

Abstract
Conventional presentations of mycosis fungoides may be diagnostically challenging, particularly in light of the controversial boundaries defining the disease. Variant presentations of this cutaneous T-cell lymphoma add a further layer of complexity, requiring a sophisticated and informed perspective when evaluating lymphoid infiltrates in the skin. Herein we discuss well-defined (WHO-EORTC) variants pagetoid reticulosis, granulomatous slack skin and folliculotropic mycosis fungoides as well as less well-defined morphologic/architectural variants, and divergent immunohistochemical presentations of this typically indolent T-cell lymphoproliferative disease.
Keywords: Cutaneous T-cell lymphoma, mycosis fungoides, pagetoid reticulosis, granulomatous slack skin, non-Hodgkin, lymphoproliferative disorders, peripheral T-cell lymphoma
Introduction
Mycosis fungoides (MF), the most common primary cutaneous T-cell lymphoma (CTCL), is a clinicopathologically distinctive neoplasm of epitheliotropic skin resident effector memory T-cells.
MF mostly affects older people, and predominantly males. The disease course is protracted and indolent although marked by discomfort and disfigurement. Most patients present with early stage disease1 that responds well to skin directed therapy with recurrences, but no impact on life expectancy compared to age, sex, and race matched controls. A quarter of patients progress to late stages, with significantly diminished overall survival (1.4–4.7 years)2.
MF classically presents with the stepwise appearance of patches, plaques and tumors on non-sun exposed skin with or without eventual extracutaneous involvement1. Microscopically, epidermotropic infiltrates of medium-sized lymphocytes with hyperconvoluted cerebriform nuclei are found. These cells may increase in size, becoming less epitheliotropic with progression to tumor stage. The tumor cells in MF express a mature T-helper memory phenotype, CD3+/CD4+/CD8−/CD45RO+, with monoclonal T-cell receptor (TCR) gene rearrangements of the α/β heterodimer.
The International Society of Cutaneous Lymphoma consensus algorithm for the diagnosis of MF relies upon clinico-pathological correlation supported by immunophenotyping and molecular gene rearrangement studies3. The diagnosis becomes difficult in the absence of classic clinical or histopathologic features. Several clinical variants of MF have been described (Table I) which may correspond to consistent pathological features. Similarly, pathologic/immunophenotypic variants of MF exist (Table II), which may correspond to distinct clinical features. The 2005 WHO-EORTC classification recognizes three variants of MF including folliculotropic MF, pagetoid reticulosis, and granulomatous slack skin1. In this review we discuss these and other unusual variants of MF highlighting their clinical and histopathologic/immunophenotypic features.
Table 1.
Select clinical variants of mycosis fungoides
| Pagetoid reticulosis |
|
|
| Folliculotropic mycosis fungoides |
| Spiky mycosis fungoides |
| Follicular mucinosis |
|
|
| Granulomatous slack skin |
|
|
| Verrucous/hyperkeratotic mycosis fungoides |
|
|
| Hypopigmented mycosis fungoides |
|
|
| Hyperpigmented mycosis fungoides |
|
|
| Poikilodermatous mycosis fungoides |
|
|
| Erythodermic mycosis fungoides |
|
|
| Bullous/vesicular mycosis fungoides |
|
|
| Vegetating/papillomatous mycosis fungoides |
|
|
| Icthyosiform mycosis fungoides |
|
|
| Pigmented purpura-like mycosis fungoides |
|
|
| Unilesional mycosis fungoides |
|
|
| Palmoplantar/acral mycosis fungoides |
|
|
| Pustular mycosis fungoides |
|
|
| Mucosal mycosis fungoides |
|
|
| Intertriginous mycosis fungoides |
Table 2.
Pathologic/immunophenotypic variants of mycosis fungoides
| Folliculotropic mycosis fungoides |
| Basaloid follicular hyperplasia |
| Cysts/comedones |
| Typical folliculotropic with or without mucinosis |
| Granulomatous folliculodestructive |
| Eosinophilic folliculitis-like |
|
|
| Syringotropic mycosis fungoides |
|
|
| Granulomatous mycosis fungoides |
| Interstitial mycosis fungoides |
| Granulomatous slack skin |
|
|
| Spongiotic mycosis fungoides |
|
|
| Mycosis fungoides with epidermal mucinosis |
|
|
| Mycosis fungoides with pseudoepitheliomatous hyperplasia/hyperkeratotic mycosis fungoides |
|
|
| Pigmented purpuric dermatosis-like mycosis fungoides |
|
|
| Transformed mycosis fungoides |
|
|
| CD8+ mycosis fungoides |
| Pagetoid reticulosis pattern |
| Interface predominant pigment-altering variants (hypo/hyperpigmented, poikiloderma) |
| Granulomatous pattern |
|
|
| CD4/CD8 double negative mycosis fungoides |
|
|
| CD4/CD8 double positive mycosis fungoides |
|
|
| CD20+ mycosis fungoides |
|
|
| Gamma delta + mycosis fungoides |
|
|
| T-follicular helper cell + mycosis fungoides |
|
|
| CD30+ mycosis fungoides |
|
|
| Cytotoxic marker (CD56 or TIA-1) + mycosis fungoides |
|
|
| CD45+ mycosis fungoides |
WHO/EORTC variants
Pagetoid reticulosis
Described by Woringer and Kolopp in 1939, it was later renamed pagetoid reticulosis (PR) because of similarity of the epidermotropic lymphocytes to intraepidermal adenocarcinomatous cells of Paget’s disease (PD) of nipple4. PR presents as a single slow-growing hyperkeratotic patch or plaque on acral skin. The clinical differential diagnosis includes solitary plaque psoriasis, Bowen’s disease, unilesional MF, and MF palmaris et plantaris. The typical indolent course does not distinguish PR from these entities, but the pathologic findings are pathognomonic, allowing appropriate treatment by localized irradiation and/or surgical excision, which result in good response although common local recurrence.
Histopathologically, PR is an exuberantly epidermotropic small-to-medium sized atypical lymphoid infiltrate spanning an acanthotic and hyperkeratotic epidermis. Sponge-like disaggregation of the epidermis describes the buck-shot spread of pagetoid lymphocytes in these lesions. Lymphocytes exhibit abundant vacuolated cytoplasm and a haloed appearance. Eosinophils are characteristically absent which may distinguish PR from unilesional or other acral presentations of MF. Superficial spreading melanoma and PD can be excluded with attention to histology. Melanoma typically exhibits irregular nests with melanin pigment, while PD may form ducts, with the “eyeliner sign” of a compressed basal layer below aggregates of Paget’s cells. Occasionally, immunohistochemistry including melanocytic differentiation markers for melanoma or cytokeratin 7 for PD is needed.
Immunophenotypically, PR expresses T-helper cell markers CD3 and CD4 and is negative for CD8. However, CD3+/CD4−/CD8+ and CD3+/CD4−/CD8− cases are well-described. Prognosis does not differ for these cases. CD30 may be positive and the Ki-67 proliferation index is high. The neoplastic T-cells of PR are derived from TCR alpha/beta positive T-cells with rare reported TCR-gamma positive cases. Rare co-expression of TCR α/β and TCR γ in PR is consistent with the suggested origin from TCR gamma/delta positive T-cells5.
Folliculotropic mycosis fungoides
Patients with folliculotropic MF (FMF) present with follicular papules, comedones, acneiform lesions, cysts or indurated plaques and tumors. The lesions are often associated with patches of alopecia (hair or eyebrows) (fig 1A) and severe pruritus with occasional erythroderma. The disease characteristically involves the face, head and neck. Most cases occur in adults, with a male predominance. Occasional cases can be seen in young children and adolescents. Clinically, FMF should be differentiated from other disorders of the pilosebaceous unit including causes of alopecia which may co-exist with FMF.
Fig 1.

Folliculotropic mycosis fungoides A. Alopetic patch on scalp revealing patterned follicle-centered erythema with collarettes of scale. B. Perifollicular erythema and scale with markedly hyperkeratotic follicular plugs in a patient with spiky folliculotropic mycosis fungoides. C. 100x magnification. Atypical lymphocytes extend exuberantly into follicular epithelium, sparing the overlying epidermis with follicular dyskeratosis typical of folliculotropic mycosis fungoides D. 100x magnification. In this photomicrograph of spiky mycosis fungoides, atypical lymphocytes are present within follicular epithelium below a hyperkeratotic column emanating from the hair follicle.
A recently reported clinicopathologic finding, “spiky MF”, is an early presentation of FMF presenting as localized or disseminated variably hyperkeratotic follicular papules (fig 1B, D) in the absence of typical lesions of FMF. Unlike FMF, a head and neck distribution and severe pruritus are not characteristic. Patients may progress to classic FMF. The clinical course is mostly indolent which may be attributed to the relatively superficially located infiltrate.
Histopathologically, FMF is a folliculocentric atypical lymphocytic infiltrate with sparing of the interfollicular epidermis (fig 1C). Mixed inflammatory cells including plasma cells and eosinophils are prominent in perivascular and periadnexal spaces. There may be syringotropism with infiltration of eccrine glands and ducts. Approximately half of FMF show mucinous degeneration of hair follicles, ranging from intercellular spaces expanded by mucinous deposits to mucin lakes compressing epithelium with follicular rupture. Cystic dilation of hair follicles with follicular plugging may be seen. Guitart et al noted five patterns of FMF including basaloid folliculolymphoid hyperplasia, granulomatous dermatitis with destruction of the follicular unit, eosinophilic folliculitis-like FMF, dilated follicular cysts or comedones, and prototypical folliculotropism in intact follicles with or without follicular mucinosis. Suppurative folliculitis has also been described. Different patterns may occur synchronously in patients, and within single biopsy samples. In one series of 38 cases, the most common folliculotropic pattern was observed in 97% of patients, with follicular mucinosis in 87%. Cystic and granulomatous patterns were seen in 37% and 26% patients respectively, while eosinophilic folliculitis-like and basaloid folliculolymphoid hyperplasia–associated MF were observed in only a few cases6.
Follicular epithelium must be present within the biopsy sample in order to diagnose FMF. A common error is failure to perform adequate deeper level sectioning in biopsies intended for evaluation for FMF, resulting in false negative diagnoses. Tiers of parakeratosis, intracorneal neutrophils and arrector pili muscle serve as clues that deeper sections will show evaluable hair follicles.
FMF may be microscopically difficult to distinguish from tumor lesions of classic mycosis fungoides involving hair-bearing skin, granulomatous mycosis fungoides with follicular involvement, folliculocentric lymphomatoid papulosis (LyP), adult T-cell leukemia/lymphoma (ATLL) with a prominently folliculocentric distribution, and T-follicle helper lymphomas which commonly present in hair-bearing skin. FMF often exhibits large cell transformation. The clinical history and exam is invaluable in providing evidence of predominantly follicular accentuation, absence of a waxing and waning course, and absent HTLV-1 serology.
Folliculocentric lymphocytic dermatoses and infections such as lichen planus, discoid lupus erythematosus, granulomatous rosacea, suppurative folliculitis, and HIV-associated eosinophilic folliculitis are common histological differentials which can usually be distinguished by clinical data. While of a controversial relationship to FMF, benign follicular mucinosis presents more in younger patients with a solitary lesion on the head and neck, and a slight perivascular and periadnexal polyclonal infiltrate of bland lymphocytes. Epitheliotropism is not significant, and the CD4:8 ratio is 1:17.
The immunophenotype of FMF is similar to classic MF with CD4+ predominance. CD8+ variants are not unusual. CD30+ blast cells and/or small clusters of CD20+ B-cells may be present. T-follicle helper markers have been described in FMF but do not imply an altered clinical course. However, the presence of notable PD-1+ or B-cells should prompt re-examination of the clinical presentation, as nodules of CD4+ small-to-medium-sized pleomorphic T-cell lymphoproliferative disorders may share the immunophenotype. Strong CD25 expression should prompt serologic evaluation for HTLV-1.
The clinical diagnosis of FMF is often delayed due to the unusual presentation. The deeper infiltrates makes them less amenable to skin directed therapies and may impart a poor treatment response. The prognosis of FMF is worse compared to classic MF with a 5-year overall survival of 75%2. Unilesional follicular MF may respond well to skin directed therapies.
Granulomatous slack skin
Granulomatous slack skin (GSS) is a rare but distinct variant of MF marked by the development of lax folds of skin in the axillae and groin. Very few cases have been reported and an association with Hodgkin’s lymphoma has been suggested8. Most reported cases are in men and have a favorable clinical course.
The histopathology shows dense diffuse granulomatous infiltrates of lymphocytes and histiocytes throughout the dermis and subcutaneous tissue. Multinucleated giant cells with peripherally located nuclei show phagocytosis of lymphocytes and elastic fibers. Epidermotropism is not prominent as in classic MF. Atypical lymphocytes have a CD3+/CD4+/CD8− phenotype.
Non-WHO/EORTC variants
Granulomatous mycosis fungoides
Granulomatous MF (GMF) presents similarly to classic MF or with granuloma annulare (GA)-like lesions.
Histologically, a range of granulomatous reaction patterns is seen, always admixed with atypical lymphocytes exhibiting focal epidermotropism. The most commonly reported pattern is of non-necrotizing epithelioid or sarcoidal granulomas9. In the authors’ study of 27 patients with GMF10, interstitial and perivascular granulomas with multinucleated giant cells and eosinophils were more common. These interstitial granulomas may have prominent histiocytic-rich central regions imparting a GA-like appearance (fig 2A). A minimum histiocytic component of 25% has been suggested for GMF, which can be confirmed with immunohistochemistry for CD68 or CD163 (fig 2B). Histiocytes may be mistaken for or overlap with large cell transformation of MF, which can be resolved immunohistochemically. Folliculotropism is common. Most cases of GMF are CD4+ but there have been a few case reports of CD8+ GMF with no clear influence on prognosis.
Fig 2.

Fig 2A. 200x magnification; granulomatous mycosis fungoides with prominent granuloma annulare-like appearance (palisading of histiocytes within papillary dermis), associated with atypical lymphocytes B. 200x magnification; an immunohistochemical stain for CD68 highlights the 25% component of histiocytes in this case C. 200x magnification; in syringotropic mycosis fungoides, a dense lymphoid infiltrate surrounds the eccrine glands and extends into glandular epithelium D. CD30 immunohistochemical stain, 200x; CD30+ mycosis fungoides with two distinct populations of lymphocytes characterized by CD30 positivity and small (intraepidermal) or large(dermal) cell size.
Some authors speculate that granuloma formation is the result of local tissue response to trauma by infiltrating malignant lymphocytes. It may also be induced by treatment with interferon alfa and bexarotene11.
Once infection has been excluded, the histopathologic differential diagnosis includes interstitial GA, granulomatous drug reaction, cutaneous Rosai-Dorfmann disease, and peripheral T-cell lymphoma e.g. angioimmunoblastic T-cell lymphoma (AITL). The history can help to elucidate a drug etiology. If T-follicle helper cell markers such as PD-1, CXCL-13 and Bcl-6 are noted with clusters of B-cells and EBER-ISH positivity, staging should be considered to evaluate for AITL. S100 and inspection for emperipolesis can help to exclude Rosai-Dorfmann disease. If interstitial GA is within the differential diagnosis, time and multiple biopsies may determine the disease classification. GSS is histologically differentiated by a more prominent and extensive diffuse histiocytic infiltrate with giant cells and loss of elastin fibers, as well as clinically distinct features.
The prognosis and response to treatment is variable but is typically worse for GMF compared to classic MF, with poor response to skin directed therapies. In contrast, patients with GSS have favorable prognosis despite resistance to therapy.
Syringotropic mycosis fungoides
Syringotropic MF (STMF) is also known as syringolymphoid hyperplasia with alopecia, syringotropic CTCL or adnexotropic T-cell lymphoma. STMF is a variant of MF with prominent tropism of the lymphocytic infiltrate to the eccrine glands, associated with syringometaplasia. It is described as a subset of FMF in the WHO-EORTC classification because of shared clinical features and frequent coexisting folliculotropism1. Patients with STMF present with multiple or solitary erythematous papules, patches and plaques with punctate follicular accentuation, often with alopecia overlying the lesions (63–70%). It has been suggested that STMF, despite overlap with FMF, may differ from FMF by a more frequent acral distribution, lower clinical stage at presentation, more common involvement of overlying epidermis and less large cell transformation. STMF has higher overall and disease specific survival, and may thus be better regarded as a distinct entity within the spectrum of adnexotropic MF.
Histopathologically, STMF is characterized by a dense lymphoid infiltrate predominantly surrounding the eccrine glands with glandular epitheliotropism and variable degrees of lymphadenoma-like syringometaplasia (fig 2C). Eccrine coils and acrosyringia may be involved by uniformly small mildly atypical lymphocytes. Epidermotropism is prominent (up to 84%), unlike in FMF where epidermotropism is the exception to the rule. Follicular involvement is seen in up to 68% of cases, and follicular mucinosis in 15–32% of patients12. Immunohistochemistry reveals a CD4+ T-helper cell immunophenotype.
The differential diagnosis for STMF includes neutrophilic eccrine hidradenitis (NEC), perniosis, conventional MF and syringometaplasia devoid of a prominent lymphocytic infiltrate, as in radiation dermatitis, ischemia and post-chemotherapy. NEC is usually distinguishable by a predominance of neutrophils. While perniosis may exhibit a deep perieccrine infiltrate, the lymphocytes do not localize to the eccrine epithelium and syringometaplasia is rare. Conventional MF shows a predominant localization to the epidermis, and when tumorous, may approach the eccrine glands and coils, but neither infiltrates them, nor results in syringometaplasia.
Mycosis fungoides with epidermal mucinosis
In 1986 Nickoloff et al noted colloidal iron positive material in zones of spongiosis in cases of patch/plaque stage MF, proffering the term “epidermal mucinosis”, in which acid mucopolysaccharides caused spongiosis by forming an osmotic gradient13. Recently, Hu et al postulated that these acid mucopolysaccharides were derived from secretory follicular keratinocytes reacting to stimulation by perifollicular T-cell-associated cytokines, after examining biopsies from 18 patients with patch-lesion MF and epidermal mucin in a spongiosis-like pattern. No clinical folliculotropism or vesiculation was identified, although histopathologic vesiculation (eosinophilic deposits in stratum corneum or droplets to large pools of non-Langerhans cell associated mucin within the spinous layers) was present14.
Further review of histopathology showed a superficial infiltrate of atypical lymphocytes, background dermal fibroplasia, and Pautrier’s microabscesses associated with widened intercellular spaces in the epidermis. In 6 biopsies with abundant epidermal mucin, follicular mucin was noted. Dermal mucin was proportional to epidermal mucin. Immunohistochemical analysis showed a CD4+ T-helper cell population with significant loss of CD5 and CD7. Colloidal iron was negative to minimally positive in 18 cases, and moderate in 2 cases.
The differential diagnosis for MF with epidermal mucinosis includes spongiotic dermatitis; this can be discerned by the presence of eosinophilic serum-crust, Langerhans cells within spongiosis, and eosinophils (more than 3 per tissue section, or fewer when spongiosis is PAS positive). Erythrodermic MF and spongiotic MF may exhibit spongiosis-like changes.
Immunophenotypic variants
In MF, the CD3+/CD4+/CD45RO+ memory T-cell phenotype is conventional. When thinking about the variants suggested in the following paragraphs, it may be useful to consider that MF is still best defined by a combination of clinical and histopathologic features, and that our knowledge of these variants is currently incomplete.
CD30+ mycosis fungoides
The majority of MF is CD30 negative by routine immunohistochemistry. MF with CD30 positivity has been described in a low number of cases of non-transformed MF [median dermal positivity 4.7% (range 0–80%), and median epidermal positivity 14%.]15 and in 40% of transformed MF16. The definition of “positive” is subjective; amongst Edinger’s 47 cases, there were only 2 cases in which more than 25% of the cells labeled for CD30, with multiple patterns of labeling noted (scattered, dermal-epidermal junction, clusters), with differing proportions of CD30+ cells in the epidermis and dermis (fig 2D).
The significance of CD30 in MF remains controversial and may differ with stage. In early stage MF, CD30 labeling greater than 50% of epidermal lymphocytes signified no difference in outcomes versus CD30-MF17. However, higher proportions of dermal CD30 have been found to be associated with higher stage at diagnosis, and along with higher epidermal CD30, were associated with higher maximum stage15. Dermal CD30 greater than the median correlated with shorter survival from onset of rash, time of biopsy and time of diagnosis, and higher dermal CD30 when assessed as a continuous variable was associated with an adverse prognosis. However, the association disappeared when CD30 was evaluated as a continuous variable with Ki-67, suggesting that it may be a surrogate marker. In transformed MF, dermal CD30 positivity has been associated with better outcomes18,19.
CD30+ MF may be difficult to distinguish from the CD30+ lymphoproliferative disorders anaplastic large cell lymphoma (ALCL) and LyP. ALCL requires >75% CD30 labeling for diagnosis, and often shows a crisp membranous and perinuclear/Golgi pattern of staining. If systemic, the classic t(2;6) translocation may be indicated by nuclear ALK staining. LyP shows a range of labeling, and requires clinical distinction. HTLV-1 serologies can help to diagnosis ATLL if there is clinical suspicion.
CD30 is currently used to determine eligibility for treatment with anti-CD30 conjugate drugs.
CD20+ mycosis fungoides
The cell-surface antigen CD20 is a transmembrane glycosylated phosphoprotein encoded by the MS4A1 gene and expressed in early stages of B-cell development. It is involved in signal transduction and cell activation and is a marker for B-cell lymphoma and leukemia.
Although CD20 is relatively specific for B-cell differentiation, atypical expression of this pan B-cell marker by T-cell lymphomas has been reported in peripheral T-cell lymphoma. Rarely, cases of CD20+ MF may be seen. CD3 is uniformly positive in such cases, although other pan-T-cell markers may be diminished. CD20 is dim to brightly positive in the same cells, best confirmed with dual-labeling techniques. Other B-cell markers CD79a and PAX5 are negative. Sequential biopsies have revealed increased expression of CD20 by neoplastic cells, associated with disease progression.
Diagnosis may be difficult in these cases, requiring a high index of suspicion and thorough evaluation. Epidermal lymphocytes may be seen in rare cases of B-cell lymphoma, therefore CD20 expression by epidermotropic neoplastic cells might lead to this consideration. The lack of other pan B-cell antigens including CD19, CD22, CD79a, PAX5 and monoclonal IgH gene rearrangements favors the diagnosis of MF in the presence of T-cell markers CD3, CD4 and clonal TCR-gamma gene rearrangement.
Aggregates of CD20+ reactive B-cells are common in classical MF, occasionally causing diagnostic confusion. They are seen more commonly in tumor and transformed tumor lesions of MF than in early stage disease. Such aggregates vary in terms of percentage of tumor mass, and have been suggested to be a marker of poor prognosis, correlating with transformation and outcomes. These aggregates are defined by their distinction from the CD3+ lesional T-cells and do not show co-labeling. Aggregates of B cells can also be seen in so-called composite lymphomas and in AITL.
Targeted therapy with anti-CD20 antibody could be considered in CD20+ MF cases that are refractory to standard therapy.
T-follicular helper (Tfh) cell predominant mycosis fungoides
T-follicular helper cells are a subgroup of T-helper lymphocytes which reside in the germinal center of the lymph node, expressing a CD3+/CD4+/CD10+/PD-1+/CXCL-13+/ICOS+/Bcl-6+ immunophenotype. While no single one of these markers is specific for Tfh cells, the expression of 3 or more is supportive. Guitart and colleagues suggested that nuclear co-labeling of Bcl-6 with cytoplasmic/membranous CD3 best demonstrates a Tfh phenotype in cutaneous lymphoma in the context of a brisk reactive B-cell and histiocytic infiltrate. In their experience, this was most suggestive in intertriginous d’emblee tumors with an aggressive clinical course20. The presence of one or more Tfh markers in MF is increasingly reported, the significance of which is not clear. Caution is warranted in the interpretation of these markers, particularly as the distinction between tumor cells and tumor microenvironment remains challenging.
PD-1 is the best studied Tfh marker. There have been conflicting observations regarding PD-1 expression in MF. In a recent study including 21 patients with MF, Bosisio et al found a complete Tfh phenotype in most cells in some MF, and a partial phenotype in a variable proportion of neoplastic cells in the majority of MF21. The finding of a Tfh phenotype was mostly concordant among biopsies from the same patient, and was not dependent on disease stage with a cut-off of at least 10% of neoplastic cells expressing at least 3 antigens21. PD-1 was the most sensitive marker, followed by ICOS, CXCL-13, Bcl-6 and CD10. It is controversial if PD-1 expression itself changes with evolution of disease. CD10 was detected on 10–40% of CD4+ T-cells in MF, using flow cytometry on skin and blood specimens22. CXCL-13 may be more commonly found in Sezary patients.
The differential diagnosis of TfH positive infiltrates in the skin includes extranodal dissemination of AITL, CD4+ SMPTCL, and Sezary syndrome. AITL in the skin may be clinically morbilliform and histopathologically comprised of bottom heavy dermal infiltrates with abundant histiocytes and B-cells, associated with EBER+ nodal disease. SMPTCL may be identified by a solitary nodular presentation with a low proliferation index. Sezary syndrome requires standard peripheral blood confirmation.
The utility of PD1 as a biomarker for therapeutic decision making is currently under evaluation.
CD8+ mycosis fungoides
Approximately 5–10% of adults with bona fide MF and up to 60% childhood MF present with a CD4−/CD8+ T-cell phenotype. Aggressive CD8+ epidermotropic T-cell lymphomas (CD8+AETCL) with loss of CD2 and CD5, retention of CD7, T-cell restricted intracellular antigen-1 (TIA-1) and or granzyme B (GZB) positivity and ulcerative clinical features have been split off from CD8+ MF, as their distinct clinicopathological and immunophenotypic features are truly divergent. The remaining CD8+ epidermotropic CTCL still considered CD8+ MF have a typically indolent chronic course, good response to topical therapies, retain CD2 and CD5, and lose CD7, with or without TIA-1 expression. The prognosis does not differ from conventional MF23. This group includes predominantly hyperpigmented, hypopigmented (fig 3), poikilodermatous (fig 4), and PR-type clinical presentations. The hypopigmented lesions are most often in children. CD4:CD8 ratios within these cases span a range of 1:1 to greater than 1:20, indicating a background of CD4+ lymphoid or dendritic cells.
Fig 3.
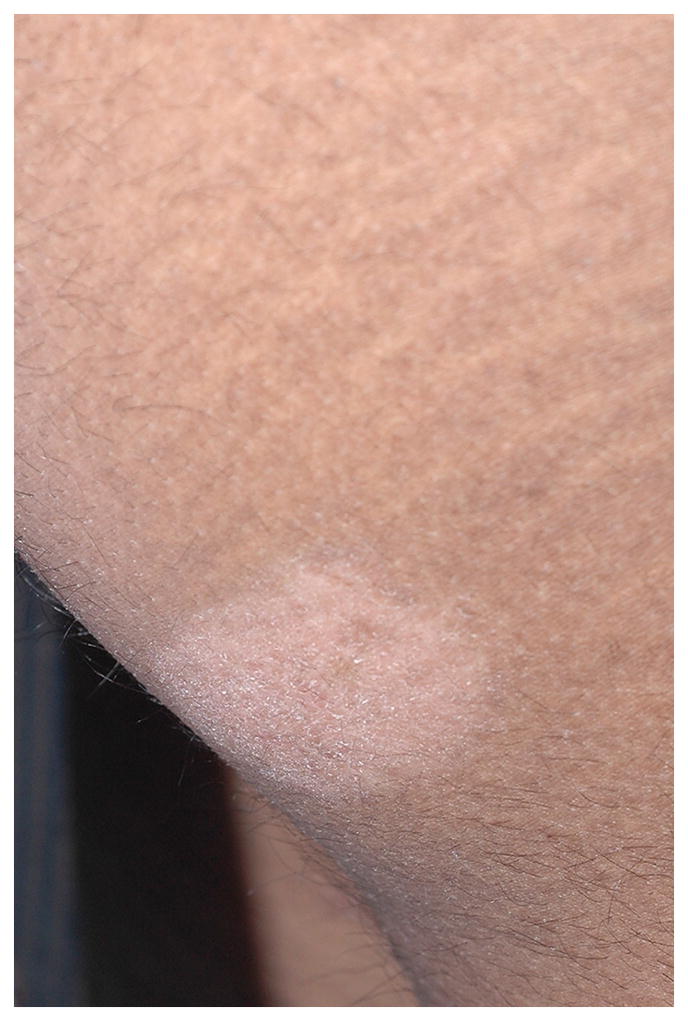
Hypopigmented mycosis fungoides; a typical hypopigmented to achromic patch on the buttock
Fig 4.
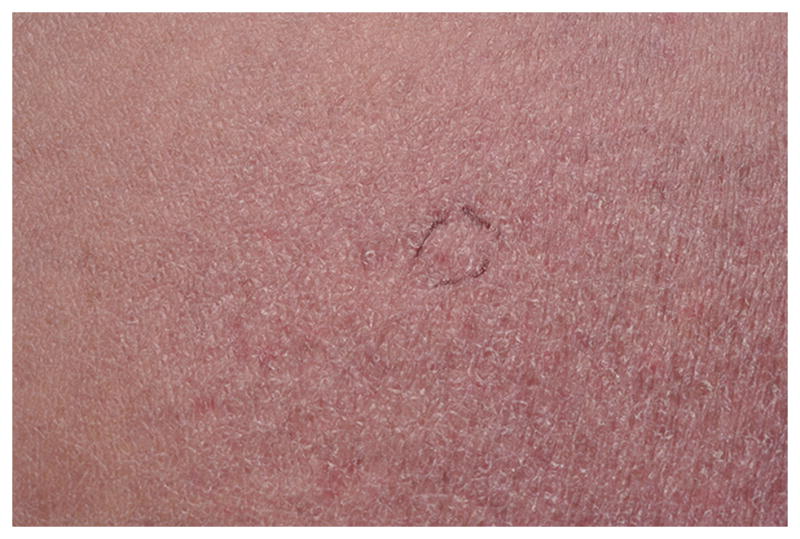
Poikilodermatous mycosis fungoides; an atrophic patch showing both hypo- and hyperpigmentation with telangiectasia
Pathophysiologically, the cytotoxic/suppressor CD8+ T-cells may cause pigmentary changes via exocytosis of cytotoxic granules (perforin, granzyme, TIA-1) or via Fas-FAS-ligand mediated cell death causing damage to the neighboring melanocytes and basilar keratinocytes with melanin displacement, and altered thickness of epidermis (fig 5A). Similar interface findings can be seen in conventional CD4+ patch/plaque stage MF which may harbor CD8+/TIA-1+/GZB-cytotoxic T-lymphocytes24. Decreased CD117 has been noted in epidermal keratinocytes of both CD8+ hypopigmented MF and vitiligo, which could relate to decreased melanocyte differentiation or loss, via cytotoxicity by melanosome antigen specific CD8+ neoplastic T-cells25.
Fig 5.

CD8+ mycosis fungoides A. 400x magnification of an acanthotic lesion with prominent basal layer tagging and marked extension of lymphocytes into mid to upper layers of epidermis. A few necrotic keratinocytes are present B. 400x magnification of an atrophic lesion with marked basilar epidermotropism. Both A and B show few remaining pigmented basilar keratinocytes, but many superficial dermal melanophages, connoting basal layer destruction and melanin dropout. C. 200x CD8 immunohistochemical stain of case in part A showing diffuse staining of all epidermotropic lymphocytes D. 200x CD7 immunohistochemical stain of case in part A shows characteristic complete absence of this pan T-cell antigen
Histopathology shows variably lichenoid, perivascular and/or perifollicular infiltrates of small to medium sized lymphocytes with hyperchromatic, irregular nuclei (fig 5B). Epidermotropism is not as distinct as in the CD4+ variant, with few to no Pautrier’s microabscesses, and more interface changes including keratinocyte apoptosis. Melanophages/melanin drop-out is pronounced. Folliculotropic patterns may be seen. A granulomatous variant has been reported. As with CD4+ MF, CD8+ MF shows loss of CD7 (fig 5C, D). T-cell receptor analysis typically reveals monoclonal alpha-beta gene rearrangements. CD30 is not often positive. CD56 is rare, and is associated with poikiloderma; histologically correlated to epidermal atrophy, basal hydropic degeneration, and telangiectasia. TIA-1 and GZB may be noted in these cases. The clinicopathologic features of poikiloderma may be related to the cytotoxic granule associated proteins in these CD8+ variants. The overall clinical behavior of this subtype is on par with classical MF.
The differential diagnosis includes morphea, lichen sclerosus, ashy dermatosis, and vitiligo. Some cases fit the described characteristics of lichenoid interface T-cell dyscrasia26, which may be best diagnosed retrospectively. Vitiligo may show more basement membrane thickening, complete loss of pigment and melanocytes, with absent lymphocytes, whereas CD8+ hypopigmented MF appears more “active” with hydropic degeneration and numerous lymphocytes. Absent T-cell receptor monoclonal gene rearrangements allows deferral of definitive diagnosis as the clinical picture develops. CD8+AETCL can be discerned by characteristic aggressive clinical features and CD45RA labeling27. CD8+ LyP type D, while epidermotropic, is characterized by waxing and waning papules with spontaneous regression. Pityriasis lichenoides et varioliformis acuta/chronica (PLEVA/PLC) and other benign dermatitis with CD8+ inflammatory cells may have monoclonal TCR gene rearrangement causing diagnostic difficulty.
CD4/CD8 double negative mycosis fungoides
In 1993 Ralfkaier et al reported a case of CD4−/CD8− “double negative” (DN) MF28. By 2012 Kempf et al noted reports of 35 cases in a literature review29. Many of these were from a 2006 series from the Middle East reporting a double negative immunophenotype in 18 (12%) of 140 patients with early MF. CD4 and CD8 expression was reported in less than 10% of intraepidermal T-cells30. 10/18 patients had an unusual clinical presentation including hypopigmented (mostly< age 18 at diagnosis), purpuric, localized, and ichthyosiform. All exhibited indolent behavior, responding to skin directed therapy. As with conventional MF, cases were CD45RO positive, and most expressed TCR-beta. Unusual features included gamma protein expression in one case and a cytotoxic phenotype in others, with 11/15 expressing TIA-1. The authors cited challenges in immunophenotyping the cases because of discordant expression of CD4 and CD8 intradermally and intraepidermally, with many cases showing some CD4 labeling within the epidermis. Immunophenotypic shift was described in two cases, with one CD8+ case becoming double negative, and one double negative case becoming CD8+.
The finding of so many cases in a Middle Eastern population caused speculation that the cell of origin may respond to a particular local antigen response, invoking similarity to “autoimmune lymphoproliferative syndrome” in which null phenotypes are known. However, Kempf et al reported a Swiss patient with a CD4−/CD8− DN MF, noted to also show PD-1 expression (CXCL13-/TIA-1--/BCL-6 minimal); suggesting that upregulation of PD-1 supported antigen stimulated T-cell activation.
Differentially, it is critical to distinguish DN MF from primary cutaneous gamma-delta T-cell lymphomas, which are typically CD4−/CD8−, gamma positive, and clinically aggressive.
TCR gamma+ mycosis fungoides
The gamma-delta complex is expressed by a small number of T-cells in peripheral tissues including skin, and can be identified by the antibody gamma M1 on formalin fixed paraffin embedded tissue (FFPE). In 1991 and 1992 Burg et al31 and Ralfkaier et al32 reported gamma-delta positive CTCLs which differed from conventional MF by exhibiting CD45RO and CD45RA positivity, and aggressive behavior. Recent analyses of TCR-gamma protein expression in large numbers of CTCL have shown that approximately 2% of conventional clinical MF cases are TCR-gamma positive. This immunophenotype does not seem to alter prognosis.
The differential diagnosis is important. Primary cutaneous gamma-delta T-cell lymphoma is an aggressive cytotoxic cutaneous T-cell lymphoma with limited therapeutic options, unlike gamma+ MF. Other cutaneous lymphomas which may express gamma-TCR include CD8+ lichenoid and epidermotropic LyP type D, which may co-express TCR Beta-F1. A subset of self-limited cytotoxic lymphocytic dermatitis, pityriasis lichenoides, in which clonal T-cell gene rearrangements are sometimes identified, may also exhibit a gamma-delta T-cell phenotype, with no apparent clinical bearing. Clinical correlation is critical in distinguishing each of these entities.
It was previously said that the absence of BetaF1 expression could serve as a surrogate for gamma positivity in the characterization of lymphoma, however, this is no longer considered adequate by many practitioners, particularly in light of the stark prognostic/diagnostic significance that this marker may impart.
TIA-1+ mycosis fungoides
T-cell restricted intracellular antigen-1 is expressed by resting and activated cytotoxic CD4 or CD8 positive T-cells and natural killer cells. A study of 74 cases of MF found that TIA-1 labeled >10% of cells in 37% of cases, with increased expression in stage T3 versus T2 overall, and increased expression in sequential biopsies in 5/8 cases. TIA-1 expression in the first diagnostic biopsies did not correlate with prognosis. TIA-1 did not correlate with blastic transformation33. As described above, CD4−/CD8− DN MF is commonly TIA-1 positive. CD8+/TIA-1+ MF, while rare, may be more common in adult females with poikilodermatous lesions and an indolent course.
Strong expression of such cytotoxic markers in MF should cause consideration of other cytotoxic lymphomas, such as CD30+ spectrum of lymphoproliferative disorders or CD8+ AETCL. The clinical picture usually allows the distinction with ease.
CD56+ mycosis fungoides
CD56 is a homophilic binding glycoprotein expressed on NK cells and rarely in MF, with 8 cases reported in the literature8,34,35. Most cases were CD8+, of younger age (6–45 years old at onset), and clinically poikilodermatous as with CD8+/CD56− MF. One case was tumor stage. All were indolent and responded well to therapy, similarly to the indolent course described in CD56+ SS and LyP.
The increased CD56+ MF in young patients mirrors CD8+ positivity in this group. It has been suggested to reflect cytotoxic responses to antigenic stimulation after more frequent first time exposure to viruses in childhood, causing activation of T-cells.
Histopathologically, CD56+ MF shows cytotoxic interface dermatitis with basal hydropic degeneration, pigment incontinence and telangiectasia. Immunophenotypically, CD7 is lost, contrasting CD8+ AETCL. One case was CD4+/CD8−36. Two cases were CD4−/CD8− double negative30. Cytotoxic mediators GZB and perforin have been positive.
CD45RA+ mycosis fungoides
While MF is typically characterized by a CD3+/CD4+/CD8−/CD45RO+ phenotype, cases of CD45RA positive CTCL have been identified. Fierro et al found that 22/215 cases (8.7%) of MF were CD45RA+. Twenty of these were conventional patch/plaque stage indolent disease37. Two cases were aggressive, suggesting another lymphoma subtype. The differential diagnosis for CD45RA+ CTCL includes CD8+AECTCL, gamma-delta TCL, and PR. Gamma M1 and clinical history can assist with the distinction.
Summary
Mycosis fungoides is the most common primary cutaneous T-cell lymphoma. It has varied clinical presentation with differing histological findings making it challenging to diagnose. Definitive diagnosis of MF is based on clinicopathological correlation, supported by immunohistochemical and molecular studies. The neoplastic cells in most cases are CD3+/CD4+/CD8−/CD45RO+ mature T-helper memory cells with a T-cell receptor of alpha/beta heterodimer. Demonstration of monoclonality of T-cell receptor and loss of pan T-cell antigens CD2, CD3 and CD5 are important adjuncts in the diagnosis. Regular follow up and repeat biopsies often help in arriving at a diagnosis in suspicious cases.
Recognition of unusual clinical and pathological variants of MF is important for early diagnosis and appropriate management. The disease course and prognosis of some of the rare immunophenotypic variants have been less well characterized. They are often misdiagnosed initially leading to delay in diagnosis. Immunophenotypic characterization of cutaneous lymphomas is important to prevent misdiagnosis and inappropriate treatment.
Practice points.
The typical immunophenotype of mycosis fungoides is CD3+/CD4+/CD8−/CD45RO+
Immunophenotyping is not usually necessary when clinical and histologic characteristics are conventional
If an aberrant histology (interface dermatitis, mucin, syringotropism) or immunophenotype is identified in an atypical lymphoid proliferation in the skin, then clinicopathologic correlation is always required
Footnotes
Conflicts of interest: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Pooja Virmani, Department of Medicine/Dermatology Service, Memorial Sloan Kettering Cancer Center, New York, USA.
Patricia L. Myskowski, Department of Medicine/Dermatology Service, Memorial Sloan Kettering Cancer Center, New York, USA.
Melissa Pulitzer, Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA.
References
- 1.Willemze R, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–3785. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 2.Agar NS, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sezary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730–4739. doi: 10.1200/JCO.2009.27.7665. [DOI] [PubMed] [Google Scholar]
- 3.Olsen E, et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC) Blood. 2007;110:1713–1722. doi: 10.1182/blood-2007-03-055749. [DOI] [PubMed] [Google Scholar]
- 4.Haghighi B, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502–510. doi: 10.1038/modpathol.3880088. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Pinilla SM, et al. TCR-gamma expression in primary cutaneous T-cell lymphomas. Am J Surg Pathol. 2013;37:375–384. doi: 10.1097/PAS.0b013e318275d1a2. [DOI] [PubMed] [Google Scholar]
- 6.Demirkesen C, Esirgen G, Engin B, Songur A, Oguz O. The clinical features and histopathologic patterns of folliculotropic mycosis fungoides in a series of 38 cases. J Cutan Pathol. 2015;42:22–31. doi: 10.1111/cup.12423. [DOI] [PubMed] [Google Scholar]
- 7.Rongioletti F, et al. Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides-associated follicular mucinosis. J Cutan Pathol. 2010;37:15–19. doi: 10.1111/j.1600-0560.2009.01338.x. [DOI] [PubMed] [Google Scholar]
- 8.Poppe H, et al. Childhood mycosis fungoides with a CD8+ CD56+ cytotoxic immunophenotype. J Cutan Pathol. 2015;42:258–264. doi: 10.1111/cup.12452. [DOI] [PubMed] [Google Scholar]
- 9.Dabski K, Stoll HL., Jr Granulomatous reactions in mycosis fungoides. Journal of surgical oncology. 1987;34:217–229. doi: 10.1002/jso.2930340402. [DOI] [PubMed] [Google Scholar]
- 10.Li JY, et al. A case-control study of clinicopathologic features, prognosis, and therapeutic responses in patients with granulomatous mycosis fungoides. J Am Acad Dermatol. 2013;69:366–374. doi: 10.1016/j.jaad.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 11.Kempf W, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization For Research and Treatment of Cancer (EORTC) Arch Dermatol. 2008;144:1609–1617. doi: 10.1001/archdermatol.2008.46. [DOI] [PubMed] [Google Scholar]
- 12.de Masson A, et al. Syringotropic mycosis fungoides: clinical and histologic features, response to treatment, and outcome in 19 patients. J Am Acad Dermatol. 2014;71:926–934. doi: 10.1016/j.jaad.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 13.Nickoloff BJ. Epidermal mucinosis in mycosis fungoides. J Am Acad Dermatol. 1986;15:83–86. doi: 10.1016/s0190-9622(86)70147-3. [DOI] [PubMed] [Google Scholar]
- 14.Hu SW, Ratech H, Naeem R, Latkowski JA, Kamino H. Mycosis fungoides with epidermal mucinosis: a variant of mycosis fungoides with a spongiosis-like pattern. J Cutan Pathol. 2015;42:730–738. doi: 10.1111/cup.12513. [DOI] [PubMed] [Google Scholar]
- 15.Edinger JT, Clark BZ, Pucevich BE, Geskin LJ, Swerdlow SH. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860–1868. doi: 10.1097/PAS.0b013e3181bf677d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arulogun SO, et al. Long-term outcomes of patients with advanced-stage cutaneous T-cell lymphoma and large cell transformation. Blood. 2008;112:3082–3087. doi: 10.1182/blood-2008-05-154609. [DOI] [PubMed] [Google Scholar]
- 17.Wu H, Telang GH, Lessin SR, Vonderheid EC. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212–216. doi: 10.1097/00000372-200006000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Barberio E, Thomas L, Skowron F, Balme B, Dalle S. Transformed mycosis fungoides: clinicopathological features and outcome. Br J Dermatol. 2007;157:284–289. doi: 10.1111/j.1365-2133.2007.08008.x. [DOI] [PubMed] [Google Scholar]
- 19.Pulitzer M, et al. Mycosis fungoides with large cell transformation: clinicopathological features and prognostic factors. Pathology. 2014;46:610–616. doi: 10.1097/PAT.0000000000000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guitart J, Gammon B. The difficulties in defining follicular T helper phenotype in cutaneous lymphomas. Am J Dermatopathol. 2013;35:691. doi: 10.1097/DAD.0b013e31827eaebd. [DOI] [PubMed] [Google Scholar]
- 21.Bosisio FM, Cerroni L. Expression of T-follicular helper markers in sequential biopsies of progressive mycosis fungoides and other primary cutaneous T-cell lymphomas. Am J Dermatopathol. 2015;37:115–121. doi: 10.1097/DAD.0000000000000258. [DOI] [PubMed] [Google Scholar]
- 22.Meyerson HJ, et al. Follicular center helper T-cell (TFH) marker positive mycosis fungoides/Sezary syndrome. Mod Pathol. 2013;26:32–43. doi: 10.1038/modpathol.2012.124. [DOI] [PubMed] [Google Scholar]
- 23.Massone C, Crisman G, Kerl H, Cerroni L. The prognosis of early mycosis fungoides is not influenced by phenotype and T-cell clonality. Br J Dermatol. 2008;159:881–886. doi: 10.1111/j.1365-2133.2008.08761.x. [DOI] [PubMed] [Google Scholar]
- 24.Vermeer MH, et al. CD8+ T cells in cutaneous T-cell lymphoma: expression of cytotoxic proteins, Fas Ligand, and killing inhibitory receptors and their relationship with clinical behavior. J Clin Oncol. 2001;19:4322–4329. doi: 10.1200/JCO.2001.19.23.4322. [DOI] [PubMed] [Google Scholar]
- 25.Singh ZN, Tretiakova MS, Shea CR, Petronic-Rosic VM. Decreased CD117 expression in hypopigmented mycosis fungoides correlates with hypomelanosis: lessons learned from vitiligo. Mod Pathol. 2006;19:1255–1260. doi: 10.1038/modpathol.3800644. [DOI] [PubMed] [Google Scholar]
- 26.Magro CM, et al. Hypopigmented interface T-cell dyscrasia: a form of cutaneous T-cell dyscrasia distinct from hypopigmented mycosis fungoides. J Dermatol. 2014;41:609–617. doi: 10.1111/1346-8138.12458. [DOI] [PubMed] [Google Scholar]
- 27.Berti E, et al. Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. A distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483–492. doi: 10.1016/S0002-9440(10)65144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ralfkiaer E, Wollf-Sneedorff A, Thomsen K, Vejlsgaard GL. Immunophenotypic studies in cutaneous T-cell lymphomas: clinical implications. Br J Dermatol. 1993;129:655–659. doi: 10.1111/j.1365-2133.1993.tb03327.x. [DOI] [PubMed] [Google Scholar]
- 29.Kempf W, et al. CD4/CD8 double negative mycosis fungoides with PD-1 (CD279) expression--a disease of follicular helper T-cells? Am J Dermatopathol. 2012;34:757–761. doi: 10.1097/DAD.0b013e31825b26d1. [DOI] [PubMed] [Google Scholar]
- 30.Hodak E, et al. CD4/CD8 double-negative epidermotropic cutaneous T-cell lymphoma: an immunohistochemical variant of mycosis fungoides. J Am Acad Dermatol. 2006;55:276–284. doi: 10.1016/j.jaad.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 31.Burg G, et al. A subcutaneous delta-positive T-cell lymphoma that produces interferon gamma. N Engl J Med. 1991;325:1078–1081. doi: 10.1056/NEJM199110103251506. [DOI] [PubMed] [Google Scholar]
- 32.Ralfkiaer E, Wollf-Sneedorff A, Thomsen K, Geisler C, Vejlsgaard GL. T-cell receptor gamma delta-positive peripheral T-cell lymphomas presenting in the skin: a clinical, histological and immunophenotypic study. Exp Dermatol. 1992;1:31–36. doi: 10.1111/j.1600-0625.1992.tb00069.x. [DOI] [PubMed] [Google Scholar]
- 33.Vermeer MH, Geelen FA, Kummer JA, Meijer CJ, Willemze R. Expression of cytotoxic proteins by neoplastic T cells in mycosis fungoides increases with progression from plaque stage to tumor stage disease. Am J Pathol. 1999;154:1203–1210. doi: 10.1016/S0002-9440(10)65372-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawada Y, et al. CD8+ CD56+ mycosis fungoides with an indolent clinical behaviour: case report and literature review. Acta Derm Venereol. 2010;90:525–526. doi: 10.2340/00015555-0831. [DOI] [PubMed] [Google Scholar]
- 35.Shiomi T, et al. Poikilodermatous mycosis fungoides with a CD8+ CD56+ immunophenotype: a case report and literature review. J Cutan Pathol. 2013;40:317–320. doi: 10.1111/cup.12067. [DOI] [PubMed] [Google Scholar]
- 36.Horst BA, Kasper R, LeBoit PE. CD4+, CD56+ mycosis fungoides: case report and review of the literature. Am J Dermatopathol. 2009;31:74–76. doi: 10.1097/DAD.0b013e31818ffbe8. [DOI] [PubMed] [Google Scholar]
- 37.Fierro MT, et al. CD45RA+ immunophenotype in mycosis fungoides: clinical, histological and immunophenotypical features in 22 patients. J Cutan Pathol. 2001;28:356–362. doi: 10.1034/j.1600-0560.2001.280704.x. [DOI] [PubMed] [Google Scholar]