

Abstract
Compelling genetic evidence links the amyloid precursor protein (APP) to Alzheimer's disease (AD) and several theories have been advanced to explain the relationship. A leading hypothesis proposes that a small amphipathic fragment of APP, the amyloid β-protein (Aβ), self-associates to form soluble aggregates that impair synaptic and network activity. Here, we used the most disease-relevant form of Aβ, protein isolated from AD brain. Using this material, we show that the synaptotoxic effects of Aβ depend on expression of APP and that the Aβ-mediated impairment of synaptic plasticity is accompanied by presynaptic effects that disrupt the excitatory/inhibitory (E/I) balance. The net increase in the E/I ratio and inhibition of plasticity are associated with Aβ localizing to synapses and binding of soluble Aβ aggregates to synapses requires the expression of APP. Our findings indicate a role for APP in AD pathogenesis beyond the generation of Aβ and suggest modulation of APP expression as a therapy for AD.
SIGNIFICANCE STATEMENT Here, we report on the plasticity-disrupting effects of amyloid β-protein (Aβ) isolated from Alzheimer's disease (AD) brain and the requirement of amyloid precursor protein (APP) for these effects. We show that Aβ-containing AD brain extracts block hippocampal LTP, augment glutamate release probability, and disrupt the excitatory/inhibitory balance. These effects are associated with Aβ localizing to synapses and genetic ablation of APP prevents both Aβ binding and Aβ-mediated synaptic dysfunctions. Our results emphasize the importance of APP in AD and should stimulate new studies to elucidate APP-related targets suitable for pharmacological manipulation.
Keywords: Alzheimer's disease, amyloid beta, amyloid precursor protein, array tomography, long-term potentiation, whole-cell patch-clamp
Introduction
Mutation, overexpression, or altered processing of the amyloid precursor protein (APP) underlie all known monogenic cases of familial Alzheimer's disease (AD) (Tanzi, 2012; Guerreiro and Hardy, 2014). Although the physiological roles of APP are not fully understood, a myriad of studies indicate that APP plays a role in synaptic plasticity, dendritic morphogenesis, and neuroprotection (Müller and Zheng, 2012). Membrane-tethered APP can act as a cell-adhesion molecule linking the presynapse and postsynapse (Soba et al., 2005) and APP has been shown to regulate synaptic vesicle proteins, synaptic transmission, and plasticity (Dawson et al., 1999; Laßek et al., 2013; Fanutza et al., 2015; Laßek et al., 2016). In the rat dentate gyrus (DG), APP expression is known to change during memory consolidation (Conboy et al., 2005) and intraventricular administration of anti-APP antibodies or antisense oligonucleotides results in profound amnesia (Doyle et al., 1990; Huber et al., 1993; Mileusnic et al., 2000). APP is a component of the presynaptic GABA-B1a receptor (GABAB1a-R) complex (Bai et al., 2008; Schwenk et al., 2016) and neuron-type-specific knock-out (KO) of APP indicates an important role for APP in GABAergic transmission and maintenance of the excitatory/inhibitory (E/I) balance (Wang et al., 2014).
APP is a complex molecule that undergoes substantial posttranslational modification and at least 10 different proteolytic fragments of APP have been identified. Several of these are suggested to be pathogenic (Neve and McPhie, 2007; Yankner and Lu, 2009; Tamayev et al., 2012; Welzel et al., 2014; Willem et al., 2015), whereas others are neuroprotective (Mockett et al., 2017). The fragment from which the precursor protein derives its name, the amyloid β-protein (Aβ), is found in the tell-tale amyloid plaques that litter the brains of individuals who die with AD. Aβ comprises a family of APP-derived peptides that share a common core of ∼30 aa (Walsh and Teplow, 2012) that are produced by the concerted action of two aspartyl proteases, β-secretase and γ-secretase (De Strooper, 2010). Aβ peptides are prone to self-associate and multiple studies indicate that certain forms of Aβ adversely affect synaptic form and function (Li et al., 2009).
The synaptotoxic activity of Aβ and the involvement of APP in synapse formation and activity are particularly relevant to AD because in vivo and postmortem studies indicate that synapse dysfunction and loss are prominent early features of AD (Scheff et al., 2006; Scheff et al., 2007; Johnson et al., 2012). Acute studies in wild-type (WT) rodents show that nonfibrillar, water-soluble Aβ from a variety of sources are potent synaptotoxins (Lambert et al., 1998; Walsh et al., 2002; Cleary et al., 2005; Lesné et al., 2006; Klyubin et al., 2008; Minkeviciene et al., 2009; Kurudenkandy et al., 2014). Furthermore, in vitro and in vivo studies demonstrate that the most disease-relevant form of nonfibrillar Aβ, Aβ extracted from the water-soluble phase of AD brain, inhibits LTP, facilitates LTD, reduces synaptic remodeling, and impairs memory consolidation (Shankar et al., 2008; Barry et al., 2011; Freir et al., 2011; Borlikova et al., 2013; Yang et al., 2017). Here, we show that the block of LTP mediated by Aβ-containing AD brain extracts is accompanied by opposing changes in excitatory and inhibitory presynaptic release probabilities and consequent disruption of the E/I balance. The net increase in the E/I ratio and inhibition of LTP require expression of APP and are associated with Aβ localizing to synapses. These findings suggest a link between Aβ toxicity and perturbation of the normal regulatory role of APP and are consistent with prior studies showing a role for APP in Aβ toxicity (White et al., 1998; Lorenzo et al., 2000; Shaked et al., 2006; Sola Vigo et al., 2009; Fogel et al., 2014; Kirouac et al., 2017). In light of these results, we suggest that downregulation of APP expression or modulation of its interaction with synaptotoxic Aβ species should be investigated as an approach to treat AD.
Materials and Methods
Reagents.
All chemicals and reagents were purchased from Sigma-Aldrich unless otherwise noted. Synthetic Aβ1-42 was synthesized and purified using reverse-phase HPLC by Dr. James I. Elliott at the ERI Amyloid laboratory (Oxford, CT). Peptide mass and purity (>99%) were confirmed by reverse-phase HPLC and electrospray/ion trap mass spectrometry. The N-terminally extended Aβ peptide, -31Aβ1-40, was prepared and purified as described previously (McDonald et al., 2015) and recombinant Aeta-α (Aη-α, APP505-611) was a gift from Drs. Willem and Haass (Ludwig-Maximillian University, Munich, Germany).
Antibodies.
The antibodies used and their sources are described in Table 1.
Table 1.
Primary and secondary antibodies
| Antibody | Type | Antigen/epitope | Dilution for IP | Concentration for WB | Concentration for ELISA | Dilution for AT | Source/reference |
|---|---|---|---|---|---|---|---|
| 3D6 | Monoclonal | Aβ1-5 | — | — | 1 μg/ml | — | Elan/(Johnson-Wood et al., 1997) |
| 6E10 | Monoclonal | Aβ6-10 | — | 1 μg/ml | — | — | BioLegend/(Kim et al., 1988) |
| 266 | Monoclonal | Aβ16-23 | — | — | 3 μg/ml | — | Elan/(Seubert et al., 1992) |
| 2G3 | Monoclonal | Aβ terminating at Val40 | — | 1 μg/ml | — | — | Elan/(Johnson-Wood et al., 1997) |
| 21F12 | Monoclonal | Aβ terminating at Ile42 | — | 1 μg/ml | 1 μg/ml | — | Elan/(Johnson-Wood et al., 1997) |
| 1C22 | Monoclonal | Aβ aggregates | — | — | 3 μg/ml | 20 μg/ml | Walsh laboratory/(Mably et al., 2015) |
| AW7 | Polyclonal | Pan anti-Aβ | 1:80 | — | — | — | Walsh laboratory/(McDonald et al., 2012) |
| 22C11 | Monoclonal | APP66-81 | — | 1 μg/ml | — | 20 μg/ml | Millipore/(Austin et al., 2009) |
| AB1543P | Polyclonal | Rabbit anti-synapsin-1 | — | — | — | 1:100 | Millipore/(Kay et al., 2013) |
| 3450P | Polyclonal | Rabbit anti-PSD95 | — | — | — | 1:50 | Cell Signaling Technology/(Kay et al., 2013) |
| A21202 | Polyclonal | Donkey anti-mouse 488 | — | — | — | 1:50 | Invitrogen |
| A21207 | Polyclonal | Donkey anti-rabbit 594 | — | — | — | 1:50 | Invitrogen |
| T6074 | Monoclonal | Anti-α-tubulin | — | 1 μg/ml | — | — | Sigma-Aldrich |
Preparation of human brain extracts.
All human specimens were obtained and used in accordance with the Partner's Institutional Review Board (Protocol: Walsh BWH 2011). Brain tissue was obtained from two individuals (referred to as AD1 and AD2) who died with AD and one individual who died free of AD (designated NC). AD1 was an 87-year-old man who had scored 23 on the Mini Mental State Examination (MMSE) 9 months before death and was designated Braak stage 4 at postmortem with pathological changes consistent with mild AD. AD2 was a 68-year-old female with end-stage AD. Three years before death, AD2 scored 23 on the MMSE, but in her last weeks, she was unable to answer questions other than to provide her first name. Upon postmortem examination, there was evidence of fulminant amyloid and neurofibrillary tangle pathology that was designated Braak stage V/VI. Neither AD1 nor AD2 had a family history of AD. NC was a 58-year-old female who died free of AD symptoms and pathology. AD1 and NC had postmortem intervals (PMIs) of 18 h and AD2 had a PMI of 12 h. Aqueous extracts of brain were prepared by homogenizing cortical tissue in a buffer that we refer to as artificial CSF base buffer (aCSF-B) containing the following (in mm): 124 NaCl, 2.8 KCl, 1.25 NaH2PO4, and 26 NaHCO3, pH 7.4. aCSF-B was the core buffer used in subsequent electrophysiology experiments. Whole frozen temporal cortex was left at 4°C until the tissue was sufficiently soft to cut. Meninges and large blood vessels were removed and gray matter dissected from white matter. The total amount of gray matter obtained was between 12 and 14 g. Two gram lots of tissue were diced using a razor blade and then homogenized in 10 ml of ice-cold aCSF-B containing 5 mm EDTA, 1 mm EGTA, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 2 μg/ml pepstatin, 120 μg/ml pefabloc, and 5 mm NaF with 25 strokes of a Dounce homogenizer (Fisher). Homogenates from 6 2 g lots were pooled and centrifuged at 198,000 × g and 4°C for 110 min in a SW 41-Ti rotor (Beckman Coulter). The upper 90% of supernatant was dialyzed using Slide-A-Lyzer G2 Dialysis Cassettes at a 2 kDa molecular weight cutoff (Fisher Scientific) against fresh aCSF-B to remove bioactive small molecules and drugs. Dialysis was performed at 4°C against a 100-fold excess of buffer with buffer changed 3 times over a 72 h period. Thereafter, extracts were divided into 2 parts: 1 portion was immunodepleted (ID) of Aβ by 3 rounds of 12 h incubations with the anti-Aβ antibody AW7 plus Protein A Sepharose (PAS) beads at 4°C (Freir et al., 2011). The second portion was treated in an identical manner, but this time incubated with preimmune serum plus PAS beads. Samples were cleared of beads and 0.5 ml aliquots stored at −80°C until used for biochemical or electrophysiological experiments. Samples were thawed only once before use.
Preparation of amyloid-derived diffusible ligands (ADDLs).
ADDLs were prepared essentially as described previously (Freir et al., 2011). Hexafluoro-2-propanol (HFIP; 222 μl) was added to 1 mg of Aβ(1-42) in a 2 ml low-binding microcentrifuge tube to produce a peptide concentration of 1 mm. The solution was split into two tubes, incubated at 37°C for 1 h, and mixed by vortexing every 10 min. The HFIP was gently evaporated under a nitrogen stream with rotation of the tube to ensure formation of an even film of peptide on the lower walls of the tube. Dried peptide films were stored over desiccant at 20°C for a minimum of 14 h. The peptide film from each tube was then dissolved in 20 μl of anhydrous DMSO (Life Technologies) and 5 μl lots of the DMSO mixture were added stepwise to 980 μl of F12 medium (Life Technologies), with vortexing between each addition. The resulting solution was incubated at 4°C for 48 h and then centrifuged at 16,000 × g for 10 min. The upper 95% of the supernatant was transferred to a new microcentrifuge tube and the protein concentration determined using the extinction coefficient ε275 = 1361 M−1 cm−1 (O'Malley et al., 2014). Aliquots were then immediately frozen on dry ice and stored at −80°C.
Characterization of ADDLs.
The size and morphology of structures present in the ADDL preparation were investigated using negative contrast electron microscopy and analytical size exclusion chromatography (SEC). Samples were stained and visualized essentially as described previously (Betts et al., 2008). An aliquot of the ADDL preparation (50 μl) was diluted 1:1 with F12 medium and then adsorbed (10 μl) onto formvar-coated copper grids (Electron Microscopy Sciences). After 1 min, 10 μl of 0.25% glutaraldehyde was added and incubated for 1 min. Thereafter, grids were wicked dry using filter paper, washed twice with MilliQ water (Millipore), and then stained with 1% uranyl acetate for 2 min. Grids were allowed to air dry for at least 10 min, stored at room temperature, and then examined using a 1200EX microscope (JEOL).
A separate aliquot of ADDLs (50 μl) was thawed and loaded on to a Superdex 75 3.2/300 PE column (GE Healthcare) eluted in PBS, pH 7.4, at 0.8 ml/min using a Shimadzu HPLC system.
Preparation of synthetic peptides used for Western blotting (WB).
Aη-α peptide was dissolved in 50 mm ammonium bicarbonate, pH 8.5, diluted to 10 ng/μl, aliquoted, and stored frozen at −80°C. Aβ1-42 and -31Aβ1-40, which are prone to aggregate, were treated to depolymerize any preexisting aggregates. Briefly, peptides were dissolved in 50 mm Tris-HCl, pH 8.5, containing 7 m guanidium-HCl (GuHCl) and 5 mm EDTA at a concentration of 1 mg/ml and incubated at room temperature overnight. Samples were then centrifuged for 30 min at 16,000 × g and chromatographed on a Superdex 75 10/300 column eluted at 0.5 ml/min with 50 mm ammonium bicarbonate, pH 8.5. The concentration of the peak fraction for each sample was determined by absorbance at 275 nm. The peptide was then diluted to 10 ng/μl, aliquoted, and stored frozen at −80°C.
Immunoprecipitation/WB(IP/WB) of Aβ in brain extracts.
Extracts were first precleared with PAS beads to minimize nonspecific interactions in the subsequent IP. One-milliliter aliquots of extracts were incubated with 15 μl of PAS beads for 1 h at 4°C with gentle shaking. PAS beads were removed by centrifugation at 4000 × g for 5 min and the supernatant was divided into 0.5 ml aliquots. Each aliquot was incubated with 10 μl of AW7 and 15 μl of PAS beads overnight at 4°C with gentle shaking. Aβ-antibody–PAS complexes were collected by centrifugation and washed as described previously (Shankar et al., 2011). The immunoprecipitated Aβ was eluted by boiling in 18 μl of 1× sample buffer (50 mm Tris, 2% w/v SDS, 12% v/v glycerol with 0.01% phenol red) and electrophoresed on hand-poured, 15-well 16% polyacrylamide Tris-tricine gels. Aη-α, Aβ1-42, and -31Aβ1-40 were run as loading controls and protein transferred onto 0.2 μm nitrocellulose at 400 mA and 4°C for 2 h. Blots were microwaved in PBS and Aβ detected using the anti-Aβ40 and anti-Aβ42 antibodies 2G3 and 21F12, respectively, and bands visualized using a Li-COR Odyssey infrared imaging system. To determine whether AW7 immunoprecipitated other APP metabolites (e.g., APPsα, N-terminally extended Aβ, or Aη peptides) from AD brain extracts, certain blots were developed with 6E10 (Table 1).
Meso Scale Discovery (MSD) Aβ immunoassays.
Samples were analyzed for Aβ content using two distinct assay formats: the Aβx-42 assay, which detects Aβ42 monomers preferentially, and the oAssay, which detects Aβ oligomers and aggregates preferentially (Mably et al., 2015; Yang et al., 2015). Immunoassays were performed using the MSD platform and reagents from Meso Scale. The Aβx-42 assay uses mAb 266 (3 μg/ml) for capture and biotinylated 21F12 (1 μg/ml) for detection and the oAssay uses mAb 1C22 (3 μg/ml) for capture and biotinylated 3D6 (1 μg/ml) for detection. Samples, standards, and blanks were loaded in duplicate and analyzed as described previously (Mably et al., 2015).
Because GuHCl effectively disaggregates high-molecular-weight Aβ species (Mably et al., 2015), samples were analyzed both with and without incubation in 5 m GuHCl. Analysis of samples in the absence of GuHCl allows the measurement of native Aβ42 monomer using the Aβx-42 assay and native Aβ aggregates using the oAssay. Analysis of samples treated with GuHCl allows detection of disassembled aggregates with Aβx-42 assay. To dissociate aggregates, 20 μl of extract was incubated overnight with 50 μl of 7 m GuHCl at 4°C. Thereafter, samples were diluted 1:10 with assay diluents so that the final GuHCl concentration was 0.5 m. Aβ standards were prepared in TBS, pH 7.4, containing 0.5 m GuHCl, 0.05% Tween 20, and 1% blocker A so that both standards and samples contained the same final concentration of GuHCl.
Mice.
All animal procedures were performed in accordance with the National Institutes of Health Policy on the Use of Animals in Research and were approved by the Harvard Medical School Standing Committee on Animals. WT C57BL/6 mice were purchased from The Jackson Laboratory. APP KO mice on a C57BL/6 background and littermate WT controls were obtained from the Young-Pearse laboratory (Callahan et al., 2017). A second line of APP KO mice were purchased from The Jackson Laboratory (APPtm1Dbo/J) (Zheng et al., 1995) and, for certain experiments, these animals were bred with WT C57BL/6 mice to generate APP+/− hemizygotes. Animals were housed in a room with a 12 h light/dark circadian cycle with ad libitum access to food and water. Mice were genotyped by PCR before use and both male and female mice were used. In certain experiments, upon completion of electrophysiological recordings, brain slices were used for WB or array tomography (AT).
Brain slice preparation.
Two- to 3-month-old male and female animals were anesthetized with isoflurane and decapitated. Brains were rapidly removed and immediately immersed in ice-cold (0–4°C) artificial CSF (aCSF). The aCSF contained the following (in mm): 124 NaCl, 3 KCl, 2.4 CaCl2, 2 MgSO4·7H2O, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose equilibrated with 95% O2 and 5% CO2, pH 7.4, 310 mOsm. Coronal brain slices (350 μm), including hippocampus (Wang et al., 2008), were prepared using a Leica VT1000 S vibratome, transferred to an interface chamber, incubated at 34 ± 5°C for 20 min, and then kept at room temperature for 1 h before recording.
LTP recording.
Brain slices were transferred to a submerged recording chamber and perfused (10 ml/min) with oxygenated (95% O2 and 5% CO2) aCSF for 10 min before electrophysiological recordings. Brian slices were visualized using an infrared and differential interference contrast (IR-DIC) camera (Hitachi) mounted on an upright Olympus microscope. Recording electrodes were pulled from borosilicate glass capillaries (Sutter Instruments) using a micropipette puller (model P-97; Sutter Instruments) with resistance of ∼2 MΩ when filled with aCSF. To induce field EPSPs (fEPSPs) in the hippocampal CA1, a tungsten wire-stimulating electrode (FHC) was placed on the Schaffer collaterals of the CA3 and a recording electrode was placed at least 300 μm away on the striatum radiatum of the CA1. The initial 10–40% slopes of fEPSPs were calculated. Test stimuli were delivered once every 20 s (0.05 Hz) and the stimulus intensity was adjusted to produce a baseline fEPSP of 30–40% of the maximal response. A stable baseline was recorded for at least 10 min before the addition of sample. Thirty minutes after application of sample, LTP was induced by theta-burst stimulation. This involved 3 trains of 4 pulses each delivered at 100 Hz 10 times, with an interburst interval of 200 ms with a 20 s interval between each train. Field potentials were recorded using a Multiclamp amplifier (model 700B; Molecular Devices) coupled to a Digidata 1440A digitizer. Signal was sampled at 10 kHz and filtered at 2 kHz and data were analyzed using Clampex 10 software (Molecular Devices).
Whole-cell patch-clamp recording.
Brain slices were prepared from male and female WT and APP KO mice (1–2 months old) as described above for LTP experiments but using a cutting solution that contained the following (in mm): 72 sucrose, 83 NaCl, 2.5 KCl, 1 NaH2PO4, 3.3 MgSO4 · 7H2O, 26.2 NaHCO3, 22 dextrose, and 0.5 CaCl2 saturated with 95% O2 and 5% CO2, pH 7.4, 310 mOsm (Wang et al., 2015). Slices were incubated in oxygenated slicing solution for 20 min and held at room temperature for a further 40 min before recording. Slices were transferred to a submerged recording chamber and perfused (10 ml/min) with oxygenated (95% O2 and 5% CO2) aCSF for 30 min at room temperature. Whole-cell recordings were made from the somata of CA1 pyramidal neurons visualized using an IR-DIC camera mounted on an upright Olympus microscope. Patch pipettes (4–7 MΩ) were filled with an internal solution containing the following (in mm): 120 Cs-gluconate, 5 MgCl2, 0.6 EGTA, 30 HEPES, 4 MgATP, 0.4 Na2GTP, 10 phosphocreatine-Tris, 5 N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314) at 290 mOsm, pH was adjusted to 7.2 with CsOH. Signal was acquired using a Multiclamp amplifier (model 700B; Molecular Devices) with Clampex 10 software (Molecular Devices), sampled at 10 kHz, and filtered at 2 kHz. Data were stored on a PC after digitization by an A/D converter (Digidata 1440A; Molecular Devices) for offline analysis. Membrane potential was corrected for the liquid junction potential of 13.7 mV. Neurons with negative resting membrane potential less than −60 mV were not analyzed. Input resistance and patching access resistance were monitored continuously during the experiment and cells that exhibited >15–20% changes in these parameters were excluded from analysis.
To preserve a relatively intact neuronal circuit, no receptor antagonists were used. Spontaneous EPSCs (sEPSCs) were collected at a membrane holding potential of −70 mV, which is close to the calculated reverse potential of GABA. To measure the excitatory and inhibitory input on the same neuron, the spontaneous IPSCs (sIPSCs) were also measured on the same neuron, but this time the holding potential was increased to 5–10 mV, a potential close to the reverse potential of excitatory input without visual negative deflection. Recorded neuronal activities were detected as described previously (Lillis et al., 2015) by custom software (DClamp: available at www.ieeg.org/?q=node/34). Integrated excitatory conductance (sEPSCs, GE) and integrated inhibitory conductance (sIPSCs, GI) were calculated as follows (Slomowitz et al., 2015):
 |
Preparation of mouse brain homogenates and detection of APP.
Certain brain slices from WT and APP KO mice were frozen immediately after completion of electrophysiological recording (see Figs. 4, 5, and 6) and stored at −80°C until analyzed. Tissue (∼0.1 mg) was homogenized in 5 volumes (w/v) of ice-cold 20 mm Tris-HCl with 150 mm NaCl and 1% TX-100 (TBS-Tx), pH 7.4, containing protease inhibitors and centrifuged at 100,000 × g and 4°C for 78 min in a TLA-55 rotor (Beckman Coulter). The upper 90% of the supernatant was removed, aliquoted, and stored at −80°C pending analysis. Ten micrograms of total protein was boiled in 1× sample buffer (62.5 mm Tris, 1% w/v SDS, 10% v/v glycerol, 0.01% phenol red, and 2% β-mercaptoethanol) for 5 min and electrophoresed on hand-poured, 15-well 10% polyacrylamide Tris-glycine gels. Gels were rinsed in transfer buffer (10% methanol, 192 mm glycine, and 25 mm Tris) and proteins were electroblotted onto 0.2 μm nitrocellulose membranes at 400 mA and 4°C for 2.5 h. Membranes were developed using the anti-APP antibody 22C11 and bands visualized using a Li-COR Odyssey infrared imaging system.
Figure 4.
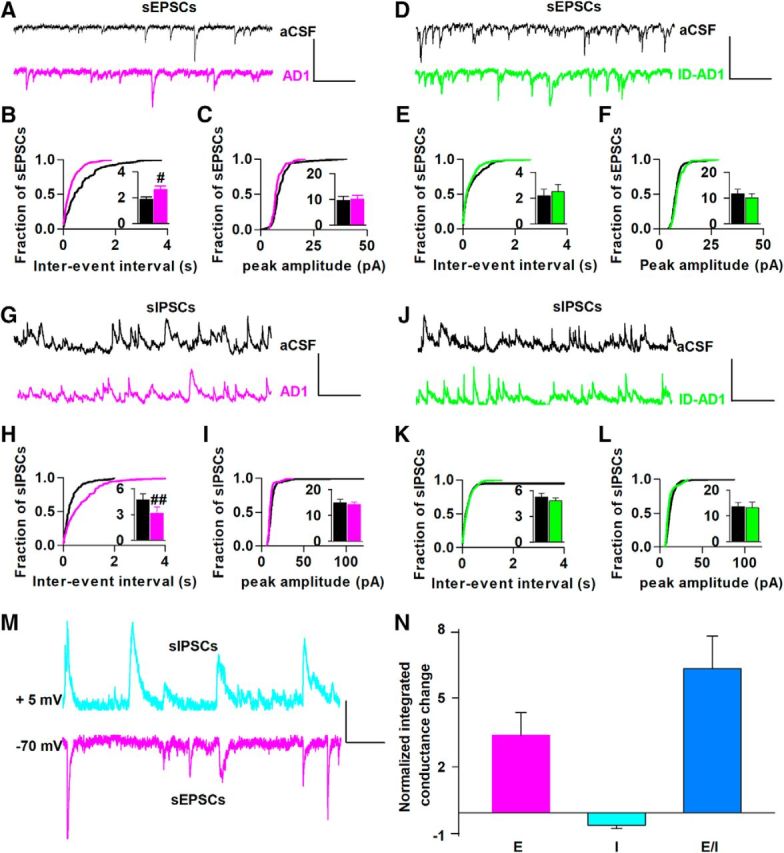
AD-brain-derived Aβ affects both excitatory and inhibitory synaptic inputs, causing disruption of the excitatory/inhibitory ratio at individual CA1 neurons. A, D, Example traces of sEPSCs before (aCSF, black) and 30 min after (AD1, magenta; ID-AD1, green) addition of sample recorded from individual pyramidal neurons in the hippocampal CA1 area of brain slices with the holding potential fixed at −70 mV. Scale bars, 20 pA, 700 ms. B, Thirty minutes of AD1 treatment decreased cumulative distributions of interevent intervals and increased mean frequency (inset; p = 1.65E-6, K-S test; p < 0.02, Student's t test; n = 7; B), but did not change the cumulative distributions or the mean value (inset) of the amplitude of sEPSCs (n = 7; C). E, F, The ID-AD1 sample had no effect on either frequency or amplitude of sEPSCs (n = 6). G, J, Example traces of sIPSCs before (aCSF, black) and 30 min after (AD1, magenta; ID-AD1, green) treatment were recorded on the same individual pyramidal neurons upon increasing the holding potential to 5 mV. Scale bars, 20 pA, 700 ms. H, 30 min of treatment with the AD1 sample increased interevent intervals and decreased mean frequency (inset) of sIPSCs (magenta) versus aCSF (black; p = 6.19E-6, K-S test; p = 0.008, Student's t test; n = 7). I, Treatment with the AD1 sample did not affect the amplitude of sIPSCs (n = 7) and the ID-AD1 sample had no effect on frequency (K) or the amplitude (L) of sIPSCs versus aCSF control (n = 7). M, Representative traces of sIPSCs and sEPSCs from the same pyramidal neuron show charge transfer measured as the area of events above threshold in the aCSF control. Scale bars, 10 pA, 200 ms. N, Integrated conductances measured between 30 and 35 min after the addition of AD1 application were normalized to the value of 5 min before the addition of AD1. Mean excitatory integrated conductance increased and mean inhibitory integrated conductance decreased upon treatment of AD1 (E: excitatory input/sEPSCs; I: inhibitory input/sIPSCs). Each slice used for each treatment was from a different animal. #p < 0.05, ##p < 0.01.
Figure 5.

Expression of APP is required for the plasticity-disrupting activity of Aβ-containing AD brain extract. A, Detergent extracts of mouse brain slices used for electrophysiology were analyzed for APP by WB with 22C11. Full-length APP was readily detected in extracts from WT littermate mice but not APP KO mice. Slices from two APP KO mice (KO1 and KO2) and two WT mice (WT1 and WT2) are shown. B, Input/output curves recorded in the hippocampal CA1 area are highly similar for both WT and APP KO mouse brain slices (p = 0.19, one-way ANOVA). Values are shown as mean ± SEM. C, LTP recorded in hippocampal CA1 was similar in brain slices from WT and APP KO mice (WT control, black circles vs APP KO control, gray hexagons, p = 0.79, comparison of the last 10 min recording using one-way ANOVA). However, the extract from AD1 brain blocked LTP in WT but not in APP KO mice brain slices. Horizontal gray bar indicates the duration in which sample was present. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF control in WT mice is shown with black circles, AD1 treatment in WT mice is shown with magenta diamonds, the aCSF control in APP KO mice is shown with gray hexagons, and AD1 treatment in APP KO mice is shown using pink upward triangles. WT slices for each treatment came from different animals; the APP KO slices came from a total of four APP KO mice. Scale bars, 0.5 mV, 15 ms. D, Comparison of average potentiation from last 10 min of LTP recording (F = 4.5, p = 0.0005, control vs AD1 in WT mice; F = 4.5, p = 0.41, control vs AD1 in APP KO mice; one-way ANOVA). Symbols correspond to those in C. E, Representative traces of averaged field recordings were collected after 5 stimulation bursts (interstimulation interval 20 ms, interburst interval 30 s) before (gray, aCSF) and 30 min after perfusion with the AD1 sample (pink) on brain slices from APP KO mouse. Scale bars, 0.5 mV, 10 ms. F, fEPSPs amplitude after two to five stimulations were normalized to the value obtained after the first stimulation. There is no significant difference between aCSF control and the presence of AD1 brain extract application (n = 5, F = 5.32, p = 0.7, one-way ANOVA). Values are shown as mean ± SEM. Each slice used for each treatment was from a different animal. ###p < 0.001.
Figure 6.

Characterization of the aqueous extract from AD2 brain, synthetic Aβ oligomers, and a second APP KO mouse line. A, Aqueous extract of AD2 was treated with either preimmune serum or AW7 antiserum. Portions of the mock-ID sample (AD2, magenta) and the AW7 ID sample (ID-AD2, green) were then analyzed by IP/WB using AW7 for IP and a combination of 2G3 and 21F12 (left) or 6E10 (right) for WB. As expected, recombinant Aη-α was detected by 6E10, but not 2G3/21F12. M, Aβ monomer. *Broad smear ∼7–8 kDa. Only nonspecific (NS) bands were detected above the16 kDa marker. B, AD2 (magenta) and ID-AD2 (green) samples were incubated with and without 5 m GuHCl and analyzed using an immunoassay that recognizes Aβ42 monomer preferentially (266-21F12b). AW7 ID reduced monomer from 6.65 ± 0.01 ng/ml to an undetectable level without GuHCl treatment. Upon treatment with GuHCl, the amount of Aβ42 increased to 46.94 ± 0.2 ng/ml in AD2 and this was reduced to 8.62 ± 0.1 ng/ml by immunodepletion. C, Size-exclusion chromatography of ADDLs revealed a prominent high-molecular-weight peak, a trail of intermediate-molecular-weight species, and a small Aβ monomer peak. D, Negative contrast electron micrograph shows mostly protofibril-like structures. Scale bar, 50 nm. E, Brain slices from WT and a second line of APP KOs were analyzed for APP by WB with 22C11. Full-length APP was readily detected in extracts from WT but not APP KO. Slices from two KO (KO1 and KO2) and two WT (WT1 and WT2) mice are shown. F, Input/output curves recorded in the hippocampal CA1 area are highly similar for WT and APP KO mouse brain slices (F = 4.6, p = 0.91, one-way ANOVA). Values are shown as mean ± SEM. Each slice used for each treatment was from a different animal.
AT imaging of mouse brain slices.
Upon completion of electrophysiology recordings, certain brain slices from WT and APP KO mice (see Figs. 4, 5, and 6) were processed for AT (Koffie et al., 2009; Pickett et al., 2016). Slices were fixed in PBS containing 4% paraformaldehyde and 2.5% sucrose at 4°C overnight. Samples were then washed 3 times (10 min each) in cold wash buffer (PBS containing 3.5% sucrose and 50 mm glycine) and the hippocampus was dissected out under a Leica Wild M3Z stereozoom microscope. Thereafter, hippocampi were dehydrated using an ethanol gradient of 50%, 70%, 95%, and 100%. Tissue was then placed into a solution of 1:1 ethanol: LR White resin (Electron Microscopy Sciences) for 5 min and then washed 3 times with LR White. Tissue was incubated overnight at 4°C in LR White, embedded in a gelatin capsule, and polymerized overnight at 53°C. Three embedded blocks per condition were cut into ribbons of 70 nm sections on an ultracut microtome (Leica) using a Jumbo Histo Diamond Knife (Diatome). Ribbons were collected on gelatin-coated glass coverslips, stained with antibodies, and imaged along the ribbon. Two ribbons per slice were collected and one was stained for PSD95 and 1C22 and the other for synapsin-1 and 1C22. Primary antibodies were 1C22 (20 μg/ml), rabbit anti-PSD95 (1:50, 3450P; Cell Signaling Technology), and rabbit anti-synapsin-1 (1:100, AB1543P; Millipore). The secondary antibodies donkey anti-mouse 488 (A21202) and donkey anti-rabbit 594 (A21207) were from Invitrogen and used at 1:50 concentrations.
Two image stacks per ribbon were collected from the stratum radiatum using a Zeiss axio Imager Z2 epifluorescent microscope with a 63× 1.4 numerical aperture Plan Apochromat objective. Images were acquired with a CoolSnap digital camera and AxioImager software with AT macros (Carl Zeiss). Images from each set of serial sections were compiled to create a 3D stack and aligned using ImageJ multistackreg macros (Kay et al., 2013). Regions of interest (10 × 10 μm) were selected, cropped, and thresholded in ImageJ (Schindelin et al., 2012; Ollion et al., 2013) (Fig. 1). Custom MATLAB macros were used to remove single slice punctuates, to count synaptic punctuates, and assess colocalization with 1C22 (a minimum of 50% overlap between 1C22 and synaptic punctuate was required to be designated as colocalization). All custom analysis macros will be freely available at http://datashare.is.ed.ac.uk after publication.
Figure 1.

Processing of AT images. Fields of 10 × 10 μm are cropped from an image stack, made into binary stacks in ImageJ, and processed in MATLAB to remove objects not found in serial slices.
Data analysis and statistical analysis.
Figures showing IP/WB and MSD Aβ immunoassay data are representative of at least two independent experiments. For electrophysiological experiments, the AD, ID-AD, and aCSF samples were coded and tested in an interleaved manner to avoid variances in animals or slice quality influencing results. Slices in each group came from different animals unless otherwise noted. Electrophysiological data were analyzed offline by pclamp 10.2 (Molecular Devices) and tested with one-way or two-way ANOVA with Bonferroni post hoc tests or Student's t tests (#p < 0.05, ##p < 0.01, and ###p < 0.001). A Kolmogorov–Smirnov (K-S) test was used to compute differences in distributions of sEPSCs and sIPSCs. AT was analyzed using SPSS version 22. A single percentage colocalization for each parameter was calculated for each slice from ∼41 regions of interest and ≈7500 synapses (∼3500 presynapses and ∼3500 postsynapses). These were analyzed with a Kruskal–Wallis with Dunn's post hoc test. Electrophysiology data are shown as means ± SEM. AT data are shown as medians ± the interquartile range, with each point representing all synapses measured within one slice. Analyses of the same sample using different slices are considered technical replicates and analysis of extracts from different AD brains are considered biological replicates.
Results
We reported previously that aqueous extracts of certain end-stage AD brains block hippocampal LTP in vivo and in vitro (Shankar et al., 2008; Barry et al., 2011; Freir et al., 2011). Here, we further investigated the mechanism of this effect and the requirement of endogenous APP.
Water-soluble extract from AD brain contains both Aβ monomers and oligomers and blocks LTP in a manner dependent on Aβ
Brain extracts were prepared as described and a portion was ID of Aβ or mock-ID with preimmune rabbit serum. Here, the mock-ID extract is referred to as the AD sample and the material depleted of Aβ as ID-AD. ID and AD samples from AD1 were analyzed using IP/WB, and MSD immunoassays that recognize either Aβ oligomers (oAssay) or Aβ42 monomers preferentially (McDonald et al., 2015). IP/WB analysis allows the capture of Aβ structures under native conditions and their detection after denaturing SDS-PAGE. We were careful to also determine whether AW7 altered APP metabolites present in AD brain extracts that contain might all or part of the Aβ sequence; that is, APPsα, N-terminally extended Aβ peptides, or so-called Aη peptides. AW7 IPs were used for WB with the C-terminal anti-Aβ antibodies 2G3 and 21F12 and the N-terminal anti-Aβ antibody 6E10. The latter, but not the former, reacts with APPsα and Aη-α peptides (Portelius et al., 2013; Welzel et al., 2014; Willem et al., 2015). 6E10 readily detected ∼4 kDa Aβ, but it did not detect any bands consistent with APPsα or Aη-α. Furthermore, direct WB analysis of AD brain extract demonstrated highly similar levels of APPs in both AW7 and mock ID extracts (not shown). Therefore, it appears that AW7 does not deplete AD extracts of non-Aβ APP metabolites that contain the N-terminal portion of Aβ (Fig. 2A). The ∼7 kDa Aβ species detected with 2G3 and 21F12 was not detected with 6E10, consistent with our prior observation that most ∼7 kDa Aβ is N-terminally truncated, a pattern we have seen in aqueous extracts of >100 AD brains (McDonald et al., 2015). Because SDS-PAGE is highly denaturing, the ∼4 and ∼7 kDa species do not necessarily reflect native Aβ species. Rather, these simply indicate that at least two different Aβ species are present. The same samples were treated with or without 5 m GuHCl and then analyzed using MSD assays. In prior studies, we found that GuHCl effectively disaggregates high-molecular-weight Aβ species such that the signal detected by our oAssay is greatly decreased, whereas the signal detected by the monomer-preferring Aβx-42 immunoassay is proportionately increased (Mably et al., 2015). A similar outcome was evident when the extract of AD1 was treated with GuHCl (Fig. 2B). Specifically, GuHCl treatment caused an ∼70% decrease in the oligomer signal and a >8-fold increase in the monomer signal. Together, these immunoassay and IP/WB results indicate that the majority of Aβ in the AD1 extract exist as labile aggregates made up of ∼4 kDa Aβ and ∼7 kDa Aβ. Importantly, AW7 ID effectively removed the large majority of the various Aβ species detected (Fig. 2A,B). For instance, AW7 ID reduced the oligomer signal from 5.1 ± 0.03 to 0.32 ± 0.12 ng/ml (Fig. 2B, left) and monomer from 3.42 ± 0.03 to 0.12 ± 0.04 ng/ml (Fig. 2B, right).
Figure 2.

Water-soluble extract of AD brain, but not normal control brain, contains both Aβ monomers and oligomers and perturbs long-term synaptic plasticity. A, Aqueous extract of AD1 was treated with either preimmune rabbit serum or with AW7 antiserum. Portions of the mock ID sample (AD1, magenta) and the AW7 ID sample (ID-AD1, green) were then analyzed by IP/WB using AW7 for IP and a combination of 2G3 and 21F12 (left) or 6E10 (right) for WB. M, Aβ monomer. *Broad smear ∼7–8 kDa. Synthetic Aβ1-42, -31Aβ1-40, and Aη-α each at 2 ng/lane were used as controls. As expected, 6E10 detected all three synthetic peptides, whereas 2G3/21F12 detected Aβ1-42 and -31Aβ1-40, but not Aη-α. Only nonspecific (NS) bands were detected above the 16 kDa marker. B, AD1 (magenta) and ID-AD1 (green) samples were incubated with and without 5 m GuHCl and analyzed using immunoassays that recognize Aβ oligomers preferentially (1C22-3D6b, left) or Aβ42 monomer (266-21F12b, right). Values shown are the mean ± SEM of duplicate measurements and are representative of two separate experiments. C, Time course plots show that the AD sample but not the ID-AD sample blocked hippocampal LTP. The gray horizontal bar indicates the time period when sample was present in the bath. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF control is shown with black circles, AD treatment is shown with magenta diamonds, and ID-AD with green downward triangles. Each slice used for each treatment was from a different animal. Scale bar, 0.2 mV, 10 ms. D, Histogram plots of the average potentiation for the last 10 min of traces shown in C. Treatment of slices with AD1 sample inhibited LTP significantly compared with the aCSF vehicle control (F = 4.26, p = 6.98E-9) and ID-AD1 treatment (F = 4.14, p = 3.56E-12); in contrast, ID-AD1 had no effect on LTP relative to the vehicle control (F = 4.23, p = 0.12, one-way ANOVA). Symbols are the same as in C. E, Time course plots showing that the brain extract from a cognitively intact non-AD control (NC) did not blocked hippocampal LTP. The gray horizontal bar indicates the time period when sample was present in the bath. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF control is shown with black circles; NC treatment is shown with gray hexagons. Each slice used for each treatment was from a different animal. Scale bar, 0.5 mV, 10 ms. F, Histogram plots of the average potentiation for the last 10 min of traces shown in E and average of last 10 min from individual experiment in every group. Treatment of slices with NC sample did not inhibit LTP compared with the aCSF vehicle control (F = 4.6, p = 0.95, one-way ANOVA). Symbols are the same as in E. Values are shown as mean ± SEM. ### p < 0.001.
For slices that received vehicle aCSF-B (control), TBS induced strong potentiation that lasted for the whole recording period (181.1 ± 10.7%, n = 17; Fig. 2C, black circles), and ID-AD1 allowed a similar response (173.6 ± 8.7%, n = 11, p = 0.12, one-way ANOVA; Fig. 2C,D, green downward triangles). Consistent with previous studies (Shankar et al., 2008; Freir et al., 2011), application of the AD1 extract (Fig. 2C,D, magenta diamonds) significantly decreased LTP compared with both the control and ID-AD1 treatment (136 ± 4.2%, n = 18, F = 4.26, p = 6.98E-9 AD1 vs control; F = 4.14, p = 3.56E-12 AD1 vs ID-AD1, one-way ANOVA). The fact that the ID-AD1 and AD1 samples are identical except that the latter contains more Aβ than the former is evidence that some form of Aβ is responsible for the block of LTP induced by the AD1 extract. To further test the Aβ dependency of the block of LTP mediated by AD1, we investigated whether an extract from a non-AD brain (NC) could impair LTP. As anticipated, the NC extract lacked appreciable levels of Aβ (data not shown) and did not impair LTP (159.54 ± 10.6% in NC, n = 8; 160.36 ± 6.26% in control, n = 8; F = 4.6, p = 0.95 one-way ANOVA; Fig. 2E,F).
Aβ-containing AD brain extract affects presynaptic release probabilities
Accumulating evidence indicates that soluble Aβ species may interact with excitatory and inhibitory presynaptic terminals, modulate neurotransmitter release, and cause synaptic dysfunction in the very early stages of AD (Nimmrich et al., 2008; Abramov et al., 2009; Kabogo et al., 2010; Parodi et al., 2010; Russell et al., 2012; Sokolow et al., 2012; Huang et al., 2013; Ripoli et al., 2013; Kurudenkandy et al., 2014). Although the effects of Aβ on LTP are well established (Klyubin et al., 2012), little is known about whether and how Aβ-containing AD extracts affect presynaptic and postsynaptic elements. To investigate effects on presynaptic release, we measured short-term synaptic facilitation (Zucker and Regehr, 2002) in slices before and 30 min after treatment with AD1 extract. Because synapse release probability is inversely correlated with synaptic facilitation (Zucker and Regehr, 2002), we used high-frequency burst stimulation (5 pulses with 20 ms intraburst stimulus interval). Application of AD1 extract induced a reduction in the short-term facilitation during burst stimulation (Fig. 3A,B). When responses were normalized based on the ratio of each fEPSP to the first response, we found that treatment with AD1 extract had no effect on the second response, but decreased the third, fourth, and fifth response significantly (p = 0.02 at third stimulation, p = 0.004 at fourth stimulation and p = 0.004 at fifth stimulation, n = 6, Student's t test, and also by group and time with two-way ANOVA, F(4,7) = 6.39, p = 0.006; Fig. 3B, magenta circles). In contrast, the slices treated with ID-AD1 yielded a pattern highly similar to that obtained with aCSF-B control (n = 7, F = 5.32, p = 0.91, two-way ANOVA, Fig. 3C,D). Therefore, Aβ in the AD extract caused a reduction in short-term synaptic plasticity associated with increased presynaptic glutamate release.
Figure 3.

The Aβ-containing water-soluble extract of AD1 perturbs short-term facilitation. A, Representative traces of averaged field recordings were collected after five stimulation bursts (interstimulation interval 20 ms, interburst interval 30 s) before (black, aCSF) and 30 min after (magenta) perfusion with the AD1 sample. The traces shown for the AD1 samples are scaled so that the first response matches that of the aCSF control. Scale bars, 0.5 mV, 10 ms. B, fEPSP amplitude after two to five stimulations normalized to the value obtained after the first stimulation. Compared with vehicle control, AD1 treatment induced a small but significant decrease in short-term synaptic facilitation after the third (p = 0.02), fourth (p = 0.004), and fifth stimulation (p = 0.004); n = 6, Student's t test. Values are shown as mean ± SEM. #p < 0.05; ##p < 0.01. C, Representative traces of averaged field recordings were collected after five stimulation bursts (interstimulation interval 20 ms, interburst interval 30 s) before (black, aCSF) and 30 min after (green) perfusion with the ID-AD1 sample. Scale bars, 0.4 mV, 10 ms. ID-AD1 treatment did not affect short-term synaptic facilitation (n = 5, F = 5.32, p = 0.91, one-way ANOVA).
Aβ-containing AD brain extract disrupts the E/I balance
To estimate the effect of Aβ on the total synaptic input at the single-neuron level, we used whole-cell voltage-clamp recordings to measure sEPSCs on CA1 pyramidal neurons before and 30 min after addition of AD extract. The holding potential was kept constant at −70 mV and sEPSCs measured before and 30 min after addition of AD1 extract; this 30 min interval was chosen to match the preincubation time used in our LTP and short-term facilitation experiments. Application of the AD1 extract decreased the interevent interval significantly (p = 1.65E-6, K-S test) and increased the mean frequency of sEPSCs (from 1.8 ± 0.2 Hz to 2.7 ± 0.3 Hz, p = 0.02, n = 7, Student's t test; Fig. 4A,B), but did not alter the sEPSC amplitude (mean amplitude from 11.7 ± 1.8 pA to 10.1 ± 1.6 pA, p = 0.65, n = 7, Student's t test; Fig. 4A,C). In contrast, the ID-AD1 sample had no effect on the frequency or the amplitude of sEPSCs (mean frequency: from 2.2 ± 0.5 Hz to 2.3 ± 0.7 Hz, mean amplitude: from 9.7 ± 1.7 pA to 10.2 ± 1.4 pA, p = 0.45, n = 6, Student's t test; Fig. 4D–F). These results indicate that the AD-brain-derived Aβ significantly augments excitatory synaptic input on CA1 pyramidal neurons.
Pyramidal neurons receive both excitatory (sEPSCs) and inhibitory (sIPSCs) inputs, GABAergic axon terminals more easily form synapses with perisomtatic regions of pyramidal cells and strongly influence the output of neurons (DeFelipe, 2002; Garcia-Marin et al., 2009). To record sIPSCs on the same neurons, we adjusted the holding potential to 5 mV, a voltage close to the calculated sEPSC reverse potential. As shown in Figure 4, G–I, the AD1 sample increased interevent intervals significantly (p = 6.19E-6, K-S test) and decreased the frequency of sIPSCs (from 4.7 ± 0.7 Hz to 3.1 ± 0.7 Hz, p = 0.008, n = 7, Student's t test) without altering sIPSC amplitude (from 14.8 ± 1.4 pA to 14.2 ± 0.9 pA, p = 0.75, n = 7, Student's t test). In contrast, the ID-AD1 sample had no effect on sIPSCs (frequency: from 5.3 ± 0.4 Hz to 4.8 ± 0.7 Hz, amplitude: from 13.6 ± 1.6 pA to 13.2 ± 2.1 pA, p = 0.21, n = 6, Student's t test; Fig. 4J–L). These results revealed that brain-derived Aβ significantly reduces GABAergic input on CA1 pyramidal cells.
To assess whether the changes of excitatory input (E) and inhibitory input (I) to the same neuron affect the E/I balance of that neuron, we calculated the integrated conductance of sEPSCs and sIPSCs over a 5 min period (Fig. 4M). Comparison of the integrated conductance before and 30 min after AD1 sample application revealed that E was increased ∼3 fold and I was decreased ∼50%; therefore, the E/I balance was increased ∼6 fold (n = 7; Fig. 4N). These results show that AD-brain-derived Aβ oppositely affects excitatory and inhibitory synaptic transmission, causing an increase in the E/I ratio. These changes, especially the reduction of GABAergic tone on individual neurons, may contribute to neuronal hyperactivity and disturb network homeostasis, thereby perturbing LTP (Wang et al., 2014; Gillespie et al., 2016).
Genetic ablation of APP occludes the effects of Aβ on LTP and presynaptic activity and normalizes the E/I balance
Multiple lines of evidence suggest that the APP may play a role in both GABAergic and glutamatergic neurotransmission (Bai et al., 2008; Kabogo et al., 2010; Pliássova et al., 2016; Schwenk et al., 2016) and separate studies impute a link between Aβ and APP (Lorenzo et al., 2000; Fogel et al., 2014; Kirouac et al., 2017). Therefore, having found that brain-derived Aβ acts on presynapses and modulates both GABA and glutamate transmission, we investigated whether APP was required for these effects. For this, we used mice null for APP (Fig. 5A). In agreement with previous studies, brain slices from APP KO and WT littermate mice exhibited similar levels of basal activity (p = 0.19, one-way ANOVA) and LTP (Fig. 5B,C; Dawson et al., 1999; Jedlicka et al., 2012). In both WT and APP KO slices treated with the aCSF-B control, TBS induced strong potentiation that lasted for the entire recording period (158.1 ± 6.3% in WT, n = 11, black circles; 151.2 ± 8.5% in APP KO, n = 9, gray hexagons; F = 4.4, p = 0.79, comparison of the last 10 min of recording using one-way ANOVA; Fig. 5C,D). In agreement with experiments shown in Figure 2, the addition of AD1 extract to WT slices decreased LTP significantly compared with addition of aCSF-B (121.8 ± 5.4% in WT + AD1, n = 7, F = 4.5, p = 0.0005, WT control vs WT + AD1, one-way ANOVA; Fig. 5C,D, magenta diamonds). However, application of the same extract to slices from APP KO mice had no effect on LTP, with the level of LTP in APP KOs indistinguishable from that of WT or APP KO treated with aCSF-B control (145.4 ± 4.2% in APP KO + AD1, F = 4.5, p = 0.41, APP KO control vs APP KO + AD1; one-way ANOVA; Fig. 5C,D, pink upward triangles). Similarly, when applied to APP KO brain slices, AD1 extract had no effect on short-term facilitation (Fig. 5E,F).
To assess the generalizability of the rescue of LTP by APP ablation, we tested the effect of an extract from a second AD brain (AD2) on another APP KO mouse line (Zheng et al., 1995). As with the AD1 extract (Fig. 2), the AD2 and ID-AD2 extracts were characterized by IP/WB and ELISA. The profiles obtained for AD2 (Fig. 6A) were similar to those of AD1 (Fig. 2A) except that AD2 contained relatively more ∼7 kDa species than AD1 (Fig. 6A,B). As seen with the first APP KO line tested (Fig. 5), brain slices from the second APP KO line (which we refer to as Zheng APP KOs; Zheng et al., 1995) exhibited similar levels of basal activity as slices from WT mice (F = 4.6, p = 0.91, one-way ANOVA; Fig. 6E,F). When AD2 extract was applied to slices from WT mice, it blocked LTP in an Aβ-dependent fashion (184.1 ± 7.7% in WT control, black circles, n = 12; 137.1 ± 7.2% in WT + AD2, n = 12; F = 4.96, p = 0.0001, one-way ANOVA, Fig. 7A,B, magenta diamonds), but had no effect on LTP elicited from APP KO mice (175.8 ± 9% in APP KO control, gray hexagons, n = 11; 169.9 ± 4% in APP KO + AD2, pink upward triangles, n = 12; F = 5.12, p = 0.56, one-way ANOVA; Fig. 7A,B).
Figure 7.

A second APP KO mouse line confirms that APP is required for the synaptic-disrupting activity of both AD brain and synthetic Aβ oligomers. A, LTP recorded in hippocampal CA1 was similar in brain slices from WT and Zheng APP KO mice. Notably, the extract from AD2 blocked LTP in brain slices from WT but not APP KO mice. Horizontal gray bar indicates the duration during when sample was present. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF control in WT mice is shown with black circles, AD2 treatment in WT mice is shown with magenta diamonds, and ID-AD2 treatment in WT slices in green downward triangles. The aCSF control in APP KO mice is shown with gray hexagons and AD2 treatment in APP KO mice is shown using pink upward triangles. WT slices for each treatment came from different animals; the APP KO slices came from a total of 6 APP KO mice. Scale bars, 0.5 mV, 15 ms. B, Comparison of average potentiation from last 10 min of LTP recording (F = 4.96, p = 0.0001, control vs AD2 in WT mice; F = 5.12, p = 0.56, control vs AD2 in APP KO mice; one-way ANOVA). Symbols correspond to those in A. C, ADDLs blocked LTP in WT, but not in APP KO, brain slices. Horizontal gray bar indicates the duration during when sample was present. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF WT slice control is shown with black circles, WT slices treated with ADDLs are shown with magenta diamonds, and vehicle is shown with green downward triangles. The aCSF control in APP KO mice is shown with gray hexagons and ADDLs treatment in APP KO mice is shown using pink upward triangles. WT slices for each treatment came from different animals; the APP KO slices came from a total of 6 APP KO mice. Scale bars, 0.7 mV, 15 ms. D, Comparison of average potentiation from last 10 min of LTP recording (F = 4.75, p = 0.0006, control vs ADDLs in WT mice; F = 4.75, p = 0.93, control vs vehicle in WT mice; F = 4.84, p = 0.07, control vs ADDLs in APP KO mice; one-way ANOVA). Symbols correspond to those in C. Each slice used for each treatment was from a different animal. ###p < 0.001.
To further examine whether the APP-dependent block of LTP by AD brain extracts was indeed mediated by Aβ and not some other AW7-reactive material, we tested if the well-established block of LTP mediated by ADDLs (Lambert et al., 1998; Wang et al., 2002; Laurén et al., 2009; Freir et al., 2011) also required expression of APP. ADDLs were prepared as described previously and then assessed using SEC and EM (Fig. 6C,D). The ADDL preparation contained a mixture of Aβ aggregates and a small amount of monomer (Fig. 6C,D). When applied to brain slices from WT mice, ADDLs (200 nm) blocked LTP (188.9 ± 11.5% in WT control, Fig. 7C,D, black circles, n = 8; 123.8 ± 6% in WT + ADDLs, Fig. 7C,D, magenta diamonds, n = 6; F = 4.75, p = 0.0007, one-way ANOVA), but had no effect on LTP elicited from APP KO slices (181.5 ± 15% in APP KO control, Fig. 7C,D, gray hexagons, n = 7; 168.1 ± 10% in APP KO + ADDLs, Fig. 7C,D, pink upward triangles, n = 7; F = 4.85, p = 0.07, one-way ANOVA; Fig. 7C,D). Therefore, it appears that the well documented plasticity-disrupting activity of both Aβ extracted from AD brains (Klyubin et al., 2008; Shankar et al., 2008; Barry et al., 2011; Freir et al., 2011; Klyubin et al., 2012) and synthetic Aβ (Lambert et al., 1998; Wang et al., 2002; Laurén et al., 2009; Freir et al., 2011) require expression of APP.
To investigate whether APP is necessary for the effect of Aβ on the E/I balance (Fig. 4), we studied the effects of Aβ on sEPSCs and sIPSCs in brains of Zheng APP KO and WT littermate mice (Fig. 8). When applied to WT slices, AD1 extract again increased mean sEPSC frequency (from 2.2 ± 0.1 Hz to 3.4 ± 0.2 Hz, p = 0.003, n = 5, Student's t test) and decreased interevent intervals, p = 6.34E-15, K-S test) without altering the amplitude of sEPSCs (mean amplitude: 17.8 ± 0.4 pA vs 18 ± 1.5 pA, p = 0.32, n = 5, Student's t test; Fig. 8A–C) and on the same neuron decreased mean sIPSC frequency (from 4.2 ± 0.8 Hz to 2.7 ± 0.4 Hz, p = 0.006, n = 5, Student's t test) and increased interevent intervals (p = 9.44E-20, K-S test), but not amplitude (mean amplitude from 20 ± 3 pA to 19.3 ± 1.3 pA, p = 0.34, n = 5, Student's t test; Fig. 8D–F). These results, which were obtained with WT mice from an entirely different colony as those used in Figure 4, nicely demonstrate the robustness of the Aβ effect (cf. Figs. 4, 8). Most importantly, when AD1 extract was applied to Zheng APP KO slices, there was no change in the frequency or amplitude of sEPSCs (mean frequency: from 2.6 ± 0.1 Hz to 2.7 ± 0.4 Hz, mean amplitude: from 15 ± 1.4 pA to 14.6 ± 0.5 pA, p = 0.14, K-S test; p = 0.26, n = 6, Student's t test; Fig. 8G–I). Similarly, sIPSCs were also unchanged (mean frequency: from 3.5 ± 0.5 Hz to 3.5 ± 0.3 Hz, mean amplitude: from 16.7 ± 1 pA to 16.4 ± 1.6 pA, p = 0.58, K-S test; p = 0.25, n = 6, Student's t test; Fig. 8J–L). Therefore, as with our LTP experiments (Figs. 5, 7), ablation of APP completely rescued the effects of Aβ on excitatory and inhibitory input on CA1 pyramidal neurons. Further, because APP KO occluded Aβ alterations on the E and I input at individual neurons, it also prevented Aβ-mediated changes in the integrated conductance of sEPSCs and sIPSCs (Fig. 8M). When AD1 extract was applied to WT slices, E increased ∼3-fold and I decreased ∼44%, resulting in an ∼5.8-fold increase in the E/I ratio. However, APP KO prevented those E/I ratio changes (p = 0.001, E/I in WT vs E/I in APP KO, one-way ANOVA; Fig. 8M). These results indicate that APP plays an important role in regulating the acute effects of Aβ on excitatory and inhibitory presynaptic release and the consequent maintenance of network homeostasis.
Figure 8.

APP KO occludes the effects of Aβ-containing AD brain extract on both EPSCs and IPSCs and rescues the disruption of E/I balance. A, D, Example traces of sEPSCs (A) and sIPSCs (D) before (aCSF, black) and 30 min after (magenta) the addition of AD1 extract on WT hippocampal brain slices. Scale bars, 20 pA, 700 ms. B, C,Treatment with AD1 extract decreased interevent intervals and increased mean frequency (inset) of sEPSCs (p = 6.34E-15, K-S test; p = 0.003, Student's t test; n = 5; B), but did not change the cumulative distributions or the mean value (inset) of the amplitude of sEPSCs (n = 5) significantly on WT slices (C). E, F, 30 min of AD1 treatment increased interevent intervals and decreased mean frequency (inset) of sIPSCs (p = 9.44E-20, K-S test; p = 0.006, Student's t test; n = 5; E), but did not affect the cumulative distributions or the mean value (inset) of the amplitude of sIPSCs (n = 5) on WT slices (F). G, J, Example traces of sEPSCs (G) and sIPSCs (J) before (aCSF, gray) and 30–40 min after the addition of AD1 extract (pink) on APP KO mice hippocampal brain slices. Scale bars, 20 pA, 700 ms. H, I, Treatment with AD1 sample affected neither frequency (I) nor amplitude (I) of sEPSCs (p = 0.14, K-S test; p = 0.26, Student's t test; n = 6) on APP KO mice. K, L, Similarly, treatment of APP KO neurons with AD1 did not change frequency (K) or the amplitude (L, p = 0.58, K-S test; p = 0.25 Student's t test; n = 6) of sIPSCs. M, Application of Aβ-containing AD brain extract changed the integrated conductance of both excitatory (E) and inhibitory (I) input to neurons significantly and disrupted the E/I balance in WT animals, but not in APP KO mice (p = 0.001, E/I in WT vs E/I in APP KO, one-way ANOVA).
Aβ binding to synapses requires APP
To further investigate the targeting of synaptic elements by Aβ and how this might be influenced by APP, we used a powerful, high-resolution microscopic technique, AT, to search for evidence of Aβ binding to synapses in the same brain slices used in our electrophysiology experiments. Upon completion of LTP recording, certain slices from the treatment groups used in Figures 2C and 5C were immediately fixed, processed, and used for AT. Sections were stained with 1C22, the same aggregate-preferring antibody (Mably et al., 2015; Pickett et al., 2016) used in our oAssay, anti-synapsin-1 (for presynapses) and anti-PSD95 (for postsynapses). Approximately 7000 synapses (∼3500 presynapses and ∼3500 postsynapses) per slice were analyzed for a total of 100,359 presynapses and 99,075 postsynapses. AT revealed significant anti-Aβ staining at synapses of slices incubated with AD1 extract, with only background staining in samples incubated with aCSF controls and ID-AD1 (Fig. 9A–C; Kruskal-Wallis test for synapsin-1, χ(4)2 = 10.844, p = 0.028; Kruskal-Wallis test for PSD95, χ(4)2 = 11.583, p = 0.021). In slices incubated with AD1 extract, 1.27 ± 0.47% of presynapses and 0.58 ± 0.19% of postsynapses stained with 1C22, whereas in slices that had been incubated with aCSF, only 0.0076 ± 0.013% of presynapses and 0.0184 ± 0.087% of postsynapses were 1C22 positive (Dunn's post hoc between AD and control for presynapses p = 0.024 and for postsynapses p = 0.010). Slices incubated with extracts immunodepeleted of Aβ exhibited similar background staining with 1C22 as the aCSF control (Fig. 9A–C). Therefore, the same treatment with AD1 extract that disrupts synaptic plasticity in an Aβ-dependent fashion (Figs. 2, 4) also leads to Aβ binding to synapses (Fig. 9A–C). Moreover, the finding that Aβ is present at more presynapses than postsynapses (Mann–Whitney U test between AD presynapses and AD postsynapses, U = 0, p = 0.004) is consistent with our results that suggest a presynaptic effect of Aβ (Figs. 4, 8).
Figure 9.

Aβ binding to synaptic terminals requires expression of APP. A, AT of hippocampi stained for synapsin-1 (presynapses), Aβ (1C22), and PSD95 (postsynapses) reveal colocalization of Aβ at synapses in slices incubated with AD1 brain extract. Images have been processed for analysis as described in the Materials and Methods and Figure 1. B, C, Amount of synaptic 1C22 staining was significantly greater in slices incubated with AD1 extract than in slices incubated with aCSF or ID-AD1 extract based on colocalization of 1C22 and synapsin 1 staining (Kruskal-Wallis test (χ(4)2 = 10.844, p = 0.028) Dunn's post hoc vs control p = 0.021; B) and 1C22 and PSD95 colocalization (Kruskal–Wallis test for PSD95, χ(4)2 = 11.583, p = 0.021; Dunn's post hoc vs control, p = 0.01; C). Importantly, when slices from APP KO mice were incubated with AD1 extract, there was no significant colocalization of 1C22 staining with either synapsin 1 (B; Dunn's post hoc vs control, p = 1.000) or PSD-95 (C; Dunn's post hoc vs control, p = 1.000). Graphs represent the medians ± the interquartile range per treatment. Each data point is derived from the analysis of ∼3500 synapses imaged per brain slice. Within each treatment group, the three slices used were from three different mice (B, C). Arrows indicate specific examples of 1C22 staining colocalizing with presynapses or postsynapses. Scale bar in A is 2 μm. #p < 0.05.
The number of Aβ-positive synapses detected here is much lower than the amount of Aβ observed at synapses when synthetic oligomers are applied to cultured hippocampal neurons (Lacor et al., 2004; Lacor et al., 2007). However, the current paradigm of applying soluble AD brain extract to intact mouse brain slices is more relevant to the in vivo situation than model systems in which Aβ is applied directly and at high concentrations to dissociated neurons (Lacor et al., 2004; Lacor et al., 2007). Indeed, it is noteworthy that the percentage of synapses positive for Aβ in the current study are consistent with our findings in APP-transgenic mouse brain, where we observed ∼1% of postsynaptic densities (PSDs) positive for Aβ distant from plaques (Koffie et al., 2009). Similarly, in human AD brain at sites distant from plaques, we detected Aβ at 0.6% of PSDs and 0.5% of presynaptic terminals (Koffie et al., 2012). Therefore, at disease relevant concentrations sufficient to disrupt plasticity, synaptic Aβ binding occurs at levels similar to that observed in human AD brain.
Importantly, when brain slices from APP KO mice were incubated with AD1 extract, little or no synaptic 1C22 staining was detected (Fig. 9A–C). These results are notable because expression of APP was found to be required for Aβ-mediated disruption of both long-term plasticity (Figs. 5, 7) and neurotransmitter release (Fig. 8). In sum, our AT data are completely congruent with the results of our electrophysiological experiments and indicate that expression of APP is required for the binding and subsequent plasticity-disrupting effects of Aβ and that these effects are largely mediated on the presynapse.
APP mediates binding of synaptotoxic Aβ to brain cells
We reasoned that, if synaptotoxic forms of Aβ bound to APP or to an APP-containing complex, then it should be possible to pretreat bioactive extracts with a source of APP to deplete the extract of activity. One possible approach would be to add exogenous recombinant APP, but APP is a transmembrane protein and its expression outside of a membrane environment in the absence of proper posttranslational modifications precludes its use. Instead, we preincubated AD2 extract with either APP-containing (WT) or APP lacking (KO) brain slices (Fig. 10A). When AD2 was preincubated with WT brain slices and then applied to a fresh WT brain slice, it was no longer capable of blocking LTP (216.4 ± 26% in WT slices + AD2, green upward triangles, n = 6; F = 4.96, p = 0.82, one-way ANOVA; Fig. 10B,C). In contrast, when AD2 extract was preincubated with APP KO slices and then applied to a fresh WT brain slice, the AD2 extract retained its ability to block LTP (210.3 ± 15% in WT control, black circles, n = 6; 146.8 ± 5.4% in APP KO +AD2, magenta diamonds, n = 6; F = 4.96, p = 0.003, one-way ANOVA; Fig. 10B,C). These results are entirely consistent with our AT experiments and provide further evidence that APP enables synaptotoxic forms of Aβ to bind to and perturb neurons.
Figure 10.

APP-expressing, but not APP-lacking, brain slices bind synaptotoxic Aβ. A, AD2 brain extract was preincubated with either 4 WT or 4 APP KO brain slices for 2 h and the resultant solutions were used to perfuse WT brain slices. B, Time course plots of LTP recorded in WT brain hippocampal CA1 show that AD2 brain extract preincubated with APP KO brains slices blocked LTP, whereas AD2 preincubated with WT brain slices allow normal LTP. Horizontal gray bar indicates the duration during when sample was present. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF control in WT mice is shown with black circles, AD2 incubated with APP KO brain slices in WT mice is shown with magenta diamonds, and AD2 preincubated with WT brain slices is in green downward triangles. Scale bars, 0.5 mV, 15 ms. C, Comparison of average potentiation from last 10 min of LTP recording (F = 4.96, p = 0.003, control vs APP KO slices with AD2; F = 4.96, p = 0.82, control vs WT slices with AD2; one-way ANOVA). Symbols correspond to those in B. Each slice used for recording for each treatment was from a different animal. ##p < 0.005.
Aβ-containing AD brain extract partially blocks LTP in APP hemizygous brain slices
To further investigate the requirement of APP for Aβ synaptotoxicity, we tested the effect of AD2 extract on brain slices from APP hemizygous mice. APP expressing WT C57BL/6 were bred with Zheng APP KO mice and the hemizygous progeny used for LTP experiments. Hemizygous mice express 50% as much APP as WT mice (Fig. 11A) and exhibit similar levels of basal activity relative to slices from WT mice (F = 4.6, p = 0.75, one-way ANOVA; Fig. 11B). The control level of LTP was also similar in WT and hemizygous brain slices (187.85 ± 5.63% in WT control, black circles, n = 9; 189.69 ± 7.19% in hemizygous brain slices, gray hexagons, n = 9; F = 4.45, p = 0.84, one-way ANOVA; Fig. 11C,D). When AD2 extract was applied to slices from WT mice, it blocked LTP to an extent comparable to that seen in previous experiments (cf. Figs. 11C,D and 7A,B; 187.85 ± 5.63% in WT control, black circles, n = 9; 136.93 ± 3.14% in WT + AD2, magenta diamonds, n = 10; F = 4.5, p = 2.67E-007, one-way ANOVA). Similarly, AD2 extract impaired LTP in APP-hemizygous mice (189.69 ± 7.19% in APP+/− control, gray hexagons, n = 9; 154.83 ± 6% in APP+/− AD2, pink upward triangles, n = 10; F = 4.49, p = 0.003, one-way ANOVA; Fig. 11C,D). Although the extent of the block in hemizygous slices was somewhat reduced compared with WT slices (F = 4.45, p = 0.84, control in WT mice vs control in APP+/− mice; F = 4.5, p = 2.67E-007, control vs AD2 in WT mice; F = 4.49, p = 0.003, control vs AD2 in WT mice; one-way ANOVA), the effect of AD2 extract on hemizygous brain slices stands in contrast to its lack of effect on APP KO slices (Fig. 7A,B). The partial attenuation of Aβ synaptotoxicity in APP-hemizygous brain indicates a gene–dose effect such that 50% of the normal level of APP is sufficient to mediate some block of LTP, but not the full block of LTP seen in APP WT mice. Further studies will be required to determine the minimal reduction in APP levels that allows full protection against the plasticity-disrupting effects of Aβ.
Figure 11.

The level of APP expression influences the plasticity-disrupting activity of Aβ-containing AD brain extract. A, Detergent extracts of from WT, APP+/−, and APP−/− mouse brain slices used for electrophysiology were analyzed for APP by WB with 22C11. Full-length APP was readily detected in extracts from WT mice and APP+/− mice, but not APP−/− mice. Ten micrograms of total protein from APP+/− slices contained a similar amount of APP as 5 μg of total protein from WT slices. B, Input/output curves recorded in the hippocampal CA1 area are highly similar for both WT and APP+/− mouse brain slices (F = 4.6, p = 0.75, one-way ANOVA). C, LTP recorded in hippocampal CA1 was similar in brain slices from WT and APP+/− mice. However, AD2 caused a stronger block of LTP in WT slices compared with APP+/− slices. Horizontal gray bar indicates the duration during when sample was present. 1, 2, Example traces from time points just before the theta burst stimulation (↑↑↑ TBS; 1) and 60 min after TBS (2), respectively. The aCSF control in WT mice is shown with black circles, AD2 treatment in WT mice is shown with magenta diamonds, aCSF control in APP+/− mice is shown with gray hexagons, and AD2 treatment in APP+/− mice is shown with pink upward triangles. Scale bars, 0.5 mV, 15 ms. Each slice used for each treatment was from a different animal. D, Comparison of average potentiation from the last 10 min of LTP recording (F = 4.45, p = 0.84, control in WT mice vs control in APP+/− mice; F = 4.5, p = 2.67E-007, control vs AD2 in WT mice; F = 4.49, p = 0.003, control vs AD2 in WT mice; one-way ANOVA). Symbols correspond to those in C. Values are shown as mean ± SEM. Each slice used for each treatment was from a different animal. ##p < 0.005, ###p < 0.0001.
Discussion
To better understand how Aβ disrupts synaptic plasticity, we combined the use of the most disease relevant form of Aβ, material extracted from human AD brain, with electrophysiological approaches and high-resolution microscopy. Consistent with prior studies, we show that extracts from the brains of individuals who died with AD block LTP (Shankar et al., 2008; Barry et al., 2011; Freir et al., 2011; Yang et al., 2017). We further show that, concomitant with the block of LTP, there is an increase in presynaptic release and disruption of E/I balance. Consistent with these synaptic effects of Aβ, we demonstrate that exogenously applied AD-brain-derived Aβ binds to synapses, with more Aβ oligomers detected on presynapses than on the postsynapses. Our finding that treatment with brain-derived Aβ enhances excitatory drive agrees well with studies showing that aggregated forms of synthetic Aβ increase EPSPs, action potentials, and membrane depolarizations (Hartley et al., 1999; Minkeviciene et al., 2009; Kurudenkandy et al., 2014). Our study is unique in that we used brain-derived Aβ and that the concentration of this material was much lower than the synthetic Aβ used in prior studies. In support of the strength of this experimental paradigm, the levels of synaptic Aβ that we observed in this study are very similar to those observed with AT in studies of human AD brain (Koffie et al., 2012).
The apparent paradox that ectopic application of Aβ causes a net increase in excitation yet impairs LTP may result because of glutamate spillover and activation of extrasynaptic or perisynaptic NR2B-enriched NMDARs, which play a major role in LTD induction (Li et al., 2011; Zhang et al., 2017). In such a scenario, synaptic depression may result from an initial increase in synaptic activation of NMDARs by glutamate, followed by synaptic NMDAR desensitization, NMDAR/AMPAR internalization, and activation of extrasynaptic NMDARs and mGluRs (Born et al., 2014). However, it is unclear why ablation of APP could recover such effects.
An alternative explanation that accounts for a role for APP in the impairment of postsynaptic efficacy is that exogenous AD-brain-derived soluble aggregates and endogenously produced monomer have differential effects. Aβ is known to be released in an activity-dependent manner (Kamenetz et al., 2003; Cirrito et al., 2005), whereas elevated Aβ levels result in depressed glutamatergic synaptic transmission and glutamate receptor endocytosis (Kamenetz et al., 2003; Hsieh et al., 2006). Therefore, it is plausible that the increase in glutamate release induced by soluble Aβ aggregates may also lead to an increase in de novo Aβ monomer production and this in turn may depress postsynaptic activity. Such a scenario would necessarily require expression of endogenous APP and explain why ablation of APP can obviate the block of LTP caused by brain-derived soluble Aβ aggregates. The fact that Aβ-treated APP hemizygous slices exhibited an attenuated block is consistent with a partial reduction in the amount of endogenous Aβ. With regard to the protection of LTP upon ablation of APP, it is important to emphasize the robust nature and generalizability of this phenomenon. We observed the same protection using two different APP KO mouse lines (Zheng et al., 1995; Callahan et al., 2017), extracts from two different AD brains, and synthetic Aβ oligomers. Both AD extracts blocked LTP in an Aβ-dependent manner when applied to WT mouse brain slices, but the same AD extracts had no effect on LTP elicited from APP KO slices. Moreover, the extent of Aβ binding to synapses was similar in two different sources of WT mice (Fig. 7B,C) and the pattern observed was reminiscent of that seen in AD brain (Pickett et al., 2016).
There is evidence that APP can act as a receptor for Aβ (Melchor and Van Nostrand, 2000; Van Nostrand et al., 2002; Yankner and Lu, 2009; Fogel et al., 2014; Kirouac et al., 2017) and that APP may mediate increased excitatory drive (Fogel et al., 2014). Specifically, Aβ was unable to promote aberrant neurotransmitter release in the absence of APP (Fogel et al., 2014). Our findings that binding of soluble Aβ aggregates to synapses requires expression of APP and that synaptotoxic Aβ can be bound by APP-expressing brain tissue, but not APP-lacking brain tissue, are consistent, but not proof, that APP may act as a receptor for Aβ. APP is known to both regulate L-type calcium channels in GABAergic neurons and interact with the pore-forming subunit Cav1.2 (Yang et al., 2009) and is a member of the GABAB-R receptor complex (Schwenk et al., 2016). In addition, there is evidence from proteomic studies indicating that APP interacts with >30 different proteins, including proteins key to synaptic vesicle turnover (Kohli et al., 2012; Del Prete et al., 2014; Laßek et al., 2014; Wilhelm et al., 2014) and proteins (such as the prion protein) implicated in binding Aβ (Bai et al., 2008; Laurén et al., 2009). Therefore, Aβ could exert an APP-dependent effect either by binding directly to APP or binding to protein complexes of which APP is a component and stabilizing member. The APP gene-dose-dependent response to Aβ that we observed is equally compatible with direct or indirect binding to APP.
So far, we have considered the effects of Aβ on synapses and a single hippocampal pathway (the Schaffer collateral), but Aβ is also thought to have network-wide effects (Palop and Mucke, 2010). For instance, Aβ-induced increases in excitatory network activity could lead to synaptic depression through homeostatic mechanisms. It is well established that acute treatment of primary neurons with bicuculline (a GABAA antagonist) increases overall neuronal activity and firing rates (Vertkin et al., 2015). However, after a few days, neuronal activity returns to control levels. By analogy, it is reasonable that the disruption of the E/I balance seen with our acute Aβ treatment may also cause both short-term local and long-lasting network effects. Given the fact that Aβ treatment increases excitatory drive and decreases inhibitory drive and that GABAergic interneurons express high levels of APP in DG (Wang et al., 2014; Del Turco et al., 2016), it is tempting to speculate that Aβ-mediated disruption of GABAergic interneurons may play a critical role in the cognitive impairment that occurs early in AD (Gillespie et al., 2016). Clearly, further studies will be required to delineate the influence of APP on both network regulation and other forms of synaptic plasticity such as LTD.
Considerable data from the study of APP transgenics implicate impairment of GABAergic interneurons as being central to the network disturbances evident in these models (Busche and Konnerth, 2015; Palop and Mucke, 2016). However, the nonphysiological expression of high levels of APP and the concomitant release of Aβ from the expressed transgene make it difficult to differentiate between effects mediated by Aβ versus APP or non-Aβ APP metabolites (Melnikova et al., 2013; Born et al., 2014; Fowler et al., 2014). Nonetheless, growing evidence suggests that GABAergic interneurons play a prominent role in homeostatic regulation of hippocampal networks, and there is compelling proteomic and physiological data that link APP and GABAB1a-R (Wang et al., 2014; Gillespie et al., 2016; Schwenk et al., 2016). Therefore, further investigations on how Aβ effects GABAB-R expression, GABAB-R-APP interactions, and whether GABAB-R KOs are resistant to Aβ are merited and may lead to a pharmacological means to attenuate Aβ synaptotoxicity. Similarly, modulation of APP expression may also offer therapeutic potential. However, whereas our results demonstrate that ablation of APP in brain slices from young (2–3 months old) mice protects against the acute synaptotoxicity of Aβ, widespread KO of APP is not recommended. APP appears to be involved in many physiological processes (Yang et al., 2009; Müller and Zheng, 2012; Del Prete et al., 2014; Laßek et al., 2014; Wang et al., 2014) and aged APP-null mice exhibit hypersensitivity to kainate-induced seizures (Steinbach et al., 1998), altered exploratory behavior, deficits in spatial memory, and impairment of LTP (Dawson et al., 1999; Seabrook et al., 1999; Ring et al., 2007). No such deficits have been reported in APP-hemizygous mice. Therefore, it may be possible to downregulate APP expression so as to maintain normal function yet attenuate Aβ synaptotoxicity. However, hemizygous reduction of APP allows only partial protection against the plasticity-disrupting effects of Aβ and further studies will be required to determine the minimal reduction in APP levels that allows a more fulsome protection.
Footnotes
This work was supported by the National Institutes of Health (Grant AG046275 to D.M.W.), Bright Focus (D.M.W.); the United States–Israel Binational Science Foundation (Grant 2013244 to D.M.W. and I.S.); Alzheimer's Research UK and the Scottish Government (Grant ARUK-SPG2013-1 to T.S.J.), Wellcome Trust-University of Edinburgh Institutional Strategic Support funds (T.S.J.); the H2020 European Research Council (ALZSYN; T.S.J.); and the Massachusetts Alzheimer's Disease Research Center (Grant AG05134). T.S.J. is a member of the FENS Kavli Network of Excellence. We thank Dr. Tiernan T. O'Malley for useful discussions and technical advice.
The authors declare no competing financial interests.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I (2009) Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 12:1567–1576. 10.1038/nn.2433 [DOI] [PubMed] [Google Scholar]
- Austin SA, Sens MA, Combs CK (2009) Amyloid precursor protein mediates a tyrosine kinase-dependent activation response in endothelial cells. J Neurosci 29:14451–14462. 10.1007/bf00267823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Markham K, Chen F, Weerasekera R, Watts J, Horne P, Wakutani Y, Bagshaw R, Mathews PM, Fraser PE, Westaway D, St George-Hyslop P, Schmitt-Ulms G (2008) The in vivo brain interactome of the amyloid precursor protein. Mol Cell Proteomics 7:15–34. 10.1074/mcp.M700077-MCP200 [DOI] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, McDonald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ (2011) Alzheimer's disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci 31:7259–7263. 10.1523/JNEUROSCI.6500-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts V, Leissring MA, Dolios G, Wang R, Selkoe DJ, Walsh DM (2008) Aggregation and catabolism of disease-associated intra-Abeta mutations: reduced proteolysis of AbetaA21G by neprilysin. Neurobiol Dis 31:442–450. 10.1016/j.nbd.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlikova GG, Trejo M, Mably AJ, McDonald JM, Sala Frigerio C, Regan CM, Murphy KJ, Masliah E, Walsh DM (2013) Alzheimer brain-derived amyloid beta-protein impairs synaptic remodeling and memory consolidation. Neurobiol Aging 34:1315–1327. 10.1016/j.neurobiolaging.2012.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born HA, Kim JY, Savjani RR, Das P, Dabaghian YA, Guo Q, Yoo JW, Schuler DR, Cirrito JR, Zheng H, Golde TE, Noebels JL, Jankowsky JL (2014) Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci 34:3826–3840. 10.1523/JNEUROSCI.5171-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Konnerth A (2015) Neuronal hyperactivity: a key defect in Alzheimer's disease? Bioessays 37:624–632. 10.1002/bies.201500004 [DOI] [PubMed] [Google Scholar]
- Callahan DG, Taylor WM, Tilearcio M, Cavanaugh T, Selkoe DJ, Young-Pearse TL (2017) Embryonic mosaic deletion of APP results in displaced Reelin-expressing cells in the cerebral cortex. Dev Biol 424:138–146. 10.1016/j.ydbio.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM (2005) Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48:913–922. 10.1016/j.neuron.2005.10.028 [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8:79–84. 10.1038/nn1372 [DOI] [PubMed] [Google Scholar]
- Conboy L, Murphy KJ, Regan CM (2005) Amyloid precursor protein expression in the rat hippocampal dentate gyrus modulates during memory consolidation. J Neurochem 95:1677–1688. 10.1111/j.1471-4159.2005.03484.x [DOI] [PubMed] [Google Scholar]
- Dawson GR, Seabrook GR, Zheng H, Smith DW, Graham S, O'Dowd G, Bowery BJ, Boyce S, Trumbauer ME, Chen HY, Van der Ploeg LH, Sirinathsinghji DJ (1999) Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90:1–13. 10.1016/S0306-4522(98)00410-2 [DOI] [PubMed] [Google Scholar]
- De Strooper B. (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev 90:465–494. 10.1152/physrev.00023.2009 [DOI] [PubMed] [Google Scholar]
- DeFelipe J. (2002) Cortical interneurons: from Cajal to 2001. Prog Brain Res 136:215–238. 10.1016/S0079-6123(02)36019-9 [DOI] [PubMed] [Google Scholar]
- Del Prete D, Lombino F, Liu X, D'Adamio L (2014) APP is cleaved by Bace1 in presynaptic vesicles and establishes a presynaptic interactome, via its intracellular domain, with molecular complexes that regulate presynaptic vesicles functions. PLoS One 9:e108576. 10.1371/journal.pone.0108576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Turco D, Paul MH, Schlaudraff J, Hick M, Endres K, Müller UC, Deller T (2016) Region-specific differences in amyloid precursor protein expression in the mouse hippocampus. Front Mol Neurosci 9:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle E, Bruce MT, Breen KC, Smith DC, Anderton B, Regan CM (1990) Intraventricular infusions of antibodies to amyloid-beta-protein precursor impair the acquisition of a passive avoidance response in the rat. Neurosci Lett 115:97–102. 10.1016/0304-3940(90)90524-D [DOI] [PubMed] [Google Scholar]
- Fanutza T, Del Prete D, Ford MJ, Castillo PE, D'Adamio L (2015) APP and APLP2 interact with the synaptic release machinery and facilitate transmitter release at hippocampal synapses. Elife 4:e09743. 10.7554/eLife.09743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel H, Frere S, Segev O, Bharill S, Shapira I, Gazit N, O'Malley T, Slomowitz E, Berdichevsky Y, Walsh DM, Isacoff EY, Hirsch JA, Slutsky I (2014) APP homodimers transduce an amyloid-beta-mediated increase in release probability at excitatory synapses. Cell Rep 7:1560–1576. 10.1016/j.celrep.2014.04.024 [DOI] [PubMed] [Google Scholar]
- Fowler SW, Chiang AC, Savjani RR, Larson ME, Sherman MA, Schuler DR, Cirrito JR, Lesné SE, Jankowsky JL (2014) Genetic modulation of soluble Abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer's disease. J Neurosci 34:7871–7885. 10.1523/JNEUROSCI.0572-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, McDonald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J (2011) Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun 2:336. 10.1038/ncomms1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marin V, Blazquez-Llorca L, Rodriguez JR, Boluda S, Muntane G, Ferrer I, Defelipe J (2009) Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front Neuroanat 3:28. 10.3389/neuro.05.028.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM, Huang Y (2016) Apolipoprotein E4 Causes age-dependent disruption of slow gamma oscillations during hippocampal sharp-wave ripples. Neuron 90:740–751. 10.1016/j.neuron.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Hardy J (2014) Genetics of Alzheimer's disease. Neurotherapeutics 11:732–737. 10.1007/s13311-014-0295-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ (1999) Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19:8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R (2006) AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52:831–843. 10.1016/j.neuron.2006.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JK, Ma PL, Ji SY, Zhao XL, Tan JX, Sun XJ, Huang FD (2013) Age-dependent alterations in the presynaptic active zone in a Drosophila model of Alzheimer's disease. Neurobiol Dis 51:161–167. 10.1016/j.nbd.2012.11.006 [DOI] [PubMed] [Google Scholar]
- Huber G, Martin JR, Löffler J, Moreau JL (1993) Involvement of amyloid precursor protein in memory formation in the rat: an indirect antibody approach. Brain Res 603:348–352. 10.1016/0006-8993(93)91261-P [DOI] [PubMed] [Google Scholar]
- Jedlicka P, Owen M, Vnencak M, Tschäpe JA, Hick M, Müller UC, Deller T (2012) Functional consequences of the lack of amyloid precursor protein in the mouse dentate gyrus in vivo. Exp Brain Res 217:441–447. 10.1007/s00221-011-2911-9 [DOI] [PubMed] [Google Scholar]
- Johnson KA, Fox NC, Sperling RA, Klunk WE (2012) Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006213. 10.1101/cshperspect.a006213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L (1997) Amyloid precursor protein processing and A β42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A 94:1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabogo D, Rauw G, Amritraj A, Baker G, Kar S (2010) ss-amyloid-related peptides potentiate K+-evoked glutamate release from adult rat hippocampal slices. Neurobiol Aging 31:1164–1172. 10.1016/j.neurobiolaging.2008.08.009 [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37:925–937. 10.1016/S0896-6273(03)00124-7 [DOI] [PubMed] [Google Scholar]
- Kay KR, Smith C, Wright AK, Serrano-Pozo A, Pooler AM, Koffie R, Bastin ME, Bak TH, Abrahams S, Kopeikina KJ, McGuone D, Frosch MP, Gillingwater TH, Hyman BT, Spires-Jones TL (2013) Studying synapses in human brain with array tomography and electron microscopy. Nat Protoc 8:1366–1380. 10.1038/nprot.2013.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Miller DL, Sapienza VJ, Chen CM, Bai C, Gundke-Iqbal I, Currie JR, Wisniewski HM (1988) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun 2:121–130. [Google Scholar]
- Kirouac L, Rajic AJ, Cribbs DH, Padmanabhan J (2017) Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer's disease. eNeuro 4: pii: ENEURO.0149-16.2017. 10.1523/ENEURO.0149-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ (2008) Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci 28:4231–4237. 10.1523/JNEUROSCI.5161-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Cullen WK, Hu NW, Rowan MJ (2012) Alzheimer's disease Abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol Brain 5:25. 10.1186/1756-6606-5-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106:4012–4017. 10.1073/pnas.0811698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, Holtzman DM, Hyman BT, Spires-Jones TL (2012) Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain 135:2155–2168. 10.1093/brain/aws127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli BM, Pflieger D, Mueller LN, Carbonetti G, Aebersold R, Nitsch RM, Konietzko U (2012) Interactome of the amyloid precursor protein APP in brain reveals a protein network involved in synaptic vesicle turnover and a close association with Synaptotagmin-1. J Proteome Res 11:4075–4090. 10.1021/pr300123g [DOI] [PubMed] [Google Scholar]
- Kurudenkandy FR, Zilberter M, Biverstål H, Presto J, Honcharenko D, Strömberg R, Johansson J, Winblad B, Fisahn A (2014) Amyloid-beta-induced action potential desynchronization and degradation of hippocampal gamma oscillations is prevented by interference with peptide conformation change and aggregation. J Neurosci 34:11416–11425. 10.1523/JNEUROSCI.1195-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL (2004) Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci 24:10191–10200. 10.1523/JNEUROSCI.3432-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 27:796–807. 10.1523/JNEUROSCI.3501-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95:6448–6453. 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laßek M, Weingarten J, Einsfelder U, Brendel P, Müller U, Volknandt W (2013) Amyloid precursor proteins are constituents of the presynaptic active zone. J Neurochem 127:48–56. 10.1111/jnc.12358 [DOI] [PubMed] [Google Scholar]
- Laßek M, Weingarten J, Acker-Palmer A, Bajjalieh SM, Müller U, Volknandt W (2014) Amyloid precursor protein knockout diminishes synaptic vesicle proteins at the presynaptic active zone in mouse brain. Curr Alzheimer Res 11:971–980. 10.2174/1567205011666141107152458 [DOI] [PubMed] [Google Scholar]
- Laßek M, Weingarten J, Wegner M, Mueller BF, Rohmer M, Baeumlisberger D, Arrey TN, Hick M, Ackermann J, Acker-Palmer A, Koch I, Müller U, Karas M, Volknandt W (2016) APP is a context-sensitive regulator of the hippocampal presynaptic active zone. PLoS Comput Biol 12:e1004832. 10.1371/journal.pcbi.1004832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457:1128–1132. 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440:352–357. 10.1038/nature04533 [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D (2009) Soluble oligomers of amyloid beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62:788–801. 10.1016/j.neuron.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638. 10.1523/JNEUROSCI.0203-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillis KP, Wang Z, Mail M, Zhao GQ, Berdichevsky Y, Bacskai B, Staley KJ (2015) Evolution of network synchronization during early epileptogenesis parallels synaptic circuit alterations. J Neurosci 35:9920–9934. 10.1523/JNEUROSCI.4007-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA (2000) Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci 3:460–464. 10.1038/74833 [DOI] [PubMed] [Google Scholar]
- Mably AJ, Kanmert D, McDonald JM, Liu W, Caldarone BJ, Lemere CA, O'Nuallain B, Kosik KS, Walsh DM (2015) Tau immunization: a cautionary tale? Neurobiol Aging 36:1316–1332. 10.1016/j.neurobiolaging.2014.11.022 [DOI] [PubMed] [Google Scholar]
- McDonald JM, Cairns NJ, Taylor-Reinwald L, Holtzman D, Walsh DM (2012) The levels of water-soluble and triton-soluble Abeta are increased in Alzheimer's disease brain. Brain Res 1450:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JM, O'Malley TT, Liu W, Mably AJ, Brinkmalm G, Portelius E, Wittbold WM 3rd, Frosch MP, Walsh DM (2015) The aqueous phase of Alzheimer's disease brain contains assemblies built from approximately 4 and approximately 7 kDa Abeta species. Alzheimers Dement 11:1286–1305. 10.1016/j.jalz.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor JP, Van Nostrand WE (2000) Fibrillar amyloid beta-protein mediates the pathologic accumulation of its secreted precursor in human cerebrovascular smooth muscle cells. J Biol Chem 275:9782–9791. 10.1074/jbc.275.13.9782 [DOI] [PubMed] [Google Scholar]
- Melnikova T, Fromholt S, Kim H, Lee D, Xu G, Price A, Moore BD, Golde TE, Felsenstein KM, Savonenko A, Borchelt DR (2013) Reversible pathologic and cognitive phenotypes in an inducible model of Alzheimer-amyloidosis. J Neurosci 33:3765–3779. 10.1523/JNEUROSCI.4251-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileusnic R, Lancashire CL, Johnston AN, Rose SP (2000) APP is required during an early phase of memory formation. Eur J Neurosci 12:4487–4495. [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H (2009) Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29:3453–3462. 10.1523/JNEUROSCI.5215-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett BG, Richter M, Abraham WC, Müller UC (2017) Therapeutic potential of secreted amyloid precursor protein APPsalpha. Front Mol Neurosci 10:30. 10.3389/fnmol.2017.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller UC, Zheng H (2012) Physiological functions of APP family proteins. Cold Spring Harb Perspect Med 2:a006288. 10.1101/cshperspect.a006288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, McPhie DL (2007) Dysfunction of amyloid precursor protein signaling in neurons leads to DNA synthesis and apoptosis. Biochim Biophys Acta 1772:430–437. 10.1016/j.bbadis.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C (2008) Amyloid beta oligomers (A beta(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 28:788–797. 10.1523/JNEUROSCI.4771-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Cochennec J, Loll F, Escudé C, Boudier T (2013) TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29:1840–1841. 10.1093/bioinformatics/btt276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley TT, Oktaviani NA, Zhang D, Lomakin A, O'Nuallain B, Linse S, Benedek GB, Rowan MJ, Mulder FA, Walsh DM (2014) Abeta dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies. Biochem J 461:413–426. 10.1042/bj20140219 [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13:812–818. 10.1038/nn.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2016) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17:777–792. 10.1038/nrn.2016.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi J, Sepúlveda FJ, Roa J, Opazo C, Inestrosa NC, Aguayo LG (2010) Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J Biol Chem 285:2506–2514. 10.1074/jbc.M109.030023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett EK, Koffie RM, Wegmann S, Henstridge CM, Herrmann AG, Colom-Cadena M, Lleo A, Kay KR, Vaught M, Soberman R, Walsh DM, Hyman BT, Spires-Jones TL (2016) Non-fibrillar oligomeric amyloid-beta within synapses. J Alzheimers Dis 53:787–800. 10.3233/JAD-160007 [DOI] [PubMed] [Google Scholar]
- Pliássova A, Lopes JP, Lemos C, Oliveira CR, Cunha RA, Agostinho P (2016) The association of amyloid-beta protein precursor with alpha- and beta-secretases in mouse cerebral cortex synapses is altered in early Alzheimer's disease. Mol Neurobiol 53:5710–5721. 10.1007/s12035-015-9491-9 [DOI] [PubMed] [Google Scholar]
- Portelius E, Olsson M, Brinkmalm G, Rüetschi U, Mattsson N, Andreasson U, Gobom J, Brinkmalm A, Hölttä M, Blennow K, Zetterberg H (2013) Mass spectrometric characterization of amyloid-beta species in the 7PA2 cell model of Alzheimer's disease. J Alzheimers Dis 33:85–93. [DOI] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Müller UC (2007) The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci 27:7817–7826. 10.1523/JNEUROSCI.1026-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoli C, Piacentini R, Riccardi E, Leone L, Li Puma DD, Bitan G, Grassi C (2013) Effects of different amyloid beta-protein analogues on synaptic function. Neurobiol Aging 34:1032–1044. 10.1016/j.neurobiolaging.2012.06.027 [DOI] [PubMed] [Google Scholar]
- Russell CL, Semerdjieva S, Empson RM, Austen BM, Beesley PW, Alifragis P (2012) Amyloid-beta acts as a regulator of neurotransmitter release disrupting the interaction between synaptophysin and VAMP2. PLoS One 7:e43201. 10.1371/journal.pone.0043201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384. 10.1016/j.neurobiolaging.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ (2007) Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68:1501–1508. 10.1212/01.wnl.0000260698.46517.8f [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Pérez-Garci E, Schneider A, Kollewe A, Gauthier-Kemper A, Fritzius T, Raveh A, Dinamarca MC, Hanuschkin A, Bildl W, Klingauf J, Gassmann M, Schulte U, Bettler B, Fakler B (2016) Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat Neurosci 19:233–242. 10.1038/nn.4198 [DOI] [PubMed] [Google Scholar]
- Seabrook GR, Smith DW, Bowery BJ, Easter A, Reynolds T, Fitzjohn SM, Morton RA, Zheng H, Dawson GR, Sirinathsinghji DJ, Davies CH, Collingridge GL, Hill RG (1999) Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38:349–359. 10.1016/S0028-3908(98)00204-4 [DOI] [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D (1992) Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature 359:325–327. 10.1038/359325a0 [DOI] [PubMed] [Google Scholar]
- Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH (2006) Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597–624). FASEB J 20:1254–1256. 10.1096/fj.05-5032fje [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14:837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Welzel AT, McDonald JM, Selkoe DJ, Walsh DM (2011) Isolation of low-n amyloid beta-protein oligomers from cultured cells, CSF, and brain. Methods Mol Biol 670:33–44. 10.1007/978-1-60761-744-0_3 [DOI] [PubMed] [Google Scholar]
- Slomowitz E, Styr B, Vertkin I, Milshtein-Parush H, Nelken I, Slutsky M, Slutsky I (2015) Interplay between population firing stability and single neuron dynamics in hippocampal networks. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, Masters CL, Müller U, Kins S, Beyreuther K (2005) Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J 24:3624–3634. 10.1038/sj.emboj.7600824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolow S, Luu SH, Nandy K, Miller CA, Vinters HV, Poon WW, Gylys KH (2012) Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer's disease cortex. Neurobiol Dis 45:381–387. 10.1016/j.nbd.2011.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola Vigo F, Kedikian G, Heredia L, Heredia F, Añel AD, Rosa AL, Lorenzo A (2009) Amyloid-beta precursor protein mediates neuronal toxicity of amyloid beta through Go protein activation. Neurobiol Aging 30:1379–1392. 10.1016/j.neurobiolaging.2007.11.017 [DOI] [PubMed] [Google Scholar]
- Steinbach JP, Müller U, Leist M, Li ZW, Nicotera P, Aguzzi A (1998) Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ 5:858–866. 10.1038/sj.cdd.4400391 [DOI] [PubMed] [Google Scholar]
- Tamayev R, Matsuda S, Arancio O, D'Adamio L (2012) beta- but not gamma-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med 4:171–179. 10.1002/emmm.201100195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE. (2012) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med pii: a006296. 10.1101/cshperspect.a006296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand WE, Melchor JP, Keane DM, Saporito-Irwin SM, Romanov G, Davis J, Xu F (2002) Localization of a fibrillar amyloid beta-protein binding domain on its precursor. J Biol Chem 277:36392–36398. 10.1074/jbc.M204676200 [DOI] [PubMed] [Google Scholar]
- Vertkin I, Styr B, Slomowitz E, Ofir N, Shapira I, Berner D, Fedorova T, Laviv T, Barak-Broner N, Greitzer-Antes D, Gassmann M, Bettler B, Lotan I, Slutsky I (2015) GABAB receptor deficiency causes failure of neuronal homeostasis in hippocampal networks. Proc Natl Acad Sci U S A 112:E3291–E3299. 10.1073/pnas.1424810112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Teplow DB (2012) Alzheimer's disease and the amyloid beta-protein. Prog Mol Biol Transl Sci 107:101–124. 10.1016/B978-0-12-385883-2.00012-6 [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539. 10.1038/416535a [DOI] [PubMed] [Google Scholar]
- Wang B, Wang Z, Sun L, Yang L, Li H, Cole AL, Rodriguez-Rivera J, Lu HC, Zheng H (2014) The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J Neurosci 34:13314–13325. 10.1523/JNEUROSCI.2848-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL (2002) Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 924:133–140. 10.1016/S0006-8993(01)03058-X [DOI] [PubMed] [Google Scholar]
- Wang Z, Neely R, Landisman CE (2015) Activation of group I and group II metabotropic glutamate receptors causes LTD and LTP of electrical synapses in the rat thalamic reticular nucleus. J Neurosci 35:7616–7625. 10.1523/jneurosci.3688-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZM, Qi YJ, Wu PY, Zhu Y, Dong YL, Cheng ZX, Zhu YH, Dong Y, Ma L, Zheng P (2008) Neuroactive steroid pregnenolone sulphate inhibits long-term potentiation via activation of alpha2-adrenoreceptors at excitatory synapses in rat medial prefrontal cortex. Int J Neuropsychopharmacol 11:611–624. 10.1017/S1461145707008334 [DOI] [PubMed] [Google Scholar]
- Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, Selkoe DJ (2014) Secreted amyloid beta-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry 53:3908–3921. 10.1021/bi5003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AR, Zheng H, Galatis D, Maher F, Hesse L, Multhaup G, Beyreuther K, Masters CL, Cappai R (1998) Survival of cultured neurons from amyloid precursor protein knock-out mice against Alzheimer's amyloid-beta toxicity and oxidative stress. J Neurosci 18:6207–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claßen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO (2014) Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344:1023–1028. 10.1126/science.1252884 [DOI] [PubMed] [Google Scholar]
- Willem M, et al. (2015) eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526:443–447. 10.1038/nature14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang Z, Wang B, Justice NJ, Zheng H (2009) Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci 29:15660–15668. 10.1523/JNEUROSCI.4104-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, O'Malley TT, Kanmert D, Jerecic J, Zieske LR, Zetterberg H, Hyman BT, Walsh DM, Selkoe DJ (2015) A highly sensitive novel immunoassay specifically detects low levels of soluble Abeta oligomers in human cerebrospinal fluid. Alzheimers Res Ther 7:14. 10.1186/s13195-015-0100-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, Selkoe DJ (2017) Large soluble oligomers of amyloid beta-protein from alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37:152–163. 10.1523/JNEUROSCI.1698-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Lu T (2009) Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem 284:4755–4759. 10.1074/jbc.R800018200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Mably AJ, Walsh DM, Rowan MJ (2017) Peripheral interventions enhancing brain glutamate homeostasis relieve amyloid beta- and TNFalpha-mediated synaptic plasticity disruption in the rat hippocampus. Cereb Cortex 27:3724–3735. 10.1093/cercor/bhw193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH (1995) beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81:525–531. 10.1016/0092-8674(95)90073-X [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]