

Abstract
Preterm infants are at risk for a broad spectrum of neurobehavioral disabilities associated with diffuse disturbances in cortical growth and development. During brain development, subplate neurons (SPNs) are a largely transient population that serves a critical role to establish functional cortical circuits. By dynamically integrating into developing cortical circuits, they assist in consolidation of intracortical and extracortical circuits. Although SPNs reside in close proximity to cerebral white matter, which is particularly vulnerable to oxidative stress, the susceptibility of SPNs remains controversial. We determined SPN responses to two common insults to the preterm brain: hypoxia-ischemia and hypoxia. We used a preterm fetal sheep model using both sexes that reproduces the spectrum of human cerebral injury and abnormal cortical growth. Unlike oligodendrocyte progenitors, SPNs displayed pronounced resistance to early or delayed cell death from hypoxia or hypoxia-ischemia. We thus explored an alternative hypothesis that these insults alter the maturational trajectory of SPNs. We used DiOlistic labeling to visualize the dendrites of SPNs selectively labeled for complexin-3. SPNs displayed reduced basal dendritic arbor complexity that was accompanied by chronic disturbances in SPN excitability and synaptic activity. SPN dysmaturation was significantly associated with the level of fetal hypoxemia and metabolic stress. Hence, despite the resistance of SPNs to insults that trigger white matter injury, transient hypoxemia disrupted SPN arborization and functional maturation during a critical window in cortical development. Strategies directed at limiting the duration or severity of hypoxemia during brain development may mitigate disturbances in cerebral growth and maturation related to SPN dysmaturation.
SIGNIFICANCE STATEMENT The human preterm brain commonly sustains blood flow and oxygenation disturbances that impair cerebral cortex growth and cause life-long cognitive and learning disabilities. We investigated the fate of subplate neurons (SPNs), which are a master regulator of brain development that plays critical roles in establishing cortical connections to other brain regions. We used a preterm fetal sheep model that reproduces key features of brain injury in human preterm survivors. We analyzed the responses of fetal SPNs to transient disturbances in fetal oxygenation. We discovered that SPNs are surprisingly resistant to cell death from low oxygen states but acquire chronic structural and functional changes that suggest new strategies to prevent learning problems in children and adults that survive preterm birth.
Keywords: anatomy and physiology, dendrite, hypoxia, ischemia, preterm brain injury, subplate neurons
Introduction
During human development, the preterm brain commonly sustains antenatal, perinatal, or postnatal disturbances in cerebral blood flow and metabolism. In 25%–50% of survivors of preterm birth, these insults contribute to a broad spectrum of unexplained neurological disabilities in motor control, vision, language, cognition, attention, learning, and social development (Vohr, 2014). These behavioral abnormalities coincide with significant impairment in cortical growth and connectivity defined by volumetric MRI studies (Ball et al., 2013; Vinall et al., 2013). With the exception of more severe cerebral injury (Ligam et al., 2009; Andiman et al., 2010; Kinney et al., 2012), most cortical volume loss appears unrelated to significant cortical oxidative damage or neuronal degeneration (Back et al., 2005; Dean et al., 2013; Back and Miller, 2014). Given that subplate neurons (SPNs) mature when the preterm cerebrum displays peak vulnerability (Volpe, 2009), insults to SPNs were proposed to contribute to aberrant cortical growth and connectivity (Volpe, 1996). SPNs are a largely transient and unique neuronal population just below cortical layer 6 (Kostovic and Rakic, 1990; Hoerder-Suabedissen et al., 2013) that dynamically integrate into developing cortical circuits to promote consolidation of functional intracortical and extracortical connectivity. SPNs generate the first axons of the corticothalamic tract (McConnell et al., 1989). These projections provide a scaffold for thalamic inputs to directly innervate cortical layer 4 (Kanold and Luhmann, 2010; Viswanathan et al., 2012). Most SPNs later undergo programmed cell death (Chun and Shatz, 1989; Torres-Reveron and Friedlander, 2007), which leaves a monosynaptic thalamocortical projection to layer 4.
Because ablation of SPNs disrupts the entry or patterning of corticothalamic projections (Ghosh et al., 1990; Kanold et al., 2003), hypoxia-ischemia (HI) was proposed to similarly trigger SPN degeneration to disrupt formation of thalamocortical afferents. After HI, neonatal rat SPNs selectively degenerated unlike other neuronal populations (McQuillen et al., 2003). However, analysis of preterm human autopsy brains found no significant loss of the total SPN population (Kinney et al., 2012). Recently, three different SPN-specific markers were used to quantify SPN vulnerability to HI in the neonatal rat. SPN degeneration was only observed in association with more severe cortical neuronal loss (Okusa et al., 2014).
Because these disparate results leave unresolved the role of SPNs in preterm brain injury, we used an instrumented preterm fetal sheep model of in utero global cerebral HI that closely replicates in a gyrencephalic brain both human cortical maturation and the spectrum of antenatal and postnatal preterm cerebral injury (Back et al., 2012; Back, 2014). We uniquely defined the dendritic arbor of preterm SPNs through DiOlistic labeling in combination with immunolabeling for complexin 3 (Cplx3), a selective SPN marker (Hoerder-Suabedissen and Molnár, 2013). We analyzed SPN responses to hypoxia (Hx) and HI. Whereas SPNs were strikingly resistant to degeneration from either insult, they displayed persistent disturbances in dendritic arbor maturation that were significantly related to the severity of the hypoxic insult. Dysmaturation of SPNs was accompanied by chronically increased SPN synaptic activity and a highly correlated reduced electrical excitability. Hence, aberrant SPN maturation appears to contribute to disturbances in preterm cortical growth and connectivity during a critical window in the evolution of cortical circuitry.
Materials and Methods
Ethics statement.
All experiments performed on animals adhere to strict protocols approved by the Oregon Health & Science University Institutional Animal Care and Use Committee.
Instrumented fetal sheep surgery.
Sterile surgeries on time-bred pregnant ewes were performed between 88 and 92 d gestational age (dGA; term 145 dGA) at Oregon Health and Science University as previously described (Hagen et al., 2014). Briefly, a midline laparotomy and hysterotomy were used in a sterile field to access each of the twin fetuses. A vinyl catheter was placed nonocclusively in a carotid, brachial, or femoral artery, and an inflatable Silastic occluder was placed around the brachiocephalic artery to control blood flow to the brain through the common carotid and vertebral arteries. In the sheep, all collateral circulation to the brain, except the spinal arteries, is provided through branches of the brachiocephalic artery. An additional catheter was sewn to the fetal skin to allow monitoring of the fetal amniotic fluid pressure. Following surgical instrumentation, the fetus was returned to the uterus, the maternal incision was closed, and the ewe was allowed to recover from surgery for 3 d after which HI studies were conducted (see Fig. 1A). Fetuses of either sex were included in the study (n = 10 female, n = 12 male).
Figure 1.
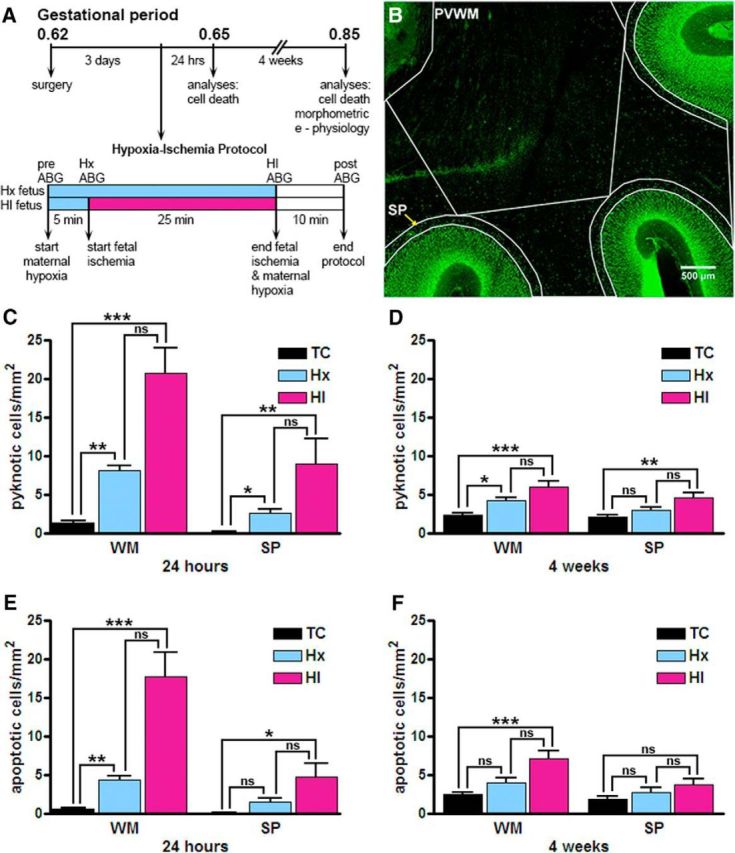
Increased cellular degeneration following fetal HI, analyzed in the WM and the SP. A, Flow chart of the experimental protocol described in Materials and Methods. B, A fetal (93 dGA, 0.65 gestation) sheep brain slice collected 24 h after exposure to maternal Hx stained for the Tbr1 cell marker (green). SP and PVWM demarcate the subplate and periventricular white matter regions, respectively, which were sampled for pyknotic and apoptotic cell death. Scale bar, 500 μm. C, D, Density of pyknotic nuclei visualized using Hoechst 33342 at 24 h (C) and 4 weeks (D) after HI. E, F, Density of apoptotic cells visualized using AC3 at 24 h (E) and 4 weeks (F) after HI. Error bars indicate SE. Overall statistical comparisons were by Kruskal–Wallis Test. *p < 0.05 (Dunn's post hoc test for multiple comparisons). **p < 0.01 (Dunn's post hoc test for multiple comparisons). ***p < 0.001 (Dunn's post hoc test for multiple comparisons).
Cerebral HI studies and physiological monitoring.
Maternal Hx was induced by supplying the pregnant ewe with a mixture of 50% room air and 50% nitrogen gas for an inhaled oxygen fraction of 10.5%. Following 5 min of maternal Hx, cerebral ischemia was initiated in one of the twin fetuses (designated HI fetus) and maintained by inflating the brachiocephalic occluder for 25 min. Four arterial blood gas (ABG) samples were collected from each fetus throughout experimentation: pre-Hx (baseline or pre-ABG), at 5 min after initiation of maternal Hx (Hx ABG), 25 min after initiation of ischemia-induction (HI or HI ABG), and 10 min after cessation of ischemia (recovery or post-ABG). Blood samples were analyzed using an ABL800 flex blood gas analyzer (Radiometer Medical, A/S RRID:SCR_014772). If the fetal arterial oxygen concentration (ctO2) fell <5.0 vol% after a 5 min exposure to maternal Hx, the maternal inhaled oxygen fraction was enriched to 14% for the duration of the experiment. Fetuses exposed exclusively to maternal Hx were designated hypoxic controls (Hx), whereas fetuses exposed to maternal Hx in combination with cerebral hypoperfusion were designated HI animals (see Fig. 1A). Each experimental twin pregnancy was comprised of a Hx and HI pair.
Tissue collection and processing.
To ensure the viability of the fetal brains for concurrent electrophysiological studies, the ewe was placed under general anesthesia and ventilated. After fetal harvest, ewes were then killed with a 12 ml intravenous bolus of a pentobarbital sodium and phenytoin sodium solution (Euthasol NAC# 10230760, Virbac). Fetal brains were harvested either 24 h (Hx, n = 5; HI, n = 4) or 4 weeks (Hx, n = 8; HI, n = 8) after exposure to maternal Hx and HI. Separate groups of appropriately age-matched normal twin fetuses were harvested to serve as true controls (TC: 4 weeks/120 dGA, n = 7; 24 h/95 dGA, n = 3) to the Hx and HI experimental groups. Fetal brains were removed from the cranial vault, and the cerebellum and brainstem were removed by cutting the cerebral peduncles at the pontopeduncular junction. The remaining cerebrum and diencephalon was then divided into anteroposterior pieces with a single coronal cut at the rostral end of the ectomarginal sulcus, caudal to the ansate (cruciate) sulcus. Next, the anterior and posterior blocks were hemisected by a single sagittal cut through the midline. The frontal block (anterior to section 0640 in the Michigan State University online sheep brain atlas: https://msu.edu/∼brains/brains/sheep/index.html) of one hemisphere (specifically, the ansate (cruciate) gyrus containing the primary motor cortex) (Simpson and King, 1911; Vanderwolf and Cooley, 1990) was used for the cortical subplate (SP) electrophysiological studies reported on in this paper, whereas corresponding regions of the frontal block, including primary motor and somatosensory cortical areas (Dinopoulos et al., 1985) of the opposite hemisphere, were used for the SP morphometric studies. The hemisphere destined for morphometric studies was subdivided into two equivalent coronal blocks (labeled A and B, where A is most anterior), and immersed in 4% PFA in 0.1 m phosphate buffer (pH 7.4, 4°C). Subsequent tissue processing of the B-block (level of frontal periventricular white matter (WM), and the head of the caudate nucleus) varied according to individual studies. This block has been analyzed extensively by us in prior studies (Riddle et al., 2006; Riddle et al., 2011) because of the predilection for cerebral WM injury at this level both in our fetal sheep preparations and in human preterm infants.
Fetal brains used for glial fibrillary acidic protein (GFAP) area fraction and Cplx3/1-1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) studies were collected 4 weeks after exposure to maternal Hx and/or HI at ∼119–123 dGA. B-blocks were removed from fixative 3 h after immersion and were cut into 200 μm coronal slices using a vibrating microtome (VT1000S, Leica Microsystems). The brain slices were stored in PBS with 0.05% sodium azide at 4°C until use. Sections were collected at equivalent intervals across the B-block to ensure systematic sampling across the entire block.
Fetal brains used for activated caspase-3 (AC3)/T-Box, Brain,1 (Tbr1) counts were collected both 24 h (TC, n = 3; Hx, n = 5; HI, n = 4) and 4 weeks (TC, n = 7; Hx, n = 3; HI, n = 6) after HI. Fetal brains were also examined for the presence of inflammation 24 h after HI using GFAP and Iba1 antisera (TC, Hx, and HI, n = 3 animals/treatment group). B-blocks were sectioned at 50 μm using a vibrating microtome and stored until further use at −20°C in cryoprotectant (30% v/v ethylene chloride, 15% m/v sucrose, 0.3 m PO4).
Immunohistochemical studies.
Antigen retrieval (Tris-EDTA with 0.05% Tween 20 at pH 9.0 for 10 min) and Triton X-100 (0.1% in 55 mm PBS, for all staining steps) were used in all immunohistochemical studies to facilitate antibody penetration of tissue, except when double-staining with DiI was necessary. AC3 (rabbit polyclonal, Cell Signaling Technology, catalog #9662, RRID:AB_331439, 1:500) and Tbr1 (chicken polyclonal, Millipore, catalog #AB2261, RRID:AB_10615497; 1:100) primary antisera were visualized with rhodamine Red-X-conjugated secondary antibodies against rabbit (Jackson ImmunoResearch Laboratories, catalog #111-295-045, RRID:AB_2338025; 1:200) or chicken (Invitrogen, catalog #A-11041, RRID:AB_2534098; 1:100), respectively, and counterstained with the nuclear marker Hoechst 33342 (Invitrogen, catalog #H3570). Cplx3 primary antibody (rabbit IgG, Synaptic Systems, catalog #122 302, RRID:AB_2281240, 1:1000) was visualized with either a FITC-conjugated secondary antibody against rabbit (Jackson ImmunoResearch Laboratories, 1:200), or a biotin-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, catalog #111-065-045, RRID:AB_2337962, 1:200) and FITC-conjugated streptavidin (Jackson ImmunoResearch Laboratories, catalog #016-010-084, RRID:AB_2337236, 1:200), and a Hoechst counterstain. A rabbit polyclonal antisera against GFAP (Dako, catalog #Z0334, RRID:AB_10013382, 1:500) and a rabbit polyclonal antisera against the Iba1 calcium binding protein expressed in microglia/macrophages (Iba1; Wako Chemicals, catalog #019-19741, RRID:AB_2665520, 1:500) were visualized with a biotin-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories, catalog #111-065-045, RRID:AB_2337962, 1:200) and FITC-conjugated streptavidin (Jackson ImmunoResearch Laboratories, catalog #016-010-084, RRID:AB_2337236, 1:200). The mouse monoclonal O4 used to visualize oligodendrocytes was a gift from Dr. Steven E. Pfeiffer (University of Connecticut Medical School, Farmington, CT), and its purification and visualization have been described previously (Bansal et al., 1989; Back et al., 2001). Antigen retrieval and detergents were not used in DiI studies to maintain cell membrane integrity. To improve antibody penetration, both primary (Cplx3) and secondary (biotinylated anti-rabbit) antibody incubations steps were extended to 6 d and conducted in PBS with 0.05% sodium azide at 4°C to avoid tissue degradation. Tertiary incubations (FITC-streptavidin) were conducted overnight at 4°C in PBS. Overnight washes were conducted after primary and secondary antibody incubation steps.
DiI bullet preparation.
DiI bullets were prepared as described in a JoVE tutorial (Seabold et al., 2010).
DiI delivery.
DiI was delivered to neurons by shooting DiI-covered tungsten bullets into Cplx3-stained tissue slices using a Helios Gene Gun System (Bio-Rad Laboratories, catalog #165-2431 and #165-2432). Air pressure was set to 180 psi for optimum DiI penetration of the tissue. Following DiI delivery, tissue sections were washed in PBS for 30 min and mounted for imaging using Vectashield Antifade Mounting Medium (Vector Laboratories, catalog #H-1000, RRID:AB_2336789). Sections were imaged within 24 h to avoid DiI diffusion out of labeled cellular membranes.
SP cell death imaging and analysis.
Image montages for Tbr1 and AC3 staining were collected using a Confocal Microscope A1 system (Nikon). All images were collected using a 20× air lens (PLAN APO, Nikon, NA = 0.8) with consistent acquisition settings maintained between samples (pinhole = 57.6 μm, 2× optical zoom, 1024 × 1024 pixel resolution per tile, 2× averaging); 405 nm and 561 nm excitation used a solid state laser source, whereas 488 nm emission used a variable argon/krypton laser set to emit at 488 nm.
Regions of interest (ROIs) were chosen to include the periventricular WM and SP. Confocal images in the Nikon *.nd2 format were opened using FIJI RRID:SCR_002285 (Schindelin et al., 2012), and Tbr1 and Hoechst staining was used in combination to define the lower boundary of the cortical mantle and the boundary between the SP and periventricular WM (see Fig. 1B), which were outlined using FIJI's ROI tool. A grid was overlaid on the image to assist in counting, and the cell-counter plugin of FIJI was used to note the presence of apoptotic, pyknotic, and Tbr1+ cells in each ROI.
GFAP area fraction analysis.
At 4 weeks after HI, WM injury was assessed in 7 brains/treatment group by imaging GFAP staining using a systematic random sampling approach with StereoInvestigator software (MicroBrightfield RRID:SCR_004314) on an upright fluorescent microscope (Zeiss AxioImager M2). Systematic random sampling image series were acquired at 20× air lens (Zeiss EC Plan-NeoFluar, NA = 0.5), with image distribution such that ∼25% of the WM from each section was imaged. Quantification of the GFAP area fraction for each image was performed as described previously (Hagen et al., 2014).
Stereological quantification of GFAP cell densities.
The density of GFAP-labeled cell somas 4 weeks after HI was estimated using the StereoInvestigator optical fractionator probe (version 2017.01.1, MBF Bioscience, RRID:SCR_004314) on a Zeiss Imager M2 upright microscope. Contours for the WM ROIs were drawn as previously described (Riddle et al., 2006). A digital sampling grid containing at least 50 sampling sites overlaid the entire ROI. Cells were counted using an APO chrom-AT 63×/1.40 NA oil objective within an 85 × 85 μm counting frame. The distance from the top of the section to the unbiased virtual counting zone was fixed at 3 μm (guard zone), and the height of the unbiased virtual counting zone (optical dissector) was set at 30 μm. The thickness of the section was measured at every sample site. Density and coefficients of error were calculated as previously described using the averages of the slices sampled from each animal (TC, n = 5; Hx, n = 3; and HI, n = 3 animals) (McNeal et al., 2016).
Neuron virtual reconstruction.
Confocal z stacks of the 200-μm-thick tissue sections were acquired at 20× (488 nm and 561 nm excitation, 2048 × 2048 pixel resolution, 1× optical zoom, pinhole = 19.2 μm, 1 μm step size, 2× averaging), such that the entire dendritic arbor of SPNs could be visualized and later reconstructed. Images were converted from *.nd2 to *.tif format using FIJI, and tiff stacks were opened using Neurolucida (MBF Bioscience, RRID:SCR_001775). Neurons were traced if they met the following criteria: (1) expression of Cplx3, (2) adequate filling of the cell body and dendritic processes with DiI, (3) presence of the entire dendritic arbor without significant process truncation, and (4) no obstruction of the neuron by other DiI-filled structures. The outline of the cell soma was traced at its widest point in the 2D plane to provide an estimate of cross-sectional area, and the dendritic arbor was traced in the x, y, and z coordinate planes. The dendritic arbor was not followed into adjacent slices, and the dendritic diameter was not studied. Using these selection criteria, a total of 658 Cplx3+ SPNs (n = 230 TC neurons; 6 fetuses, 4 female, 2 male; n = 217 Hx neurons, 8 fetuses, 3 female, 5 male; n = 211 HI neurons, 8 fetuses, 3 female, 5 male) were examined. To estimate arbor complexity, a value was computed by Neurolucida software by the following equation: [Sum of the terminal orders + Number of terminals] × [Total dendritic length/Number of primary dendrites] (Pillai et al., 2012).
Fetal ovine ansate (cruciate) gyrus acute slice preparation.
Methods for preparation of brain slices and subsequent patch-clamp recording were similar to our previous reports in rodents (Andrade and Rossi, 2010; Mohr et al., 2010), nonhuman primates (Welsh et al., 2011; Mohr et al., 2013), and fetal sheep (McClendon et al., 2014) All standard salts were from Sigma Chemicals, and kynurenic acid was from Abcam.
After rapid removal of the brain, frontal forebrain was rapidly isolated and immersed in ice-cold (0°C–2°C), low-sodium, artificial cerebrospinal fluid (ACSF) composed of the following (in mm): 234 sucrose, 3.2 KCl, 17 NaH2PO4, 6 MgSO4, 10 d-glucose, 27.7 NaHCO3, and 0.216 CaCl2, and transported to the electrophysiology laboratory (total transport time was ∼5 min). The ansate (cruciate) gyrus was microdissected from the surrounding cortex and mounted perpendicular to the WM in a slicing chamber filled with ice-cold (0°C–2°C), low sodium ACSF. Horizontal slices (225 μm) were made with a ceramic razor blade mounted in a vibrating microtome (Vibratome, Oxford Instruments). Slices were incubated in warmed (33 ± 1°C), normal ACSF (in mm as follows: 124 NaCl, 26 NaHCO3, 1 NaH2PO4, 2.5 KCl, 2.5 CaCl2, 2 MgCl2, and 10 d-glucose, and bubbled with 95%O2/5% CO2, pH 7.4) for 1 h after brain collection and subsequently held in room temperature ACSF until used. Kynurenic acid (1 mm) was included in the holding solution (to block glutamate receptors to reduce potential excitotoxic damage) but was omitted from other experimental solutions.
Electrophysiology.
Slices were placed in a submersion chamber and viewed with a Hitachi KP-M2RN CCD camera (Kokuasi Electric) mounted on an Olympus BX-51 microscope equipped with a 60× 0.9 N.A. water-immersion objective (Olympus) and modified Dodt contrast enhancement optics. Slices were perfused with warmed (32°C–34°C) ACSF at a rate of ∼3 ml/min. The SPN layer was ∼1.5 mm from the edge of the slice, adjacent to the WM, and was visually identified using DIC. Patch-clamp recordings (using a Molecular Devices Multiclamp 700-B amplifier in voltage- and current-clamp configurations) were made with patch pipettes constructed from thick-walled borosilicate glass capillaries and filled with an internal solution containing the following (in mm): 132.3 K gluconate, 7.7 KCl, 4 NaCl, 0.5 CaCl2, 10 HEPES, 5 EGTA, 4 MgATP, and 0.5 Na2GTP. Solutions were pH adjusted to 7.2–7.3 with KOH. The internal solution also contained 0.125% neurobiotin in ∼50% of the recordings for histology. Since previous studies found that neurobiotin can affect electrical excitability at higher concentrations (Schlösser et al., 1998), we initially analyzed neuronal recording properties with varying concentrations of neurobiotin. When 0.5% neurobiotin was included in the recording electrode, electrical excitability was clearly altered (data not shown). However, no significant effects were detected at 0.125%, and all reported studies were done with concentrations of neurobiotin of ≤0.125%. Electrode resistance was 3.5–5.5 mΩ. Cells were rejected if access resistance was >10 mΩ.
Recording protocol.
After establishing whole-cell voltage-clamp configuration with stable access resistance (assessed by −5 mV voltage steps), the amplifier was switched to current-clamp mode. For each cell in the three experimental groups, a constant current was injected to bring Vm to −65 mV (TC = −66.0 ± 18.1 pA, n = 36; Hx = −19.3 ± 5.7 pA, n = 53; HI = −31.1 ± 6.5 pA, n = 52 cells). Cell excitability was determined by injecting a series of currents (300 ms duration), from −40 pA to 125 pA in 15 pA increments per step (12 current injection steps), and recording the voltage response. If none of the depolarizing current injections evoked action potentials, an additional current injection protocol was implemented, starting at 125 pA and increased by 25 pA increment steps to 250 pA (6 total steps). To quantify general excitability, plots of the number of action potentials evoked by a given current injection were generated for each cell (see Fig. 9G), and the area under the resultant curve (AUC) was determined and used for a single, summary value of cellular excitability to be compared with various ischemic/metabolic parameters (see Fig. 9H–J). Subsequently, the amplifier was switched to voltage-clamp configuration (Vh = −65 mV), and an I–V curve was constructed by implementing a series of 10 voltage steps from −85 mV to 5 mV in 10 mV increments, each lasting 50 ms. Voltage and current responses were acquired at 20 kHz, filtered at 10 kHz, and their magnitude and kinetics were analyzed with pClamp software (version 10.6, Molecular Devices) and IGOR (Wavemetrics, RRID:SCR_000325). After completion of the I–V protocols, the holding potential was changed to Vh = −80 mV and spontaneous synaptic activity was recorded at 5 kHz, filtered at 2 kHz for 3–5 min. The kinetics and magnitude of the spontaneous synaptic currents were analyzed with pClamp. For correlation analysis between synaptic current frequencies and metabolic parameters, two HI cells (of 51 total) were excluded from analysis as aberrant outliers because their frequency was >3 SDs higher than the mean for all cells.
Figure 9.
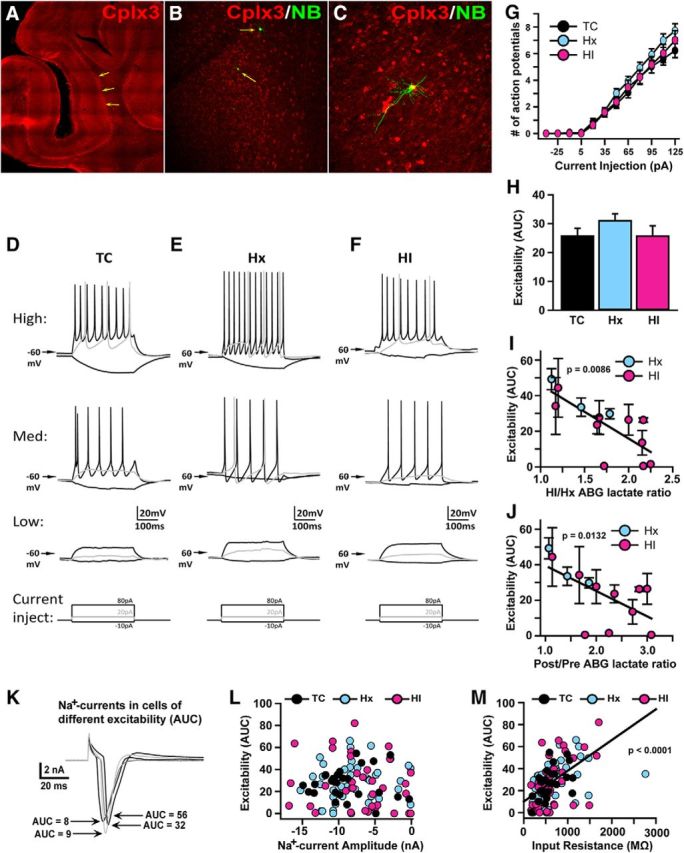
SPN excitability is directly associated with hypoxemia-induced lactate accumulation. A–C, Images show increasingly high-magnification views of a Cplx3-stained cortical slices from which SPN recordings were made. Arrows point to cells that were recorded with neurobiotin included in the recording electrode (B), showing the location of recorded cells within the densely Cplx3-stained SP (A, B), and typical SPN morphology and colocalization with Cplx3 (C). D–F, Representative current-clamp recordings from high (top row), medium (middle row), and low (bottom row) excitable cells from each group of animals: TC (D), Hx (E), and HI (F). Square traces beneath the recordings represent the current injection protocol used to elicit the responses shown. G, Plot of mean number of action potentials elicited for each level of current injection for the 3 groups of cells. H, Plot of mean AUC (i.e., area under individual cell action potential plots, as in G) for each group of cells. I, J, Plots of mean excitability (AUC) for each fetus (n = 3 Hx, 10 HI; mean ± SEM) versus its ABG lactate ratio for the HI/Hx (I) and Post/Pre (J) periods. K, Representative traces from voltage-clamped (−65 mV) cells showing the sodium current elicited by a depolarizing step from −65 to 15 mV in cells with varying levels of excitability (AUC), demonstrating that low excitability (D, E, bottom traces) is not due to lack of Na+ channels or ability to generate large Na+ currents. Initial upward transient deflection and subsequent transient downward deflection are, respectively, the capacitance transient and Na+ current elicited by the voltage step. L, Plot of excitability (AUC) versus Na+ current (evoked by depolarization, as in K) amplitude across individual cells, showing no significant relationship between the two variables. M, Plot of excitability (AUC) versus input resistance (determined by measuring the steady-state current response to a small, 5 mV, hyperpolarizing step) across individual cells, showing a significant positive association between the variables.
Postrecording immunohistochemistry for Cplx3.
To confirm recordings in SPNs, a subset of cells were filled with neurobiotin (Vector Laboratories, catalog #SP-1120, RRID:AB_2313575) and labeled for Cplx3. Four cells were recorded from each slice, in the same laminar position. The first and fourth cells, which defined the outer boundaries of the recording site, were filled with neurobiotin. Immediately after termination of the recording from the fourth cell, the two neurobiotin-loaded cells were visualized with fluorescence optics on the patch-clamp microscope. After confirming the location of the recorded cells within the SP, slices were immersion fixed overnight at 4°C in phosphate-buffered 2% PFA, 15% picric acid, pH 7.4, and processed for immunohistochemical detection of neurobiotin with FITC-conjugated streptavidin, and Cplx3, as described above, visualized with anti-rabbit Rhodamine Red X-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, catalog #111-295-045, RRID:AB_2338025; 1:200).
Statistical analyses.
All data analyses were performed with either SAS/STAT (SAS Institute RRID:SCR_008567) or Prism 4 statistical software (GraphPad Software RRID:SCR_002798). Data are expressed as mean ± SEM. Comparisons of group means for cell counts and area fractions among age-matched TC, Hx control, and HI animals were performed with the Kruskal–Wallis test (one-way ANOVA) followed by the Dunn post hoc test for multiple pairs. Comparisons of group means for dendritic morphology and electrophysiological parameters among age-matched TC, Hx, and HI animals were performed with the SAS/STAT GLIMMIX mixed-effects model, which tested and controlled for any effects of ewe, fetus, or gender, followed by the Bonferroni post hoc test for multiple treatment pairs. No effects of ewe, fetus, or gender were observed unless otherwise noted. Sholl (radius × treatment group) and branch order (branch order × treatment group) analyses of dendritic morphology parameters between age-matched TC, Hx, and HI animals were performed by two-way ANOVA, followed by Bonferroni post hoc tests. A value of p < 0.05 was considered statistically significant.
Results
Fetal HI generates acute cell degeneration in cerebral WM and SP
Because human SPNs are highly enriched in the preterm brain during the period of increased risk for cerebral white matter injury (WMI) (Volpe, 2009), we quantified cell degeneration in the SP in a preterm fetal sheep model of diffuse WMI (Back et al., 2012) induced by brachiocephalic artery occlusion and/or maternal Hx at 0.65 gestation (preterm human equivalent ∼24–28 weeks gestation). At 24 h (n = 3 TC, 5 Hx, 4 HI) and 4 weeks (n = 7 TC, 3 Hx, 6 HI) after the insult, cell death was quantified at the level of the frontal cerebral WM, a region that is particularly susceptible to cerebral WMI in human and sheep. Cellular degeneration was identified by the presence of AC3 staining or condensed, pyknotic nuclei visualized with Hoechst 33342.
At 24 h survival, the WM displayed a modest increase in cell death in response to fetal Hx and a more pronounced increase from HI, as measured by pyknotic nuclei (Fig. 1C: p < 0.0001, Kruskal–Wallis test; TC 1.35 ± 0.33 vs Hx 8.16 ± 0.58, p < 0.01; TC vs HI 20.79 ± 3.18, p < 0.001, Dunn's post hoc test for multiple pairs) and AC3+/apoptotic cells (Fig. 1E: p < 0.0001; TC 0.63 ± 0.14 vs Hx 4.42 ± 0.51, p < 0.01; TC vs HI 17.71 ± 3.22, p < 0.001). A similar, but more moderate, response was also observed in the SP for pyknotic nuclei (Fig. 1C: p < 0.0013; TC 0.14 ± 0.08 vs Hx 2.61 ± 0.55, p < 0.05; TC vs HI 9.04 ± 3.20, p < 0.01) and apoptotic nuclei (Fig. 1E: p < 0.0113; TC 0.08 ± 0.05 vs Hx 1.48 ± 0.53, not significant; TC vs HI 4.80 ± 1.72, p < 0.05).
After a 4 week survival, a low level of delayed cell degeneration was seen in the WM and SP in response to either insult (Fig. 1D,F). In response to fetal Hx, a modest increase in cell death was seen in the WM (Fig. 1D; pyknosis TC 2.40 ± 0.31 vs Hx 4.30 ± 0.35, p < 0.05; Fig. 1F; apoptosis TC 2.55 ± 0.26 vs Hx 4.05 ± 0.64, not significant). A similar response was seen for HI (Fig. 1D; pyknosis TC 2.40 ± 0.31 vs HI 6.06 ± 0.74, p < 0.001; Fig. 1F; apoptosis TC 2.55 ± 0.26 vs HI 7.15 ± 0.95, p < 0.001). Like the WM, the incidence of pyknosis in the SP also declined in the Hx group (Fig. 1D; TC 2.13 ± 0.32 vs Hx 3.00 ± 0.40, not significant) but remained significantly different in the HI group (Fig. 1D; TC 2.13 ± 0.32 vs HI 4.60 ± 0.62, p < 0.01). In contrast to the WM, apoptosis failed to reach significance in the SP 4 weeks after exposure to Hx or HI (Fig. 1F; TC 1.96 ± 0.33 vs Hx 2.80 ± 0.64 or HI 3.75 ± 0.77, not significant). Like the WM, no significant differences in the degree of pyknosis or apoptosis were observed between Hx versus HI in the SP. Hence, cell death in the WM was accompanied by a low level of cell death in the preterm SP in response to Hx or HI.
Cplx3 discretely localizes to fetal ovine neurons
We next asked whether degenerating SPNs were present in the WM or SP in response to Hx or HI. We first tested several potential SPN markers, Nurr1, Tbr1, and Cplx3, in preterm fetal ovine brain at 0.65 and 0.85 gestation. Unlike the robust staining observed in neonatal rodents (Wang et al., 2010), Nurr1 staining gave inconsistent results at both dGAs in the fetal ovine forebrain (data not shown). Although we obtained reproducible Tbr1 staining at both dGAs, Tbr1 localized nonselectively to neurons both in deeper cortical layers as well as the SP layer and subjacent WM (Fig. 2A,B). By contrast, Cplx3 discretely localized to the SP and scattered interstitial neurons in the WM at both dGAs (Fig. 2C,D), similar to that previously reported in rat (Hoerder-Suabedissen and Molnár, 2013). To confirm the localization of Cplx3 in the fetal ovine SP, the neuronal marker Tbr1 was covisualized with Cplx3. All Cplx3+ cells were also Tbr1+ (Fig. 2E), but not all Tbr1+ cells were Cplx3+. Although both Cplx3 and Tbr1 were robustly expressed in SPNs, the discrete localization of Cplx3 to the SP identified it as a selective SPN marker in fetal ovine brain consistent with prior findings in rodents (Hoerder-Suabedissen and Molnár, 2013) and human (Wang et al., 2010; Molnár and Clowry, 2012).
Figure 2.
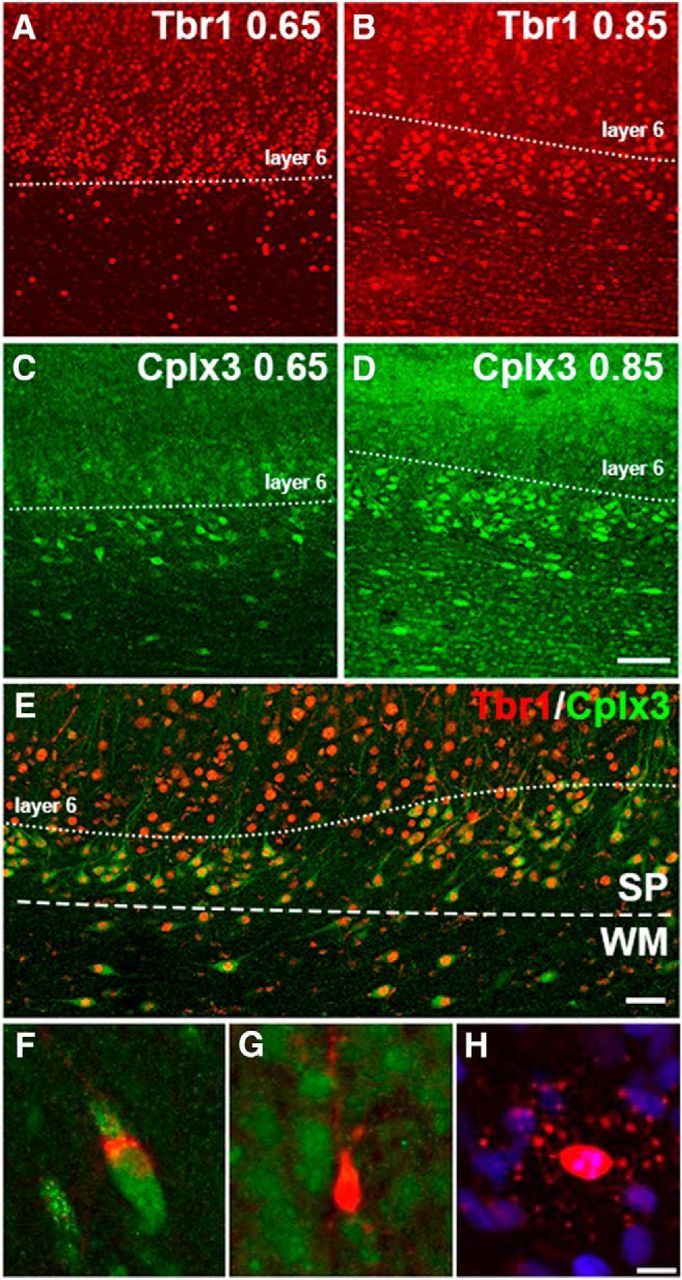
Cplx3+ SPNs are also Tbr1+. Tbr1 labeling of the cortex and SP layers at both 0.65 gestation (A) and 0.85 gestation (B). Unlike Tbr1, Cplx3 label is more confined to the SP at both 0.65 gestation (C) and 0.85 gestation (D). Sections stained for Cplx3 and Tbr1 were visualized at 0.85 gestation and examined for colocalization of both markers within cells of the WM, SP, and cortex. Whereas Tbr1 (red) labeling extends into deep cortical layers above the SP, all Cplx3+ SPNs also contain a Tbr1+ nucleus (E). Scale bar, 50 μm. F–H, Morphologies of the degenerating cells found in the SP, cortex, and WM. F, Apoptotic Tbr1+ neuron found in the SP: green represents Tbr1; red represents AC3. G, Apoptotic Tbr1− neuron found deep within the cortex: green represents Tbr1; red represents AC3. H, Apoptotic oligodendrocyte with a pyknotic nucleus: red represents AC3; blue represents Hoechst. Nearly all the degenerating cells displayed the pattern of AC3 labeling observed in H and could be identified as oligodendrocytes. Scale bar, 5 μm.
Degenerating cells in the SP zone are not Tbr1+ SPNs.
To determine the magnitude and extent of SPN degeneration, we next sought to combine staining for AC3 with a SPN marker. It was not feasible to covisualize AC3 and Cplx3 because both were detected with a rabbit antisera. Consequently, we pursued an alternative approach to quantify staining for AC3 and Tbr1 in the SP (Fig. 2F,G). Surprisingly, at 24 h after exposure to Hx or HI, colocalization of Tbr1 and AC3 was not observed in the SP. We confirmed this result by quantifying Tbr1-labeled cells that had a pyknotic nucleus visualized with Hoechst 33342. As summarized in Table 1, only 3.7% of pyknotic nuclei in the SP were Tbr1+ in the Hx group. In 4 week survivors, the percentage of degenerating Tbr1+ SP cells was still very low or negligible as defined by pyknotic nuclei (TC = 0%, Hx = 10%, and HI = 8.3%) or AC3-labeled cells (TC = 2.2%, Hx = 0%, and HI = 1.5%). In cerebral WM, the percentage of degenerating Tbr1+ cells was also similarly low for both the 24 h and 4 week survivors. Hence, SPN degeneration did not appear to substantially contribute to the observed cellular degeneration in either the preterm fetal SP or WM.
Table 1.
Degeneration of SPNs and interstitial WM neurons was rarely detected at 24 h or 4 weeks after maternal Hx or HIa
| 24 h |
4 weeks |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pyknotic |
Apoptotic |
Pyknotic |
Apoptotic |
|||||||||
| TC | Hx | HI | TC | Hx | HI | TC | Hx | HI | TC | Hx | HI | |
| WM | ||||||||||||
| Total cells counted | 135 | 877 | 1583 | 68 | 487 | 1329 | 337 | 233 | 645 | 356 | 215 | 755 |
| No. colocalized Tbr1 | 6 | 19 | 24 | 7 | 0 | 3 | 1 | 8 | 24 | 3 | 0 | 4 |
| % colocalization | 4.4 | 2.2 | 1.5 | 10.3 | 0 | 0.23 | 0.30 | 3.4 | 3.7 | 0.84 | 0 | 0.53 |
| SP | ||||||||||||
| Total cells counted | 3 | 27 | 61 | 1 | 12 | 37 | 43 | 30 | 84 | 46 | 28 | 67 |
| No. colocalized Tbr1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 7 | 1 | 0 | 1 |
| % colocalization | 0 | 3.7 | 0 | 0 | 0 | 0 | 0 | 10 | 8.3 | 2.2 | 0 | 1.5 |
aData are total counts of pyknotic nuclei or apoptotic nuclei in the SP or periventricular WM. A small percentage of degenerating cells were labeled with Tbr1 (for details of the counting protocol, see Materials and Methods).
Degenerating cells in the SP layer are oligodendrocyte lineage cells
Because we did not identify either early or delayed degeneration of Tbr1-labeled neurons in the SP, we tested an alternative hypothesis that the degenerating cells might be glial in origin. Because we previously detected widespread degeneration of O4-antibody labeled late oligodendrocyte progenitors (preOLs) in fetal cerebral WM at 0.65 gestation in response to HI (Riddle et al., 2006), we determined whether preOLs might also degenerate in the SP in response to HI or Hx. Staining for the O4 antibody in combination with Hoechst 33342 detected numerous degenerating O4-labeled cells in the SP after HI (Fig. 3A–E), as well as in cerebral WM and the cortex. Scattered foci of degenerating preOLs were also detected throughout the WM after Hx (Fig. 3G). Hence, preterm WMI was accompanied by degeneration of preOLs, but neuronal degeneration was rarely observed in the SP or adjacent cerebral cortex.
Figure 3.
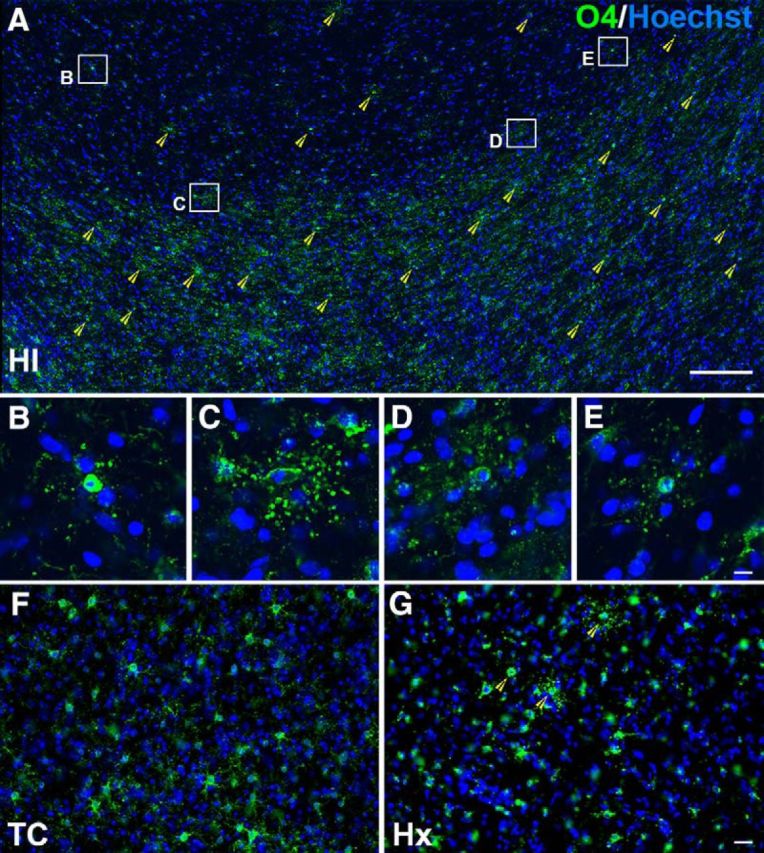
Degenerating cells in the cortex, SP, and WM are predominantly oligodendrocytes. A, Section of cortex and WM from a 94 dGA (0.65 gestation) sheep brain 24 h after HI. Green O4 stain represents cells of the oligodendrocyte lineage; blue Hoechst stain allows identification of degenerating nuclei (yellow arrowheads). Four degenerating cells from the cortex (B) and SP (C–E) are depicted in enlarged insets (B–E). F, Healthy O4-labeled cells in the WM of a 94 dGA (0.65 gestation) sheep brain TC. G, Foci of degenerating oligodendrocytes are seen throughout the WM of a 94 dGA (0.65 gestation) sheep brain 24 h after exposure to maternal Hx. Scale bars: A, 200 μm; B–E, 10 μm; F, G, 30 μm.
WMI from HI is not accompanied by SPN inflammation
Periventricular WMI from HI in preterm fetal sheep and human is accompanied by persistent reactive astrogliosis and microgliosis (Riddle et al., 2011; Buser et al., 2012). We first determined whether an acute inflammatory response was observed in the cerebral WM adjacent to the SP layer in response to HI or Hx. Fetal (93 dGA, 0.65 gestation) brains were analyzed at 24 h after HI with the astrocyte marker GFAP (Fig. 4A–F) and the macrophage/microglial marker Iba1 (Fig. 4G–L). Despite the presence of periventricular WMI with preOL degeneration, we did not observe increased astroglial or micoglial/macrophage reactivity in the superficial WM adjacent to the SP.
Figure 4.
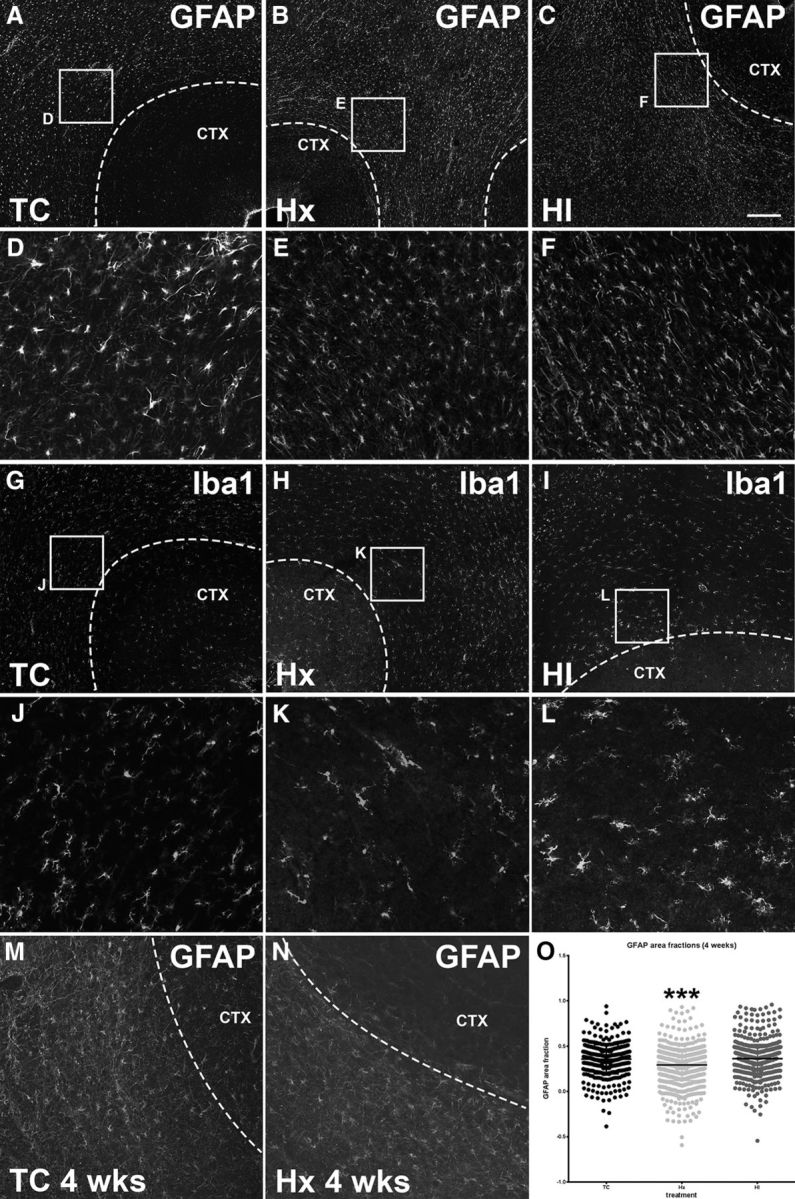
Astrocyte and microglial morphology and density are comparable across treatment groups both at 24 h and 4 weeks after exposure to Hx and/or ischemia. A, A normal fetal (93 dGA, 0.65 gestation) sheep brain slice (TC) stained for the astrocyte marker GFAP. The region bounded by the white box is enlarged in D. B, A fetal (93 dGA, 0.65 gestation) sheep brain slice collected 24 h after exposure to maternal Hx stained for the astrocyte marker GFAP. The region bounded by the white box is enlarged in E. C, A fetal (93 dGA, 0.65 gestation) sheep brain slice collected 24 h after exposure to both maternal Hx and fetal ischemia (HI) stained for the astrocyte marker GFAP. The region bounded by the white box is enlarged in F. Comparable sections from TC, Hx, and HI animals, respectively, are stained for the macrophage/microglial marker Iba1 in G–I, and shown enlarged in K, L. M, A normal fetal (120 dGA, 0.85 gestation) sheep brain slice (TC) stained for the astrocyte marker GFAP. N, A fetal (120 dGA, 0.85 gestation) sheep brain slice collected 4 weeks after exposure to maternal Hx stained for the astrocyte marker GFAP. O, GFAP was quantified in the WM at 120 dGA, 4 weeks after exposure to Hx and/or ischemia using areal fraction analysis of section images. No significant difference was seen between TC versus HI groups. The Hx group was significantly lower than both TC and HI groups. ***p < 0.001. Overall statistical comparisons were by Kruskal–Wallis Test; Dunn's post hoc test was used for multiple comparisons. Scale bars: A–C, G–I, 200 μm; D–F, J–L, 50 μm; M, N, 100 μm.
Similarly, at 4 weeks after HI or Hx, there were no significant differences in GFAP-labeled cells as determined by area fraction analysis in the TC versus HI groups (Fig. 4O), whereas GFAP labeling was significantly lower in the Hx group relative to both TC and HI groups (Fig. 4O; p < 0.001 Dunn's post hoc for multiple comparisons). The density of GFAP-labeled cells, quantified by stereology, did not differ among the three groups (ANOVA F(2,8) = 1.048, p = 0.3943). Hence, neither HI nor Hx appeared to promote acute or chronic enhanced glial reactivity adjacent to the SP.
SPN basal dendrite maturation is disrupted in response to fetal Hx or HI
We previously found that HI-induced WMI in the preterm fetal brain was accompanied by disturbances in neuronal maturation in the cerebral cortex (Dean et al., 2013) and caudate nucleus (McClendon et al., 2014). Although SPNs displayed significant resistance to cell death in the setting of WMI, we determined whether they might display disturbances in dendritic arborization in response to Hx or HI. Because SPNs display a wide variety of morphological subtypes, this heterogeneity precluded the unambiguous identification of SPNs through the widely used Golgi-impregnation technique. We used an alternative approach where Cplx3 immunohistochemistry was combined with DiOlistics to specifically label SPNs by gene gun delivery of DiI to permit 3D reconstruction of dendritic arbors visualized by confocal microscopy (Fig. 5A–C, top row). Unlike the Golgi stain, the use of DiI not only allowed us to select the Cplx3-immunoreactive SP population, but it also allowed us to visualize and trace the complete apical arbors (Fig. 5A–C, bottom row) as well as the basal dendritic trees (Fig. 5A–C, third row). The four criteria for selection of SPNs for morphometric analysis are described in Materials and Methods.
Figure 5.

A–C, Top row, Maximum projection images of confocal z stacks of representative SPNs colabeled with Cplx3 (green) and DiI (red) from control (A, TC), hypoxic (B, Hx), and HI brains (C, HI). Second row, The 3D reconstructions of the soma and full basal and apical dendritic arbors of the representative neurons in the top row were obtained by tracing in Neurolucida confocal z stacks collected at a 1 μm step size. Branch order rank is indicated by color: bright yellow represents first order; white represents second order; purple/hot pink represents third order; bright green represents fourth order; cyan blue represents fifth order; orange represents sixth order; slate gray represents seventh order; salmon pink represents eighth order; forest green represents ninth order; bright blue represents 10th order. Third row, Neurolucida tracings of only the basal dendritic arbors and soma of the representative TC, Hx, and HI neurons. Fourth row, Neurolucida tracings of only the apical dendritic arbors and soma of the representative TC, Hx, and HI neurons.
The basal and apical dendrites of a total of 658 Cplx3+/DiI+ SPNs subjacent to the primary motor and somatosensory cortices (n = 230 TC, 217 Hx, and 211 HI cells; n = 6 TC, 8 Hx, and 8 HI fetal brains) were traced and quantified by Neurolucida after a 4 week survival (∼119–123 dGA, 0.85 gestation). For multiple measures of basal dendritic complexity, SPNs from the controls (TC) generally displayed greater complexity than either the Hx or HI groups (Figs. 5, 6). Although not evident from most global measures of complexity, significant differences in SPN basal dendrites were also observed between the Hx and HI groups when complexity metrics were explored by branch order.
Figure 6.
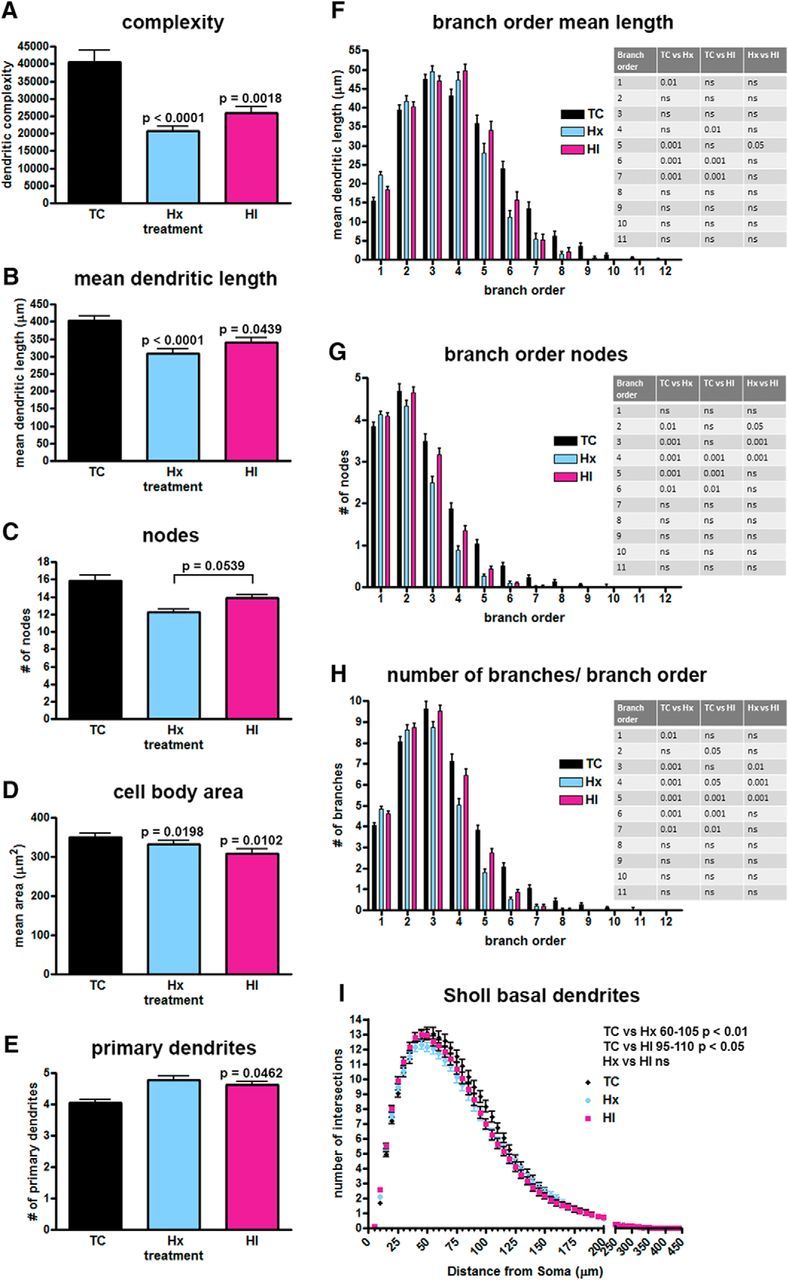
Decreased basal dendritic complexity found in SPNs after exposure to maternal Hx or in combination with ischemia (HI). Paired comparisons are made to normal controls (TC) unless designated by brackets. A, Computed complexity index for basal dendrites. B, Mean basal dendritic length. C, Total number of nodes (branch points). D, Mean cell body area. E, Number of primary basal dendrites. F, Mean basal dendritic length by branch order. G, Basal dendritic nodes (branch points) by branch order. H, Number/quantity of dendritic branches by branch order. I, Sholl analysis, number of dendritic intersections of Sholl sphere. Black bars/symbols represent true normal controls (TC). Blue bars/symbols represent maternal Hx only (hypoxic control, Hx). Red bars/symbols represent HI. Error bars indicate SE. p values were calculated using Bonferroni's post hoc test for multiple comparisons after GLIMMIX procedure for global metrics. For branch order and Sholl analyses (F–I), p-values were calculated using Bonferroni's post-hoc test for multiple comparisons following 2-way ANOVA.
To estimate the effect of Hx or HI on arbor maturation, an overall complexity value was calculated using Neurolucida (see Materials and Methods). In response to both Hx and HI, SPN basal dendrites displayed a significant reduction in overall complexity relative to controls (Fig. 6A; TC 40,470 ± 3441 vs Hx 20,640 ± 1531, mean ± SEM, df = 2, 652, t = −4.25, Bonferroni p = 0.0001; TC 40,470 ± 3441 vs HI 25,960 ± 1798, t = −3.45, p = 0.0018). No significant differences (t = −0.32, p = 1.0000) were present between the HI and Hx groups.
Although several factors contribute to the calculation of the global complexity metric, for our population of SPNs, the dendritic length of the branches had the most impact on the complexity in the Hx and HI groups. The mean dendritic length was significantly reduced for the Hx and HI groups versus controls (Fig. 6B; TC 402.30 ± 13.50 vs Hx 308.90 ± 12.27, mean ± SEM, df = 2, 652, t = −4.24, Bonferroni p = 0.0001; TC 402.30 ± 13.50 vs HI 339.80 ± 13.25, t = −2.45, p = 0.0439). Branch order analysis revealed that the overall reduction in dendritic length was mostly related to reduced maturation of the sixth- and seventh-order basal dendritic branches, which decreased significantly in length in both Hx and HI groups (Fig. 6F).
Although a trend toward fewer total branch points (nodes) was present globally in SPNs exposed to Hx and HI versus control, these groups failed to reach statistical significance (Fig. 6C). However, when nodes were examined on the basis of individual branch orders, the decline in the quantity of branch points was significant for branch orders 2–6 for the Hx group and branch orders 4–6 for the HI group (Fig. 6G). Consistent with the decline in nodes for the Hx and HI groups, there was also a significant decrease in the number of branches for branch orders 4–7 for both the Hx and HI groups versus controls (Fig. 6H). No differences were seen in the number of total branches or endings (terminals) between the Hx and HI groups versus controls (data not shown).
The complex responses of the SPNs was further illustrated by analysis of soma size and the related number of primary dendrites. The mean area of the cell soma was lower in both Hx and HI animals versus controls (Fig. 6D; TC 350.50 ± 9.34 vs Hx 333.20 ± 8.93, mean ± SEM, df = 2, 652, t = −2.72, Bonferroni p = 0.0198; TC 350.50 ± 9.34 vs HI 307.80 ± 11.67, t = −2.94, p = 0.0102). No differences were observed between soma size of the Hx and HI groups (t = 0.28, p = 1.0000). It was notable that changes in complexity of the basal dendritic arbors to Hx or HI were variable and related to distance from the soma. Despite the smaller soma size of the HI SPNs, the number of primary basal dendrites projecting from the soma of the Hx and HI SPNs tended to increase versus control but only reached statistical significance in the HI SPNs (Fig. 6E; TC 4.06 ± 0.10 vs Hx 4.79 ± 0.11 mean ± SEM, df = 2, 644, t = 1.76, Bonferroni p = 0.2377; TC 4.06 ± 0.10 vs HI 4.62 ± 0.10, t = 2.43, p = 0.0462). This finding suggested that an increase in primary dendrites in concert with an overall decline in dendritic complexity might potentially produce a null difference in Sholl intersections between controls and treated groups because multiple simple arbors can produce as many intersections as a few complex arbors. Nevertheless, the significant decline in basal dendritic complexity of the Hx and HI SPNs relative to controls did produce significant differences in the number of observed intersections in the Sholl analysis (Fig. 6I; TC vs Hx radii 60–105, p < 0.01; TC vs HI radii 95–110, p < 0.05; Hx vs HI, not significant).
SPN apical dendrite maturation is disrupted in response to fetal Hx but not HI
Among the total population of SPNs studied, 99.5% of them had a single apical dendrite oriented toward the pial surface (TC = 100%, Hx = 99.1%, and HI 99.5%). In contrast to the basal dendritic arbors, the apical dendritic arbors of the HI group generally displayed no differences in morphology compared with controls. Both the control and HI groups tended to be more complex than the Hx group for multiple global measures of complexity but failed to reach statistical significance for any of these metrics (Fig. 7). Specifically, Hx SPNs had fewer branch points (Fig. 7B; TC 8.71 ± 0.32 vs Hx 7.09 ± 0.26 mean ± SEM, df = 2, 641, t = −0.61, Bonferroni p = 1.0000; Hx 7.09 ± 0.26 vs HI 8.18 ± 0.27 t = −0.42, p = 1.0000), endings (Fig. 7C; TC 10.00 ± 0.34 vs Hx 8.52 ± 0.28 mean ± SEM, df = 2, 641, t = −0.57, Bonferroni p = 1.0000; Hx 8.52 ± 0.28 vs HI 9.68 ± 0.28 t = −0.09, p = 1.0000), and total number of branches (Fig. 7A; TC 18.37 ± 0.64 vs Hx 15.61 ± 0.54 mean ± SEM, df = 2, 641, t = −0.41, Bonferroni p = 1.0000; Hx 15.61 ± 0.54 vs HI 17.85 ± 0.56 t = 0.02, p = 1.0000), which resulted in lower complexity values for the Hx and HI groups (Fig. 7D; TC 60,950 ± 6206 vs Hx 39,170 ± 3244 mean ± SEM, df = 2, 649, t = −1.79, Bonferroni p = 0.2208; Hx 39,170 ± 3244 vs HI 49,560 ± 4116 t = 0.32, p = 1.0000). However, branch order analyses of nodes (Fig. 7G) and total number of branches per branch order (Fig. 7F) revealed that the tendency toward reduced complexity of SPNs in the Hx group was related to significant changes in branch orders 3–6 for nodes and 4–7 for numbers of branches. No significant differences in the apical dendritic arbors were observed among the three groups for total dendritic length or mean dendritic length when considered as a whole or by branch order (data not shown). Although the individual indices of complexity (nodes, endings, and total branch number) failed to reveal significant differences in the apical arbor between the TC and HI groups, a Sholl analysis (Fig. 7E) demonstrated that the number of intersections increased in the HI group at 25–50 μm from the soma relative to controls (p < 0.01) and 30–60 μm (p < 0.01) from the soma relative to the Hx group. A significant decline in the number of intersections was observed between TC and Hx groups at 100–120, and 135 μm (p < 0.05) from the soma.
Figure 7.

A trend toward decreased apical dendritic complexity was seen in SPNs after exposure to maternal Hx but failed to reach significance in global measures. Dendritic differences only become apparent when analyzed by branch order and Sholl intersections. A, Total number of apical dendritic branches. B, Total number of nodes (branch points). C, Total number of endings (terminals). D, Computed complexity index for apical dendrites. E, Sholl analysis, number of dendritic intersections of Sholl sphere. F, Number/quantity of dendritic branches by branch order. G, Basal dendritic nodes (branch points) by branch order. Black bars/symbols represent true normal controls (TC). Blue bars/symbols represent maternal Hx only (hypoxic control, Hx). Red bars/symbols represent HI. Error bars indicate SE. p values were calculated using Bonferroni's post hoc test for multiple comparisons after GLIMMIX procedure for global metrics (not significant). For branch order and Sholl analyses (E–G), p-values were calculated using Bonferroni's post-hoc test for multiple comparisons following 2-way ANOVA.
SPN complexity is directly related to fetal systemic oxygenation
As noted above, we often paradoxically observed that the HI group was less significantly affected than the Hx group. To evaluate this paradox, we instrumented 13 sets of twins to collect ABG values on both Hx and HI fetuses (4 females/9 males in each experimental group). Blood samples were collected (Fig. 8A) at the start of the experiment (pre-ABG), 5 min after the onset of maternal Hx (Hx-ABG), 25 min later at the end of Hx or HI (HI-ABG), and 10 min after the end of Hx or HI (post-ABG). The mean oxygen content, lactate, and glucose values of the 13 twin pairs (Fig. 8B–D, blue [Hx] and red [HI] lines) were compared with the mean values of the 8 HI fetuses used in this study to analyze SP anatomy (Fig. 8B–D, black [HI-SP] lines). Figure 8B shows that all fetal groups had the same mean baseline (pre-ABG) values for oxygen content. Five minutes into maternal Hx (Hx ABG), all fetal groups respond with a drop in oxygen content (vol %). Twenty-five minutes later (HI ABG), the two fetal HI groups mounted a compensatory response that elevated their oxygen content back to baseline levels, whereas the Hx twins continued to display reduced oxygen content until maternal Hx was terminated.
Figure 8.
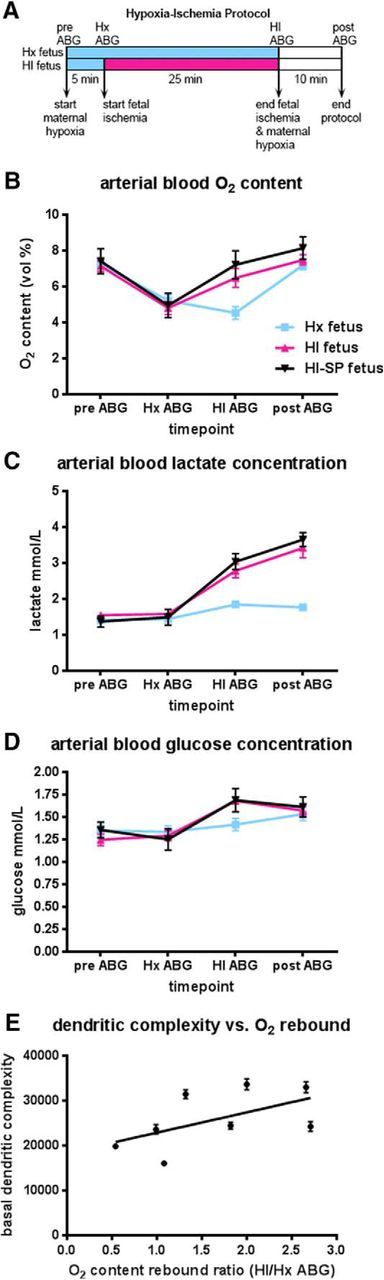
Fetal ABG values for Hx and HI groups during experimental paradigm. A, Flow chart of the experimental protocol described in Materials and Methods. B, Fetal oxygen content (vol %) varies with changes in maternal oxygenation and fetal ischemia (both HI groups red and black lines) while remaining constant in the fetuses exposed only to maternal Hx (Hx group blue line). C, Fetal lactate concentration increases markedly in HI groups (red and black lines) by the end of the ischemic period and continues to rise 10 min after the cessation of fetal ischemia and maternal Hx compared with fetuses exposed only to maternal Hx (Hx group blue lines). D, Fetal glucose concentration rises markedly in HI groups (red and black lines) compared with Hx group (blue line). E, Basal dendritic complexity is positively correlated with the fetal rebound ratio defined as the HI-ABG oxygen content value divided by the Hx-ABG oxygen content value for HI-SP fetuses.
To better understand the source of the rise in mean oxygen content values, we examined hemoglobin, hematocrit, oxygen saturation (sO2%), and partial pressure (PaO2 mmHg) values. Whereas there were no differences in hemoglobin or hematocrit levels across the groups, oxygen saturation and partial pressure values had a corresponding rise in the HI group relative to the Hx (Table 2). Hence, this compensatory response of the HI group to the severity of the HI insult resulted in normalization of the fetal systemic hypoxemia and appeared to be related to the generally greater complexity of the SPNs in the HI group relative to the Hx. To test this hypothesis, we analyzed the complexity of the basal dendritic arbors of the SPNs for 8 HI animals for which blood gas values were available to calculate an oxygen content rebound ratio, which was defined as the ratio of the HI-ABG/Hx-ABG oxygen content values (Fig. 8E). Among HI animals, basal dendritic complexity was positively associated with the oxygen content rebound ratio (F(1,208) = 3.81, p = 0.05) such that, as the ratio increased, so did basal dendritic complexity.
Table 2.
The ABG values of 13 twin pairs (one Hx and one HI fetus/pair) are compared with the ABG values of the 8 HI fetuses used to study the SP in this paper (HI-SP)
| Treatment |
p-value |
||||||
|---|---|---|---|---|---|---|---|
| ABG parameter | ABG time point | Hx (n = 12) | HI (n = 11) | HI-SP (n = 8) | Hx versus HI | Hx versus HI-SP | HI versus HI-SP |
| O2 content (vol %) | pre-ABG | 7.3 ± 0.8 | 7.1 ± 1.0 | 7.4 ± 2.0 | NS | NS | NS |
| Hx-ABG | 5.2 ± 1.1 | 4.8 ± 1.2 | 5.0 ± 2.0 | NS | NS | NS | |
| HI-ABG | 4.5 ± 1.2 | 6.5 ± 1.7 | 7.2 ± 2.2 | <0.01 | <0.001 | NS | |
| post-ABG | 7.2 ± 0.9 | 7.5 ± 0.9 | 8.1 ± 1.8 | NS | NS | NS | |
| Lactate (mm) | pre-ABG | 1.4 ± 0.1 | 1.5 ± 0.2 | 1.4 ± 0.5 | NS | NS | NS |
| Hx-ABG | 1.4 ± 0.1 | 1.6 ± 0.2 | 1.5 ± 0.6 | NS | NS | NS | |
| HI-ABG | 1.8 ± 0.3 | 2.8 ± 0.6 | 3.0 ± 0.6 | <0.001 | <0.001 | NS | |
| post-ABG | 1.8 ± 0.3 | 3.4 ± 0.9 | 3.7 ± 0.6 | <0.001 | <0.001 | NS | |
| Glucose (mm) | pre-ABG | 1.4 ± 0.2 | 1.2 ± 0.2 | 1.4 ± 0.3 | NS | NS | NS |
| Hx-ABG | 1.3 ± 0.2 | 1.3 ± 0.2 | 1.3 ± 0.3 | NS | NS | NS | |
| HI-ABG | 1.4 ± 0.2 | 1.7 ± 0.1 | 1.7 ± 0.4 | <0.05 | NS | NS | |
| post-ABG | 1.5 ± 0.2 | 1.6 ± 0.2 | 1.6 ± 0.3 | NS | NS | NS | |
| Hematocrit (%) | pre-ABG | 28 ± 2.3 | 28 ± 1.9 | 27 ± 2.7 | NS | NS | NS |
| Hx-ABG | 28 ± 3.2 | 28 ± 2.4 | 27 ± 2.6 | NS | NS | NS | |
| HI-ABG | 29 ± 2.6 | 30 ± 2.7 | 29 ± 2.6 | NS | NS | NS | |
| Hemoglobin (g/dl) | pre-ABG | 9.1 ± 0.8 | 9.0 ± 0.6 | 8.7 ± 0.9 | NS | NS | NS |
| Hx-ABG | 9.0 ± 1.1 | 9.0 ± 0.8 | 8.6 ± 0.9 | NS | NS | NS | |
| HI-ABG | 9.2 ± 0.9 | 10 ± 0.9 | 9.4 ± 0.9 | NS | NS | NS | |
| post-ABG | 9.1 ± 0.8 | 9.3 ± 0.7 | 8.8 ± 0.8 | NS | NS | NS | |
| sO2 (%) | pre-ABG | 59 ± 2.9 | 59 ± 8.0 | 62 ± 11 | NS | NS | NS |
| Hx-ABG | 43 ± 8.1 | 40 ± 11 | 42 ± 13 | NS | NS | NS | |
| HI-ABG | 36 ± 10 | 50 ± 15 | 57 ± 18 | <0.05 | <0.001 | NS | |
| post-ABG | 59 ± 4.3 | 60 ± 10 | 69 ± 9.8 | NS | NS | NS | |
| paO2 (mmHg) | pre-ABG | 22 ± 1.3 | 22 ± 2.6 | 23 ± 3.0 | NS | NS | NS |
| Hx-ABG | 17 ± 2.0 | 17 ± 2.7 | 17 ± 2.0 | NS | NS | NS | |
| HI-ABG | 16 ± 2.8 | 20 ± 3.6 | 22 ± 4.6 | <0.05 | <0.001 | NS | |
| post-ABG | 22 ± 1.6 | 23 ± 2.9 | 26 ± 3.9 | NS | <0.01 | <0.05 | |
| paCO2 (mmHg) | pre-ABG | 48 ± 2.9 | 48 ± 2.7 | 47 ± 4.2 | NS | NS | NS |
| Hx-ABG | 46 ± 2.6 | 46 ± 3.3 | 44 ± 5.8 | NS | NS | NS | |
| HI-ABG | 47 ± 2.6 | 45 ± 2.7 | 44 ± 7.8 | NS | NS | NS | |
| post-ABG | 48 ± 2.5 | 48 ± 3.5 | 46 ± 4.2 | NS | NS | NS | |
| pH | pre-ABG | 7.4 ± 0.02 | 7.4 ± 0.02 | 7.4 ± 0.02 | NS | NS | NS |
| Hx-ABG | 7.4 ± 0.02 | 7.4 ± 0.02 | 7.4 ± 0.03 | NS | NS | NS | |
| HI-ABG | 7.4 ± 0.02 | 7.4 ± 0.02 | 7.3 ± 0.04 | NS | <0.001 | <0.001 | |
| Post-ABG | 7.4 ± .02 | 7.4 ± .03 | 7.3 ± .02 | <0.005 | <0.001 | <0.05 | |
SPN excitability and synaptic activity are directly related to fetal systemic lactic acidosis
To define how HI and Hx affect SPN signaling, we conducted current- and voltage-clamp recordings from cells in the SP region (Figs. 9, 10). Cells were confirmed to be neuronal by their ability to fire action potentials and/or the presence of synaptic currents (Figs. 9D–F, 10A,B). These neurons were all located in the region of highest density of Cplx3-labeled cells and had dendritic morphologies similar to those observed from morphometric DiOlistic labeling (Figs. 5, 9A–C). Of the 34 cells that were filled with neurobiotin and survived removal of the recording electrode and subsequent fixation, 80% were confirmed to be immunoreactive for Cplx3. Those cells that did not colocalize with Cplx or for which staining was ambiguous were not analyzed.
Figure 10.

SPN synaptic activity is directly related to hypoxemia-induced glucose and lactate accumulation. A, B, Representative traces showing spontaneous synaptic currents (SSCs; A, B, downward deflections; B, inset, time expanded exemplar) in voltage-clamped SPNs with low-frequency (A) and high-frequency (B) synaptic activity. Given the intracellular and extracellular solution composition and holding potential (−85 mV), both GABAA and glutamate receptor-mediated SSCs will be downward (for details, see Materials and Methods). And the kinetics of individual events suggest a mixed population of GABAA and glutamate receptor-mediated SSCs (data not shown), although no pharmacological confirmation was conducted. C, Plot of mean SSC frequency across groups of cells showing no significant difference in SSC frequency across conditions. D, E, Plots of mean SSC frequency for each fetus (n = 3 Hx, 10 HI; mean ± SEM) versus its ABG lactate ratio (Post/Pre; D) and glucose ratio (HI/Hx; E) showing significant positive associations across animals. F, Plot of input resistance (as in Fig. 9M) versus SSC frequency across individual cells in all conditions, showing a significant negative association.
To quantify overall neuronal excitability, current-clamp recordings were conducted, and voltage responses to a series of hyperpolarizing and depolarizing current injections were recorded (Fig. 9D–F). SPNs did not exhibit hyperpolarization-activated potentials or rebound spiking, but the vast majority of SPNs in each condition (TC = 92%, Hx = 96%, and HI = 92%) exhibited action potentials in response to depolarizing current injections (Fig. 9D–F). Excitability (defined as the number of action potentials fired for a given depolarizing current magnitude, and quantified by the area under the input–output curve for each cell: AUC; for details, see Materials and Methods) varied considerably across individual cells, but there were no significant differences across conditions (p = 0.3310; Fig. 9D–H).
However, excitability (AUC) was significantly negatively associated with the lactate rebound ratio (Fig. 9I) defined as the HI-ABG value/Hx-ABG value (df = 1, 56, t = −2.72, p = 0.0086) and the lactate recovery ratio defined as the post-ABG value/pre-ABG baseline value (Fig. 9J; df = 1, 52, t = −2.57, p = 0.0132). The observed differences in excitability were not due to differences in the properties of the Na+ channels that generate action potentials because there was no significant relationship between excitability and the Na+ current magnitude assessed under voltage clamp (Fig. 9K,L). Instead, differences in excitability were significantly positively associated with differences in resting input resistance (df = 1, 132, t = 7.18, p < 0.0001; Fig. 9M), suggesting that differences in excitability are due to differences in electrical shunting (Heigele et al., 2016).
Although recording solutions were optimized for recording action potentials under physiological ionic conditions, upon switching to voltage-clamp (Vh = −80 mV), spontaneous synaptic currents were clearly resolvable (Fig. 10A,B). Given that our recording solutions result in the reversal potential for glutamate-gated and GABA receptor-gated currents being ∼0 mV and −60 mV, respectively, both synaptic currents would be inward at Vh = −80 mV, and so all analyses likely reflect the activity of both GABAergic and glutamatergic synapses. Similar to SPN excitability, the frequency of synaptic currents in SPNs was widely variable across cells, but there were no significant differences across conditions (df = 2, 125, F = 2.71, p = 0.0706; Fig. 10A–C). However, synaptic current frequency was significantly positively associated with the lactate recovery ratio (Post/Pre; df = 1, 53, t = 2.05, p = 0.0451; Fig. 10D) and the glucose ratio (HI/Hx; df = 1, 57, t = 2.25, p = 0.0283; Fig. 10E). Given that variation in cellular input resistance appears to explain differences in SPN excitability (Fig. 9M), we determined whether variation in synaptic activity across cells could account for the differences in input resistance. Indeed, the frequency of synaptic currents was significantly negatively associated with cellular input resistance (df = 1, 125, t = −3.25, p = 0.0015; Fig. 10F). Thus, SPN synaptic activity and electrical excitability (which is likely determined by the level of spontaneous synaptic activity) are directly related to hypoxemia-induced changes in levels of glucose and lactate.
Discussion
We demonstrated that transient disturbances in fetal oxygenation can trigger chronic dysmaturation of SPNs without significant SPN degeneration during a critical window in the establishment of cortical connectivity. Under conditions of moderately severe Hx or HI, SPNs and interstitial WM neurons were markedly more resistant to degeneration than preOLs. PreOL degeneration appeared to account for almost all cell death in the SP and adjacent WM in response to HI or Hx. Future studies are needed to determine whether preOL degeneration in the SP zone may also contribute to SPN dysmaturation through disruption of trophic interactions between preOLs and SPNs at the level of their dendritic or axonal arbors. Moreover, preOL dysmaturation in chronic WM lesions may disrupt axonal inputs to SPNs (e.g., thalamocortical afferents), which may further contribute to aberrant cortical connectivity.
The paucity of SPN degeneration is consistent with the response of preterm fetal ovine cortical pyramidal neurons (Dean et al., 2013) and caudate medium spiny neurons (McClendon et al., 2014), where minimal neuronal degeneration also occurred in response to HI. More severe cerebral insults appear to be required for SPN degeneration in rodents (Okusa et al., 2014; Jantzie et al., 2015; Mikhailova et al., 2017) or human (Folkerth et al., 2008; Andiman et al., 2010; Kinney et al., 2012; Pogledic et al., 2014), in which case neuronal degeneration is also observed in other gray matter structures. Similarly, rodent models of excitotoxic WM injury generate necrotic lesions that are accompanied by extensive neuronal degeneration and gyral abnormalities (Marret et al., 1995). The resistance of SPNs to degeneration does not appear related to a lower level of SP ischemia because fetal ovine cortex and WM sustain similar blood flow during ischemia and reperfusion (Riddle et al., 2006; McClure et al., 2008). Hence, both the selective vulnerability of preOLs (Back and Rosenberg, 2014) and the resistance of SPNs appear to reflect cell-specific responses to a similar magnitude of ischemia.
We identified a new form of SPN vulnerability related to disturbances in dendritic development, synaptic activity, and neuronal excitability. The long-term maturation of basal and apical dendritic arbors of SPNs was associated with transient disruption of fetal oxygenation 1 month earlier. Moreover, electrophysiological studies of SPNs from chronic survivors found increased synaptic activity and a resultant shunting inhibition of electrical excitability, both associated with fetal hypoxemia-induced changes. Serial fetal blood gas measurements demonstrated that the preterm ovine fetus responds to maternal Hx with a broad range of systemic oxygen levels. In fetuses exposed only to Hx, fetal systemic oxygen levels remain low throughout the period of maternal Hx and only return to baseline levels with the cessation of maternal Hx. In contrast to their Hx-only twin, the fetal systemic oxygen levels of the HI twin rebounds in a compensatory response to the added ischemic insult before the cessation of maternal Hx and the onset of reperfusion. Although we expected that HI would more adversely impact SPN maturation, we actually observed that SPNs in the HI group were more similar to control than Hx neurons. We attribute this paradoxical difference to the compensatory response of the fetus to HI. In contrast to fetal exposure to Hx, animals that sustained HI displayed significantly higher systemic oxygen levels during ischemia, which suggested that HI promotes a fetal physiological response to cerebral hypoxemia that extracts more circulating oxygen (Brew et al., 2014). Consistent with the apparent inability of the Hx fetus to mount a protective response to hypoxemia, SPN arborization was significantly more affected. Moreover, the severity of the metabolic insult, as measured by lactic acidemia, was also significantly associated with SPN dendritic dysmaturation, as defined electrophysiologically. The association with lactate is consistent with the notion that lactic acidosis is a major consequence of hypoxemia (Volpe, 2008). Hence, our morphometric and electrophysiology studies similarly support that chronic SPN dysmaturation is associated with the severity of fetal systemic hypoxemia and lactic acidemia, which are indicators of global fetal metabolic stress. A limitation of our in utero preparation is the lack of feasibility to measure fetal cerebral oxygenation or glucose metabolites, such as lactate. Although our data found significant associations between measures of SPN activity and systemic lactate levels, we cannot conclude that cerebral lactate levels are mechanistically linked to SPN dysmaturation. Systemic hypoxemia may contribute to SPN dysmaturation through one or more potential mechanisms that include aberrant expression of genes that stabilize dendritic arbors (Curristin et al., 2002; Koleske, 2013) or regulation of inflammatory mediators shown to play a role in elaboration of the dendritic arbor or regulation of synaptic activity (Stellwagen, 2011; Pribiag and Stellwagen, 2014).
Although the response of SPNs to isolated acute or chronic Hx has not been studied in rodents, neonatal rodents respond to intermittent Hx or severe chronic Hx with disturbances in WM gliogenesis and myelination (Schwartz et al., 2004; Back et al., 2006; Salmaso et al., 2012; Fancy et al., 2014; Scafidi et al., 2014; Juliano et al., 2015). Chronic Hx disrupts myelination through Hx inducible factor that couples angiogenesis and preOL maturation (Yuen et al., 2014). However, chronic Hx does not trigger preOL degeneration in neonatal mice. Similarly, unilateral acute HI is not associated with preOL degeneration in the contralateral hypoxic hemisphere in neonatal rats (Back et al., 2002; Segovia et al., 2008). In contrast, we observed that both preterm fetal HI and Hx triggered preOL degeneration. Hence, there appear to be species-associated differences in the response to Hx between rodents and sheep that may also be related to differences in adaptation to the in utero versus extrauterine environment of the WM.
Although SPN maturation has not been specifically studied, chronic Hx also results in a variety of disturbances in gray matter maturation. Chronic Hx in mice reduces cortical volume, triggers neuronal apoptosis (Banasiak et al., 2000), and induces a delay in cortical maturation (Salmaso et al., 2014) that appears to involve disturbances in neurogenesis and disruptions in interneuron maturation, connectivity, and activity (Komitova et al., 2013). Prolonged exposure to hypoxemia, induced by restricting uteroplacental flow for 6 or 12 h in utero in ∼0.6 gestation preterm fetal sheep resulted in reductions in cortical, cerebellar, and hippocampal volume, and reductions in Purkinje cell arborization and spine density after 1 week (Rees et al., 1999), but with sparse cell death. Similarly, chronic Hx in full-term neonatal piglets disrupts cortical growth and reduces neurogenesis (Morton et al., 2017).
Systemic inflammation induced by endotoxin or other inflammatory mediators is commonly associated with perinatal brain injury that results in cell death (Hagberg et al., 2015) and maturational disturbances in WM and gray matter (Dean et al., 2011; Favrais et al., 2011). In contrast, we did not observe that acute Hx resulted in significant acute or chronic inflammation. Our findings are consistent with multiple studies of chronic Hx in mice, where neither gray nor WM inflammation mediated by reactive astrocytes or microglial was observed (for review, see Back and Rosenberg, 2014). Although we found no evidence that our models of HI or Hx activated glial inflammation in the SP or adjacent subcortical WM, there remains a potential role for inflammatory mediators as a synergistic contributor to SPN dysmaturation that might be identified by a more detailed analysis of intermediate recovery periods.
The timing of insults to the preterm SP may be a critical factor that defines the magnitude of disruption to cortical connectivity. SPN dendritic maturation in rodents and carnivores involves both initial progressive growth and regressive events in which the arbor remodels. SPNs initially have large arbors that contact all cortical layers, but later most SPN connections are restricted to layer 4 or below (Friauf et al., 1990; Hoerder-Suabedissen and Molnár, 2013). The reduction in SPN arbors in response to fetal ovine hypoxemia is consistent with disruption of SPN maturation during a phase of progressive arbor growth. Hence, the timing of hypoxemia may have variable impact depending on the phase of growth of the SPN arbors.
SPN dysmaturation may have several potential consequences for maturation of the cortex, thalamus, and their connections. Thalamic inputs to the SP and SPN projections to cortical layer 4 may be particularly affected as may be local interneuron projections to layer 4. In human preterm survivors, MRI-defined reductions in cortical and thalamic growth have been extensively described (Nagasunder et al., 2011; Ball et al., 2015; Loh et al., 2017) and may be related to SPN-mediated disturbances in neuronal maturation in these structures. Consistent with this notion, we observed that the fetal ovine cortex displays progressive volume loss in response to HI that is accompanied by reduction in the dendritic arbor of cortical pyramidal neurons (Dean et al., 2013). Alternatively, in the setting of more severe insults, primary injury to the thalamus (Pierson et al., 2007; Ligam et al., 2009) or cortex (Andiman et al., 2010) may disrupt projections to the SP with secondary disturbances in SPN maturation.
Human preterm survivors who sustain Hx or HI are at increased risk for a broad spectrum of cognitive and neurobehavioral disabilities (Volpe, 2008). Although our findings are derived from an in utero model, they suggest that persistent SPN dysmaturation may also contribute to these chronic neurobehavioral deficits when sustained by transient hypoxemia postnatally in preterm neonates. The particular sensitivity of fetal SPNs to transient disturbances in oxygen delivery in utero is clinically possible in several settings. During human pregnancy, transient or chronic intrauterine hypoxemia may arise from maternal, placental, or fetal causes (Hutter et al., 2010). Transient or chronic episodes of hypoxemia are also commonly encountered in some preterm neonates in association with critical illness, cardiopulmonary disease, or sepsis (Martin et al., 2011). Although experimental hypoxic preconditioning studies have demonstrated the neuroprotective effects of a transient hypoxic exposure to subsequent neonatal HI (Bergeron et al., 2000), our studies suggest that transient cerebral hypoxemia may also contribute to adverse neurodevelopmental outcomes by disrupting cortical connectivity at a critical window in development through persistent functional disturbances in activity of SPNs or other neuronal populations.
The magnitude of SPN dysmaturation was significantly related to the nadir of systemic Hx and lactic acidosis, which suggests a role in dendritic arborigenesis. Transient Hx in vitro disrupts long-term neuronal activity and connectivity defined with multielectrode arrays (le Feber et al., 2017). Hypoxic conditions disrupt neurite extension and axon path finding through stabilization of Hx-inducible factor 1 (Pocock and Hobert, 2008; Miyake et al., 2015). Recurrent moderate hypoxemia in neonatal mice caused a wide array of behavioral disturbances (Juliano et al., 2015) but may also be protective (Bouslama et al., 2015).
The minimum duration of transient oxygen desaturations that may be deleterious requires further study. A deleterious role for mild chronic hypoxemia is supported by the recent BOOST trials, which found that infants maintained at chronically lower oxygen saturations of 85%–89% had an increased risk of death relative to infants administered oxygen to target saturations of 91%–95%. Another potential benefit of the avoidance of low oxygen states during pregnancy or in preterm neonates may be to reduce the risk of abnormal cortical connectivity related to SPN dysmaturation. Future studies are needed to define critical developmental windows when isolated or recurrent hypoxic events maximally impact the maturation of SPNs and potentially other neuronal populations. It will also be important to determine whether SPN dysmaturation may be reversible or treatable via strategies that promote neuronal maturation and cerebral growth, such as environmental enrichment, optimized neonatal nutrition, or reduced neonatal stress (Back and Miller, 2014).
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS045737 and NS054044.
The authors declare no competing financial interests.
References
- Andiman SE, Haynes RL, Trachtenberg FL, Billiards SS, Folkerth RD, Volpe JJ, Kinney HC (2010) The cerebral cortex overlying periventricular leukomalacia: analysis of pyramidal neurons. Brain Pathol 20:803–814. 10.1111/j.1750-3639.2010.00380.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade AL, Rossi DJ (2010) Simulated ischaemia induces Ca2+-independent glutamatergic vesicle release through actin filament depolymerization in area CA1 of the hippocampus. J Physiol 588:1499–1514. 10.1113/jphysiol.2010.187609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA. (2014) Cerebral white and gray matter injury in newborns: new insights into pathophysiology and management. Clin Perinatol 41:1–24. 10.1016/j.clp.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Miller SP (2014) Brain injury in premature neonates: a primary cerebral dysmaturation disorder? Ann Neurol 75:469–486. 10.1002/ana.24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rosenberg PA (2014) Pathophysiology of glia in perinatal white matter injury. Glia 62:1790–1815. 10.1002/glia.22658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC (2001) Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21:1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Tam J, Xanthoudakis S, Arvin KL, Holtzman DM (2002) Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci 22:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Mallinson RA, O'Malley JP, Wallen LD, Frei B, Morrow JD, Petito CK, Roberts CT Jr, Murdoch GH, Montine TJ (2005) Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol 58:108–120. 10.1002/ana.20530 [DOI] [PubMed] [Google Scholar]
- Back SA, Craig A, Luo NL, Ren J, Akundi RS, Ribeiro I, Rivkees SA (2006) Protective effects of caffeine on chronic hypoxia-induced perinatal white matter injury. Ann Neurol 60:696–705. 10.1002/ana.21008 [DOI] [PubMed] [Google Scholar]
- Back SA, Riddle A, Dean J, Hohimer AR (2012) The instrumented fetal sheep as a model of cerebral white matter injury in the preterm infant. Neurotherapeutics 9:359–370. 10.1007/s13311-012-0108-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball G, Srinivasan L, Aljabar P, Counsell SJ, Durighel G, Hajnal JV, Rutherford MA, Edwards AD (2013) Development of cortical microstructure in the preterm human brain. Proc Natl Acad Sci U S A 110:9541–9546. 10.1073/pnas.1301652110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball G, Pazderova L, Chew A, Tusor N, Merchant N, Arichi T, Allsop JM, Cowan FM, Edwards AD, Counsell SJ (2015) Thalamocortical connectivity predicts cognition in children born preterm. Cereb Cortex 25:4310–4318. 10.1093/cercor/bhu331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasiak KJ, Xia Y, Haddad GG (2000) Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol 62:215–249. 10.1016/S0301-0082(00)00011-3 [DOI] [PubMed] [Google Scholar]
- Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE (1989) Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res 24:548–557. 10.1002/jnr.490240413 [DOI] [PubMed] [Google Scholar]
- Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR (2000) Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol 48:285–296. 10.1002/1531-8249(200009)48:3%3C285::AID-ANA2%3E3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- Bouslama M, Adla-Biassette H, Ramanantsoa N, Bourgeois T, Bollen B, Brissaud O, Matrot B, Gressens P, Gallego J (2015) Protective effects of intermittent hypoxia on brain and memory in a mouse model of apnea of prematurity. Front Physiol 6:313. 10.3389/fphys.2015.00313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew N, Walker D, Wong FY (2014) Cerebral vascular regulation and brain injury in preterm infants. Am J Physiol Regul Integr Comp Physiol 306:R773–R786. 10.1152/ajpregu.00487.2013 [DOI] [PubMed] [Google Scholar]
- Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo NL, Ren J, Struve J, Sherman LS, Miller SP, Chau V, Hendson G, Ballabh P, Grafe MR, Back SA (2012) Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol 71:93–109. 10.1002/ana.22627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun JJ, Shatz CJ (1989) Interstitial cells of the adult neocortical white matter are the remnant of the early generated subplate neuron population. J Comp Neurol 282:555–569. 10.1002/cne.902820407 [DOI] [PubMed] [Google Scholar]
- Curristin SM, Cao A, Stewart WB, Zhang H, Madri JA, Morrow JS, Ment LR (2002) Disrupted synaptic development in the hypoxic newborn brain. Proc Natl Acad Sci U S A 99:15729–15734. 10.1073/pnas.232568799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JM, van de Looij Y, Sizonenko SV, Lodygensky GA, Lazeyras F, Bolouri H, Kjellmer I, Huppi PS, Hagberg H, Mallard C (2011) Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. Ann Neurol 70:846–856. 10.1002/ana.22480 [DOI] [PubMed] [Google Scholar]
- Dean J, McClendon E, Hansen K, Azimi-Zonooz A, Chen K, Riddle A, Gong X, Sharifnia E, Hagen M, Ahmad T, Leigland L, Hohimer A, Kroenke C, Back S (2013) Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med 5:168ra167. 10.1126/scitranslmed.3004669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinopoulos A, Karamanlidis AN, Papadopoulos G, Antonopoulos J, Michaloudi H (1985) Thalamic projections to motor, prefrontal, and somatosensory cortex in the sheep studied by means of the horseradish peroxidase retrograde transport method. J Comp Neurol 241:63–81. 10.1002/cne.902410106 [DOI] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Baranzini SE, Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S, Rowitch DH (2014) Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat Neurosci 17:506–512. 10.1038/nn.3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelièvre V, Gressens P (2011) Systemic inflammation disrupts the developmental program of white matter. Ann Neurol 70:550–565. 10.1002/ana.22489 [DOI] [PubMed] [Google Scholar]
- Folkerth RD, Trachtenberg FL, Haynes RL (2008) Oxidative injury in the cerebral cortex and subplate neurons in periventricular leukomalacia. J Neuropathol Exp Neurol 67:677–686. 10.1097/NEN.0b013e31817e5c5e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friauf E, McConnell SK, Shatz CJ (1990) Functional synaptic circuits in the subplate during fetal and early postnatal development of cat visual cortex. J Neurosci 10:2601–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Antonini A, McConnell SK, Shatz CJ (1990) Requirement for subplate neurons in the formation of thalamocortical connections. Nature 347:179–181. 10.1038/347179a0 [DOI] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P (2015) The role of inflammation in perinatal brain injury. Nat Rev Neurol 11:192–208. 10.1038/nrneurol.2015.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen MW, Riddle A, McClendon E, Gong X, Shaver D, Srivastava T, Dean JM, Bai JZ, Fowke TM, Gunn AJ, Jones DF, Sherman LS, Grafe MR, Hohimer AR, Back SA (2014) Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PLoS One 9:e112800. 10.1371/journal.pone.0112800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigele S, Sultan S, Toni N, Bischofberger J (2016) Bidirectional GABAergic control of action potential firing in newborn hippocampal granule cells. Nat Neurosci 19:263–270. 10.1038/nn.4218 [DOI] [PubMed] [Google Scholar]
- Hoerder-Suabedissen A, Molnár Z (2013) Molecular diversity of early-born subplate neurons. Cereb Cortex 23:1473–1483. 10.1093/cercor/bhs137 [DOI] [PubMed] [Google Scholar]
- Hoerder-Suabedissen A, Oeschger FM, Krishnan ML, Belgard TG, Wang WZ, Lee S, Webber C, Petretto E, Edwards AD, Molnár Z (2013) Expression profiling of mouse subplate reveals a dynamic gene network and disease association with autism and schizophrenia. Proc Natl Acad Sci U S A 110:3555–3560. 10.1073/pnas.1218510110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter D, Kingdom J, Jaeggi E (2010) Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int J Pediatr 2010:401323. 10.1155/2010/401323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzie LL, Corbett CJ, Firl DJ, Robinson S (2015) Postnatal erythropoietin mitigates impaired cerebral cortical development following subplate loss from prenatal hypoxia-ischemia. Cereb Cortex 25:2683–2695. 10.1093/cercor/bhu066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano C, Sosunov S, Niatsetskaya Z, Isler JA, Utkina-Sosunova I, Jang I, Ratner V, Ten V (2015) Mild intermittent hypoxemia in neonatal mice causes permanent neurofunctional deficit and white matter hypomyelination. Exp Neurol 264:33–42. 10.1016/j.expneurol.2014.11.010 [DOI] [PubMed] [Google Scholar]
- Kanold PO, Luhmann HJ (2010) The subplate and early cortical circuits. Annu Rev Neurosci 33:23–48. 10.1146/annurev-neuro-060909-153244 [DOI] [PubMed] [Google Scholar]
- Kanold PO, Kara P, Reid RC, Shatz CJ (2003) Role of subplate neurons in functional maturation of visual cortical columns. Science 301:521–525. 10.1126/science.1084152 [DOI] [PubMed] [Google Scholar]
- Kinney HC, Haynes RL, Xu G, Andiman SE, Folkerth RD, Sleeper LA, Volpe JJ (2012) Neuron deficit in the white matter and subplate in periventricular leukomalacia. Ann Neurol 71:397–406. 10.1002/ana.22612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ. (2013) Molecular mechanisms of dendrite stability. Nat Rev Neurosci 14:536–550. 10.1038/nrn3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komitova M, Xenos D, Salmaso N, Tran KM, Brand T, Schwartz ML, Ment L, Vaccarino FM (2013) Hypoxia-induced developmental delays of inhibitory interneurons are reversed by environmental enrichment in the postnatal mouse forebrain. J Neurosci 33:13375–13387. 10.1523/JNEUROSCI.5286-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostovic I, Rakic P (1990) Developmental history of the transient subplate zone in the visual and somatosensory cortex of the macaque monkey and human brain. J Comp Neurol 297:441–470. 10.1002/cne.902970309 [DOI] [PubMed] [Google Scholar]
- le Feber J, Erkamp N, van Putten MJ, Hofmeijer J (2017) Loss and recovery of functional connectivity in cultured cortical networks exposed to hypoxia. J Neurophysiol 118:394–403. 10.1152/jn.00098.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligam P, Haynes RL, Folkerth RD, Liu L, Yang M, Volpe JJ, Kinney HC (2009) Thalamic damage in periventricular leukomalacia: novel pathologic observations relevant to cognitive deficits in survivors of prematurity. Pediatr Res 65:524–529. 10.1203/PDR.0b013e3181998baf [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh WY, Anderson PJ, Cheong JL, Spittle AJ, Chen J, Lee KJ, Molesworth C, Inder TE, Connelly A, Doyle LW, Thompson DK (2017) Neonatal basal ganglia and thalamic volumes: very preterm birth and 7-year neurodevelopmental outcomes. Pediatr Res. Advance online publication. Retrieved Aug. 30, 2017. doi: 10.1038/pr.2017.161. 10.1038/pr.2017.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P (1995) Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol 54:358–370. 10.1097/00005072-199505000-00009 [DOI] [PubMed] [Google Scholar]
- Martin RJ, Wang K, Köroğlu O, Di Fiore J, Kc P (2011) Intermittent hypoxic episodes in preterm infants: do they matter? Neonatology 100:303–310. 10.1159/000329922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon E, Chen K, Gong X, Sharifnia E, Hagen M, Cai V, Shaver DC, Riddle A, Dean JM, Gunn AJ, Mohr C, Kaplan JS, Rossi DJ, Kroenke CD, Hohimer AR, Back SA (2014) Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann Neurol 75:508–524. 10.1002/ana.24100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure MM, Riddle A, Manese M, Luo NL, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Vinecore K, Roberts CT, Hohimer AR, Back SA (2008) Cerebral blood flow heterogeneity in preterm sheep: lack of physiological support for vascular boundary zones in fetal cerebral white matter. J Cereb Blood Flow Metab 28:995–1008. 10.1038/sj.jcbfm.9600597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell SK, Ghosh A, Shatz CJ (1989) Subplate neurons pioneer the first axon pathway from the cerebral cortex. Science 245:978–982. 10.1126/science.2475909 [DOI] [PubMed] [Google Scholar]
- McNeal DW, Brandner DD, Gong X, Postupna NO, Montine TJ, Keene CD, Back SA (2016) Unbiased stereological analysis of reactive astrogliosis to estimate age-associated cerebral white matter injury. J Neuropathol Exp Neurol 75:539–554. 10.1093/jnen/nlw032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM (2003) Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci 23:3308–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailova A, Sunkara N, McQuillen PS (2017) Unbiased quantification of subplate neuron loss following neonatal hypoxia-ischemia in a rat model. Dev Neurosci 39:171–181. 10.1159/000460815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake S, Muramatsu R, Hamaguchi M, Yamashita T (2015) Prolyl hydroxylase regulates axonal rewiring and motor recovery after traumatic brain injury. Cell Death Dis 6:e1638. 10.1038/cddis.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr C, Brady JD, Rossi DJ (2010) Young age and low temperature, but not female gender delay ATP loss and glutamate release, and protect Purkinje cells during simulated ischemia in cerebellar slices. Neuropharmacology 58:392–403. 10.1016/j.neuropharm.2009.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr C, Kolotushkina O, Kaplan JS, Welsh J, Daunais JB, Grant KA, Rossi DJ (2013) Primate cerebellar granule cells exhibit a tonic GABAAR conductance that is not affected by alcohol: a possible cellular substrate of the low level of response phenotype. Front Neural Circuits 7:189. 10.3389/fncir.2013.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry G (2012) Cerebral cortical development in rodents and primates. Prog Brain Res 195:45–70. 10.1016/B978-0-444-53860-4.00003-9 [DOI] [PubMed] [Google Scholar]
- Morton PD, Korotcova L, Lewis BK, Bhuvanendran S, Ramachandra SD, Zurakowski D, Zhang J, Mori S, Frank JA, Jonas RA, Gallo V, Ishibashi N (2017) Abnormal neurogenesis and cortical growth in congenital heart disease. Sci Transl Med 9:eaah7029. 10.1126/scitranslmed.aah7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasunder AC, Kinney HC, Blüml S, Tavaré CJ, Rosser T, Gilles FH, Nelson MD, Panigrahy A (2011) Abnormal microstructure of the atrophic thalamus in preterm survivors with periventricular leukomalacia. AJNR Am J Neuroradiol 32:185–191. 10.3174/ajnr.A2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusa C, Oeschger F, Ginet V, Wang WZ, Hoerder-Suabedissen A, Matsuyama T, Truttmann AC, Molnár Z (2014) Subplate in a rat model of preterm hypoxia-ischemia. Ann Clin Transl Neurol 1:679–691. 10.1002/acn3.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson CR, Folkerth RD, Billiards SS, Trachtenberg FL, Drinkwater ME, Volpe JJ, Kinney HC (2007) Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol 114:619–631. 10.1007/s00401-007-0295-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AG, de Jong D, Kanatsou S, Krugers H, Knapman A, Heinzmann JM, Holsboer F, Landgraf R, Joëls M, Touma C (2012) Dendritic morphology of hippocampal and amygdalar neurons in adolescent mice is resilient to genetic differences in stress reactivity. PLoS One 7:e38971. 10.1371/journal.pone.0038971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock R, Hobert O (2008) Oxygen levels affect axon guidance and neuronal migration in Caenorhabditis elegans. Nat Neurosci 11:894–900. 10.1038/nn.2152 [DOI] [PubMed] [Google Scholar]
- Pogledic I, Kostovic I, Fallet-Bianco C, Adle-Biassette H, Gressens P, Verney C (2014) Involvement of the subplate zone in preterm infants with periventricular white matter injury. Brain Pathol 24:128–141. 10.1111/bpa.12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H, Stellwagen D (2014) Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology 78:13–22. 10.1016/j.neuropharm.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Rees S, Breen S, Loeliger M, McCrabb G, Harding R (1999) Hypoxemia near mid-gestation has long-term effects on fetal brain development. J Neuropathol Exp Neurol 58:932–945. 10.1097/00005072-199909000-00004 [DOI] [PubMed] [Google Scholar]
- Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Hohimer AR, Back SA (2006) Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci 26:3045–3055. 10.1523/JNEUROSCI.5200-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle A, Dean J, Buser J, Gong X, Maire J, Chen K, Ahmad T, Chen V, Nguyen T, Kroenke C, Hohimer A, Back S (2011) Histopathological correlates of MRI-defined chronic perinatal white matter injury. Ann Neurol 70:493–507. 10.1002/ana.22501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Silbereis J, Komitova M, Mitchell P, Chapman K, Ment LR, Schwartz ML, Vaccarino FM (2012) Environmental enrichment increases the GFAP+ stem cell pool and reverses hypoxia-induced cognitive deficits in juvenile mice. J Neurosci 32:8930–8939. 10.1523/JNEUROSCI.1398-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Tomasi S, Vaccarino FM (2014) Neurogenesis and maturation in neonatal brain injury. Clin Perinatol 41:229–239. 10.1016/j.clp.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M, Szigeti-Buck K, Coman D, Huang Y, McCarter RJ Jr, Hyder F, Horvath TL, Gallo V (2014) Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 506:230–234. 10.1038/nature12880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlösser B, ten Bruggencate G, Sutor B (1998) The intracellular tracer Neurobiotin alters electrophysiological properties of rat neostriatal neurons. Neurosci Lett 249:13–16. 10.1016/S0304-3940(98)00366-8 [DOI] [PubMed] [Google Scholar]
- Schwartz ML, Vaccarino F, Chacon M, Yan WL, Ment LR, Stewart WB (2004) Chronic neonatal hypoxia leads to long term decreases in the volume and cell number of the rat cerebral cortex. Semin Perinatol 28:379–388. 10.1053/j.semperi.2004.10.009 [DOI] [PubMed] [Google Scholar]
- Seabold GK, Daunais JB, Rau A, Grant KA, Alvarez VA (2010) DiOLISTIC labeling of neurons from rodent and non-human primate brain slices. J Vis Exp 6:1–5. 10.3791/2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia K, McClure M, Moravec M, Luo N, Wang Y, Gong X, Riddle A, Craig A, Struve J, Sherman L, Back S (2008) Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol 63:517–526. 10.1002/ana.21359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson S, King JL (1911) Localisation of the motor area in the sheep. Q J Exp Physiol 4:53–65. 10.1113/expphysiol.1911.sp000083 [DOI] [Google Scholar]
- Stellwagen D. (2011) The contribution of TNFalpha to synaptic plasticity and nervous system function. Adv Exp Med Biol 691:541–557. 10.1007/978-1-4419-6612-4_57 [DOI] [PubMed] [Google Scholar]
- Torres-Reveron J, Friedlander MJ (2007) Properties of persistent postnatal cortical subplate neurons. J Neurosci 27:9962–9974. 10.1523/JNEUROSCI.1536-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderwolf C, Cooley R (1990) The sheep brain: a photographic series, Ed 2 London, Ontario: Kirby. [Google Scholar]
- Vinall J, Grunau RE, Brant R, Chau V, Poskitt KJ, Synnes AR, Miller SP (2013) Slower postnatal growth is associated with delayed cerebral cortical maturation in preterm newborns. Sci Transl Med 5:168ra168. 10.1126/scitranslmed.3004666 [DOI] [PubMed] [Google Scholar]
- Viswanathan S, Bandyopadhyay S, Kao JP, Kanold PO (2012) Changing microcircuits in the subplate of the developing cortex. J Neurosci 32:1589–1601. 10.1523/JNEUROSCI.4748-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohr BR. (2014) Neurodevelopmental outcomes of extremely preterm infants. Clin Perinatol 41:241–255. 10.1016/j.clp.2013.09.003 [DOI] [PubMed] [Google Scholar]
- Volpe JJ. (1996) Subplate neurons: missing link in brain injury of the premature infant? Pediatrics 97:112–113. [PubMed] [Google Scholar]
- Volpe JJ. (2008) Neurology of the newborn. Philadelphia: Saunders. [Google Scholar]
- Volpe JJ. (2009) Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 8:110–124. 10.1016/S1474-4422(08)70294-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WZ, Hoerder-Suabedissen A, Oeschger FM, Bayatti N, Ip BK, Lindsay S, Supramaniam V, Srinivasan L, Rutherford M, Møllgård K, Clowry GJ, Molnár Z (2010) Subplate in the developing cortex of mouse and human. J Anat 217:368–380. 10.1111/j.1469-7580.2010.01274.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Han VZ, Rossi DJ, Mohr C, Odagiri M, Daunais JB, Grant KA (2011) Bidirectional plasticity in the primate inferior olive induced by chronic ethanol intoxication and sustained abstinence. Proc Natl Acad Sci U S A 108:10314–10319. 10.1073/pnas.1017079108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, Zahed H, Maltepe E, Rowitch DH (2014) Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 158:383–396. 10.1016/j.cell.2014.04.052 [DOI] [PMC free article] [PubMed] [Google Scholar]