

Abstract
Thalassemia and sickle cell disease (SCD) are disorders of hemoglobin that affect millions of people worldwide. The carrier states for these diseases arose as common, balanced polymorphisms during human history because they afforded protection against severe forms of malaria. These complex, multisystem diseases are reviewed here with a focus on current standards of clinical management and recent research findings. The importance of a comprehensive, multidisciplinary and lifelong system of care is also emphasized.
Keywords: Sickle cell disease, Thalassemia, Thalassaemia, Clinical care, Standard care, Outcome
Introduction
Thalassemia and sickle cell disease (SCD) are disorders of hemoglobin that affect millions of people worldwide. The carrier states for these diseases arose as common, balanced polymorphisms during human history because they afforded protection against severe forms of malaria. These complex, multisystem diseases are reviewed here with a focus on current standards of clinical management and recent research findings. The importance of a comprehensive, multidisciplinary and lifelong system of care is also emphasized.
Thalassemia
Thalassemia is the name for a group of disorders of hemoglobin characterized by diminished or absent production of alpha or beta globin chains, the two protein subunits of the hemoglobin molecule. This inherited disorder derives its name from the Greek “Thalassa” (sea) and “Haema” (blood), referring to its discovery among people living around the Mediterranean Sea. It also occurs in high frequencies in parts of Africa, the Asian sub-continent, southeast Asia and the Middle East. Thalassemia is mainly divided into alpha thalassemia (defective or absent alpha chain synthesis) and beta thalassemia (defective or absent beta chain synthesis). This section on thalassemia will largely focus on the clinical management of these patients with a brief introduction to the molecular biology underlying the different forms of thalassemia.
Molecular Basis and Classification
The alpha globin gene cluster is on chromosome 16 and the beta globin gene cluster is on chromosome 11. These gene clusters include regulatory regions as well as distinct genes for embryonic, fetal (for β-like globins), and adults versions of the α-like and β-like globins. Thalassemic mutations can affect any of these genes, but the most common forms of thalassemia occur due to defects in the α and β globin genes and their related regulatory sequences. The main subtypes of alpha and beta thalassemia are presented in Tables 3.1 and 3.2.
Table 3.1.
Common genotypes and basic classification of alpha thalassemia
| Genotype | α-globin gene numbera | Name | Phenotype |
|---|---|---|---|
| αα/αα | 4 | Normal state | None |
| αα/α– | 3 | Silent carrier | None (values for Hb and MCV may be near the lower limits of normal) |
| – –/αα or α–/α– | 2 | Thalassemia trait | Thalassemia minor: asymptomatic, mild microcytic anemia |
| – –/α– | 1 | Hb H disease | Thalassemia intermedia: mild to moderate microcytic anemia |
| – –/αCSα | 1 | Hb H-Constant Spring | Thalassemia intermedia: moderate to severe microcytic anemia |
| – –/– – | 0 | Alpha thalassemia major | Thalassemia major: hydrops fetalis |
Number of normal alpha globin genes
Abbreviations: αα a chromosome with a normal complement (2) of α-globin genes, α– a chromosome with 1 of the 2 α-globin genes deleted, – – a chromosome with both α-globin genes deleted, αα a chromosome with the thalassemic Hb Constant Spring mutation affecting 1 of the 2 α-globin genes
Table 3.2.
Common genotypes and basic classification of beta thalassemia
| Common genotypes | Name | Phenotype |
|---|---|---|
|
| ||
| β/β | Normal | None |
|
| ||
| β/β0 | Beta thalassemia trait | Thalassemia minor: asymptomatic, mild microcytic hypochromic anemia |
| β/β+ | ||
|
| ||
| β+/β+ | Beta thalassemia intermedia | Variable severity |
| β+/β0 | Mild to moderate anemia | |
| βE/β+ | Possible extramedullary hematopoiesis | |
| βE/β0 | Iron overload | |
|
| ||
| β0/β0 | Beta thalassemia major (Cooley’s Anemia) | Severe anemia |
| Transfusion dependence | ||
| Extramedullary hematopoiesis | ||
| Iron overload | ||
Abbreviations: β0 a β-thalassemia mutation that eliminates globin transcription or translation, β+ a β-thalassemia mutation that decreases globin transcription or translation, βE the β-globin mutation that results in Hb E
Alpha thalassemia is usually caused by deletions of one or more of the 4 alpha globin genes (2 on each chromosome). Small deletions removing one alpha globin gene (α+) are more commonly seen in people of African ancestry. Larger deletions of DNA removing both alpha globin genes (α0) on each chromosome are more commonly seen in Southeast Asia. Alpha globin point mutations and structural variants are less common but well-known forms of alpha thalassemia.
Beta thalassemia results mostly from over 200 point mutations, but thalassemic deletions also occur less commonly. These point mutations affect gene regulation and expression to varying degrees [1]. Beta globin is normally synthesized from two beta globin genes (one on each copy of chromosome 11). If a mutation abolishes the expression of beta-globin, it is classified as a beta-zero (β0) thalassemia allele, while reduced or partial expression is classified as a beta-plus (β+) thalassemia allele. As shown in Table 3.2, clinically important thalassemia variants can also occur due to interactions of structural beta globin variants (especially Hb E, βE) with β0 or β+ thalassemia mutations.
The Globin Gene Server (http://globin.cse.psu.edu) database of hemoglobin variants continues to be updated on a regular basis with newly identified mutations [2].
Clinical Presentation
The clinical phenotype or severity varies considerably, mainly depending on the type and number of genetic defects of the alpha or beta globin genes and regulatory regions.
In alpha thalassemia, the clinical severity is primarily based on the number of alleles affected (Table 3.1). Many forms of alpha thalassemia are asymptomatic or mild. Individuals who have the alpha thalassemia silent carrier state (α-/αα) or trait (--/αα or α-/α-) are asymptomatic. Thalassemia trait produces microcytosis, hypochromia and mild anemia. These individuals do not encounter any problems, but awareness of their condition is important to prevent injudicious use of iron therapy to treat their anemia and facilitate genetic and reproductive counseling. Most patients with classical hemoglobin (Hb) H disease (--/α-) are generally transfusion-independent except occasional need during periods of illnesses or surgery. A form of Hb H disease that is more severe is Hb H-Constant Spring. In this variant, a non-deletional alpha globin gene that disrupts the stop codon is co-inherited with a two-gene deletion alpha thalassemia (--/αCSα) (Table 3.1). This is an important cause of Hb H disease, especially in Southeast Asia [3]. These patients are more likely to be transfusion-dependent and have other clinical complications (reviewed below). Homozygous alpha thalassemia major (--/--) produces a hydrops fetalis phenotype.
In beta thalassemia, the clinical severity is also a function of the number and type (β0 or β+) of genetic defects. Beta chain deficiency results in an excess of unbound alpha globin chains (due to lack of beta chains to pair with to make Hb A; α2β2). These excess alpha globin chains precipitate within and damage erythroid precursors and red blood cells causing ineffective erythropoiesis and hemolysis, respectively [4, 5]. Patients with beta thalassemia trait or minor (β/β+ or β/β0) are asymptomatic and have a mild, hypochromic, microcytic anemia. Patients with beta thalassemia intermedia have a quite variable phenotype that ranges between the asymptomatic (minor) phenotype and the transfusion-dependent (major) phenotype. Much of this variability depends on the particular combination of mutations (e.g., β+/β+, β0/β+, βE/β+, βE/β0) and alpha globin gene number. Thalassemia intermedia patients typically present around 4–6 years of age with mild to moderate anemia and require intermittent transfusions, but less often than patients with thalassemia major. Children with beta thalassemia major present in infancy, corresponding to the decline in fetal hemoglobin production in infancy. The typical presentation is failure to thrive, progressive anemia that becomes transfusion-dependent, and hepatosplenomegaly as a manifestation of extramedullary hematopoiesis.
It is important to note that thalassemia mutations can be co-inherited with other mutations that cause thalassemia or other types of hemoglobinopathies. As such, it is necessary to know the alpha globin and beta globin genotypes in all patients with thalassemia, because the phenotype depends on the joint interaction of the alpha and beta globin genotypes. For example, the phenotype of patients with a single beta thalassemia mutation is made more severe by the coinheritance of alpha thalassemia triplications. The sickle-β-thalassemia syndromes are forms of SCD discussed elsewhere in this chapter.
Diagnosis
Newborn screening is an important way to identify thalassemia, especially in high-risk populations, before symptoms appear. Common methods of diagnosis in the newborn and later life are the Hb separation techniques, such as gel-based electrophoresis (especially isoelectric focusing), high-performance liquid chromatography, and capillary electrophoresis. A diagnosis of alpha thalassemia is often inferred by Hb separation techniques by decreased levels of Hb A2 or decreased ratios of a variant beta-globin to normal Hb A (e.g., decreased Hb S: Hb A ratio). The presence of Hb Barts in the newborn period, Hb H, or Hb Constant-Spring specifically indicate the presence of alpha thalassemia. Beta thalassemia is often inferred by raised levels of Hb A2 of Hb F. The absence of Hb A indicates homozygous or compound heterozygous beta thalassemia major.
Given the genetic complexity of thalassemia and the limitations of Hb separation techniques, DNA-based testing is key to identify the specific mutations underlying alpha and beta thalassemia. Newer techniques to identify fetal DNA in maternal circulation can permit prenatal diagnosis. Such screening can be beneficial to prevent hydrops fetalis and related maternal complications [6].
Preventive Care and Screening Measures (Table 3.3)
Table 3.3.
Standard care and screening guidelines for thalassemia intermedia and major
| Assessment | Ages | Frequency | Comments |
|---|---|---|---|
| Bone mineral density | ≥10 | Yearly | DEXA scan or quantitative CT |
| Tanner stage | 10–20 | Yearly | Perform yearly starting at age 10 and continuing until breast or gonadal Tanner Stage 5 or age 20. |
| Liver iron content | All | Yearly | MRI method (R2, T2*) or liver biopsy |
| Cardiac T2* | ≥ 10 | Yearly (see comment) | To be performed when available for patients with biochemical evidence of iron overload or age 10 years or older |
| Cardiac studies | ≥ 10 | Yearly | Echocardiography and/or cardiac function by MRI; indicate the need to assess for pulmonary hypertension when ordering an echocardiogram |
| CBC plus differential | All | Yearly (minimum) | If transfused, preceding each transfusion |
| Blood chemistries | All | Yearly | BUN, creatinine, calcium, magnesium, phosphorus, and zinc |
| LFTs | All | Yearly | ALT, AST, total bilirubin, albumin |
| Ferritin | All | Yearly (minimum) | If transfused, preceding each transfusion |
| HIV | All | Yearly | Only for transfused participants starting at the first transfusion |
| Hepatitis testing | All | Yearly | Only for transfused participants starting at the first transfusion. HBV serology. HCV serology and PCR |
| Plasma ascorbate | All | Yearly | 12 h fast |
| Serum glucose | ≥10 | Yearly | 12 h fast |
| Endocrine panel I | ≥6 | Yearly | TSH, free T4, parathyroid hormone, 25-hydroxy vitamin D, and 1,25 dihydroxy vitamin D levels |
| Endocrine panel II | ≥10 | Yearly | Testosterone (males only), FSH and LH (males and females), and estradiol (females only) |
| Ophthalmology and Audiology testing | Children and Adults | Yearly | All patients undergoing chelation. Auditory testing necessary for those on desferroxamine |
Growth and Development
Children with thalassemia intermedia and major can have growth abnormalities from chronic anemia, a hypermetabolic state, endocrine abnormalities, poor nutrition, and iron overload, among others. Short stature can be seen in over 30% of children with thalassemia major [7, 8]. Assessment of growth with height velocity, weight, along with pubertal assessment during clinic visits is essential. This should be at a minimum of every 6 months. Involvement of nutritionist will be beneficial.
Endocrine Studies
Abnormalities involving gonadal function, glucose intolerance, hypoparathyroidism and hypothyroidism have been noted with increasing frequency. Many of these endocrine abnormalities are the consequence of iron overload. This necessitates monitoring of pituitary hormones, along with testing for thyroid function, glucose tolerance and micronutrients (calcium, folate, Vitamin D, phosphate). Annual monitoring of these is recommended and, if abnormal, it warrants involvement of an endocrinologist for expert care.
Iron Monitoring
Iron loading rate and iron burden are monitored based on volume of pRBC in mL/kg/year transfused, serum ferritin and liver Iron concentration. Serum ferritin is an inexpensive and easily monitored marker of body iron stores, but it underestimates the body iron and liver iron concentration [9]. It is also increased in states of inflammation and suppressed by deficiency of vitamin C. Serum ferritin should be tested every 3 months to monitor the trend among chronically transfused patients and less frequently in patients who receive fewer transfusions. Liver Iron concentration (LIC) of ≥5 mg/g dry weight is associated with increased morbidity and mortality in patients with thalassemia [10, 11]. MRI methods to measure LIC have largely supplanted serial liver biopsies. Regular monitoring of LIC (at least yearly for chronically transfused patients) will inform the initiation and intensity of iron chelation therapy. Serum ferritin and/or LIC do not reliably predict the cardiac iron overload. Non-transferrin bound iron and labile plasma iron are other markers being studied to correlate with the toxicity of circulating iron [12].
Cardiac Studies
Cardiac pathology, especially due to cardiac iron overload, has become the most common cause of death in thalassemia. Cardiac iron burden and risk of heart failure can be estimated by MRI-based techniques (e.g., T2*) [13]. Cardiac T2* measurements are recommended at least yearly in chronically transfused patients, and less frequently in non-transfusion dependent patients. Cardiac MRI screening should be commenced around 8–10 years of age, or sooner if there is concern for severe iron overload, especially if compliance with chelation is poor. Monitoring of ventricular function is also important and can be done by functional cardiac MRI, echocardiography or both. Pulmonary hypertension has also been reported in patients with thalassemia, which is thought to arise from iron overload and factors such as splenectomy, thrombocytosis and activation of coagulation factors. Monitoring for pulmonary hypertension can be done by echocardiography, and regular transfusion therapy, iron chelation, and hydroxyurea have shown to be protective against pulmonary hypertension [14].
Infection
While screening and closer monitoring have helped in preventing morbidity and mortality due to iron overload, infection-related deaths in developing and developed countries still occur. Some of the important risk factors include chronic transfusion therapy, use of central venous lines, iron overload, splenectomy and immune abnormalities [8, 15]. Hepatitis C still remains one of the important transfusion related infection in thalassemia patients in North America, as the incidence of HIV and Hepatitis B have reduced [16]. Yearly screening for HIV and viral hepatitis infections is necessary for regularly transfused patients.
Bone Mineral Density
Reduced bone mineral density (Z-score ≤ 2.0) occurs in over 30% of individuals with thalassemia syndromes despite adequate transfusion therapy. Greater age, males with Collagen type 1A polymorphism, endocrine abnormalities, lower weight, poor nutrition, reduced physical activity and increased bone turnover are some of the risk factors [17, 18]. Annual DEXA scans will help diagnose and institute early management.
Ophthalmologic and Auditory Screening
Children and adults on iron chelation therapy are recommended to get yearly eye assessments whilst on iron chelation therapy though it is unclear if lens opacities are due to underlying thalassemia or iron chelation [19]. Yearly audiograms are also recommended for patients receiving chelation therapy, especially with deferoxamine.
Medical and Surgical Management of Patients with Thalassemia
Current, most effective, and evidence-based management strategies will be highlighted here, including chronic transfusion therapy, iron chelation, induction of fetal Hb, and splenectomy.
Transfusion Therapy
An early and appropriate transfusion program is a pillar of care for children with beta thalassemia major and in some children with beta thalassemia intermedia. Transfusion is beneficial because it alleviates symptomatic anemia, suppresses ineffective erythropoiesis, improves growth and development, reduces iron loading from increased gastrointestinal absorption, and prolongs life [16, 20, 21].
The decision to begin chronic transfusion therapy is often based on clinical characteristics due to complex and incompletely understood genotype-phenotypes correlations in thalassemia [22]. Chronic transfusion is considered in thalassemia patients when the hemoglobin concentration is consistently below 6–7 g/dL, the anemia is symptomatic, quality of life is poor, growth and development is faltering, or there is troublesome extramedullary hematopoiesis. Early initiation of transfusions may also minimize the risk of alloimmunization [20, 23].
The usual transfusion goal is to maintain a pre-transfusion hemoglobin level between 9 and 10 g/dL [20, 21, 24]. Once this goal is reached, transfusions can usually be given every 3–4 weeks, typically using 8–15 mL/kg of packed red blood cells (PRBC). Leukodepleted PRBCs should be used to minimize febrile non-hemolytic transfusion reactions and lessen the risk of cytomegalovirus transmission and alloimmunization. Irradiation of blood products is not necessary unless hematopoietic stem cell transplant is planned in the near future. A central venous catheter can facilitate transfusions.
Detailed documentation of pre-transfusion hemoglobin levels, transfusion reactions, alloimmunization, and volume of all PRBCs received is necessary for high quality care. While chronic (and intermittent) transfusions have remarkably improved the survival and quality of life, of individuals with thalassemia, transfusions do come at a cost. In developing countries, the transfusion-transmitted infections (primarily hepatitis B and C) remain an important cause of morbidity and mortality, and transfusional hemosiderosis affects all transfused individuals.
Fetal Hemoglobin (Hb F) Inducing Agents
In beta thalassemia, increased gamma globin chain synthesis (that is, increased Hb F production) decreases the degree of alpha chain to beta-like chain imbalance. This is associated with increased red cell life span, decreased ineffective erythropoiesis and hemolysis, and thereby decreases the severity of beta thalassemia [25, 26]. Several genetic polymorphisms are associated with high Hb F levels. Pharmacologic agents can also augment Hb F production, such as demethylating agents histone deacetylase (HDAC) inhibitors, and hydroxyurea.
Demethylating Agents
5-azacytidine and decitabine are cytidine nucleoside analogs that become incorporated into DNA where they sequester DNA methyltransferases (DNMTs) and result in DNA hypomethylation, thereby leading to the expression of previously silenced genes, such as the gamma globin genes. Since Ley and DeSimone first reported the use of 5-azacytidine, a cytosine analogue in a patient with severe beta thalassemia, multiple smaller cohort studies have since been reported [27]. Initial studies identified the potential for toxicity, including mutagenicity, myelosuppression, and others. More recent studies using decitabine (5-aza-2′-deoxycytidine), an analogue of azacytidine, have shown clinical benefits with better safety profile. A pilot study in 2011 by Oliveri et al. for Thalassemia Clinical Research Network treated 5 patients with beta thalassemia intermedia with subcutaneous decitabine given at 0.2 mg/kg two times per week for 12 weeks. They noticed an increase in total hemoglobin from 7.88 to 9.04 g/dL and absolute fetal hemoglobin from 3.64 to 4.29 g/dL. They also reported favorable changes in red blood cell indices of hemolysis. Major side effects noted was an elevation in platelet count [28]. This is an additional concern for thalassemia patients post-splenectomy, who have thrombocytosis and a higher risk for venous and arterial thromboembolism.
Histone Deacetylase (HDAC) Inhibitors
Butyrates and short chain fatty acid derivatives regulate gene expression by inhibition of HDAC, which increases core histone acetylation, modifies chromatin structure, and affects the rate of transcription. Perrine and colleagues demonstrated that butyrates could activate gamma globin gene expression. This discovery was predicated on the observation that infants born to mothers with diabetes had high levels of butyrates and a delayed fetal to adult hemoglobin switch. A subsequent study of butyrates in patients with thalassemia and SCD patients demonstrated increases in fetal Hb production [25]. Though multiple smaller studies have shown some benefit, it is thought that butyrate, in addition to inducing gamma globin expression, also increases alpha globin expression in thalassemia, thus minimizing the beneficial effects in beta thalassemia. This is contrary to the effect seen in SCD where in addition to augmentation of gamma globin mRNA levels, the alpha globin mRNA levels decreased in response to butyrate [29]. Among other newer and ongoing pilot studies, an oral butyrate derivative (HQK-1001) that does not have HDAC2 inhibitory activity has been shown to stimulate gamma globin expression [30].
Erythropoietin
Erythropoietin preparations have shown to increase total hemoglobin levels, thereby decreasing transfusion requirements. While its use may be beneficial in beta thalassemia intermedia [31], inability of erythropoietin or darbepoetin to increase fetal hemoglobin might limit its use in beta thalassemia major patients [32]. There is potential for its use along with hydroxyurea in patients with low erythropoietin levels [21].
Hydroxyurea
Hydroxyurea is a ribonucleotide reductase inhibitor that can induce Hb F production [33]. Hydroxyurea has been shown to increase the total hemoglobin level in beta thalassemia intermedia. A long term study in Iran of its use in patients with beta thalassemia showed dramatic response in 149 of 163 patients with 83 of 106 transfusion-dependent patients becoming transfusion-free [34]. The retrospective OPTIMAL CARE study in spite of its limitations, noted some decrease in complication rates if hydroxyurea was used along with transfusion and iron chelation in thalassemia intermedia patients [35]. Unlike its benefits in SCD where improved red blood cell deformability, reduced oxidative stress and reduced hemolysis were noted, its beneficial effects in thalassemia is limited [10].
Hydroxyurea is often started at a dose of 8–10 mg/kg/day and escalated as tolerated to 20 mg/kg/day. Predictors of good response to hydroxyurea are younger age, higher pretreatment hemoglobin level, associated alpha thalassemia, Hb E/beta thalassemia, and history of splenectomy [36].
Iron Chelation Therapy
Iron overload occurs from chronic transfusion therapy and from increased gastrointestinal absorption in thalassemia patients with ineffective erythropoiesis. Poorly transfused individuals can absorb around 3–5 mg/day or more of iron through their gut. Hemolysis and chronic hypoxia can further increase this gut absorption by decreasing hepcidin. Each 100–200 mL/kg body weight of PRBC delivers about 116–232 mg of iron/kg body weight/year. Therefore, it is transfusion therapy that most rapidly increases the iron overload.
Generally, chelation is initiated for transfusion-dependent thalassemia patients between 2 and 4 years of age, after 200 mL/kg of PRBC have been transfused, with a serum ferritin level > 1000–1500 ng/mL and an LIC >3–7 mg Fe/g dry weight as measured by liver biopsy or by noninvasive hepatic R2 or T2* MRI methods [12, 21]. The regimen of any chelator should be adjusted individually to achieve an iron excretion rate that is commensurate with the rate of ongoing iron loading.
There are three main iron chelators in clinical practice: subcutaneous (or intravenous) deferoxamine, oral deferiprone and oral deferasirox (Table 3.4). Current recommendations in children are to begin with an oral chelator such as deferasirox due to its advantages of high oral bioavailability, longer half-life of 12–18 h enabling once daily dosing, and ability to better chelate cardiac iron. Adverse effects like skin rash, nausea, and diarrhea can be mitigated by starting at 20 mg/kg per day and slowly escalating up to 30–40 mg/kg/day. Serum creatinine, proteinuria and liver function should be monitored monthly during deferasirox treatment.
Table 3.4.
Iron chelators
| Agent | Route half-life | Classificationa | Dose and regimen | Common or typical adverse effects |
|---|---|---|---|---|
| Deferoxamine | IV or SQ 8–10 min |
Hexadentate | 30–40 mg/kg/day over 8–12 h 5–7 day/week |
Anaphylaxis Local irritation Retinopathy Hearing loss Renal dysfunction Hepatic dysfunction |
| Deferasirox | Oral 12–18 h |
Tridentate | 20–40 mg/kg/day Once daily |
Diarrhea Rash Proteinuria Renal dysfunction Hepatic dysfunction− |
| Deferiprone | Oral 1.5–4 h |
Bidentate | 75–100 mg/kg/day 3 divided doses daily |
Arthralgia Arthropathy Agranulocytosis Renal dysfunction Hepatic dysfunction |
Indicates the number of coordination sites of the Fe atom that each molecule of the chelator can bind with; Fe has 6 coordination sites
Transfusion history and serum ferritin are not reliable indicators for iron overload in children with non-transfusion dependent thalassemia intermedia [37]. Non-transfusion dependent thalassemia patients have increased enteral absorption of iron that is not accounted for when considering only transfusions as a source of iron loading. Also, serum ferritin is known to underestimate iron burden in thalassemia intermedia. The THALASSA study investigated the use of deferasirox at 5–10 mg/kg/day in 166 patients with non-transfusion-dependent thalassemia and iron overload (LIC ≥5 mg Fe/g dry weight and serum ferritin levels >300 ng/mL). Compared to placebo, deferasirox significantly reduced LIC, mean serum ferritin levels with a manageable toxicity profile [12, 38].
Combination therapy with deferoxamine and deferasirox or deferiprone has been reported as an option for patients requiring rapid reduction in their cardiac and/or systemic iron burden [12].
Splenectomy
Splenomegaly with hypersplenism is a common problem in thalassemia intermedia and major. The main cause of splenomegaly is extramedullary hematopoiesis. Due to hypersplenism, chronic hemolysis is exacerbated, thereby worsening the anemia and increasing transfusion requirements. Splenectomy is indicated in a small group of patients with annual transfusion requirements exceeding 200–250 mL/kg, with hypersplenism with or without the presence of splenomegaly-related complications such as cytopenias, pain or risk of rupture [21, 23]. Splenectomy should only be strongly considered if it is likely to improve anemia and minimize extramedullary hematopoiesis and growth failure. In addition to risk of post-splenectomy sepsis, multiple studies have highlighted an increased risk of other post-splenectomy complications in thalassemia, including pulmonary hypertension, heart failure, thrombosis, cholelithiasis, leg ulcers, osteoporosis, brain infarcts, and others [23, 35, 39]. Therefore, the benefits of splenectomy should we weighed against the increased risk of post-splenectomy complications. Overwhelming post-splenectomy infection is a clearly documented risk, and splenectomy should be avoided in children less than 5 years of age, when possible, because pneumococcal bacteremia is more common in young children. Immunizations prior to surgery are necessary against pneumococcus, Haemophilus influenzae type b, and meningococcus. Post-splenectomy antibiotic prophylaxis should be prescribed for at least several years after splenectomy (if not life-long).
Cure
Hematopoietic stem cell transplantation and gene therapy are discussed in other chapters in this book.
Long Term Outcomes
The life expectancy of patients with thalassemia has improved dramatically over the past 50 years (Fig. 3.1). Improved survival has mainly been achieved by early diagnosis of the disease, intensified transfusion regimens (“hyper-transfusion”), early introduction (especially in the first decade of life) and intensification of chelation therapy, and reduction or elimination of HCV and HIV infection. In developed nations, complication-free survival rates higher than 90% have been documented in children and young adults [40]. Early mortality has not been eliminated, and cardiac disease (heart failure, dysrhythmia, and myocardial infarction) is the most common cause of death. Other common morbidities, and less common causes of mortality, include infection, cirrhosis, thrombosis, malignancy, diabetes, and other endocrinopathies. Many of these conditions, especially cardiac disease, are directly related to iron overload, highlighting the need for adequate iron chelation therapy. Even despite good, contemporary therapy, health-related quality of life is decreased in patients with thalassemia, indicating that ongoing improvements in therapy and support are needed [40–42].
Fig. 3.1.

Improving survival in thalassemia major. Complication-free survival for patients with thalassemia major cared for in Italy is shown by birth cohort (1970–74, 1975–79, 1980–84, 1985–97). From: Borgna-Pignatti C, et al. Ann N Y Acad Sci. 2005;1054(1):40–7. Reprinted with permission from John Wiley and Sons
In conclusion, while significant disparities remain between developed and developing nations, improvements in outcomes over time along with continued research into better therapies with potential for cure–gene therapy and HSCT--will certainly change the face of this common genetic disease.
Sickle Cell Disease
Sickle cell disease (SCD) is the name for a group of disorders caused by a structural abnormality of hemoglobin (Hb) called sickle Hb (Hb S). Since the discovery of sickle-shaped cells in 1910 by Herrick [43], many advances have been made in understanding the genetics and pathophysiology of SCD which is now beginning to guide the development of specific treatments for this condition. SCD is widespread amongst people of African descent as well as in the Middle East, Indian subcontinent, and other Mediterranean regions. This section on SCD will largely focus on the diagnosis and standard clinical management of patients and the associated complications.
Molecular Basis, Pathophysiology and Diagnosis
Hb S is the result of a single amino acid substitution (glutamic acid to valine) at the sixth position of the mature beta globin protein, the gene for which is on chromosome 11. This abnormal hemoglobin is inherited in an autosomal recessive fashion, and SCD can occur due to homozygosity for the Hb S mutation (βS/βS) or from compound heterozygous states with certain other β-globin variants (βS/βC, βS/β0 or βS/β+) (Table 3.5).
Table 3.5.
Common forms of SCD in order of decreasing relative frequency
| Genotype | Name | Main Hbs | Hb (g/dL)a | MCV (fL)a,b | Retic (%)a | Severityc | |
|---|---|---|---|---|---|---|---|
| βSβS | SS | Sickle cell anemia | S > F | 6–9 | nl | 10–25 | 4+ |
| βSβC | SC | Sickle-Hb C disease | S ≈C | 9–12 | nl or ↓ | 5–10 | 2+ |
| βSβ+ | Sβ;+ | Sickle-β+-thalassemia | S > A | 10–13 | ↓ | 5–10 | 1–2+ |
| βSβ0 | Sβ0 | Sickle-β0-thalassemia | S > F | 6–9 | ↓ | 10–25 | 4+ |
Common range of laboratory values in un-treated state (e.g., no hydroxyurea)
Does not consider effect of co-inherited alpha thalassemia or therapeutic effect of hydroxyurea
Population-based generalization
Abbreviations: nl normal, ↓ decreased, β+ a β-thalassemia mutation that eliminates globin transcription or translation, β+ a β-thalassemia mutation that decreases globin transcription or translation, βE the β-globin mutation that results in Hb S, βC the β-globin mutation that results in Hb C
Hb S is normally functional and soluble when oxygenated, but it forms insoluble polymers upon deoxygenation (“sickling”). This process deforms the red blood cell (RBC), leading to progressive cytoskeletal and other cellular damage that dramatically shortens the lifespan of the RBC. The result is a chronic, partially compensated, hemolytic anemia. This process of RBC “sickling” is initially reversible, but recurrent cycles of oxygenation and deoxygenation lead to cumulative and permanent membrane damage that gives rise to the hallmark irreversibly sickled cell. The RBCs in SCD also participate in abnormal adhesive interactions with platelets, leukocytes [44] and endothelial cells, which leads to stasis in post-capillary venules [45]. Stasis exacerbates the deoxygenation-polymerization process, leading to vaso-occlusion. The consequence is ongoing ischemia-reperfusion injury in nearly all organs, punctuated by acute vaso-occlusive events, leading to chronic organ damage and shortened life-expectancy of affected individuals.
SCD in developed countries is primarily diagnosed at birth by newborn screening for hemoglobinopathies. Common methods of diagnosis in the newborn period and later life are the Hb separation techniques, such as gel-based electrophoresis (especially isoelectric focusing), high-performance liquid chromatography, and capillary electrophoresis. Table 3.5 shows the relative abundance of the main Hbs in different forms of SCD. Like the thalassemias, genetic testing is being increasingly used to provide a definitive diagnosis, and also to identify clinically important phenotypic modifiers, such as alpha globin gene number and genetic determinants of Hb F production.
Prenatal diagnosis using chorionic villi sampling (10–12 weeks of pregnancy), amniotic fluid sampling (15–20 weeks) or fetal blood (17 weeks onwards) is also possible. This invasive testing comes with a small but significant risk to the fetus and mother. Newer non-invasive techniques using cell-free fetal DNA in maternal blood allows earlier (6 weeks) and less risky testing [46].
Screening Tests and Prevention of Complications
Bacterial Infection
Infection still poses significant risk for children with SCD, especially in less developed countries. Invasive pneumococcal disease, especially, results from functional asplenia due to progressive ischemic involution of the spleen. Immunizations are essential for children with SCD, as they have over 100-fold risk for invasive pneumococcal infection [47]. All children should complete the routine pneumococcal protein-conjugate vaccine (PCV13) series during infancy and also receive the 23-valent pneumococcal polysaccharide vaccine (PPSV-23) starting at 2 years of age and a booster dose at 5 years of age (some recommend a booster dose every 5 years thereafter). Other recommended vaccines include meningococcal protein-conjugate vaccine (MCV-4) starting at 2 years of age (and consider booster doses every 5 years). Vaccination against H. influenzae type b is also critical due to hyposplenism. Hepatitis B virus and yearly influenza vaccines are also prudent [48, 49].
Antibiotic prophylaxis should be commenced at 1–2 months of age as risk for infection from functional asplenia coincides with falling fetal hemoglobin. Children with Hb SS and Hb Sβ0 should receive oral penicillin: 125 mg twice daily under 3 years of age and 250 mg twice daily over 3 years of age. Prophylactic penicillin is usually stopped at 5 years of age [50], but there are recent reports of invasive pneumococcal disease from non-vaccine serotypes in older children with SCD [51, 52], so ongoing vigilance will be needed. Penicillin prophylaxis may not be needed for young children with Hb SC disease due to the delayed occurrence of hyposplenism [53].
In addition to above, any child or adult with SCD should be evaluated urgently for fever with prompt use of empiric intravenous broad-spectrum antibiotics. The management should also include a complete blood count and a blood culture. Presence of a central venous catheter in these individuals poses an additional risk. Very young age, toxic clinical appearance, laboratory values significantly different from baseline, and respiratory or cardiovascular instability require hospitalization. Most children with fever, however, can be safely managed as outpatients following empiric antibiotic therapy.
Ongoing education and reminders for patients, families and providers of the risk of sepsis is essential in preventing infection related morbidity and mortality. Combined with immunization, antibiotic prophylaxis, and empiric antibiotic treatment for fever episodes have largely eliminated death from infection in developed countries.
Acute Splenic Sequestration
This remains a potentially life-threatening complication in children with SCD. It occurs in younger patients, usually less than 5 years of age, before spleen involution is complete. Potentially reversible sequestration of red blood cells can occur suddenly with acute anemia, hypovolemia, splenomegaly, thrombocytopenia and reticulocytosis. Prompt detection of an enlarged spleen by parents who are taught splenic palpation along with urgent medical evaluation for this finding can usually prevent fatal episodes. A complete blood count and the presence of splenomegaly can help in differentiating this from transient aplastic crisis, which is characterized by reticulocytopenia and a normal or increased platelet count. For moderate to severe episodes, supportive measures with oxygen and fluid resuscitation (if necessary) and judicious PRBC transfusions can be lifesaving.
While acute splenic sequestration usually occurs in younger Hb SS or Hb Sβ0 patients (<5 years of age) before splenic involution is complete, splenic sequestration can occur in older Hb SC and Hb Sβ+ patients, in whom splenic involution is delayed. These adolescents and young adults often present with left upper quadrant pain from accompanying splenic infarction. The increasing use of hydroxyurea and chronic transfusion therapy can delay or partially reverse splenic involution, so the occurrence of splenic sequestration may also be delayed in such treated patients.
Primary Prevention of Stroke
In children with Hb SS and Hb Sβ0, screening for increased risk of overt stroke is performed using transcranial Doppler (TCD) ultrasonography at least yearly starting at 2 years of age until at least 16 years of age. This detects abnormally increased cerebral arterial blood flow velocities that indicate an increased risk of overt stroke. The randomized STOP Trial (Stroke Prevention Trial in Sickle Cell Anemia) demonstrated that chronic transfusions reduced the rate of first overt stroke by 92% in patients with abnormal TCDs. Recently, early, unpublished results from the TWiTCH study (Transcranial Doppler with Transfusions Changing to Hydroxyurea; clinicaltrials.gov: NCT01425307) suggest that hydroxyurea appears to be non-inferior to chronic transfusion therapy (for TCD velocities, not MRI findings or stroke) for patients without severe vasculopathy by MRA after at least 12 months of chronic transfusions. So, chronic transfusions may not have to be indefinite therapy for primary stroke prevention.
Health Maintenance and Screening (Table 3.6)
Table 3.6.
Health maintenance and screening for individuals with SCDa
| Health topic | Recommendations |
|---|---|
| Prevention of infection | Administer oral penicillin prophylaxis (125 mg for age <3 years and 250 mg for age ≥3 years) twice daily until age 5 in all children with HbSS or and HbSβ0-thalassemia Discontinue prophylactic penicillin in children with HbSS or and HbSβ0-thalassemia at age 5 unless they have had a splenectomy or invasive pneumococcal infection. When discontinuing penicillin prophylaxis at age 5, it is important to assure that the child has completed the recommended pneumococcal vaccination series, and if not, complete the series immediately Penicillin prophylaxis may not be necessary for children with HbSC disease and HbSβ+-thalassemia unless they have had a splenectomy Assure that people of all ages with SCD have been vaccinated against Streptococcus pneumoniae, Haemophilus influenzae type b, and the meningococcus Remind people with SCD, their families, and caregivers to seek immediate medical attention whenever fever (temperature greater than 101.3°F or 38.5°C) occurs, due to the risk for severe bacterial infections |
| Renal disease | Screen all individuals with SCD, beginning by age 10, for proteinuria. If the result is negative, repeat screening annually. If the result is positive, perform a first morning void urine albumin-creatinine ratio and if abnormal, consult with or refer to a nephrologist |
| Heart disease | Consider annual echocardiography for adults with SCD Routine echocardiography screening is not recommended for children with SCD Routine ECG screening is not recommended in children and adults with SCD |
| Hypertension | In adults with SCD, screen for hypertension and treat to lower systolic blood pressure ≤140 and diastolic blood pressure ≤90 according to national guidelines In children with SCD, measure blood pressure, and evaluate and treat hypertension following recommendations from national guidelines |
| Retinopathy | Refer individuals with SCD to an ophthalmologist for a dilated eye examination to evaluate for retinopathy beginning at age 10 For people with a normal dilated retinal examination, re-screen at 1–2 year intervals Refer people with suspected retinopathy to a retinal specialist |
| Stroke | In children with HbSS and HbSβ0-thalassemia, screen annually with TCD according to methods employed in the STOP studies, beginning at age 2 and continuing until at least age 16 In children with abnormal (non-imaging TAMMV >200 cm/s in dICA, bifurcation or MCA) TCD results, begin chronic transfusion therapy aimed at preventing stroke In children with genotypes other than SCA (e.g., HbSβ+-thalassemia or HbSC), do not perform screening with TCD Consider screening MRI for detection of silent cerebral infarction (SCI) in children and adults with HbSS and HbSβ0-thalassemia For children with SCI, offer chronic transfusion therapy for prevention of recurrent infarction |
| Lung disease | In children and adults with SCD, assess for signs and symptoms of respiratory problems (such as asthma, COPD, restrictive lung disease, or obstructive sleep apnea) by history and physical examination In children and adults with SCD found to have signs or symptoms of respiratory problems by history and/or physical examination, further assessment, which includes pulmonary function tests, is recommended to determine the cause and develop a plan to address the problem Do not screen asymptomatic children and adults with pulmonary function tests |
| Contraception | Progestin-only contraceptives (pills, injections, and implants), levonorgestrel IUDs, and barrier methods have no restrictions or concerns for use in women with SCD |
Adapted (modified and extended) from Evidence-Based Management of Sickle Cell Disease, Expert Panel Report 2014, National Heart, Lung and Blood Institute
Growth and Development
Children with SCD can have poor growth in comparison to normal African American children. Pubertal and skeletal maturation delay has been noted in these patients [54, 55]. Chronic anemia, poor nutrition, increased metabolic rate and renal loss of nutrients have been posited to lead to growth failure in children with SCD. Contemporary management of SCD with early hydroxyurea, monitoring and supplementation of nutrients (such as zinc and vitamin D) and appropriate use of age-, sex-, and ethnicity-matched growth charts can help maintain normal or near normal growth in most patients, thereby potentially improving quality of life [56, 57].
Blood Counts and Serum Chemistries
Monitoring of hematological and biochemical parameters is recommended during clinic visits. The following laboratory studies are recommended at least once or twice yearly: complete blood count, reticulocyte count, differential leukocyte count (if taking hydroxyurea therapy), renal and liver function tests, urinalysis, urine microalbumin and protein quantitation, and ferritin (for a history of transfusions). Routine monitoring will help determine baseline (steady-state) blood count values and detect early evidence renal dysfunction, iron overload, and other complications associated with SCD.
Electrocardiogram and Echocardiography
Routine screening with electrocardiography in asymptomatic children with SCD is currently not recommended. While there is evidence that adults with SCD who have a tricuspid regurgitant velocity (TRV) equal to or greater than 2.5 m/s are at risk of early mortality [58], similar evidence in children is lacking. Given the potential for cardiac or pulmonary disease associated with elevated TRV, we think it is still prudent to screen adolescents by echocardiography before transition to adult medical care. Children with signs or symptoms of cardiopulmonary disease should be tested accordingly.
TCD Ultrasonography
Annual screening with TCD ultrasonography can detect high risk of overt stroke and direct the initiation of chronic transfusion programs for primary prevention of stroke. Please refer to the section on primary prevention of stroke earlier in this chapter.
MRI of Brain and Silent Cerebral Infarction (SCI)
Routine magnetic resonance imaging (MRI) of brain in children with Hb SS is currently not recommended by the NHLBI guidelines [59]. However, silent cerebral infarcts (SCI), which can only be detected by a screening MRI (or incidentally on an MRI for another reason) are associated with neurocognitive deficits, poor school performance, and increased risk for stroke [60, 61]. Students with SCD who have SCI and decreased cognitive function can be eligible for specific educational resources for which they would not otherwise be eligible. Similar resources are available for young adults who need vocational support or assistance in college. Therefore, the benefits of an MRI of the brain performed without anesthesia or sedation to identify SCI very likely outweigh the few risks of the procedure.
The recently completed Silent Cerebral Infarct Transfusion Trial (SIT Trial) [61] showed that chronic transfusions for children with Hb SS and SCI who are not at high risk of stroke by TCD criteria: (1) decrease the incidence of recurrent or progressive cerebral infarction; (2) decrease the frequency of pain, chest syndrome, AVN and priapism; and (3) improve quality of life. This comes at the cost of an increase in the likelihood of iron overload (ferritin >1500 ng/mL). As such, we believe it would be judicious to perform at least one screening MRI for SCI in young school-aged children and consider it for older individuals with SCD as well.
Immunizations
Immunizations are a vital component of standard medical care for individuals with SCD. Please refer to the section on bacterial infection earlier in this chapter.
Educational Attainment
As a group, children with SCD face educational difficulties from a combination of factors, such as chronic anemia, cerebral infarction, school absenteeism from recurrent illnesses, and hospital visits, limited parenteral education, and low socio-economic status has been associated with poor academic performance [62, 63]. This highlights the importance of psychosocial, school and occupational intervention in SCD patients as a key component of standard care.
Management of Specific Complications
Acute Painful Episodes
The acute painful episode of SCD, often called vaso-occlusive crisis (VOC), is thought to be the consequence of ischemia and infarction of bone, bone marrow, and other tissues. This can manifest in infants (until about 3 years of age) as dactylitis, which is vaso-occlusive ischemia and infarction of phalanges and metacarpals that produces painful swelling. In toddlers and older children, acute SCD pain can involve extremities, back or chest wall. Pain can be local to one part of the limb, regional (involving multiple contiguous sites), or abdominal. Physical signs (edema, induration, erythema) are only present in about 15% of episodes. Triggers of pain are often not obvious, but known triggers include infection, inflammation, cooling of the skin, and psychological stress.
The management of painful episodes includes analgesia with a combination NSAIDs and opiates that is individualized to the patient and degree of pain. Non-pharmacological adjuncts can also be used (warm compresses, relaxation, massage). It is important to avoid over- and under-treatment of pain. The goal of fluid therapy is to correct dehydration, maintain normal hydration, and avoid over-hydration. Incentive spirometry is needed to prevent acute chest syndrome (ACS), and clinicians need to remain vigilant for the development of fever and ACS. Supportive care also includes laxatives for opiate-related constipation and anti-pruritics for opiate-related pruritus. Acute transfusion is not indicated for uncomplicated pain.
There is ongoing research into drugs that alter cell adhesion, blood viscosity, inflammation, and platelet activity that may prove useful in the treatment or prevention of acute painful episodes.
Acute Chest Syndrome (ACS)
ACS is a non-specific term for an acute pulmonary illness in an individual with SCD. It is defined clinically by a new, radiographic pulmonary infiltrate in conjunction with fever and respiratory signs and symptoms, such as cough, tachypnea, dyspnea, increased work of breathing, hypoxia/desaturation, and chest pain. ACS is not apparent in 30–60% of patients at the time of hospitalization for pain or fever. Incentive spirometry, therefore, is an important adjunct for patients with thoracic or abdominal pain that has been shown to prevent the development of ACS [64]. Common or known antecedents or triggers of ACS include infection (viruses, Mycoplasma, Chlamydia, S. pneumoniae), pulmonary vascular occlusion, hypoventilation/atelectasis, pulmonary edema, bronchospasm, and surgery or general anesthesia. Pre-operative simple transfusion has been shown to prevent post-operative ACS. Prior ACS, younger age, and asthma are known risk factors for the development of ACS in general [48, 65].
Management of ACS is primarily supportive, including empiric antibiotic therapy with a cephalosporin (e.g., ceftriaxone) and a macrolide (e.g., azithromycin). Transfusion is often not needed for young children with mild disease. Simple transfusion can be given for hypoxemia or acute exacerbation of chronic anemia. Exchange transfusion is indicated for severe disease, such as rapid clinical deterioration, widespread pulmonary infiltrates, and hypoxemia despite supplemental oxygen. Other principles of supportive care include correction of hypoxemia, maintenance of normal hydration, adequate analgesia, bronchodilators for wheezing, and support of ventilation for severe disease.
Overt Stroke
TCD screening programs and chronic transfusion therapy for primary prevention of stroke is discussed earlier in this chapter. Without primary prevention, unselected children with Hb SS have a 10% risk of overt stroke by 18 years of age. Although overt stroke is now much less common in developed countries, it can still occur despite the availability of screening programs. Acute stroke should be suspected in the event of focal neurologic deficits, weakness, asymmetric face, changes in speech, or altered mental state. Principles of management for acute stroke include rapid triage, supplemental oxygen, and avoidance of delays to transfusion. Erythrocytapheresis appears to be associated with better long-term outcomes than simple transfusion [66]. Simple transfusion can be given if the Hb concentration is <9 g/dL as a temporizing measure for any delay in erythrocytapheresis. If the diagnosis of stroke is strongly suspected by history and examination, transfusion therapy should not be delayed by imaging. The goal of exchange transfusion should be an end-procedure hematocrit of 28–30%, and Hb S percentage <30%, and a fraction of cells remaining (FCR) of <0.3.
The risk of recurrent stroke is high (50–90%), and chronic transfusions are the mainstay of secondary stroke prevention, reducing the rate to 20% [67]. Because chronic transfusions are burdensome, need to be given indefinitely, and carry the added risk of iron overload, investigation of alternative therapies is ongoing. Stem cell transplantation is indicated in this setting (discussed elsewhere in this book). Hydroxyurea has also been studied for secondary stroke prevention in the SWiTCH Trial. This showed that chronic transfusions and chelation is better than hydroxyurea and phlebotomy for the prevention of recurrent stroke and resolution of iron overload.
Acute Anemic Events
Patients with SCD have a chronic, partially compensated hemolytic anemia. Patients may also experience acute exacerbations of this chronic anemia, referred to as acute anemic events (AAEs). AAEs commonly occur as a consequence of parvovirus B19 infection, acute splenic sequestration, or a “hyperhemolytic” event accompanying other complications of the disease. It is now clear that the degree of chronic anemia and the frequency of AAEs are important risk factors, in particular, for silent cerebral ischemia and infarction [60, 68–70]. Given the association of AAEs with cerebral ischemia, transfusion to correct at least the acute component of anemia might be neuroprotective during AAEs, even in the absence of “symptomatic anemia” based on cardiopulmonary signs and symptoms.
Priapism
Priapism occurs in up to 75% of males with SCD by 20 years of age. It is defined as painful, purposeless and persistent erection of penis [71]. Patients can have brief, intermittent (“stuttering”) episodes of priapism or prolonged, severe episodes (usually defined as lasting >4 h). Priapism is recurrent in about 90% of patients, especially in patients with the Hb SS genotype [72]. Home management includes advice to patients to initiate oral hydration, warm compresses or showers, exercise, voiding, oral analgesics and pseudoephedrine. Patients should be instructed to seek urgent care for any priapism lasting >4 h. Principles of care for prolonged priapism include IV analgesia, IV hydration, oral pseudoephedrine (if not recently given), and Urologic consultation for aspiration and irrigation. Acute transfusion not shown to be helpful, and there are old reports that exchange transfusion may be harmful [73]. Surgical management with shunts has shown to have less favorable outcome with erectile dysfunction and should be avoided if possible [71]. Erectile dysfunction and subsequent sexual, reproductive issues are troublesome adverse effects of prolonged priapism [74]. Prevention of recurrent priapism can be attempted by optimizing disease-modifying therapies (hydroxyurea, chronic transfusions). There are also a few reports of successful prevention with leuprolide or bicalutamide.
Disease-modifying Therapies
Hydroxyurea
Hydroxyurea is a ribonucleotide reductase inhibitor that induces Hb F synthesis. Higher concentrations of Hb F in RBCs are beneficial because further polymerization of Hb S fibers is prevented when Hb F is incorporated into a growing polymer. This is the primary beneficial mechanism of hydroxyurea in SCD. Secondary mechanisms of action include improved blood rheology, reduced adhesive interactions or RBCs with other cells, increased total hemoglobin concentration, and decreases in platelet, reticulocyte and neutrophil counts. This is an FDA approved medication for adults with SCD. There are abundant data supporting its benefit in SCD in reducing the frequency of painful episodes, hospitalizations, ACS, and transfusions, all of which likely prevents some chronic organ damage and decreases mortality [75–77].
Current trends in practice include the early and pre-symptomatic prescription of hydroxyurea to young children, based on the results of BABY HUG trial and other investigations. The current NHLBI recommendation is to offer hydroxyurea to children with Hb SS beginning at 9 months of age and older irrespective of clinical status [59, 76]. The side effect profile of hydroxyurea is quite favorable, mainly including mild, dose-related neutropenia and thrombocytopenia [76, 78]. There seems to be no increase in malignancy in children or adults taking hydroxyurea, even for extended periods of therapy. Likewise, there is no evidence of impaired growth or development in children treated with hydroxyurea. Long term monitoring studies are on-going [56, 76]. Table 3.7 reviews dosing and monitoring guidelines for hydroxyurea.
Table 3.7.
Hydroxyurea dosing and monitoring
| Phase of therapy | Dosing regimen | Monitoring |
|---|---|---|
| Initiation | 20–25 mg/kg/day in one daily dose | Baseline: CBC, differential, reticulocyte count, Hb F, creatinine, LFTs Monthly: CBC, differential, reticulocyte count Monitor for toxicitya |
| Dose escalation | Increase dose by 3–5 mg/kg/day every 4–6 weeks to maximum tolerated dose (MTD) MTD is achieved when the absolute neutrophil count is between 1.5 and 3.0 × 109/L |
Monthly: CBC, differential, reticulocyte count Monitor for toxicitya |
| Maintenance | Continue MTD dose | Every 3 months: CBC, differential, reticulocyte count, Hb F Every 6 months: creatinine, LFTs Monitor for toxicitya |
Common criteria for toxicity include: ANC <1.0 × 109/L; platelet count <80 × 109/L; absolute reticulocyte count <80 × 109/L if hemoglobin <9.0 g/dL; Hemoglobin <5 g/dL or >20% below baseline; serum creatinine >1.0 mg/dL; ALT that is double the patient’s baseline or three times the ULN for ALT. If toxicity occurs, consider stopping hydroxyurea for 4–7 days or until toxicity resolves. Hydroxyurea can then be resumed at the same dose or a dose decreased by 2.5–5 mg/kg/day
Chronic Transfusions
Regularly scheduled transfusions of Hb S-negative PRBCs to maintain the percentage of Hb S in the blood <30% can effectively prevent most complications of SCD and minimize the chronic anemia. Common indications for transfusions are listed in Table 3.8. “Top-up” or simple transfusions are usually given in acute anemic states when the hemoglobin is 2–3 g/dL or more below baseline hemoglobin. In patients with SCD who require general anesthesia for surgery, a pre-operative simple transfusion to raise the total hemoglobin concentration to 10 g/dL can decrease the frequency of post-operative complications such as ACS and VOC [79]. Routine transfusion for minor elective procedures (e.g., myringotomy, circumcision) is not required, especially in those whose HbS level is below 30% with chronic transfusion therapy [80].
Table 3.8.
Common indications for transfusion in SCD
| Episodic transfusions for acute management | Chronic transfusions for long-term prevention |
| Aplastic crisis | Stroke (primary and secondary prevention) |
| Acute splenic sequestration | Frequent painful episodes |
| Acute chest syndrome | Frequent or severe ACS |
| Acute stroke (exchange transfusion) | Other severe or chronic complications |
| Pre-operative preparation (prevention of ACS) |
Abbreviation: ACS acute chest syndrome
Chronic indefinite transfusion therapy is currently the treatment of choice for prevention of secondary stroke [81] and in the prevention of recurrent infarction in children with silent cerebral infarction [61]. Once initiated, it should be performed every 3–4 weeks to maintain pre-transfusion (maximum) Hb S of <30%, which usually achieves a nadir hemoglobin concentration of 9–10 g/dL. Complications of transfusions include febrile and allergic transfusion reactions, hemolytic transfusion reactions, transfusion-transmitted infections, alloimmunization, and iron overload. Detailed documentation of pre-transfusion hemoglobin levels, transfusion reactions, alloimmunization, and volume of all PRBCs received is necessary for high quality care. Iron chelation therapy is reviewed in the section above on thalassemia and in Table 3.4. Transfusion using phenotype-matched (ABO, C, c, D, E, e, and K-matched) PRBCs has the potential to prevent over 50% of alloantibodies in SCD patients [82].
Cure
Hematopoietic stem cell transplantation and gene therapy are discussed in other chapters in this book.
Long-Term Outcomes
With contemporary management in developed nations, almost all children (>95%) with SCD will survive to adulthood (Fig. 3.2) [83, 84]. This excellent overall survival is the result of a many incremental advancements, including newborn screening, prophylactic penicillin, protein-conjugate vaccines, advances in supportive care, and the increased use of disease-modifying treatments (hydroxyurea, chronic transfusions, and stem cell transplantation). The burden of mortality in SCD has now shifted to adults, and the transition to adult medical care is a high-risk period for death. There are a number of possible explanations for this vulnerability, including a flawed interface between pediatric and adult medical care and the gradual accumulation of SCD-related chronic organ injury during childhood that becomes manifest in young adulthood. Long-term survival estimates (beyond childhood) are less accurately known, but median survival is estimated to be over 50 years for individuals with Hb SS and Hb Sβ0. The survival of individuals with Hb SC and Hb Sβ+ approach that of the general population. Especially for those with Hb SS and Hb Sβ0, advances in gene therapy and hematopoietic transplantation hold the potential for even greater improvements in survival and quality of life.
Fig. 3.2.
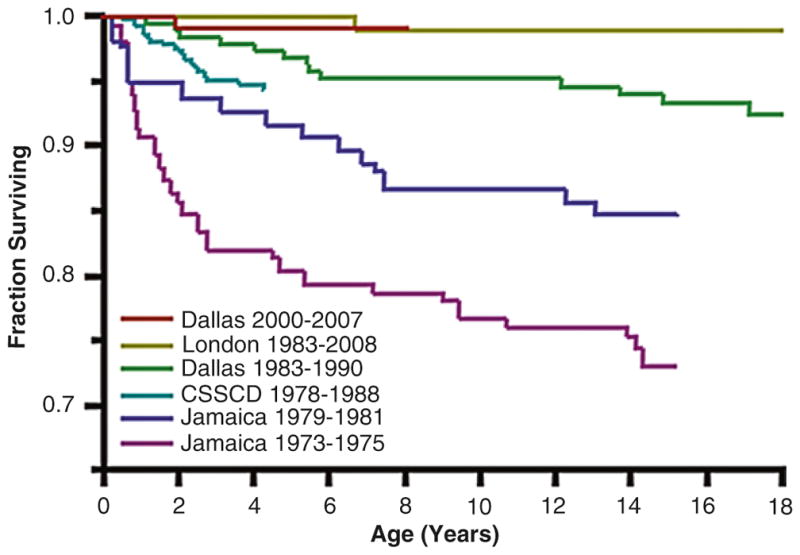
Improving survival in sickle cell disease (SCD). Overall survival curves for patients with sickle cell anemia (Hb SS) and sickle-β0-thalassemia spanning by years of birth are shown for large SCD cohorts in the US, UK and Jamaica. Figure adapted from: Quinn et al. Blood. 2010;115:3447–3452
Contributor Information
Satheesh Chonat, Emory University School of Medicine and Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, 2015 Uppergate Drive, Atlanta, GA 30322, USA.
Charles T. Quinn, Hematology, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45220, USA
References
- 1.Chui DH, Hardison R, Riemer C, et al. An electronic database of human hemoglobin variants on the World Wide Web. Blood. 1998;91(8):2643–2644. [PubMed] [Google Scholar]
- 2.Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42(Database issue):D1063–9. doi: 10.1093/nar/gkt911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laig M, Pape M, Hundrieser J, et al. The distribution of the Hb constant spring gene in Southeast Asian populations. Hum Genet. 1990;84(2):188–190. doi: 10.1007/BF00208939. [DOI] [PubMed] [Google Scholar]
- 4.Voon HPJ, Vadolas J. Controlling alpha-globin: a review of alpha-globin expression and its impact on beta-thalassemia. Haematologica. 2008;93(12):1868–1876. doi: 10.3324/haematol.13490. [DOI] [PubMed] [Google Scholar]
- 5.Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood. 2000;96(7):2606–2612. [PubMed] [Google Scholar]
- 6.Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood Reviews. 2003;17(1):43–53. doi: 10.1016/s0268-960x(02)00061-9. [DOI] [PubMed] [Google Scholar]
- 7.De Sanctis V, Soliman AT, Elsedfy H, et al. Growth and endocrine disorders in thalassemia: The international network on endocrine complications in thalassemia (I-CET) position statement and guidelines. Indian J Endocrinol Metab. 2013;17(1):8–18. doi: 10.4103/2230-8210.107808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cappellini MD, Cohen A. Guidelines for the management of transfusion dependent thalassemia (TDT) Thalassaemia International Federation. 2014:1–253. [PubMed] [Google Scholar]
- 9.Pakbaz Z, Fischer R, Fung E, et al. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer. 2007;49(3):329–332. doi: 10.1002/pbc.21275. [DOI] [PubMed] [Google Scholar]
- 10.Musallam KM, Taher AT, Cappellini, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood. 2013;121(12):2199–2212. doi: 10.1182/blood-2012-10-408021. [DOI] [PubMed] [Google Scholar]
- 11.Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood. 1997;89(3):739–761. [PubMed] [Google Scholar]
- 12.Aydinok Y, Kattamis A, Viprakasit V. Current approach to iron chelation in children. British Journal of Haematology. 2014;165(6):745–755. doi: 10.1111/bjh.12825. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter JP, He T, Kirk P, Roughton M, Anderson LJ. On T2* Magnetic Resonance and Cardiac Iron. Circulation. 2011;123(14):1519–1528. doi: 10.1161/CIRCULATIONAHA.110.007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karimi M, Musallam KM, Cappellini MD, et al. Risk factors for pulmonary hypertension in patients with β thalassemia intermedia. European Journal of Internal Medicine. 2011;22(6):607–610. doi: 10.1016/j.ejim.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Vento S, Cainelli F, Cesario F. Infections and thalassaemia. Lancet Infect Dis. 2006;6(4):226–233. doi: 10.1016/S1473-3099(06)70437-6. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham MJ. Complications of β-thalassemia major in North America. Blood. 2004;104(1):34–39. doi: 10.1182/blood-2003-09-3167. [DOI] [PubMed] [Google Scholar]
- 17.Vogiatzi MG, Macklin EA, Fung EB, et al. Bone disease in thalassemia: a frequent and still unresolved problem. J Bone Miner Res. 2009;24(3):543–557. doi: 10.1359/jbmr.080505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voskaridou E, Terpos E. New insights into the pathophysiology and management of osteoporosis in patients with beta thalassaemia. British Journal of Haematology. 2004;127(2):127–139. doi: 10.1111/j.1365-2141.2004.05143.x. [DOI] [PubMed] [Google Scholar]
- 19.Ford JM, Rojkjaer L. Comment to: Development of lens opacities with peculiar characteristics in patients affected by thalassemia major on chelating treatment with deferasirox. Haematologica 2008;93:e9–10. Haematologica. 2008;93(6):e49. doi: 10.3324/haematol.12944. discussion e50. [DOI] [PubMed] [Google Scholar]
- 20.Rund D, Rachmilewitz E. β-Thalassemia. N Engl J Med. 2005;353(11):1135–1146. doi: 10.1056/NEJMra050436. [DOI] [PubMed] [Google Scholar]
- 21.Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479–3488. doi: 10.1182/blood-2010-08-300335. [DOI] [PubMed] [Google Scholar]
- 22.Weatherall DJ. Phenotype|[mdash]|genotype relationships in monogenic disease: lessons from the thalassaemias. Nature Reviews Genetics. 2001;2(4):245–255. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]
- 23.Taher AT, Musallam KM, Karimi M, Cappellini MD. Contemporary approaches to treatment of beta-thalassemia intermedia. YBLRE. 2012;26:S24–S27. doi: 10.1016/S0268-960X(12)70008-5. [DOI] [PubMed] [Google Scholar]
- 24.Piomelli S, Graziano J, Karpatkin M, et al. Chelation Therapy, Transfusion Requirement, And Iron Balance In Young Thalassemic Patients*. Ann N Y Acad Sci. 1980;344(1):409–417. doi: 10.1111/j.1749-6632.1980.tb33680.x. [DOI] [PubMed] [Google Scholar]
- 25.Perrine SP, Ginder GD, Faller DV, et al. A Short-Term Trial of Butyrate to Stimulate Fetal- Globin-Gene Expression in the β-Globin Disorders. N Engl J Med. 1993;328(2):81–86. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 26.Musallam KM, Sankaran VG, Cappellini MD, et al. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood. 2012;119(2):364–367. doi: 10.1182/blood-2011-09-382408. [DOI] [PubMed] [Google Scholar]
- 27.Ley TJ, DeSimone J, Anagnou NP, et al. 5-azacytidine selectively increases gamma-globin synthesis in a patient with beta+ thalassemia. N Engl J Med. 1982;307(24):1469–1475. doi: 10.1056/NEJM198212093072401. [DOI] [PubMed] [Google Scholar]
- 28.Olivieri NF, Saunthararajah Y, Thayalasuthan V, et al. A pilot study of subcutaneous decitabine in β-thalassemia intermedia. Blood. 2011;118(10):2708–2711. doi: 10.1182/blood-2011-03-341909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fathallah H, Taher A, Bazarbachi A, Atweh GF. Differences in response to fetal hemoglobin induction therapy in β-thalassemia and sickle cell disease. Blood Cells, Molecules, and Diseases. 2009;43(1):58–62. doi: 10.1016/j.bcmd.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inati A, Kahale M, Perrine SP, et al. A phase 2 study of HQK-1001, an oral fetal haemoglobin inducer, in β-thalassaemia intermedia. British Journal of Haematology. 2014;164(3):456–458. doi: 10.1111/bjh.12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singer ST, Vichinsky EP, Sweeters N, Rachmilewitz E. Darbepoetin alfa for the treatment of anaemia in alpha- or beta- thalassaemia intermedia syndromes. British Journal of Haematology. 2011;154(2):281–284. doi: 10.1111/j.1365-2141.2011.08617.x. [DOI] [PubMed] [Google Scholar]
- 32.Perrine SP, Pace BS, Faller DV. Targeted fetal hemoglobin induction for treatment of beta hemoglobinopathies. Hematology/Oncology Clinics of North America. 2014;28(2):233–248. doi: 10.1016/j.hoc.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Platt OS, Orkin SH, Dover G, Beardsley GP. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. Journal of Clinical …. 1984;74(2):652–656. doi: 10.1172/JCI111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karimi M, Darzi H, Yavarian M. Hematologic and Clinical Responses of Thalassemia Intermedia Patients to Hydroxyurea During 6 Years of Therapy in Iran. J Pediatr Hematol Oncol. 2005;27(7):380–385. doi: 10.1097/01.mph.0000174386.13109.28. [DOI] [PubMed] [Google Scholar]
- 35.Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892. doi: 10.1182/blood-2009-09-243154. [DOI] [PubMed] [Google Scholar]
- 36.Karimi M, Cohan N, De Sanctis V, Mallat NS, Taher A. Guidelines for Diagnosis and Management of Beta-Thalassemia Intermedia. Pediatr Hematol Oncol. 2014;31(7):583–596. doi: 10.3109/08880018.2014.937884. [DOI] [PubMed] [Google Scholar]
- 37.Taher A, Rassi El F, Isma’eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93(10):1584–1586. doi: 10.3324/haematol.13098. [DOI] [PubMed] [Google Scholar]
- 38.Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92(11):1485–1493. doi: 10.1007/s00277-013-1808-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cappellini, Robbiolo L, Bottasso BM. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. British Journal of …. 2000;111(2):467–473. doi: 10.1046/j.1365-2141.2000.02376.x. [DOI] [PubMed] [Google Scholar]
- 40.Borgna-Pignatti C, Cappellini MD, Stefano P, et al. Survival and Complications in Thalassemia. Ann N Y Acad Sci. 2005;1054(1):40–47. doi: 10.1196/annals.1345.006. [DOI] [PubMed] [Google Scholar]
- 41.Caocci G. Health related quality of life in Middle Eastern children with beta-thalassemia. 2012:1–7. doi: 10.1186/1471-2326-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. The Lancet. 2000;355(9220):2051–2052. doi: 10.1016/S0140-6736(00)02357-6. [DOI] [PubMed] [Google Scholar]
- 43.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. 1910;6:517–521. [PMC free article] [PubMed] [Google Scholar]
- 44.Epstein FH, Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 45.Wood A, Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340(13):1021–1030. doi: 10.1056/NEJM199904013401307. [DOI] [PubMed] [Google Scholar]
- 46.Hoppe CC. Prenatal and newborn screening for hemoglobinopathies. Int Jnl Lab Hem. 2013;35(3):297–305. doi: 10.1111/ijlh.12076. [DOI] [PubMed] [Google Scholar]
- 47.Adamkiewicz TV, Sarnaik S, Buchanan GR, et al. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. The Journal of Pediatrics. 2003;143(4):438–444. doi: 10.1067/S0022-3476(03)00331-7. [DOI] [PubMed] [Google Scholar]
- 48.Quinn CT. Sickle cell disease in childhood: from newborn screening through transition to adult medical care. Pediatric Clinics of North America. 2013;60(6):1363–1381. doi: 10.1016/j.pcl.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Booth C, Inusa B, Obaro SK. Infection in sickle cell disease: A review. International Journal of Infectious Diseases. 2010;14(1):e2–e12. doi: 10.1016/j.ijid.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 50.Falletta JM, Woods GM, Verter JI, et al. Discontinuing penicillin prophylaxis in children with sickle cell anemia. Prophylactic Penicillin Study II. The Journal of Pediatrics. 1995;127(5):685–690. doi: 10.1016/s0022-3476(95)70154-0. [DOI] [PubMed] [Google Scholar]
- 51.McCavit TL, Quinn CT, Techasaensiri C, Rogers ZR. Increase in invasive Streptococcus pneumoniae infections in children with sickle cell disease since pneumococcal conjugate vaccine licensure. The Journal of Pediatrics. 2011;158(3):505–507. doi: 10.1016/j.jpeds.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCavit TL, Xuan L, Zhang S, Flores G, Quinn CT. Hospitalization for invasive pneumococcal disease in a national sample of children with sickle cell disease before and after PCV7 licensure. Pediatr Blood Cancer. 2012;58(6):945–949. doi: 10.1002/pbc.23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lane PA, O’Connell JL, Lear JL, et al. Functional asplenia in hemoglobin SC disease. Blood. 1995;85(8):2238–2244. [PubMed] [Google Scholar]
- 54.Zemel BS, Kawchak DA, Ohene-Frempong K, Schall JI, Stallings VA. Effects of delayed pubertal development, nutritional status, and disease severity on longitudinal patterns of growth failure in children with sickle cell disease. Pediatr Res. 2007;61(5 Pt 1):607–613. doi: 10.1203/pdr.0b013e318045bdca. [DOI] [PubMed] [Google Scholar]
- 55.Platt OS, Rosenstock W, Espeland MA. Influence of sickle hemoglobinopathies on growth and development. N Engl J Med. 1984;311(1):7–12. doi: 10.1056/NEJM198407053110102. [DOI] [PubMed] [Google Scholar]
- 56.Rana S, Houston PE, Wang WC, et al. Hydroxyurea and growth in young children with sickle cell disease. PEDIATRICS. 2014;134(3):465–472. doi: 10.1542/peds.2014-0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dekker LH, Fijnvandraat K, Brabin BJ, van Hensbroek MB. Micronutrients and sickle cell disease, effects on growth, infection and vaso-occlusive crisis: a systematic review. Pediatr Blood Cancer. 2012;59(2):211–215. doi: 10.1002/pbc.24163. [DOI] [PubMed] [Google Scholar]
- 58.Klings ES, Machado RF, Barst RJ, et al. An Official American Thoracic Society Clinical Practice Guideline: Diagnosis, Risk Stratification, and Management of Pulmonary Hypertension of Sickle Cell Disease. Am J Respir Crit Care Med. 2014;189(6):727–740. doi: 10.1164/rccm.201401-0065ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.NHLBI. Evidence-based management of sickle cell disease: expert panel report, 2014. PEDIATRICS. 2014;134(6):e1775–e1775. [Google Scholar]
- 60.Quinn CT, McKinstry RC, Dowling MM, et al. Acute silent cerebral ischemic events in children with sickle cell anemia. JAMA Neurol. 2013;70(1):58–65. doi: 10.1001/jamaneurol.2013.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeBaun MR, Gordon M, McKinstry RC, et al. Controlled Trial of Transfusions for Silent Cerebral Infarcts in Sickle Cell Anemia. N Engl J Med. 2014;371(8):699–710. doi: 10.1056/NEJMoa1401731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith KE, Patterson CA, Szabo MM, Tarazi RA, Barakat LP. Predictors of academic achievement for school-age children with sickle cell disease. Advances in School Mental Health Promotion. 2013;6(1):5–20. doi: 10.1080/1754730X.2012.760919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.King AA, Rodeghier MJ, Panepinto JA, et al. Silent cerebral infarction, income, and grade retention among students with sickle cell anemia. Am J Hematol. 2014;89(10):E188–92. doi: 10.1002/ajh.23805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bellet PS, Kalinyak KA, Shukla R, Gelfand MJ, Rucknagel DL. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N Engl J Med. 1995;333(11):699–703. doi: 10.1056/NEJM199509143331104. [DOI] [PubMed] [Google Scholar]
- 65.DeBaun MR, Rodeghier M, Cohen R, et al. Factors predicting future ACS episodes in children with sickle cell anemia. Am J Hematol. 2014 doi: 10.1002/ajh.23819. n/a–n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hulbert ML, Scothorn DJ, Panepinto JA, et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. The Journal of Pediatrics. 2006;149(5):710–712. doi: 10.1016/j.jpeds.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 67.Scothorn DJ, Price C, Schwartz D, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. The Journal of Pediatrics. 2002;140(3):348–354. doi: 10.1067/mpd.2002.122498. [DOI] [PubMed] [Google Scholar]
- 68.Dowling MM, Quinn CT, Plumb P, et al. Acute silent cerebral ischemia and infarction during acute anemia in children with and without sickle cell disease. Blood. 2012;120(19):3891–3897. doi: 10.1182/blood-2012-01-406314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dowling MM, Quinn CT, Rogers ZR, Buchanan GR. Acute silent cerebral infarction in children with sickle cell anemia. Pediatr Blood Cancer. 2010;54(3):461–464. doi: 10.1002/pbc.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bernaudin F, Verlhac S, Arnaud C, et al. Chronic, acute anemia and eICA stenosis are independent risk factors for silent cerebral infarcts in sickle cell anemia. Blood. 2014 doi: 10.1182/blood-2014-09-599852. [DOI] [PubMed] [Google Scholar]
- 71.Adeyoju AB, Olujohungbe ABK, Morris J, et al. Priapism in sickle-cell disease; incidence, risk factors and complications - an international multicentre study. BJU Int. 2002;90(9):898–902. doi: 10.1046/j.1464-410x.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 72.Madu AJ, Ubesie A, Ocheni S, et al. Priapism in Homozygous Sickle Cell Patients: Important Clinical and Laboratory Associations. Med Princ Pract. 2014;23(3):259–263. doi: 10.1159/000360608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siegel JF, Rich MA, Brock WA. Association of sickle cell disease, priapism, exchange transfusion and neurological events: ASPEN syndrome. J Urol. 1993;150(5 Pt 1):1480–1482. doi: 10.1016/s0022-5347(17)35817-2. [DOI] [PubMed] [Google Scholar]
- 74.Smith-Whitley K. Reproductive issues in sickle cell disease. Blood. 2014;124(24):3538–3543. doi: 10.1182/blood-2014-07-577619. [DOI] [PubMed] [Google Scholar]
- 75.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS) Blood. 2010;115(12):2354–2363. doi: 10.1182/blood-2009-05-221333. [DOI] [PubMed] [Google Scholar]
- 76.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG) Lancet. 2011;377(9778):1663–1672. doi: 10.1016/S0140-6736(11)60355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Lobo CLC, Pinto JFC, Nascimento EM, et al. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. British Journal of Haematology. 2013;161(6):852–860. doi: 10.1111/bjh.12323. [DOI] [PubMed] [Google Scholar]
- 78.Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115(26):5300–5311. doi: 10.1182/blood-2009-04-146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of Sickle Cell Disease. JAMA. 2014;312(10):1033. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 80.Fu T, Corrigan NJ, Quinn CT, Rogers ZR, Buchanan GR. Minor elective surgical procedures using general anesthesia in children with sickle cell anemia without pre-operative blood transfusion. Pediatr Blood Cancer. 2005;45(1):43–47. doi: 10.1002/pbc.20283. [DOI] [PubMed] [Google Scholar]
- 81.Ware RE, Helms RW SWiTCH Investigators. Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) Blood. 2012;119(17):3925–3932. doi: 10.1182/blood-2011-11-392340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Castro O, Sandler SG, Houston-Yu P, Rana S. Predicting the effect of transfusing only phenotype-matched RBCs to patients with sickle cell disease: theoretical and practical implications. Transfusion. 2002;42(6):684–690. doi: 10.1046/j.1537-2995.2002.00126.x. [DOI] [PubMed] [Google Scholar]
- 83.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115(17):3447–3452. doi: 10.1182/blood-2009-07-233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92(7):905–912. doi: 10.3324/haematol.10937. [DOI] [PubMed] [Google Scholar]