

Abstract
Exposure to cocaine-associated contextual cues contributes significantly to relapse. Extinction of these contextual associations, which involves a new form of learning, reduces cocaine-seeking behavior; however, the molecular mechanisms underlying this process remain largely unknown. We report that extinction, but not acquisition, of cocaine conditioned place preference (CPP) in male mice increased Cav1.2 L-type Ca2+ channel mRNA and protein in postsynaptic density (PSD) fractions of the hippocampus, a brain region involved in drug–context associations. Moreover, viral-mediated deletion of Cav1.2 in the dorsal hippocampus attenuated extinction of cocaine CPP. Molecular studies examining downstream Cav1.2 targets revealed that extinction recruited calcium/calmodulin (Ca2+/CaMK)-dependent protein kinase II (CaMKII) to the hippocampal PSD. This occurred in parallel with an increase in phosphorylation of the AMPA GluA1 receptor subunit at serine 831 (S831), a CaMKII site, along with an increase in total PSD GluA1. The necessity of S831 GluA1 was further demonstrated by the lack of extinction in S831A GluA1 phosphomutant mice. Of note hippocampal GluA1 levels remained unaltered at the PSD, but were reduced near the PSD and at perisynaptic sites of dendritic spines in extinction-resistant S831A mutant mice. Finally, conditional knock-out of Cav1.2 in dopamine D1 receptor (D1R)-expressing cells resulted in attenuation of cocaine CPP extinction and lack of extinction-dependent changes in hippocampal PSD CaMKII expression and S831 GluA1 phosphorylation. In summary, we demonstrate an essential role for the hippocampal Cav1.2/CaMKII/S831 GluA1 pathway in cocaine CPP extinction, with data supporting contribution of hippocampal D1R-expressing cells in this process. These findings demonstrate a novel role for Cav1.2 channels in extinction of contextual cocaine-associated memories.
SIGNIFICANCE STATEMENT Continued drug-seeking behavior, a defining characteristic of cocaine addiction, can be precipitated by contextual cues, yet the molecular mechanisms required for extinction of these context-specific memories remain poorly understood. Here, we have uncovered a novel and selective role of the Cav1.2 L-type Ca2+ channel and its downstream signaling pathway in the hippocampus that mediate extinction of cocaine conditioned place preference (CPP). We additionally provide evidence that supports a role of Cav1.2 within dopamine D1 receptor-expressing cells of the hippocampus for extinction of cocaine CPP. Therefore, these findings reveal a previously unknown role of Cav1.2 channels within the hippocampus and in D1 receptor-expressing cells in extinction of cocaine-associated memories, providing a framework for further exploration of mechanisms underlying extinction of cocaine-seeking behavior.
Keywords: CaMKII, Cav1.2, cocaine, dopamine D1 receptor, extinction, GluA1
Introduction
Addiction to drugs of abuse such as cocaine results in part from exceptionally strong, maladaptive associations formed between contextual cues and the rewarding properties of the drug (Kelley, 2004; Koob and Volkow, 2010; Perry et al., 2014). These drug-associated contextual memories are long lasting, highly resistant to extinction, and contribute to the high rate of relapse in cocaine-dependent individuals, as well as the reinstatement of cocaine-seeking behavior in rodents (Marchant et al., 2015; Venniro et al., 2016). Therefore, identifying mechanisms that can facilitate extinction and diminish relapse may aid in treating addiction. Human and rodent studies implicate the hippocampus, a critical substrate of learning and memory, as a key brain region for the development and maintenance of cocaine–context associations (Koob and Volkow, 2010; Kutlu and Gould, 2016). Studies modeling extinction training in rodents have revealed that extinction recruits a new learning process (Bouton, 2004; Vianna et al., 2004; Lattal et al., 2006) at the level of synaptic and molecular changes within brain reward regions (Nic Dhonnchadha et al., 2013) including the hippocampus, which is particularly involved in the extinction of contextual aspects of drug-associated memories (Chambers and Self, 2002; Szalay et al., 2013). However, the molecular mechanisms within the hippocampus that mediate contextual extinction remain largely unknown.
L-type Ca2+ channels (LTCCs) are well known for their role in hippocampal-dependent learning and memory (Moosmang et al., 2005; Krug et al., 2014; Temme et al., 2016; Kabir et al., 2017a). In particular, LTCCs have been implicated in mediating cocaine-associated behaviors (Licata and Pierce, 2003; Chartoff et al., 2006; Anderson et al., 2008; Giordano et al., 2010; Schierberl et al., 2011; Berger and Bartsch, 2014; Martinez-Rivera et al., 2017) and, recently, extinction of cocaine conditioned place preference (CPP) (Degoulet et al., 2016).
LTCCs are key mediators of activity-dependent molecular processes with activation and synaptic localization of the calcium/calmodulin (Ca2+/CaMK)-dependent protein kinase II (CaMKII) subunits α and β representing its central downstream targets (Rose et al., 2009; Ma et al., 2012; Murphy et al., 2014; Striessnig et al., 2014). CaMKIIα/β are critically important for hippocampal-dependent contextual learning (Shonesy et al., 2014) and contribute to the molecular mechanisms of cocaine's action (Robison, 2014). CaMKIIα promotes glutamatergic signaling via phosphorylation at serine 831 (S831) on the GluA1 subunit of AMPA receptors (Mammen et al., 1997), a posttranslational modification that modulates channel conductance (Derkach et al., 1999; Kristensen et al., 2011). AMPA receptors play a central role in mediating cocaine-induced synaptic and behavioral plasticity (Pierce and Wolf, 2013; Wolf, 2016), including extinction of cocaine-associated memories, within brain reward regions including the nucleus accumbens (NAc) and prefrontal cortex (PFC) (Self et al., 2004; Peters et al., 2009; Rothwell et al., 2011; Nic Dhonnchadha et al., 2013); however, the role of hippocampal AMPA receptors in the extinction of cocaine contextual learning remains unknown.
One mechanism that regulates LTCC channel activity and signaling involves the activation of dopamine D1 receptors (D1Rs) (Surmeier et al., 1995; Liu and Graybiel, 1996; Giordano et al., 2010; Navakkode et al., 2012). Growing evidence indicates that D1Rs contribute to extinction of learned behaviors (Abraham et al., 2014; Singewald et al., 2015), including extinction of cocaine CPP (Brenhouse et al., 2010; Fricks-Gleason et al., 2012; Brenhouse et al., 2015; Abraham et al., 2016).
Together, these findings suggest that the extinction of cocaine contextual memories may involve activation of a Cav1.2-regulated signaling pathway within hippocampal D1R-expressing cells. To test this hypothesis, a combination of genetic, molecular, and pharmacological approaches were used to assess the role and cell-type specificity of Cav1.2 channels in CaMKII and GluA1 levels in hippocampal PSD fractions after cocaine CPP extinction. We provide evidence for a selective role of a Cav1.2 channel-mediated mechanism in the hippocampus in extinction of cocaine CPP, with data supporting that this occurs via Cav1.2 signaling in D1R-expressing cells of the hippocampus.
Materials and Methods
Animals
Adult (>postnatal day 60) male wild-type (WT) mice (C57BL/6J; The Jackson Laboratory) as well as transgenic and mutant mice on the C57BL/6J background were used for all experiments. Viral-vector-mediated deletion of Cav1.2 was achieved using homozygous cacna1c floxed (Cav1.2fl/fl) mice (Moosmang et al., 2005). Mice expressing a serine to alanine substitution at position 831 of GluA1 (S831A mutants) were generated as described previously (Lee et al., 2010) and bred in the Weill Cornell Medicine facilities. Mice with conditional knock-out of Cav1.2 in CaMKII-expressing cells (CaMK2cre, Cav1.2fl/fl) and their WT littermates (CaMK2cre, Cav1.2+/+) were generated by crossing Cav1.2fl/fl mice with mice expressing Cre recombinase under the transcriptional control of the CaMK2a promoter (Camk2a-Cre T29-1; The Jackson Laboratory). Mice with conditional knock-out of Cav1.2 in D1R-expressing cells (D1cre, Cav1.2fl/fl) and their WT littermates (D1cre, Cav1.2+/+) were generated by crossing Cav1.2fl/fl mice with mice expressing Cre recombinase under the transcriptional control of the D1R promoter [D1cre, Tg(Drd1-cre)EY262Gsat/Mmucd, GENSAT]. For each mouse line, male and female Cre+/−, Cav1.2fl/+ mice were bred to generate Cre+/−, Cav1.2+/+ and Cre+/−, Cav1.2fl/fl experimental mice (herein referred to as CaMK2cre, Cav1.2+/+, CaMK2cre, Cav1.2fl/fl and D1cre, Cav1.2+/+, D1cre, Cav1.2fl/fl). Mutant mice were indistinguishable from WT mice in weight, development, and general health. Mice were maintained under climate-controlled conditions on a 12 h light/dark cycle with ad libitum access to food and water. All procedures were approved by the Weill Cornell Medicine Institutional Animal Care and Use Committee and conducted in accordance with the 2011 Eighth Edition of the National Institutes of Health's Guidelines for the Care and Use of Laboratory Animals.
Drugs
Cocaine hydrochloride (Sigma-Aldrich) was dissolved in 0.9% saline at a concentration of 1 mg/ml and injected intraperitoneally at 0.01 ml/g body weight for a final dose of 10 mg/kg. Ca2+/calmodulin-dependent protein kinase II inhibitor N-[2-[N-(4-chlorocinnamyl)-N-methylaminomethyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide phosphate salt (KN93; Sigma-Aldrich) was dissolved in 0.9% saline containing 1.5% DMSO and 1.5% Tween 80 at a concentration of 15 μg/μl and microinjected into the dorsal hippocampus at a volume of 0.2 μl/hemisphere.
Stereotaxic viral vector delivery
Bilateral brain-region-specific deletion of cacna1c was achieved using stereotaxic surgical procedures as described previously (Lee et al., 2012a). Adeno-associated viral vectors expressing either GFP (AAV-GFP) or GFP and Cre recombinase (AAV-Cre) were delivered into the respective target regions of cacna1c floxed mice using coordinates adopted from the Mouse Brain Atlas (Paxinos and Franklin, 2004). To target the dorsal hippocampus, either AAV2/2-GFP or AAV2/2-Cre was microinjected at a volume of 0.2 μl/hemisphere 1.4 mm posterior, 2.0 mm ventral, and ±1.1 mm lateral to bregma at a 10° angle. To target the ventral hippocampus, either AAV2/5-GFP or AAV2/5-Cre was microinjected at a volume of 0.5 μl/hemisphere 3.5 mm posterior, 4.2 mm ventral, and ±2.8 mm lateral to bregma at a 4° angle. For each microinjection site, the needle was left in place for an additional 5 min after the infusion to ensure complete delivery of the viral vector. Behavioral experiments began after a minimum of 3 weeks to allow for maximal gene deletion.
Guide cannulae implantation
For delivery of the CaMKII inhibitor KN93 into the dorsal hippocampus, bilateral guide cannulae were implanted as described previously (Schierberl et al., 2011). Briefly, chronic indwelling, intracranial stainless-steel guide cannulae (5 mm in length; CMA/Microdialysis, model CMA/7) were implanted stereotaxically in mice under anesthesia (ketamine, 100 mg/ml; xylazine, 20 mg/ml) over the dorsal hippocampus at coordinates 1.9 mm posterior, 0.8 mm ventral, and ±1.3 mm lateral to bregma (Paxinos and Franklin, 2004). Cannulae were secured to the skull with dental acrylic resin. Sham cannulae (33 gauge, 5 mm stainless-steel stylets) were cut flush with the 24 gauge cannulae, inserted until the time of drug microinfusions, and replaced after microinjections. After surgery, mice were allowed a minimum of 7 d recovery before beginning behavioral experiments. Microinfusions were delivered into the dorsal hippocampus using microinjectors (33 gauge) that extended 1 mm beyond the cannulae, connected by flexible polyethylene tubing to a microinjection system, and mounted with a 5 μl Hamilton syringe. KN93 (0.2 μl/hemisphere) was microinjected bilaterally into the dorsal hippocampus immediately before each extinction session. Accurate bilateral cannulae placement was confirmed in 50 μm paraformaldehyde-perfused sections by cresyl violet staining. Mice with inaccurate targeting on either side of the dorsal hippocampus were eliminated from the study.
CPP behavioral protocol
Cocaine CPP was performed as described previously (Tropea et al., 2008) using a biased protocol. Cocaine preference was assessed using a three-chamber place preference apparatus (Med Associates Inc.). The behavioral protocols implemented in this study are outlined in Figure 1A. During the first preconditioning testing session (baseline test, day 1), mice were initially placed in the central gray chamber for a 1 min acclimation period, followed by free access to all three chambers for 20 min. Time spent in each chamber was recorded. During the acquisition training period (days 2–4), in the morning session, each mouse was given a systemic injection of cocaine (10 mg/kg, i.p.) and confined for 20 min to the chamber that the mouse preferred less during the baseline test, after which it was returned to its home cage. Six hours later (afternoon session), each mouse was given a systemic injection of saline (0.01ml/g body weight) and confined to the opposite chamber for 20 min and then returned to its home cage. For experiments involving a saline control group, mice were injected with saline in both chambers during the morning and afternoon sessions. For the acquisition test session (day 5), mice were placed in the central chamber without drug treatment for a 1 min acclimation period before being allowed free access to all three chambers. Cocaine preference for all testing sessions was calculated as time spent in the cocaine-paired chamber minus time spent in the saline-paired chamber and is reported as a preference score (in seconds). Mice were defined as having acquired cocaine CPP when the average cocaine preference score of the cocaine-treated group was significantly higher than the preference score at baseline. To assess extinction (“CPP extinction” group in Fig. 1A), mice were allowed free access to all three chambers (beginning on day 6) without drug treatment, and cocaine preference was evaluated as above. This extinction protocol was repeated every 24 h until the average preference score of mice within the cocaine extinction control group was significantly lower than the preference score on the acquisition test day. To achieve consistent extinction across experiments and within 4 d of extinction training, a second separate investigator not previously exposed to the mice during cocaine pairing and acquisition conducted the extinction training sessions. For the “CPP no extinction” group (Fig. 1A), mice remained in their home cage for 4 d after acquisition testing. To test maintenance of cocaine CPP, mice were given free access to all three chambers of the cocaine CPP apparatus. For molecular studies with the “CPP no extinction” group, mice were killed after 4 d of home cage exposure without reexposure to the CPP chamber.
Figure 1.
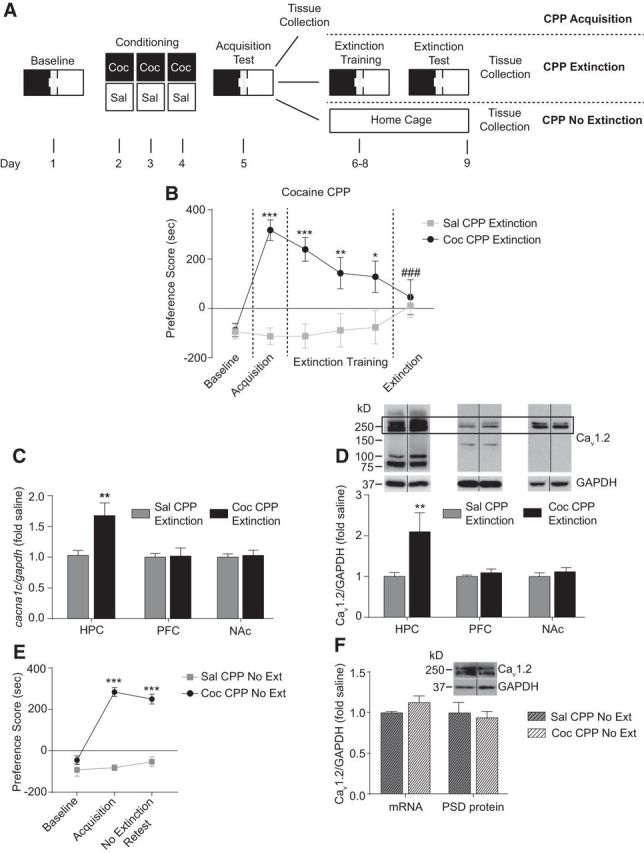
Extinction of cocaine CPP increases Cav1.2 mRNA and protein in the hippocampus. A, Experimental timeline of the CPP protocol indicating the “CPP acquisition,” “CPP extinction,” and “CPP no extinction” groups used for behavioral and molecular studies. B, Cocaine-treated WT mice demonstrate robust acquisition of cocaine CPP as shown by significantly higher preference scores on acquisition versus baseline testing. After extinction training, these mice exhibit significantly lower preference scores on the extinction test day compared with the acquisition test day, indicating extinction of cocaine CPP. Control mice treated with saline did not show any changes in preference score when tested after acquisition and extinction training. *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline test; ###p < 0.001 compared with acquisition test. C, Extinction of cocaine CPP increased Cav1.2 (cacna1c) mRNA significantly in the hippocampus, but not in the PFC or NAc, compared with saline control mice. **p < 0.01. D, Extinction of cocaine CPP increased Cav1.2 protein levels significantly in the PSD-enriched fraction from the hippocampus, but not the PFC or NAc compared with saline controls. For quantitation of Cav1.2 protein, the doublet surrounding the predicted full-length 250 kDa protein was used. **p < 0.01. E, Cocaine-treated mice demonstrate robust CPP acquisition and sustained preference score in the absence of extinction training when retested 4 d later compared with baseline. Control mice treated with saline did not show any change in preference scores after testing on acquisition and a retest 4 d later. ***p < 0.001 compared with baseline test. F, Cav1.2 mRNA and PSD protein were unaltered in mice that underwent cocaine CPP acquisition after 4 d of home cage exposure without extinction training. Data are presented as mean ± SEM.
Fluorescence immunohistochemistry
Fluorescence immunohistochemistry was performed to confirm injection placement of the AAV as described previously (Lee et al., 2012a). After the final cocaine CPP extinction session, mice were anesthetized with an injection of euthasol (150 mg/kg, i.p.) before transcardial perfusion with 4% paraformaldehyde (PFA). Brains were dissected and postfixed overnight in 4% PFA, followed by cryoprotection in 30% sucrose in 0.1 m phosphate buffer (PB) at 4°C for 72 h. Forty-micrometer sections spanning the injection site region were obtained using a sliding microtome. Sections were incubated in anti-chicken GFP (1:5000; Aves Laboratories) for 18 h at 4°C, rinsed in PB, and incubated with donkey Alexa Fluor 488 (1:300; Thermo Fisher Scientific) antibody for 1 h at room temperature before mounting. Sections were imaged using an epifluorescence microscope (Leica DM550B with Leica Application Suite Advanced Fluorescence 3.0.0 build 8134 software).
Subcellular fractionation
Subcellular fractionation was performed using a previously established protocol (Knackstedt et al., 2010) and as we have described previously (Bavley et al., 2017; Kabir et al., 2017c). One hour after the final extinction trial, mice were rapidly decapitated and whole brains were quickly removed. Fresh brains were sectioned in the coronal plane using a brain block (Zivic Instruments). For each mouse, the hippocampus was bilaterally dissected with microforceps and the PFC and NAc were bilaterally dissected with a 17-gauge stainless steel stylet. Tissue was immediately frozen on dry ice and subsequent processing steps were performed at 4°C. Once all tissue samples were collected, a region was homogenized using a Teflon pestle (Pyrex) in cold sucrose buffer containing protease and phosphatase inhibitors. After centrifuging the homogenate at 1000 × g for 10 min, the supernatant was centrifuged at high speed (12,000 × g) for 20 min. The pellet was resuspended in HEPES/EDTA buffer and centrifuged at high speed for 20 min. The supernatant was saved as the cytoplasmic fraction. To obtain the synaptosomal fraction, the pellet was resuspended in 1% SDS buffer and saved. For generation of the PSD fraction, the synaptosomal pellet was resuspended in 0.5% Triton X-100 and incubated for 15 min before being centrifuged at 12,000 × g. The pellet, containing the Triton X-100-insoluble PSD fraction, was further resuspended in 1% Triton X-100 plus 1% SDS buffer and incubated for 1 h. This solution was centrifuged at 10,000 × g for 15 min and the supernatant was saved as the Triton X-100-insoluble PSD fraction.
Western immunoblotting
Protein concentration was determined by a BCA assay (Pierce) and absorbance was read on a plate reader at 570nm (iMark Absorbance Microplate Reader; Bio-Rad Laboratories). Twenty to 30 μg of protein was separated by 10% SDS-PAGE and transferred to PVDF membranes. Membranes were blocked in 5% nonfat dry milk and then incubated 12–24 h at 4°C in a primary antibody.
Blots were incubated with the following primary antibodies at a dilution of 1:1000; Cav1.2 (Alomone Laboratories, catalog #AGP-001, RRID:AB_11219156), pan-CaMKII (Cell Signaling Technology, catalog #3362, RRID:AB_2067938), phospho-CaMKII at threonine 286/287 (P-CaMKII T286/T287; Cell Signaling Technology, catalog #3361S, RRID:AB_2275070), GluA1 (Millipore, catalog #AB1504, RRID:AB_2113602), phospho-GluA1 at serine 831 (P-GluA1 S831; Millipore, catalog #AB5847, RRID:AB_92077), or 1:10,000;GAPDH (Abcam, catalog #ab22555, RRID:AB_447153). The Cav1.2 antibody used has been validated for its specificity using full-brain Cav1.2 knock-out mice in our recent study (Bavley et al., 2017). Two bands surrounding the predicted 250 kDa full-length Cav1.2 protein were identified as Cav1.2-specific variants. Proteins were quantified at the following molecular weights: Cav1.2 at 250 kDa, CaMKIIβ and P-CaMKIIβ at 60 kDa, CaMKIIα and P-CaMKIIα at 50 kDa, GluA1 and P-GluA1 S831 at 100 kDa, and GAPDH at 36 kDa. Membranes were incubated in horseradish peroxidase-linked IgG conjugated secondary antibody at 1:5000 at room temperature for 1 h before development using chemiluminescence solution (Western Lighting; PerkinElmer Life Science). Prestained standards (Bio-Rad) were used for protein size determination. Blots were processed on a medical film processor (Konica Minolta Medical and Graphic, model SRX-101A) and films were scanned with a Xerox DocuMate 510 scanner. Protein bands were quantified as mean optical intensity using ImageJ software and normalized to GAPDH. Normalized optical density values from each sample were used to calculate the percentage fold change for each drug condition compared with the control group (set to 1.0).
Quantitative real-time PCR (qPCR)
To examine mRNA levels in mice after cocaine CPP, we performed qPCR as described previously (Schierberl et al., 2011). Mice were decapitated and regions were dissected and immediately frozen as performed for cellular subfractionation described above. mRNA was extracted from one hemisphere of the hippocampus with the RNeasy Mini Kit (Qiagen). cDNA was synthesized from resulting purified RNA using the High-Capacity RNA-to-cDNA kit (Applied Biosystems). Cacna1c levels were examined using the mRNA-specific primers (QuantiTect Primer assay QT00150752; Qiagen) on an ABI PRISM 7000 Sequence Detection System with SYBR Green PCR Master Mix (Applied Biosystems). Amplification was performed for 40 cycles (95°C for 15 s, 60°C for 30 s, and 72°C for 30 s), followed by an extension period (72°C for 10 min). Cycle threshold (Ct) values for target genes were normalized to the housekeeping gene gapdh (QuantiTect Primer assay QT01658692; Qiagen). Each experiment was performed in triplicate and values were averaged. For mRNA data analysis, the ΔCt method was used (Livak and Schmittgen, 2001). Briefly, experimental Ct values were normalized to gapdh values using the following formula: ΔCt = Ct (cacna1c) − Ct (gapdh). Expression levels were calculated relative to controls using the following formula: ΔΔCt = ΔCt (treated) − ΔCt (control average). The final expression levels were obtained using the formula 2−ΔΔCt.
Electron microscopic immunocytochemistry
Immunocytochemical studies were performed in S831A phosphomutant mice to examine the subcellular distribution of GluA1 in dendritic spines after cocaine CPP extinction. Mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and transcardially perfused with normal saline containing 2% heparin, followed by 30 ml of 3.75% acrolein and 2% PFA in PB (Milner et al., 2011). Brains were removed and postfixed for 30 min in 1.85% acrolein and 2% PFA in PB. Forty-micrometer sections containing the dorsal hippocampus (1.10 mm posterior to bregma through 2.06 mm posterior to bregma) were sectioned on a vibratome (VT1000 S; Leica) and stored in cryoprotectant solution (30% sucrose and 30% ethylene glycol in PB) at −20°C.
Before immunocytochemistry, sections were rinsed in PB. To ensure identical labeling conditions between experimental groups for quantitative immunocytochemistry, the sections were coded with hole punches, pooled in single containers, and then processed together through all immunocytochemical procedures (Pierce et al., 1999). The sections were processed for immunocytochemistry using a preembedding protocol as described previously (Milner et al., 2011). To remove excess aldehydes, sections were incubated in 1% sodium borohydride for 30 min. Sections were then sequentially rinsed in PB and 0.1 m Tris-saline (TS), followed by a 30 min incubation in a blocking solution of 0.5% bovine serum albumin (BSA) in TS. Next, sections were rinsed in TS and then incubated in rabbit anti-GluA1 (1:150; Millipore, catalog #AB1504, RRID:AB_2113602) antisera for 24 h at room temperature and 24 h at 4°C. Sections were then processed for immunogold labeling of GluA1 as described previously (Beckerman et al., 2013). Sections were rinsed in PB with 0.001% gelatin and 0.08% BSA and then incubated overnight at 4°C in an anti-rabbit secondary antiserum conjugated to 1 nm colloidal gold particles (1:50; Electron Microscopy Sciences). The nanogold-conjugated IgG was fixed by incubating sections in 2% glutaraldehyde and the gold particles were enhanced using a Silver IntenSEM kit (RPN491; GE Healthcare) for 7 min.
Sections were postfixed in 2% osmium tetroxide for 1 h, dehydrated, and flat embedded in Embed-812 (Electron Microscopy Sciences) between two sheets of Aclar plastic (Milner et al., 2011). Ultrathin sections (70 nm) through the dorsal hippocampus were cut with a diamond knife (Electron Microscopy Sciences) on a Leica EM UC6 ultratome and collected on 400-mesh, thin-bar copper grids (Electron Microscopy Sciences). Grids were then counterstained with uranyl acetate and Reynold's lead citrate (Milner et al., 2011). Sections were examined on a CM10 electron microscope (FEI).
Ultrastructural data analysis
An investigator blinded to the experimental conditions collected and analyzed all electron microscopic data. Sections were first examined at low magnification (3400×) to select fields having good morphological preservation and the presence of silver-intensified immunogold (SIG) particle labeling for GluA1, which were visible as black, electron-dense particles. From these fields, images were randomly photographed at 13,500×. These images were obtained exclusively at the plastic–tissue interface to ensure even antibody tissue penetration (Milner et al., 2011). Profiles were identified by defined morphological criteria (Peters et al., 1991). Dendritic profiles contained regular arrays of microtubules and were generally found postsynaptic to axon terminals. Dendritic spines were postsynaptic to an axon terminal, exhibited postsynaptic densities, sometimes contained spine apparati, and were devoid of mitochondria.
Single-labeled dendritic spines in the dorsal hippocampus at least 55 μm distal to the stratum radiatum region of CA1 or within the dentate gyrus (n = 50, area examined for each type of profile in each subregion per mouse) were collected from three animals per experimental condition using established methods (Milner et al., 2011). The subcellular localization of GluA1 SIG particles was defined as either on the plasmalemma (touching the membrane), near the plasmalemma (within 50 nm, but not touching the plasma membrane), or cytoplasmic. The distribution of GluA1 was assessed in synaptic (PSD), perisynaptic (within 50 nm of the PSD), and nonsynaptic (>70 nm from the PSD and perisynaptic regions) areas of dendritic spines (Marques-Lopes et al., 2017). The numbers of SIG particles in each subcellular compartment were compared statistically by genotype.
Statistics
All CPP behavioral data were analyzed by two-way repeated-measures (RM) ANOVA using a within-subjects factorial design followed by the Bonferroni–Dunn post hoc test to compare main effects of drug condition and testing session. For all mRNA and Western analyses, data were first analyzed for normality using the Shapiro–Wilk normality test. All normally distributed experimental data were analyzed by a parametric independent-samples t test. Non-normally distributed data were analyzed with a nonparametric test (Mann–Whitney). Statistical analyses were conducted using GraphPad Prism version 6.0 for Mac and were considered to be statistically significant for values of p ≤ 0.05. Details of each statistical analysis are listed below.
Experimental procedures
Experiment 1. Effect of acquisition of cocaine CPP on Cav1.2 mRNA and protein levels in the hippocampus.
A factorial design that included the within-subject factors of day (baseline and acquisition) and drug condition (saline or cocaine) was used to assess the acquisition of cocaine CPP following the behavioral protocol outlined in Figure 1A (“CPP acquisition” group). After behavior, a between-subject factor of drug condition (saline or cocaine) was used to assess the effect of cocaine CPP acquisition on levels of Cav1.2 mRNA or PSD protein within the hippocampus. The following numbers of mice were used for analysis of behavior, mRNA levels, and protein levels: n = 7 saline and n = 9 cocaine.
Experiment 2. Effect of extinction of cocaine CPP on Cav1.2 mRNA and PSD protein levels in the hippocampus, PFC, and NAc.
A factorial design that included the within-subject factors of day (baseline, acquisition, and extinction test) and drug condition (saline or cocaine) was used to assess extinction after acquisition of cocaine CPP (Fig. 1A: “CPP extinction” group, n = 16 saline- and n = 16 cocaine-treated mice). After behavior, a between-subject factor of drug condition (saline or cocaine) was used to assess the effect of cocaine CPP extinction on levels of Cav1.2 mRNA or PSD protein in the hippocampus, PFC, and NAc. The following numbers of mice were used for molecular studies: hippocampus mRNA: saline, n = 9 and cocaine, n = 8; hippocampus protein: saline, n = 12 and cocaine, n = 13; PFC mRNA: saline, n = 9 and cocaine, n = 7; PFC protein: saline, n = 5 and cocaine, n = 9; NAc mRNA: saline, n = 10 and cocaine, n = 6; and NAc protein: saline, n = 8 and cocaine, n = 9.
Experiment 3. Effect of home cage exposure without extinction training on maintenance of cocaine CPP.
A factorial design that included the within-subject factors of day (baseline, acquisition, and no extinction retest) and drug condition (saline or cocaine) was used to assess acquisition and maintenance of CPP after a home cage exposure period of 4 d without extinction training. The following numbers of mice were used for behavior: saline, n = 8 and cocaine, n = 10.
Experiment 4. Effect of home cage exposure without extinction training on Cav1.2 mRNA and PSD protein levels in the hippocampus.
A factorial design that included the within-subject factors of day (baseline and acquisition) and drug condition (saline or cocaine) was used to assess acquisition of cocaine CPP (Fig. 1A: “CPP no extinction” group; saline, n = 7 and cocaine, n = 8). A between-subject factor of drug condition (saline or cocaine) was used to assess the effect of 4 d of home cage exposure without extinction training on levels of Cav1.2 mRNA and PSD protein in the hippocampus. The following numbers of mice per drug condition were used for molecular analyses: saline, n = 6 and cocaine, n = 8 for mRNA analysis; and saline, n = 6 and cocaine, n = 8 for protein analysis.
Experiment 5. Effect of deleting Cav1.2 in the dorsal hippocampus or ventral hippocampus on extinction of cocaine CPP behavior.
Cav1.2 deletion in the dorsal hippocampus and ventral hippocampus was achieved with site-specific microinjection of AAV2/2 (dorsal hippocampus) or AAV2/5 (ventral hippocampus) expressing Cre-GFP or GFP (control) in cacna1c floxed mice. In this experiment, a factorial design that included the within-subject factors of day (baseline, acquisition, and extinction test) and viral treatment (AAV-GFP or AAV-Cre) was used to assess the effect of Cav1.2 knock-down on acquisition and extinction of cocaine CPP. The following numbers of mice were used for behavioral analysis for each region: dorsal hippocampus: AAV-GFP, n = 9 and AAV-Cre, n = 10; ventral hippocampus: AAV-GFP, n = 6, AAV-Cre, n = 7.
Experiment 6. Effect of deleting Cav1.2 in the dorsal hippocampus or ventral hippocampus on maintenance of cocaine CPP without extinction training.
In this experiment, a factorial design that included the within-subject factors of day (baseline, acquisition, and no extinction retest) and viral treatment (AAV-GFP or AAV-Cre) was used to assess the effect of Cav1.2 knock-down within the dorsal hippocampus or ventral hippocampus on acquisition of cocaine CPP and maintenance of CPP behavior without extinction training. The following numbers of mice were used for behavioral analysis for each region: dorsal hippocampus: AAV-GFP, n = 8 and AAV-Cre, n = 6; ventral hippocampus: AAV-GFP, n = 7 and AAV-Cre, n = 8.
Experiment 7. Effect of extinction of cocaine CPP on total and phosphorylated CaMKIIα/β protein levels in the hippocampus.
Levels of CaMKIIα, P-CaMKIIα T286, CaMKIIβ, and P-CaMKIIβ T287 were examined in saline- and cocaine-extinguished mice. In this experiment, a between-subject factor of drug condition (saline or cocaine) was used to assess the effect of cocaine CPP extinction on PSD protein within the hippocampus. The following numbers of mice was used for analysis of each protein: P-CaMKIIα and CaMKIIα: saline n = 16, cocaine n = 16; CaMKIIβ: saline n = 6, cocaine n = 8; and P-CaMKIIβ: saline n = 8, cocaine n = 10.
Experiment 8. Effect of home cage exposure without extinction training on total and phosphorylated CaMKIIα/β protein levels in the hippocampus.
A between-subject factor of drug condition (saline or cocaine) was used to assess the effect of home cage exposure on levels of CaMKIIα, P-CaMKIIα T286, and P-CaMKIIβ T287 PSD protein in the hippocampus. The following numbers of mice per drug condition was used for analysis of each protein: P-CaMKIIα and P-CaMKIIβ: saline, n = 6 and cocaine, n = 8; CaMKIIα: saline, n = 6 and cocaine, n = 7.
Experiment 9. Effect of pharmacological inhibition of CaMKIIα/β in the dorsal hippocampus on extinction of cocaine CPP.
The necessity of CaMKII activity within the dorsal hippocampus was tested by local infusion of KN93, a CaMKIIα/β inhibitor, or vehicle into the dorsal hippocampus of C57BL/6J mice during extinction training. In this experiment, a within-subject factor of day (baseline and acquisition) and treatment condition (vehicle or KN93) was used to confirm that both groups designated to the treatment conditions acquired cocaine CPP, followed by a between-subject factor of treatment condition (vehicle or KN93) to assess the effect of CaMKIIα/β inhibition in the dorsal hippocampus on CPP behavior on the final extinction test. The following numbers of mice were used for behavioral analysis: vehicle, n = 7 and KN93, n = 7.
Experiment 10. Effect of extinction of cocaine CPP on protein levels of total and phosphorylated GluA1 at site S831 in the hippocampus.
Levels of GluA1 and phosphorylated GluA1 at S831 (P-GluA1 S831) in hippocampal PSD fractions and GluA1 in hippocampal cytoplasmic fractions were first examined in saline- and cocaine-extinguished mice. In this experiment, a between-subject factor of drug condition (saline or cocaine) was used to assess the effect of cocaine CPP extinction on levels of protein in the PSD (saline, n = 13 and cocaine, n = 13–14) and cytoplasmic (saline, n = 8 and cocaine, n = 6) fraction in addition to mRNA levels (saline, n = 9 and cocaine, n = 10).
Experiment 11. Effect of home cage exposure without extinction training on P-GluA1 S831 protein levels in the hippocampus.
A between-subject factor of drug condition (saline or cocaine) was used to assess the effect of home cage exposure on levels of PSD P-GluA1 S831 protein in the hippocampus. The following numbers of mice per drug condition were used for behavioral analyses to confirm acquisition of cocaine CPP: saline, n = 6; cocaine, n = 8. The following numbers of mice per drug condition were used for analysis of protein levels: saline, n = 6; cocaine, n = 8.
Experiment 12. Effect of loss of phosphorylation of GluA1 at site S831 on extinction of cocaine CPP and GluA1 levels in the PSD of the hippocampus.
The necessity of S831 phosphorylation for cocaine CPP extinction was tested in S831A phosphomutant mice. In this experiment, the within-subject factors of day (baseline, acquisition, and extinction test) and genotype (WT or S831A mutants) were used to assess the effect of disruption of the S831 phosphorylation site on cocaine CPP extinction (WT, n = 13 and S831A, n = 15). After extinction, GluA1 levels were measured in hippocampal PSD and synaptosomal fractions. In this experiment, a between-subject factor of drug condition (saline or cocaine) was used to assess the effect of loss of GluA1 S831 phosphorylation on GluA1 levels in PSD and synaptosomal fractions (WT, n = 13; S831A, n = 13–14).
Experiment 13. Effect of loss of phosphorylation of GluA1 at site S831 on subcellular localization of GluA1 protein in the CA1 and dentate gyrus of the hippocampus.
After the final cocaine CPP extinction test, S831A GluA1 mutant mice and WT littermates were killed and their brains were processed for analysis by electron microscopy to measure GluA1 levels in the CA1 and the dentate gyrus (on and near the membrane of synaptic, perisynaptic, and nonsynaptic sites). In this experiment, a between-subject factor of genotype (WT or S831A mutant) was used to assess the effect of disruption of the S831 phosphorylation site on subcellular localization of GluA1 for each subcellular compartment within the hippocampal CA1 and dentate gyrus after cocaine CPP extinction (WT: n = 3, 50 fields examined per mouse; S831A: n = 3, 50 fields examined per mouse).
Experiment 14. Effects of knock-out of Cav1.2 in CaMKII-expressing cells on acquisition and extinction of cocaine CPP.
CaMK2cre, Cav1.2fl/fl mice and CaMK2cre, Cav1.2+/+ littermates were tested in cocaine CPP extinction behavior. In this experiment, within-subject factor of day (baseline, acquisition, and extinction test) and genotype (CaMK2cre, Cav1.2+/+ or CaMK2cre, Cav1.2fl/fl) was used to assess the effect of knock-out of Cav1.2 in CaMKII-expressing cells on cocaine CPP acquisition and extinction behavior (CaMK2cre, Cav1.2+/+ mice, n = 13; CaMK2cre, Cav1.2fl/fl mice, n = 14).
Experiment 15. Effects of knock-out of Cav1.2 in D1R-expressing cells on acquisition and extinction of cocaine CPP in addition to changes in hippocampal protein levels of Cav1.2-regulated targets.
D1cre, Cav1.2+/+ mice and D1cre, Cav1.2fl/fl littermates were tested in cocaine CPP extinction behavior using within-subject factors of day (baseline, acquisition, and extinction test) and genotype (D1cre, Cav1.2+/+ or D1cre, Cav1.2fl/fl) to assess the effect of knock-out of Cav1.2 in D1R-expressing cells on cocaine CPP acquisition and extinction behavior (D1cre, Cav1.2+/+ mice, n = 13; D1cre, Cav1.2fl/fl mice, n = 7). After the final cocaine CPP extinction session, PSD fractions of the hippocampus and NAc were used to measure protein levels of CaMKIIα, P-CaMKIIα T286, CaMKIIβ, P-CaMKIIβ T287, GluA1 and P-GluA1 S831. In this experiment, a between-subject factor of genotype (D1cre, Cav1.2+/+ or D1cre, Cav1.2fl/fl) was used to assess the effect of knock-out of Cav1.2 in D1R-expressing cells on levels of proteins in the hippocampus and NAc PSD (D1cre, Cav1.2+/+ mice, n = 9–17; D1cre, Cav1.2fl/fl mice, n = 10) was used for the treatment groups.
Results
Extinction but not acquisition of cocaine CPP increases Cav1.2 in the hippocampus
Mice were trained and tested for cocaine CPP acquisition as outlined in Figure 1A (“CPP acquisition” group). Cocaine-conditioned mice exhibited robust acquisition of cocaine CPP, represented by a significantly higher preference score for the cocaine-associated context during the acquisition test compared with the baseline test (data not shown; two-way RM-ANOVA, significant interaction test day × drug condition, F(1,13) = 27.93, p = 0.0001). Hippocampal Cav1.2 mRNA and Cav1.2 protein levels in PSD fractions were measured in brain tissue collected 1 h after the acquisition session to assess gene expression and synaptic Cav1.2, respectively. After acquistion of cocaine CPP, no significant difference in Cav1.2 mRNA (saline CPP: 1.00 ± 0.09 vs cocaine CPP: 0.995 ± 0.04, t0.1100 = 14; p = 0.9140, unpaired t test) or Cav1.2 PSD protein levels (saline CPP: 1.00 ± 0.07 vs cocaine CPP: 0.94 ± 0.06, t(0.6549) = 13; p = 0.5240, unpaired t test) was observed in the hippocampus.
In a second cohort, mice were trained and tested for cocaine CPP acquisition, followed by 4 d of extinction training (Fig. 1A: “CPP extinction” group) until they demonstrated significantly lower preference for the cocaine-associated context compared with that on the acquisition test day (Fig. 1B; two-way RM-ANOVA, significant interaction, test day × drug condition, F(5,150) = 6.998, p < 0.0001). Brain tissue was collected 1 h after the last extinction session. After extinction, there were significantly higher levels of Cav1.2 mRNA (Fig. 1C; t(2.421) = 12; p = 0.0085, unpaired t test) and PSD Cav1.2 protein (Fig. 1D; p = 0.0080, U = 30.0, Mann–Whitney) in the hippocampus.
Next to determine whether the increase in Cav1.2 mRNA and protein is specific to extinction, we examined levels of Cav1.2 in the hippocampus of mice that underwent home cage exposure without extinction training for 4 d after the acquisition of cocaine CPP, a time period equivalent to that of extinction training. First, we confirmed that mice maintain CPP at this time point (Fig. 1E; two-way RM-ANOVA, significant interaction, test day × drug condition, F(2,32) = 38.73, p < 0.0001). Examination of Cav1.2 levels in a separate cohort (Fig. 1A:“CPP no extinction” group) that acquired cocaine CPP (cocaine CPP: baseline = 48.32 ± 48.36 s, acquisition = 572.6 ± 59.01 s vs saline CPP: baseline = −38.6 ± 61.35 s, acquisition = 83.14 ± 72.77 s, two-way RM-ANOVA, significant interaction, test day × drug condition, F(1,13) = 21.97, p = 0.0004) and returned to their home cage environment for 4 d without extinction training did not exhibit altered levels of Cav1.2 mRNA (Fig. 1F; t(1.354) = 12; p = 0.1583, unpaired t test) or Cav1.2 PSD protein levels (Fig. 1F; t(0.4459) = 12; p = 0.6636, unpaired t test) compared with saline control mice. These results indicate that extinction training increases hippocampal Cav1.2 mRNA levels selectively, with subsequent increases in Cav1.2 at synaptic sites in the hippocampus after extinction.
In addition, the increase in Cav1.2 mRNA and protein after extinction training was specific to the hippocampus. After extinction of cocaine CPP, no difference in levels of Cav1.2 were seen in the PFC or NAc (Fig. 1C: PFC mRNA, t(0.1486) = 14; p = 0.8840, NAc mRNA, t(0.3038) = 14; p = 0.7658; Fig. 1D: PFC protein, t(1.099) = 12; p = 0.2932, NAc protein, t(0.8456) = 15; p = 0.4111, unpaired t test), brain regions implicated in extinction of cocaine seeking behavior. Therefore, extinction training of cocaine CPP increases hippocampal Cav1.2 mRNA and Cav1.2 protein selectively within PSD fractions.
Spatiotemporal deletion of Cav1.2 in the dorsal hippocampus results in a deficit in extinction of cocaine CPP
Because we observed an increase in Cav1.2 protein in the hippocampus after extinction, we next tested the necessity of hippocampal Cav1.2 channels for extinction of cocaine CPP by deleting Cav1.2 locally. We targeted the dorsal hippocampus because it has been shown to be necessary for aspects of cocaine-associated memories (Meyers et al., 2003; Meyers et al., 2006; Ramirez et al., 2009), including extinction of cocaine-seeking behavior (Szalay et al., 2013). In addition, we targeted the ventral hippocampus because it has been shown to be required for extinction of cocaine self-administration (Chambers and Self, 2002). We generated focal deletion of Cav1.2 in the dorsal hippocampus, targeted within the CA1 subregion, by bilateral stereotactic delivery of AAV-Cre in adult Cav1.2fl/fl mice (Fig. 2A). Virus expression typically spread from 1.2 mm through 1.7 mm posterior to bregma (Fig. 2A). This resulted in significantly lower levels of Cav1.2 mRNA in CA1 compared with control AAV-GFP-injected mice (mRNA fold change compared with controls: AAV-GFP, n = 7, 1.0 ± 0.11 vs AAV-Cre, n = 4, 0.65 ± 0.09: t(2.325) = 10; p = 0.0424, unpaired t test). Deleting Cav1.2 in the dorsal hippocampus had no effect on acquisition of cocaine CPP. Both AAV-GFP- and AAV-Cre-injected mice demonstrated significantly higher preference for the cocaine-associated context compared with the baseline test day (Fig. 2B). However, AAV-Cre injected mice showed a significant deficit in the extinction of cocaine CPP compared with AAV-GFP injected controls (Fig. 2B; two-way RM-ANOVA, significant interaction, test day × viral injection, F(2,34) = 9.074, p = 0.0007), demonstrating that Cav1.2 in the dorsal hippocampus is not required for the acquisition of cocaine CPP, but is necessary for extinction of cocaine CPP. This deficit was not due to alterations in locomotor activity because viral vector injections had no effect on basal locomotion (AAV-GFP, n = 10, 5273 ± 181 cm vs AAV-Cre, n = 11, 4842 ± 379 cm, F(1,19) = 0.9930, p = 0.3332, unpaired t test). In addition, the deletion of Cav1.2 in the dorsal hippocampus had no effect on maintenance of CPP behavior because a separate cohort of mice demonstrated sustained CPP scores when tested again 4 d after acquisition of cocaine CPP but without receiving extinction training (Fig. 2C; two-way RM-ANOVA, main effect of day, F(2,24) = 98, p < 0.0001; no main effect of viral treatment, F(1,12) = 0.0020, p = 0.9649).
Figure 2.
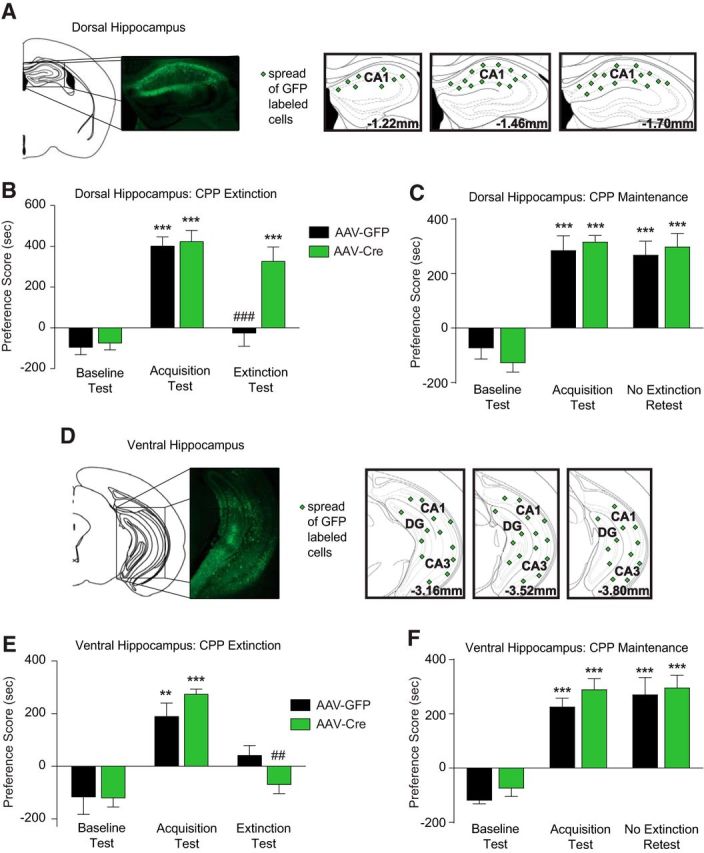
Focal deletion of Cav1.2 in the dorsal hippocampus induces a deficit in extinction of cocaine CPP. A, D, Representative images of green fluorescent protein (GFP)-positive cells and representations of coronal sections showing spread of AAV expression, as indicated by green symbol labeling, resulting from AAV bilaterally microinjected into dorsal hippocampus (A) or ventral hippocampus (D) of cacna1cfl/fl mice. B, Deletion of Cav1.2 in the dorsal hippocampus had no effect on acquisition of cocaine CPP, but attenuated extinction significantly compared with mice injected with AAV-GFP. C, Deletion of Cav1.2 in the dorsal hippocampus had no effect on maintenance of cocaine CPP when tested 4 d after the acquisition test. E, Deletion of Cav1.2 in the ventral hippocampus had no effect on acquisition or extinction of cocaine CPP. F, Deletion of Cav1.2 in the ventral hippocampus had no effect on maintenance of cocaine CPP. ***p < 0.001 compared with baseline test; #p < 0.05, ###p < 0.001 compared with acquisition test. Data are presented as mean ± SEM.
Contrary to the dorsal hippocampus, bilateral deletion of Cav1.2 in the ventral hippocampus, which resulted in AAV-Cre expression in the ventral CA1, CA3, and DG from 3.1 mm through 3.8 mm posterior to bregma (Fig. 2D), had no effect on CPP acquisition or extinction (Fig. 2E; two-way RM-ANOVA, main effect of day, F(2,22) = 17.01, p < 0.0001; no main effect of viral treatment, F(1,11) = 0.0195, p = 0.8916). Similar to the dorsal hippocampus, deletion of Cav1.2 in the ventral hippocampus had no effect on maintenance of CPP behavior without extinction (Fig. 2F; two-way RM-ANOVA, main effect of day, F(2,26) = 95.2, p < 0.0001; no main effect of viral treatment, F(1,13) = 1.062, p = 0.3215).
Together, these results demonstrate that Cav1.2 channels, specifically in the dorsal hippocampus, and not the ventral hippocampus, are essential for extinction of cocaine CPP, whereas Cav1.2 channels in neither of these subregions are necessary for the acquisition of cocaine CPP or maintenance of CPP tested 4 d later.
CaMKII α/β activity in the dorsal hippocampus is essential for extinction of cocaine CPP
Due to its localized positioning at the synapse (Di Biase et al., 2008; Tippens et al., 2008; Kabir et al., 2017b), Cav1.2 initiates a multitude of intracellular signaling cascades after channel activation (Schierberl et al., 2011; Simms and Zamponi, 2014; Mullins et al., 2016; Kabir et al., 2017a). One key downstream target of Cav1.2 channels is CaMKII (Wu et al., 2001; Schierberl et al., 2011; Wheeler et al., 2012), which is involved in extinction learning (Szapiro et al., 2003; Kimura et al., 2008; Myskiw et al., 2014; de Carvalho Myskiw et al., 2014). We first examined whether CaMKII levels were altered at synaptic locations in the hippocampus after extinction of cocaine CPP. Protein levels of the α and β CaMKII isoforms, as well as their phosphorylated active forms, were measured in hippocampal PSD fractions. We found that total CaMKIIα, as well as the phosphorylated form at the T286 site, were increased in PSD fractions (Fig. 3A; CaMKIIα: t(2.341) = 30; p = 0.0261, unpaired t test; P-CaMKIIα T286: t(3.371) = 18; p = 0.0014, unpaired t test). Examination of CaMKIIβ protein revealed a trending increase in total protein and significantly higher phosphorylated CaMKIIβ at site T287 compared with saline controls (Fig. 3A; total CaMKIIβ: t(1.936) = 12; p = 0.0768, unpaired t test; P-CaMKIIβ T287: t(2.756) = 16; p = 0.014, unpaired t test). This increase in CaMKII protein within the PSD fraction of the hippocampus appeared to be dependent on extinction training because mice that acquired cocaine CPP followed by 4 d of home cage exposure without extinction training (“CPP no extinction” group outlined in Fig. 1A) had lower levels of CaMKIIα protein (t(2.691) = 11; p = 0.0210, unpaired t test), P-CaMKIIα protein (t(2.405) = 12; p = 0.0332, unpaired t test), and trending lower levels of P-CaMKIIβ T287 (t(1.813) = 12; p = 0.0950, unpaired t test) compared with saline control mice (Fig. 3B).
Figure 3.
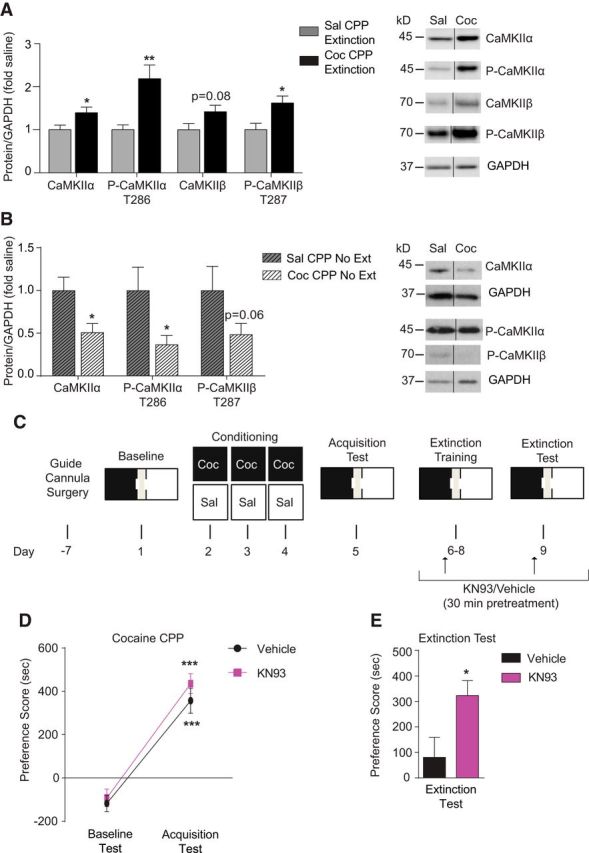
Pharmacological inhibition of CaMKIIα/β in the dorsal hippocampus induces a deficit in extinction of cocaine CPP. A, Total and phosphorylated CaMKIIα, as well as phosphorylated CaMKIIβ, in the PSD-enriched fraction from the hippocampus were significantly higher after extinction of cocaine CPP compared with saline control mice. Total CaMKIIβ trended higher but did not reach statistical significance. B, Total and phosphorylated CaMKIIα in the PSD-enriched fraction from the hippocampus were significantly lower in mice that acquired cocaine CPP followed by home cage exposure without extinction training compared with saline control mice. Phosphorylated CaMKIIβ trended lower but did not reach statistical significance. *p < 0.05, **p < 0.01 compared with saline group. C, Timeline of surgery, CPP conditioning, acquisition test, and microinjection of the CaMKII α/β inhibitor KN93 or vehicle before extinction training. D, Similar acquisition of cocaine CPP was observed within each designated group before treatment during extinction training. ***p < 0.001 compared with baseline test. E, Infusion of KN93 into the dorsal hippocampus of mice (n = 7) before the each extinction training session attenuated extinction compared with vehicle-infused mice (n = 7). *p < 0.05 compared with vehicle group. Data are presented as mean ± SEM.
Next, to test directly whether CaMKII activity is essential for extinction of cocaine CPP, mice were bilaterally infused with the CaMKIIα/β inhibitor KN93 (Sumi et al., 1991) in the dorsal hippocampus during extinction training, as outlined in Figure 3C. Mice were split randomly into two treatment groups that would subsequently receive vehicle or KN93 during extinction. Each group was then trained to acquire cocaine CPP. Both groups demonstrated increased preference scores during the acquisition test compared with baseline test (Fig. 3D; two-way RM-ANOVA, main effect of day F(1,12) = 81.42, p < 0.0001; no main effect of treatment assignment, F(1,12) = 2.394, p = 0.1477). After acquisition, vehicle or KN93 was infused into the dorsal hippocampus during extinction training (Fig. 3C). KN93 attenuated extinction significantly compared with vehicle treated mice (Fig. 3E; t(2.4740) = 12; p = 0.0293, unpaired t test), demonstrating the necessity of CaMKIIα/β within the dorsal hippocampus for extinction of cocaine CPP.
Phosphorylation of GluA1 at S831 is increased in the hippocampus and is essential for extinction of cocaine CPP
One key target of active CaMKII is phosphorylation of the AMPA receptor GluA1 subunit at S831 (Mammen et al., 1997), a key posttranslational modification necessary for hippocampal-dependent learning and memory (Olivito et al., 2016). Using a phosphospecific antibody, we found increased levels of phosphorylated GluA1 at S831 (P-GluA1 S831) in the hippocampal PSD of cocaine-extinguished WT mice (Fig. 4A; t(3.416) = 25; p = 0.0022, unpaired t test). This increase in P-GluA1 S831 was dependent on extinction training because mice that acquired cocaine CPP followed by 4 d of home cage exposure without extinction training (“CPP no extinction” group outlined in Fig. 1A) demonstrated no difference in P-GluA1 S831 protein compared with saline control mice (Fig. 4B; t(0.23) = 12; p = 0.8220, unpaired t test). We also examined levels of total GluA1 because changes in cell surface levels of this protein have been observed after cocaine-associated behavior (Wolf, 2016). Extinction increased total GluA1 protein levels significantly in hippocampal PSD fractions compared with saline controls (Fig. 4A; t(2.376) = 24; p = 0.0258, unpaired t test). No difference in hippocampal GluA1 mRNA levels (saline extinction, n = 9, 1.00 ± 0.14 vs cocaine extinction, n = 10, 1.05 ± 0.36, t(0.1241) = 17; p = 0.9027, unpaired t test) and no difference in GluA1 protein levels in the cytoplasmic fraction of the hippocampus was observed (Fig. 4C; t(0.6552) = 12; p = 0.5247, unpaired t test), suggesting that the observed increase in PSD GluA1 levels after extinction are not due to increased gene expression and could involve changes in GluA1 subcellular localization.
Figure 4.

S831A GluA1 mutant mice have a deficit in extinction of cocaine CPP and lower levels of GluA1 in synaptosomal fractions but not at the hippocampal PSD. A, Total and phosphorylated GluA1 at S831 were significantly higher in the PSD-enriched fraction of the hippocampus after extinction of cocaine CPP compared with saline control mice. *p < 0.05, **p < 0.01. B, Phosphorylated GluA1 at S831 was not altered in the PSD-enriched fraction of the hippocampus in mice that underwent cocaine CPP acquisition followed by home cage exposure without extinction training compared with saline control mice. C, Total GluA1 was not altered in the cytoplasmic fraction of the hippocampus after extinction of cocaine CPP compared with saline control mice. D, S831A mutant mice displayed no difference in acquisition of cocaine CPP, but demonstrated an extinction deficit compared with WT mice. *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline test; #p < 0.05 compared with acquisition test. E, After extinction, there was no difference in levels of GluA1 in PSD fractions generated from the hippocampus of S831A mutant mice compared with WT mice, whereas synaptosomal fractions had significantly lower levels of GluA1 in mutants compared with WT mice. ***p < 0.001. Data are presented as mean ± SEM.
To further investigate the necessity of GluA1 S831 phosphorylation for extinction of cocaine context learning, mice harboring a serine to alanine substitution at the S831 site of GluA1 (Lee et al., 2010) and their WT littermates were trained and tested for acquisition and extinction of cocaine CPP. Although S831A mutant and WT mice had similar CPP acquisition scores, S831A mutant mice differed from WT mice in their sustained high preference for the cocaine-associated context during extinction testing (Fig. 4D; two-way RM-ANOVA, significant interaction, test day × genotype, F(2,52) = 6.475, p = 0.0031). Therefore, S831 GluA1 phosphorylation is necessary for cocaine CPP extinction, but is not required for the acquisition of the initial cocaine context-associated memory.
Next, to determine whether phosphorylation of GluA1 S831 influences the increase in hippocampal PSD GluA1 levels observed after extinction (Fig. 4A), we measured levels of GluA1 in PSD fractions from S831A GluA1 mutants after the last extinction session. We found no difference in levels of PSD GluA1 protein between S831A mutant and WT littermates (Fig. 4E), suggesting that the increase in GluA1 at the hippocampal PSD is not necessary for extinction of cocaine CPP.
In addition to being located at the PSD, GluA1 is also present within other compartments, including the plasma membrane of perisynaptic and nonsynaptic sites (Richmond et al., 1996; Passafaro et al., 2001). Therefore, we probed synaptosomal fractions containing these components to determine whether GluA1 levels were affected in S831A GluA1 mutants after cocaine CPP extinction. S831A mutants had significantly lower levels of GluA1 protein within synaptosomal fractions compared with WT mice after extinction training (Fig. 4E, t(5.048) = 10; p = 0.0005, unpaired t test), demonstrating that GluA1 is decreased outside the PSD after CPP extinction in S831A mutants. No difference in basal GluA1 levels was observed in hippocampal synaptosomal fractions from S831A mutants compared with WT littermates (data not shown), which is supported by previous findings demonstrating no difference in GluA1 levels in crude membrane fractions (Lee et al., 2010).
S831A mutation is associated with decreases in perisynaptic GluA1 in dendritic spines of hippocampal CA1 neurons
The reduction in synaptosomal GluA1 after extinction prompted us to further probe the subcellular localization of GluA1 in dendritic spines of hippocampal pyramidal neurons. We used high-resolution immunoelectron microscopy to detect the presence of GluA1 on and near select compartments within dendritic spines, as indicated in Figure 5B, of the stratum radiatum of the CA1 subregion and the dentate gyrus of the dorsal hippocampus (Fig. 5A).
Figure 5.
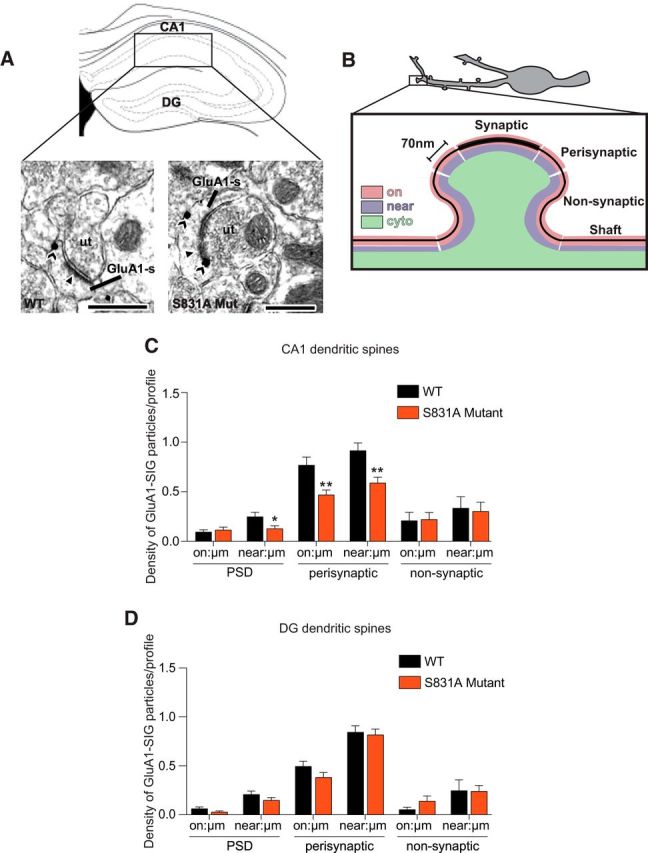
S831A GluA1 mutant mice have lower levels of GluA1 near the PSD and on and near the plasma membrane of perisynaptic sites of dendritic spines in the hippocampal CA1 subregion after impaired cocaine CPP extinction. A, Representative electron micrographs from stratum radiatum of CA1 of WT and S831A mutant mice after cocaine CPP extinction showing GluA1 SIG particle labeling in spines (sp) contacted by unlabeled axon terminals (ut). Scale bars, 500nm. B, Schematic figure demonstrating the criteria used to apportion SIG particles when quantifying GluA1 localization in dendritic spines by electron microscopy. C, In dendritic spines, significantly lower densities of GluA1 SIG particles were seen near, but not on the plasma membrane of the PSD in S831A mice compared with WT mice. Moreover, significantly lower densities of GluA1 SIG particles were observed on and near the plasma membrane of perisynaptic sites in S831A mice compared with WT mice. No difference in density of GluA1 SIG particles was observed on or near the plasma membrane of nonsynaptic sites. D, No difference in densities of GluA1 SIG particles were observed at synaptic or perisynaptic sites of the dentate gyrus of S831A mice and WT mice *p < 0.05, **p < 0.01. Data are presented as mean ± SEM.
Consistent with the Western blot analysis (Fig. 4D), electron microscopy revealed that there was no difference in levels of GluA1 SIG particles on the PSD of spines in the CA1 of S831A mutants compared with WT littermates after CPP extinction (Fig. 5C). However, there were significantly lower levels of GluA1 SIG particles near the PSD (Fig. 5C, t(2.1683) = 298; p = 0.0309, unpaired t test), as well as both on and near the plasma membrane at perisynaptic sites of S831A mutants compared with WT littermates (Fig. 5C, on: t(3.0791) = 298; p = 0.0023, unpaired t test; near: t(3.2915) = 298; p = 0.0011, unpaired t test). There were no differences in levels of GluA1 SIG particles on or near the plasma membrane of nonsynaptic sites of dendritic spines between the two genotypes (Fig. 5C). The altered levels of GluA1 in S831A mutants was specific to the CA1 because examination of spines of the dentate gyrus revealed no difference in levels of GluA1 particles both on and near the PSD, as well as on and near perisynaptic or nonsynaptic sites (Fig. 5D). These results indicate that GluA1 is reduced near the PSD and at perisynaptic areas of dendritic spines of CA1 neurons in S831 mutant mice with impaired cocaine CPP extinction.
Conditional knock-out of Cav1.2 in D1R-expressing but not glutamatergic neurons results in a deficit in extinction of cocaine CPP with corresponding decreases in PSD CaMKII and P-GluA1 S831 in the hippocampus
To identify potential cell-type-specific contributions of Cav1.2 to extinction of cocaine CPP, we first examined the role of Cav1.2 in glutamatergic neurons, a cell type in which Cav1.2 is highly expressed at postsynaptic sites (Di Biase et al., 2008; Kabir et al., 2017b). We generated the CaMK2cre, Cav1.2fl/fl mouse strain in which Cav1.2 is conditionally deleted in glutamatergic neurons of the forebrain, resulting in an ∼70% decrease in Cav1.2 mRNA in the hippocampus and other forebrain regions (Lee et al., 2012b). CaMK2cre, Cav1.2fl/fl mice and CaMK2cre, Cav1.2+/+ littermates were tested in the acquisition and extinction of cocaine CPP. Interestingly, knock-out of Cav1.2 in glutamatergic neurons had no effect on acquisition or extinction of cocaine CPP because CaMK2cre, Cav1.2fl/fl mice demonstrated similar acquisition and extinction as that seen in CaMK2cre, Cav1.2+/+ littermates (Fig. 6A; two-way RM-ANOVA, main effect of day, F(2,50) = 42.45, p < 0.0001, no main effect of genotype, F(1,25) = 0.0073, p = 0.9326).
Figure 6.
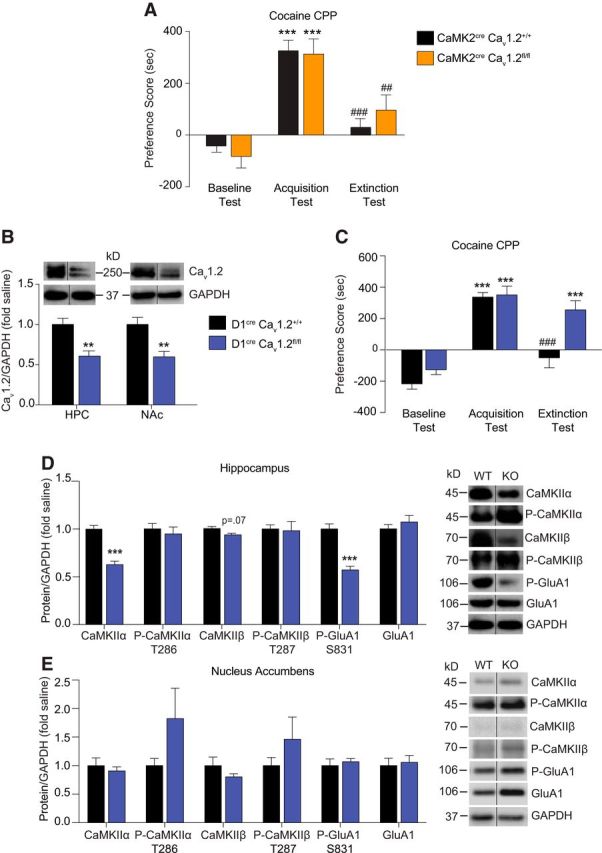
Mice with selective deletion of Cav1.2 in D1R-expressing cells have a deficit in extinction of cocaine CPP and lower levels of CaMKIIα and P-GluA1 S831, but not total GluA1 at the hippocampal PSD. A, CaMK2cre, Cav1.2+/+ mice and CaMK2cre, Cav1.2fl/fl mice displayed similar acquisition and extinction of cocaine CPP. ***p < 0.001 compared with baseline; #p < 0.05, ###p < 0.001 compared with acquisition. B, Validation of the D1cre, Cav1.2fl/fl mouse line. Cav1.2 protein levels in hippocampal and NAc PSD fractions were significantly lower in D1cre, Cav1.2fl/fl mice compared with D1cre, Cav1.2+/+ mice. **p < 0.01. C, Compared with D1cre, Cav1.2+/+ mice, D1cre, Cav1.2fl/fl mice displayed similar acquisition of cocaine CPP, but demonstrated a deficit in extinction of cocaine CPP. ***p < 0.001 compared with baseline test; ###p < 0.001 compared with acquisition test. D, After cocaine CPP extinction test, D1cre, Cav1.2fl/fl mice had significantly lower protein levels of CaMKIIα and P-GluA1 S831 in the PSD-enriched fraction of the hippocampus compared with D1cre, Cav1.2+/+ mice, with no difference in GluA1 levels. ***p < 0.001. E, After the cocaine CPP extinction test, there were no significant changes in the proteins examined in NAc PSD fractions of D1cre, Cav1.2fl/fl mice compared with D1cre, Cav1.2+/+ mice. Data are presented as mean ± SEM.
Because recent studies have highlighted a role for D1Rs in the extinction of cocaine CPP (Fricks-Gleason et al., 2012; Abraham et al., 2014, 2016), particularly within the hippocampus for memory of contextual cocaine-paired stimuli (Xie et al., 2014), we next generated the D1cre, Cav1.2fl/fl mouse strain in which Cav1.2 is conditionally deleted in D1R-expressing cells. The efficacy of knock-out in the hippocampus was revealed by lower levels of Cav1.2 protein in D1cre, Cav1.2fl/fl mice compared with D1cre, Cav1.2+/+ mice (Fig. 6B, t(3.537) = 24; p = 0.0017, unpaired t test). D1cre, Cav1.2fl/fl and D1cre, Cav1.2+/+ mice were trained and tested for CPP acquisition and extinction. Although D1cre, Cav1.2fl/fl mice acquired cocaine CPP to the same degree as D1cre, Cav1.2+/+ mice, D1cre, Cav1.2fl/fl mice were unable to extinguish cocaine CPP (Fig. 6C; two-way RM-ANOVA, significant interaction, test day × genotype, F(2,36) = 5.191, p = 0.0105). These results demonstrate that Cav1.2 channels in D1R-expressing cells are required for extinction of cocaine-associated memories.
Next, to investigate whether the molecular changes observed in the hippocampus after extinction (Figs. 3A, 4A) are mediated by Cav1.2 channels in D1R-expressing cells, we measured levels of CaMKII and GluA1 proteins in hippocampal PSD fractions of D1cre, Cav1.2+/+ mice and D1cre, Cav1.2fl/fl mice after cocaine CPP extinction training. D1cre, Cav1.2fl/fl mice had significantly lower levels of total CaMKIIα (Fig. 6D, t(6.788) = 24; p < 0.0001, unpaired t test) and a trend toward lower levels of CaMKIIβ (Fig. 6D, t(2.027) = 12; p = 0.0655, unpaired t test). Interestingly, no difference in P-CaMKIIα T286 or P-CaMKIIβ T287 between the genotypes was observed. Examination of GluA1 in hippocampal PSD fractions revealed significantly lower P-GluA1 S831 in D1cre, Cav1.2fl/fl mice (t(6.062) = 23; p < 0.0001, unpaired t test), but no difference in total GluA1 levels (Fig. 6D, t(0.9258) = 24; p = 0.3638, unpaired t test). Therefore, the above results demonstrated that Cav1.2 channels in D1R-expressing cells are necessary for hippocampal GluA1 phosphorylation at S831 after extinction, but do not appear to mediate the increase in levels of total GluA1 at the PSD, further supporting the finding in S831A GluA1 phosphomutant mice that S831 phosphorylation does not mediate the increase in PSD GluA1 levels in the hippocampus after extinction (Fig. 4C). There was no difference in basal levels of any of the proteins tested in PSD fractions of the hippocampus between genotypes (CaMKIIα, P-CaMKIIα T286, CaMKIIβ, P-CaMKIIβ T287, GluA1, and P-GluA1 S831; data not shown).
Because D1R-expressing cells in the NAc are key mediators of important behavioral effects of cocaine (Smith et al., 2013), including extinction of cocaine CPP (Calipari et al., 2016), we investigated the effect of loss of Cav1.2 in D1R-expressing cells in the NAc on CaMKII and GluA1 levels. Western blot confirmed the efficacy of Cav1.2 knock-out in the NAc with lower Cav1.2 protein in the PSD fraction of D1cre, Cav1.2fl/fl mice compared with D1cre, Cav1.2+/+ mice (Fig. 6B; t(3.093) = 15; p = 0.0074, unpaired t test). As opposed to the hippocampus, there was no significant difference in levels of CaMKIIα, CaMKIIβ, GluA1, or their phosphorylated forms in PSD fractions isolated from the NAc (Fig. 6D). Therefore, together, the above data suggest that Cav1.2-mediated changes in levels of CaMKII and P-GluA1 S831 within D1R-expressing cells of the hippocampus are necessary for extinction of cocaine CPP.
Discussion
Here, we describe a previously unknown role for Cav1.2 channels in the dorsal hippocampus in extinction of cocaine-associated memories. In addition, we show that Cav1.2/CaMKII-mediated phosphorylation of GluA1 at S831 in the hippocampus is essential for extinction of cocaine CPP. By conditionally deleting Cav1.2 in D1R-expressing cells, we further demonstrate a cell-type-specific role for Cav1.2 channels in extinction of cocaine CPP, hippocampal CaMKII expression, and phosphorylation of GluA1 at S831. These results provide the first illustration that a signaling pathway in the hippocampus involving Cav1.2 channels, CaMKII, and S831 GluA1 phosphorylation plays an essential role in extinguishing contextual cocaine-associated memories, with data supporting the contribution of hippocampal D1R-expressing cells.
Cav1.2 channels, dorsal hippocampus, and extinction of cocaine-associated memories
Extinction learning is thought to involve new learning that overrides, as opposed to erases, original memories (Bouton, 2004). Extinction of cocaine memories, as well as extinction of fear memories, which share overlapping mechanisms (Peters et al., 2009; Tipps et al., 2014), requires activation of new gene expression and protein synthesis (Cammarota et al., 2003; Vianna et al., 2004; Lattal et al., 2006; Szalay et al., 2013). We found extinction-specific increases in hippocampal Cav1.2 mRNA and PSD protein with no change in levels after acquisition, suggesting that newly synthesized Cav1.2 contributes to extinction learning. In addition, viral-mediated deletion of Cav1.2 in the dorsal hippocampus attenuates extinction of cocaine CPP with no effect on acquisition. The lack of a role for dorsal hippocampal Cav1.2 in acquisition of cocaine CPP is intriguing because Cav1.2 channels are highly involved in hippocampal-dependent learning and memory (Moosmang et al., 2005; Krug et al., 2014; Temme et al., 2016; Kabir et al., 2017a), suggesting that Cav1.2 may play a differential role in cocaine-associated versus other forms of hippocampal-dependent learning. Our data further suggest that distinct hippocampal mechanisms are most likely recruited for the acquisition versus extinction of cocaine-associated contextual memories.
Although studies examining hippocampal mechanisms involved in acquisition versus extinction of cocaine CPP are limited, few studies have suggested that acquisition and extinction recruit distinct transcriptional mechanisms (Quirk and Mueller, 2008). For example, a recent study on the role of the transcriptional regulator histone deacetylase 3 (HDAC3) in cocaine CPP revealed that dorsal hippocampal HDAC3 plays a selective role in extinction of cocaine CPP, but has no role in acquisition (Alaghband et al., 2017). Similarly, a study examining changes in microRNAs (miRNAs) that are involved in posttranscriptional regulation of gene expression via silencing mRNAs, has found that miRNA profiles in the hippocampus of rats after extinction of cocaine CPP differs compared with that seen after acquisition of cocaine CPP (Chen et al., 2013), suggesting that these phases of CPP are recruiting distinct molecular pathways. Although the regulatory relationship between these particular HDAC3 and miRNA-mediated mechanisms and Cav1.2 is unknown, these prior studies are nonetheless consistent with the present finding that distinct signaling pathways contribute to the extinction and acquisition of cocaine CPP.
Our finding that Cav1.2 channels within the dorsal, but not ventral, hippocampus are required for extinction of cocaine CPP is supported by previous results showing the necessity of dorsal hippocampus activity for extinction of cocaine-associated memories (Szalay et al., 2011; Szalay et al., 2013; Castilla-Ortega et al., 2016; McGlinchey and Aston-Jones, 2017) and extinction of fear memories (Corcoran and Maren, 2004; Corcoran et al., 2005; Ji and Maren, 2005). Furthermore, LTCCs within the dorsal hippocampus are particularly required for extinction of contextual fear memories (de Carvalho Myskiw et al., 2014). The observed lack of a role for Cav1.2 channels in the ventral hippocampus for extinction of cocaine CPP, a region required for extinction of cocaine self-administration (Chambers and Self, 2002), may reflect disparate circuitry involved in classical conditioning versus instrumental behavior.
Cav1.2, CaMKII, S831 GluA1 phosphorylation, and extinction of cocaine contextual memories
CaMKIIα/β serve as key downstream LTCC targets (Ma et al., 2012; Li et al., 2016) that transduce activity-dependent signals critical for various aspects of learning/memory (Hell, 2014; Shonesy et al., 2014), cocaine extinction (Rich et al., 2016), and fear extinction learning (Szapiro et al., 2003; Kimura et al., 2008; Radwanska et al., 2011). Here, we find that cocaine CPP extinction increases total and phosphorylated CaMKIIα/β protein in hippocampal PSD fractions and pharmacological inhibition of CaMKIIα/β in the dorsal hippocampus attenuates extinction. Interestingly, the increase in T286 phospho-CaMKIIα, the most characterized active form of CaMKIIα, is not attenuated in the hippocampus of D1cre, Cav1.2fl/fl mice, potentially a reflection of the limited population of D1R-expressing cells within the hippocampus (discussed below) (Gangarossa et al., 2012). Alternatively, CaMKII activity may be modulated by other phosphorylation sites such as S331, which was shown recently to mediate extinction of cocaine self-administration in the NAc (Rich et al., 2016).
CaMKIIα activity increases phosphorylation of GluA1 at site S831 (Barria et al., 1997; Mammen et al., 1997), resulting in potentiation of single-channel conductance of GluA1 homomeric and heteromeric AMPA receptors (Derkach et al., 1999; Oh and Derkach, 2005; Kristensen et al., 2011), a process implicated in synaptic activity associated with hippocampal-dependent learning (Kessels and Malinow, 2009). Phosphorylation of S831 has been linked to learning and memory (Whitlock et al., 2006; Crombag et al., 2008; Lee et al., 2013) and cocaine-related behaviors (Anderson et al., 2008; Kim et al., 2009; Schierberl et al., 2011). Here, we show that extinction of cocaine CPP is associated with an increase in phosphorylated S831 in hippocampal PSD fractions and demonstrate the necessity of S831 for extinction, with no role in acquisition of cocaine CPP using phosphomutant mice. In addition, lower levels of phosphorylated S831 in the hippocampus, but not the NAc, of extinction-deficient D1cre, Cav1.2fl/fl mice suggests that hippocampal Cav1.2-induced phosphorylation of GluA1 at S831 may be involved selectively in extinction of contextual cocaine memories.
Phosphorylation of S831 has been suggested to increase trafficking of GluA1 to the cell surface (Shepherd and Huganir, 2007). Increase in cell surface GluA1, particularly in the NAc, has been shown by several groups of investigators to be a critical step in mediating cocaine's long-term effects, particularly via formation of Ca2+-permeable GluA1 homomeric AMPARs (Wolf, 2016), a mechanism that we have demonstrated to require S831 (Martinez-Rivera et al., 2017). Although no studies on hippocampal GluA1 trafficking in extinction of cocaine behaviors exist, the necessity of GluA1 trafficking to the cell surface in the NAc has been demonstrated for extinction of cocaine self-administration behavior (Sutton et al., 2003). Here, using Western blots and electron microscopy, we found that S831 is not necessary for the increase in GluA1 at the hippocampal PSD, which is consistent with a previous in vitro study showing that the S831A mutation does not alter CaMKII-dependent or activity-dependent synaptic delivery of GluA1 (Hayashi et al., 2000). However, using electron microscopy, we found that S831 is necessary for trafficking of GluA1 near the PSD and near or on the plasma membrane of perisynaptic sites, suggesting that these particular pools of GluA1 may be essential for extinction of cocaine CPP. This is supported by studies demonstrating that perisynaptically localized GluA1 is important for expression of hippocampal LTP and modulation of synaptic strength (Guire et al., 2008; Yang et al., 2010), cellular processes known to be associated with extinction of cocaine-associated memories (del Olmo et al., 2006). Future studies will address the necessity of nonsynaptic GluA1 in the hippocampus for extinction of cocaine CPP and the role of Cav1.2 channels therein.
Hippocampal D1Rs, Cav1.2, and extinction of cocaine contextual memories
Growing evidence suggests that dopamine signaling via D1Rs plays a key role in extinction learning (El-Ghundi et al., 2007; Singewald et al., 2015), including extinction of cocaine-associated memories (Fricks-Gleason et al., 2012; Abraham et al., 2016). Although D1Rs are expressed throughout the brain, dorsal hippocampal D1Rs have been shown to play a role in extinction of contextual fear memories (Fiorenza et al., 2012; Menezes et al., 2015), in addition to drug context-induced reinstatement of self-administration (Xie et al., 2014). D1R-expressing cells are found within the hippocampus, located most densely within the stratum radiatum of CA1 and CA3 and the granule layer of the dentate gyrus (Gangarossa et al., 2012), and are suggested to be part of an extinction circuit, with mechanisms initiated within the CA1 subregion (Sil'kis, 2008). Our electron microscopy findings in S831A GluA1 phosphomutant mice support a role for CA1 in Cav1.2-mediated extinction of cocaine CPP.
We found that deleting Cav1.2 in D1R-expressing cells of the brain results in an impairment in the extinction of cocaine CPP. We also showed that CaMKII and phosphorylation of GluA1 at S831 are lower in the hippocampus, but not the NAc, of extinction-deficient D1R-cell-specific Cav1.2 knock-out mice. Although our molecular data suggest that the Cav1.2/CaMKII/S831 GluA1 signaling pathway in D1R-expressing cells of the hippocampus may mediate extinction of cocaine CPP, Cav1.2 is lacking in all D1R-expressing cells in the mouse model used, allowing for the possibility that Cav1.2 in D1R cells within other D1R-rich regions are involved in this behavior.
Previous studies support the interaction of D1Rs, CaMKII, and GluA1 activity because pharmacological activation of D1R produces a CaMKII-mediated increase in surface expression of GluA1 (Gao et al., 2006; Anderson et al., 2008; Schierberl et al., 2011) and total S831 phosphorylation (Anderson et al., 2008; Schierberl et al., 2011). Although D1Rs (Gangarossa et al., 2012) and Cav1.2 (Hell et al., 1993) are both expressed in hippocampal glutamatergic neurons, the precise cell type within the hippocampus that contributes to extinction remains to be addressed, given that a recent study has classified D1R-expressing cells as primarily GABAergic interneurons (Puighermanal et al., 2017). Cav1.2 is also present in several GABAergic interneuron subtypes within the hippocampus (Vinet and Sík, 2006; Dedic et al., 2017); however, low or undetectable levels of CaMKIIα within inhibitory neurons of the hippocampus (Jones et al., 1994; Liu et al., 2014) raise the possibility that both glutamatergic and GABAergic cell types within the hippocampus may be recruited during extinction of cocaine CPP, a subject for future studies.
In summary, this study is the first to report that Cav1.2 channels play a critical role in the dorsal hippocampus for the extinction of cocaine-associated memories, with molecular data supporting a contribution of Cav1.2-activated CaMKII/S831 GluA1 within D1R-expressing cells of the hippocampus. Therefore, this study sheds light on a new role of Cav1.2 channels in extinction of contextual cocaine-associated memory and advances our understanding of a dopamine- and Ca2+-dependent molecular pathway mediating extinction learning. These findings merit further exploration to better understand hippocampal mechanisms that contribute to addictive behavior as studied using animal models.
Footnotes
This work was supported by the National Institutes of Health (Grant R01 DA029122 to A.M.R., Grant T32DA039080 to C.E.B. and T.A.M., Grant T32DA7274 to K.C.S., Grant F31DA032169 to A.S.L., Grant HL135498 to M.J.G., Grant HL136520 to T.A.M. and M.J.G., Grant DA08259 to T.A.M., Grant HL098351 to T.A.M., and Grant R01 NS03615 to R.L.H.); The Paul Fund (A.M.R.); and the Neuroanatomy EM Core in The Feil Family Brain and Mind Research Institute (T.A.M.). We thank Charlotte Bavley for helpful suggestions in manuscript preparation, Marie Monfils for advice in establishing the cocaine CPP extinction protocol, and Nii Addy for help with statistical analyses.
The authors declare no competing financial interests.
References
- Abraham AD, Neve KA, Lattal KM (2014) Dopamine and extinction: a convergence of theory with fear and reward circuitry. Neurobiol Learn Mem 108:65–77. 10.1016/j.nlm.2013.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham AD, Neve KA, Lattal KM (2016) Activation of D1/5 dopamine receptors: a common mechanism for enhancing extinction of fear and reward-seeking behaviors. Neuropsychopharmacology 41:2072–2081. 10.1038/npp.2016.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaghband Y, Kwapis JL, López AJ, White AO, Aimiuwu OV, Al-Kachak A, Bodinayake KK, Oparaugo NC, Dang R, Astarabadi M, Matheos DP, Wood MA (2017) Distinct roles for the deacetylase domain of HDAC3 in the hippocampus and medial prefrontal cortex in the formation and extinction of memory. Neurobiol Learn Mem 145:94–104. 10.1016/j.nlm.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC (2008) CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci 11:344–353. 10.1038/nn2054 [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T (1997) Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem 272:32727–32730. 10.1074/jbc.272.52.32727 [DOI] [PubMed] [Google Scholar]
- Bavley CC, Fischer DK, Rizzo BK, Rajadhyaksha AM (2017) Cav1.2 channels mediate persistent chronic stress-induced behavioral deficits that are associated with prefrontal cortex activation of the p25/Cdk5-glucocorticoid receptor pathway. Neurobiol Stress 7:27–37. 10.1016/j.ynstr.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman MA, Ogorodnik E, Glass MJ (2013) Acute morphine associated alterations in the subcellular location of the AMPA-GluR1 receptor subunit in dendrites of neurons in the mouse central nucleus of the amygdala: comparisons and contrasts with other glutamate receptor subunits. Synapse 67:692–704. 10.1002/syn.21673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SM, Bartsch D (2014) The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 357:463–476. 10.1007/s00441-014-1936-3 [DOI] [PubMed] [Google Scholar]
- Bouton ME. (2004) Context and behavioral processes in extinction. Learn Mem 11:485–494. 10.1101/lm.78804 [DOI] [PubMed] [Google Scholar]
- Brenhouse HC, Dumais K, Andersen SL (2010) Enhancing the salience of dullness: behavioral and pharmacological strategies to facilitate extinction of drug-cue associations in adolescent rats. Neuroscience 169:628–636. 10.1016/j.neuroscience.2010.05.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenhouse HC, Thompson BS, Sonntag KC, Andersen SL (2015) Extinction and reinstatement to cocaine-associated cues in male and female juvenile rats and the role of D1 dopamine receptor. Neuropharmacology 95:22–28. 10.1016/j.neuropharm.2015.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Peña CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, Deisseroth K, Nestler EJ (2016) In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci U S A 113:2726–2731. 10.1073/pnas.1521238113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota M, Bevilaqua LR, Kerr D, Medina JH, Izquierdo I (2003) Inhibition of mRNA and protein synthesis in the CA1 region of the dorsal hippocampus blocks reinstallment of an extinguished conditioned fear response. J Neurosci 23:737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla-Ortega E, Blanco E, Serrano A, Ladrón de Guevara-Miranda D, Pedraz M, Estivill-Torrús G, Pavón FJ, Rodríguez de Fonseca F, Santín LJ (2016) Pharmacological reduction of adult hippocampal neurogenesis modifies functional brain circuits in mice exposed to a cocaine conditioned place preference paradigm. Addict Biol 21:575–588. 10.1111/adb.12248 [DOI] [PubMed] [Google Scholar]
- Chambers RA, Self DW (2002) Motivational responses to natural and drug rewards in rats with neonatal ventral hippocampal lesions: an animal model of dual diagnosis schizophrenia. Neuropsychopharmacology 27:889–905. 10.1016/S0893-133X(02)00365-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoff EH, Pliakas AM, Carlezon WA Jr (2006) Microinjection of the L-type calcium channel antagonist diltiazem into the ventral nucleus accumbens shell facilitates cocaine-induced conditioned place preferences. Biol Psychiatry 59:1236–1239. 10.1016/j.biopsych.2005.09.024 [DOI] [PubMed] [Google Scholar]
- Chen CL, Liu H, Guan X (2013) Changes in microRNA expression profile in hippocampus during the acquisition and extinction of cocaine-induced conditioned place preference in rats. J Biomed Sci 20:96. 10.1186/1423-0127-20-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Maren S (2004) Factors regulating the effects of hippocampal inactivation on renewal of conditional fear after extinction. Learn Mem 11:598–603. 10.1101/lm.78704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Desmond TJ, Frey KA, Maren S (2005) Hippocampal inactivation disrupts the acquisition and contextual encoding of fear extinction. J Neurosci 25:8978–8987. 10.1523/JNEUROSCI.2246-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombag HS, Sutton JM, Takamiya K, Lee HK, Holland PC, Gallagher M, Huganir RL (2008) A necessary role for GluR1 serine 831 phosphorylation in appetitive incentive learning. Behav Brain Res 191:178–183. 10.1016/j.bbr.2008.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Carvalho Myskiw J, Furini CR, Benetti F, Izquierdo I (2014) Hippocampal molecular mechanisms involved in the enhancement of fear extinction caused by exposure to novelty. Proc Natl Acad Sci U S A 111:4572–4577. 10.1073/pnas.1400423111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedic N, Pöhlmann ML, Richter JS, Mehta D, Czamara D, Metzger MW, Dine J, Bedenk BT, Hartmann J, Wagner KV, Jurik A, Almli LM, Lori A, Moosmang S, Hofmann F, Wotjak CT, Rammes G, Eder M, Chen A, Ressler KJ, Wurst W, Schmidt MV, Binder EB, Deussing JM (epub ahead of print July 11, 2017) Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry. 10.1038/mp.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degoulet M, Stelly CE, Ahn KC, Morikawa H (2016) L-type Ca(2)(+) channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug-associated contextual memory. Mol Psychiatry 21:394–402. 10.1038/mp.2015.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Olmo N, Miguéns M, Higuera-Matas A, Torres I, García-Lecumberri C, Solís JM, Ambrosio E (2006) Enhancement of hippocampal long-term potentiation induced by cocaine self-administration is maintained during the extinction of this behavior. Brain Res 1116:120–126. 10.1016/j.brainres.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR (1999) Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A 96:3269–3274. 10.1073/pnas.96.6.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase V, Obermair GJ, Szabo Z, Altier C, Sanguesa J, Bourinet E, Flucher BE (2008) Stable membrane expression of postsynaptic CaV1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. J Neurosci 28:13845–13855. 10.1523/JNEUROSCI.3213-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Ghundi M, O'Dowd BF, George SR (2007) Insights into the role of dopamine receptor systems in learning and memory. Rev Neurosci 18:37–66. [DOI] [PubMed] [Google Scholar]
- Fiorenza NG, Rosa J, Izquierdo I, Myskiw JC (2012) Modulation of the extinction of two different fear-motivated tasks in three distinct brain areas. Behav Brain Res 232:210–216. 10.1016/j.bbr.2012.04.015 [DOI] [PubMed] [Google Scholar]
- Fricks-Gleason AN, Khalaj AJ, Marshall JF (2012) Dopamine D1 receptor antagonism impairs extinction of cocaine-cue memories. Behav Brain Res 226:357–360. 10.1016/j.bbr.2011.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangarossa G, Longueville S, De Bundel D, Perroy J, Hervé D, Girault JA, Valjent E (2012) Characterization of dopamine D1 and D2 receptor-expressing neurons in the mouse hippocampus. Hippocampus 22:2199–2207. 10.1002/hipo.22044 [DOI] [PubMed] [Google Scholar]
- Gao C, Sun X, Wolf ME (2006) Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem 98:1664–1677. 10.1111/j.1471-4159.2006.03999.x [DOI] [PubMed] [Google Scholar]
- Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE, Rajadhyaksha AM (2010) Molecular switch from L-type Ca v 1.3 to Ca v 1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci 30:17051–17062. 10.1523/JNEUROSCI.2255-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA (2008) Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci 28:6000–6009. 10.1523/JNEUROSCI.0384-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287:2262–2267. 10.1126/science.287.5461.2262 [DOI] [PubMed] [Google Scholar]
- Hell JW. (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81:249–265. 10.1016/j.neuron.2013.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123:949–962. 10.1083/jcb.123.4.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J, Maren S (2005) Electrolytic lesions of the dorsal hippocampus disrupt renewal of conditional fear after extinction. Learn Mem 12:270–276. 10.1101/lm.91705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG, Huntley GW, Benson DL (1994) Alpha calcium/calmodulin-dependent protein kinase II selectively expressed in a subpopulation of excitatory neurons in monkey sensory-motor cortex: comparison with GAD-67 expression. J Neurosci 14:611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir ZD, Martínez-Rivera A, Rajadhyaksha AM (2017a) From gene to behavior: L-Type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics 14:588–613. 10.1007/s13311-017-0532-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir ZD, Che A, Fischer DK, Rice RC, Rizzo BK, Byrne M, Glass MJ, De Marco Garcia NV, Rajadhyaksha AM (2017b) Rescue of impaired sociability and anxiety-like behavior in adult cacna1c-deficient mice by pharmacologically targeting eIF2alpha. Mol Psychiatry 22:1096–1109. 10.1038/mp.2017.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir ZD, Lee AS, Burgdorf CE, Fischer DK, Rajadhyaksha AM, Mok E, Rizzo B, Rice RC, Singh K, Ota KT, Gerhard DM, Schierberl KC, Glass MJ, Duman RS, Rajadhyaksha AM (2017c) Cacna1c in the prefrontal cortex regulates depression-related behaviors via REDD1. Neuropsychopharmacology 42:2032–2042. 10.1038/npp.2016.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AE. (2004) Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron 44:161–179. 10.1016/j.neuron.2004.09.016 [DOI] [PubMed] [Google Scholar]
- Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61:340–350. 10.1016/j.neuron.2009.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Ahn SM, Go BS, Wang JQ, Choe ES (2009) Alterations in AMPA receptor phosphorylation in the rat striatum following acute and repeated cocaine administration. Neuroscience 163:618–626. 10.1016/j.neuroscience.2009.06.054 [DOI] [PubMed] [Google Scholar]
- Kimura R, Silva AJ, Ohno M (2008) Autophosphorylation of alphaCaMKII is differentially involved in new learning and unlearning mechanisms of memory extinction. Learn Mem 15:837–843. 10.1101/lm.1049608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW (2010) Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci 30:7984–7992. 10.1523/JNEUROSCI.1244-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35:217–238. 10.1038/npp.2009.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF (2011) Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci 14:727–735. 10.1038/nn.2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, Witt SH, Backes H, Dietsche B, Nieratschker V, Shah NJ, Nöthen MM, Rietschel M, Kircher T (2014) A genome-wide supported variant in CACNA1C influences hippocampal activation during episodic memory encoding and retrieval. Eur Arch Psychiatry Clin Neurosci 264:103–110. 10.1007/s00406-013-0428-x [DOI] [PubMed] [Google Scholar]
- Kutlu MG, Gould TJ (2016) Effects of drugs of abuse on hippocampal plasticity and hippocampus-dependent learning and memory: contributions to development and maintenance of addiction. Learn Mem 23:515–533. 10.1101/lm.042192.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Radulovic J, Lukowiak K (2006) Extinction: [corrected] does it or doesn't it? The requirement of altered gene activity and new protein synthesis. Biol Psychiatry 60:344–351. 10.1016/j.biopsych.2006.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Gonzales KL, Lee A, Moosmang S, Hofmann F, Pieper AA, Rajadhyaksha AM (2012a) Selective genetic deletion of cacna1c in the mouse prefrontal cortex. Mol Psychiatry 17:1051. 10.1038/mp.2012.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Ra S, Rajadhyaksha AM, Britt JK, De Jesus-Cortes H, Gonzales KL, Lee A, Moosmang S, Hofmann F, Pieper AA, Rajadhyaksha AM (2012b) Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Mol Psychiatry 17:1054–1055. 10.1038/mp.2012.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103:479–489. 10.1152/jn.00835.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Song B, Kim J, Park K, Hong I, An B, Song S, Lee J, Park S, Kim J, Park D, Lee CJ, Kim K, Shin KS, Tsien RW, Choi S (2013) GluA1 phosphorylation at serine 831 in the lateral amygdala is required for fear renewal. Nat Neurosci 16:1436–1444. 10.1038/nn.3491 [DOI] [PubMed] [Google Scholar]
- Li B, Tadross MR, Tsien RW (2016) Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351:863–867. 10.1126/science.aad3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata SC, Pierce RC (2003) The roles of calcium/calmodulin-dependent and Ras/mitogen-activated protein kinases in the development of psychostimulant-induced behavioral sensitization. J Neurochem 85:14–22. [DOI] [PubMed] [Google Scholar]
- Liu FC, Graybiel AM (1996) Spatiotemporal dynamics of CREB phosphorylation: transient versus sustained phosphorylation in the developing striatum. Neuron 17:1133–1144. 10.1016/S0896-6273(00)80245-7 [DOI] [PubMed] [Google Scholar]
- Liu X, Liu Y, Zhong P, Wilkinson B, Qi J, Olsen CM, Bayer KU, Liu QS (2014) CaMKII activity in the ventral tegmental area gates cocaine-induced synaptic plasticity in the nucleus accumbens. Neuropsychopharmacology 39:989–999. 10.1038/npp.2013.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Ma H, Cohen S, Li B, Tsien RW (2012) Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep 33:97–101. 10.1042/BSR20120099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL (1997) Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem 272:32528–32533. 10.1074/jbc.272.51.32528 [DOI] [PubMed] [Google Scholar]
- Marchant NJ, Kaganovsky K, Shaham Y, Bossert JM (2015) Role of corticostriatal circuits in context-induced reinstatement of drug seeking. Brain Res 1628:219–232. 10.1016/j.brainres.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Lopes J, Tesfaye E, Israilov S, Van Kempen TA, Wang G, Glass MJ, Pickel VM, Iadecola C, Waters EM, Milner TA (2017) Redistribution of NMDA receptors in estrogen-receptor-beta-containing paraventricular hypothalamic neurons following slow-pressor angiotensin II hypertension in female mice with accelerated ovarian failure. Neuroendocrinology 104:239–256. 10.1159/000446073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Rivera A, Hao J, Tropea TF, Giordano TP, Kosovsky M, Rice RC, Lee A, Huganir RL, Striessnig J, Addy NA, Han S, Rajadhyaksha AM (epub ahead of print February 14 2017) Enhancing VTA Cav1.3 L-type Ca2+ channel activity promotes cocaine and mood-related behaviors via overlapping AMPA receptor mechanisms in the nucleus accumbens. Mol Psychiatry. 10.1038/mp.2017.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey EM, Aston-Jones G (epub ahead of print July 11 2017) Dorsal hippocampus drives context-induced cocaine seeking via inputs to lateral septum. Neuropsychopharmacology. 10.1038/npp.2017.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes J, Alves N, Borges S, Roehrs R, de Carvalho Myskiw J, Furini CR, Izquierdo I, Mello-Carpes PB (2015) Facilitation of fear extinction by novelty depends on dopamine acting on D1-subtype dopamine receptors in hippocampus. Proc Natl Acad Sci U S A 112:E1652–E1658. 10.1073/pnas.1502295112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Neisewander JL (2003) Dorsal, but not ventral, hippocampal lesions disrupt cocaine place conditioning. Neuroreport 14:2127–2131. 10.1097/00001756-200311140-00023 [DOI] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Speer CM, Neisewander JL (2006) Dorsal hippocampus inhibition disrupts acquisition and expression, but not consolidation, of cocaine conditioned place preference. Behav Neurosci 120:401–412. 10.1037/0735-7044.120.2.401 [DOI] [PubMed] [Google Scholar]
- Milner TA, Waters EM, Robinson DC, Pierce JP (2011) Degenerating processes identified by electron microscopic immunocytochemical methods. Methods Mol Biol 793:23–59. 10.1007/978-1-61779-328-8_3 [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T (2005) Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 25:9883–9892. 10.1523/JNEUROSCI.1531-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins C, Fishell G, Tsien RW (2016) Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89:1131–1156. 10.1016/j.neuron.2016.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell'Acqua ML (2014) AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7:1577–1588. 10.1016/j.celrep.2014.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myskiw JC, Izquierdo I, Furini CR (2014) Modulation of the extinction of fear learning. Brain Res Bull 105:61–69. 10.1016/j.brainresbull.2014.04.006 [DOI] [PubMed] [Google Scholar]
- Navakkode S, Sajikumar S, Korte M, Soong TW (2012) Dopamine induces LTP differentially in apical and basal dendrites through BDNF and voltage-dependent calcium channels. Learn Mem 19:294–299. 10.1101/lm.026203.112 [DOI] [PubMed] [Google Scholar]
- Nic Dhonnchadha BÁ, Lin A, Leite-Morris KA, Kaplan GB, Man HY, Kantak KM (2013) Alterations in expression and phosphorylation of GluA1 receptors following cocaine-cue extinction learning. Behav Brain Res 238:119–123. 10.1016/j.bbr.2012.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA (2005) Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat Neurosci 8:853–854. 10.1038/nn1476 [DOI] [PubMed] [Google Scholar]
- Olivito L, Saccone P, Perri V, Bachman JL, Fragapane P, Mele A, Huganir RL, De Leonibus E (2016) Phosphorylation of the AMPA receptor GluA1 subunit regulates memory load capacity. Brain Struct Funct 221:591–603. 10.1007/s00429-014-0927-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Piëch V, Sheng M (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4:917–926. 10.1038/nn0901-917 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K (2004) The mouse brain in stereotaxic coordinates. Boston: Elsevier Academic. [Google Scholar]
- Perry CJ, Zbukvic I, Kim JH, Lawrence AJ (2014) Role of cues and contexts on drug-seeking behaviour. Br J Pharmacol 171:4636–4672. 10.1111/bph.12735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Palay S, Webster H (1991) The fine structure of the nervous system. New York: OUP. [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ (2009) Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem 16:279–288. 10.1101/lm.1041309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JP, Kurucz OS, Milner TA (1999) Morphometry of a peptidergic transmitter system: dynorphin B-like immunoreactivity in the rat hippocampal mossy fiber pathway before and after seizures. Hippocampus 9:255–276. 10.1002/(SICI)1098-1063(1999)9:3%3C255::AID-HIPO6%3E3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- Pierce RC, Wolf ME (2013) Psychostimulant-induced neuroadaptations in nucleus accumbens AMPA receptor transmission. Cold Spring Harb Perspect Med 3:a012021. 10.1101/cshperspect.a012021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puighermanal E, Cutando L, Boubaker-Vitre J, Honoré E, Longueville S, Hervé D, Valjent E (2017) Anatomical and molecular characterization of dopamine D1 receptor-expressing neurons of the mouse CA1 dorsal hippocampus. Brain Struct Funct 222:1897–1911. 10.1007/s00429-016-1314-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D (2008) Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 33:56–72. 10.1038/sj.npp.1301555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwanska K, Medvedev NI, Pereira GS, Engmann O, Thiede N, Moraes MF, Villers A, Irvine EE, Maunganidze NS, Pyza EM, Ris L, Szymańska M, Lipiński M, Kaczmarek L, Stewart MG, Giese KP (2011) Mechanism for long-term memory formation when synaptic strengthening is impaired. Proc Natl Acad Sci U S A 108:18471–18475. 10.1073/pnas.1109680108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DR, Bell GH, Lasseter HC, Xie X, Traina SA, Fuchs RA (2009) Dorsal hippocampal regulation of memory reconsolidation processes that facilitate drug context-induced cocaine-seeking behavior in rats. Eur J Neurosci 30:901–912. 10.1111/j.1460-9568.2009.06889.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MT, Abbott TB, Chung L, Gulcicek EE, Stone KL, Colangelo CM, Lam TT, Nairn AC, Taylor JR, Torregrossa MM (2016) Phosphoproteomic analysis reveals a novel mechanism of CaMKIIalpha regulation inversely induced by cocaine memory extinction versus reconsolidation. J Neurosci 36:7613–7627. 10.1523/JNEUROSCI.1108-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond SA, Irving AJ, Molnar E, McIlhinney RA, Michelangeli F, Henley JM, Collingridge GL (1996) Localization of the glutamate receptor subunit GluR1 on the surface of living and within cultured hippocampal neurons. Neuroscience 75:69–82. 10.1016/0306-4522(96)00217-5 [DOI] [PubMed] [Google Scholar]
- Robison AJ. (2014) Emerging role of CaMKII in neuropsychiatric disease. Trends Neurosci 37:653–662. 10.1016/j.tins.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Rose J, Jin SX, Craig AM (2009) Heterosynaptic molecular dynamics: locally induced propagating synaptic accumulation of CaM kinase II. Neuron 61:351–358. 10.1016/j.neuron.2008.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Kourrich S, Thomas MJ (2011) Synaptic adaptations in the nucleus accumbens caused by experiences linked to relapse. Biol Psychiatry 69:1124–1126. 10.1016/j.biopsych.2010.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q, Garraway SM, Hofmann F, Moosmang S, Striessnig J, Inturrisi CE, Rajadhyaksha AM (2011) Cav1.2 L-type Ca(2)(+) channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Ca(v)1.3 channels. J Neurosci 31:13562–13575. 10.1523/JNEUROSCI.2315-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW, Choi KH, Simmons D, Walker JR, Smagula CS (2004) Extinction training regulates neuroadaptive responses to withdrawal from chronic cocaine self-administration. Learn Mem 11:648–657. 10.1101/lm.81404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL (2007) The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 23:613–643. 10.1146/annurev.cellbio.23.090506.123516 [DOI] [PubMed] [Google Scholar]
- Shonesy BC, Jalan-Sakrikar N, Cavener VS, Colbran RJ (2014) CaMKII: a molecular substrate for synaptic plasticity and memory. Prog Mol Biol Transl Sci 122:61–87. 10.1016/B978-0-12-420170-5.00003-9 [DOI] [PubMed] [Google Scholar]
- Sil'kis IG. (2008) The role of dopamine-dependent negative feedback in the hippocampus-basal ganglia-thalamus-hippocampus loop in the extinction of responses. Neurosci Behav Physiol 38:399–405. 10.1007/s11055-008-0057-4 [DOI] [PubMed] [Google Scholar]
- Simms BA, Zamponi GW (2014) Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82:24–45. 10.1016/j.neuron.2014.03.016 [DOI] [PubMed] [Google Scholar]
- Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ (2015) Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149:150–190. 10.1016/j.pharmthera.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Lobo MK, Spencer S, Kalivas PW (2013) Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol 23:546–552. 10.1016/j.conb.2013.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P (2014) L-type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3:15–38. 10.1002/wmts.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H (1991) The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun 181:968–975. 10.1016/0006-291X(91)92031-E [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC Jr, Nairn AC, Greengard P (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14:385–397. 10.1016/0896-6273(95)90294-5 [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D, Karanian DA, Monteggia LM, Neve RL, Self DW (2003) Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421:70–75. 10.1038/nature01249 [DOI] [PubMed] [Google Scholar]
- Szalay JJ, Morin ND, Kantak KM (2011) Involvement of the dorsal subiculum and rostral basolateral amygdala in cocaine cue extinction learning in rats. Eur J Neurosci 33:1299–1307. 10.1111/j.1460-9568.2010.07581.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay JJ, Jordan CJ, Kantak KM (2013) Neural regulation of the time course for cocaine-cue extinction consolidation in rats. Eur J Neurosci 37:269–277. 10.1111/ejn.12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I (2003) The role of NMDA glutamate receptors, PKA, MAPK, and CaMKII in the hippocampus in extinction of conditioned fear. Hippocampus 13:53–58. 10.1002/hipo.10043 [DOI] [PubMed] [Google Scholar]
- Temme SJ, Bell RZ, Fisher GL, Murphy GG (2016) Deletion of the mouse homolog of CACNA1C disrupts discrete forms of hippocampal-dependent memory and neurogenesis within the dentate gyrus. eNeuro 3. 10.1523/ENEURO.0118-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippens AL, Pare JF, Langwieser N, Moosmang S, Milner TA, Smith Y, Lee A (2008) Ultrastructural evidence for pre- and postsynaptic localization of Cav1.2 L-type Ca2+ channels in the rat hippocampus. J Comp Neurol 506:569–583. 10.1002/cne.21567 [DOI] [PubMed] [Google Scholar]
- Tipps ME, Raybuck JD, Lattal KM (2014) Substance abuse, memory, and post-traumatic stress disorder. Neurobiol Learn Mem 112:87–100. 10.1016/j.nlm.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea TF, Kosofsky BE, Rajadhyaksha AM (2008) Enhanced CREB and DARPP-32 phosphorylation in the nucleus accumbens and CREB, ERK, and GluR1 phosphorylation in the dorsal hippocampus is associated with cocaine-conditioned place preference behavior. J Neurochem 106:1780–1790. 10.1111/j.1471-4159.2008.05518.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venniro M, Caprioli D, Shaham Y (2016) Animal models of drug relapse and craving: From drug priming-induced reinstatement to incubation of craving after voluntary abstinence. Prog Brain Res 224:25–52. 10.1016/bs.pbr.2015.08.004 [DOI] [PubMed] [Google Scholar]
- Vianna MR, Coitinho AS, Izquierdo I (2004) Role of the hippocampus and amygdala in the extinction of fear-motivated learning. Curr Neurovasc Res 1:55–60. 10.2174/1567202043480170 [DOI] [PubMed] [Google Scholar]
- Vinet J, Sík A (2006) Expression pattern of voltage-dependent calcium channel subunits in hippocampal inhibitory neurons in mice. Neuroscience 143:189–212. 10.1016/j.neuroscience.2006.07.019 [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW (2012) Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149:1112–1124. 10.1016/j.cell.2012.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF (2006) Learning induces long-term potentiation in the hippocampus. Science 313:1093–1097. 10.1126/science.1128134 [DOI] [PubMed] [Google Scholar]
- Wolf ME. (2016) Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci 17:351–365. 10.1038/nrn.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A 98:2808–2813. 10.1073/pnas.051634198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Wells AM, Fuchs RA (2014) Cocaine seeking and taking: role of hippocampal dopamine D1-like receptors. Int J Neuropsychopharmacol 17:1533–1538. 10.1017/S1461145714000340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Zhou Q (2010) Perisynaptic GluR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc Natl Acad Sci U S A 107:11999–12004. 10.1073/pnas.0913004107 [DOI] [PMC free article] [PubMed] [Google Scholar]