape” fashion dependent on plasma glucose levels and related to metabolic states.
SIGNIFICANCE STATEMENT The lateral hypothalamic area (LHA) to ventral tegmental area (VTA) projection is an important neural pathway involved in balancing whole-body energy states and reward. We found that the excitatory synaptic inputs to both orexin- and melanin-concentrating hormone expressing LHA neurons projecting to the VTA were suppressed by leptin, a peptide hormone derived from adipocytes that signals peripheral energy status to the brain. Interestingly, energy states seem to affect how leptin regulates synaptic transmission since both the depletion of energy induced by acute food deprivation and excessive storage of energy by high-fat diet feeding dampen the suppressive effect of leptin on synaptic transmission. Together, these data show that leptin regulates synaptic transmission and might be important for maintaining energy homeostasis.
Keywords: fasting, lateral hypothalamus, leptin, obesity, pathway specific, synaptic transmission
Introduction
Synaptic transmission controls brain activity and behaviors, including food intake. Leptin, an adipocyte-derived catabolic hormone (Zhang et al., 1994), plays an instrumental role in maintaining energy homeostasis. Mutations in the leptin gene or that of its receptor cause massive hyperphagia and morbid obesity (Bray and York, 1997; Friedman and Halaas, 1998). However, the specific mechanisms by which leptin regulates brain function remain elusive (Flier and Maratos-Flier, 2017). Recent work has demonstrated that leptin may achieve its function by regulating synaptic transmission in the brain (Thompson and Borgland, 2013; Lee et al., 2015), including the lateral hypothalamic area (LHA; Goforth et al., 2014). However, the precise cell types, neural projections, and underlying signaling mechanisms mediating this effect remain poorly understood. Furthermore, both circulating leptin levels and leptin sensitivity of neurons vary significantly according to whole-body energy status (Ahima et al., 1996; Bjørbaek et al., 1998; Enriori et al., 2006; Hebebrand et al., 2007), and it is unclear how metabolic states may influence the regulatory effect of leptin on synaptic transmission.
The LHA has been recognized as a “feeding center” and is positioned to integrate energy status and metabolic signals to control food intake (Hoebel and Teitelbaum, 1962; Grossman et al., 1978; Jennings et al., 2013; Sheng et al., 2014). It is well established that LHA neurons are major targets for central leptin signaling (Leinninger et al., 2009, 2011; Domingos et al., 2013; Goforth et al., 2014; Laque et al., 2015), and it has been suggested that they may play a role in regulating the reward-associated ventral tegmental area (VTA) dopaminergic (DA) signaling and reward (Harris et al., 2005; Borgland et al., 2006; Narita et al., 2006; Leinninger et al., 2009; Domingos et al., 2013; Sheng et al., 2014). However, the complex wiring pattern and neuronal heterogeneity of the LHA (Liu et al., 2015; Mickelsen et al., 2017) has complicated the discovery of precise neural mechanisms mediating leptin function. Two major subtypes of neuropeptide-containing neurons in the LHA are orexin/hypocretin and melanin-concentrating hormone (MCH) neurons, and both are proposed to be capable not only of mediating energy sensing, but also of affecting food intake, body weight, and reinforcement of reward behaviors (Shimada et al., 1998; Hara et al., 2001; Harris et al., 2005; Narita et al., 2006). Previous studies mainly focused on the GABAergic system, showing that systematic activation of LHA GABAergic neurons induces acute eating-related behaviors (Leinninger et al., 2009; Barbano et al., 2016; Nieh et al., 2016). Leptin receptor (LepR)-expressing GABAergic neurons have been shown to play a major role in mediating impact of leptin on food intake/body weight (Leinninger et al., 2009; Vong et al., 2011). However, disruption of either GABA or glutamate release from LepR-expressing neurons causes obesity in mice (Xu et al., 2012, 2013), suggesting that both excitatory and inhibitory synaptic transmission are involved in mediating leptin function in the brain. Synaptic modulation by leptin in defined neuronal circuitries, particularly in the LHA-to-VTA projection pathway, has not been previously studied. Interestingly, both orexin and MCH receptors are expressed in the VTA (Hervieu et al., 2000; Marcus et al., 2001; Saito et al., 2001; Korotkova et al., 2003). Therefore, it is conceivable that the LHA-to-VTA-projecting orexin and MCH neurons could influence VTA DA neuronal function and hence mediate the function of leptin in motivated and reward-associated food intake.
We found in this study that leptin profoundly suppresses excitatory synaptic strengths onto the LHA-to-VTA-projecting orexin and MCH neurons via distinct signaling mechanisms. Remarkably, both depletion of energy induced by acute food deprivation and excessive storage of energy by diet-induced obesity (DIO) dampened the regulatory effects of leptin, suggesting that this mechanism is sensitive to whole-body energy status. Collectively, our data implicate a specific synaptic signaling mechanism and neurocircuitry by which leptin may serve to maintain energy homeostasis.
Materials and Methods
Animals
Five- to 12-week-old male wild-type mice (C57BL/6 background) and 4- to 7-week-old B6 db/db mice (stock #000697, The Jackson Laboratory; RRID:IMSR_JAX:000697; Coleman, 1978) were used. Mice were group housed except when otherwise stated and maintained on a 12 h light/dark cycle (lights on at 6:00 A.M.) with food and water available ad libitum. Studies were approved by the Rutgers University Animal Care and Use Committee and followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Retrograde labeling
Mice (5–6 weeks old) were anesthetized using isoflurane and placed in a stereotactic frame (catalog #M1900, KOPF). Standard serological and injection procedures were followed as described previously (Steininger et al., 2004). Red or green RetroBeads (100 nl; LumaFluor) were injected unilaterally into the VTA using the following coordinates: anteroposterior, −3.0 mm; mediolateral, ±0.35 mm; and dorsoventral, −4.3 mm. Animals were allowed to recover in their home cages for at least 7 d to allow adequate retrograde transportation of the beads to the soma of LHA neurons. Injection sites were verified in all animals.
Brain slice electrophysiology
Mice were anesthetized and decapitated, and brains were removed and quickly immersed in cold (4°C) oxygenated cutting solution containing the following (in mm): 50 sucrose, 2.5 KCl, 0.625 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 2.5 glucose, pH 7.3 with NaOH. Coronal hypothalamic or VTA slices, 300 μm in thickness, were cut using a vibratome (catalog #VT 1200S, Leica). Brain slices were collected in artificial CSF (ACSF) and bubbled with 5% CO2 and 95% O2. The ACSF contained the following (in mm): 125 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 2.5 glucose, pH 7.3 with NaOH. After at least 1 h of recovery, slices were transferred to a recording chamber and constantly perfused with bath solution (30°C) at a flow rate of 2 ml/min. For recordings made from fasted animals, the glucose concentrations of the cutting and ACSF solutions were adjusted to 0.7 mm. Whole-cell patch-clamp recordings were performed as described previously with modifications (Domingos et al., 2013). Briefly, patch pipettes with a resistance of 4∼6 MΩ were made from borosilicate glass (World Precision Instruments) with a puller (catalog #PC-10, Narishige) and filled with a pipette solution containing the following (in mm): 126 K-gluconate, 4 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Na2-GTP, and 10 phosphocreatine, pH adjusted to 7.2 with KOH for current clamp; and 90 K-gluconate, 40 CsCl, 1.8 NaCl, 1.7 MgCl2, 3.5 KCl, 0.05 EGTA, 10 HEPES, 2 Mg-ATP, 0.4 Na2-GTP, 10 phosphocreatine, and 5 QX314, pH adjusted to 7.2 with CsOH for voltage clamp. Dextran Alexa Fluor 594 or 488 was included in the intracellular recording solution for cell labeling. After whole-cell patch clamp was achieved, spontaneous EPSCs (sEPSCs) or miniature EPSCs (mEPSCs) were recorded under voltage clamp at −70 mV in the presence of picrotoxin (PTX; 50 μm) and d-APV (50 μm) with or without tetrodotoxin (TTX; 1 μm), respectively. To monitor evoked EPSCs (eEPSCs), the stimulating electrode was placed at least 125 μm laterally from the recorded neuron. The extracellular stimulation was performed using a model 2100 Isolated Pulse Stimulator (A-M Systems). Evoked EPSCs were recorded in voltage clamp at −70 or +40 mV in the presence of PTX (50 μm) to obtain specific AMPA receptor (AMPAR) and NMDA receptor (NMDAR) components, respectively. Input resistance and series resistance were monitored throughout the experiments, and recordings were rejected if series resistance increased >25 MΩ. Drugs were applied via perfusion into the bath recording solution after at least 10 min of baseline control recording. All data were sampled at 5 kHz and analyzed off-line using Clampfit 10.2 software(Molecular Devices). For graphical representation, the stimulus artifacts of evoked EPSCs were removed. Orexin-A, MCH, PTX, TTX, d-APV, and QX-314 were obtained from Tocris Bioscience; orexin (catalog #H-003-30; RRID:AB_2315019) and MCH (catalog #H-070-47; RRID:AB_10013632) antibodies were obtained from Phoenix Pharmaceuticals; and Dorsomorphin was purchased from BioVision.
High-fat diet-induced conditioned place preference in mice
The conditioned place preference (CPP) and locomotor activity protocols were adapted and modified from those provided in another study (Kanoski et al., 2014). All animals were implanted with a bilateral cannula targeting the LHA region. After 14 d of recovery, animals were put on restricted food intake (2.5 g/d) of control chow for 7 extra days before the CPP training session. Behavioral experiments were performed during the dark phase (lights off at 6:00 P.M.). The CPP apparatus consists of two spatially distinct compartments with different wall patterns and floor textures, and the apparatus was placed in a SmartCage home-cage system (AfaSci). To obtain the initial preference scores, baseline activity was recorded in all animals for 10 min, during which time the door between the two compartments was left open to allow animals to explore both sides freely. The less preferred compartment was subsequently paired with 5 g of 45% kcal/fat diet (catalog #D12451, Research Diets), in five aliquots (∼1 g) that were scattered around. Meanwhile, the initially preferred chamber was subsequently paired with no food. During training sessions, mice were restricted to one side of the box, alternating between the high-fat diet (HFD)-paired and the no food-paired side for 20 min each on 7 d. On the test day, animals received bilateral injections of leptin or saline 1.5 h before the test. Mice were placed in the apparatus with access to both compartments for 15 min. No food was presented on either side. The final preference score of each animal was compared with its baseline. Leptin (R&D Systems) was reconstituted in sterile 20 mm Tris-HCl, pH 8.0, and injected with 0.025 μg/side in a 50 nl volume bilaterally. All data are presented as the mean ± SEM. Statistical comparisons before and after the application of leptin were made using paired two-tailed Student's t tests.
Intra-LHA cannula implantation and leptin infusion
Five- to 6-week-old male C57BL/6 wild-type mice were anesthetized using isoflurane and placed in a stereotaxic apparatus (catalog #M1900, KOPF). After exposing the skull, a bilateral guide cannula with a dummy injector (PlasticsOne) was inserted and affixed into the area above the designated coordinates from LHA (anteroposterior, −1.25 mm; mediolateral, ±1.1 mm; dorsoventral, −5.1 mm). Mice were individually housed for 2 weeks to allow recovery from the surgery, during which daily food intake and body weight were monitored. During the CPP training, the dummy injector was removed and replaced by an injector (with 0.1 mm extra in length), and 50 nl of sterile PBS was injected per site 1.5 h before each training session to allow adaptation of the animals. On the test day, animals were injected with either PBS or leptin (25 ng in a 50 nl volume per site). After the CPP experiment, animals were perfused and injection sites were confirmed.
Energy state manipulation in mice
Acute food deprivation.
Chow was removed from the home cage at 6:00 P.M. and electrophysiological recordings were performed beginning at 10:00 A.M. the next day. Cardiac blood was collected to measure the blood glucose or plasma leptin levels before decapitation (see below).
High-fat diet-induced obesity.
Wild-type littermate mice (postnatal day 22) were randomly separated into HFD or control chow groups. HFD (45% kcal/fat; catalog #D12451) and control chow (10% kcal/fat; catalog #D12450B) were obtained from Research Diets. Animals were fed either an HFD or control chow for 6 weeks before electrophysiological recordings. At 4 weeks after HFD or control chow feeding, the injection of retrograde microfluorescent beads into the VTA was performed. Body weight was measured every week.
Blood glucose and leptin levels measurement
Blood was taken from the left ventricle of the heart before decapitation. Plasma glucose level was measured immediately by a blood glucose monitoring system (FreeStyle, Abbott). Protease inhibitors were added into each blood sample. Serum was separated by centrifugation at 4°C and stored at −80°C until assayed. Serum leptin levels were measured by a commercial ELISA kit with 96-well plate assay, according to the protocol provided by the manufacturer (EZML-82K Mouse Leptin ELISA, EMD Millipore). The appropriate detection range for the leptin assay was 0.23 to 30 ng/ml, and the lowest detection level was 0.05 ng/ml.
Experimental design and statistical analysis
The effect of leptin on synaptic transmission was evaluated before and after the bath application of 100 nm leptin in the same neurons recorded in hypothalamic brain slices; therefore, these experiments were not performed blindly. In all cases, three or more animals were used for each parameter collected. Individual sample sizes for slice patch-clamp recording (n = number of neurons; labeled in each figure) are reported separately for each experiment. All data are presented as the mean ± SEM. Statistical comparisons before and after the application of leptin were made using paired two-tailed Student's t tests. Statistical comparisons for different groups were made using unpaired two-tailed Student's t tests. Statistical differences for HFD treatment and body weights were performed using one-way ANOVA and post hoc Bonferroni test.
Results
Leptin suppresses excitatory synaptic drive onto LHA-to-VTA-projecting orexin and MCH neurons
We used an established retrograde tracing approach (Katz et al., 1984; Köbbert et al., 2000; Lammel et al., 2008; Brown and Hestrin, 2009) to label LHA neurons forming synapses within the VTA. Briefly, we injected microfluorescent RetroBeads into the VTA and identified retrograde labeled neuronal cell bodies in the LHA (Fig. 1A). We then conducted immunohistochemistry (IHC) and determined that 30.78 ± 5.48% and 23.61 ± 4.15% of the retrograde labeled neurons express orexin or MCH, respectively. To determine whether leptin affects the excitatory synaptic drive onto these subtype- and pathway-identified LHA neurons, we performed whole-cell patch-clamp recordings and identified the neurochemistry of the recorded cells by post hoc IHC (Fig. 1B). The average membrane capacitances were 51.3 ± 5.1 and 64.2 ± 5.9 pF, and the input resistances were as follows: 803.9 ± 52.4 and 410.9 ± 32.7 MΩ, in LHA-to-VTA orexin (n = 15/15, cells/mice) and MCH (n = 16/16, cells/mice) neurons, respectively. LHA-to-VTA-projecting non-orexin or MCH neurons had significantly different capacitance and resistance parameters (27.1 ± 4.8 and 3.018 ± 0.394 × 103 MΩ, respectively; n = 11/11, cells/mice), suggesting that these neurochemically defined LHA-to-VTA neural subtypes possess different intrinsic membrane properties.
Figure 1.
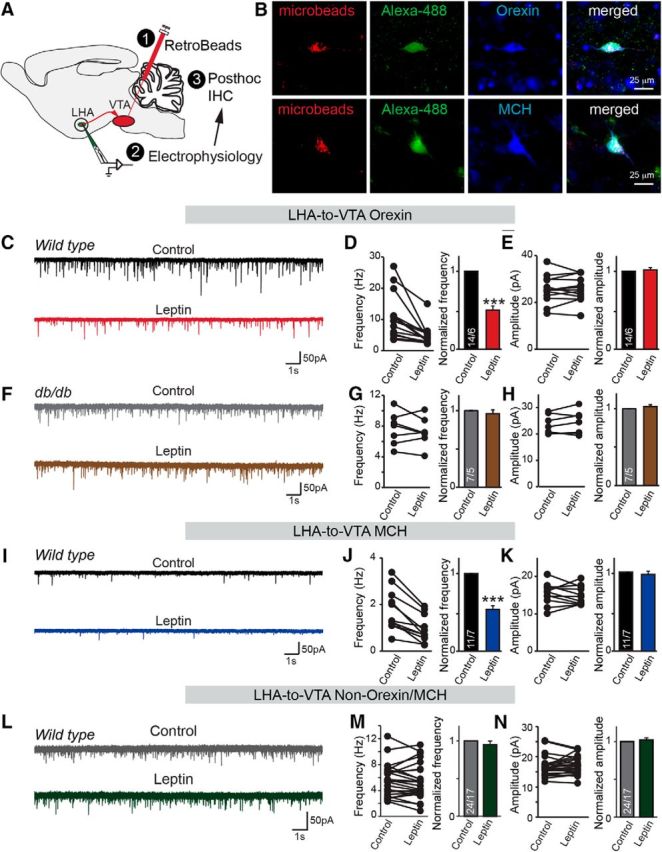
Spontaneous excitatory synaptic transmission in a defined LHA-to-VTA neuronal circuitry is suppressed by leptin. A, Diagram showing the combination of neural tracing for LHA-to-VTA projection and post hoc identification of the neurochemical subtype. B, Images of recorded LHA-to-VTA-projecting neurons. Cells containing retrograde beads were recorded and labeled with Alexa Fluor 488, and post hoc IHC for orexin or MCH. C, Sample traces of sEPSCs in LHA-to-VTA-projecting orexin neurons before and after bath application of leptin (100 nm). D, E, Pooled data showing the effect of leptin on the frequency and amplitude of sEPSCs in orexin neurons. F, Sample traces of sEPSCs in LHA-to-VTA-projecting orexin neurons in leptin receptor mutant db/db mice before and after bath application of leptin. G, H, Pooled data showing the effect of leptin on the frequency and amplitude of sEPSCs in orexin neurons from db/db mice. I, Sample traces of sEPSCs in LHA-to-VTA-projecting MCH neurons before and after bath application of leptin. J, K, Pooled data showing the effect of leptin on the frequency and amplitude of sEPSCs in MCH neurons. L, Representative traces of sEPSCs in non-orexin/MCH neurons before and after the application of leptin. M, N, Pooled data. Data are the mean ± SEM; the numbers of cells/animals analyzed are indicated in bars. Paired two-tailed Student's t tests were used: ***p < 0.001.
We first examined pharmacologically isolated sEPSCs in the presence of PTX, which revealed significant differences in basal excitatory synaptic strength onto MCH and orexin neurons projecting to the VTA. Specifically, the frequency of sEPSCs recorded from orexin neurons was approximately fivefold higher than those from MCH neurons (10.6 ± 1.9 and 1.8 ± 0.3 Hz, n = 14/6 and 11/7 cells/mice, in orexin and MCH neurons, respectively, p < 0.001); the amplitudes of sEPSCs recorded from orexin neurons were significantly greater than those in MCH neurons as well (25.6 ± 1.5 pA in orexin neurons vs 15.4 ± 0.9 Hz in MCH neurons; p < 0.001). Strikingly, bath application of leptin (100 nm) significantly decreased the frequency, but not the amplitude, of sEPSCs in both orexin and MCH neurons projecting to the VTA, suggesting that, despite the intrinsic differences in basal synaptic strength, both subtypes were regulated by leptin (Fig. 1C–E,I–K). Moreover, the effects of leptin on synaptic regulation were abolished in LepR-deficient db/db mice (Fig. 1F–H), suggesting that the specificity of leptin function depends on the successful activation of leptin receptors and their downstream signaling (Fig. 1F–H). Interestingly, leptin had no effect on excitatory synaptic transmission of the remaining cell types, which were identified as negative for both orexin and MCH (i.e., non-orexin/MCH), suggesting that this effect may be cell type specific (Fig. 1L–N).
To consolidate the decrease in excitatory synaptic strength onto MCH and orexin neurons projecting to the VTA, we investigated the effect of leptin on eEPSCs elicited by field stimulation with a bipolar stimulation electrode placed ∼100–150 μm away from the recorded neurons. AMPAR- and NMDAR-mediated eEPSCs were recorded at a holding potential of −70 or +40 mV, respectively, in the presence of PTX (Fig. 2A,B). Remarkably, both AMPAR- and NMDAR EPSCs in LHA-to-VTA-projecting orexin and MCH neurons were suppressed by leptin, while the AMPA/NMDAR EPSC ratios were unchanged (Fig. 2C–H).
Figure 2.

Evoked excitatory synaptic transmission in defined LHA-to-VTA neuronal circuitry is suppressed by leptin. A, B, Sample traces of evoked AMPAR and NMDAR responses recorded from LHA-to-VTA orexin (A) or MCH (B) neurons before and after bath application of leptin (in orexin neurons: AMPAR-eEPSCs reduced from 193.7 ± 30.4 to 95.4 ± 22.8 pA; NMDAR-eEPSCs reduced from 17.2 ± 2.1 to 6.0 ± 1.8 pA; AMPAR/NMDAR ratio reduced from 14.3 ± 2.2 to 13.9 ± 1.9; in MCH neurons: AMPAR-eEPSCs went from 119.4 ± 25.4 to 73.0 ± 24.3 pA; NMDAR-eEPSCs went from 29.6 ± 3.9 to 18.1 ± 3.4 pA; AMPAR/NMDAR ratio went from 5.2 ± 1.3 to 5.3 ± 1.5). C–E, Pooled data in LHA-to-VTA orexin neurons before and after leptin treatment. F–H, Pooled data in LHA-to-VTA MCH neurons. Data are the mean ± SEM; numbers of cells/animals analyzed are indicated in bars. Paired two-tailed Student's t tests were used: ***p < 0.001.
Leptin regulates excitatory synaptic transmission in LHA-to-VTA-projecting MCH and orexin neurons via a presynaptic mechanism and distinct signaling cascades
The reduction of both AMPAR- and NMDAR-mediated EPSCs by leptin in LHA-to-VTA-projecting neurons suggests a presynaptic molecular mechanism. To test this, we recorded mEPSCs. A significant reduction (∼50%) in the frequency, but not the amplitude, of mEPSCs was observed in both orexin and MCH neurons (Fig. 3A–F), which is highly suggestive of reduction in presynaptic release probability. Therefore, we conducted paired-pulse ratio (PPR) analysis of the eEPSCs. A decreased release probability usually can be demonstrated by an increase in the ratio between the second and first responses of paired eEPSCs (Zucker and Regehr, 2002). Indeed, we observed a dramatic increase in the PPR of eEPSCs on both MCH and orexin neurons after the application of leptin (Fig. 3G–J). Consistent with the idea that leptin has no regulatory effects in db/db mice as well as in LHA-to-VTA non-orexin/MCH, the PPRs of eEPSCs show no difference in the absence or presence of leptin (Fig. 3K–N). Collectively, these results provide strong evidence that leptin acts to reduce excitatory synaptic drive onto LHA MCH and orexin neurons projecting to the VTA by a presynaptic mechanism. This observation is consistent with previous reports showing that LHA orexin and MCH neurons do not express LepRs (Leinninger et al., 2009) and are therefore likely regulated by presynaptic nerve terminals originating from various brain regions (González et al., 2016).
Figure 3.
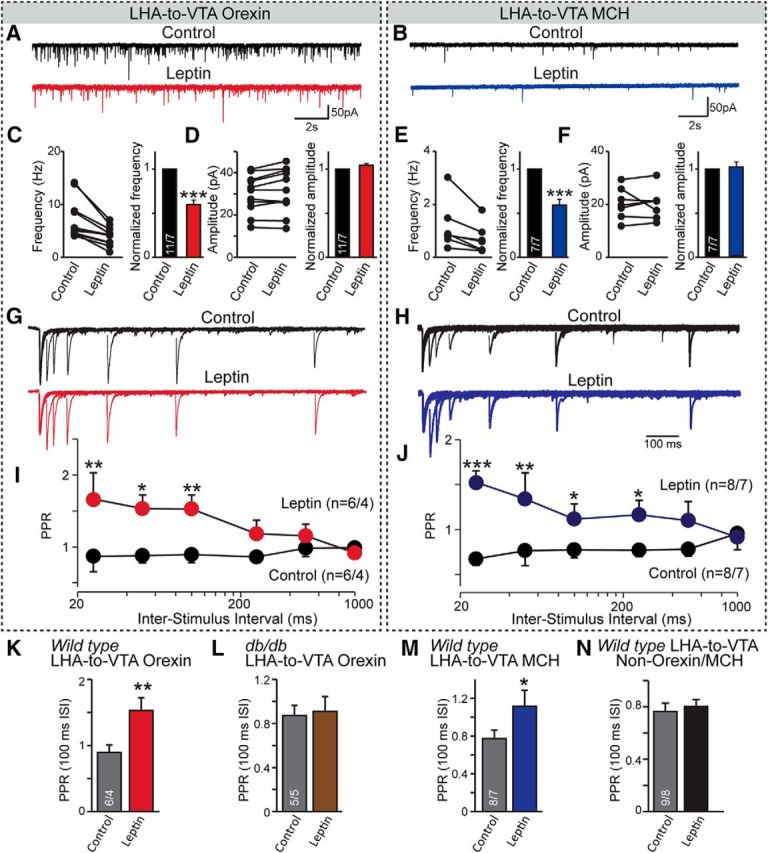
Presynaptic effects of leptin in LHA-to-VTA-projecting orexin and MCH neurons. A, B, Sample traces of mEPSCs from LHA-to-VTA-projecting orexin (A) and MCH (B) neurons before and after bath application of leptin (100 nm). C–F, Pooled data showing the effect of leptin on the frequency and amplitude (both absolute and normalized values) in orexin or MCH neurons. G, H, Sample traces of evoked EPSCs at different intervals in LHA-to-VTA orexin (G) and MCH (H) neurons. I, J, Pooled data showing the PPRs before and after the application of leptin in orexin (I) or MCH (J) neurons. Two-way ANOVA with Bonferroni post hoc test. K–N, PPRs of evoked EPSCs at an interstimulus interval (ISI) of 100 ms, in LHA-to-VTA-projecting orexin wild-type (K), db/db (L), MCH (M), and non-orexin/MCH (N) neurons. Student's t tests were used for statistical analysis. Data are presented as the mean ± SEM; numbers of neurons recorded/animals used are indicated in each graph. *p < 0.05; **p < 0.01; ***p < 0.001.
To identify the effect of leptin on the overall activity of LHA-to-VTA-projecting neurons, we examined spontaneous action potential (sAP) firing in the target cells. Among the eight orexin cells patched, six had sAPs and all six had their firing frequencies reduced by leptin application (Fig. 4A,B), while none of the MCH neurons that were patched had spontaneous firing, with a resting potential at −61.6 ± 1.0 (n = 5/3, cells/mice). Interestingly, when the synaptic transmissions were blocked by AMPA, NMDA, and GABAA receptor blockers, leptin no longer suppressed orexin neuronal firing (Figure 4C,D), suggesting that leptin likely reduces the neuronal activity of LHA-to-VTA orexin neurons largely via suppressing their excitatory inputs.
Figure 4.

Leptin suppresses sAPs in the LHA-to-VTA neurons. A, Sample traces of sAP firing recorded from LHA-to-VTA-projecting orexin neurons before and after bath perfusion of leptin (100 nm). B, Pooled data showing the frequency of sAPs were reduced by leptin from 4.2 ± 1.1 to 1.6 ± 0.6 Hz. C, Sample traces of the effects of leptin on sAPs in LHA-to-VTA orexin neurons in the presence of synaptic blockers. D, Pooled data showing the frequency of sAPs were unchanged. The frequency was 4.2 ± 0.9 before vs 4.4 ± 0.9 Hz after leptin application. Data are presented as the mean ± SEM. Numbers in bars indicate the number of cells/animals analyzed. Paired two-tailed Student's t tests were used to define the statistical significance before and after leptin application. ***p < 0.001.
We then sought to examine the intracellular signaling cascades involved in leptin regulation of synaptic transmission. It has been shown that both the phosphoinositide 3-kinase (PI3K)–phospholipase C (PLC) and AMP-activated protein kinase (AMPK) pathways mediate neuronal regulation by leptin in hypothalamic neurons (Williams et al., 2011; Lee et al., 2015). To define the possible involvement of these pathways, we used pharmacological intervention to impede either PI3K–PLC or AMPK signaling cascades. Remarkably and unexpectedly, we found that these pathways act in a nonredundant and cell type-specific manner to mediate the effect of leptin. Specifically, we found that blocking the AMPK pathway occluded the suppressive effect of leptin on synaptic transmission in MCH neurons but not in orexin neurons (Fig. 5A–D), whereas PLC signaling appeared to be specifically required for orexin neurons (Fig. 5E–H). Together, these data, although not extensive, suggest that leptin is likely engaging distinct cell signaling mechanisms to mediate its effect on synaptic transmission, suggesting a possible molecular basis for the differential regulation of MCH and orexin neurons by leptin.
Figure 5.

Leptin affects synaptic inputs to LHA-to-VTA-projecting orexin and MCH neurons via distinct cell-signaling pathways. A, B, Sample traces of mEPSCs recorded from LHA-to-VTA-projecting orexin or MCH neurons in the presence of AMPK inhibitor Dorsomorphin, before and after bath perfusion of leptin (100 nm). C, D, Pooled data of frequencies of mEPSCs in the presence or absence of leptin in LHA-to-VTA-projecting orexin or MCH neurons. E, F, Sample traces of mEPSCs of LHA-to-VTA-projecting orexin or MCH neurons in the presence of PLC inhibitor U-73122, before and after leptin application. G, H, Pooled data of frequencies of mEPSCs in the presence or absence of leptin in either orexin or MCH neurons. Data are presented as the mean ± SEM. Numbers in bars indicate the number of cells/animals analyzed. Paired two-tailed Student's t tests were used to define the statistical significance before and after leptin application. **p < 0.01; ***p < 0.001.
Intra-LHA leptin blocks the expression of HFD-induced CPP
Next, we tested the behavioral relevance of leptin signaling in the LHA. We found that HFD induced a significant preference shift, as measured by CPP behavioral assay. The infusion of leptin into the LHA significantly dampened the preference associated with HFD (Fig. 5A,B). To gain more information about the possible downstream effects of LHA-to-VTA-projecting neuronal excitability changes, we evaluated the functions of MCH and orexin in the VTA DA neurons. By using whole-cell recording in VTA-containing brain slices, we found that the sAP firings in DA neurons were reliably facilitated by exogenous orexin, while MCH produced notable heterogeneous effects (both facilitation and suppression were observed), which is consistent with the high degree of neuronal heterogeneity in the VTA (Fig. 6C–H). Therefore, these data suggest that the suppression of synaptic strength onto orexin and MCH neurons by leptin may have downstream functional consequences on VTA DA neuronal function. Together, these data suggest that MCH and orexin inputs to VTA can modulate DA neurons and leptin tone in the LHA and can reduce reward-guided behaviors.
Figure 6.
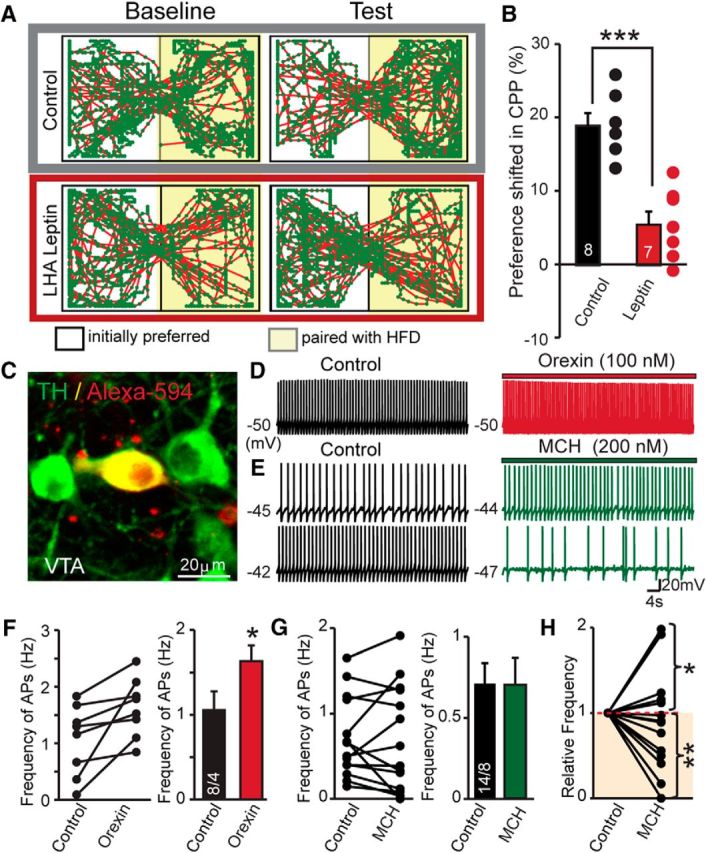
Intra-LHA leptin reduces HFD reward and the effects of orexin and MCH peptides on VTA dopaminergic neurons. A, Representative locomotor activity in CPP chamber after pairing with HFD. B, Summary of the preference shift of CPP expression in intra-LHA infusion of vehicle or leptin. C, Representative confocal image showing the recorded VTA DA neurons loaded with dextran Alexa Fluor 594 after post hoc IHC for tyrosine hydroxylase (TH). D, Representative traces of sAPs in VTA DA neurons before and after the application of orexin. E, Traces of spontaneous APs in VTA DA neurons before and after the application of MCH; the top traces demonstrate a facilitatory effect of MCH, while the bottom traces display an inhibitory effect of MCH in another cell. F, Pooled data showing the effects of orexin on VTA DA neuronal AP firing. Left, Individual changes in the frequencies of APs from individual cells. Right, Average values. G, Pooled data showing the effects of MCH on VTA DA neuronal firings. Left, The changes of frequencies of AP individual cells. Right, Average values. H, Normalized frequencies of APs before and after the application of MCH. Two different responses (facilitation and inhibition) were observed. Data are presented as the mean ± SEM; numbers of cells/animals analyzed are indicated in bar graphs. Paired two-tailed Student's t tests were used: *p < 0.05; **p < 0.01; ***p < 0.001.
Altered energy states impair synaptic regulation by leptin
It is well established that food deprivation (i.e., low plasma glucose and leptin levels) results in reduced leptin levels (Ahima et al., 1996), while excessive energy storage causes hyperleptinemia and impairs leptin signaling, often termed “leptin resistance” (Bjørbaek et al., 1998; Enriori et al., 2006); however, the interplay been energy status and the effect of leptin on synaptic transmission has not been addressed. We hypothesized that altered sensitivity to leptin might manifest at the level of synaptic transmission under obese or energy-deprived conditions, respectively. DIO (Enriori et al., 2007) and acute fasting were used to test the influence of energy state on the regulatory effects of leptin on synaptic transmission.
We first asked whether “insensitivity” (i.e., leptin resistance) was present at the level of synaptic transmission in the diet-induced obese state. As expected, wild-type animals maintained on an HFD for 6 weeks (starting from postnatal day 22) exhibited significantly increased body weight, blood glucose levels, as well as plasma leptin levels when compared with animals maintained on control chow (Fig. 7A–C). We recorded synaptic transmission in the LHA-to-VTA-projecting orexin and MCH neurons in DIO and control animals in the presence of leptin. Remarkably, the suppressive effect of leptin on mEPSCs was blunted by DIO in MCH neurons (Fig. 7J–L), while leptin was still somewhat effective, but to a lesser extent (∼12% vs ∼50%), in orexin neurons compared with those from control chow-fed mice (Fig. 7D–I). These data suggest that leptin “resistance” could be manifest at the level of synaptic transmission, at least within the LHA-to-VTA MCH and orexin neurocircuitry.
Figure 7.
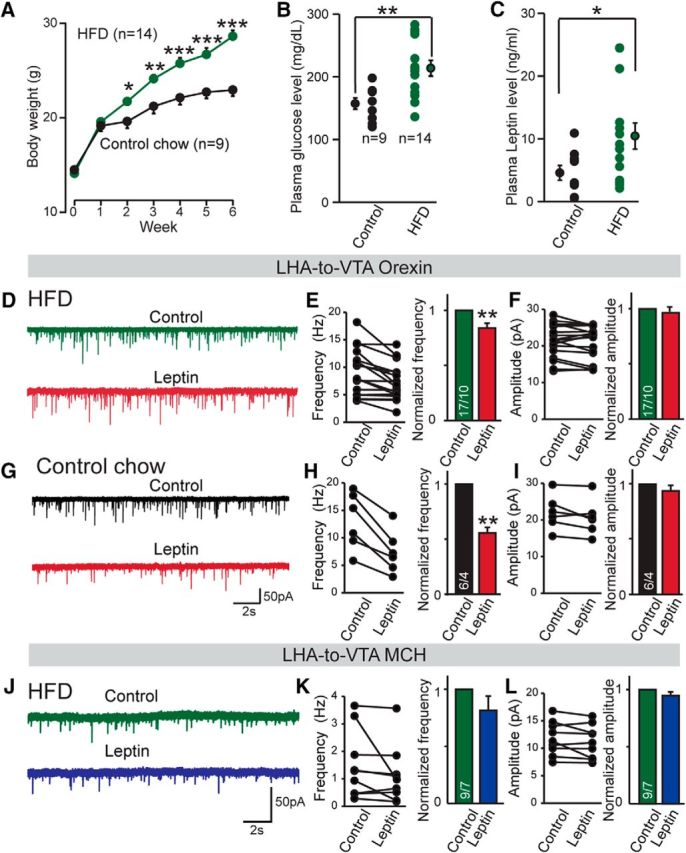
Diet-induced obesity attenuates the suppressive effects of leptin on synaptic transmission in the LHA-to-VTA circuitry. A, Summary of the body weights of mice fed with control chow, HFD, or control diet for 6 weeks. One-way ANOVA was performed to compare the HFD and control chow feeding groups: *p < 0.05; **p < 0.01; ***p < 0.001. B, C, Blood glucose (B) and plasma leptin (C) levels of mice fed control chow or HFD. C, Plasma leptin levels. D, Representative traces of mEPSCs in LHA-to-VTA-projecting orexin neurons in the presence or absence of leptin in HFD-fed mice. E, F, Pooled data. G, Representative traces of mEPSCs of LHA-to-VTA orexin neurons before and after exogenous leptin treatment from control chow-fed mice. H, I, Pooled data. J, Representative traces of mEPSCs of LHA-to-VTA MCH neurons before and after exogenous leptin treatment from mice fed with HFD. K, L, Pooled data. Data are presented as the mean ± SEM; numbers of cells/animals analyzed in bars indicate number of cells analyzed. Student's t tests were used: *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we tested the effect of leptin on synaptic transmission during energy depletion induced by 16 h of overnight fasting. As expected, both the plasma glucose and leptin levels dropped significantly in fasted animals (Fig. 8A,B). To maintain hypoglycemic conditions, we lowered the glucose levels in the bath solution to 0.7 mm when cutting brain slices and performing electrophysiological recordings (Hardie et al., 2012). The frequency of mEPSCs in LHA-to-VTA-projecting orexin neurons of fasted mice was significantly increased when compared with controls with ad libitum food supply (Fig. 8C), demonstrating their intrinsic ability to be altered by energy state. In contrast, food deprivation did not change the frequency of mEPSCs on MCH neurons (Fig. 8D), supporting our initial observation that the regulation of basal excitatory synaptic drive onto orexin and MCH neurons may be differentially regulated. However, contrary to our expectation, we found that food deprivation blunted the effect of leptin on excitatory synaptic strength onto both orexin and MCH neurons (Fig. 8E–H). Interestingly, sensitivity to synaptic regulation by leptin could be selectively rescued in orexin, but not MCH, neurons by increasing the glucose level from 0.7 to 2.5 mm (Fig. 8I–L). Together, these data suggest that diminished sensitivity to leptin is manifest at the level of synaptic transmission and further support the hypothesis that leptin may use distinct signaling mechanisms to regulate orexin and MCH neurons.
Figure 8.
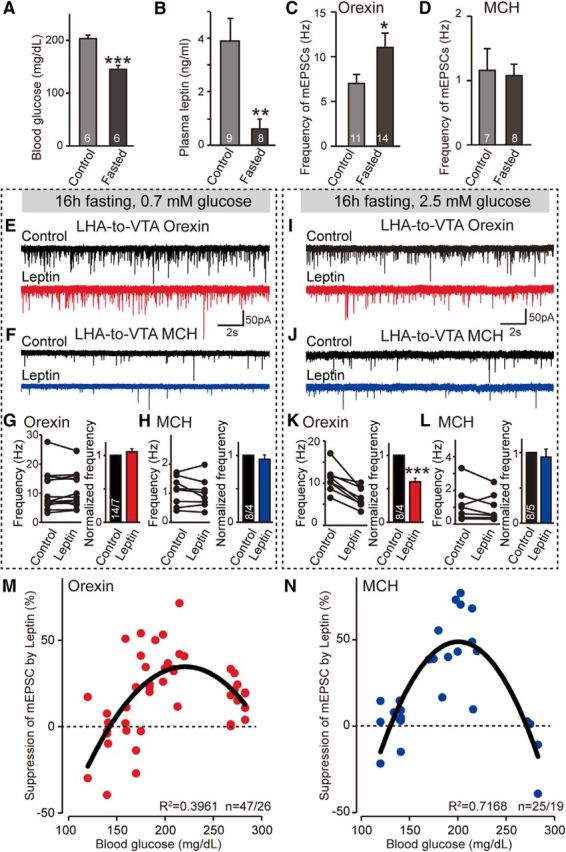
Impact of acute energy deprivation on the regulatory effect of leptin in the LHA-to-VTA circuitry. A, B, Blood glucose (A) and leptin (B) levels in control and fasted animals before electrophysiological analysis. C, D, Pooled data of the frequencies of mEPSCs in LHA-to-VTA-projecting orexin (C) and MCH (D) neurons before and after energy deprivation. E, F, Sample traces of mEPSCs of LHA-to-VTA-projecting orexin (E) or MCH (F) neurons of fasted mice under low-glucose (0.7 mm) conditions in the presence or absence of leptin. G, H, Pooled data showing the effect of leptin on the mEPSC frequency of orexin (G) or MCH (H) LHA-to-VTA neurons in the 0.7 mm glucose recording condition. I, J, Sample traces of mEPSCs from fasted animals recorded in 2.5 mm glucose before and after the application of leptin. K, L, Pooled data. M, N, The percentage of suppression of mEPSCs by leptin in animals as a function of blood glucose level in orexin (M) and MCH (N) neurons. Black lines represent polynomial fittings of the datasets (Excel, Microsoft). Values of the correlation coefficients (R2) are indicated in each plot. Note that the fittings are meant to provide the trend of the synaptic regulation of leptin as a function of glucose levels. Data are presented as the mean ± SEM; n numbers number of cells/animals analyzed are indicated in the bars. Paired two-tailed Student's t tests were used: *p < 0.05; **p < 0.01; and ***p < 0.001.
It is somewhat surprising that under both energy-deficient and energy-excessive conditions leptin regulation of synaptic transmission in the LHA-to-VTA neurocircuitry was compromised, since leptin resistance has traditionally been reported under conditions of chronic energy excess (Bjørbaek et al., 1998; Enriori et al., 2006); however, this has been recently disputed (Myers, 2015; Ottaway et al., 2015). To test for the existence of a dynamic relationship between energy state and the effect of leptin, we plotted the suppression in mEPSC frequency as a function of blood glucose levels. Surprisingly, we observed an inverted “U-shape” relationship (Fig. 8M,N), demonstrating that extremes of energy states (i.e., fasted and obese), impair leptin function. Collectively, these results reveal that opposite extremes of energy status affect synaptic regulation by leptin in LHA-to-VTA-projecting MCH and orexin neurons.
Discussion
Leptin is an adipocyte-derived, energy state-signaling factor, and mutations in the genes encoding leptin or its receptor cause massive hyperphagia and morbid obesity in rodents and humans, demonstrating that the neural circuitry mediating the effects leptin are nonredundant and indispensable (Friedman, 2016). This study shows that leptin suppresses excitatory synaptic strength onto LHA orexin and MCH neurons projecting to the VTA via distinct signaling mechanisms and that this effect is regulated by whole-body energy state (i.e., as measured by blood glucose level).
Modulation of LHA-to-VTA neuronal circuitry function may regulate food intake and reward
Since reward-related feeding is considered an important component for maintaining a positive energy balance (Liu et al., 2015), we focused on the LHA-to-VTA neural pathways and found that both orexin and MCH alter VTA DA neuronal firing in a distinct manner: orexin increases firing while MCH can exert both facilitation and inhibition of action potential firing (Fig. 6). A previous report showed that orexin consistently increased the firing of A10 DA neurons, but possibly not MCH neurons (Korotkova et al., 2003), likely due to biphasic modulatory effects of DA neurons by MCH (Fig. 6C,E,F) or VTA cellular heterogeneity. Combined with previous evidence showing that orexin increases excitatory strengths onto VTA DA neurons and intra-VTA administration of orexin reinstates drug-seeking behavior (Harris et al., 2005; Borgland et al., 2006; Sheng et al., 2014), we believe that VTA-projecting orexin and MCH neurons can directly modulate DA neuron activity, at least partially, via the release of orexin and MCH. Therefore, we propose that the excitatory synaptic inputs onto the LHA-to-VTA-projecting orexin and MCH neurons are likely important mediators for food reward and, possibly, other reward-associated behaviors. Indeed, it has been shown that the activation of LHA orexin neurons is strongly linked to preference for cues associated with drug and food reward (Harris et al., 2005), while the activation of MCH neurons in the LHA has been shown to influence the CPP for sucrose and induce DA release (Domingos et al., 2013). Through the infusion of leptin into the LHA, we were able to suppress the expression of reward-guided behavior (Fig. 6).
LHA orexin- and MCH-expressing neurons are also capable of releasing glutamate and/or GABA (Ziegler et al., 2002; Jego et al., 2013; Schöne et al., 2014; Chee et al., 2015). Indeed, the manipulation of both glutamatergic and GABAergic subpopulations of LHA neurons was shown to affect food intake and/or reward (Barbano et al., 2016; Nieh et al., 2016). Therefore, the reduction of excitatory synaptic drive onto these groups of neurons will likely affect their downstream targets via the release of classical neurotransmitters. Moreover, both MCH and orexin neuropeptides have been demonstrated to be important for regulating food intake as well as reward behavior (Shimada et al., 1998; Hara et al., 2001; Harris et al., 2005; Narita et al., 2006). Indeed, central administration of MCH and orexin causes an increase in feeding (Qu et al., 1996; Rossi et al., 1997; Sakurai et al., 1998), and transgenic animal models with overexpression of MCH become obese (Ludwig et al., 2001). In contrast, genetic deletion of MCH leads to hypophagia and a lean phenotype (Shimada et al., 1998), and genetic ablation of orexin neurons also causes hypophagia (Hara et al., 2001). In addition to the shared orexigenic property, the two neuronal subtypes exert distinct functions in the brain. For example, orexin neurons are capable of energy sensing and promote wakefulness (Hara et al., 2001), while MCH neurons are necessary in maintaining rapid eye movement sleep (Jego et al., 2013). Therefore, we believe that defining the underlying synaptic and circuitry level mechanisms provide essential insight that may be harnessed to combat metabolic dysregulation and pathologic food intake behavior.
Leptin regulation of synaptic transmission via a presynaptic mechanism
Leptin has profound effects on both hypothalamic and extrahypothalamic neuronal activity including regulation of neuronal firing, excitatory synaptic transmission, inhibitory synaptic transmission, and long-term synaptic plasticity (Pang and Han, 2012; Williams and Elmquist, 2012). Within the LHA, it has been shown that leptin may indirectly inhibit a subset of orexin neurons via leptin receptor long isoform (LepRb)-expressing neurotensin neurons as well as increasing the ATP− sensitive potassium conductance of orexin neurons (Goforth et al., 2014). We focused on the LHA orexin and MCH neurons that specifically project to the VTA, and demonstrated that leptin significantly decreases excitatory synaptic strength onto these neurons. This regulatory effect is specific to LepR signaling because leptin failed to regulate synaptic transmission in db/db mice. Although both LHA orexin and MCH neurons do not express LepRb (Leinninger et al., 2009), we provide the following strong evidence that leptin acts to directly regulate presynaptic nerve terminals: (1) the frequency but not the amplitude of mEPSCs (Fig. 3A–F) and sEPSCs (Fig. 1C–K) was significantly reduced; (2) both AMPAR- and NMDAR-mediated EPSCs were reduced with no change in the AMPA/NMDA EPSC ratio (Fig. 2); and (3) we observed a dramatic increase in the PPRs (Fig. 3G–J). These results are consistent with prior reports demonstrating that leptin mediates the suppression of excitatory synaptic transmission via a presynaptic mechanism in both the VTA (Thompson and Borgland, 2013) and arcuate nucleus (Lee et al., 2015).
Both LHA MCH and orexin neurons receive presynaptic inputs from diverse but overlapping brain regions (González et al., 2016). However, our data, although not definitive, suggest that the origins of these synaptic inputs to LHA-to-VTA orexin, MCH, or non-orexin/MCH neurons are likely to be different because of the following: (1) the basal excitatory synaptic input strengths are different (Fig. 1C,D,I,J); (2) changes in PPR are induced by leptin (Fig. 3K–N); and (3) leptin regulates excitatory synaptic inputs likely via different intracellular signaling presynaptically (Fig. 5). Determination of the origins of the excitatory inputs from defined brain regions to these VTA-projecting LHA neurons underlying leptin regulation requires further detailed investigation.
Energy states affect how leptin regulates synaptic transmission
While synaptic regulation by leptin has been reported extensively under normal energy states (Harvey et al., 2006; Irving et al., 2006; Solovyova et al., 2009; Thompson and Borgland, 2013; Lee et al., 2015), before this study little information was available regarding the regulation of synaptic transmission by leptin under conditions mimicking perturbed energy homeostasis. Here, we provide direct evidence that energy status influences the effect of leptin on synaptic transmission and demonstrate that this effect occurs differentially among specific cell types in the LHA-to-VTA neurocircuitry. Previous studies suggest that acute calorie deprivation produces an increase of sEPSCs in orexin neurons, likely caused by increased excitatory synapse formation onto these neurons (Horvath and Gao, 2005), while obesity causes a shift in excitation–inhibition balance in arcuate nuclei neurons (Pinto et al., 2004; Horvath et al., 2010). Beyond these structural modifications associated with energy states, we found that under both calorie deprivation (hypoleptinemia and/or hypoglycemia) and DIO (hyperleptinemia), the regulatory effect of leptin on synaptic transmission was significantly diminished. Specifically, under acute calorie deprivation, leptin produced no suppressive effect on mEPSCs when we recorded using low-glucose conditions. Interestingly, orexin-expressing but not MCH-expressing LHA-to-VTA-projecting neurons regained sensitivity to leptin when glucose level was increased. It is possible that the acute calorie deprivation provided some permissive conditions for the differential regulation of leptin on these neurons. Nevertheless, the specific contributions of hypoglycemia and/or hypoleptinemia to the diminished leptin regulation under acute calorie deprivation conditions needs further investigation.
Obese individuals normally have elevated circulating leptin, and it was generally believed that they have leptin resistance (Bjørbaek et al., 1998; Enriori et al., 2006); however, recent data show that both the peripheral and central organs show reduced, but not absent, responses to leptin (Ottaway et al., 2015). We found that the regulatory effect of leptin on synaptic transmission was significantly impaired under HFD-fed conditions (i.e., elevated body weight, hyperleptinemia, hyperglycemia), particularly among orexin-expressing neurons. When plotted, the synaptic response to leptin as a function of the measured plasma glucose level under fasting, control chow-feeding, or HFD-feeding conditions, we observed an inverted “U-shape” relationship, further suggesting that extremes of whole-body energy status may impair synaptic regulation by leptin.
In summary, here we show that leptin regulates fast excitatory synaptic transmission in LHA-to-VTA-projecting orexin and MCH neurons via a presynaptic mechanism governed by distinct signaling cascades, an effect that is most pronounced under normal energy conditions (i.e., euleptinemia or euglycemia). These data suggest that leptin is important for signaling energy fluxes, such that imbalanced synaptic regulation could contribute to excess energy intake and an obese phenotype. Therefore, targeting the synaptic mechanisms dysregulated by leptin resistance may provide new insight for therapeutic strategies aimed to correct pathologies associated with altered food intake behavior.
Footnotes
This research was supported by Robert Wood Johnson Foundation Grant 67038, the New Jersey Health Science Foundation, the United States–Israel Binational Science Foundation, and the Sinsheimer Foundation. The Pang laboratory is supported by National Institutes of Health (NIH) Grants AA-023797 and DA-039686. The Bello laboratory is supported by US Department of Agriculture-National Institute of Food and Agriculture Grant NJ-06156 and NIH Grant AT-008933. We thank Dr. Guohui Li for his help during the initial stage of this project. We thank Juliet Gotthardt for helping with the measurement of plasma leptin levels; and Isadora Zhang, Vincent Mirabella, and Drs. Nicola Francis (Rutgers University), Kevin Williams (University of Texas Southwestern Medical Center, Dallas, TX), and Martin Myers (University of Michigan, Ann Arbor, MI) for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS (1996) Role of leptin in the neuroendocrine response to fasting. Nature 382:250–252. 10.1038/382250a0 [DOI] [PubMed] [Google Scholar]
- Barbano MF, Wang HL, Morales M, Wise RA (2016) Feeding and reward are differentially induced by activating GABAergic lateral hypothalamic projections to VTA. J Neurosci 36:2975–2985. 10.1523/JNEUROSCI.3799-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS (1998) Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell 1:619–625. 10.1016/S1097-2765(00)80062-3 [DOI] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A (2006) Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron 49:589–601. 10.1016/j.neuron.2006.01.016 [DOI] [PubMed] [Google Scholar]
- Bray GA, York DA (1997) Clinical review 90: leptin and clinical medicine: a new piece in the puzzle of obesity. J Clin Endocrinol Metab 82:2771–2776. 10.1210/jcem.82.9.4224 [DOI] [PubMed] [Google Scholar]
- Brown SP, Hestrin S (2009) Intracortical circuits of pyramidal neurons reflect their long-range axonal targets. Nature 457:1133–1136. 10.1038/nature07658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee MJ, Arrigoni E, Maratos-Flier E (2015) Melanin-concentrating hormone neurons release glutamate for feedforward inhibition of the lateral septum. J Neurosci 35:3644–3651. 10.1523/JNEUROSCI.4187-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman DL. (1978) Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia 14:141–148. 10.1007/BF00429772 [DOI] [PubMed] [Google Scholar]
- Domingos AI, Sordillo A, Dietrich MO, Liu ZW, Tellez LA, Vaynshteyn J, Ferreira JG, Ekstrand MI, Horvath TL, de Araujo IE, Friedman JM (2013) Hypothalamic melanin concentrating hormone neurons communicate the nutrient value of sugar. Elife 2:e01462. 10.7554/eLife.01462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Cowley MA (2006) Leptin resistance and obesity. Obesity (Silver Spring) 14 [Suppl 5]:254S–258S. 10.1038/oby.2006.319 [DOI] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA (2007) Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5:181–194. 10.1016/j.cmet.2007.02.004 [DOI] [PubMed] [Google Scholar]
- Flier JS, Maratos-Flier E (2017) Leptin's physiologic role: does the emperor of energy balance have no clothes? Cell Metab 26:24–26. 10.1016/j.cmet.2017.05.013 [DOI] [PubMed] [Google Scholar]
- Friedman J. (2016) The long road to leptin. J Clin Invest 126:4727–4734. 10.1172/JCI91578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395:763–770. 10.1038/27376 [DOI] [PubMed] [Google Scholar]
- Goforth PB, Leinninger GM, Patterson CM, Satin LS, Myers MG Jr (2014) Leptin acts via lateral hypothalamic area neurotensin neurons to inhibit orexin neurons by multiple GABA-independent mechanisms. J Neurosci 34:11405–11415. 10.1523/JNEUROSCI.5167-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JA, Iordanidou P, Strom M, Adamantidis A, Burdakov D (2016) Awake dynamics and brain-wide direct inputs of hypothalamic MCH and orexin networks. Nat Commun 7:11395. 10.1038/ncomms11395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SP, Dacey D, Halaris AE, Collier T, Routtenberg A (1978) Aphagia and adipsia after preferential destruction of nerve cell bodies in hypothalamus. Science 202:537–539. 10.1126/science.705344 [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T (2001) Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30:345–354. 10.1016/S0896-6273(01)00293-8 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262. 10.1038/nrm3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Aston-Jones G (2005) A role for lateral hypothalamic orexin neurons in reward seeking. Nature 437:556–559. 10.1038/nature04071 [DOI] [PubMed] [Google Scholar]
- Harvey J, Solovyova N, Irving A (2006) Leptin and its role in hippocampal synaptic plasticity. Prog Lipid Res 45:369–378. 10.1016/j.plipres.2006.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebebrand J, Muller TD, Holtkamp K, Herpertz-Dahlmann B (2007) The role of leptin in anorexia nervosa: clinical implications. Mol Psychiatry 12:23–35. 10.1038/sj.mp.4001909 [DOI] [PubMed] [Google Scholar]
- Hervieu GJ, Cluderay JE, Harrison D, Meakin J, Maycox P, Nasir S, Leslie RA (2000) The distribution of the mRNA and protein products of the melanin-concentrating hormone (MCH) receptor gene, slc-1, in the central nervous system of the rat. Eur J Neurosci 12:1194–1216. 10.1046/j.1460-9568.2000.00008.x [DOI] [PubMed] [Google Scholar]
- Hoebel BG, Teitelbaum P (1962) Hypothalamic control of feeding and self-stimulation. Science 135:375–377. 10.1126/science.135.3501.375 [DOI] [PubMed] [Google Scholar]
- Horvath TL, Gao XB (2005) Input organization and plasticity of hypocretin neurons: possible clues to obesity's association with insomnia. Cell Metab 1:279–286. 10.1016/j.cmet.2005.03.003 [DOI] [PubMed] [Google Scholar]
- Horvath TL, Sarman B, García-Cáceres C, Enriori PJ, Sotonyi P, Shanabrough M, Borok E, Argente J, Chowen JA, Perez-Tilve D, Pfluger PT, Brönneke HS, Levin BE, Diano S, Cowley MA, Tschöp MH (2010) Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc Natl Acad Sci U S A 107:14875–14880. 10.1073/pnas.1004282107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving AJ, Wallace L, Durakoglugil D, Harvey J (2006) Leptin enhances NR2B-mediated N-methyl-D-aspartate responses via a mitogen-activated protein kinase-dependent process in cerebellar granule cells. Neuroscience 138:1137–1148. 10.1016/j.neuroscience.2005.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, Friedman J, Burdakov D, Adamantidis AR (2013) Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci 16:1637–1643. 10.1038/nn.3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD (2013) The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341:1517–1521. 10.1126/science.1241812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanoski SE, Alhadeff AL, Fortin SM, Gilbert JR, Grill HJ (2014) Leptin signaling in the medial nucleus tractus solitarius reduces food seeking and willingness to work for food. Neuropsychopharmacology 39:605–613. 10.1038/npp.2013.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Burkhalter A, Dreyer WJ (1984) Fluorescent latex microspheres as a retrograde neuronal marker for in vivo and in vitro studies of visual cortex. Nature 310:498–500. 10.1038/310498a0 [DOI] [PubMed] [Google Scholar]
- Köbbert C, Apps R, Bechmann I, Lanciego JL, Mey J, Thanos S (2000) Current concepts in neuroanatomical tracing. Prog Neurobiol 62:327–351. 10.1016/S0301-0082(00)00019-8 [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE (2003) Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J Neurosci 23:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J (2008) Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57:760–773. 10.1016/j.neuron.2008.01.022 [DOI] [PubMed] [Google Scholar]
- Laque A, Yu S, Qualls-Creekmore E, Gettys S, Schwartzenburg C, Bui K, Rhodes C, Berthoud HR, Morrison CD, Richards BK, Münzberg H (2015) Leptin modulates nutrient reward via inhibitory galanin action on orexin neurons. Mol Metab 4:706–717. 10.1016/j.molmet.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DK, Jeong JH, Chun SK, Chua S Jr, Jo YH (2015) Interplay between glucose and leptin signalling determines the strength of GABAergic synapses at POMC neurons. Nat Commun 6:6618. 10.1038/ncomms7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Münzberg H, Myers MG Jr (2009) Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab 10:89–98. 10.1016/j.cmet.2009.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Opland DM, Jo YH, Faouzi M, Christensen L, Cappellucci LA, Rhodes CJ, Gnegy ME, Becker JB, Pothos EN, Seasholtz AF, Thompson RC, Myers MG Jr (2011) Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab 14:313–323. 10.1016/j.cmet.2011.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Mukherjee D, Haritan D, Ignatowska-Jankowska B, Liu J, Citri A, Pang ZP (2015) High on food: the interaction between the neural circuits for feeding and for reward. Front Biol 10:165–176. 10.1007/s11515-015-1348-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, Maratos-Flier E (2001) Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 107:379–386. 10.1172/JCI10660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK (2001) Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol 435:6–25. 10.1002/cne.1190 [DOI] [PubMed] [Google Scholar]
- Mickelsen LE, Kolling FW, Chimileski BR, Fujita A, Norris C, Chen K, Nelson CE, Jackson AC (2017) Neurochemical heterogeneity among lateral hypothalamic hypocretin/orexin and melanin-concentrating hormone neurons identified through single-cell gene expression analysis. eNeuro 4:0013–17.2017. 10.1523/ENEURO.0013-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG., Jr (2015) Leptin keeps working, even in obesity. Cell Metab 21:791–792. 10.1016/j.cmet.2015.05.017 [DOI] [PubMed] [Google Scholar]
- Narita M, Nagumo Y, Hashimoto S, Narita M, Khotib J, Miyatake M, Sakurai T, Yanagisawa M, Nakamachi T, Shioda S, Suzuki T (2006) Direct involvement of orexinergic systems in the activation of the mesolimbic dopamine pathway and related behaviors induced by morphine. J Neurosci 26:398–405. 10.1523/JNEUROSCI.2761-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieh EH, Vander Weele CM, Matthews GA, Presbrey KN, Wichmann R, Leppla CA, Izadmehr EM, Tye KM (2016) Inhibitory input from the lateral hypothalamus to the ventral tegmental area disinhibits dopamine neurons and promotes behavioral activation. Neuron 90:1286–1298. 10.1016/j.neuron.2016.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaway N, Mahbod P, Rivero B, Norman LA, Gertler A, D'Alessio DA, Perez-Tilve D (2015) Diet-induced obese mice retain endogenous leptin action. Cell Metab 21:877–882. 10.1016/j.cmet.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Han W (2012) Regulation of synaptic functions in central nervous system by endocrine hormones and the maintenance of energy homoeostasis. Biosci Rep 32:423–432. 10.1042/BSR20120026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL (2004) Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304:110–115. 10.1126/science.1089459 [DOI] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E (1996) A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 380:243–247. 10.1038/380243a0 [DOI] [PubMed] [Google Scholar]
- Rossi M, Choi SJ, O'Shea D, Miyoshi T, Ghatei MA, Bloom SR (1997) Melanin-concentrating hormone acutely stimulates feeding, but chronic administration has no effect on body weight. Endocrinology 138:351–355. 10.1210/endo.138.1.4887 [DOI] [PubMed] [Google Scholar]
- Saito Y, Cheng M, Leslie FM, Civelli O (2001) Expression of the melanin-concentrating hormone (MCH) receptor mRNA in the rat brain. J Comp Neurol 435:26–40. 10.1002/cne.1191 [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, et al. (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585. 10.1016/S0092-8674(00)80949-6 [DOI] [PubMed] [Google Scholar]
- Schöne C, Apergis-Schoute J, Sakurai T, Adamantidis A, Burdakov D (2014) Coreleased orexin and glutamate evoke nonredundant spike outputs and computations in histamine neurons. Cell Rep 7:697–704. 10.1016/j.celrep.2014.03.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z, Santiago AM, Thomas MP, Routh VH (2014) Metabolic regulation of lateral hypothalamic glucose-inhibited orexin neurons may influence midbrain reward neurocircuitry. Mol Cell Neurosci 62:30–41. 10.1016/j.mcn.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E (1998) Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396:670–674. 10.1038/25341 [DOI] [PubMed] [Google Scholar]
- Solovyova N, Moult PR, Milojkovic B, Lambert JJ, Harvey J (2009) Bi-directional modulation of fast inhibitory synaptic transmission by leptin. J Neurochem 108:190–201. 10.1111/j.1471-4159.2008.05751.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger TL, Kilduff TS, Behan M, Benca RM, Landry CF (2004) Comparison of hypocretin/orexin and melanin-concentrating hormone neurons and axonal projections in the embryonic and postnatal rat brain. J Chem Neuroanat 27:165–181. 10.1016/j.jchemneu.2004.02.007 [DOI] [PubMed] [Google Scholar]
- Thompson JL, Borgland SL (2013) Presynaptic leptin action suppresses excitatory synaptic transmission onto ventral tegmental area dopamine neurons. Biol Psychiatry 73:860–868. 10.1016/j.biopsych.2012.10.026 [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Elmquist JK (2012) From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci 15:1350–1355. 10.1038/nn.3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Sohn JW, Donato J Jr, Lee CE, Zhao JJ, Elmquist JK, Elias CF (2011) The acute effects of leptin require PI3K signaling in the hypothalamic ventral premammillary nucleus. J Neurosci 31:13147–13156. 10.1523/JNEUROSCI.2602-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, O'Brien WG 3rd, Lee CC, Myers MG Jr, Tong Q (2012) Role of GABA release from leptin receptor-expressing neurons in body weight regulation. Endocrinology 153:2223–2233. 10.1210/en.2011-2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Kim ER, Zhao R, Myers MG Jr, Munzberg H, Tong Q (2013) Glutamate release mediates leptin action on energy expenditure. Mol Metab 2:109–115. 10.1016/j.molmet.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432. 10.1038/372425a0 [DOI] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP (2002) Distribution of vesicular glutamate transporter mRNA in rat hypothalamus. J Comp Neurol 448:217–229. 10.1002/cne.10257 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]