

Abstract
Pathological hallmarks of Alzheimer's disease (AD) include amyloid-β (Aβ) plaques, neurofibrillary tangles, and reactive gliosis. Glial cells offer protection against AD by engulfing extracellular Aβ peptides, but the repertoire of molecules required for glial recognition and destruction of Aβ are still unclear. Here, we show that the highly conserved glial engulfment receptor Draper/MEGF10 provides neuroprotection in an AD model of Drosophila (both sexes). Neuronal expression of human Aβ42arc in adult flies results in robust Aβ accumulation, neurodegeneration, locomotor dysfunction, and reduced lifespan. Notably, all of these phenotypes are more severe in draper mutant animals, whereas enhanced expression of glial Draper reverses Aβ accumulation, as well as behavioral phenotypes. We also show that the signal transducer and activator of transcription (Stat92E), c-Jun N-terminal kinase (JNK)/AP-1 signaling, and expression of matrix metalloproteinase-1 (Mmp1) are activated downstream of Draper in glia in response to Aβ42arc exposure. Furthermore, Aβ42-induced upregulation of the phagolysosomal markers Atg8 and p62 was notably reduced in draper mutant flies. Based on our findings, we propose that glia clear neurotoxic Aβ peptides in the AD model Drosophila brain through a Draper/STAT92E/JNK cascade that may be coupled to protein degradation pathways such as autophagy or more traditional phagolysosomal destruction methods.
SIGNIFICANCE STATEMENT Alzheimer's disease (AD) and similar dementias are common incurable neurodegenerative disorders in the aging population. As the primary immune responders in the brain, glial cells are implicated as key players in the onset and progression of AD and related disorders. Here we show that the glial engulfment receptor Draper is protective in a Drosophila model of AD, reducing levels of amyloid β (Aβ) peptides, reversing locomotor defects, and extending lifespan. We further show that protein degradation pathways are induced downstream of Draper in AD model flies, supporting a model in which glia engulf and destroy Aβ peptides to reduce amyloid-associated toxicity.
Keywords: amyloid, autophagy, Draper, Drosophila, glia
Introduction
One neuropathological feature of Alzheimer's disease (AD) is the formation of extracellular deposits of insoluble fibrillar amyloid-β (Aβ) peptides (Holtzman et al., 2011; Nelson et al., 2012). These Aβ aggregates may adversely affect CNS function by sequestering protective molecules or by releasing small Aβ oligomers under certain physiological conditions (Rosenblum, 2014; Tipping et al., 2015; Jucker and Walker, 2015). Soluble Aβ oligomers can directly promote synaptic dysfunction (Selkoe, 2012; Rosenblum, 2014; Tipping et al., 2015) and may be an early causal factor in AD-related cognitive phenotypes. Although the precise mechanisms of Aβ-induced toxicity are enigmatic, there is compelling genetic and pharmacological evidence from model systems that reducing Aβ levels is largely protective (Lashuel et al., 2002; Wang et al., 2010; Miners et al., 2011; Baruch et al., 2016), emphasizing the need to understand how brain Aβ levels are normally kept in check.
Genome-wide association studies and genetic analyses of AD models support the notion that glial cells are coupled to AD pathogenesis. For example, mutations in the microglial receptor Trem2 (triggering receptor expressed on myeloid cells 2) are associated with increased AD risk (Guerreiro et al., 2013; Jonsson et al., 2013). The immunoreceptor CD33/Siglec-3 is another well described AD susceptibility locus. CD33 mutations are associated with reduced microglial internalization of Aβ and increased fibrillar amyloid accumulation in vivo (Bradshaw et al., 2013; Griciuc et al., 2013; Malik et al., 2013). Unsurprisingly, one prominent area of AD research has focused on glial phagocytic function; multiple glial cell types (e.g., microglia and astrocytes) have the capacity to reduce Aβ levels and influence AD progression (Wyss-Coray et al., 2003; Herber et al., 2007; Demattos et al., 2012; Hong et al., 2016; Pomilio et al., 2016; Wes et al., 2016). Understanding precisely how glia recognize, internalize, and degrade amyloid-like peptides will provide additional molecular and genetic targets to pursue for disease treatment.
Drosophila offers a powerful in vivo model to rapidly explore Aβ-induced toxicity. Transgenic expression of human Aβ1–42 fragment (Aβ42) results in age-dependent presentation of multiple molecular and behavioral phenotypes, including Aβ accumulation, neuronal hyperexcitability, defective mitochondrial function, locomotor dysfunction, and reduced lifespan (Finelli et al., 2004; Crowther et al., 2005; Iijima et al., 2008; Wentzell and Kretzschmar, 2010; Ping et al., 2015), mirroring the cellular and behavioral decline observed in other organisms (Bayer and Wirths, 2008; Woodruff-Pak, 2008; Götz and Ittner et al., 2008; Rivest, 2009). Interestingly, electron microscopy reveals that when human Aβ42 is expressed in Drosophila neurons, Aβ is detected in glia, suggesting that fly glia internalize secreted Aβ peptides in vivo (Iijima et al., 2008). The molecules involved in glial engulfment of human Aβ peptides in Drosophila, however, have never been explored.
The highly conserved engulfment receptor Draper is required for glial clearance of apoptotic cells and degenerating neuronal projections in flies during development and in the adult CNS (MacDonald et al., 2006; Tasdemir-Yilmaz and Freeman, 2014). Draper signals through a tyrosine kinase-signaling cascade to drive cytoskeletal remodeling, alter glial gene expression, and promote phagocytic activity (MacDonald et al., 2006, 2013; Ziegenfuss et al., 2008). The vertebrate orthologs of Draper (MEGF10 and Jedi) and downstream effectors (e.g., Fyn-related kinase, GULP, Rac1) are also implicated in glial immunity, including glial clearance of degenerating neurons (Osada et al., 2009; Wu et al., 2009; Linnartz et al., 2010; Chung et al., 2013; Iram et al., 2016), indicating that the core components of glial engulfment machinery are conserved across species. Interestingly, overexpression of MEGF10 in cultured HeLa cells enhances engulfment of bath-applied Aβ (Singh et al., 2010), but the role of Draper/MEGF10 in the context of AD has never been explored in vivo.
Here, we show that human Aβ42arc activates Draper-dependent signaling cascades in adult Drosophila glia, including signal transducer and activator of transcription (Stat92E) and c-Jun N-terminal kinase (JNK)/AP-1-mediated transcription, matrix metalloproteinase-1 (Mmp1) upregulation, and induction of protein degradation pathways. Glial inhibition of Draper exacerbates molecular and behavioral Aβ42arc-associated phenotypes, whereas overexpression of Draper/MEGF10 partially rescues these defects, providing the first in vivo evidence that Draper offers protection against Aβ42-induced toxicity.
Materials and Methods
Fly strains.
The following Drosophila strains were used: repo-Gal4 (MacDonald et al., 2006), elavc155-Gal4 (BL458), elav-Gal4 (BL8765), Appl-Gal4 (obtained from Doris Kretzschmar, OHSU; RRID:BDSC_32040), UAS-Aβ42arc (BL33773 and BL33774), UAS-Draper-I (Logan et al., 2012), UAS-DraperRNAi (MacDonald et al., 2006), drprΔ5rec9 (Neukomm et al., 2014), UAS-JraRNAi (BL31595, RRID:BDSC_3159), UAS-kayakRNAi (BL31391, RRID:BDSC_31391), UAS-stat92eRNAi (VDRC 43866), TRE-GFP, and 10xStat92E-dGFP (Bach et al., 2007). UAS-MEGF10 transgenic flies were generated by subcloning mouse MEGF10 (cDNA clone MGC:100091, IMAGE:30620548) from pEGFP-N3 (kind gift from B. Carter, Vanderbilt University, Nashville, TN) into the pUAST vector and transgenic flies were generated using standards methods by BestGene.
Brain dissection and immunostaining.
Adult fly brains of either sex were dissected, fixed, and stained using previously described methods (Purice et al., 2016). Fly heads were fixed in 4% paraformaldehyde + 0.01% Triton X-100 for 16 min while rocking at room temperature, then washed with PBST-1 (PBS containing 0.01% Triton X-100) for 3 × 2 min at room temperature. Brains were dissected in ice-cold PBST-1, transferred to microcentrifuge tubes containing 4% paraformaldehyde + 0.1% Triton X-100, fixed while rocking at room temperature for 20 min, and then washed with PBST-2 (PBS containing 0.1% Triton X-100) for 3 × 2 min at room temperature. For immunostaining, brains were blocked in Sea Block blocking buffer (ThermoFisher Scientific) for 15 min at room temperature, followed by incubation in primary antibodies diluted in PBST-2 overnight at 4°C. Brains were then washed in PBST-2 for 4 × 30 min at room temperature, incubated in secondary antibodies diluted in PBST-2 for 2 h at room temperature in the dark, and washed in PBST-2 for 4 × 30 min in the dark. After the final wash, any remaining liquid was removed from the brains with a glass pipette and replaced with VECTASHIELD Mounting Media (Vector Laboratories; RRID:AB_2336789).
Confocal imaging.
After sitting for 1 h overnight at 4°C in the dark, brains for each single experiment were mounted on microscope slides under a single no. 1.5 cover glass in VECTASHIELD before imaging on a Zeiss LSM 700 laser scanning microscope equipped with a Zeiss 40× 1.4 numerical aperture oil-immersion lens. On rare occasions, a brain was visibly damaged in the central region from the dissection or transfer process; these brains were excluded from analysis.
Antibodies.
Primary antibodies were used at the following dilutions: chicken anti-GFP (1:1000; ThermoFisher Scientific, A10262; RRID:AB_2534023), mouse anti-Aβ 6E10 (1:500; BioLegend, 39320), rabbit anti-dAtg8 (Shelly et al., 2009; 1:1000; kind gift from Sara Cherry, University of Pennsylvania, Philadelphia, Pennsylvania), rabbit anti-Ref(2)P (Wyers et al., 1995; 1:1000; kind gift from Tor Erik Rusten, University of Oslo, Norway), guinea pig anti-Draper (Shklover et al., 2015; 1:1000; kind gift from Estee Kurant, Israel Institute of Technology), mouse anti-MMP1 (Developmental Studies Hybridoma Bank, 14A3D2; RRID:AB_579782, 3A6B4; RRID:AB_579780, 3B8D12; RRID:AB_579781, 5H7B11; RRID:AB_579779) at 1:50 used at 1:1:1:1 ratio. Secondary antibodies: AlexaFluor 488 anti-chicken or anti-mouse (1:400; Life Technologies, 703-545-155; RRID:AB_2340375), RhodoRed anti-mouse (1:400; Life Technologies, 715-295-150; RRID:AB_2340831), AlexaFluor 647 anti-guinea pig (1:400; Life Technologies,706-605-148; RRID:AB_2340476). Amyloid fibrillary deposits were detected by Thioflavin T (ThT) staining using previously described methods (Marcora et al., 2014). Briefly, immunostained brains were incubated in 1:1 solution of water–ethanol containing 1% ThT (Sigma-Aldrich) for 20 min, washed in 1:1 solution of ethanol–1× PBS, and mounted for confocal imaging on a Zeiss LSM 700 microscope as described above.
Aggregate and fluorescence intensity quantification.
To quantify Aβ aggregates, fluorescence intensity was measured in a region of interest in the cortex region adjacent to the antennal lobes on each side of the brain. Fluorescence of Atg8, p62 (Ref2), GFP (for TRE-GFP or 10XSTAT92E-dGFP reporters), or Mmp1 was quantified by measuring the fluorescence intensity of three different regions of interest at standardized positions in or around the antennal lobe. Specifically, twenty-four 1 μm sections were included in each quantification analysis. All image analyses were performed using Volocity Analysis software (PerkinElmer; RRID:SCR_002668).
Lifespan and climbing assay.
For lifespan analysis, flies were collected at a density of 20 flies per vial. At least three vials were used per treatment (total of 60 flies per genotype). Flies were passed to fresh vials every 3 d, and the number of dead flies was recorded. Locomotor function of flies was assessed using a negative geo-taxis assay as reported by Bahadorani et al. (2008) with slight modifications. Briefly, 20 flies/trial were tapped to the bottom of an empty fly vial. After 7 s, the number of flies that climbed a minimum vertical distance of 8 cm was recorded. Each trial was repeated three times.
Quantitative RT-PCR.
Total RNA was extracted from 15 to 20 fly heads using Trizol (Invitrogen) according to the manufacturer's instructions and quantified by Nanodrop Spectrophotometer. 100 ng of total RNA was then subjected to DNA digestion using DNase I (Ambion), immediately followed by reverse transcription using the Superscript II system (Invitrogen) with oligo(dT) primers. Quantitative PCR was performed on an ABI 7500 Fast Real-Time PCR machine using TaqMan master mix (Applied Biosystems) and the following TaqMan assays: Ribosomal Protein L32 (Applied Biosystems, standard Dm02151827_g1), Draper-I custom assay: F-primer, 50-TGTGATCATGGTTACGGAGGAC-30; R-primer, 50-CAGCCGGGTGGGCAA-30; probe, 50-CGCCTGCGATATAA-30.
Sectioning.
Paraffin serial sections were prepared as described in Sunderhaus et al. (2016). Briefly, whole flies were fixed in Carnoy solution and washed in an ethanol series, followed by methylbenzoate. Sections were cut at 7 μm thickness and imaged on a Zeiss LSM 700 laser scanning microscope. Neurodegenerative phenotypes were analyzed as described by Sunderhaus et al. (2016). The number of vacuoles in the central brain per section were quantified.
Experimental design and statistical analyses.
Quantified data presented in all figures are shown as mean ± SEM. Statistical details for each experiment are included in Figure legends, including number of flies used (N), statistical test (one-way ANOVA or unpaired, two-tailed Student's t test), and post hoc analysis (e.g., Tukey's test). For the lifespan assays, log rank (Mantel–Cox) test was used for analyses. P value for each test is indicated in the legends along with the value of the corresponding degrees of freedom (df), Student's t distribution, or ANOVA F distribution.
Data availability.
Relevant transgenic strains and data relevant to this paper are available from the corresponding author.
Results
Neuronal expression of Aβ42arc triggers Draper upregulation in glia
To establish an in vivo model for exploring the role of Draper in Aβ-mediated pathogenesis, we first used three different pan-neuronal drivers (elavc155-Gal4, elav-Gal4, and Appl-Gal4) to express a highly pathogenic form of human Aβ42 that carries the “Arctic” Swedish mutation Glu22Gly (Aβ42arc; Martin et al., 1995) fused to the N-terminal signal sequence of Drosophila argos for secretion (Casas-Tinto et al., 2011). We compared Aβ peptide levels by immunostaining, as well as the lifespan of each genotype (Fig. 1B–D). Based on this analysis, we subsequently opted to use the elav-Gal4 driver for the majority of our experiments as this driver induced moderate Aβ-related phenotypes that would allow us to discern enhancement and suppression following genetic manipulations. We also confirmed that a subset of elav-Gal4-driven Aβ42arc puncta colabeled with ThT, a marker of fibrillar amyloid plaques (Fig. 1E).
Figure 1.
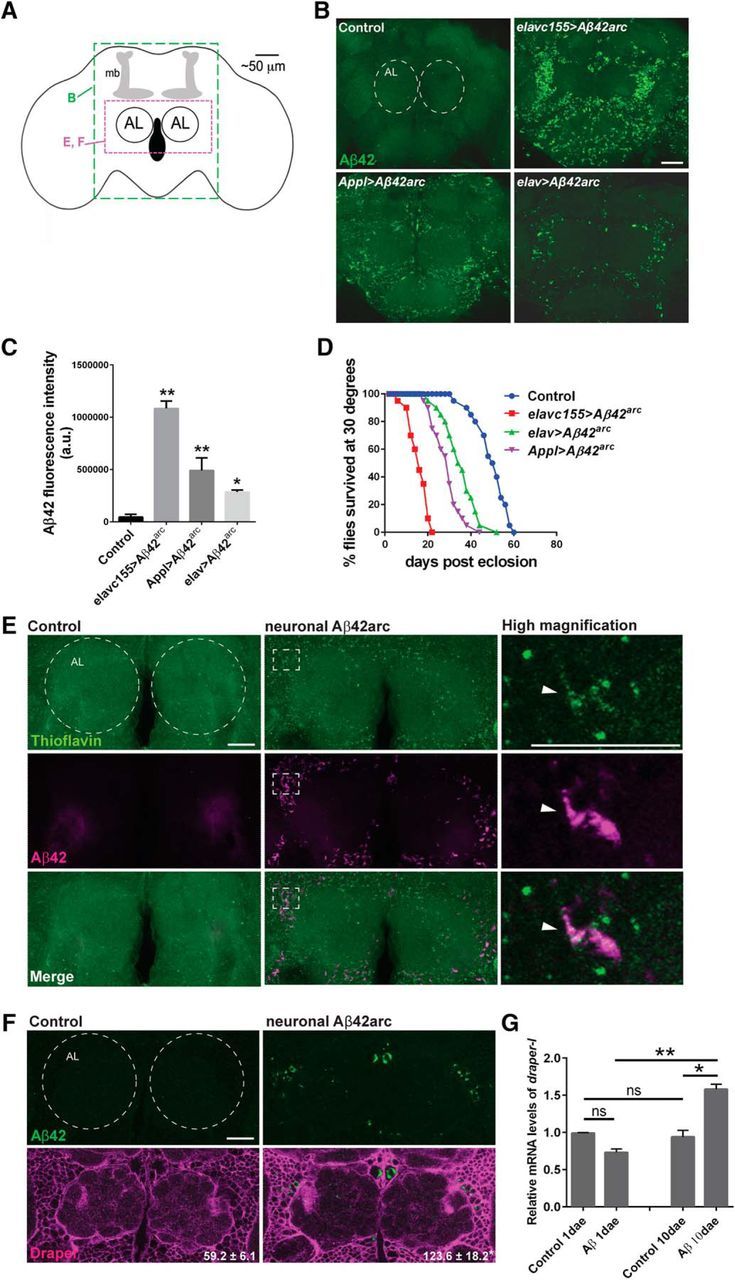
Glial Draper is upregulated in response to neuronal expression of human Aβ42arc. A, Schematic of the adult Drosophila brain showing relative location of the antennal lobes (AL) and mushroom bodies (mb). Green dotted box corresponds to brain region shown in B. Magenta dotted box shows brain region shown in E and F, and remaining confocal images throughout the paper. B, Representative confocal z-stack projections of adult brains expressing human Aβ42arc under the control of various neuronal drivers. Genotypes: control (elav-Gal4/+), elavc155>Aβ42arc (elavc155-Gal4/+; UAS-Aβ42arc/+), Appl>Aβ42arc (Appl-Gal4/+; UAS-Aβ42arc/+), elav>Aβ42arc (elav-Gal4/+; UAS-Aβ42arc/+); 7-d-old flies immunostained with anti-Aβ. Scale bar, 20 μm. Dotted circles outline the antennal lobes. C, Quantification of Aβ42 fluorescence intensity in central brains of genotypes in A; mean ± SEM. N ≥ 19; F(3,80) = 40.99; *p < 0.05 and ****p < 0.0001; one-way ANOVA with Tukey's post hoc test. D, Lifespan analysis: N = 3 groups of 20 flies, df = 3, p < 0.0001 for each Aβ42arc-expressing strain compared with Control, log-rank (Mantel–Cox) test. E, Z-stack projections of the antennal lobe regions of control (elav-Gal4/+) or neuronal Aβ-expressing (elav-Gal4/+; UAS-Aβ42arc/+) flies immunostained for Aβ (magenta) and incubated with ThT (green). Dotted circles outline the antennal lobes. White dotted rectangle indicates region showing high-magnification images of a single confocal slice. Arrowhead points to representative example of Aβ and ThT puncta colocalization. F, Single confocal slice images of the antennal lobe regions of 10-d-old flies immunostained with anti-Aβ (green) and anti-Draper (magenta). Dotted circles outline the antennal lobes. Quantification of cortical Draper fluorescence intensity in control and Aβ-expressing brains written as mean ± SEM, N ≥ 16, t = 2.809, df = 38; **p = 0.0078; unpaired t test. G, Quantitative PCR analysis of draper-I transcript levels in control and neuronal Aβ-expressing flies 1 and 10 dae; mean ± SEM plotted. N = 20; **p < 0.01, ***p < 0.001, n.s., not significant; one-way ANOVA with Tukey's post hoc test. Scale bar, 20 μm. Genotypes: (elav-Gal4/+) and (elav-Gal4/+; UAS-Aβ42arc/+).
In adult flies, glia upregulate the engulfment receptor Draper at both the mRNA and protein level as they respond to degenerating axons following olfactory nerve axotomy (MacDonald et al., 2006; Doherty et al., 2009; Etchegaray et al., 2012; Logan et al., 2012). We wondered whether Draper might similarly be upregulated following exposure to human Aβ42arc peptides and performed Draper immunostaining on Aβ42arc-expressing flies (elav-Gal4/+; UAS-Aβ42arc/+) 10 d after eclosion (10 dae). We detected a significant increase in Draper in the central brain of 10-d-old adult Aβ42arc animals by immunostaining compared with age-matched controls (Fig. 1F), as well as increased draper-I transcript expression by quantitative PCR (Fig. 1G). Thus, within the first ∼10 d of adulthood, neuronal expression of Aβ42arc in neurons triggers induction of Draper transcript and protein, suggesting that a Draper-dependent innate immune program is elicited in adult Drosophila glia in response to human Aβ42arc.
Loss of Draper enhances Aβ42arc-associated phenotypes
To determine whether Draper alters Aβ42-related defects, we expressed Aβ42arc pan-neuronally in draper-null mutant flies and assessed several molecular and behavioral phenotypes. First, we detected a striking increase in Aβ42 levels in the cortical region of the adult brain in draper-depleted flies (Fig. 2A). Notably, we detect more Aβ “puncta” in the antennal lobe neuropil in draper mutants compared with Draper-expressing flies (Fig. 2A, white arrowheads). Next, we assessed locomotor activity by performing negative geotaxis climbing assays. Although neuronal expression of Aβ42arc or loss of draper alone resulted in a partial climbing phenotype, this defect was notably exacerbated when Aβ42arc was expressed in draper mutant flies (Fig. 2B). Similarly, neuronal expression of Aβ42arc in a draper mutant background reduced adult lifespan, by ∼50% compared with draper mutants or Aβ42arc expression alone (Fig. 2C).
Figure 2.

Loss of draper enhances Aβ42arc levels, neurodegeneration, locomotor dysfunction, and reduced lifespan in human Aβ42arc-expressing flies. A, Representative single confocal images of Aβ immunostaining (green) in the antennal lobe regions of the central brain (2 dae) following neuronal expression of Aβ42arc in wild-type (elav-Gal4/+; UAS-Aβ42arc/+) and draper-null (elav-Gal4/+; UAS-Aβ42arc,draper Δ5/ draper Δ5) background. White dotted circles outline the antennal lobes. Magenta circle shows representative region of interest used for quantifying Aβ levels. Arrowheads point to visible Aβ42 accumulation in the central region of the antennal neuropil of draper mutants. Single confocal slice located approximately mid-way through the anterior/posterior boundaries of the antennal lobes. Quantification of Aβ42 fluorescence intensity in the cortex region adjacent to neuropil regions written as mean ± SEM. N = 20, t = 7.823, df = 38; ****p < 0.0001; unpaired t test. B, Quantification of locomotor climbing index of following genotypes: control (elav-Gal4/+), neuronal Aβ42arc (elav-Gal4/+;UAS-Aβ42arc/+), draper-null (draperΔ5/ draperΔ5), and neuronal Aβ42arc, draper-null flies (elav-Gal4/+; UAS-Aβ42arc,draperΔ5/ draperΔ5); mean ± SEM plotted. N = 3 groups of 20 7-d-old flies; F(3,8) = 71.91; **p < 0.01, ****p < 0.0001; one-way ANOVA with Tukey's post hoc test. C, Lifespan analysis of the genotypes shown in B. N = 3 groups of 20 flies, df = 3. Control versus neuronal Aβ42arc flies: p < 0.0001. Control versus draper-null flies: p < 0.0001. Neuronal Aβ42arc versus neuronal Aβ42arc, draper-null flies: p < 0.0001; log rank (Mantel–Cox) test. D, Quantification of vacuoles in central brain region of 7-d-old flies. N ≤ 10 flies for each genotype; mean ± SEM plotted. F(3,57) = 25.34; *p < 0.05, ****p < 0.0001; one-way ANOVA with Tukey's post hoc test. Genotypes same as in B and C. E, Representative images of 7 μm paraffin sections used for quantification in D. Arrowheads point to vacuoles indicating neurodegeneration. Asterisks indicate antennal lobe region. re, Retina. Scale bar, 20 μm.
Neuronal expression of Aβ42 or Aβ42arc induces neurodegeneration in adult Drosophila (Iijima et al., 2004, 2008). Thus, we wondered if loss of Draper would exacerbate Aβ42-related degeneration. Using vacuolar formation as a readout for neuronal integrity, we found that degeneration induced by neuronal expression of Aβ42arc was enhanced in draper mutant animals (Fig. 2D,E). Thus, these findings indicate that Draper reduces the overall Aβ42arc load in the adult brain and suggest that loss of Draper enhances Aβ42-associated neurodegeneration, locomotor dysfunction, and shortened lifespan.
Glial expression of Aβ42 phenocopies neuronal Aβ42 expression in adult flies
APP is robustly expressed in neurons but also detected in mammalian oligodendrocytes, astrocytes, and microglia (Cahoy et al., 2008; Oberstein et al., 2015). Moreover, under certain conditions of CNS stress and injury, the mammalian APP gene is selectively targeted in glial cells through signaling of the cytokine transforming growth factor-β1 and, subsequently, activation of the glial Smad transcription factors and the zinc finger nuclear factor CTCF (Amara et al., 1999; Burton et al., 2002; Lesné et al., 2003). It is unclear, however, how neuronal versus glial sources of Aβ peptides differentially contribute to AD pathology. Thus, we asked whether expression of Aβ42arc in glia phenocopied the defects observed following neuronal expression of Aβ42arc. Indeed, in flies expressing glial Aβ42arc, we detected robust Aβ immunostaining (Fig. 3A–C), a subset of which colabeled with ThT (Fig. 3A). In addition, glial expression of Aβ42arc inhibited locomotor activity, as assessed by a negative geotaxis climbing assay (Fig. 3D), and lifespan was reduced by ∼50% compared with control animals (Fig. 3E). Next, to knockdown glial Draper, we coexpressed UAS-draperRNAi and UAS-Aβ42arc with repo-Gal4. Notably, we detected a significant increase in Aβ42 throughout the central brain (Fig. 3B) and observed a modest further reduction in locomotor function (Fig. 3D) and lifespan (Fig. 3E). The fact that Aβ42 levels were increased when UAS-draperRNAi was coexpressed with UAS-Aβ42arc argues against Gal4 titration occurring due to the introduction of an additional UAS transgene; we also confirmed that Aβ42 levels were not significantly reduced when cytosolic GFP (UAS-GFP) was coexpressed under the control of repo-Gal4 (Fig. 3F). Together, these findings suggest that despite the cellular source of Aβ42 (neuronal versus glial), resultant molecular and behavioral defects are similar in adult flies, and also further bolster glial Draper as a protective molecule that minimizes Aβ42-related phenotypes.
Figure 3.
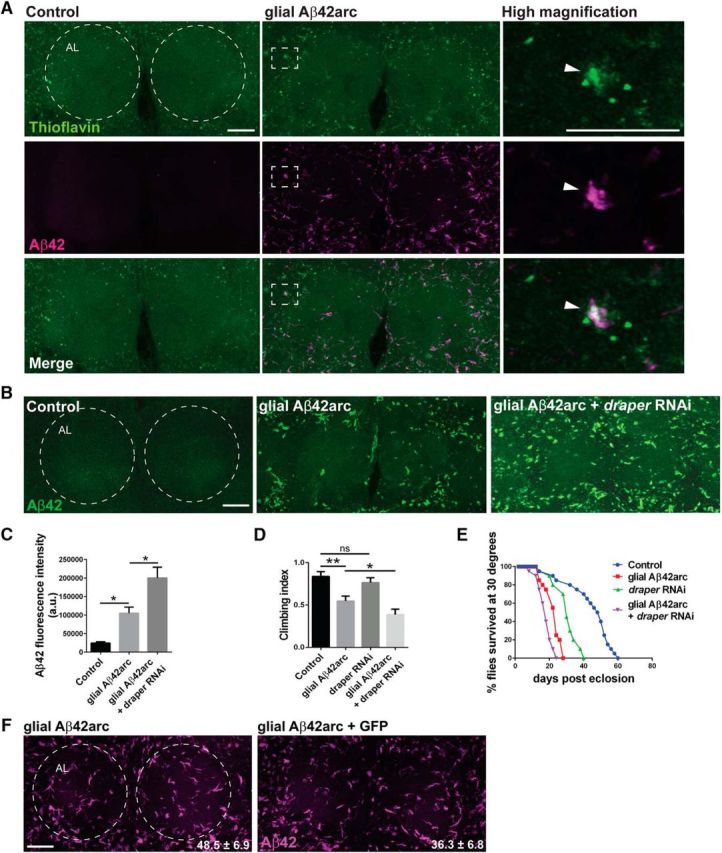
Glial expression of Aβ42arc phenocopies neuronal Aβ42arc expression. A, Z-stack projections of the antennal lobe regions of control (repo-Gal4/+) or glial Aβ-expressing (UAS-Aβ42arc/+; repo-Gal4/+) flies immunostained for Aβ (magenta) and incubated with ThT (green). Dotted circles outline the antennal lobes (AL). White dotted rectangle indicates region shown in high-magnification images of single confocal slices. Arrowhead points to colocalization of glial Aβ and ThT. Scale bars, 20 μm. B, Images of anti-Aβ staining in driver Control (repo-Gal4/+), glial Aβ42arc (UAS-Aβ42arc/+; repo-Gal4/+), and glial Aβ42arc + draper RNAi flies (UAS-Aβ42arc/draperRNAi; repo-Gal4/+); 7-d-old flies. Circles outline the AL. C, Quantification of Aβ42 fluorescence intensity in experiments depicted in B; mean ± SEM plotted. N ≥ 18; F(2,57) = 21.92; **p < 0.01; one-way ANOVA with Tukey's post hoc test; a.u., arbitrary units. D, Locomotor assay, 15-d-old flies, mean ± SEM plotted. N = 3 groups of 20 flies, F(3,8) = 37.14, *p < 0.05, **p < 0.01, n.s., nonsignificant; one-way ANOVA with Tukey's post hoc test. E, Lifespan analysis: N = 3 groups of 20 flies; df = 3; Control versus glial Aβ flies: p < 0.0001; Control versus glial Aβ + draperRNAi flies: p < 0.0001; Control versus draperRNAi flies: p < 0.0001; glial Aβ versus glial Aβ + draperRNAi flies: p < 0.0001; log rank (Mantel–Cox) test. Scale bar, 20 μm. Genotypes same as in D. F, Representative confocal z-stack projections of brains expressing glial Aβ42arc alone (UAS-Aβ42arc/+; repo-Gal4) or coexpressing glial Aβ42 arc and cytosolic GFP (UAS-Aβ42arc/+; repo-Gal4/UAS-GFP); 8-d-old flies immunostained for Aβ42. Dotted circle outlines AL. Quantification of Aβ42 fluorescence intensity written as mean ± SEM. N ≥ 16, t = 1.258, df = 31; unpaired t test; p = 0.2177. Scale bar, 20 μm.
Overexpression of glial Draper reverses Aβ-related phenotypes
Next, to determine whether boosting Draper levels could conversely rescue defects associated with human Aβ42 overexpression, we coexpressed UAS-Draper-I and UAS-Aβ42arc with repo-Gal4. Overexpression of Draper resulted in a significant reduction of Aβ immunofluorescence throughout the central brain cortex (Fig. 4A,B,D), extended adult lifespan (Fig. 4E), and significantly enhanced the climbing of Aβ42arc-expressing flies in negative geotaxis assays (Fig. 4F). Next, we asked whether glial expression of MEGF10, the mammalian homolog of Draper, could similarly reverse any defects in AD model flies. MEG10 is required for glial engulfment of neuronal projections and apoptotic neurons in the developing mammalian nervous system (Wu et al., 2009; Chung et al., 2013; Iram et al., 2016). MEGF10 also shares basic structural features with Draper, including EGF-like motifs in the extracellular domain and intracellular immunoreceptor tyrosine-based activation motifs, which couple to conserved Syk tyrosine kinases that are required for phagocytic function (Logan et al., 2012; Scheib et al.,, 2012). Finally, in ced-1 mutant worms, defective clearance of apoptotic cells is partially rescued by overexpression of human MEGF10 (Hamon et al., 2006), further highlighting the functional conservation of this family of receptors across species. We used repo-Gal4 to coexpress mouse MEGF10 and Aβ42arc and, as with Draper overexpression, we saw a significant reduction in Aβ immunostaining (Fig. 4A,C,D). Notably, we observed a very modest shift in the survival curve of MEGF10-expressing flies (Fig. 4E), and detected a trend in climbing index reversal, but this did not reach significance (Fig. 4F). It remains to be determined whether the weaker behavioral rescue observed with MEGF10 overexpression is due to differences in the extracellular region or an altered capacity for MEGF10 to couple to intracellular signaling pathways within Drosophila cells. Nonetheless, these results suggest that upregulating glial Draper/MEGF10 offers protection against Aβ-related molecular and behavioral phenotypes and also argues against the notion that enhancement of Aβ-related defects observed in draper mutants (Figs. 2, 3) are exclusively due to additive parallel effects.
Figure 4.

Overexpression of Draper/MEGF10 reverses Aβ accumulation, locomotor defects, and reduced lifespan in Aβ42arc-expressing flies: A–C, Z-stack images of anti-Aβ stained brains expressing glial Aβ42arc (UAS-Aβ42arc/+; repo-Gal4/+), coexpressing glial Aβ42arc and Draper (UAS-Aβ42arc/UAS-Draper-I; repo-Gal4/+), or glial Aβ42arc and mouse MEGF10 (UAS-Aβ42arc/+; repo-Gal4/UAS-MEGF10); 7-d-old flies. Dotted circles outline antennal lobes. D, Quantification of Aβ42 fluorescence intensity for experiments shown in A–C; mean ± SEM plotted. N ≥ 16; F(2,49) = 7.907; *p < 0.05, ***p < 0.001; one-way ANOVA with Tukey's post hoc test; n.s., not significant; a.u., arbitrary units. E, Lifespan analysis: N = 3 groups of 20 flies; df = 5; glial Aβ versus glial Aβ + Draper flies: p < 0.0001; glial Aβ versus glial Aβ + MEGF10 flies: p < 0.05; Control versus Draper: p < 0.001; Control versus MEGF10: p < 0.0001; log rank (Mantel–Cox) test. Genotypes: Control (repo-Gal4/+), glial Aβ42arc (UAS-Aβ42arc/+; repo-Gal4/+), Draper (UAS-Draper-I/+), MEGF10 (UAS-MEGF10/+), glial Aβ42arc + Draper (UAS-Aβ42arc/UAS-Draper-I; repo-Gal4/+), and glial Aβ42arc + MEGF10 (UAS-Aβ42arc/+; repo-Gal4/UAS-MEGF10). F, Negative geotaxis climbing assay results (20-d-old flies); mean ± SEM plotted. N = 3 groups of 20 flies; F(5,12) = 14.97; **p < 0.01; n.s., not significant; one-way ANOVA with Tukey's post hoc test. Genotypes same as in E. Scale bar, 20 μm.
JNK, Stat92e, and Mmp1 are activated upon Aβ42 exposure and influence Aβ42 levels in the brain
Following acute nerve injury in adult flies, two highly conserved transcription factors are activated downstream of Draper within responding glia: (1) Stat92E and (2) the heterodimer AP-1, which consists of Jra (homolog of c-Jun) and kayak (homolog of c-fos). These factors, in turn, target the draper locus to upregulate Draper levels as the glial cells become highly migratory and phagocytic to clear degenerating axons. Because we found that Draper was upregulated at the protein and transcript levels in Aβ42arc-expressing flies (Fig. 1F,G), we wondered whether these two transcriptional cascades are similarly activated in glia upon Aβ exposure. Both Stat92E and AP-1 activity can be readily monitored with well characterized in vivo reporter transgenes (MacDonald et al., 2013). First, we assessed activation of Stat92E by expressing human Aβ42arc with the neuronal elav-Gal4 driver in flies that carried the 10XStat92E-GFP reporter, which contains 10 Stat92E binding sites driving expression of destabilized GFP (dGFP). In Aβ-expressing flies, we observed a significant increase in dGFP levels (Fig. 5A,B) in a glial pattern in and around the antennal lobes (Doherty et al., 2009). We observed a similar significant increase in dGFP levels following glial-driven expression of Aβ (16.74 ± 3.3 in control animals vs 29.8 ± 5.1 following glial expression of Aβ42arc; p < 0.05). Next, we assessed activation of the TRE-GFP reporter, which contains multiple AP-1 binding sites controlling expression of GFP. In control flies, some basal TRE-GFP activation is apparent in the antennal lobes, but the GFP signal within and around the antennal lobes is significantly enhanced in Aβ-expressing flies (Fig. 5C,D). We also observed enhanced AP-1 activity following expression of human Aβ42arc in glia (41.1 ± 6.7 in controls vs 77.6 ± 14.3 following glial expression of Aβ42arc; p < 0.05). To determine whether Stat92E and AP-1, like Draper, influence Aβ levels in the Drosophila brain, we performed RNAi-mediated knockdown of Stat92E, Jra, and Kayak specifically in glia while coexpressing human Aβ42arc. Interestingly, glial depletion of Stat92E, Jra, or Kayak resulted in higher levels of Aβ in central brain (Fig. 5G,H). Finally, activation of the glial Draper receptor following acute axon injury also triggers transcriptional upregulation of the secreted matrix metalloproteinase Mmp1 (Purice et al., 2017). Notably, proper Mmp1 induction postinjury requires JNK/AP-1 and Stat92e signaling in responding glial cells (Purice et al., 2017). We discovered that neuronal expression of Aβ42arc also promotes a striking increase in Mmp1 levels by immunostaining in the central brain, and this upregulation is blocked in draper mutant flies (Fig. 5E,F). Collectively, these results suggest that glial Stat92E/AP-1 signaling cascades are engaged when exposed to Aβ42 peptides, perhaps in part driving the expression and release of Mmp1. Moreover, these transcriptional pathways influence Aβ levels in the CNS.
Figure 5.

Expression of human Aβ42arc promotes STAT92E and AP-1 signaling and upregulates Mmp1. A–D, Representative images (merged projection of 2 confocal sections) of single antennal lobes (7-d-old flies) showing GFP expressed in flies (expressing driver control or neuronal Aβ42arc) that carry the STAT92E transcriptional reporter 10XSTAT92E-GFP (A, B) or the JNK/AP-1 reporter TRE-GFP (C, D). Scale bar, 20 μm. Quantification of GFP written as mean ± SEM. Statistical data for A and B: N ≥ 16; t = 2.739; df = 40; *p < 0.05; unpaired t test. Statistical data for C and D: N ≥ 16; t = 2.174; df = 30; *p < 0.05; unpaired t test. Genotypes: A and B: Control (elav-Gal4/+; 10xStat92E-dGFP/+), neuronal Aβ42arc (elav-Gal4/+; UAS-Aβ42arc/10xStat92E-dGFP); C, D, Control (elav-Gal4/+; TRE-GFP/+), neuronal Aβ42arc (elav-Gal4/+; UAS-Aβ42arc/ TRE-GFP). E, Representative single confocal images of Mmp1 immunostaining in the antennal lobe regions of 7-d-old control flies and flies expressing neuronal Aβ42arc in wild-type or draper-null background. Genotypes: Control (elav-Gal4/+), neuronal Aβ42arc (elav-Gal4/+; UAS-Aβ42arc/+), neuronal Aβ42arc, draper-null flies (elav-Gal4/+; UAS-Aβ42arc,draper Δ5/ draper Δ5). F, Quantification of Mmp1 fluorescence intensity in E; mean ± SEM, plotted. N ≥ 15; F(2,15) = 13.27; **p < 0.01; one-way ANOVA with Tukey's post hoc test. G, Representative confocal images of antennal lobe regions in brains immunostained for Aβ42. Flies (7-d-old) coexpressed Aβ42arc and the noted RNAi constructs in glial cells. Control (UAS-Aβ42arc/+; repo-Gal4/+), Jra RNAi (UAS-Aβ42arc/ JraRNAi; repo-Gal4/+), Kayak RNAi (UAS-Aβ42arc/ KayakRNAi; repo-Gal4/+), and Stat92E RNAi (UAS-Aβ42arc/ Stat92ERNAi; repo-Gal4/+). H, Quantification of Aβ42 fluorescence intensity in experiment shown in G; mean ± SEM plotted. N ≥ 18; F(3,76) = 4.097; *p < 0.05; one-way ANOVA with Tukey's post hoc test; a.u., arbitrary units. Scale bar, 20 μm. Dotted circles in A, E, and G outline antennal lobes (AL).
Aβ42 induces Draper-dependent activation of Atg8 and p62
Increased activation of protein destruction pathways [ubiquitin-proteasome system (UPS), autophagy, etc.] has been reported in AD animal models and human AD postmortem tissue (Hong et al., 2014; Chen et al., 2015). Interestingly, autophagy signaling is upregulated in the developing Drosophila salivary gland in a Draper-dependent manner (McPhee and Baehrecke, 2010; Lin et al., 2017), although a connection between glial Draper/MEGF10 and autophagy has never been explored in the adult brain. We assessed expression of two markers that are associated with increased protein degradation (including autophagy), Atg8, and p62, in the central brain following neuronal expression of Aβ42arc. We observed little Atg8 or p62 staining in the central brain region of control animals, but detected a significant increase of both markers following neuronal expression of Aβ42arc (Fig. 6A–D). High-magnification images reveal that many Atg8/p62 puncta were either colocalized or juxtaposed to Aβ staining (Fig. 6A,C). Intriguingly, Atg8 and p62 upregulation was significantly reduced in draper mutants expressing neuronal Aβ42arc (Fig. 6A–D), suggesting that neuronal Aβ42 alters the activity of protein degradation pathways in the adult brain, with a significant portion being Draper-dependent. Given our current analysis, it is not clear which puncta reside inside neurons versus glia. However, we favor the model that Aβ42 peptides released from neurons are engulfed by glia in a Draper-dependent manner and shuttled through protein destruction pathways (UPS, autophagy, etc.) that include Atg8- and/or p62-positive compartments to promote destruction of these notoriously toxic protein fragments.
Figure 6.

Human Aβ42 expression induces Draper-dependent activation of Atg8 and p62 in the adult fly brain. A, Representative confocal images of brains immunostained for Atg8 (green) and Aβ42 (magenta). Area of high-magnification image is indicated with dotted white box. Circles outline antennal lobes (AL). Control (elav-Gal4/+), neuronal Aβ42arc (elav-Gal4/UAS-Aβ42arc), and neuronal Aβ42arc; draper (elav-Gal4/+; UAS-Aβ42arc,draperΔ5/ draperΔ5). B, Quantification of Atg8 fluorescence in A. Total Atg8 fluorescence intensity in the cortical regions adjacent to the antennal lobe neuropil was measured; mean ± SEM, plotted. N ≥ 15; F(2,45) = 44.97; ***p < 0.001, ****p < 0.0001; one-way ANOVA with Tukey's post hoc test; a.u., arbitrary units. C, Representative images of brains immunostained for p62 (green) and Aβ42 (magenta). Regions corresponding to high-magnification images are indicated with dotted white box. Control (elav-Gal4/+), neuronal Aβ42arc (elav-Gal4/UAS-Aβ42arc), and neuronal Aβ42arc; draper (elav-Gal4/+; UAS-Aβ42arc,draperΔ5/ draperΔ5). D, Quantification of p62 fluorescence shown in C. Total p62 fluorescence was quantified from the cortical regions adjacent to the antennal lobes; mean ± SEM, plotted. N ≥ 13; F(2,39) = 44.70; ****p < 0.0001; n.s., not significant; one-way ANOVA with Tukey's post hoc test; a.u., arbitrary units. Scale bars, 20 μm. Atg8- and p62-positive structures range from 0.1 to 1.5 μm. Average neuronal cell body size in the central brain is 3.1 ± 0.4 μm. All confocal images are 24 μm projections.
Discussion
Defective glial function is strongly implicated in the progression of AD (Akiyama et al., 2000), although the precise mechanisms that underlie glial contributions to disease pathogenesis are still unclear. Microglia and astrocytes can engulf amyloid peptides, likely through an array of cell surface receptors that act in concert to recognize and internalize Aβ in its various forms (Gold and El Khoury, 2015; Eugenin et al., 2016; Ries and Sastre, 2016), and, notably, many studies have identified AD susceptibility loci that encode factors required for proper glial phagocytic function (Hollingworth et al., 2011; Golde et al., 2013; Hickman and El Khoury, 2014). Here, we show that the conserved glial engulfment receptor Draper/MEGF10 provides protection in an in vivo Drosophila model of AD. Expression of pathogenic human Aβ42arc induces activation of a Draper/AP-1/STAT92E pathway in glia, which is known to trigger expression of the secreted metalloproteinase Mmp1 and promote glial phagocytic activity in response to acute neural injury (MacDonald et al., 2013; Doherty et al., 2014). We also show that loss of Draper exacerbates Aβ42-induced phenotypes, including locomotor dysfunction and reduced longevity, while overexpression of glial Draper, or the homologous mammalian receptor MEGF10, partially rescues most of these phenotypes. Finally, our work also indicates that Aβ42 expression triggers protein destruction pathways, largely in a Draper-dependent manner. Together, our findings suggest a mechanism whereby glia rely on the Draper receptor to activate a canonical Draper/JNK/STAT92E signaling cascade, internalize amyloid peptides, and perhaps shuttle them through Atg8/p62 protein degradation pathways, which attenuates Aβ42-induced CNS dysfunction.
Glia can detect fibrillar forms of Aβ through various cell-surface pattern recognition receptors, such as SCARA1 (scavenger receptor-A1), CD36, and Toll-like receptors (Malm et al., 2015). Soluble Aβ can also be internalized by microglia through fluid phase pinocytosis (Mandrekar et al., 2009; Li et al., 2013). In Caenorhabditis elegans the homolog of the Draper receptor, CED-1, recognizes necrotic cells by cell surface exposure of phosphatidylserine (Li and Zhou, 2016). Similarly, during Drosophila embryogenesis, two candidate ligands (Pretaporter and DmCaBP1) are presented by apoptotic cells and are likely bound by glial Draper to promote engulfment of dying neurons (Kuraishi et al., 2009; Okada et al., 2012). Thus, although Draper seems to be a requisite factor for glial recruitment to damaged neurons in the adult fly brain, it can serve as a true engulfment recognition receptor. Forced expression of the MEGF10 receptor in HeLa cells enhances phagocytosis of FITC-conjugated Aβ42 (Singh et al., 2010), raising the intriguing possibility that this family of receptors may contribute to recognition of extracellular Aβ. The extracellular domain (ECD) of Draper closely resembles the ECDs of the related mammalian receptors MEGF10, MEGF11, and Jedi; all contain multiple (>10) epidermal growth factor-like (EGF-like) domains (Wu et al., 2009). Notably, low density lipoprotein receptor-related receptor 1 (LRP1), a large endocytic receptor expressed by various cell types including glial cells, neurons, and vascular cells, binds directly to Aβ to promote uptake (Kanekiyo and Bu, 2014). Like Draper and its mammalian homologs, LRP1 possesses multiple EGF-like repeats in the ECD. It remains to be determined whether Draper and the related receptors can also directly bind Aβ, or cooperatively through coreceptors or additional ligands, to facilitate glial engulfment of neurotoxic aggregates or soluble peptides.
A recent study revealed that in the developing mouse brain, the MEGF10 receptor recognizes apoptotic neurons that have become decorated with the secreted complement factor C1q, allowing astrocytes to phagocytically clear them from the CNS (Iram et al., 2016). Aβ can similarly be opsonized by secreted complement factors (Wyss-Coray and Rogers, 2012), and indeed the complement pathway is gaining recognition as a key player in AD pathogenesis (Wyss-Coray and Rogers, 2012; Hong et al., 2016). Together, these observations raise the intriguing possibility that complement may also direct glial recognition of amyloid via the MEGF10/Jedi family of engulfment receptors. Finally, Pearce et al. (2015) have recently shown that Drosophila glia internalize mutant human Huntingtin aggregates in a Draper-dependent manner in the adult brain, which implicates Draper as potential broad player in the transfer of the pathogenic protein aggregates in various neurodegenerative disorders.
In addition to their shared capacity to internalize amyloid peptides, mammalian microglia and astrocytes both release cytokines in response to Aβ, which is likely a robust trigger for microglial recruitment to Aβ deposit sites (Malm et al., 2015; Guillot-Sestier and Town, 2013). In the adult Drosophila CNS, Draper, the downstream transcription factors AP-1 and STAT92E, and their transcriptional target Mmp1 are all upregulated in response to acute nerve injury and are also all required for ensheathing glia to properly infiltrate neuropil regions and, subsequently, clear degenerating axonal debris (Logan et al., 2012; MacDonald et al., 2013; Doherty et al., 2014; Purice et al., 2017). Here, we show that Draper expression, AP-1/Stat92e activity, and Mmp1 are also enhanced in Aβ42arc-expressing flies. Mmps are indeed well known for accommodating cell migration in various cell types and contexts and, notably, have been coupled to AD symptomology in other models. Mmp expression is induced upon exposure to soluble and fibrillar Aβ in microglia and astrocytes (Deb et al., 2003; Yan et al., 2006; Ito et al., 2007), and Mmp overexpression in AD model mice attenuates Aβ oligomer formation and rescues cognitive defects (Fragkouli et al., 2014; Kaminari et al., 2017). It is still unclear to what extent migratory responses are elicited in Drosophila glia in response to Aβ and how Mmp1 specifically contributes to Aβ processing and glial morphogenic responses in the adult fly brain. Future experiments that couple in vivo live imaging and single glial cell labeled clones will hopefully reveal how the Draper/Mmp1 pathway governs glial recruitment to sites of Aβ expression and glial recognition/internalization/degradation of Aβ peptides.
Enhanced autophagy is one viable mechanism by which both neurons and glia manage excessive amyloid peptides and other noxious proteins (Ling et al., 2009; Piras et al., 2016). Moreover, dysfunctional autophagy has indeed been implicated as a contributing factor to AD (Boland et al., 2008). Here, we show that human Aβ42 peptides enhance expression of Atg8 and p62 in the adult Drosophila brain in a Draper-dependent manner, although the mechanistic significance of this finding is still unclear. Together, Atg8 and p62 promote ubiquitination of damaged cargo fated for destruction through autophagy (and other) cascades (Wurzer et al., 2015). Increased expression of Atg8 and p62 in AD model flies may indicate an accumulation of autophagosomes resulting from enhanced activation of glial autophagy. However, it is important to note that a build up of autophagosomes could instead reflect suppression of a downstream step in the pathway (e.g., phagosome maturation, lysosomal fusion or degradation; Mizushima et al., 2010). Finally, although Atg8 and p62 are often described as autophagy markers, these molecules can also be coupled to other degradation/destruction pathways (Galluzzi et al., 2017). More focused experiments, including genetic perturbation of autophagy regulators, will be essential to define precisely how autophagic flux and other protein degradation pathways are altered in glia in response to Aβ42 overexpression and how Draper informs these events.
AD is marked by age-dependent progressive accumulation of extracellular aggregates in the brain and, notably, aged glia display reduced phagocytic function (Floden and Combs, 2011; Malm et al., 2015). Subtle deterioration of glial autophagy-lysosomal destruction pathways occurs normally with age and may enhance the aged brain's susceptibility to AD and related neurodegenerative disorders (Gorojod et al., 2015; Nikoletopoulou et al., 2015). Our laboratory has recently shown that glial Draper levels decrease with age in adult Drosophila and that aged ensheathing glia are slow to phagocytically clear degenerating axons in vivo (Purice et al., 2016). Based on the high functional conservation between Draper and the MEGF10 family of receptors in glial engulfment responses (MacDonald et al., 2006; Wu et al., 2009; Chung et al., 2013) and their coupling to downstream signaling pathways (Osada et al., 2009; Wu et al., 2009; Linnartz et al., 2010), we anticipate that our work will provide new molecular insight into glial contributions to AD progression and potentially provide a new cohort of molecular targets for therapeutic intervention.
Footnotes
This work was supported by the Oregon Partnership for Alzheimer's Research Awards (A.R.), National Institutes of Health Grants R01 NS079387 (M.A.L.), R21 AG056747 (M.A.L.), and R21 NS084112 (M.A.L. and S.D.S.); the Medical Research Foundation of Oregon (S.D.S. and M.A.L.); the Fred W. Fields Foundation (S.D.S. and M.A.L.); and the Ken and Ginger Harrison Term Professor Award (M.A.L.).We to thank Tor Erik Rusten (University of Oslo, Norway), Estee Kurant (Israel Institute of Technology), Doris Kretzschmar (OHSU, Portland, OR), Bruce Carter (Vanderbilt University, Nashville, TN), the Bloomington Drosophila Stock Center at Indiana University (Bloomington, IN), the Vienna Drosophila Resource Center (Vienna, Austria), and the Developmental Studies Hybridoma Bank (University of Iowa) for flies, plasmids, and antibodies; Marlène Cassar and Elizabeth Sunderhaus with assistance in paraffin sectioning and vacuole analysis; and Philip Copenhaver and Doris Kretzschmar for helpful comments on the paper.
The authors declare no competing financial interests.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421. 10.1016/S0197-4580(00)00124-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara FM, Junaid A, Clough RR, Liang B (1999) TGF-β1, regulation of alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Brain Res Mol Brain Res 71:42–49. 10.1016/S0169-328X(99)00158-8 [DOI] [PubMed] [Google Scholar]
- Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, Baeg GH (2007) GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns 7:323–331. 10.1016/j.modgep.2006.08.003 [DOI] [PubMed] [Google Scholar]
- Bahadorani S, Bahadorani P, Phillips JP, Hilliker AJ (2008) The effects of vitamin supplementation on Drosophila life span under normoxia and under oxidative stress. J Gerontol A Biol Sci Med Sci 63:35–42. 10.1093/gerona/63.1.35 [DOI] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, Kertser A, David E, Amit I, Schwartz M (2016) PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat Med 22:135–137. 10.1038/nm.4022 [DOI] [PubMed] [Google Scholar]
- Bayer TA, Wirths O (2008) Review on the APP/PS1KI mouse model: intraneuronal Aβ accumulation triggers axonopathy, neuron loss and working memory impairment. Genes Brain Behav 7:6–11. 10.1111/j.1601-183X.2007.00372.x [DOI] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 28:6926–6937. 10.1523/jneurosci.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A; Alzheimer Disease Neuroimaging Initiative, Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL (2013) CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci 16:848–850. 10.1038/nn.3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton T, Liang B, Dibrov A, Amara F (2002) Transforming growth factor-beta-induced transcription of the Alzheimer beta-amyloid precursor protein gene involves interaction between the CTCF-complex and Smads. Biochem Biophys Res Commun 295:713–723. 10.1016/S0006-291X(02)00725-8 [DOI] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P (2011) The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet 20:2144–2160. 10.1093/hmg/ddr100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Kondo K, Motoki K, Homma H, Okazawa H (2015) Fasting activates macroautophagy in neurons of Alzheimer's disease mouse model but is insufficient to degrade amyloid-beta. Sci Rep 5:12115. 10.1038/srep12115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. 10.1038/nature12776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, Lomas DA (2005) Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience 132:123–135. 10.1016/j.neuroscience.2004.12.025 [DOI] [PubMed] [Google Scholar]
- Deb S, Wenjun Zhang J, Gottschall PE (2003) β-Amyloid induces the production of active, matrix-degrading proteases in cultured rat astrocytes. Brain Res 970:205–213. 10.1016/S0006-8993(03)02344-8 [DOI] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML (2012) A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer's disease mice. Neuron 76:908–920. 10.1016/j.neuron.2012.10.029 [DOI] [PubMed] [Google Scholar]
- Doherty J, Logan MA, Taşdemir OE, Freeman MR (2009) Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci 29:4768–4781. 10.1523/JNEUROSCI.5951-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Sheehan AE, Bradshaw R, Fox AN, Lu TY, Freeman MR (2014) PI3K signaling and Stat92E converge to modulate glial responsiveness to axonal injury. PLoS Biol 12:e1001985. 10.1371/journal.pbio.1001985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray JI, Timmons AK, Klein AP, Pritchett TL, Welch E, Meehan TL, Li C, McCall K (2012) Draper acts through the JNK pathway to control synchronous engulfment of dying germline cells by follicular epithelial cells. Development 139:4029–4039. 10.1242/dev.082776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenín J, Vecchiola A, Murgas P, Arroyo P, Cornejo F, von Bernhardi R (2016) Expression pattern of scavenger receptors and amyloid-β phagocytosis of astrocytes and microglia in culture are modified by acidosis: implications for Alzheimer's disease. J Alzheimers Dis 53:857–873. 10.3233/JAD-160083 [DOI] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M (2004) A model for studying Alzheimer's Aβ42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci 26:365–375. 10.1016/j.mcn.2004.03.001 [DOI] [PubMed] [Google Scholar]
- Floden AM, Combs CK (2011) Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J Alzheimers Dis 25:279–293. 10.3233/JAD-2011-101014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkouli A, Tsilibary EC, Tzinia AK (2014) Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer's 5xFAD mice. Neurobiol Dis 70:179–189. 10.1016/j.nbd.2014.06.021 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, et al. (2017) Molecular definitions of autophagy and related processes. The EMBO J 36:1811–1836. 10.15252/embj.201796697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold M, El Khoury J (2015) β-Amyloid, microglia, and the inflammasome in Alzheimer's disease. Semin Immunopathol 37:607–611. 10.1007/s00281-015-0518-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Streit WJ, Chakrabarty P (2013) Alzheimer's disease risk alleles in TREM2 illuminate innate immunity in Alzheimer's disease. Alzheimers Res Ther 5:24. 10.1186/alzrt178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorojod RM, Alaimo A, Porte Alcon S, Pomilio C, Saravia F, Kotler ML (2015) The autophagic- lysosomal pathway determines the fate of glial cells under manganese- induced oxidative stress conditions. Free Radic Biol Med 87:237–251. 10.1016/j.freeradbiomed.2015.06.034 [DOI] [PubMed] [Google Scholar]
- Götz J, Ittner LM (2008) Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci 9:532–544. 10.1038/nrn2420 [DOI] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE (2013) Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78:631–643. 10.1016/j.neuron.2013.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, et al. (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117–127. 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Town T (2013) Innate immunity in Alzheimer's disease: a complex affair. CNS Neurol Disord Drug Targets 12:593–607. 10.2174/1871527311312050008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon Y, Trompier D, Ma Z, Venegas V, Pophillat M, Mignotte V, Zhou Z, Chimini G (2006) Cooperation between engulfment receptors: the case of ABCA1 and MEGF10. PLoS One 1:e120. 10.1371/journal.pone.0000120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Mercer M, Roth LM, Symmonds K, Maloney J, Wilson N, Freeman MJ, Morgan D, Gordon MN (2007) Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol 2:222–231. 10.1007/s11481-007-9069-z [DOI] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J (2014) TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol 88:495–498. 10.1016/j.bcp.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, et al. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 43:429–435. 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM (2011) Alzheimer's disease: the challenge of the second century. Sci Transl Med 3:77sr1. 10.1126/scitranslmed.3002369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Huang HC, Jiang ZF (2014) Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer's disease. Neurol Res 36:276–282. 10.1179/1743132813Y.0000000288 [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y (2004) Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proc Natl Acad Sci U S A 101:6623–6628. 10.1073/pnas.0400895101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y (2008) Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One 3:e1703. 10.1371/journal.pone.0001703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iram T, Ramirez-Ortiz Z, Byrne MH, Coleman UA, Kingery ND, Means TK, Frenkel D, El Khoury J (2016) Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci 36:5185–5192. 10.1523/JNEUROSCI.3850-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Kimura K, Haneda M, Ishida Y, Sawada M, Isobe K (2007) Induction of matrix metallo-proteinases (MMP3, MMP12 and MMP13) expression in the microglia by amyloid beta stimulation via the PI3K/Akt pathway. Exp Gerontol 42:532–537. 10.1016/j.exger.2006.11.012 [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, et al. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368:107–116. 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2015) Neurodegeneration: amyloid-β pathology induced in humans. Nature 525:193–194. 10.1038/525193a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminari A, Giannakas N, Tzinia A, Tsilibary EC (2017) Overexpression of matrix metalloproteinase-9 (MMP-9) rescues insulin-mediated impairment in the 5XFAD model of Alzheimer's disease. Sci Rep 7:683. 10.1038/s41598-017-00794-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Bu G (2014) The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front Aging Neurosci 6:93. 10.3389/fnagi.2014.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraishi T, Nakagawa Y, Nagaosa K, Hashimoto Y, Ishimoto T, Moki T, Fujita Y, Nakayama H, Dohmae N, Shiratsuchi A, Yamamoto N, Ueda K, Yamaguchi M, Awasaki T, Nakanishi Y (2009) Pretaporter, a Drosophila protein serving as a ligand for Draper in the phagocytosis of apoptotic cells. The EMBO J 28:3868–3878. 10.1038/emboj.2009.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ (2002) New class of inhibitors of amyloid-beta fibril formation: implications for the mechanism of pathogenesis in Alzheimer's disease. J Biol Chem 277:42881–42890. 10.1074/jbc.M206593200 [DOI] [PubMed] [Google Scholar]
- Lesné S, Docagne F, Gabriel C, Liot G, Lahiri DK, Buée L, Plawinski L, Delacourte A, MacKenzie ET, Buisson A, Vivien D (2003) Transforming growth factor-â1 potentiates amyloid-â generation in astrocytes and in transgenic mice. J Biol Chem 278:18408–18418. 10.1074/jbc.M300819200 [DOI] [PubMed] [Google Scholar]
- Li HQ, Chen C, Dou Y, Wu HJ, Liu YJ, Lou HF, Zhang JM, Li XM, Wang H, Duan S (2013) P2Y4 receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol Cell Biol 33:4282–4293. 10.1128/MCB.00544-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhou Z (2016) How are necrotic cells recognized by their predators? Worm 5:e1120400. 10.1080/21624054.2015.1120400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Rodrigues FSLM, Kary C, Contet A, Logan M, Baxter RHG, Wood W, Baehrecke EH (2017) Complement-related regulates autophagy in neighboring cells. Cell. 170:158–171.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM (2009) Aβ42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One 4:e4201. 10.1371/journal.pone.0004201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz B, Wang Y, Neumann H (2010) Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int J Alzheimers Dis 2010:587463. 10.4061/2010/587463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan MA, Hackett R, Doherty J, Sheehan A, Speese SD, Freeman MR (2012) Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat Neurosci 15:722–730. 10.1038/nn.3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR (2006) The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50:869–881. 10.1016/j.neuron.2006.04.028 [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Doherty J, Hackett R, Freeman MR (2013) The c-Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ 20:1140–1148. 10.1038/cdd.2013.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, Estus S (2013) CD33 Alzheimer's risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 33:13320–13325. 10.1523/JNEUROSCI.1224-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Jay TR, Landreth GE (2015) The evolving biology of microglia in Alzheimer's disease. Neurotherapeutics 12:81–93. 10.1007/s13311-014-0316-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE (2009) Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci 29:4252–4262. 10.1523/JNEUROSCI.5572-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcora MS, Fernández-Gamba AC, Avendaño LA, Rotondaro C, Podhajcer OL, Vidal R, Morelli L, Ceriani MF, Castaño EM (2014) Amyloid peptides ABri and ADan show differential neurotoxicity in transgenic Drosophila models of familial British and Danish dementia. Mol Neurodegener 9:5. 10.1186/1750-1326-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BL, Schrader-Fischer G, Busciglio J, Duke M, Paganetti P, Yankner BA (1995) Intracellular accumulation of beta-amyloid in cells expressing the Swedish mutant amyloid precursor protein. J Biol Chem 270:26727–26730. 10.1074/jbc.270.45.26727 [DOI] [PubMed] [Google Scholar]
- McPhee CK, Baehrecke EH (2010) The engulfment receptor Draper is required for autophagy during cell death. Autophagy 6:1192–1193. 10.4161/auto.6.8.13474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Barua N, Kehoe PG, Gill S, Love S (2011) Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol 70:944–959. 10.1097/NEN.0b013e3182345e46 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326. 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, Duyckaerts C, Frosch MP, Haroutunian V, Hof PR, Hulette CM, Hyman BT, Iwatsubo T, Jellinger KA, Jicha GA, Kövari E, et al. (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. 10.1097/NEN.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukomm LJ, Burdett TC, Gonzalez MA, Züchner S, Freeman MR (2014) Rapid in vivo forward genetic approach for identifying axon death genes in Drosophila. Proc Natl Acad Sci U S A 111:9965–99670. 10.1073/pnas.1406230111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V, Papandreou ME, Tavernarakis N (2015) Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ 22:398–407. 10.1038/cdd.2014.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein TJ, Spitzer P, Klafki HW, Linning P, Neff F, Knölker HJ, Lewczuk P, Wiltfang J, Kornhuber J, Maler JM (2015) Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol Dis 73:24–35. 10.1016/j.nbd.2014.08.031 [DOI] [PubMed] [Google Scholar]
- Okada R, Nagaosa K, Kuraishi T, Nakayama H, Yamamoto N, Nakagawa Y, Dohmae N, Shiratsuchi A, Nakanishi Y (2012) Apoptosis-dependent externalization and involvement in apoptotic cell clearance of DmCaBP1, an endoplasmic reticulum protein of Drosophila. J Biol Chem 287:3138–3146. 10.1074/jbc.M111.277921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada Y, Sunatani T, Kim IS, Nakanishi Y, Shiratsuchi A (2009) Signaling pathway involving GULP, MAPK and Rac1 for SR-BI-induced phagocytosis of apoptotic cells. J Biochem 145:387–394. 10.1093/jb/mvn176 [DOI] [PubMed] [Google Scholar]
- Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR (2015) Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun 6:6768. 10.1038/ncomms7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping Y, Hahm ET, Waro G, Song Q, Vo-Ba DA, Licursi A, Bao H, Ganoe L, Finch K, Tsunoda S (2015) Linking Aβ42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer's model. PLoS Genet 11: e1005025. 10.1371/journal.pgen.1005025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A (2016) Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4:22. 10.1186/s40478-016-0292-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomilio C, Pavia P, Gorojod RM, Vinuesa A, Alaimo A, Galvan V, Kotler ML, Beauquis J, Saravia F (2016) Glial alterations from early to late stages in a model of Alzheimer's disease: evidence of autophagy involvement in Aβ internalization. Hippocampus 26:194–210. 10.1002/hipo.22503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purice MD, Speese SD, Logan MA (2016) Delayed glial clearance of degenerating axons in aged Drosophila is due to reduced PI3K/Draper activity. Nat Commun 7:12871. 10.1038/ncomms12871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purice MD, Ray A, Münzel EJ, Pope BJ, Park DJ, Speese SD, Logan MA (2017) A novel Drosophila injury model reveals severed axons are cleared through a Draper/MMP-1 signaling cascade. eLife 6:e23611. 10.7554/eLife.23611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries M, Sastre M (2016) Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci 8:160. 10.3389/fnagi.2016.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S. (2009) Regulation of innate immune responses in brain. Nat Rev Immunol 9:429–439. 10.1038/nri2565 [DOI] [PubMed] [Google Scholar]
- Rosenblum WI. (2014) Why Alzheimer trials fail: removing soluble oligomeric beta amyloid is essential, inconsistent, and difficult. Neurobiol Aging 35:969–974. 10.1016/j.neurobiolaging.2013.10.085 [DOI] [PubMed] [Google Scholar]
- Scheib JL, Sullivan CS, Carter BD (2012) Jedi-1 and MEGF10 signal engulfment of apoptotic neurons through the tyrosine kinase Syk. J Neurosci 32:13022–13031. 10.1523/JNEUROSCI.6350-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. (2012) Preventing Alzheimer's disease. Science 337:1488–1492. 10.1126/science.1228541 [DOI] [PubMed] [Google Scholar]
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S (2009) Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30:588–598. 10.1016/j.immuni.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shklover J, Mishnaevski K, Levy-Adam F, Kurant E (2015) JNK pathway activation is able to synchronize neuronal death and glial phagocytosis in Drosophila. Cell Death Dis 6:e1649. 10.1038/cddis.2015.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TD, Park SY, Bae JS, Yun Y, Bae YC, Park RW, Kim IS (2010) MEGF10 functions as a receptor for the uptake of amyloid-β. FEBS Lett 584:3936–3942. 10.1016/j.febslet.2010.08.050 [DOI] [PubMed] [Google Scholar]
- Sunderhaus ER, Kretzschmar D (2016) Mass histology to quantify neurodegeneration in Drosophila. J Vis Exp 118:e54809. 10.3791/54809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir-Yilmaz OE, Freeman MR (2014) Astrocytes engage unique molecular programs to engulf pruned neuronal debris from distinct subsets of neurons. Genes Dev 28:20–33. 10.1101/gad.229518.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping KW, van Oosten-Hawle P, Hewitt EW, Radford SE (2015) Amyloid fibres: inert end-stage aggregates or key players in disease? Trends Biochem Sci 40:719–727. 10.1016/j.tibs.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CM, Devries S, Camboni M, Glass M, Martin PT (2010) Immunization with the SDPM1 peptide lowers amyloid plaque burden and improves cognitive function in the APPswePSEN1(A246E) transgenic mouse model of Alzheimer's disease. Neurobiol Dis 39:409–422. 10.1016/j.nbd.2010.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzell J, Kretzschmar D (2010) Alzheimer's disease and tauopathy studies in flies and worms. Neurobiol Dis 40:21–28. 10.1016/j.nbd.2010.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wes PD, Sayed FA, Bard F, Gan L (2016) Targeting microglia for the treatment of Alzheimer's disease. GLIA 64:1710–1732. 10.1002/glia.22988 [DOI] [PubMed] [Google Scholar]
- Woodruff-Pak DS. (2008) Animal models of Alzheimer's disease: therapeutic implications. J Alzheimers Dis 15:507–521. 10.3233/JAD-2008-15401 [DOI] [PubMed] [Google Scholar]
- Wu HH, Bellmunt E, Scheib JL, Venegas V, Burkert C, Reichardt LF, Zhou Z, Fariñas I, Carter BD (2009) Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci 12:1534–1541. 10.1038/nn.2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S (2015) Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. eLife 4:e08941. 10.7554/eLife.08941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F, Petitjean AM, Dru P, Gay P, Contamine D (1995) Localization of domains within the Drosophila ref(2)P protein involved in the intracellular control of sigma rhabdovirus multiplication. J Virol 69:4463–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease: a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2:a006346. 10.1101/cshperspect.a006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457. 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, Xiao Q, Hsu FF, Turk JW, Xu J, Hsu CY, Holtzman DM, Lee JM (2006) Matrix metalloproteinase-9 degrades amyloid-β fibrils in vitro and compact plaques in situ. J Biol Chem 281:24566–24574. 10.1074/jbc.M602440200 [DOI] [PubMed] [Google Scholar]
- Ziegenfuss JS, Biswas R, Avery MA, Hong K, Sheehan AE, Yeung YG, Stanley ER, Freeman MR (2008) Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signaling. Nature 453:935–939. 10.1038/nature06901 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Relevant transgenic strains and data relevant to this paper are available from the corresponding author.