

Abstract
With the advent of technology, the role of nanomaterials in medicine has grown exponentially in the last few decades. The main advantage of such materials has been exploited in drug delivery applications, due to their effective targeting that in turn reduces systemic toxicity compared to the conventional routes of drug administration. Even though these materials offer broad flexibility based on targeting tissue, disease, and drug payload, the demand for more effective yet highly biocompatible nanomaterial-based drugs is increasing. While therapeutically improved and safe materials have been introduced in nanomedicine platforms, issues related to their degradation rates and bio-distribution still exist, thus making their successful translation for human use very challenging. Researchers are constantly improving upon novel nanomaterials that are safer and more effective not only as therapeutic agents but as diagnostic tools as well, making the research in the field of nanomedicine ever more fascinating. In this review stress has been made on the evolution of nanomaterials that have been approved for clinical applications by the United States Food and Drug Administration Agency (FDA).
Keywords: Drug delivery, Inorganic nanomaterial, Polymeric nanomaterial, Liposomes, Clinical trials, FDA approval
Introduction
The fascinating world of nanomaterials and their various applications is not new. As early as 2500 BC, natural asbestos was used to increase the reinforcement of a ceramic mixture for decorative purposes [1]. The famous Lycurgus cup made by the Romans in the 4th century AD displayed different colors during the day and when illuminated from inside at night [2]. This gave the first definite evidence into the synthesis and application of gold colloids. Mesopotamians in the 9th century AD introduced silver and/or copper glazed ceramics [3]. It was not until the famous statement, “there is plenty of room at the bottom” from Feynman in 1959 [4] that brought nanotechnology back into focus of the modern scientific world and exactly a decade after, the term “nanotechnology” was coined [5]. Over the years, application of nanomaterials in biomedicine has vastly increased and with the introduction of inorganic and polymeric materials further enhanced their role for drug delivery and sustained drug release. Figure 1 outlines important dates in the evolution of nanomaterials from 2500 BC until today from their discovery/invention to different applications. Not all the listed nanomaterials in Figure 1 have been approved for clinical use.
Figure 1.

Timeline showing the evolution of important nanomaterials to date.
Although nanomaterials have found their applications in different fields of research for long, their role in medicine is new and emerging. Due to their small sizes, nanomaterials take a sweet spot that correlates with the sub-micron entities of the biological world, making them especially appropriate for interactions at that scale. Nanomaterials display many distinctive physicochemical properties that differ extensively from their corresponding bulk materials. It is mainly their size-related properties that dictate their physicochemical uniqueness and make them exceptional for various biological applications, e.g. drug delivery, tissue engineering, targeted drug delivery, bio-microelectromechanical systems (bioMEMS), biosensors, microfluidics, and diagnostics [6]. Due to the extraordinary opportunity it offers, nanomaterial-based drug delivery has emerged as the mainstream application of nanotechnology in medicine [7]. In addition to enhancing targeted delivery and controlled release of drugs, nanomaterials assist in improving their circulation time and biodistribution, solubility, intracellular delivery, and crossing biological membranes [8]. Nanomaterials in medicine have been traditionally used for drug delivery applications only; however, new nanoscale platforms have been developed that offer a diagnostic application in addition to their therapeutic ability [9]. Multifunctional nanomaterials have been developed that combine therapeutic, targeting, and imaging capabilities for advanced drug delivery systems and are gaining more and more attention from the research community [10].
In this review, we attempt to outline the evolution of nanomaterials for drug delivery applications over the last few decades. The stress has been made on those materials that have been approved for therapeutic purposes by the FDA. Carbon based nanomaterials for biomedical and tissue engineering applications have been discussed elsewhere [11]. In the first section, nanomaterials of inorganic origin including gold, magnetic, and silica-based materials for drug delivery are discussed. The second section details the applications of polymer-based nanomaterials, where the development and role of dendrimers, polymeric micelles, nanogels, and polymeric nanoparticles are discussed. Lipid-based nanomaterials are explored in the next section, and the last section entails a summary of some of the nanomaterials that have been approved by the FDA for human use.
1. Inorganic Nanomaterials
Over the last few decades, scientists have actively explored the synthesis of inorganic nanoparticles (INPs) for applications in various fields. In this section, we primarily focus on the synthesis of INPs for diagnostic and therapeutic applications, which require precision engineering of the nanoparticle properties. Figure 2 outlines important applications of some INPs in diagnostics and drug delivery. The extensive research in the field has provided ample understanding to control the attributes at the nano-bio interface, which has led to numerous successful clinical trials and translations. INPs have also been exploited for their optical properties, which arise due to the quantum size effect, and have been shown to be modulated by control over size for application as effective imaging and contrast agents [12]. Similarly, various chemical modulations have been carried out on their surface, such as PEGylation [13], charge modulation, ligand conjugation, and inclusion of stimuli-responsive moieties and small-molecule probes for improved drug efficacy [14]. We further discuss the advances in the synthesis and surface modification of these INPs that have facilitated their successful drug delivery applications.
Figure 2.
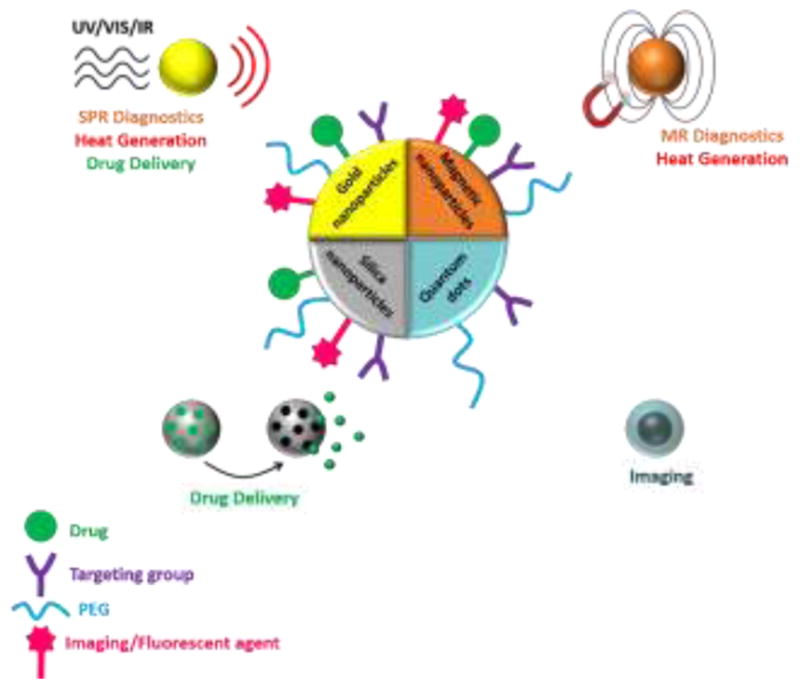
Illustration of diverse applications of INPs in biomedicine for targeted drug delivery, diagnostics and imaging (through surface plasmon resonance [SPR] or magnetic resonance [MR]), and photodynamic and photothermal therapies. Polyethylene glycol (PEG) is used as a stabilizing agent. Ultraviolet (UV), visible (Vis), or infrared (IR) wavelengths are used to trigger the desired phenomenon.
1.1 Gold nanoparticles (AuNPs)
First scientific insight into the property of AuNPs came from Michael Faraday in 1850 [15], where through synthesis he showed the ability to obtain a ruby red colloidal solution of AuNPs from yellow HAuCl4 gold salt. However, medicinal application of gold salt was not proposed until 1890, as bacteriostatic against tuberculosis [16]. Subsequently, gold was also found to be effective in the treatment of Rheumatoid arthritis in 1927 [16]. This led to prominence of nanogold as therapeutic agent towards various Rheumatoid diseases.
AuNPs have been widely explored for applications in medicine due to (i) biocompatible nature, (ii) precise control over their size distribution, (iii) modification in shape that includes spheres, nanorods, and cubes, among others, and (iv) ease of modulating surface chemistry through conjugation with various polymers, antibodies, small molecules therapeutics and molecular probes [17–19].
There are diverse methods for the synthesis of AuNPs, mostly using HAuCl4 as the precursor salt. Most of the methods for the preparation of AuNPs involve reduction of Au3+ ions to Au0, using various reducing agents at different concentrations to control the size of AuNPs. Turkevich et al. introduced citrate salts as the reducing agent for synthesis of AuNPs in 1951 [20]. Researchers have constantly been modifying the Turkevich method to further increase the precision of the size. Furthermore, thiols have also been widely explored as the reducing agent for the synthesis of AuNPs. Shiffrin-Brust used thiols in bi-phasic solvent along with sodium borohydride and phase transfer reagents to yield 2–4 nm thiol-coated AuNPs [21]. Processes to functionalize AuNPs with different water-soluble surface groups by ligand exchange have been exploited by using thiolated ligands.
In 2000, Murray et al. developed post-synthesis ligand substitution on AuNP surface [22]. This method has been extensively modified over the years to have a definitive control on the biophysicochemical interactions of these nanoparticles for low nonspecific protein adsorption [23], active targeting of the tumor [24], and enhanced cellular uptake [25]. In the last two decades, researchers have exploited ligand exchange to conjugate nucleic acids and entrapment of toxic drugs with controlled release. With latest developments in therapeutics, AuNPs have been widely pursued as effective delivery agents for small interfering RNA (siRNA)- and DNA-based enzymes (DNAzymes). Kataoka et al. modified the surface of PEGylated AuNPs with hydroxychloroquine for efficient endosomal escape and increased siRNA distribution [26]. Researchers have shown delivery of RNA to regulatory T cells, glioblastoma, and mesenchymal stem cells along with various other therapeutic targets [27–29]. Broadening their use in drug delivery applications, Tang et al. have reported the use of AuNPs-stabilized capsules (AuNPSCs) as an efficient protein delivery system [30]. AuNPSCs consisted of an oil core and were stabilized by amino acids (HKRK) conjugated cationic gold nanoparticles. These NPSCs fused directly with the cell membrane and efficiently delivered proteins such as green fluorescent protein (GFP) and caspase-3 into the cytosol. Further, Ray et al. have demonstrated the versatility of the AuNPSC-mediated delivery system by intracellular targeting of the proteins to the nucleus [31]. In a recent work, Mout et al. used programmed assembly of AuNPs with modified Cas9 protein for direct cytosolic delivery and efficient gene editing [32].
AuNPs also exhibit interesting absorption and scattering properties, which can be modulated by control over their size and shape. SPR property has been exploited for numerous in vivo theranostic applications. Gold nanorods (AuNRs) have been quite extensively used for these applications as their length and width can be modulated to obtain SPR bands in the near-infrared (NIR) regions to allow better contrast in biological tissues [33]. Oldenburg et al. synthesized gold nanoshells (AuNSs) through silica nanoparticle-templated growth of gold, which have an absorption band in the NIR region and their optical properties can be tuned by controlling thickness of the gold coating [34]. The photothermal property of the gold nanostructures also allows thermal ablation of solid tumors if the AuNSs are excited by NIR and they have shown promising results in clinical studies towards FDA approval under the name of AuroLase [35].
Understanding of size, shape, and surface properties of AuNPs has culminated into various clinical trials as drug carriers and imaging agents; but a supplementary understanding of their cytotoxicity, bio-distribution, and clearance from the body is required for improving their clinical translation [36].
1.2 Iron oxide nanoparticles (IONPs)
IONPs have been used since the 1960s as targeted imaging and therapeutic agents. Meyers et al. used an external horseshoe magnet to show accumulation of IONPs as contrast agent in the vascular and lymphatic system of dogs [37]. Application of IONPs in drug delivery was explored by Widder et al. in 1979, by encapsulating IONPs in albumin microspheres along with a chemotherapeutic agent to establish an in vivo delivery method [38]. Since then encapsulated IONPs have been explored for site-specific delivery and entered Phase I clinical trial in 1996 for delivery of epirubicin for treatment of advanced cancer [39]. However, the trial was unsuccessful as most the IONPs went to the liver.
IONPs have emerged as a successful class of nanoparticles in clinical translation as numerous IONPs are approved by the FDA for therapeutics and imaging. Magnetite (Fe3O4) nanoparticles have attained such eminence due to (i) extremely low cytotoxicity, (ii) magnetic responsiveness and its tunability, (iii) controlled size and surface modication, and (iv) contrast agent for magnetic resonance imaging (MRI) [40].
IONPs have different magnetic property than bulk magnetite. These nanoparticles exhibit single-domain magnetic property in the sub-100 nm range, which has maximum coercivity as compared to the bulk magnetite that is multi-domain [41]. Further reduction in the size of IONPs decreases their magnetic anisotropy energy. When thermal energy equals the anisotropy energy, it results in random flipping of the magnetic moment and the nanoparticles exhibit super paramagnetic nature[42]. These superparamagnetic IONPs (SPIONs) heat up when placed in an alternating electric field (hyperthermia) [43]. Subsequently, these particles have been used in vivo for targeting and thermal ablation of tumors. Recently, Espinosa et al. synthesized 20-nm iron oxide nanocube, which were heated with alternating magnetic fields and NIR radiation to show complete ablation of solid tumor in vivo due to increased heating power that they termed as dual-mode activation [44]. These SPIONs have also been extensively used in stem cell engineering for their controlled differentiation, efficient homing, and long-term tracking in cell-based therapies. Researches have used polyethylene conjugated (PEGylated) SPIONs that were internalized by the mesenchymal stem cells (MSCs) to guide them to the site of injury using external magnetic field. This technique was further exploited by Xu et al. to track the MSCs post-engraftment [45].
Surface moieties on the IONPs play an eminent role in their bio-distribution, cytotoxicity, cellular uptake, and clearance. The rapid degradation of IONPs results in release of Fe2+/3+ ions that alters levels of ferritin, and inflammatory cytokines and several other reactive oxygen species (ROS)-dependent proteins in cells, increasing their cytotoxicity [46]. Thus, efficient surface coating of these IONPs plays a very important role in their physicochemical property in vivo. For most applications, IONPs are coated with hydrophilic polymers for controlled degradation, as well as decrease in immunogenicity and opsonization. Sakhulkhu et al. analyzed the protein corona on the SPIONs coated with polyvinyl alcohol (PVA) and dextran along with the effect of charge modulation. They have shown that dextran has less adsorption of protein as compared to PVA. Further, they found that electrostatic charge played an important role and the negatively charged PVA SPIONs had less protein adsorption (22%) compared to 36% and 41% on positive and neutral PVA-coated SPIONs [47].
In 2009, FDA approved ‘Ferumtoxyl’, SPIONs for the treatment of anemia patients with chronic kidney disease. These particles are coated with polyglucose sorbitol carboxymethylether that controls their degradation and improves their pharmacokinetics. In further studies, Zanganeh et al. have shown that Ferumtoxyl could polarize macrophage in tumor tissue to pro-inflammatory M1 phenotype, which could constrain the proliferation of subcutaneous adenocarcinomas [48]. They also showed the tendency of the SPIONs to inhibit liver metastasis up to 6 times if Ferumtoxyl was pre-injected in vivo [48]. Currently, these materials are being actively explored for imaging the progression of type 1 diabetes and response of the host to the therapies. SPIONs-based imaging has also been effective in distinguishing non-diabetic patients from the ones with recent onset of diabetes due to enhanced accumulation of SPIONs in pancreases [49]; this discovery led the way for drug delivery to pancreas for diabetes.
Due to the small sizes of IONPs, macrophages and monocytes generally uptake them by phagocytosis or macro-pinocytosis depending on their surface functionality. These IONPs-loaded macrophages have been actively pursued for in vivo imaging and as drug delivery agents for diseases and injuries that involve enhanced macrophage accumulation. Macrophage-assisted IONP imaging has been explored for various cancers by taking advantage of tumor-associated macrophages. They have also been explored for imaging in myocardial infarction, myocarditis, aortic aneurysm, and atherosclerosis.
1.3 Silica nanoparticles (SiNPs)
SiNPs provide a new modality to the inorganic nanoparticles due to their rapid degradation in vivo, regulated pore sizes (2–10 nm) for drug encapsulation [50], incorporation of metals for theranostic applications [51], and ease of camouflage by chemical conjugations [52].
Since their invention in the 1960s, as catalyst due to their large surface to volume ratios, they have been actively modified to enhance their absorption properties. In 1968, Stöber et al. proposed synthesis of solid SiNPs by a process based on hydrolysis in ammonium oxide of silyl ethers [53]. This method has been adapted by many laboratories to synthesize SiNPs with sizes from 50–3000 nm. Further, researchers have recognized the niche realm of SiNPs in drug delivery since the synthesis of mesoporous silica nanoparticles (MSNPs) in 1990’s [54]. They have filled the gap that wouldn’t otherwise be covered by metallic or other inorganic nanomaterials with regards to their degradability, biocompatibility and drug release rates.
MSNPs provide an interesting and alternative route to drug delivery as they have nano-pores that can encapsulate hydrophobic drugs for efficient delivery, unlike other inorganic nanoparticles. The pore size of MSNPs can be modulated by using various templates, surfactant concentrations, pH, and solvents during the synthesis; this control allows sustained degradation of the particles to deliver various payloads [50]. MSNPs have also been explored as stimuli-responsive drug release systems, where various chemical entities on the surface of MSNPs can be used to control the release of encapsulated drug by a trigger reaction [55]. Essentially, release rates of vancomycin- and adenosine triphosphate (ATP) from mesoporous silica nanosphere-based drug delivery systems was controlled by using disulfide bond reducing molecules, such as dithiothretol (DTT) and mercaptoethanol (ME), as release triggers. This concept is termed as gatekeeping drug delivery system. In the past years, various systems such as AuNPs, IONPs, CdS-NPs, and polymers have been used as gatekeepers for controlled release [52, 56].
MSNPs have hydrophilic surface, which is attributed to the presence of hydroxyl groups that can be further modified using (3-aminopropyl) triethoxysilane (APTES) to replace with more versatile amine groups. Since early 2000s, many research groups have functionalized MSNPs with antibodies, nucleic acids, and cell membranes to control the biodistribution and reduce the systemic toxicity of MSNPs. In 2012, Parodi et al. used advanced techniques to show enhanced delivery of doxorubicin using cell membrane-cloaked MSNPs [52]. They demonstrated that MSNPs coated with leucocyte membranes (termed as ‘cloaking’) have reduced cytotoxicity and they efficiently extravasated into the blood vasculature and accumulated near the tumor site in vivo. Recently, physicochemical properties of PEGylated SiNPs have been further exploited by Kim et al. [57]. They used Cornell dots, which completed Phase I trial and is currently under FDA-investigational new drug (IND) for actively targeting melanoma by conjugating αvβ3-integrin-targeting peptides to induce ferroptosis in cancer-bearing mice. As researchers proceed to understand the fate of SiNPs and actively pursue their interactions at the nano-bio interface, we can foresee several clinical translations in the near future.
1.3 Quantum dots (QDs)
In the 1980s, researchers discovered that confined SiNPs emitted red light upon illumination with laser [58]. This led to further research in the field of inorganic nanomaterials to produce a range of light-emitting fluorescent INPs known as QDs. Quantum dots (QD) are very small semiconductor particles, only several nanometres in size, so small that their optical and electronic properties differ from those of larger particles. They are crystalline in nature and comprise of two different periodic group elements with the size varying in the range of 2–10 nm [59]. Understanding of their material composition and size modulation has led to the development of a broad spectrum of fluorescent QDs, with high quantum yield and low photobleaching. Thus, QDs have found various applications in photodynamic therapy, in vivo imaging, and tracking drug biodistribution [59–61].
Over the years, QDs have been widely explored in theranostics alone and as an adjunct with wide variety of nanomaterials [62]. Cai et al. used pH-responsive ZnO-QDs coordinated with doxorubicin for targeted drug delivery using hyaluronic acid conjugation [63]. They have shown efficient anti-cancer effect of the ZnO-QDs due to dual effect of Zn2+ and doxorubicin.
QDs have been explored for various applications in vitro and in vivo but they are yet to be approved in clinical trials due to their potential cytotoxicity [64]. This is due to two main issues with QDs: 1.) Use of heavy metals in their composition, and 2.) their clearance from the body. Oh et al. analyzed the published literature to elucidate the dominant role of surface properties and size of the QDs on the cytotoxicity [65]. They also assessed the dataset to understand the role of different core-shell QDs and their surface coating and their half maximal inhibitory concentration (IC50) value. This sequential data-mining gives researchers a broad database to comprehend the plethora of work done on QDs for designing more particles that can be used for translation in drug delivery applications and as theranostic agents. To minimize the cytotoxic effect, researchers are exploring Cd-free QDs [66], which have come up as less cytotoxic nanoparticles along with advances in more efficient polymeric coating of the QDs [67, 68].
2. Polymeric nanomaterials
Among various nanotherapeutics, polymeric nanoparticles stand out as a versatile class of nanocarriers that allow controlled drug delivery to diseased tissues [69]. The first generation of polymeric nanoparticles are to encapsulate the drug molecules and have a sustained-release in time. To manage efficient drug delivery to the target site, various strategies have been developed in the second-generation polymeric nanoparticles by incorporating stimuli-responsive properties (e.g., pH, temperature, or light activation). Third-generation polymeric nanocarriers are represented by multi-functionalities, such as targeting and multi-drug release properties [70]. In this section, we discuss four main types of polymeric nanomaterials: polymer-drug conjugates, micelles, nanogels, and dendrimers.
2.1 Polymer-drug conjugates (polymeric prodrugs)
Polymer-drug conjugates, containing a water-soluble biocompatible polymer backbone and hydrophobic therapeutic agents (Figure 3a), are one of the most explored polymeric drug delivery platforms [71, 72]. Herein, therapeutic agents are covalently bonded to the polymer backbone via a stimuli-responsive spacer (e.g., disulfide, hydrazone, peptide, azo, etc.), which prevents undesirable drug release in blood circulation [73]. Compared to the other conventional polymer based platforms, these systems generally have enhanced stability, low toxicity, higher drug-loading capacity, and prolonged drug release behaviors [74].
Figure 3. Schematic illustration of various potential approaches for triggered drug delivery using polymer based nanoparticles.

(a) Polymer-drug conjugates [119], (b) Polymeric nanoparticles [120], (c) A dual-pH-sensitive polymeric micelles from self-assembling of a block copolymer-doxorubicin conjugate [92] and (d) thermo-sensitive shelf-crosslinked micelles [97], (e) FA- and PEG- functionalized nanogels for multi-drug delivery [104], and (f) thermo-responsive dendrimers [121]. Illustrations adapted with permissions from Royal Society of Chemistry for [121], Nature Publishing Group for [119], American Society of Neuroradiology for [120], American Chemical Society for [92] and [104], and Elsevier for [97].
Fast cellular internalization and ineffective drug delivery to cancer tissues are two of the main challenges of polymer conjugates. Wang et al. developed a new endosomal acid-stimulating polymer-drug conjugate platform, poly(2-(methacryloyloxy)ethyl choline phosphate)-b-poly(2-methoxy-2-oxoethyl methacrylate-hydrazide doxorubicin) (PCP-DOX) [75]. The drug was released from the polymer backbone inside cancer cells by the cleavage of pH-sensitive hydrazone spacer-drug linkage and successfully translocated to the nucleus. Similarly, Wu et al. created a pH/reduction dual-responsive charge-conversional polymeric prodrug decorated with reduction-responsive disulfide units and acid-labile anticancer drugs for efficient co-delivery of doxorubicin and demethylcantharidin for effective cancer therapy [76]. Under acidic pH and in the presence of glutathione (GSH), polymeric nanoparticles exhibited significantly improved cellular uptake and low cytotoxicity. These findings indicated that the prodrug nanoparticle systems provide a promising approach for combination therapy in cancer treatment.
2.2 Polymeric Nanoparticles
The concept of polymeric nanoparticles for the delivery of therapeutic agents was first proposed by Speiser and his team in 1969 [77]. They studied polyacrylic nanoparticles for oral drug administration, which was the first attempt to develop nanoparticles for vaccination purposes using tetanus toxoid and human immunoglobulin [77, 78]. Since then polymeric nanoparticles have been engineered from biocompatible and biodegradable natural or synthetic polymers for various drug delivery systems and the efforts of Robert Langer have been at the forefront of popularizing such platforms [79, 80]. Depending on the preparation method of the nanoparticles, either nanocapsules or nanospheres can be obtained. Nanocapsules have a vesicular-type structure consisting of an inner core surrounded by a polymer membrane or coating, while nanospheres have matrix-type structure in which therapeutic agents are homogeneously dispersed (Figure 3b). When compared with other nanoparticulate platforms, such as micelles and liposomes, polymeric nanoparticles tend to be more stable, especially in biologic fluids after their administration [81].
The most outstanding candidates for designing of polymeric nanoparticle-based drug delivery platforms are poly(glycolic acid) (PGA), poly(lactic acid) (PLA), poly(lactic-co-glycolic acid) (PLGA), poly(ε-caprolactone) (PCL), and poly(methyl-methacrylate) (PMMA), as synthetic polymers, as well as chitosan, alginate, gelatin, collagen, dextran, heparin, and albumin, as natural polymers. Among the synthetic polymers utilized to date, PLGA-based nanoparticles have received one of the most considerable attention due to their attractive characteristics of biocompatibility and drug release tunability. The greatest advantage of this polymer is that their properties, such as molecular weight, drug release rate, hydrophobicity, and biodegradability can be regulated by manipulating lactide-to-glycolide (L:G) ratio and molecular weight (Mw) of PLGA [82]. For instance, Gumusderelioglu et al. studied the effect of the L:G ratio variations from 70:30 to 90:10 on mitomycin-C release [83]. The increased drug release amounts and rates were observed corresponding to an increase in the glycolide content. Moreover, after three months, PLGA 90:10 and 70:30 nanoparticles released 30 and 70 wt.% of drug, respectively. Among the natural polymers, chitosan-based nanoparticles have emerged as one of the most popular biopolymers used in nanomedicine by virtue of their outstanding merits such as excellent biocompatibility and biodegradability, low toxicity, mucoadhesivity, hemostatic and antimicrobial activity, simple preparation method, and easy functionalization [84–86]. The key advantage of chitosan-based nanoparticles is their strong affinity for negatively charged cell surfaces and in vivo site-specific drug delivery. The release of therapeutic agents from chitosan-based nanoparticles is dependent upon polymer erosion, polymer-drug interactions, and their swelling potential, which are affected by the pH of the medium [87, 88]. In a recent study, Wu et al. designed both recombinant human interleukin-2 (rhIL-2) and doxorubicin-loaded chitosan-based nanoparticles for the enhanced anticancer activity [89]. Folic acid (FA) was conjugated onto the amino groups of chitosan to obtain active targeting ability. In this system, in vitro acid-responsive release of doxorubicin and prolonged release of rhIL-2 were managed and in vivo improved anticancer activity and reduced side-effect were achieved.
2.3 Micelles
Polymeric micelles, first introduced as therapeutic systems by Bader et al in 1984 [90], have a spherical core-shell structure, which was formed via self-assembling of amphiphilic block copolymers in aqueous solutions. The hydrophobic core creates a cargo space for entrapment of hydrophobic therapeutic agents, whereas the hydrophilic shell protects the formed micelle against aggregation and prolongs circulation time in blood [91]. Although polymeric micellar drug delivery systems seem to be promising, most of these systems show burst drug release and lack targeting ability. To address this challenge, stimuli-responsive polymeric micellar structures have been developed in the late 1980s (Figure 3c–d).
Cancer cells have the ability to grow in an acidic tumor microenvironment and researchers have been trying to create polymeric micelle nanocarriers that change their surface charge in the intracellular acidic conditions. Dai et al. developed a simple method to design a dual-pH-responsive nanocarrier platform from self-assembly of a block copolymer, poly(2-(diisopropylamino)-ethyl methacrylate-b-poly(4-formyl phenyl methacrylate-co-polyethylene glycol monomethyl ether methacrylate) (PDPA-b-P(FPMA-co-OEGMA)), that responds to two different pH values [92]. This dual pH-responsive behavior not only led to a pH-triggered improved cellular uptake, but also permitted highly efficient drug release in target tissues. Another innovative approach to develop an alternative micellar nanocarrier platform was recently proposed by Xu et al. [93]. They designed novel GSH responsive polymeric micelles based on PEG-polycarbonate-PEG triblock copolymer. Disulfide bonds on the polymer chain promoted GSH-triggered drug release inside the tumor cells, where GSH is enriched.
It is known that the ROS levels in the tumor cells are 100 times higher than healthy cells [94]. In the presence of ROS, copolymers can be converted into more hydrophilic components [95]. By utilizing this approach, the dissociated polymers can easily pass into the endosomal membrane of tumor cells and release drugs. Recently, in the work by Yu et al., ROS-responsive micelles based on poly(ethylene glycol)-b-poly(diethyl sulfide) (mPEG-PS) copolymers were developed for specific tumor cytosolic drug delivery [94]. The disassembling of micelles was induced by the hydrophobic to hydrophilic conversion of the mPEG-PS core in response to ROS-induced oxidation in vitro. Similarly, dual redox-sensitive micelles for ROS and GSH were developed for cancer treatment. The encapsulated drugs inside the micelles were not only released in response to the increased levels of ROS and GSH in tumor cells, but also could deeply penetrate into tumor layers [96].
The stability of the conventional micelles in the systemic circulation is crucial for their use in drug delivery. To enhance stability, chemical crosslinking of polymeric micelles has been developed by Ding and Liu in 1998 [97]. In a recent work by the Shi et al. in 2015, triblock thermosensitive polymer-based micelles were synthesized and interfacially crosslinked with pH-responsive hydrazone groups [98]. The interfacially crosslinked micelles provided high drug loading capacity and enhanced drug retention under physiological conditions. A recent study by Chung et al introduced self-assembling micellar complexes conjugated with organic tea extracts for cancer therapy [99].
While the current investigations on stimuli-responsive polymeric micelles are encouraging, their function and fate in vivo is still not understood properly, particularly, how these responsive micellar architectures behave in a human body. In addition, the structural instability and challenges in controlling drug release in biological environments need to be comprehensively addressed.
2.4 Nanogels
Nanogels are hydrophilic or amphiphilic polymeric particles that absorb water and swell [100]. They were developed in the 1990s. They are physically or chemically crosslinked polymeric hydrogel networks with sizes of around 100–200 nm that show swelling behavior with high water-retaining capacity [101]. Compared to other nanocarriers, nanogels possess several significant advantages, such as higher drug loading capacity, enhanced stability, control over particle size, larger surface area, and increased stimuli-responsiveness to pH, temperature, redox conductions, and enzymes [102, 103].
Combination chemotherapy with multiple anticancer drugs has opened up a new strategy to promote synergistic effects, defeat multidrug resistance, and minimalize side effects. In this context, pioneering researchers have been trying to develop multi-stimuli-responsive nanogels for the co-encapsulation of hydrophilic and hydrophobic drugs (Figure 3e). Jin et al. synthesized new multi-stimuli-responsive nanogels, allowing for selective release of hydrophobic and hydrophilic drug molecules [104]. The hydrophilic drug was covalently bonded to the nanogel through a redox-sensitive disulfide spacer. The hydrophobic drug was encapsulated into nanogels through hydrophobic interactions. The release of hydrophobic drug was triggered by temperature, pH, and UV light whereas the hydrophilic drug was released in the presence of a redox reagent. Nanogel-based multi-stimuli-responsive dual-drug delivery platforms offer a great potential especially for combination chemotherapy [105].
Tumor site sensitive charge conversional nanoparticles are smart platforms for effective drug delivery and enhanced tumor uptake. Nanogels can be easily converted from a negatively-charged state to a positively-charged state in the tumor extracellular acidic conditions, which promote the efficiency of drug release in the targeted area [106]. Du et al. developed a drug-loaded acid-sensitive charge conversional nanogels to promote drug release and cellular internalization triggered by the extracellular pH [107]. Poly(2-aminoethyl methacrylate hydrochloride) (PAMA) nanogels were prepared and then decorated with 2,3-dimethylmaleic anhydride (DMMA) to create negatively charged nanoparticles. They showed that both the release of the drugs and cellular internalization of the designed nanogels were improved in the tumor extracellular environment.
Although many studies have shown the efficacy and safety of nanogels as potential targeting vehicles, only few nanogel-based drug delivery systems have reached clinical trials. We believe that innovations in the design of nanogels together with the investigation of various parameters, which determine nanogels’ behavior in vivo, e.g. toxicity, clearance, and cost to develop will help to carry them from bench to bedside.
2.5 Dendrimers
Dendrimers are spherical structures of highly branched synthetic polymeric macromolecules with well-defined monodisperse nanostructures having sizes in the range of 1–100 nm [108–110]. The precise controllability over the size and chemistry of dendrimers makes them a platform of choice for an ideal class of drug delivery nanocarriers. The drugs can be physically encapsulated into void spaces or covalently bonded with functional groups on the surface [111].
In general, dendrimer-based drug nanocarriers, introduced in the early 1990s, had problems in their precise control of their drug release at the targeted region [112]. A solution to this limitation is to develop stimuli-responsive dendrimers. Zhu et al. introduced pH-responsive cis-aconityl bond between doxorubicin anticancer drug and PEGylated polyamidoamine (PAMAM) dendrimers for targeted delivery [113]. Doxorubicin-conjugated dendrimers by cis-aconityl pH-cleavable bonds showed prolonged acid-triggered drug release with 5% and 60% of the total DOX over 96 hours at pH 7.4 and 5.5, respectively. Moreover, it was confirmed that pH-responsive cis-aconityl bonds facilitated enhanced cellular uptake and tumor accumulation due to the effective drug release after cellular internalization. Similarly, Guo et al. successfully designed thermosensitive codendrimers using PAMAM and oligoethylene dendron (PAMAM-co-OED) [114]. The lower critical solution temperature (LCST), the critical temperature below which the components of a mixture are miscible for all compositions, of PAMAM-co-OED codendrimers was 38.2 °C, which is higher than normal tissue temperature. When codendrimers reach a cancerous tissue, their drug release rate can be controlled by localized heating, resulting in enhanced therapeutic efficacy [115]. Figure 3f shows an example of a temperature-responsive dendrimer.
The presence of large numbers of terminal end units in the dendrimer networks enables them to achieve multifunctional abilities. Pu et al. immobilized targeting molecules (biotin) and anti-tumor drug doxorubicin on the surface of poly(L-glutamic acid) dendrimers with polyhedral oligomeric silsesquioxane cores via acid-cleavable hydrazone linkage [116]. In vitro and in vivo studies verified that drugs were released in a pH-dependent manner and doxorubicin-dendrimer conjugates had enhanced tumor inhibition activity with lower systemic toxicity.
Although dendrimer-based nanocarriers have made significant improvements over the past two decades, they are rarely used as drug delivery carriers due to the complexity in their synthesis process and the lack of mass production methods [117]. In addition, only small amounts of drugs or targeting moieties can be linked to the dendrimers without altering their properties, such as increased polydispersity and poor water solubility.
Given their biocompatiblity and low toxicity rates, polymeric nanomaterials have a huge market potential and a number of them have recently entered into clinical trials [118]. It is believed that the novel interdisciplinary approaches in chemistry, polymer physics, materials science, bioengineering, and nanotechnology will overcome current challenges and lead to their fast translation into the new clinical applications.
3. Lipid-based nanomaterials
Liposomes are composed of an aqueous core surrounded by a phospholipid bilayer resulting in the formation of amphiphilic and thermodynamically stabilized vesicles [122]. They are mostly made up of single or multiple phospholipids such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine, and phosphatidylglycerol along with a stabilizer, usually cholesterol, to increase their stability [123]. The use of lipid-based nanomaterials for drug delivery dates back to 1965 when Bangham et al. introduced the use of swollen phospholipid systems followed by the introduction of a variety of enclosed phospholipid bilayer structures, which were initially termed “bangosomes” and then “liposomes” [124]. Initially, liposomes were designed with aims such as improving permeation of hydrophilic drugs, protection of peptides/protein-based drugs against harsh conditions, improved bioavailability along with passive delivery of the molecules, and reduced toxicity and side effects to overcome different barriers faced by the drug molecules inside the complex in vivo environment [125, 126]. The targeting ability and drug release rates of liposomes inside the body is dependent on the type of lipids and ligands used, as well as the size, lamellarity, and surface properties as outlined in Figure 4.
Figure 4.

Schematic illustration showing various modifications of liposomes in terms of size, surface ligand, chemical groups, and the trigger for releasing the drug load.
3.1 Liposomal synthesis
The basic structure of the liposomes is very simple and their drug loading efficacy is affected by the method of their synthesis. The properties of liposomes such as their size, stability, drug loading capacity, and release kinetics can be tuned by varying the bilayer composition and the cholesterol content. One of the most notable features of the liposomal systems is their ability to incorporate both hydrophilic and hydrophobic moieties and deliver them to the site of action through extravascular and vascular routes of administration [127, 128]. Hydrophilic drugs are encapsulated in the aqueous core of the liposomes while lipophilic drugs are incorporated in the lipid bilayer and amphiphilic drugs are partitioned at the surface of the bilayers [129]. Liposome synthesis is very complex and involves strategies to avoid problems associated with synthesis, drug retention, release, and stability both in vitro and in vivo due to the composition and fluidic nature of the bilayer membrane and the bilayer sensitivity to temperature variations while interacting with blood components after administration and during long term storage resulting in cargo leakage [130, 131]. Developing novel synthesis techniques to control size, lamellarity, and tunable surface properties improved the potential of liposomes as a promising delivery system. The very first generation of small-size unilamellar vesicles of 100-nm size, large unilamellar vesicles of 200 to 800 nm in diameter, and multilamellar vesicles with a diameter of 500 nm to 5 microns consisting of multiple concentric bilayers, were synthesized between in early 70s [132]. Since the late 1970s methods such as electroformation, freeze-drying, hydration, and double emulsion processes were opteded to fabricate liposomes. Other techniques especially those based on microfluidics have been extensively developed. These techniques emerged in the late 1980s, include extrusion, double emulsion, droplet emulsion transfer, pulsed jetting, transient membrane ejection, and hydrodynamic focusing [133]. Among these, micro-hydrodynamic focusing was used to fabricate monodisperse liposomes with highly controllable size and encapsulation efficiency through buffer-organic phase flow rate ratios [134]. Another recent advancement in microfluidic-based synthesis is the NanoAssemblr platform and the NanoAssemblr scale up platform, which can produce liposomes with high precision and reproducibility both at laboratory and clinical scales [135].
3.2 Chemo-responsive liposomes
Physicochemical properties of the drugs greatly influence their incorporation and retention inside liposomes, which also govern their release profiles [136]. Previously reported loading methods result in low encapsulation efficiency due to limited quantity of the aqueous core since it is difficult to incorporate hydrophilic drugs into a bilayer, which is similar to the cell membrane and thus restricts the permeability of hydrophilic molecules [137]. Similarly, highly lipophilic drugs such as paclitaxel were also difficult to incorporate into liposomes [138]. A transmembrane chemical potential gradient method was developed for overcoming the problems associated with drug loading. Ammonium sulfate-based chemical gradients were used to precipitate the drugs in the inner core of liposome which resulted in high encapsulation efficiency of the drug [139]. Since many of the existing drugs are weak bases by nature, this method greatly improved their loading efficiency. Drug retention was also improved either by selecting drugs that could precipitate inside the liposomes like doxorubicin, or by linking the drugs with polyanions, or by converting acidic drugs to weak basic pro-drugs that could easily be released inside the body.
3.3 Long-circulating liposomes
Another major problem faced by the conventional liposomes is their short circulatory half-life and rapid clearance from the systemic circulation by the mononuclear phagocytic system (MPS) in the liver and spleen or opsonization by plasma proteins [140, 141]. MPS blocking through pre-dosing of empty liposomes, was a technique initially used to improve the circulatory half-life of liposomes [142–144]. Conventional delivery of liposomes relies on endocytic pathway that has certain shortcomings such as limited endosomal escape and slow degradation in lysosomes. Newer methods include the fusion between liposomes and the cell membrane. This is achieved by changing the types of phospholipids, for example, by incorporating egg phosphatidylcholine and sphingomyelin and the addition of cholesterol to the liposome structure. Long-circulating liposomes coated with PEG chains were first described by Allen and Chonn in 1987 [145]. Depending on the length and density of PEG chains over the surface of the liposome, the circulatory half-life of the liposomes can be modified, resulting in the formation of sterically stable or stealth liposomes. These stealth liposomes proved their practical application in the treatment of Kaposi’s sarcoma in HIV patients [146].
3.4 Targeted liposomes
The use of liposomes functionalized with targeting moieties that can adhere to the cells of interest has been extensively explored. Among these targeting moieties, antibodies, folic acid or folates, and transferrin have shown promising applications as they are capable of binding to target cell receptors for targeted delivery for cancer treatment, since many tumor cells overexpress folate and transferrin receptors [147]. In other studies, antibody-labeled liposomes capable of crossing the blood brain barrier and targeting brain tumors as well as peptide-labeled liposomes for anti-angiogenic activity were developed to avoid tumor growth and metastasis [148, 149]. Also, macrophage-targeting liposomes for cardiac repair after myocardial infarction, lymphatic drug delivery and treatment against infectious diseases have also been successfully fabricated.
Therapeutic outcomes of liposomal drug delivery could be improved, if the release is triggered by some intrinsic mechanism or from an external source when they reach the target site. This remote triggering is beneficial in cancer treatment based on various local triggers available at or inside the tumor site. Various triggering mechanisms showing improved outcomes have been investigated and reported. These mechanisms can be classified as physiology-dependent (pH- or enzyme-based) and external stimuli-dependent (such as light, temperature, magnetic field, and ultrasound). The use of pH-sensitive liposomes dates back to 1984 [150]. These liposomes were designed to release their cargo in acidic environments around tumors where the pH drops to around 5.0. These liposomes were also used for delivery in acidic environment of endocytic vesicles. Enzyme-sensitive liposomes rely on the presence of specific enzymes at tumor or inflammation sites. Enzymes such as phospholipase and alkaline phosphatase destabilize the lipid bilayer, resulting in the release of cargo. Thermo-sensitive liposomes were initially introduced by Yatvin and Weinstein in 1978 [151]. These liposomes have a phase transition temperature (TM) below which they are in gel state and stable while they go through fluid phase when the temperature becomes higher than TM, which occurs around 41–42 °C.
Liposomes have also been designed for combination therapy either by simultaneous co-release or sequential independent release of multiple drugs loaded inside. In one particular work, transforming growth factor-β (TGF-β) inhibitor and interleukin-2 (IL-2) were co-loaded in liposomes and were gradually released over 7 days, which significantly delayed tumor growth and increased the survival time of tumor bearing mice [152]. The new generation of liposomes were designed to provide multimodal imaging as well as controlled drug release. Li et al. fabricated a multifunctional liposomal system loaded with doxorubicin, which could be imaged by MRI, NIR fluorescence, positron emission tomography (PET), and single photon emission computed tomography (SPECT) [153].
Enveloped liposomes have been developed for increasing the stability of liposomes with improved oral pharmacokinetics and pharmacodynamics and targeted delivery of anticancer drugs. Liposomes are enveloped with polymeric coats to achieve these aims through oral routes, ocular administration, and intranasal drug delivery.
Most recently, attempts have been made to overcome the immunogenicity related to different nanoparticles by cloaking them with cell membranes of human immune system components such as white blood cells (WBCs), red blood cells (RBCs), as well as platelets, to synthesize biomimetic cloaked nanoparticles or liposome-like vesicles, “leukosomes” [52, 154].
4. FDA approval and clinical trials
Over the last three decades, researchers have been actively exploring the field of nanomedicine to improve the current standard of therapeutics. The rapid progress in the field stems from the immense potential of these nanoparticles to increase the drug abundance at the target site, decrease systemic toxicity, improve drug solubility, and as imaging and contrast agents along with various other clinical advantages. This active development in nanomedicine also demands sustained clinical translation and commercialization of these technologies, currently regulated by the FDA. Over the past two decades, the FDA has approved clinical translation or for human use of numerous nanomaterial-based drugs with over 200 new products in clinical studies. In this section, we discuss the formulation and evolution over the years of those nanomaterials that have passed these regulations and have made it to the commercial market for human use.
Compositions of the nanomaterial-based drug play a vital role in FDA approval. Based on the constituent of these clinically approved nanomedicine and consistency with the above-mentioned divisions in this review, we can categorize them as (i) inorganic, (ii) polymeric, and (iii) liposomal. Polymeric and liposome-based nanomaterials have been a major component of the nanomaterial based drugs in clinical translation with highest FDA-approved drugs. However, over the last decade several inorganic nanomaterial-based drugs, which include nanocrystals and IONPs, have been clinically approved. We discuss their current status and translation in the following section.
4.1 Inorganic
Inorganic based nanomaterials play a vital role in therapeutics for iron deficiency and as contrast agents in imaging. SPIONs have been a core component of inorganic nanomedicine [159]. Feridex (1996), GastroMARK (2001), and Feraheme (2009) are polymer-coated SPIONs that were FDA-approved as MRI contrast enhancers for imaging of cancers. However, in 2008 Feridex and in 2012 GastroMARK were withdrawn due to cytotoxicity [155].
IONPs have also been used for sustained release of iron to replenish deficiency in anemia patients. These nanomedicines have been coated with biodegradable polymers, which allows increased biocompatibility and sustained degradation of the iron core for the treatment of anemia. Dextran-coated SPIONs, Feraheme, approved in 2009 by FDA has been used for the treatment of anemia and chronic kidney disease [160].
IONPs have long been explored for use in cancer therapy due to their paramagnetic property and use as hyperthermia agents in alternating magnetic fields. NanoTherm, 15-nm SPIONs coated with aminosilane, has been recently approved in Europe for selective ablation of glioblastoma and has entered clinical trials in US for localized ablation of prostate cancer [161]. AuroLase is under phase I trial for thermal ablation of metastatic lung tumors. It consists of PEGylated gold-silica nanoshell that is optically active in the NIR region of the spectrum [162]. Recently, Hafnium oxide nanoparticles, NBTXR3, under phase II/III trials, has also shown utility in treating squamous cell carcinoma by electromagnetic radiation-induced cell death when injected locally [163]. The field of inorganic nanomedicine has shown some promisinge advancements with multiple nanomedicine formulations under clinical trials as imaging and for cancer diagnostics, photodynamic therapy of tumor and contrast agents.
4.2 Polymer-based
Polymers have been an important constituent of clinically available nanomaterials. The varied use of polymeric nanomaterials can be associated with the tunability of chemically and biologically derived polymers. In Section 2, we have classified polymeric nanomedicine into various categories and discussed their properties based on their chemical and biological moieties.
Amongst polymers, PEG has been the most diversely used for conjugation with various proteins and peptides. PEGylation of the biological drugs has resulted in a compelling increase in the stability of biologics, reduced immunogenicity, and prolonged circulation time. Adagen, a PEGylated adenosine deaminase enzyme was the first FDA-approved PEGylated drug in 1990 for severe combined immunodeficiency disease [164]. Over the last three decades, FDA has approved numerous PEGylated biologics for treating various diseases. Amongst these, Neulasta (2002), a PEGylated granulocyte-stimulating factor, has been very successful in the clinic for febrile neutropenia [165]. Recently, Plegridy (2014) and Adynovate (2015) have been FDA-approved and are PEGylated biologics for the treatment of multiple sclerosis (MS) and hemophilia, respectively [166, 167]. The field has been continuously progressing with researchers considering more polymers to further increase the stability and half-life of the biologics. For example, zwitterionic polymers have shown promising results over PEG but the research in the field is still in its early stages.
Degradable biopolymers have also shown success in clinical translation over the years. Copaxone, a random polypeptide consisting of L-glutamate, L- lysine, L- alanine, and L-tyrosine with a MW of ~6.4 kDa, was FDA-approved in 1996 for MS. In 2002, FDA approved Eligard, which consists of Leuprolide acetate and PLGA, for prostate cancer treatment [156]. Currently, Opaxio, a biodegradable conjugate of paclitaxel and polyglutamic acid is under clinical phase III trial for drug delivery to head and neck cancer and has been approved for the status of infant drug by FDA[168]. Another paclitaxel-based PEG-co-PLA nanomedicine is under phase III trial, Genexol-PM, has shown lower toxicity in the treatment of non-small cell lung cancer (NSCLC) and metastatic breast cancer [169]. CRLX-101, a cyclodextrin-conjugated camptothecin, is under clinical phase I/II trial for rectal cancer [170]. New targeted polymeric nanomaterials are also being introduced into clinical trials. BIND-014, which constitutes of docetaxel entrapped within a polymeric hydrophobic core and a PEGylated shell that also incorporates prostate-specific membrane antigen moieties, entered phase II/III trials for targeted drug delivery to NSCLC [171].
Protein-drug conjugated nanomedicines have also come up as novel biodegradable delivery vehicles in contrast to chemically synthesized polymers. Abraxane, an albumin-paclitaxel nanoconjugate was approved by FDA in 2005 for efficient drug delivery to breast cancer [172]. This nanoconjugate was shown to have improved drug solubility and efficient tumor delivery. Recently, Abraxane has been approved for treatments of NSCLC (2012) and for late stage pancreatic cancer (2014) [173]. Currently, pharmaceutical companies have been actively working in the field of protein conjugated nanotherapeutics and their primary focus is towards composition and method of administration of the nanomedicine. Recently, ABI-009 entered phase II trials, another albumin-based nanoparticle but bound to rapamycin for treating bladder cancer and cancers with mTOR mutation [174].
4.3 Liposomes
As discussed earlier, liposomes are phospholipid bilayers that serve as a boundary between inner and outer aqueous phase. They have been used as carriers of either encapsulated hydrophilic drugs in the inner aqueous core or hydrophobic drugs entrapped in the phospholipid bilayer. Due to their ability to shield chemically toxic drugs from undesirable effects and reduce their systemic toxicity, they have had tremendous success in clinical trials. Currently, several liposome-based nanomedicine formulations are approved by FDA, as described below.
Liposomal nanomaterials have made an important contribution in cancer therapy and in the treatment of some infectious diseases. FDA approved AmBisome in 1997, which consists of Amphotericin B encapsulated in the liposome for treatment of fungal infections [175]. Arikace, another actively pursued liposome-cloaked Amikacin, is a potent antibiotic for treatment of multidrug resistant gram-negative bacteria. This inhalable nanomedicine completed phase II clinical trials for the treatment of cystic fibrosis patients with pseudomonas lung infection and is actively pursued for approval from the FDA for non-tuberculous mycobacterial lung disease [176].
Liposomal nanomedicine has also been shown to improve the pharmacokinetics of the encapsulated drugs along with an enhanced accumulation of drug near a solid tumor. These properties of liposome nanomedicines can be attributed to the biofunctionalization of liposome with PEG, which increases their circulation time and allowing them to be retained near solid tumors having poor lymphatic drainage – which is attributed to enhanced permeability and retention (EPR) near the tumor site [127]. After Doxil, a PEGylated liposome-based cancer therapeutic introduced in 1995, the field of nanomedicine was actively pursued, which resulted in the subsequent approval of DaunoXome (1996), DepoCyt (1996), Marqibo (2012), and recently approved Onivyde (2015).
The limiting factor in the approval of nanomaterial-based drugs has been their passive targeting abilities based on EPR. As we further understand the physicochemical properties of these nanotherapeutics, active targeting-based nanomedicine has shown promising results in clinical studies. Currently, human epidermal growth factor receptor 2 (HER2)-targeted liposomal nanomedicine MM-302 (Merrimack Pharmaceuticals) is under clinical phase II/III trials. These liposomes are expected to show improved efficacy towards HER2-positive breast cancer and further improve the drug efficacy [177]. MBP-426 and SGT53, other nanotherapeutic agents under clinical phase I/II trials, are assumed to bind to transferrin receptor (TfR) for active targeting of liposomal encapsulated Oxaliplatin and wild-type p53 DNA, respectively [178]. Another liposomal nanomedicine under clinical phase III trial is ThermoDOX, which is a stimuli-responsive and thermosensitive drug delivery vehicle for treatment of hepatocellular carcinoma [179]. These trials will reinforce the need to improve active targeting of liposomal nanomedicines to gain improved drug efficacy and decrease the systemic toxicity of the drug. In a new approach Celator pharmaceuticals is considering combinatorial nanomedicine with CPX-351 under phase III trial. This drug is a synergistic combination of cytarabine and daunorubicin encapsulated in liposome for treatment of acute myeloid leukemia [180].
Challenges in Clinical Approvals
Although many nanomaterial-based drugs have been approved by the FDA, the success rate is not as one would expect. The number is not impressive because the development of methods and standard protocols required for their efficacy, toxicity, and safety testing for clinical use is still in need of standardization [181]. Without accepted scientific approaches for assessing safety, translation of nanomaterial-based therapeutics may prove challenging. There is a need of a systematic approach, for nanoparticle synthesis, material optimization, screening, and identification of novel drug targets to maximize nanomaterials gaining successful clinical translation and ultimately an FDA approval.
A substantial amount of time and effort has been invested by researchers over the years toward development of protocols where reproducible batches of nanoparticles can be synthesized consistently. The inability to translate nanomedicine platforms from academic reports to industrial scaling processes that comply with GMP and regulatory contexts for human use is still a challenge [182]. Several techniques, including particle replication in non-wetting template (PRINT) [183] and coaxial turbulent jet mixer [184] have been introduced in the market, promising efficient bulk nanoparticle synthesis. There is still a need for more robust technologies that could further help in large-scale production, which would support clinical translations.
In addition, controlling in vivo nanoparticle bio-distribution and targeted delivery has posed a major challenge in their clinical translation. Figure 5 shows recent developments in screening and assessing bio-distribution of barcoded nanoparticles in vivo by Dahlman et. al. [185]. Such studies are expected to provide insight on organ distribution of nanoparticles and potentially pave the path towards faster drug trials. Also, the development of organ-on-a-chip platforms, which are biomimetic in vitro models of their human counterparts, in evaluating pharmacokinetics and pharmacodynamics of nanoparticles in pre-clinical trials is an evolving field and will help a great deal in toxicity studies of nanoparticle based therapeutics [186].
Figure 5.

DNA barcoded nanoparticles for high throughput in vivo nanoparticle delivery [185]. (A) Using high-throughput fluidic mixing, nanoparticles are formulated to carry unique DNA barcodes. (B) Many nanoparticles can be formulated in a single day; each nanoparticle chemical structure carries a distinct barcode. Particles are then combined and administered simultaneously to mice. Tissues are then isolated, and delivery is quantified by sequencing the barcodes. In this example, nanoparticle 1 delivers to the lungs, nanoparticle 2 delivers to the liver, and nanoparticle N delivers to the heart. (C) This DNA barcode system enables multiplexed nanoparticle-targeting studies in vivo, improving upon the current practice, which relies on in vitro nanoparticle screening to identify lead candidates. Printed with permission from United States National Academy of Sciences.
Conclusions
The field of nanomedicine has brought forth many exciting new platforms for drug delivery. In addition to inorganic and polymeric nanomaterials, liposome-based nanotherapeutics have garnered much interest over the last few decades for excellent delivery of drugs, proteins, and genetic materials. Numerous nanomaterial-based novel methods have evolved over time for improving the successful clinical translation against plethora of diseases ranging from cancer to genetic disorders. Tremendous amount of scientific and clinical data is currently available and well understood over the years to build a core foundation for optimal design of nanomaterials to tune their bio-physicochemical characteristics.
We believe that the future of nanomedicine lies in advancing multicomponent nanomaterials that target different pathologies at the same time. In most cases, there are complications associated with a chronic disease. It will be an important move forward to have universal nanomedicine platforms that address the main disease together with the medical complications associated with the same. Also, with the introduction of novel stimuli-responsive nanomaterials [187], we envision similar products in the market that will release the encapsulated drugs on demand. Another area that we feel the field might also benefit from is the design of particles that enable a dynamic control over their biodegradation after being introduced into the human body. Moreover, an area of interest that has not seen much progress in the last two decades is drug targeting across the BBB. We foresee conjugation of nanomaterials with novel chemical groups or stimuli-responsive materials to enhance the BBB crossing to address treatment options for various neuronal diseases. In the upcoming years, we also believe the use of nanoparticles to deliver gene-editing tools (e.g. CRISPR/Cas9) [188] safely into cells to be properly established and used efficiently. In addition, organ-on-a-chip systems will likely help expedite interaction, efficacy, and toxicology studies of nanomaterials before going for in vivo investigations or clinical trials [186, 189].
In the near future, we expect an increase in the FDA approvals of nanomedicines for human use. Although researchers have explored many unconventional strategies including discovery of new drug targets and mechanisms, rapid nanomedicine screening, combinatorial therapy, and immunotherapy and immune-modulation along with functionalization of various antibody and targeting moieties, there is still a lacuna in terms of new approvals from FDA. The current research in nanomedicine not only calls for new ideas but also demands researchers to consider long term and structured approach to pave way for successful clinical translation and fast FDA approval. The recommendations put forward by Hofman-Amtenbrink et al. [182] are expected to expedite FDA approvals that will result in an increased footprint of nanomaterials in the pharmaceutical market.
Table 1.
| Name/Type of Nanomaterial | Year of Approval/Disease | Nature of Nanomaterial | Mechanism of Delivery/Targeting |
|---|---|---|---|
| Doxil® (Liposome) | 1995-AIDS/Karposi’s sarcoma, 2005-Ovarian cancer, 2008-Multiple myeloma | Doxorubicin hydrochloride encapsulated in PEGylated stealth liposome (100 nm) | Accumulation of liposome by passive targeting |
| Abelcet® (Lipid-Drug conjugate) | 1995-Fungal infections | 1:1 complex of Amphotericin B with DMPC and DMPG (7:3), ribbon-like structures of a bilayered membrane | Reduce the toxicity of Amphotericin B |
| DaunoXome® (Liposome) | 1996-AIDS/Karposi’s sarcoma | Liposome encapsulating Daunorubicin citrate (45 nm) | Accumulation of liposome by passive targeting and sustained release of Daunorubicin |
| Copaxone® (Polymer conjugate) | 1996 -Multiple sclerosis | Random copolymer of L-lysine, L-tyrosine, L-alanine, and L-glutamate | Polymer with controlled molecular weight, clearance characteristics and due to resemblance to myelin it “decoys” an autoimmune response |
| AmBisome® (Liposome) | 1997-Systemic fungal infections | Liposome encapsulating Amphotericin B (60–70 nm) | Selective release of the drug from liposome to fungal cell with minimal cellular uptake |
| DepoCyt® (Liposome) | 1999, 2007-Lymphomatous malignant meningitis | Liposome encapsulating Cytarabine | Releases the drug into the cerebral spinal fluid which results in extended half-life and prolonged exposure drug retention |
| Visudyne® (Liposome) | 2000-Age-related macular degeneration | Liposome encapsulating Verteporfin | Supports the absorption of verteporfin to lipoproteins which carries it to the eyes where it is activated by shining light |
| Venofer® (Magnetic) | 2000-Iron deficiency in chronic kidney disease | Complex of polynuclear iron (III)-hydroxide in sucrose | Increased and prolonged dosage |
| Renagel® (Polymer conjugate) | 2000-Chronic kidney disease | Poly(allylamine hydrochloride) crosslinked with epichlorohydrin | Binds to dietary phosphate and prevents its absorption |
| PegIntron® (Polymer conjugate) | 2001-Hepatitis C | PEG-conjugated IFNα-2β protein | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| Pegasys® (Polymer conjugate) | 2002-Hepatitis B and C | PEG-conjugated IFNα-2β protein | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| Neulasta® (Polymer conjugate) | 2002-Febrile neutropenia, non-myeloid malignancies, prophylaxis | PEG-conjugated Filgrastim (granulocyte colony-stimulating factor) | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| Eligard® (Polymer conjugate) | 2002-Prostate cancer | Leuprolide acetate incorporated in nanoparticles of PLGH copolymer (DL-lactide/glycolide) | Controlled delivery of payload with longer circulation time |
| Somavert® (Polymer conjugate) | 2003-Acromegaly | PEG-conjugated pegvisomant for injection, an analog of human growth hormone | PEG covalent conjugation increases the stability of GH receptor antagonist. |
| Macugen® (Polymer conjugate) | 2004-Age related macular and neovascular degeneration | PEG-conjugated anti-vascular endothelial growth factor aptamer | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| DepoDur® (Liposome) | 2004-For treatment of chronic pain | Morphine sulfate encapsulated in multi-vesicular liposomes (~20 μm) | Sustained release post the administration in the epidural |
| Abraxane® (Polymer-Drug conjugate) | 2005-Metastatic breast cancer, 2012-Metastatic non-small cell lung cancer, 2013-Metastatic adenocarcinoma of the pancreas | Albumin-conjugated with paclitaxel to form 130nm particle | Albumin is a carrier of endogenous hydrophobic molecules and help endothelial transcytosis of protein-bound and unbound plasma constituents through binding to the cell-surface |
| Mircera® (Polymer conjugate) | 2007-Anemia associated with chronic renal failure in adults | PEG-conjugated erythropoietin receptor activator | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| Cimzia® (Polymer conjugate) | 2008-Crohn’s Disease 2009-Rheumatoid Arthritis 2012-Psoriatic Arthritis 2013-Ankylosing Spondylitis |
PEG-conjugated tumor necrosis factor alpha (TNF-α) inhibitor (Certolizumab) | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| Feraheme™ (Magnetic) | 2009-Deficiency anemia and iron deficiency in chronic kidney disease | Ferumoxytol SPION with polyglucose sorbitol carboxymethylether | Polymeric coating allows sustained release of Fe2+, decreasing number of doses |
| Marqibo® (Liposome) | 2012-Acute lymphoblastic leukemia | Liposome encapsulating Vincristine sulfate (100 nm) | Enhanced efficacy and reduced toxicity of bare drug |
| Plegridy® (Polymer conjugate) | 2014-Multple sclerosis | PEG-conjugated IFNβ-1α | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
| Onivyde® (Liposome) | 2015-Pancreatic cancer | PEG-conjugated liposome nanoparticle encapsulating Irinotecan | Enhanced efficacy, improved circulation time which allows accumulation in tumor site by EPR and reduced toxicity of bare drug |
| Adynovate® (Polymer conjugate) | 2015-Hemophilia | PEG-conjugated antihemophilic factor (recombinant) | PEG covalent conjugation increases the drug hydrodynamic radius, retention time without effecting the target site of protein |
Acknowledgments
The authors acknowledge funding from the National Institutes of Health (AR057837, DE021468, D005865, AR068258, AR066193, EB022403, EB021148) and the Presidential Early Career Award for Scientists and Engineers (PECASE). Y.S.Z. acknowledges the National Cancer Institute of the National Institutes of Health Pathway to Independence Award (K99CA201603). The authors further acknowledge funding from Swiss National Science Foundation (SNSF) to S.H., TUBITAK – Turkey to A.B.O., Henri Benedictus postdoctoral fellowship from Belgian American Educational Foundation (BAEF) and King Baudouin Foundation (KBS) to S.S.
Biographies
 Dr. Shabir Hassan completed his Bachelors in Chemistry, Biology and English from University of Kashmir and his Masters in Biotechnology from University of Pune. After a year of research in molecular biology through LSZGS of UZH/ETH Zurich, he moved to a spectroscopy lab for his PhD. He used ultrafast laser spectroscopy to study protein folding, aggregation and photothermal effects in proteins in real time. After his PhD, he won the Swiss National Science Foundation (SNSF) award to carry out his research at Harvard Medical School in drug and metabolite delivery applications of novel nanoparticles targeting diseases and other medical conditions.
Dr. Shabir Hassan completed his Bachelors in Chemistry, Biology and English from University of Kashmir and his Masters in Biotechnology from University of Pune. After a year of research in molecular biology through LSZGS of UZH/ETH Zurich, he moved to a spectroscopy lab for his PhD. He used ultrafast laser spectroscopy to study protein folding, aggregation and photothermal effects in proteins in real time. After his PhD, he won the Swiss National Science Foundation (SNSF) award to carry out his research at Harvard Medical School in drug and metabolite delivery applications of novel nanoparticles targeting diseases and other medical conditions.
 Gyan Prakash is a research assistant at Whitehead Institute for Biomedical Research. He received his B.Tech in Chemical Science and Technology from Indian Institute of Technology (IITG). He then worked at University of Massachusetts, Amherst and Harvard Medical School on engineering nanoparticle surface for evaluating their bio-physicochemical properties and further understanding the role of hydrophobic moieties on nanoparticles in eliciting immune response. He believes that continuous evolution of antibiotic resistant bacteria and cancer cells confronts the scientific world to evolve current therapeutic strategies by looking at more networked view and finding new drug targets and delivery strategies with a vision for clinical translation.
Gyan Prakash is a research assistant at Whitehead Institute for Biomedical Research. He received his B.Tech in Chemical Science and Technology from Indian Institute of Technology (IITG). He then worked at University of Massachusetts, Amherst and Harvard Medical School on engineering nanoparticle surface for evaluating their bio-physicochemical properties and further understanding the role of hydrophobic moieties on nanoparticles in eliciting immune response. He believes that continuous evolution of antibiotic resistant bacteria and cancer cells confronts the scientific world to evolve current therapeutic strategies by looking at more networked view and finding new drug targets and delivery strategies with a vision for clinical translation.
 Dr. Saghi Saghazadeh received her PhD in Engineering Sciences from Catholic University of Louvain (UCL), Belgium in 2015. Her PhD research was mainly focused on Layer-by-layer Assembled Nanotubes for Drug Delivery Applications for which she received 4 years FRIA scholarship from FNRS-Fund for Scientific Research (Belgium). Awarded with Henri Benedictus Fellowship from Belgian American Educational Foundation (BAEF, Belgium) in 2016, she joined Brigham and Women’s Hospital, Harvard Medical School as a postdoctoral research fellow. Saghi’s research is focused on the interface between material science and life science with a particular interest in polymer chemistry, cell-material interactions, and drug delivery.
Dr. Saghi Saghazadeh received her PhD in Engineering Sciences from Catholic University of Louvain (UCL), Belgium in 2015. Her PhD research was mainly focused on Layer-by-layer Assembled Nanotubes for Drug Delivery Applications for which she received 4 years FRIA scholarship from FNRS-Fund for Scientific Research (Belgium). Awarded with Henri Benedictus Fellowship from Belgian American Educational Foundation (BAEF, Belgium) in 2016, she joined Brigham and Women’s Hospital, Harvard Medical School as a postdoctoral research fellow. Saghi’s research is focused on the interface between material science and life science with a particular interest in polymer chemistry, cell-material interactions, and drug delivery.
 Dr. Ayca Bal Ozturk received her Bachelor, Master and PhD degree in Chemical Engineering from Istanbul University, Turkey in 2006, 2009 and 2015, respectively. In her PhD thesis work she centered on developing the targeted polymeric nanoparticles for the cancer therapy. In 2016, she joined the lab of Prof. Ali Khademhosseini at Harvard Medical School, Harvard-MIT’s Division of Health Sciences and Technology as postdoctoral scientist for one year. Her research interests include mainly nanostructured smart materials, design and synthesis of innovative micro/nanoscale biomaterials as well as regulating stem-cell differentiation with the conclusive purpose of generating tissue-engineered organs, designing the next generation nanoparticles for efficient targeted drug delivery.
Dr. Ayca Bal Ozturk received her Bachelor, Master and PhD degree in Chemical Engineering from Istanbul University, Turkey in 2006, 2009 and 2015, respectively. In her PhD thesis work she centered on developing the targeted polymeric nanoparticles for the cancer therapy. In 2016, she joined the lab of Prof. Ali Khademhosseini at Harvard Medical School, Harvard-MIT’s Division of Health Sciences and Technology as postdoctoral scientist for one year. Her research interests include mainly nanostructured smart materials, design and synthesis of innovative micro/nanoscale biomaterials as well as regulating stem-cell differentiation with the conclusive purpose of generating tissue-engineered organs, designing the next generation nanoparticles for efficient targeted drug delivery.
 Muhammad Farhan Sohail received his Doctor of Pharmacy in 2009 and recently completed his PhD in Pharmacy with specialization in nanomedicine from Quiad-i-Azam University, Islamabad, Pakistan. He has worked as visiting scientist in Biomaterials Innovation Research Center at Brigham and Women’s Hospital, Harvard Medical School. Presently he is working as research associate in Functional Nanomaterials Research Group at LUMS and QAU, Pakistan. His research interest lies in designing biocompatible polymeric, metallic and lipid based functional nanocarriers targeting cancer and infectious disease.
Muhammad Farhan Sohail received his Doctor of Pharmacy in 2009 and recently completed his PhD in Pharmacy with specialization in nanomedicine from Quiad-i-Azam University, Islamabad, Pakistan. He has worked as visiting scientist in Biomaterials Innovation Research Center at Brigham and Women’s Hospital, Harvard Medical School. Presently he is working as research associate in Functional Nanomaterials Research Group at LUMS and QAU, Pakistan. His research interest lies in designing biocompatible polymeric, metallic and lipid based functional nanocarriers targeting cancer and infectious disease.
 Dr. Jungmok Seo is a scientist at the Center for Biomaterials, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. He served as postdoctoral research fellow at Brigham and Women’s Hospital and Harvard Medical School. His research has been focused on the development of functional systems for bio-integrative applications using a nature-inspired approach. Particularly, is interested in the micro-/nano-fabrication, nanomaterial synthesis, and surface engineering for the tissue engineering. He is working on the development of high- throughput cell/tissue stimulation system to investigate the 2D and 3D cellular behaviors and responses under the engineered microenvironment with delivered genes and drugs.
Dr. Jungmok Seo is a scientist at the Center for Biomaterials, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. He served as postdoctoral research fellow at Brigham and Women’s Hospital and Harvard Medical School. His research has been focused on the development of functional systems for bio-integrative applications using a nature-inspired approach. Particularly, is interested in the micro-/nano-fabrication, nanomaterial synthesis, and surface engineering for the tissue engineering. He is working on the development of high- throughput cell/tissue stimulation system to investigate the 2D and 3D cellular behaviors and responses under the engineered microenvironment with delivered genes and drugs.
 Dr. Mehmet R. Dokmeci received his B.S. (with distinction) and M.S. degrees from the University of Minnesota, Minneapolis and his Ph.D. degree from the University of Michigan, Ann Arbor, all in Electrical Engineering. Dr. Dokmeci is currently an Instructor at Brigham and Women’s Hospital, Harvard Medical School. He has also worked at Corning-Intellisense Corporation for 4 years, developing MEMS-based products for the telecommunications and life science industries. He has extensively published in refereed journals and conferences in the areas of BioMEMS, medical devices, biomaterials, and sensors. He is the author of 104 technical journal articles and 112 conference publications.
Dr. Mehmet R. Dokmeci received his B.S. (with distinction) and M.S. degrees from the University of Minnesota, Minneapolis and his Ph.D. degree from the University of Michigan, Ann Arbor, all in Electrical Engineering. Dr. Dokmeci is currently an Instructor at Brigham and Women’s Hospital, Harvard Medical School. He has also worked at Corning-Intellisense Corporation for 4 years, developing MEMS-based products for the telecommunications and life science industries. He has extensively published in refereed journals and conferences in the areas of BioMEMS, medical devices, biomaterials, and sensors. He is the author of 104 technical journal articles and 112 conference publications.
 Dr. Shrike Zhang received his Ph.D. from Georgia Institute of Technology in the Wallace H. Coulter Department of Biomedical Engineering. He is currently an Instructor of Medicine and Associate Bioengineer in the Division of Engineering in Medicine at Brigham and Women’s Hospital, Harvard Medical School, and Wyss Institute for Biologically Inspired Engineering at Harvard University. Dr. Zhang’s research is focused on innovating medical engineering technologies to recreate functional biomimetic tissues, including 3D bioprinting, organs-on-chips, medical devices, biomedical imaging, and biosensing. He has received >20 international, national, or regional awards for recognition of his research excellence.
Dr. Shrike Zhang received his Ph.D. from Georgia Institute of Technology in the Wallace H. Coulter Department of Biomedical Engineering. He is currently an Instructor of Medicine and Associate Bioengineer in the Division of Engineering in Medicine at Brigham and Women’s Hospital, Harvard Medical School, and Wyss Institute for Biologically Inspired Engineering at Harvard University. Dr. Zhang’s research is focused on innovating medical engineering technologies to recreate functional biomimetic tissues, including 3D bioprinting, organs-on-chips, medical devices, biomedical imaging, and biosensing. He has received >20 international, national, or regional awards for recognition of his research excellence.
 Prof. Ali Khademhosseini is Professor of Medicine at Harvard Medical School and Director of the Biomaterials Innovation Research Center at Brigham and Women’s Hospital, Harvard Medical School. He is also an Associate Faculty at the Wyss Institute for Biologically Inspired Engineering and a PI at Japan’s World Premier International-Advanced Institute for Materials Research at Tohoku University where he directs a satellite laboratory. He is recognized as a leader in combining micro- and nano-engineering approaches with advanced biomaterials for regenerative medicine applications. He has received more than 40 awards, authored over 500 journal papers and 50 books/chapters.
Prof. Ali Khademhosseini is Professor of Medicine at Harvard Medical School and Director of the Biomaterials Innovation Research Center at Brigham and Women’s Hospital, Harvard Medical School. He is also an Associate Faculty at the Wyss Institute for Biologically Inspired Engineering and a PI at Japan’s World Premier International-Advanced Institute for Materials Research at Tohoku University where he directs a satellite laboratory. He is recognized as a leader in combining micro- and nano-engineering approaches with advanced biomaterials for regenerative medicine applications. He has received more than 40 awards, authored over 500 journal papers and 50 books/chapters.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heiligtag FJ, Niederberger M. Mater Today. 2013;16:262–271. [Google Scholar]
- 2.Freestone I, Meeks N, Sax M, Higgitt C. Gold Bull. 2007;40:270–277. [Google Scholar]
- 3.Hill DV, Speakman RJ, Glascock MD, Neff H. The Technology of Mesopotamian Ceramic Glazes. ACS Sym Ser. 2007:422–446. [Google Scholar]
- 4.Nat Nanotechnol. 2009;4:781–781. doi: 10.1038/nnano.2009.356. [DOI] [PubMed] [Google Scholar]
- 5.Sandhu A. Nat Nanotechnol. 2006;1:87–87. [Google Scholar]
- 6.Gourley PL. Biotechnol Prog. 2005;21:2–10. doi: 10.1021/bp0498239. [DOI] [PubMed] [Google Scholar]
- 7.Spencer DS, Puranik AS, Peppas NA. Curr Opin Chem Eng. 2015;7:84–92. doi: 10.1016/j.coche.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh R, Lillard JW., Jr Exp Mol Pathol. 2009;86:215–223. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanza GM. Nat Nanotechnol. 2015;10:301–302. doi: 10.1038/nnano.2015.61. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Li B, Ren X, Li S, Ma Y, Cui S, Gu Y. Biomaterials. 2012;33:8461–8476. doi: 10.1016/j.biomaterials.2012.08.034. [DOI] [PubMed] [Google Scholar]
- 11.Shin SR, Li YC, Jang HL, Khoshakhlagh P, Akbari M, Nasajpour A, Zhang YS, Tamayol A, Khademhosseini A. Adv Drug Del Rev. 2016;105(Part B):255–274. doi: 10.1016/j.addr.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nam J, Won N, Bang J, Jin H, Park J, Jung S, Jung S, Park Y, Kim S. Adv Drug Deliv Rev. 2013;65:622–648. doi: 10.1016/j.addr.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Davis FF. Adv Drug Deliv Rev. 2002;54:457–458. doi: 10.1016/s0169-409x(02)00021-2. [DOI] [PubMed] [Google Scholar]
- 14.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayat MA. Colloidal gold: principles, methods, and applications. Academic Press; San Diego: 1989. [Google Scholar]
- 16.Thakor AS, Jokerst J, Zavaleta C, Massoud TF, Gambhir SS. Nano Lett. 2011;11:4029–4036. doi: 10.1021/nl202559p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dreaden EC, Austin LA, Mackey MA, El-Sayed MA. Ther Deliv. 2012;3:457–478. doi: 10.4155/tde.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chithrani BD, Ghazani AA, Chan WC. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 19.Mout R, Moyano DF, Rana S, Rotello VM. Chem Soc Rev. 2012;41:2539–2544. doi: 10.1039/c2cs15294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turkevich J, Stevenson PC, Hillier J. Discuss Faraday Soc. 1951:55-&. [Google Scholar]
- 21.Perala SR, Kumar S. Langmuir. 2013;29:9863–9873. doi: 10.1021/la401604q. [DOI] [PubMed] [Google Scholar]
- 22.Templeton AC, Wuelfing WP, Murray RW. Acc Chem Res. 2000;33:27–36. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]
- 23.Moyano DF, Saha K, Prakash G, Yan B, Kong H, Yazdani M, Rotello VM. ACS Nano. 2014;8:6748–6755. doi: 10.1021/nn5006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi CH, Alabi CA, Webster P, Davis ME. Proc Natl Acad Sci U S A. 2010;107:1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirn S, Semmler-Behnke M, Schleh C, Wenk A, Lipka J, Schaffler M, Takenaka S, Moller W, Schmid G, Simon U, Kreyling WG. Eur J Pharm Biopharm. 2011;77:407–416. doi: 10.1016/j.ejpb.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perche F, Yi Y, Hespel L, Mi P, Dirisala A, Cabral H, Miyata K, Kataoka K. Biomaterials. 2016;90:62–71. doi: 10.1016/j.biomaterials.2016.02.027. [DOI] [PubMed] [Google Scholar]
- 27.Gamrad L, Rehbock C, Westendorf AM, Buer J, Barcikowski S, Hansen W. Sci Rep. 2016;6:28709. doi: 10.1038/srep28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong L, Qiu J, Sun W, Yang J, Shen M, Wang L, Shi X. Biomater Sci. 2017 doi: 10.1039/c6bm00708b. [DOI] [PubMed] [Google Scholar]
- 29.Yu M, Lei B, Gao C, Yan J, Ma PX. Nano Res. 2016;10:49–63. [Google Scholar]
- 30.Tang R, Kim CS, Solfiell DJ, Rana S, Mout R, Velazquez-Delgado EM, Chompoosor A, Jeong Y, Yan B, Zhu ZJ, Kim C, Hardy JA, Rotello VM. ACS Nano. 2013;7:6667–6673. doi: 10.1021/nn402753y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ray M, Tang R, Jiang ZW, Rotello VM. Bioconj Chem. 2015;26:1004–1007. doi: 10.1021/acs.bioconjchem.5b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mout R, Ray M, Yesilbag Tonga G, Lee Y-W, Tay T, Sasaki K, Rotello VM. ACS Nano. 2017 doi: 10.1021/acsnano.7b02884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boisselier E, Astruc D. Chem Soc Rev. 2009;38:1759–1782. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 34.Oldenburg SJ, Averitt RD, Westcott SL, Halas NJ. Chem Phys Lett. 1998;288:243–247. [Google Scholar]
- 35.Lal S, Clare SE, Halas NJ. Acc Chem Res. 2008;41:1842–1851. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- 36.Hassan S, Singh AV. J Nanosci Nanotechnol. 2014;14:402–414. doi: 10.1166/jnn.2014.8747. [DOI] [PubMed] [Google Scholar]
- 37.Meyers PH, Nice CM, Jr, Meckstroth GR, Becker HC, Moser PJ, Goldstein M. Am J Roentgenol Radium Ther Nucl Med. 1966;96:913–921. doi: 10.2214/ajr.96.4.913. [DOI] [PubMed] [Google Scholar]
- 38.Widder KJ, Marino PA, Morris RM, Howard DP, Poore GA, Senyei AE. Eur J Cancer Clin Oncol. 1983;19:141–147. doi: 10.1016/0277-5379(83)90409-1. [DOI] [PubMed] [Google Scholar]
- 39.Lubbe AS, Bergemann C, Huhnt W, Fricke T, Riess H, Brock JW, Huhn D. Cancer Res. 1996;56:4694–4701. [PubMed] [Google Scholar]
- 40.Santhosh PB, Ulrih NP. Cancer Lett. 2013;336:8–17. doi: 10.1016/j.canlet.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 41.Akbarzadeh A, Samiei M, Davaran S. Nanoscale Res Lett. 2012;7 doi: 10.1186/1556-276X-7-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laurent S, Forge D, Port M, Roch A, Robic C, Vander Elst L, Muller RN. Chem Rev. 2008;108:2064–2110. doi: 10.1021/cr068445e. [DOI] [PubMed] [Google Scholar]
- 43.Jordan A, Wust P, Fahling H, John W, Hinz A, Felix R. Int J Hyperthermia. 1993;9:51–68. doi: 10.3109/02656739309061478. [DOI] [PubMed] [Google Scholar]
- 44.Espinosa A, Di Corato R, Kolosnjaj-Tabi J, Flaud P, Pellegrino T, Wilhelm C. ACS nano. 2016;10:2436–2446. doi: 10.1021/acsnano.5b07249. [DOI] [PubMed] [Google Scholar]
- 45.Xu C, Miranda-Nieves D, Ankrum JA, Matthiesen ME, Phillips JA, Roes I, Wojtkiewicz GR, Juneja V, Kultima JR, Zhao W, Vemula PK, Lin CP, Nahrendorf M, Karp JM. Nano Lett. 2012;12:4131–4139. doi: 10.1021/nl301658q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soenen SJ, Parak WJ, Rejman J, Manshian B. Chem Rev. 2015;115:2109–2135. doi: 10.1021/cr400714j. [DOI] [PubMed] [Google Scholar]
- 47.Sakulkhu U, Mahmoudi M, Maurizi L, Salaklang J, Hofmann H. Sci Rep. 2014;4:5020. doi: 10.1038/srep05020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE. Nat Nanotechnol. 2016;11:986–994. doi: 10.1038/nnano.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veiseh O, Tang BC, Whitehead KA, Anderson DG, Langer R. Nat Rev Drug Discov. 2015;14:45–57. doi: 10.1038/nrd4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao Y, Chen Y, Ji X, He X, Yin Q, Zhang Z, Shi J, Li Y. ACS Nano. 2011;5:9788–9798. doi: 10.1021/nn2033105. [DOI] [PubMed] [Google Scholar]
- 51.Lee JE, Lee N, Kim T, Kim J, Hyeon T. Acc Chem Res. 2011;44:893–902. doi: 10.1021/ar2000259. [DOI] [PubMed] [Google Scholar]
- 52.Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E. Nat Nanotechnol. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stober W, Fink A, Bohn E. J Colloid Interface Sci. 1968;26:62. [Google Scholar]
- 54.Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC, Beck JS. Nature. 1992;359:710–712. [Google Scholar]
- 55.Lai CY, Trewyn BG, Jeftinija DM, Jeftinija K, Xu S, Jeftinija S, Lin VS. J Am Chem Soc. 2003;125:4451–4459. doi: 10.1021/ja028650l. [DOI] [PubMed] [Google Scholar]
- 56.Slowing II, Vivero-Escoto JL, Wu CW, Lin VSY. Adv Drug Del Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 57.Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, Monette S, Pauliah M, Gonen M, Zanzonico P, Quinn T, Wiesner U, Bradbury MS, Overholtzer M. Nat Nanotechnol. 2016;11:977–985. doi: 10.1038/nnano.2016.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brus LE. J Chem Phy. 1984;80:4403–4409. [Google Scholar]
- 59.Michalet X, Pinaud F, Bentolila L, Tsay J, Doose S, Li J, Sundaresan G, Wu A, Gambhir S, Weiss S. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He X, Ma N. Colloid Surf B. 2014;124:118–131. doi: 10.1016/j.colsurfb.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 61.Alivisatos AP, Gu W, Larabell C. Annu Rev Biomed Eng. 2005;7:55–76. doi: 10.1146/annurev.bioeng.7.060804.100432. [DOI] [PubMed] [Google Scholar]
- 62.Singh AV, Khare M, Gade WN, Zamboni P. Autoimmune Dis. 2012;2012:160830. doi: 10.1155/2012/160830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cai X, Luo Y, Zhang W, Du D, Lin Y. ACS Appl Mater Interfaces. 2016;8:22442–22450. doi: 10.1021/acsami.6b04933. [DOI] [PubMed] [Google Scholar]
- 64.Yong KT, Law WC, Hu R, Ye L, Liu L, Swihart MT, Prasad PN. Chem Soc Rev. 2013;42:1236–1250. doi: 10.1039/c2cs35392j. [DOI] [PubMed] [Google Scholar]
- 65.Oh E, Liu R, Nel A, Gemill KB, Bilal M, Cohen Y, Medintz IL. Nat Nanotechnol. 2016;11:479–486. doi: 10.1038/nnano.2015.338. [DOI] [PubMed] [Google Scholar]
- 66.Xu G, Zeng S, Zhang B, Swihart MT, Yong KT, Prasad PN. Chem Rev. 2016;116:12234–12327. doi: 10.1021/acs.chemrev.6b00290. [DOI] [PubMed] [Google Scholar]
- 67.Johnson CM, Pate KM, Shen Y, Viswanath A, Tan R, Benicewicz BC, Moss MA, Greytak AB. J Colloid Interface Sci. 2015;458:310–314. doi: 10.1016/j.jcis.2015.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deng D, Chen Y, Cao J, Tian J, Qian Z, Achilefu S, Gu Y. Chem Mater. 2012;24:3029–3037. [Google Scholar]
- 69.Hu CMJ, Fang RH, Luk BT, Zhang L. Nanoscale. 2014;6:65–75. doi: 10.1039/c3nr05444f. [DOI] [PubMed] [Google Scholar]
- 70.Kaur S, Prasad C, Balakrishnan B, Banerjee R. Biomater Sci. 2015;3:955–987. doi: 10.1039/c5bm00002e. [DOI] [PubMed] [Google Scholar]
- 71.Duncan R. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 72.Duncan R. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 73.Vicent MJ, Duncan R. Trends Biotechnol. 2006;24:39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 74.Pang X, Jiang Y, Xiao Q, Leung AW, Hua H, Xu C. J Control Release. 2016;222:116–129. doi: 10.1016/j.jconrel.2015.12.024. [DOI] [PubMed] [Google Scholar]
- 75.Wang W, Wang B, Ma X, Liu S, Shang X, Yu X. Biomacromolecules. 2016 doi: 10.1021/acs.biomac.6b00455. [DOI] [PubMed] [Google Scholar]
- 76.Wu Y, Zhou D, Zhang Q, Xie Z, Chen X, Jing X, Huang Y. Biomacromolecules. 2016;17:2650–2661. doi: 10.1021/acs.biomac.6b00705. [DOI] [PubMed] [Google Scholar]
- 77.Kreuter J. Int J Pharm. 2007;331:1–10. doi: 10.1016/j.ijpharm.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 78.Birrenbach G, Speiser PP. J Pharm Sci. 65:1763–1766. doi: 10.1002/jps.2600651217. [DOI] [PubMed] [Google Scholar]
- 79.Langer R, Folkman J. Nature. 1976;263:797–800. doi: 10.1038/263797a0. [DOI] [PubMed] [Google Scholar]
- 80.Cao Y, Langer R. Proc Natl Acad Sci USA. 2008;105:13203–13205. doi: 10.1073/pnas.0806582105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cagel M, Grotz E, Bernabeu E, Moretton MA, Chiappetta DA. Drug Discov Today. 2016 doi: 10.1016/j.drudis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 82.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. J Control Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 83.Alexis F. Polym Int. 2005;54:36–46. [Google Scholar]
- 84.Gan Q, Wang T. Colloids Surf B. 2007;59:24–34. doi: 10.1016/j.colsurfb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 85.Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z. Adv Drug Del Rev. 2008;60:1650–1662. doi: 10.1016/j.addr.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 86.Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. J Control Release. 2004;100:5–28. doi: 10.1016/j.jconrel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 87.Rajitha P, Gopinath D, Biswas R, Sabitha M, Jayakumar R. Expert Opin Drug Deliv. 2016:1–18. doi: 10.1080/17425247.2016.1178232. [DOI] [PubMed] [Google Scholar]
- 88.Dutta PK. New book series: composite polymer material. Springer; 2016. Chitin and Chitosan for Regenerative Medicine. [Google Scholar]
- 89.Wu J, Tang C, Yin C. Acta Biomater. 2017;47:81–90. doi: 10.1016/j.actbio.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 90.Bader H, Ringsdorf H, Schmidt B. Angew Makromol Chem. 1984;123:457–485. [Google Scholar]
- 91.Cheng C-C, Chang F-C, Kao W-Y, Hwang S-M, Liao L-C, Chang Y-J, Liang M-C, Chen J-K, Lee D-J. Acta Biomater. 2016;33:194–202. doi: 10.1016/j.actbio.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 92.Mao J, Li Y, Wu T, Yuan C, Zeng B, Xu Y, Dai L. ACS Appl Mater Interfaces. 2016 doi: 10.1021/acsami.6b04247. [DOI] [PubMed] [Google Scholar]
- 93.Xu Z, Liu S, Kang Y, Wang M. ACS Biomater Sci Eng. 2015;1:585–592. doi: 10.1021/acsbiomaterials.5b00119. [DOI] [PubMed] [Google Scholar]
- 94.Yu L-Y, Su G-M, Chen C-K, Chiang Y-T, Lo C-L. Biomacromolecules. 2016;17:3040–3047. doi: 10.1021/acs.biomac.6b00916. [DOI] [PubMed] [Google Scholar]
- 95.Huo M, Yuan J, Tao L, Wei Y. Polym Chem. 2014;5:1519–1528. [Google Scholar]
- 96.Chiang Y-T, Yen Y-W, Lo C-L. Biomaterials. 2015;61:150–161. doi: 10.1016/j.biomaterials.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 97.Chang C, Wei H, Wu D-Q, Yang B, Chen N, Cheng S-X, Zhang X-Z, Zhuo R-X. Int J Pharm. 2011;420:333–340. doi: 10.1016/j.ijpharm.2011.08.038. [DOI] [PubMed] [Google Scholar]
- 98.Shi Y, van Nostrum CF, Hennink WE. ACS Biomater Sci Eng. 2015;1:393–404. doi: 10.1021/acsbiomaterials.5b00006. [DOI] [PubMed] [Google Scholar]
- 99.Chung JE, Tan S, Gao SJ, Yongvongsoontorn N, Kim SH, Lee JH, Choi HS, Yano H, Zhuo L, Kurisawa M, Ying JY. Nat Nanotechnol. 2014;9:907–912. doi: 10.1038/nnano.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kabanov AV, Vinogradov SV. Angew Chem Int Ed. 2009;48:5418–5429. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu H-Q, Wang C. Langmuir. 2016 [Google Scholar]
- 102.Chen W, Hou Y, Tu Z, Gao L, Haag R. J Control Release. 2016 doi: 10.1016/j.jconrel.2016.10.032. [DOI] [PubMed] [Google Scholar]
- 103.Soni KS, Desale SS, Bronich TK. J Control Release. 2015 doi: 10.1016/j.jconrel.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jin S, Wan J, Meng L, Huang X, Guo J, Liu L, Wang C. ACS Appl Mater Interfaces. 2015;7:19843–19852. doi: 10.1021/acsami.5b05984. [DOI] [PubMed] [Google Scholar]
- 105.Karimi M, Eslami M, Sahandi-Zangabad P, Mirab F, Farajisafiloo N, Shafaei Z, Ghosh D, Bozorgomid M, Dashkhaneh F, Hamblin MR. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8:696–716. doi: 10.1002/wnan.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Angew Chem Int Ed. 2014;53:12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- 107.Du JZ, Sun TM, Song WJ, Wu J, Wang J. Angew Chem Int Ed. 2010;49:3621–3626. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]
- 108.Tomalia DA, Reyna L, Svenson S. Biochem Soc Trans. 2007;35:61–67. doi: 10.1042/BST0350061. [DOI] [PubMed] [Google Scholar]
- 109.Esfand R, Tomalia DA. Drug Discov Today. 2001;6:427–436. doi: 10.1016/s1359-6446(01)01757-3. [DOI] [PubMed] [Google Scholar]
- 110.Gillies ER, Frechet JM. Drug Discov Today. 2005;10:35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- 111.Hsu HJ, Bugno J, Lee, Hong S. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016 doi: 10.1002/wnan.1409. [DOI] [PubMed] [Google Scholar]
- 112.Zhao Y, Guo Y, Li R, Wang T, Han M, Zhu C, Wang X. Sci Rep. 2016;6 doi: 10.1038/srep28983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhu S, Hong M, Tang G, Qian L, Lin J, Jiang Y, Pei Y. Biomaterials. 2010;31:1360–1371. doi: 10.1016/j.biomaterials.2009.10.044. [DOI] [PubMed] [Google Scholar]
- 114.Guo Y, Zhao Y, Zhao J, Han M, Zhang A, Wang X. Bioconj Chem. 2013;25:24–31. doi: 10.1021/bc300560p. [DOI] [PubMed] [Google Scholar]
- 115.Chung JE, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T. J Control Release. 1999;62:115–127. doi: 10.1016/s0168-3659(99)00029-2. [DOI] [PubMed] [Google Scholar]
- 116.Pu Y, Chang S, Yuan H, Wang G, He B, Gu Z. Biomaterials. 2013;34:3658–3666. doi: 10.1016/j.biomaterials.2013.01.082. [DOI] [PubMed] [Google Scholar]
- 117.Jo H, Jun H-W, Shin J, Lee S. Biomedical Engineering: Frontier Research and Converging Technologies. Springer; 2015. [Google Scholar]
- 118.Devarajan PV, Jain S. Targeted drug delivery: concepts and design. Springer; 2015. [Google Scholar]
- 119.Zhang Y, Yang C, Wang W, Liu J, Liu Q, Huang F, Chu L, Gao H, Li C, Kong D. Sci Rep. 2016;6 doi: 10.1038/srep21225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Suffredini G, East J, Levy LM. Am J Neuroradiol. 2014;35:1246–1253. doi: 10.3174/ajnr.A3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang H, Huang Q, Chang H, Xiao J, Cheng Y. Biomater Sci. 2016;4:375–390. doi: 10.1039/c5bm00532a. [DOI] [PubMed] [Google Scholar]
- 122.Gregoriadis G. Trends Biotechnol. 1995;13:527–537. doi: 10.1016/S0167-7799(00)89017-4. [DOI] [PubMed] [Google Scholar]
- 123.Pattni BS, Chupin VV, Torchilin VP. Chem Rev. 2015;115:10938–10966. doi: 10.1021/acs.chemrev.5b00046. [DOI] [PubMed] [Google Scholar]
- 124.Deamer DW. FASEB J. 2010;24:1308–1310. doi: 10.1096/fj.10-0503. [DOI] [PubMed] [Google Scholar]
- 125.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 126.Patil YP, Jadhav S. Chem Phys Lipids. 2014;177:8–18. doi: 10.1016/j.chemphyslip.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 127.Allen TM, Cullis PR. Adv Drug Deliv Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 128.Slingerland M, Guchelaar H-J, Gelderblom H. Drug Discov Today. 2012;17:160–166. doi: 10.1016/j.drudis.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 129.Eloy JO, de Souza MC, Petrilli R, Barcellos JPA, Lee RJ, Marchetti JM. Colloid Surf B. 2014;123:345–363. doi: 10.1016/j.colsurfb.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 130.Bozzuto G, Molinari A. Int J Nanomed. 2015;10:975–999. doi: 10.2147/IJN.S68861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gabizon A, Papahadjopoulos D. Proc Natl Acad Sci U S A. 1988;85:6949–6953. doi: 10.1073/pnas.85.18.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weissmann G, Bloomgarden D, Kaplan R, Cohen C, Hoffstein S, Collins T, Gotlieb A, Nagle D. Proc Natl Acad Sci U S A. 1975;72:88–92. doi: 10.1073/pnas.72.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carugo D, Bottaro E, Owen J, Stride E, Nastruzzi C. Sci Rep. 2016;6:25876. doi: 10.1038/srep25876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jahn A, Vreeland WN, Gaitan M, Locascio LE. J Am Chem Soc. 2004;126:2674–2675. doi: 10.1021/ja0318030. [DOI] [PubMed] [Google Scholar]
- 135.Balbino TA, Aoki NT, Gasperini AA, Oliveira CL, Azzoni AR, Cavalcanti LP, Lucimara G. Chem Eng J. 2013;226:423–433. [Google Scholar]
- 136.Zucker D, Marcus D, Barenholz Y, Goldblum A. J Control Release. 2009;139:73–80. doi: 10.1016/j.jconrel.2009.05.036. [DOI] [PubMed] [Google Scholar]
- 137.Gubernator J. Expert Opin Drug Deliv. 2011;8:565–580. doi: 10.1517/17425247.2011.566552. [DOI] [PubMed] [Google Scholar]
- 138.Koudelka S, Turanek J. J Control Release. 2012;163:322–334. doi: 10.1016/j.jconrel.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 139.Lasic D, Čeh B, Stuart M, Guo L, Frederik P, Barenholz Y. Biochim Biophys Acta. 1995;1239:145–156. doi: 10.1016/0005-2736(95)00159-z. [DOI] [PubMed] [Google Scholar]
- 140.Juliano RL. Adv Drug Del Rev. 1988;2:31–54. [Google Scholar]
- 141.Ishida T, Harashima H, Kiwada H. Biosci Rep. 2002;22:197–224. doi: 10.1023/a:1020134521778. [DOI] [PubMed] [Google Scholar]
- 142.van Rooijen N, van Kesteren-Hendrikx E. Methods Enzymol. 2003;373:3–16. doi: 10.1016/s0076-6879(03)73001-8. [DOI] [PubMed] [Google Scholar]
- 143.Afergan E, Epstein H, Dahan R, Koroukhov N, Rohekar K, Danenberg HD, Golomb G. J Control Release. 2008;132:84–90. doi: 10.1016/j.jconrel.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 144.Verma JN, Wassef NM, Wirtz RA, Atkinson CT, Aikawa M, Loomis LD, Alving CR. Biochim Biophys Acta. 1991;1066:229–238. doi: 10.1016/0005-2736(91)90191-a. [DOI] [PubMed] [Google Scholar]
- 145.Allen T, Chonn A. FEBS Lett. 1987;223:42–46. doi: 10.1016/0014-5793(87)80506-9. [DOI] [PubMed] [Google Scholar]
- 146.Cainelli F, Vallone A. Biol Targets Ther. 2009;3:385. doi: 10.2147/btt.2009.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang X, Lu W. J Bioequiv Availab. 2016;8:013–014. [Google Scholar]
- 148.Janssen A, Schiffelers R, Ten Hagen T, Koning G, Schraa A, Kok R, Storm G, Molema G. Int J Pharm. 2003;254:55–58. doi: 10.1016/s0378-5173(02)00682-8. [DOI] [PubMed] [Google Scholar]
- 149.Vieira DB, Gamarra LF. Int J Nanomed. 2016;11:5381–5414. doi: 10.2147/IJN.S117210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Connor J, Yatvin MB, Huang L. Proc Natl Acad Sci U S A. 1984;81:1715–1718. doi: 10.1073/pnas.81.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Science. 1978;202:1290–1293. doi: 10.1126/science.364652. [DOI] [PubMed] [Google Scholar]
- 152.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Limon PL. Nat Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li S, Goins B, Zhang L, Bao A. Bioconjug Chem. 2012;23:1322–1332. doi: 10.1021/bc300175d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc Natl Acad Sci U S A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Anselmo AC, Mitragotri S. AAPS J. 2015;17:1041–1054. doi: 10.1208/s12248-015-9780-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Alconcel SNS, Baas AS, Maynard HD. Polym Chem. 2011;2:1442–1448. [Google Scholar]
- 157.Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. Nanomedicine. 2013;9:1–14. doi: 10.1016/j.nano.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Nat Rev Cancer. 2016 doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sun C, Lee JS, Zhang M. Adv Drug Deliv Rev. 2008;60:1252–1265. doi: 10.1016/j.addr.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lu M, Cohen MH, Rieves D, Pazdur R. Am J Hematol. 2010;85:315–319. doi: 10.1002/ajh.21656. [DOI] [PubMed] [Google Scholar]
- 161.Maier-Hauff K, Ulrich F, Nestler D, Niehoff H, Wust P, Thiesen B, Orawa H, Budach V, Jordan A. J Neurooncol. 2011;103:317–324. doi: 10.1007/s11060-010-0389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Maggiorella L, Barouch G, Devaux C, Pottier A, Deutsch E, Bourhis J, Borghi E, Levy L. Future Oncol. 2012;8:1167–1181. doi: 10.2217/fon.12.96. [DOI] [PubMed] [Google Scholar]
- 164.Vellard M. Curr Opin Biotechnol. 2003;14:444–450. doi: 10.1016/s0958-1669(03)00092-2. [DOI] [PubMed] [Google Scholar]
- 165.Med Lett Drugs Ther. 2002;44:44–45. [PubMed] [Google Scholar]
- 166.Ing M, Gupta N, Teyssandier M, Maillere B, Pallardy M, Delignat S, Lacroix-Desmazes S A consortium. Cell Immunol. 2016;301:40–48. doi: 10.1016/j.cellimm.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 167.Hu X, Miller L, Richman S, Hitchman S, Glick G, Liu S, Zhu Y, Crossman M, Nestorov I, Gronke RS, Baker DP, Rogge M, Subramanyam M, Davar G. J Clin Pharmacol. 2012;52:798–808. doi: 10.1177/0091270011407068. [DOI] [PubMed] [Google Scholar]
- 168.Galic VL, Wright JD, Lewin SN, Herzog TJ. Expert Opin Investig Drugs. 2011;20:813–821. doi: 10.1517/13543784.2011.576666. [DOI] [PubMed] [Google Scholar]
- 169.Ahn HK, Jung M, Sym SJ, Shin DB, Kang SM, Kyung SY, Park JW, Jeong SH, Cho EK. Cancer Chemother Pharmacol. 2014;74:277–282. doi: 10.1007/s00280-014-2498-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Keefe SM, Hoffman-Censits J, Cohen RB, Mamtani R, Heitjan D, Eliasof S, Nixon A, Turnbull B, Garmey EG, Gunnarsson O, Waliki M, Ciconte J, Jayaraman L, Senderowicz A, Tellez AB, Hennessy M, Piscitelli A, Vaughn D, Smith A, Haas NB. Ann Oncol. 2016;27:1579–1585. doi: 10.1093/annonc/mdw188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Von Hoff DD, Mita MM, Ramanathan RK, Weiss GJ, Mita AC, LoRusso PM, Burris HA, 3rd, Hart LL, Low SC, Parsons DM, Zale SE, Summa JM, Youssoufian H, Sachdev JC. Clin Cancer Res. 2016;22:3157–3163. doi: 10.1158/1078-0432.CCR-15-2548. [DOI] [PubMed] [Google Scholar]
- 172.Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Int J Nanomedicine. 2009;4:99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Saif MW. J O P. 2013;14:686–688. doi: 10.6092/1590-8577/2028. [DOI] [PubMed] [Google Scholar]
- 174.McKiernan JM, Lascano D, Ahn J, Ghandour R, Pak JS, RoyChoudhury A, Chang S, DeCastro GJ, Desai N. J Clin Oncol. 2015;33:TPS4576–TPS4576. [Google Scholar]
- 175.Walsh TJ, Finberg RW, Arndt C, Hiemenz J, Schwartz C, Bodensteiner D, Pappas P, Seibel N, Greenberg RN, Dummer S, Schuster M, Holcenberg JS. N Engl J Med. 1999;340:764–771. doi: 10.1056/NEJM199903113401004. [DOI] [PubMed] [Google Scholar]
- 176.Clancy JP, Dupont L, Konstan MW, Billings J, Fustik S, Goss CH, Lymp J, Minic P, Quittner AL, Rubenstein RC, Young KR, Saiman L, Burns JL, Govan JR, Ramsey B, Gupta R G. Arikace Study. Thorax. 2013;68:818–825. doi: 10.1136/thoraxjnl-2012-202230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Geretti E, Leonard SC, Dumont N, Lee H, Zheng J, De Souza R, Gaddy DF, Espelin CW, Jaffray DA, Moyo V, Nielsen UB, Wickham TJ, Hendriks BS. Mol Cancer Ther. 2015;14:2060–2071. doi: 10.1158/1535-7163.MCT-15-0314. [DOI] [PubMed] [Google Scholar]
- 178.Camp ER, Wang C, Little EC, Watson PM, Pirollo KF, Rait A, Cole DJ, Chang EH, Watson DK. Cancer Gene Ther. 2013;20:222–228. doi: 10.1038/cgt.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Koning GA, Eggermont AM, Lindner LH, ten Hagen TL. Pharm Res. 2010;27:1750–1754. doi: 10.1007/s11095-010-0154-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lancet JE, Cortes JE, Hogge DE, Tallman MS, Kovacsovics TJ, Damon LE, Komrokji R, Solomon SR, Kolitz JE, Cooper M, Yeager AM, Louie AC, Feldman EJ. Blood. 2014;123:3239–3246. doi: 10.1182/blood-2013-12-540971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Grainger DW, Bae YH. J Control Release. 2015;220:569–570. doi: 10.1016/j.jconrel.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 182.Hofmann-Amtenbrink M, Grainger DW, Hofmann H. Nanomed Nanotechnol Biol Med. 11:1689–1694. doi: 10.1016/j.nano.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 183.Xu J, Wong DH, Byrne JD, Chen K, Bowerman C, DeSimone JM. Angew Chem Int Ed Engl. 2013;52:6580–6589. doi: 10.1002/anie.201209145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lim JM, Swami A, Gilson LM, Chopra S, Choi S, Wu J, Langer R, Karnik R, Farokhzad OC. ACS Nano. 2014;8:6056–6065. doi: 10.1021/nn501371n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dahlman JE, Kauffman KJ, Xing Y, Shaw TE, Mir FF, Dlott CC, Langer R, Anderson DG, Wang ET. Proc Natl Acad Sci U S A. 2017;114:2060–2065. doi: 10.1073/pnas.1620874114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bhise NS, Ribas J, Manoharan V, Zhang YS, Polini A, Massa S, Dokmeci MR, Khademhosseini A. J Control Release. 2014;190:82–93. doi: 10.1016/j.jconrel.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Carling CJ, Viger ML, Huu VA, Garcia AV, Almutairi A. Chem Sci. 2015;6:335–341. doi: 10.1039/c4sc02651a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Staahl BT, Benekareddy M, Coulon-Bainier C, Banfal AA, Floor SN, Sabo JK, Urnes C, Munares GA, Ghosh A, Doudna JA. Nat Biotechnol. 2017;35:431–434. doi: 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhang YS, Zhang Y-N, Zhang W. Drug Discov Today [Google Scholar]