

Abstract
Synthetic organic chemists have a long-standing appreciation for transition metal cyclopentadienyl complexes, of which many have been used as catalysts for organic transformations. Much less well known are the contributions of the benzo-fuzed relative of the cyclopentadienyl ligand, the indenyl ligand, whose unique properties have in many cases imparted differential reactivity in catalytic processes toward the synthesis of small molecules. In this review, we will present examples of indenylmetal complexes in catalysis and compare their reactivity to their cyclopentadienyl analogues, wherever possible.
Keywords: organic synthesis, organometallic chemistry, cyclopentadienyl, indenyl, coordination chemistry
Graphical Abstract
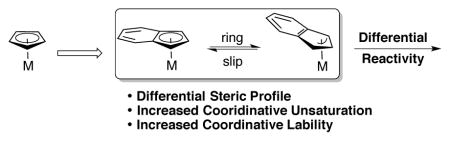
1. Introduction
Ever since Wilkinson, Woodward, and Fischer elucidated the bonding structure of ferrocene in 1952,[1] the cyclopentadienyl (Cp) ligand has been seen as a cornerstone of modern organometallic chemistry. While this ligand exhibits unique electronic properties and a fascinating bonding motif[2] that has been implemented in many scaffolds for catalysis,[3] it often behaves as a three-coordinate spectator ligand that does not directly participate in reactions. However, when an additional benzo ring is fused to the Cp ring to form a metal indenyl (Ind) complex, a noticeable acceleration of fundamental organometallic reactions and catalysis is observed.
This effect was first seen by Hart-Davis and Mawby while they were studying the rate of ligand-induced migratory insertion reactions of coordinatively saturated 18-electron molybdenum carbonyl complexes (Scheme 1).[4] An increase in rate of around an order of magnitude was observed when comparing the indenyl complex to the analogous cyclopentadienyl complex. The origin for this increase was independent of the identity of the phosphine ligand used to trigger migratory insertion and could not be explained by the difference in electronic structure of the indenyl moiety, as the Ind complex exhibited nearly identical carbonyl stretching frequencies as the Cp complex. A negative entropy of activation (ΔS‡ = −24.2 cal/mol*K in n-hexane with P(nBu)3 as ligand) implies an associative mechanism for this reaction. Coordination by an exogenous ligand via an associative mechanism would necessitate the intermediacy of a high-energy 20-electron coordination complex. Therefore, the authors proposed a mechanism whereby the η5-indenyl ligand slips into an η3-allyl coordination mode to accommodate the additional ligand necessary to induce migratory insertion. X-ray crystal structures of indenyl complexes have provided direct evidence of ring slippage by correlation to the degree of folding of the indenyl ligand.[5] The additional stabilization of the fused benzo ring on the indenide facilitates this ring slippage, and therefore accelerates the rate of the reaction.
Scheme 1.
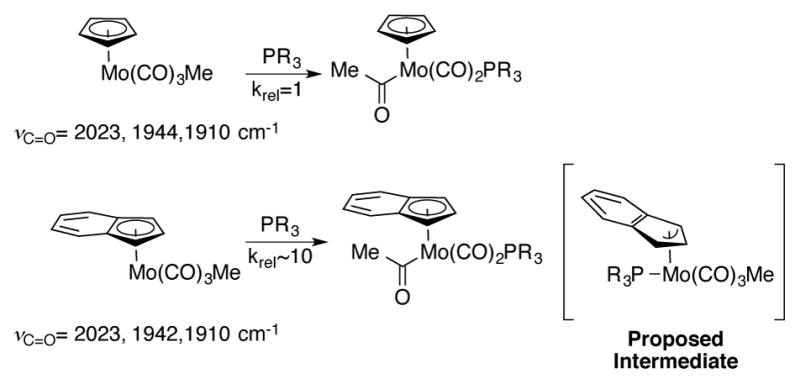
Rate acceleration of migratory insertion reactions observed with indenylmolybdenum carbonyl complexes
Similar effects in rate acceleration have also been observed for CO[6] and ethylene[7] ligand substitution reactions. In one such study by Basolo and coworkers,[8] the observed rate of carbonyl substitution by triphenylphosphine on a coordinatively saturated indenylrhodium(I) complex was observed to be 108 times faster than with the analogous cyclopentadienylrhodium complex [Eq. (1)]. Again, the increase in rate is attributed to the ease in which the metal is able to
| (1) |
adopt an η3 coordination geometry in an associative substitution mechanism.
Many chemists have noted the special properties the indenyl ligand imbues a coordination complex and have used these compounds to increase the rates of catalytic reactions. The purpose of this review is to present these cases where these indenylmetal complexes have been used in the context of catalysis, particularly for the synthesis of small, organic molecules. The use of indenylmetal complexes for polymerization reactions, including the work of Brintzinger,[9] is outside the scope of this review, and has been reviewed elsewhere.[3],[10]
2. Titanium, Zirconium, and Rare Earth Metals
Due to the success of chiral indenyltitanium and -zirconium complexes in the context of stereoselective olefin polymerization, it is unsurprising that researchers would utilize similar complexes for the catalytic transformation of small molecules. In 1992, Halterman and coworkers disclosed an asymmetric olefin migration reaction of cyclohexane 1 using chiral bis(indenyl) titanium complex 2 (Scheme 2).[11] One of the disadvantages of using planar chiral Ziegler-Natta olefin polymerization catalyst 3 is that it must be resolved from a racemic mixture of complexes. On the other hand, the synthesis of catalyst 2 is completely diastereoselective due to the chirality already present on its backbone. The bulky indenyl moieties prefer to orient themselves away from the isopropyl groups on the cyclohexane ring, which results in the complex seen in Scheme 2. When substrate 1 is subjected to 2 mol% of catalyst 2 along with 8 mol% of lithium aluminum hydride, quantitative conversion and a 74% ee of alkene 4 could be obtained. A strong temperature dependence was observed; higher temperatures led to significant erosion of enantioselectivity. Although a direct comparison of reactivity to a cyclopentadienyl complex was not made in this publication, previous studies[12] have indicated that terminal olefins that contain α-branching similar to 1 have been problematic olefin isomerization substrates for achiral Cp2TiCl2. The Halterman group have synthesized other bis(indenyl)titanium and zirconium complexes with chiral backbones, including binapththyl-based complex 5 (Figure 1).[13] This complex was used to study the catalytic asymmetric epoxidation of electronically unbiased olefins, though low turnovers and ee’s were observed with this system.
Scheme 2.
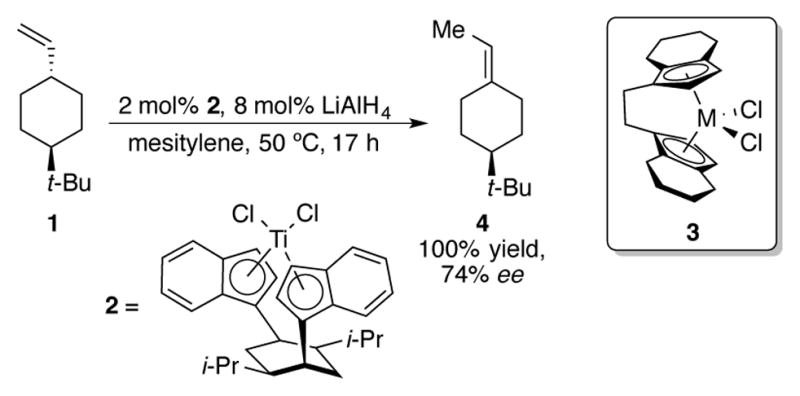
Asymmetric olefin isomerization with chiral bis(indenyl)titanium catalyst 2
Figure 1.
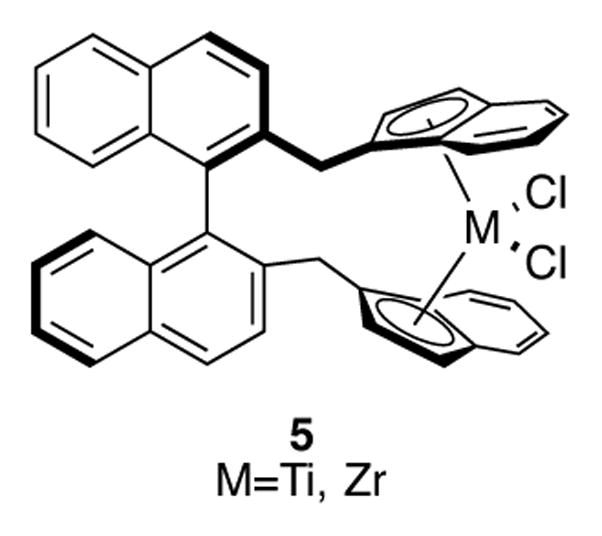
Binaphthyl-based complex 5.
Menthyl-derived indenylzirconium complexes like 6 have been shown to be an excellent catalyst for the stereoregular polymerization of polypropylene (Scheme 3).[14] Realizing that these complexes would be a readily available source of chiral information for zirconium-catalyzed asymmetric carboalumination (ZACA) reaction, Negishi et al. observed good enantioselectivities of 8 for the addition of Me3Al to terminal alkenes 7.[15–17] A subsequent publication noted that carboalumination of longer chain organometallics such as Et3Al and nPr3Al proceed to even higher enantioselectivities (>90% ee).[18] The proposed rationale for the stereochemical outcome of this reaction is that the zirconium complex enforces the preferential coordination of the olefin face seen in intermediate 9 by placing the olefin substituent away from its bulky indenyl ligands. Interestingly, attempted methylalumination of 1-octene with Cp2ZrCl2 resulted solely in products related to β-hydride elimination. The authors speculate that the bulky indenyl ligands prevent this side reaction from occurring. In addition to simple olefins, the Negishi group has also extended their studies of the ZACA reaction toward allyl alcohols,[19] 1,4-dienes,[20] and 1,4-pentenynes.[21]
Scheme 3.
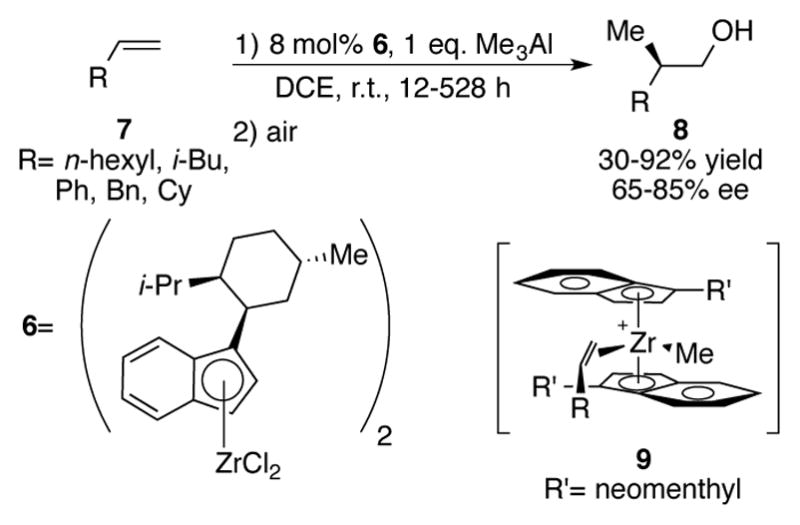
Negishi’s Zr-catalyzed asymmetric carboalumination (ZACA)
Wipf and Ribe noted a significant increase in rate as well as a slight increase in enantioselectivity of ZACA reactions when 1 equivalent of water was added to the reaction.[22] The origin of this rate increase was attributed to the in situ partial hydrolysis of trimethylaluminum reagent. Using methylaluminoxane (MAO) instead of trimethylaluminum was also observed to increase reaction rate, albeit to a lesser extent.
The ZACA process can be used to stereoselectively construct cyclic products when α,ω-dienes are used as substrates (Scheme 4). Waymouth and coworkers subjected 1,5-hexadiene 10 to modified ZACA conditions, resulting in a 62% yield of cyclopentane 11 as a 2:1 ratio of trans:cis diastereomers and in a 79% ee for the major diastereomer.[23] Tris(pentafluorophenyl)borane[24] was added as a co-catalyst to abstract a methyl group from catalyst precursor 12.[25] Mechanistically, after the first intermolecular migratory insertion reaction, alkylzirconium intermediate 13 is primed to perform a second intramolecular carbometallation reaction, furnishing the observed product. A reversal in diastereoselectivity (3:7 trans:cis) is observed when Cp*2ZrCl2 (Cp*=1,2,3,4,5-pentamethylcyclopentadienyl) is used as a catalyst in the same reaction.
Scheme 4.
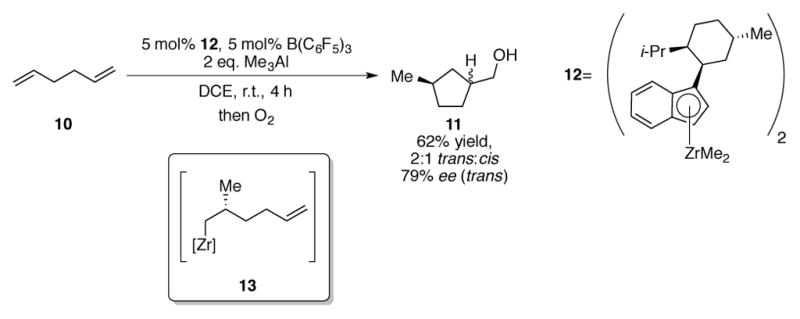
Zr-catalyzed asymmetric carboalumination (ZACA) of α,ω-dienes
Waymouth et al. have also used ZACA catalysts for desymmetrizing ring-opening reactions of oxanorbornene derivatives 14 (Scheme 5).[26] They discovered that the nature of the zirconocene derivative was crucial to both the absolute configuration and enantioselectivity of the overall process. bis(Neoisomenthylindenyl) 16 catalyzed the ring opening of 14 in a 64% yield and an excellent 96% ee. Catalyst diastereomer bis(neomenthylindenyl) 6 delivered the antipode of 15 in a −52% ee. In comparison, catalysis with achiral titanocene Cp2TiCl2 required higher reaction temperatures and provided 15 in a low 16% yield. Whether the improved reactivity of catalysts 16 and 6 is due to the indenyl effect or due to the difference in reactivity between a first and second row transition metal is unclear.
Scheme 5.
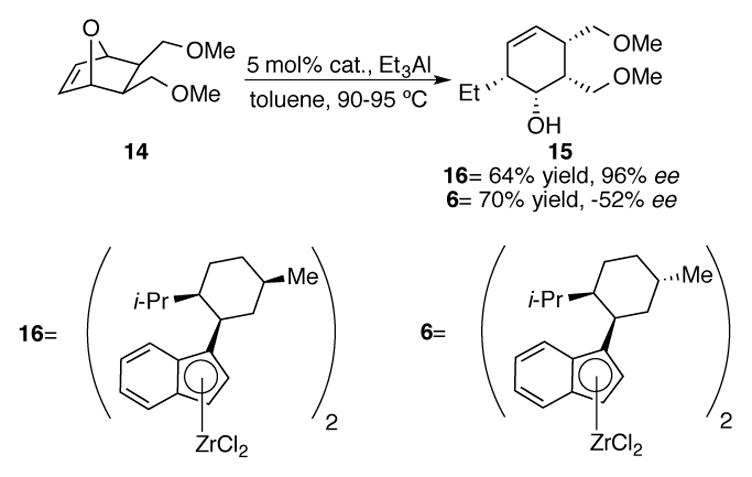
Asymmetric desymmetrization of oxanorbornenes
Negishi broadened the scope of alkylaluminum species that can be used by reporting the tandem hydroalumination/ZACA of terminal olefins (Scheme 6).[27] After hydroalumination of a terminal olefin by diisobutylaluminum hydride (DIBAL-H), the less sterically encumbered alkyl chain transmetallates to zirconium in preference to the sterically bulky isopropyl groups on aluminum. While this reaction displays good yields and excellent enantioselectivities, the drawbacks include long reaction times necessary to generate the diisobutylalkylaluminum reagent and the requirement that only terminal olefins can be used for hydroalumination. Nevertheless, this method was used to synthesize the vitamin E side chain 17[28] in four steps and 45% overall yield from 4-methyl-1-pentene.
Scheme 6.
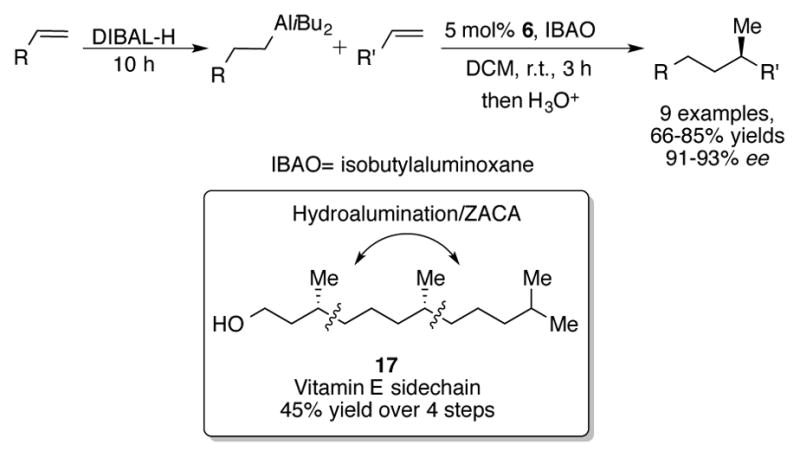
Tandem hydroalumination/ZACA reaction
In addition to oxidation to primary alcohols and protonolysis, alkylaluminum intermediates obtained from ZACA processes can be used for one-pot Pd- and Cu-catalyzed cross-coupling reactions.[29] The ability to append on olefins via cross-coupling enables an iterative ZACA/cross-coupling approach to the synthesis of complex molecules. An impressive example of this strategy was the utilization of multiple, consecutive ZACA/cross-coupling reactions for the enantioselective synthesis of phthioceranic acid 18, which contains seven deoxypropionate subunits (Scheme 7).[30] ZACA with ent-6 on 1-octadecene 19 resulted in alkylaluminum intermediate 20, which could be directly cross-coupled with vinyl bromide after transmetallation to zinc. A second ZACA reaction followed by oxidation constructed the second tertiary stereocenter in 22 in a 44% yield over two steps. Primary alcohol 22 was isolated in a greater than 50:1 d.r. and 97% ee. Bromination of 22 to 23 completed the first key fragment of the synthesis of 18.
Scheme 7.
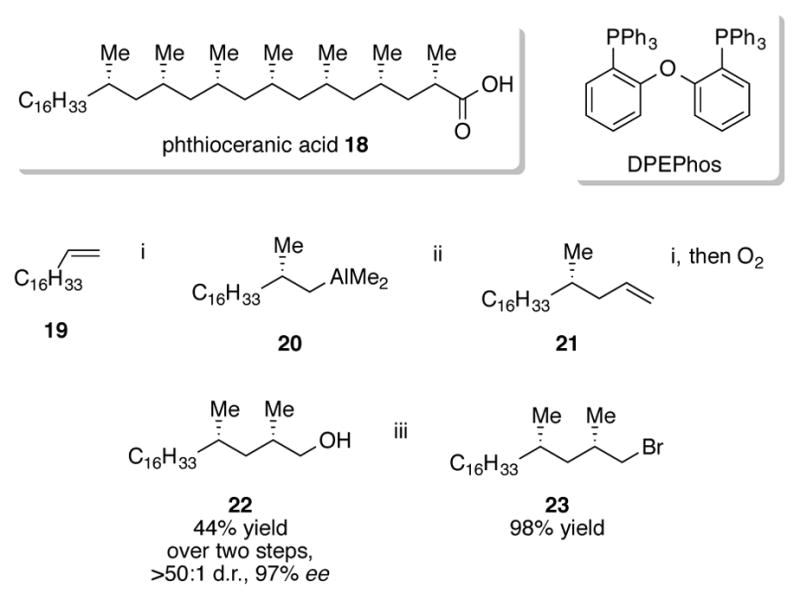
Iterative ZACA reactions for the total synthesis of phthioceranic acid 18
Conditions: i. 1 mol% [(+)-(NMI)2ZrCl2] ent-6, 1.5 eq. AlMe3, 1 eq. H2O, DCM, 0 °C; ii. 1.2 eq. Zn(OTf)2, 3 mol% [PdCl2(DPEPhos),] 6 mol% DIBAL-H, 6 eq. vinyl bromide, DMF, r.t., iii. NBS, PPh3, DCM, 0 °C.
The second key fragment was constructed in a similar fashion (Scheme 8). ZACA with 6 and Negishi cross-coupling furnished 24. This alkene was then subjected to another carboalumination reaction, followed by in situ oxidation to alcohol 25. This alcohol was isolated in a 34% overall yield over two steps and in >30:1 d.r.. Iodination of 25, lithium-halogen exchange, and Negishi cross-coupling of vinyl bromide introduces the terminal alkene necessary for the final carboalumination reaction of 26. ZACA of 26 and bromination of the resulting alcohol results in the construction and isolation of the second key intermediate 27 towards the synthesis of phthioceranic acid. Bromide 27 could be isolated in greater than 50:1 d.r. and greater than 99% ee.
Scheme 8.
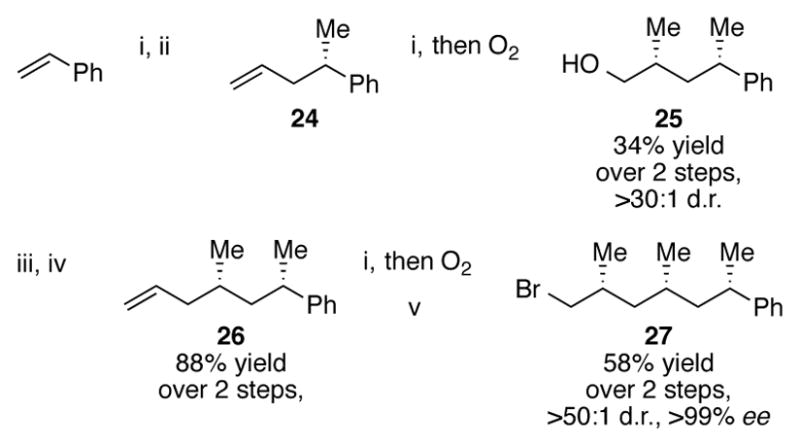
Second key fragment in the construction of phthioceranic acid 18
Conditions: i. 2 mol% [(−)-(NMI)2ZrCl2] 6, 2–3 eq. AlMe3, 1 eq. H2O, DCM, 0 °C; ii. 1.2 eq. Zn(OTf)2, 3 mol% [PdCl2(DPEPhos),] 6 mol% DIBAL-H, 6 eq. vinyl bromide, DMF, r.t., iii. PPh3, I2, imidazole, DCM; iv. tBuLi, Et2O, −78°C then ZnBr2, THF, −78 °C to 0 °C then 5 mol% [PdCl2(DPEPhos),] 10 mol% DIBAL-H, 4 eq. vinyl bromide, 0 °C to r.t., v. NBS, PPh3, DCM, 0 °C.
The two fragments were then brought together using stereospecific copper-catalyzed SN2 substitution reactions (Scheme 9). Fragment 28, which can be synthesized in three steps from acetylacetone, undergoes sp3-sp3 cross-coupling of its tosylate with the in situ-formed Grignard reagent of 23 with complete inversion of configuration. After functional group interconversion to tosylate 30, a second copper-catalyzed coupling using the alkylmagnesium of 27 was performed with complete stereospecificity. Oxidation of the phenyl group to a carboxylic acid completes this highly convergent synthesis of phthioceranic acid 18 in a 45% yield over two steps. The Negishi group and others have also used the strategies described in this synthesis for the total and partial synthesis of other natural products with deoxypropionate subunits.[31]
Scheme 9.
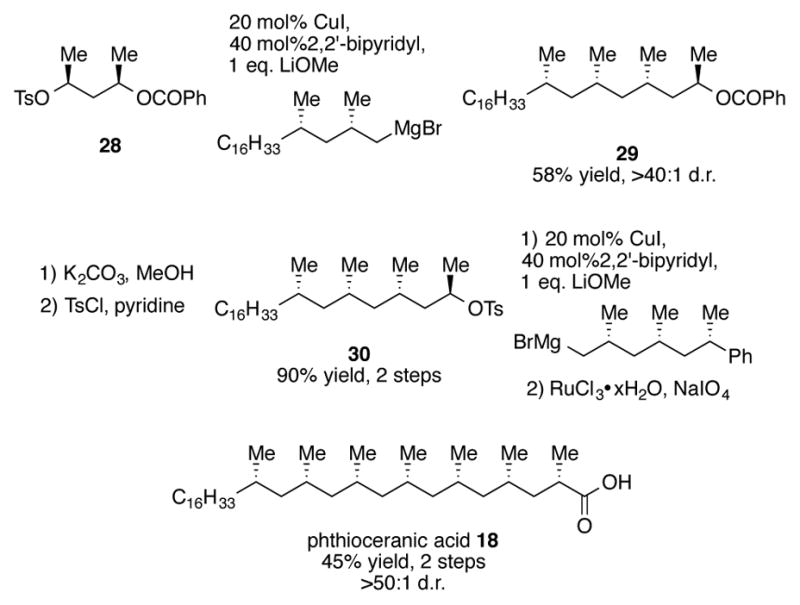
Endgame of Negishi’s total synthesis of phthioceranic acid 18
Zirconocene complex 31 (Figure 2) was found by Lipshutz et al. to be a highly active and regioselective catalyst for the methylalumination of terminal alkynes.[32] Carbon-carbon bond formation occurs selectively on the internal position of a terminal alkyne so that steric interactions between substrate and catalyst are minimized. Though not explicitly measured, the authors noted a significant faster rate of reaction when using 31 compared to bis(tetrahydroindenyl)zirconocene 3.
Figure 2.
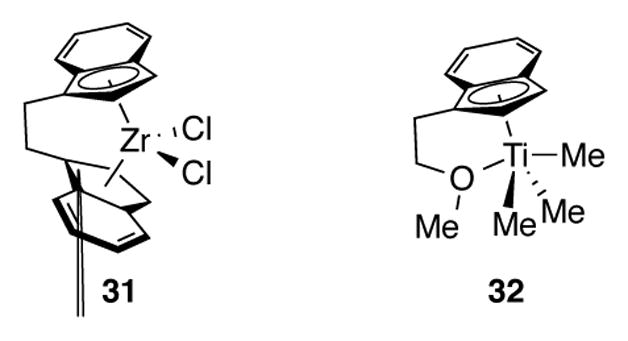
Zirconium complex 31 and Titanium Complex 32
The Doye research group has shown that (Ind)2TiMe2 is a particularly active and selective general catalyst for the intermolecular hydroamination of alkynes.[33],[34] They were able to demonstrate a broad substrate scope for this reaction; both symmetrical and unsymmetrical alkynes reacted efficiently and with high regioselectivity (Scheme 10). The imine/enamine mixture formed from hydroamination was immediately reduced with sodium cyanoborohydride to simplify reaction analysis. In general, sterically hindered primary alkylamines tended to react the most efficiently, giving the highest yields in the shortest reaction times (Scheme 10a). Anilines were also effective nucleophiles, despite their sluggish reaction times. Unhindered primary alkylamines proved to be the poorest substrates for hydroamination. For example, hydroamination of tolane with benzyl amine only proceeded to about 30% conversion after 48 hours. The authors were able to cut down the reaction times and improve the yields of hydroaminations with sterically unhindered amines by performing a slow addition of the amine over four hours. By performing this adjustment, the yield of tolane hydroamination by benzylamine increased to 67%. Hydroamination of unsymmetrical 1-phenyl-1-propyne proceeded with good (3:1) to excellent (>99:1) anti-Markovnikov regioselectivity for a variety of amine nucleophiles (Scheme 10b). In contrast, lower selectivities are observed for primary alkylamine additions to 1-phenyl-1-propyne with Cp*2TiMe2 as a catalyst (Scheme 11).[35] Furthermore, Cp*TiMe2 is completely unsuitable for the hydroamination of terminal alkynes, a reaction the indenyl complex can catalyze in high yields (Scheme 10c). The dominant regioisomer obtained was highly dependent on the nature of the nucleophile. High anti-Markovnikov hydroamination of terminal alkynes was observed with sterically bulky tert-butyl amine whereas Markovnikov addition dominated with anilines. However, phenylacetylene acceptors favored anti-Markovnikov addition regardless of the amine employed.
Scheme 10.
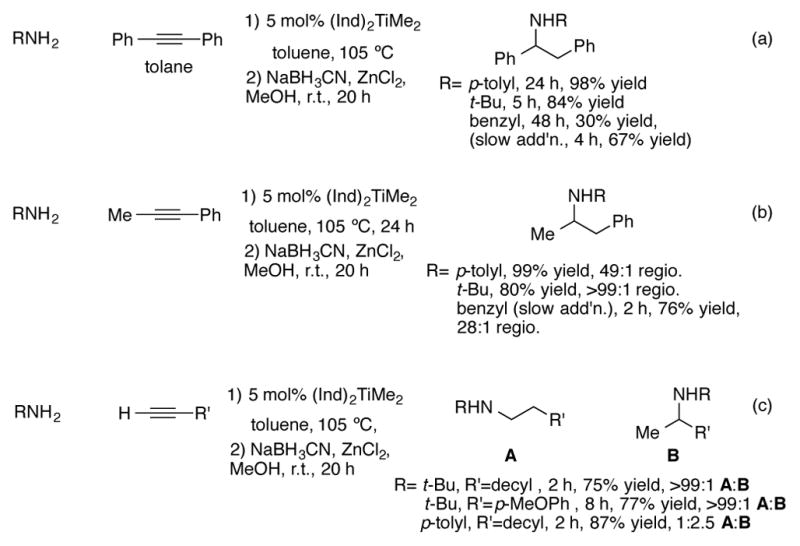
Catalytic intermolecular hydroamination of alkynes with (Ind)2TiMe
Scheme 11.
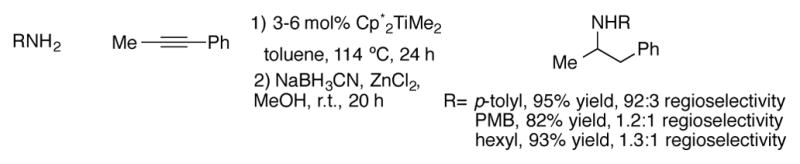
Hydroamination of unsymmetrical alkynes with Cp*2TiMe2
Buil et al. have demonstrated that indenyltitanium half-sandwich complexes containing tethered ethers 32 (Figure 2) are incredibly active alkyne hydroamination catalysts (5 mol% of complex, full conversion after 15 minutes with some substrates).[36]
The Doye group was also able to show that alkyne hydroamination can also be performed intramolecularly to form dihydro-[37] and tetrahydroisoquinolines[38] (Scheme 12). Following ring closure to the dihydroisoquinoline, 1-benzoyldihydroisoquinoline 33 can be isolated after oxidation by palladium on carbon. Alternatively, the dihydroisoquinoline can be reduced under standard sodium cyanoborohydride conditions to afford the tetrahydroisoquinoline 34.
Scheme 12.
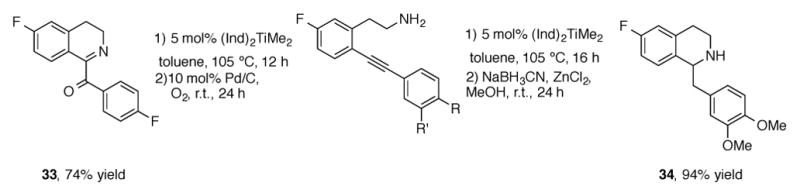
Catalytic intramolecular hydroamination for the synthesis of heterocycles
Using aryl cyclopropyl alkynes, the Doye group performed a one-pot intermolecular hydroamination/cyclopropylimine rearrangement reaction to synthesize pyrrolidines after reduction (Scheme 13).[39] The regioselectivity of addition to these unsymmetrical alkenes is excellent, >98:2 favoring the anti-Markovnikov product. Heating the imine with a catalytic amount of Brønsted acid promotes the cyclopropylimine rearrangement. After reduction, the pyrrolidine products can be isolated in modest to excellent yields.
Scheme 13.
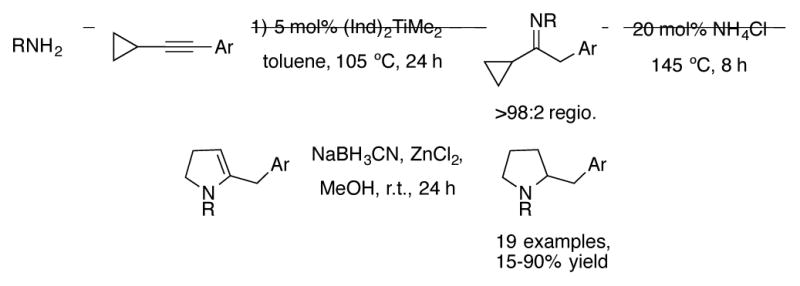
Catalytic intermolecular hydroamination/cyclopropylimine rearrangement for the construction of pyrrolidines
(Ind)2TiMe2 has also been shown to be an efficient catalyst for intramolecular alkene hydroamination (Scheme 14).[40] Using primary amine 35 as a test substrate, the Doye group showed that (Ind)2TiMe2 is a much more active catalyst when compared to Cp2TiMe2, but less active than known hydroamination catalyst Ti(NMe2)4.[41] The diphenylmethyl backbone of 35 was necessary for successful pyrrolidine formation; this moiety provides an essential Thorpe-Ingold effect to affect ring closure and without it, only recovered starting material is observed. Six-membered piperidine rings can also be constructed using this methodology, and (Ind)2TiMe2 proved to be more active for the 6-exo-trig cyclization than either Cp2TiMe2 or Ti(NMe2)4.
Scheme 14.

Catalytic intramolecular hydroamination of alkenes
In addition to titanium, indenyl complexes of rare earth metals have been used for intramolecular hydroamination of alkenes. Taking inspiration from the seminal report on cyclopentadienyllanthanide hydroamination by Marks,[42] researchers have prepared and tested a number of indenyl complexes of ytterbium,[43] yttrium,[43b,44] lutetium,[44a,45] and dysprosium.[44a] Although direct comparisons between cyclopentadienyl and indenyl complexes are limited, Vitanova and coworkers reported a substantial increase in rate of hydroamination using chiral indenylyttrium complex 38 when compared to complex 37 (Scheme 15).[44a] Unfortunately, the diastereo- and enantioselectivity for this desymmetrative hydroamination reaction was modest.
Scheme 15.
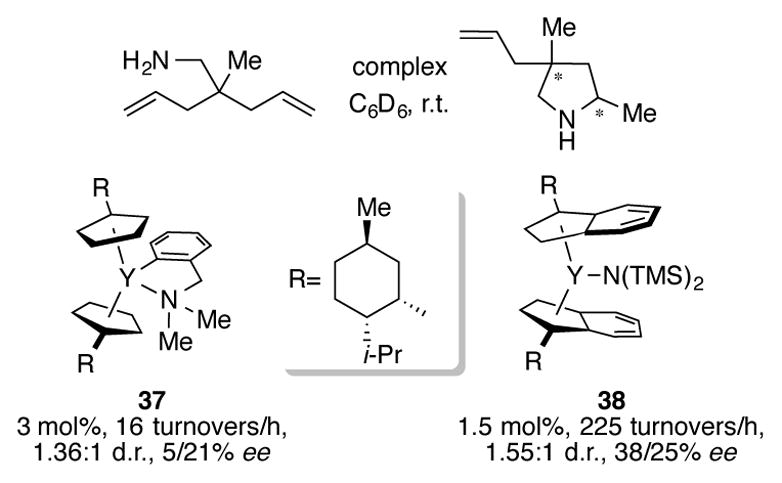
Desymmetrative hydroamination using chiral yttrium complexes
(Ind)2TiMe2 is also a catalyst for the regioselective hydroaminomethylation of alkenes under comparatively mild conditions (Scheme 16).[46] Heating a mixture of N-methylaniline 39 and 1-octene 40 in toluene with a desired titanium catalyst, a mixture of branched 41 and linear 42 is obtained. Whereas Ti(NMe2)4 provides a low yield of 41/42, (Ind)2TiMe2 efficiently delivers the hydroaminomethylated product in high yield and high regioselectivity for the branched product. Furthermore, the catalyst can perform hydroaminomethylation at a lower temperature. Styrene-based substrates tended to give lower regioselectivities (~85:15 branched:linear). The mechanism for this reaction is postulated to be similar to the mechanism proposed for tantalum-catalyzed hydroaminomethylation.[47] C-H activation of the N-methyl group of 37 results in titanaaziridine intermediate 43, which then undergoes regioselective olefin insertion to form titanacycle 44. N-Alkyl substrates other than methyl failed to react, presumably due to their more sterically demanding nature.
Scheme 16.
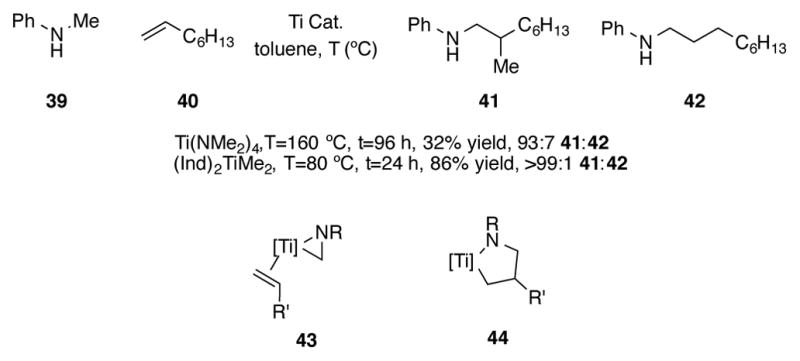
Catalytic intermolecular hydroaminomethylation of alkenes
3. Iron and Ruthenium
In 1993, the Trost group reported the ruthenium-catalyzed redox isomerization of allylic alcohols.[48] The scope of this reaction was originally explored with CpRu(PPh3)2Cl, and while the authors noted the exquisite chemoselectivity of this complex, they also observed sluggish reactivity of 1,2-disubstituted allylic alcohols. As seen in entry 1 of Scheme 17, ketone 45 could only be isolated in 23% yield after 9 hours using CpRu(PPh3)2Cl, with the remainder of the mass balance being 1-phenyl-1,3-heptadiene resulting from acid-catalyzed elimination. When the authors switched their catalyst to (Ind)Ru(PPh3)2Cl, full conversion of the allylic alcohol could be obtained in only two hours in 83% yield of 45. A similar increase in rate was detected in the redox isomerization reaction resulting in ketone 46. In this case, even after 24 hours, the CpRu catalyst could not catalyze the reaction to complete conversion, and starting material was recovered from the reaction mixture. On the other hand, catalysis utilizing (Ind)Ru(PPh3)2Cl was complete in 3 hours, and ketone 46 was isolated in an 81% yield.
Scheme 17.
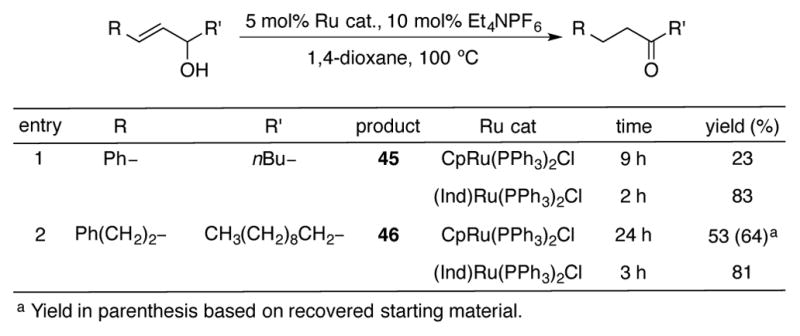
Redox isomerization of 1,2-disubstituted allylic alcohols
Rate acceleration with (Ind)Ru(PPh3)2Cl was also observed in the case of cyclic allylic alcohol 47 (Scheme 18a). A reaction that would normally require 24 hours with the CpRu catalyst was complete in only 3 hours and in an improved isolated yield of ketone 48. It is important to mention that while (Ind)Ru(PPh3)2Cl does display an increased reactivity, sometimes this advantage is accompanied by decreased chemoselectivity for allylic alcohol isomerization. While the allylic alcohol in 49 was successfully isomerized to ketone 51, significant isomerization of the terminal olefin to 50 was also observed (Scheme 18b). The isomerization of terminal olefins is not observed with CpRu(PPh3)2Cl. Therefore, it is important to be mindful of functional group reactivity trends when choosing an appropriate catalyst for the redox isomerization, as this factor can be crucial to the success of the reaction.
Scheme 18.
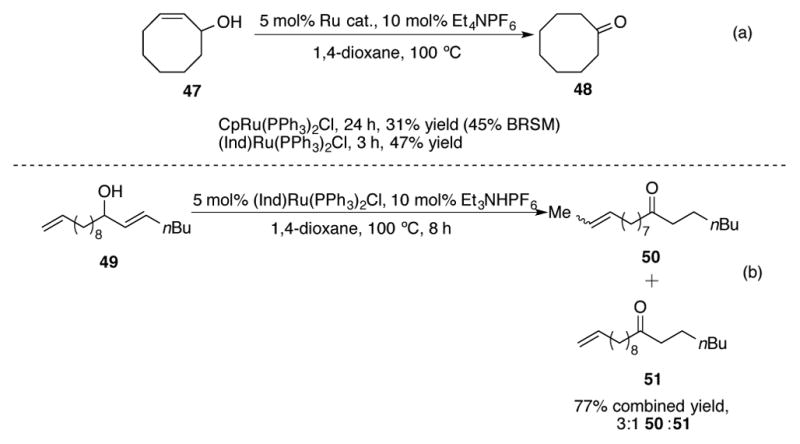
(a) Redox isomerization of a cyclic allylic alcohol (b) chemoselectivity of (Ind)Ru(PPh3)2Cl
After the success of allylic alcohols, Trost and Livingston extended the ruthenium-catalyzed redox isomerization reaction to propargyl alcohols.[49] Again, (Ind)Ru(PPh3)2Cl proved to be catalyst of choice for this transformation, isomerizing propargyl alcohol 52 to enal 53 to full conversion in 30 min (Scheme 19a). By comparison, CpRu(PPh3)2Cl required a full 4 h to reach full conversion. The catalyst loading of (Ind)Ru(PPh3)2Cl could be dropped to 1 mol% and 1 mol% In(OTf)3 co-catalyst without affecting conversion; 53 was obtained in an 83% yield.
Scheme 19.

(a) Redox isomerization of propargyl alcohol 52 (b) Deuterium labeling study and proposed mechanism of ruthenium-catalyzed redox isomerization of propargyl alcohols
The mechanism of this reaction was probed using deuterium-labeled substrate 54 (Scheme 19b). Enal 55 was isolated with complete deuterium incorporation at the α-position of its aldehyde. Based on this evidence, the authors propose a mechanism where (Ind)Ru(PPh3)2Cl is activated by the indium co-catalyst to form cationic ruthenium complex 56. After phosphine dissociation and bidentate propargyl alcohol coordination, 57 undergoes a 1,2-hydride shift where one of the carbinol protons migrates to the alkyne. Protodemetallation of vinylruthenium intermediate 58 to the enal completes the catalytic cycle and regenerates the catalyst.
To demonstrate the synthetic utility of the redox isomerization reaction, the Trost group applied this methodology to the total syntheses of leukotriene B4 (Scheme 20a)[49b] and adociacetylene B (Scheme 20b).[50] With 5 mol% of ruthenium catalyst, 4-en-2-yn-1-ol 59 could be isomerized to dienal 60 in a 92% yield - an impressive display of catalyst chemoselectivity considering the functionality on the molecule. The synthesis of leukotriene B4 could then be completed with an additional five steps. In the synthesis of adociacetylene B, the symmetric advanced intermediate 61 underwent two redox isomerizations with 10 mol% of catalyst to provide dialdehyde 62 in good yield. Using (S,S)-Prophenol 63, an asymmetric alkynylation of both aldehydes was performed in excellent diastereo- and enantioselectivity. After desilylation, adociacetylene B was synthesized in only four steps (longest linear sequence).
Scheme 20.

(a) Using redox isomerization in the total synthesis of leukotriene B4 (b) Concise total synthesis of adociacetylene B via double redox isomerization/Prophenol-catalyzed zinc alkynylation
Redox isomerization can also be combined in tandem with Michael addition reactions in order to increase molecular complexity and simplify multistep processes. For example, substituted indoles and furans can be included in the reaction mixture to synthesize β-substituted ketones (Scheme 21a).[51] An asymmetric intermolecular conjugate addition of methylene bis(sulfones) can also be executed in one pot following the ruthenium step (Scheme 21b).[52] Bidentate 1,3- bis(diphenylphosphino)propane(dppp) is added in order to prevent interference of the organocatalytic step by the ruthenium catalyst. Using these conditions, carbocycles with enantioselectivities up to 97% ee can be obtained.
Scheme 21.
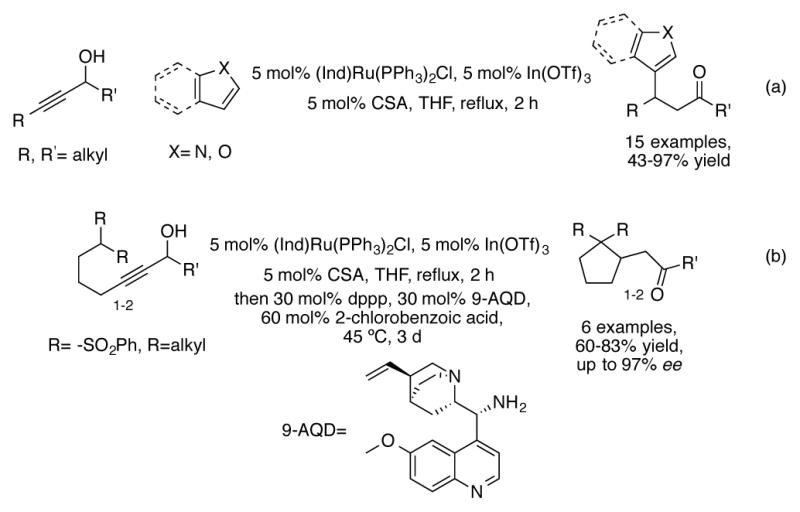
(a) Redox isomerization/conjugate addition of heteroarenes (b) Redox isomerization/Asymmetric intramolecular conjugate addition
(Ind)Ru(PPh3)2Cl is an efficient catalyst for the ring expansion of alkynylcyclopropanols 64 (Scheme 22).[53] Depending on the functional group attached to the alkyne, either cyclobutanone 65 or cyclopentenone 66 is observed as the major product. The authors attribute this mechanistic dichotomy to the degree in which the substrate can stabilize the buildup of positive charge at the alkynyl carbon adjacent to the cyclopropane.
Scheme 22.
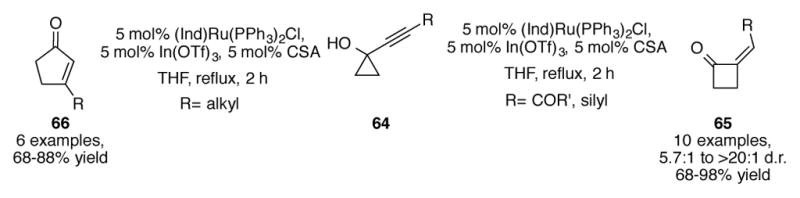
Ruthenium-catalyzed ring expansion of alkynylcyclopropanols
(Ind)Ru(PPh3)2Cl has also been used as an effective catalyst for the dynamic kinetic resolution of secondary alcohols in conjunction with Pseudomonas cepacia lipase (PCL; Scheme 23).[54]
Scheme 23.
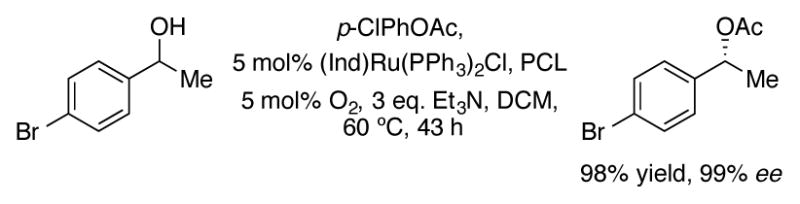
Dynamic kinetic resolution of secondary alcohols with (Ind)Ru(PPh3)2Cl and PCL
Related indenyl complex (Ind)Ru(C≡CPh)(PPh3)2 can perform head-to-head dimerization of terminal alkynes to form enynes in modest E to Z ratios (Scheme 24).[55]
Scheme 24.
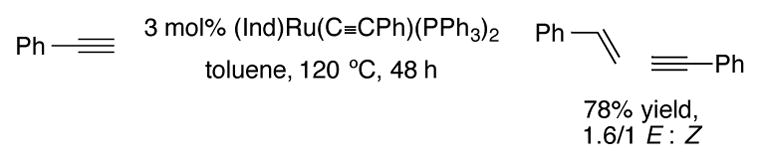
Head-to-head dimerization of phenylacetylene
Itoh and coworkers have noted an interesting trend in the product distributions of ruthenium-catalyzed reactions of 1,6-heptadiynes with strained bicyclic alkenes (Scheme 25). [56] When dimethyl dipropargylmalonate 67 is reacted with an excess of norbornene and chloro(η5-indenyl)ruthenium(1,5-cycloocatdiene) 70, polycyclic 68 is formed as the primary reaction product, with minor amounts of diene 69. However, when indenyl complex 70 is exchanged for its cyclopentadienyl analogue 71, more of the diene is formed in proportion to 68. A reversal in selectivity is seen with the more electron-donating ligand Cp* (η5-pentamethylcyclopentadienyl). With complex 72, diene 69 is favored in roughly a 3:1 ratio. The difference in product distributions can be explained by the degree in which a given ruthenium complex can stabilize a ruthenacyclopentatriene intermediate like 73. Such intermediates have been proposed for other ruthenium-mediated transformations.[57] Ruthenacyclopentatrienes are a resonance form of a ruthenacyclopentadiene, and an increase in the number of vacant orbitals around a transition metal complex increases its ability to π-bond with a coordinated ligand. Therefore, an increase in the degree of cyclopentatriene character is observed in more coordinatively unsaturated intermediates such as η3-indenyl complex 73. Intermediate 73 can be seen to be a bis(carbene) complex that can perform two successive cyclopropanation reactions via a metathesis-like [2+2] cycloaddition with the strained norbornene. Reductive elimination generates the observed cyclopropane. Because the Cp* ligand has less of a propensity to slip into a η3 coordination mode, more cyclopentadiene character is observed in intermediate 74, and therefore the olefin insertion product (i.e. 69) dominates as the major reaction product.
Scheme 25.
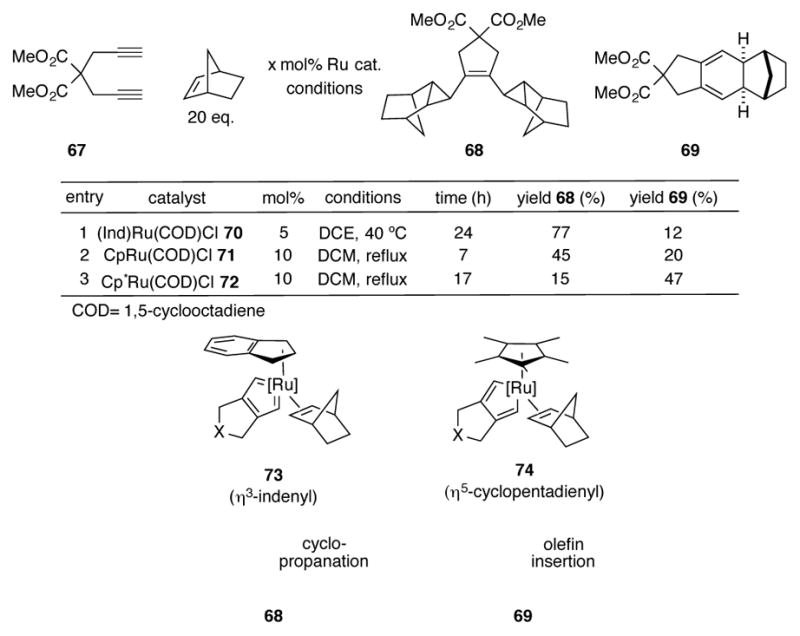
Ruthenium-catalyzed reactions of 1,6-heptadiynes with norbornene
Indenyl complex 70 has also been studied by Alvarez et al. for [2+2] and [4+2] cycloaddition reactions of alkynes (Scheme 26).[58] For the indenylruthenium-catalyzed [4+2] cycloaddition reaction, the authors noted a stronger dependence of the electronics and steric properties of the alkyne on product yield. In previous work, Trost and coworkers have shown that CpRu(COD)Cl 71 provides much higher yields of the [4+2] cycloaddition products across a diverse range of alkynes.[59]
Scheme 26.
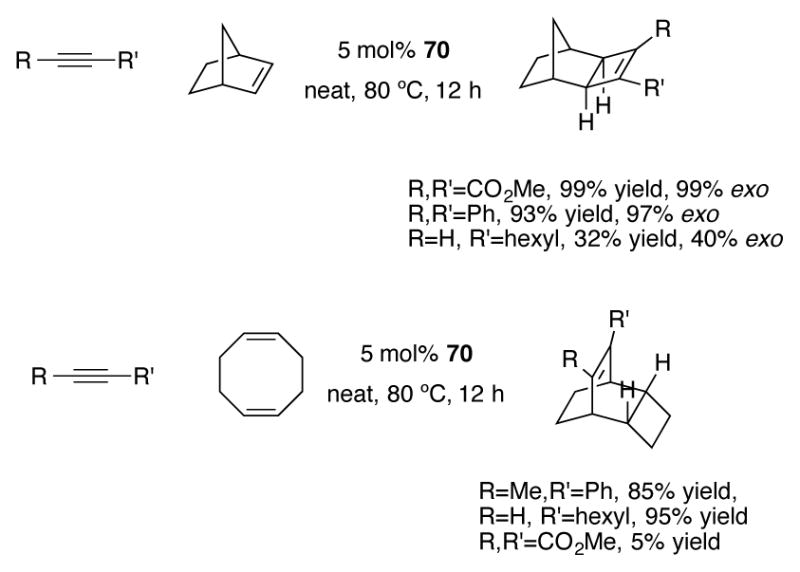
Indenylruthenium-catalyzed [2+2] and [4+2] reactions
Kündig and coworkers have reported that ruthenium catalysts 75 containing bidentate C2-symmetric chiral phosphinite ligands are excellent catalysts for asymmetric Diels-Alder cycloaddition reactions (Scheme 27).[60] In their studies, they observed an interesting reversal of selectivity of the reaction between cyclopentadiene and acrolein that depended on whether a Cp or indenyl ligand was incorporated on the catalyst scaffold. Using (S,S)-Cp-75, the endo diastereomer 78 was favored in a 70:30 ratio over the exo diastereomer 77. On the other hand, (R,R)-Ind-75 reversed the diastereoselectivity of the process, favoring the exo isomer in a 71:29 ratio and in 85% ee. The authors attribute this reversal in selectivity to the increased steric demands of the indenyl ligand. Ind-75 also proved to be a more reactive complex, catalyzing the Diels-Alder reaction between cyclopentadiene and methacrolein to completion within 3 hours. By comparison, Cp-75 only reached 60% conversion after 5 hours. Related complexes Cp- and Ind-76 are excellent catalysts for asymmetric intramolecular Diels-Alder reactions. [61] In some of the intramolecular cases, Ind-76 delivered higher enantioselectivities than Cp-76, albeit with comparable catalytic rates (Scheme 27).
Scheme 27.
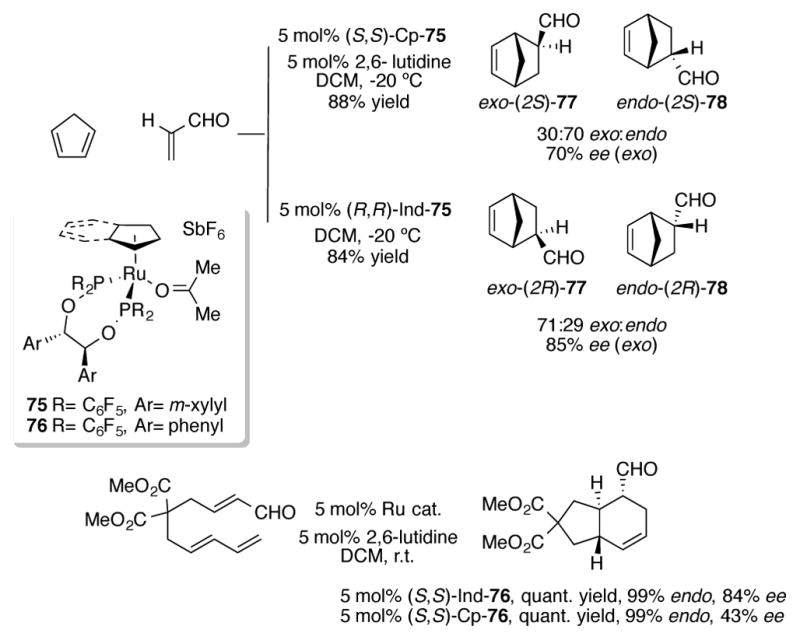
Ruthenium-catalyzed asymmetric Diels-Alder reaction between cyclopentadiene and acrolein
Although indenyl variants of complex 75 and 76 have been shown to be effective catalysts for cycloaddition reactions, the bidentate nature of their phosphinite ligands limits their use to reactions requiring single-point coordination to a Lewis acidic metal center. Recently, the Trost group introduced a novel class of chiral catalyst 79 that contains a chiral sulfoxide bound to its indenyl ring via a peri-naphthalene tether (Scheme 28).[62] The fact that this catalyst has a maximum of three possible sites around ruthenium for substrate coordination opens up a broader range of synthetic possibilities for chiral ruthenium catalysis. Therefore, when enyne 80 is subjected to 10 mol% of 79 and one equivalent of water, a 1:10 ratio of diene 81 to alcohol 82 is obtained. Alcohol 82, a product which hadn’t been observed under CpRu catalysis, could be isolated in an 80% yield and 84:16 e.r.. Catalyst 79 could also perform an asymmetric redox isomerization/C-H insertion reaction of acrylate 83. Though the reaction proceeds to 43% conversion in THF, carbocycle 84 was observed to have a promising 90:10 e.r..
Scheme 28.
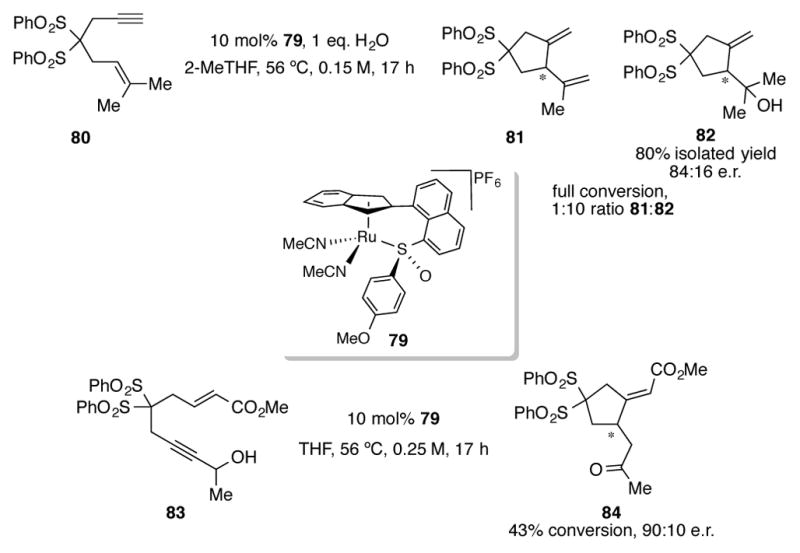
Indenylruthenium complex 79 with a tethered chiral sulfoxide developed by Trost
In 2012, the Nolan group reported that 1-phenylindenylruthenium complex 85[63] was an exceptionally active catalyst for the transfer hydrogenation of aldehydes, ketones, and imines (Scheme 29).[64] Complex 85, which can be synthesized in one high yielding step from commercially available RuCl2(PPh3)3, outperformed CpRu(PPh3)2Cl, (Ind)Ru(PPh3)2Cl, and Shvo’s catalyst 86 in the transfer hydrogenation of benzophenone. This increase in activity cannot be explained by the indenyl effect alone, as (Ind)Ru(PPh3)2Cl actually performed worse than either CpRu(PPh3)2Cl or 85 in the transfer hydrogenation reaction.
Scheme 29.
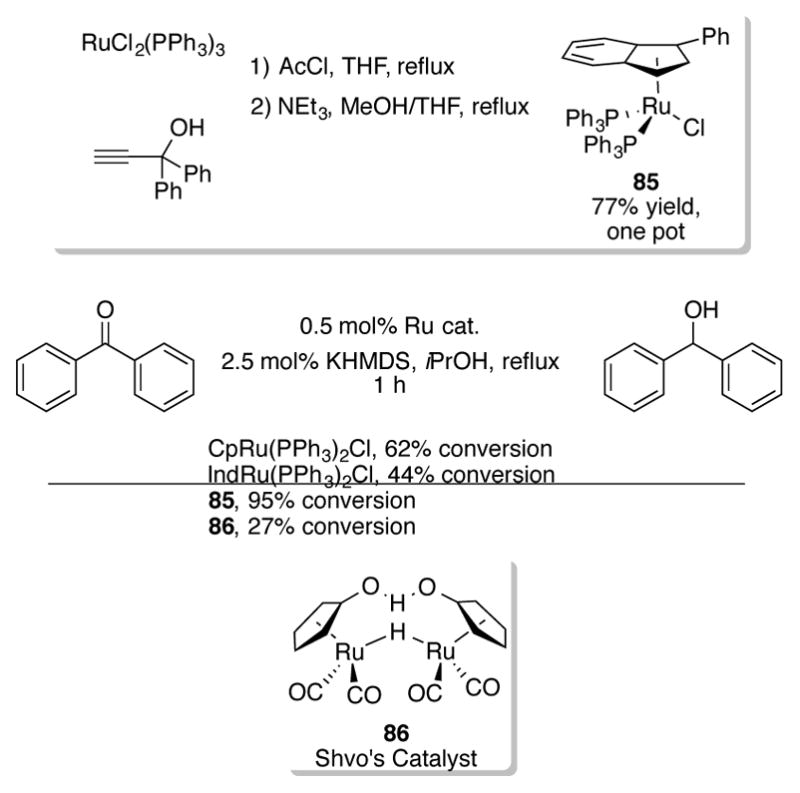
Synthesis of 85 and comparison of its activity toward transfer hydrogenation
One example of an asymmetric transfer hydrogenation has been reported with an indenylruthenium complex 87 bearing a chiral bidentate PHOX ligand in 76% ee (Scheme 30),[65] though this complex was inferior in terms of activity and selectivity when compared to other complexes.
Scheme 30.
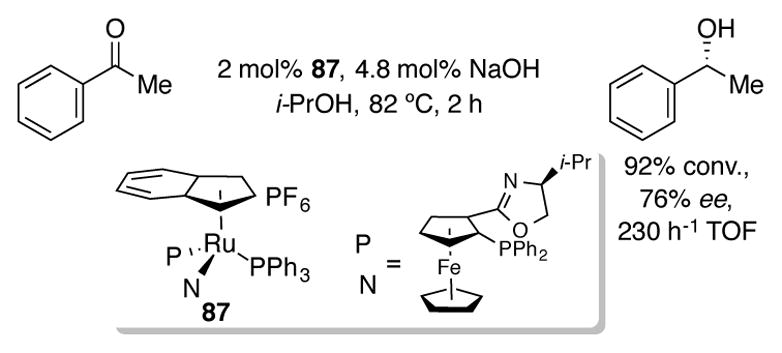
Asymmetric transfer hydrogenation with indenylruthenium PHOX complex
The phenyl substitution on complex 85 proved to be differential for the ruthenium-catalyzed redox isomerization of allylic alcohols at room temperature (Scheme 31).[66] CpRu(PPh3)2Cl and (Ind)Ru(PPh3)2Cl were completely inactive in isomerizing 88 at room temperature. Trost et al. showed that elevated temperatures are necessary for redox isomerizations with these complexes (see Scheme 17). On the other hand, 85 completely isomerized 88 to ketone 89 at room temperature in one hour. A range of terminal, 1,1-disubtituted, and 1,2-disubstituted secondary allylic alcohols were successfully isomerized to their corresponding ketones. Trisubstituted olefins failed to react for steric reasons. Mechanistic studies and DFT calculations point to the likelihood of a π-oxo-allyl complex 90 as the key intermediate of the reaction, which was originally proposed by Trost.[48] Related cationic complex 91 proved to be ideal for the isomerization of primary allylic alcohols under base-free conditions,[67] the stereoselective isomerization of terminal to internal alkenes,[67] and for the dynamic kinetic resolution of secondary alcohols.[68]
Scheme 31.
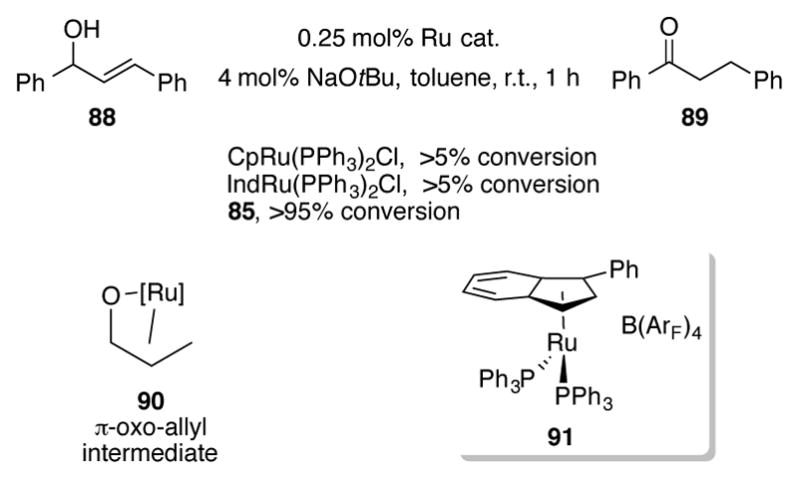
Redox isomerization of allyl alcohols at room temperature with 85
Complex 85 can also be used to perform chemoselective reductions of carboxylic acids.[69] Using 1 mol% of 85 with two equivalents of phenylsilane, excellent yields of primary alcohols 92–95 could be obtained without affecting any reducible functionalities (Scheme 32). Other silanes (PMHS, Ph2SiH2, Ph2MeSiH, PhMe2SiH, etc.) were ineffective reducing agents. A silyl ester was postulated to be an intermediate in the catalytic cycle. Similar complexes to a proposed catalytic intermediate of this transformation, 96, has been synthesized independently and used for pyridine-directed catalytic C-H borylation of arenes[70] and for heteroarene-directed H-D exchange reactions.[71]
Scheme 32.
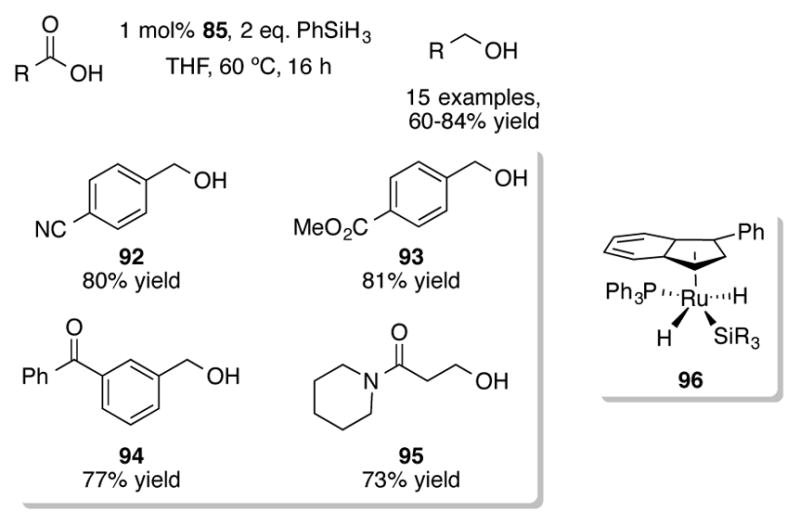
Chemoselective silane reduction of carboxylic acids with 85
Bauer et al. investigated complex 97 for catalytic activity toward OH exchange with OR of terminal propargyl alcohols (Scheme 33).[72] Typically, good isolated yields could be obtained when primary aliphatic alcohols were used as the nucleophiles for propargylation. Using more sterically restricting secondary alcohols as nucleophiles resulted in lower yields. This reaction is proposed to proceed through a ruthenium allenylidene intermediate.
Scheme 33.
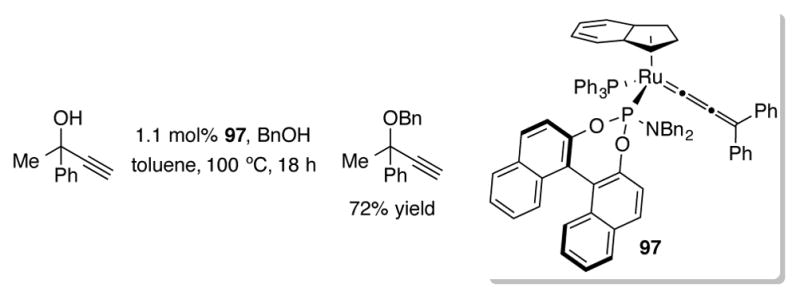
Propargyl alcohol substitution with allenylidene complex 97
There is a single report of indenyliron catalysis in the literature, also published by the Bauer group.[73] In their studies on using Lewis acidic iron complexes for Mukaiyama aldol reactions to form β-siloxyester 98, they observed a significant increase in catalytic rate going from Cp-phosphinoxazoline (PHOX) complex 99 to indenyl-PHOX complex 100 (Scheme 34). Unfortunately, no enantioinduction was observed with either 99 or 100. It was also discovered that achiral 101 could serve as an efficient catalyst for this transformation. Using 100, both aryl and alkyl aldehydes were effective substrates for the Mukaiyama aldol reaction.
Scheme 34.
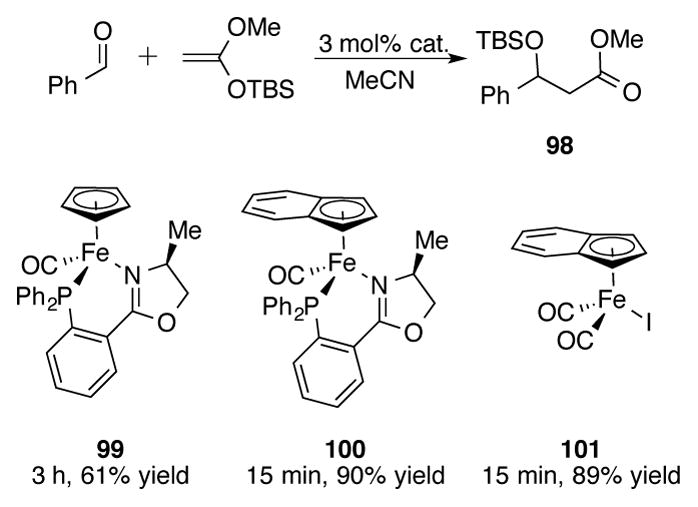
Iron-catalyzed Mukaiyama aldol reaction of benzaldehyde
4. Cobalt, Rhodium, and Iridium
The earliest examples of catalysis with group-9 indenyl transition metal complexes were reported while studying ligand effects for [2+2+2] cycloaddition reactions of alkynes. Abdulla and coworkers reported that (Ind)Rh(COD) 102 catalyzed the cyclotrimerization of dimethylacetylenedicarboxylate (DMAD) to hexacarbomethoxybenzene at an initial rate approximately ten times that of CpRh(COD) 103.[74] Whereas cyclotrimerizations with CpRh complexes required 24 hour reaction times, reactions with 102 were complete within 2 hours and produced no polymeric byproducts. Rate studies by Borrini et al. measuring the reactivity of various Cp and indenylrhodium cyclotrimerization catalysts agree with Abdulla’s observations.[75] These indenylrhodium complexes were shown to be active even at room temperature or below, albeit with significantly reduced turnover numbers. Again, increased catalytic activity was linked to the increased facility of slippage of the indenyl ligand. Computational studies by Orian on the effect of the indenyl ligand in cyclotrimerization reactions are in agreement with this line of reasoning.[76]
Despite exhibiting significant rate differences, 102 and 103 provide similar regiosisomeric mixtures when unsymmetrical alkynes are subject to [2+2+2] cycloaddition (Scheme 35).[74] For example, cyclotrimerization of ethyl propiolate with either 102 or 103 results in a 1:1 mixture of triesters 104 and 105 (Scheme 35a). Indenyl complex 102 can catalyze the cyclotrimerization of ethyl propiolate and propargyl alcohol 106 in an 83% yield of lactone 107, which was observed to be the sole product (Scheme 35b). Based on the product distributions of Scheme 35a and b, rhodacyclopentadiene 108 is most likely catalyst intermediate for these reactions. Propargyl alcohol 106 was also tested for [2+2+2] cycloaddition with DMAD to ester 109 (Scheme 35c), although competitive formation of hexacarbomethoxybenzene 110 was also observed.
Scheme 35.
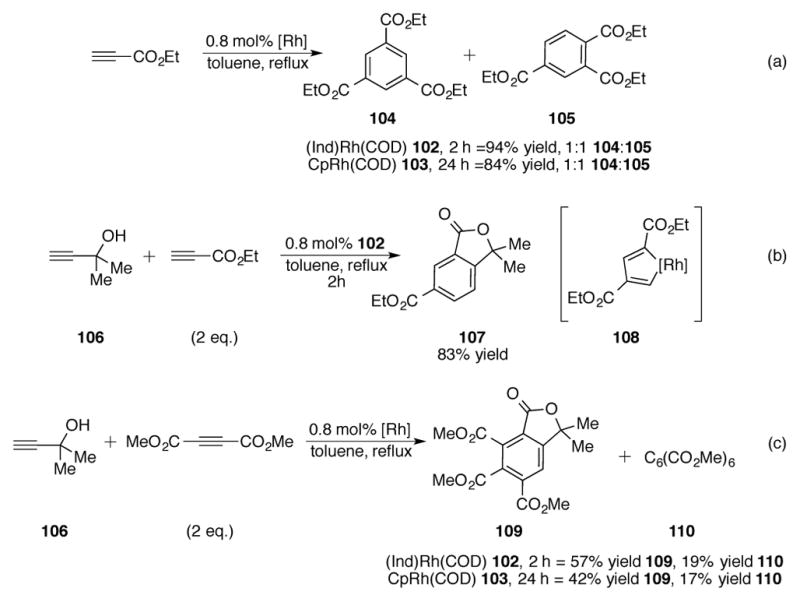
(a) Cyclotrimerization of ethyl propiolate (b) and (c) Cyclotrimerization with mixtures of unsymmetrical alkynes
In 1995, the Heller group reported the cobalt-catalyzed [2+2+2] cycloaddition of acetylene and nitriles in water (Scheme 36).[77] Both (Ind)Co(COD) 111 and CpCo(COD) 112 were established to be competent catalysts for this reaction, with no apparent significant differences in reactivity or selectivity of 2-phenylpyridine 113 over benzene.
Scheme 36.
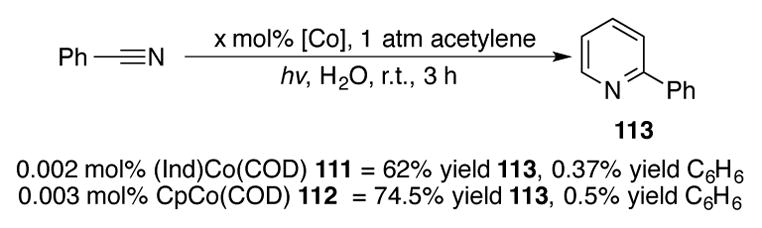
[2+2+2] cycloaddition under cobalt catalysis to make 2-substituted pyridines
However, planar chiral 1-neomenthylindenylcobalt complexes reminiscent of the ZACA complexes developed by Negishi (vide supra) proved to be differential for an atroposelective variant of this reaction (Scheme 37).[78] Using nitrile 114, a low 10% yield but promising 64% ee was obtained of 115 using indenylcobalt complex 119 as a catalyst for heterocyclotrimerization. Note that CpCo complexes 116 and 117 were less selective catalysts. The authors attribute the low yields of this reaction to the bulkiness of the complex; unsubstituted achiral CpCo(COD) 112 could catalyze this transformation in a 71% yield. Fortunately, by tethering the two alkynes together in the same substrate, as in naphthyl 120, excellent yields and enantioselectivities of tetrahydroquinolines 121 were obtained with 118 (Scheme 38).
Scheme 37.
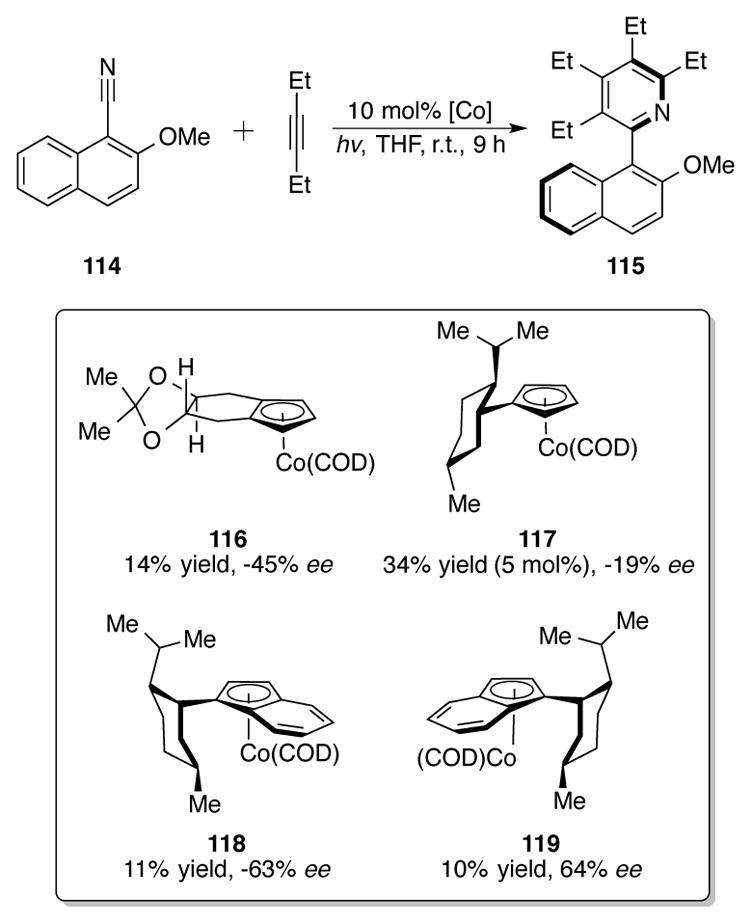
Intermolecular atroposelective syntheisis of pyridines with chiral cobalt catalysts
Scheme 38.
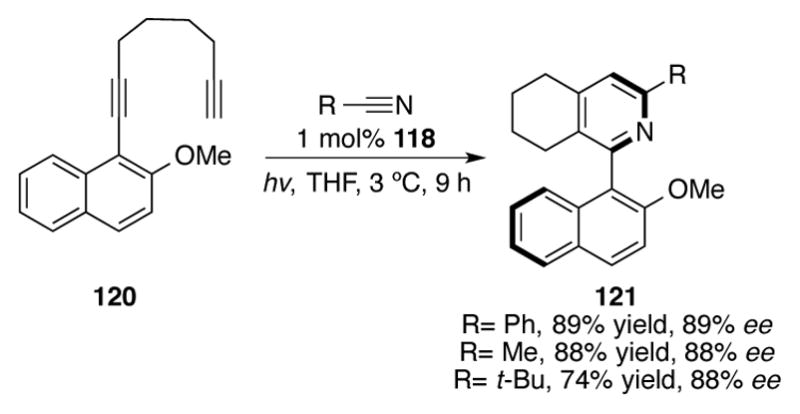
Intramolecular atroposelective syntheisis of pyridines with chiral cobalt catalysts
Cobalt complexes 118 and 119 deliver high enantioselectivities in the [2+2+2] cycloaddition of alkynylphosphine oxide 122 (Scheme 39).[79] Biaryl 123 can be recrystallized to >99% ee, and after reduction, phosphine 124 can be used as an effective ligand for palladium-catalyzed asymmetric hydrosilylation of alkenes. Chiral helicenes can also be constructed with this [2+2+2] reaction using catalyst 118, though in low (20% ee) enantioselectivity.[80]
Scheme 39.
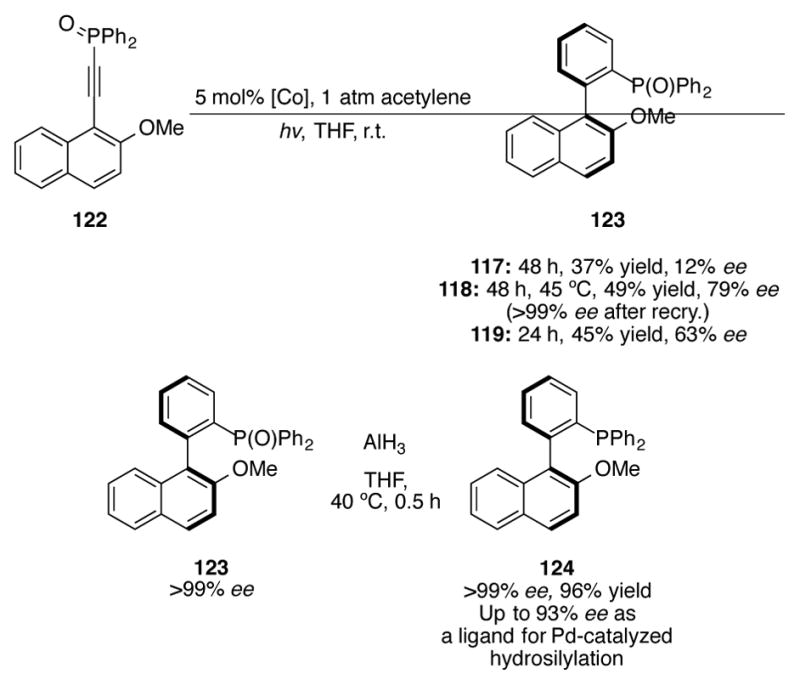
[2+2+2] cycloaddition of alkynylphosphine oxide 122
Chiral 1-menthylindenyl ligands were examined for rhodium-catalyzed asymmetric olefin hydrogenation of itaconic acid (Scheme 40).[81] Though both complex 125 and planar chiral 126 exhibited excellent reactivity, chiral succinic acid 128 was isolated in low ee. These two complexes can also be used for branched-selective hydroformylation of styrene to 2-phenylpropanal, but no enantioselectivity was observed.
Scheme 40.
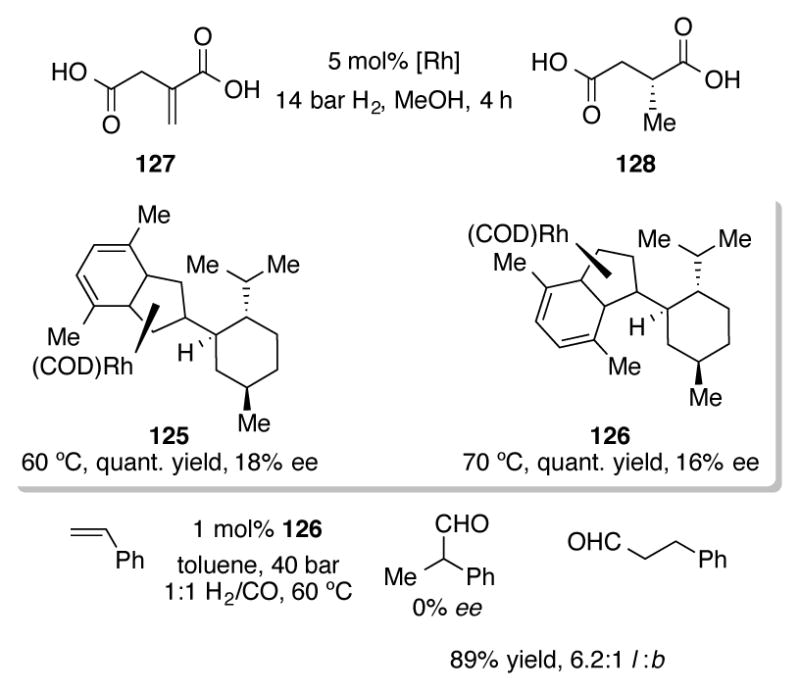
Asymmetric hydrogenation of itaconic acid 127 with chiral indenylrhodium complexes 125 and 126
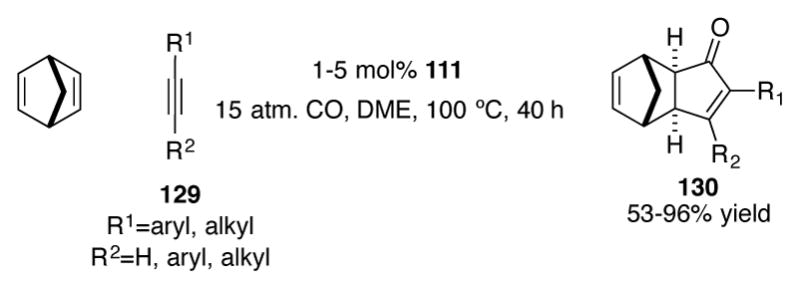
(Ind)Co(COD) 111 has also been used for catalytic intermolecular Pauson-Khand reactions of norbornadiene (Scheme 41).[82] Both internal and terminal alkynes 129 are effective substrates for this reaction; cyclopentenones 130 can be isolated in good to excellent yields with complete exo selectivity. Neither CpCo(COD) 112 nor CpCo(CO)2 provided any of the desired product under the standard reaction conditions, underscoring the necessity of the indenyl ligand for this transformation.
Scheme 41.
Co-catalyzed intermolecular Pauson-Khand reactions
The Milstein group found that the indenyl ligand was also essential for observing catalytic activity in the intermolecular rhodium-catalyzed hydroacylation of olefins (Scheme 42).[83] With benzaldehyde as a substrate under 1000 psi of ethylene at 100 °C in benzene, turnover rates of up to ca. 4 h−1 were achieved with (Ind)Rh(C2H4)2 as the catalyst. Propiophenone was the sole product detected by NMR; no decarbonylation of benzaldehyde by the catalyst was observed by 13C NMR under these conditions. This complex could also catalyze the addition of methyl formate to ethylene, although the reaction was much slower (2–3 turnovers after 24 hours). CpRh(C2H4)2, (acac)Rh(C2H4)2, and (Ph3P)2RuCl2 were all inactive catalysts under the above reaction conditions.
Scheme 42.

Hydroacylation of ethylene with (Ind)Rh(C2H4)2
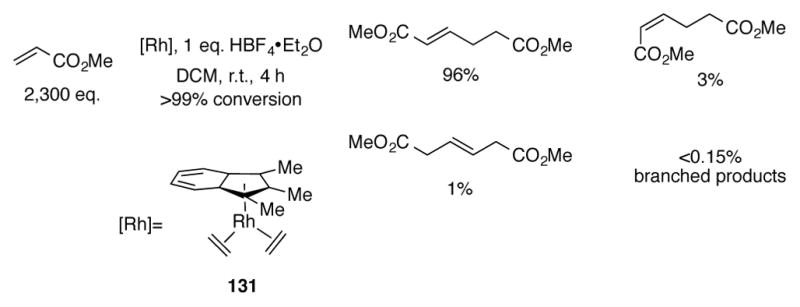
Indenylrhodium complex 131 displays increased reactivity and catalyst lifetimes for the tail-to-tail dimerization of methyl acrylate (Scheme 43).[84] The catalytic acid is added to generate the catalytically active RhIII hydride complex. Turnover frequencies of 11 min−1 could be achieved with 131 under these conditions, nearly twice that of Cp*Rh(C2H4)2, with excellent selectivity for the linear product dimethyl (E)-hex-2-enedioate. Indenylrhodium complexes can also dimerize methyl vinyl ketone with excellent trans selectivity albeit at drastically reduced rates (0.1–3 min−1), whereas Cp*Rh(C2H4)2 completely failed to produce dimers.
Scheme 43.
Rh-catalyzed tail-to-tail dimerization of methyl acrylate
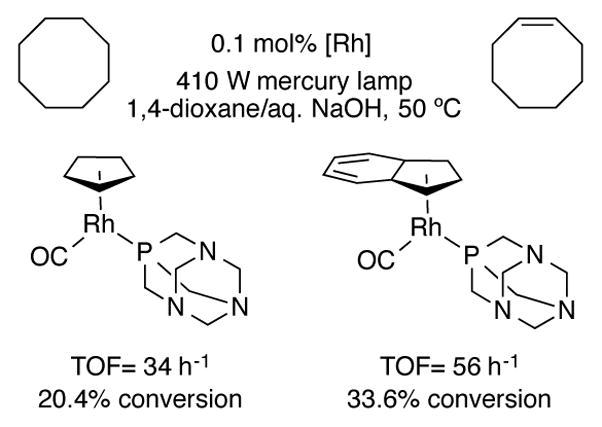
Indenylrhodium complexes have also displayed increased turnover frequencies for the photocatalytic acceptorless dehydrogenation of isopropanol, octane, and cyclooctane (Scheme 44).[85]
Scheme 44.
Acceptorless dehydrogenation of cyclooctane
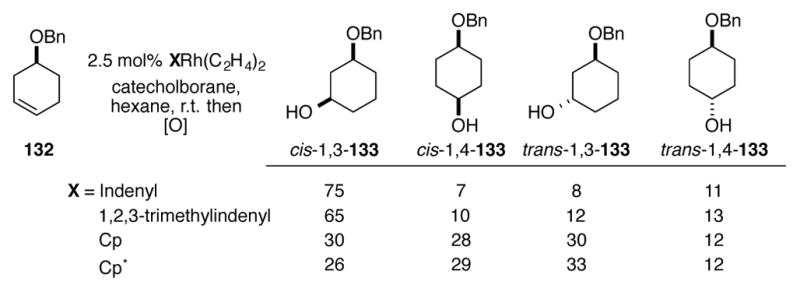
During their studies of substrate-directed hydroboration reactions, the Fu research group noted that rhodium complexes containing indenyl ligands tended to give higher ratios of cis isomers of 133 for the hydroboration of alkene 132 compared to their related Cp analogs (Scheme 45).[86] The amount of benzyl ether-directed cis hydroboration is directly correlated to the ability of a Cp or indenyl ring to slip, indicating that the additional open coordination site provided by ring slippage is required for substrate binding. Brinkman and coworkers have shown that even greater cis selectivities for this substrate can be obtained with CF3-substituted indenyl ligands under either rhodium or iridium catalysis.[87] If the benzyl ether is exchanged for a TBS ether, or if a more Lewis basic solvent such as THF is used, the directing ability of the substrate is greatly reduced, even with (Ind)Rh(C2H4)2.
Scheme 45.
RhI-directed hydroboration of 132
While studying the stoichiometric borylation of metal arene complex Cp*(PMe3)Ir(H)(Ph) with HBPin (Pin=pinacol), the Smith research group noted that the major metal-containing product of this reaction, Cp*(PMe3)Ir(H)(BPin) 134, could catalyze the C-H borylation of benzene to up to three turnovers.[88] Additionally, 134 was found to be more regioselective for meta borylation[89] than Cp*Rh(η4-C6Me6) originally developed by Hartwig for the C-H borylation of alkanes.[90] These two pieces of data led Smith et al. to pursue other iridium complexes for arene C-H borylation.[91] Mechanistic studies of the Ir-catalyzed reaction indicated that the Cp* ligand was disassociating during the catalytic cycle. Indeed, when Cp* was replaced with a more labile ligand, as in (η6-mesitylene)Ir(BPin)3, turnover numbers increased dramatically in comparison to 134 when used in conjunction with two catalyst equivalents of PMe3. However, because the synthesis of (η6-mesitylene)Ir(BPin)3 was low yielding, the authors decided to look for an alternative pre-catalyst that can be readily synthesized in high yields and contains an equally labile ligand as an η6-coordinated arene.
(Ind)Ir(COD) 135 can be synthesized in one step from indenyllithium and commercially-available [IrCl(COD)]2 in a 86% yield.[92] Used in conjunction with bidentate phosphine 1,2-bis(diphenylphosphino)ethane (dppe), 2 mol% 135 can catalyze the C-H borylation of benzene in high yields and in only two hours (Scheme 46a). The catalyst loading for this transformation can be reduced to as little as 0.02 mol% of iridium when using 1,1-bis(dimethylphosphino)ethane (dmpe) as a ligand; this represents a turnover number of 4500, which is a 1000-fold increase over pre-catalyst 134. Complete meta borylation of 1,3-dibromobenzene can be achieved (Scheme 46b) and 136 can be isolated in 92% yield. Notable is the fact that the reactive aryl bromide bonds are unaffected by the iridium catalyst under the reaction conditions. One-pot C-H borylation/oxidation with this catalyst system has been performed on multigram scale.[93] This meta-selective borylation reaction can also be used in conjunction with palladium-catalyzed amidation reactions in high yields (Scheme 46c).[94] The borylation and amidation steps can be performed in one pot, but filtration of the reaction mixture through silica prior to oxidation proved to be crucial in order to obtain high yields of phenol 137. Although (Ind)Ir(COD) 135/phosphine catalyst system has been largely supplanted by the Miyaura/Hartwig[Ir(COD)(OMe)]2/4,4′-di-tert-butyl-2,2′-bipyridine catalyst system which can catalyze arene meta C-H borylations at room temperature,[95] there is a notable exception for particularly electron-rich heteroaromatics such as 2,5-dimethylthiophene (Scheme 46d).[96] Under the standard Miyaura/Hartwig reaction conditions, no borylation is observed after 20 hours. However, the 135/dmpe system can successfully catalyze the borylation of 2,5-dimethylthiophene to 138 in a 97% yield.
Scheme 46.
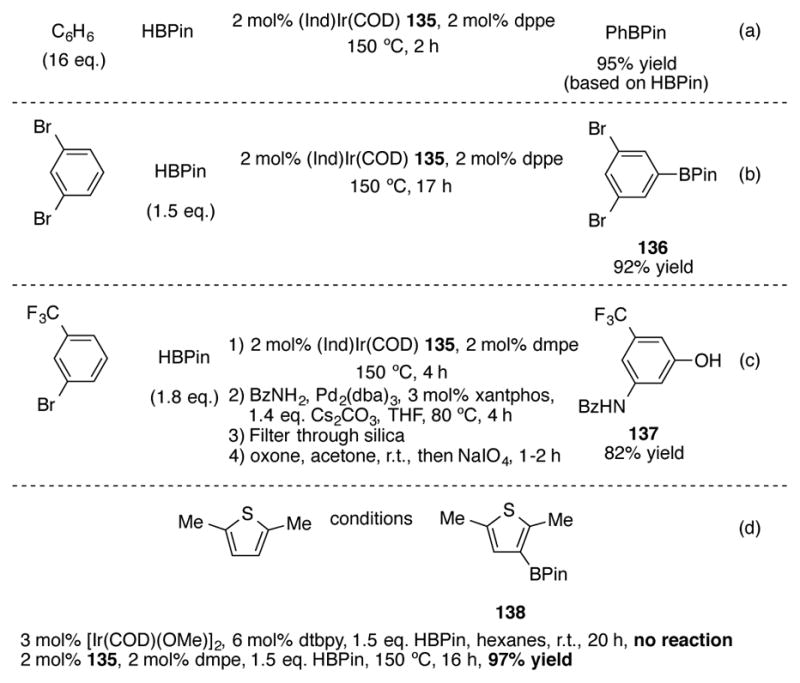
(Ind)Ir(COD)-catalyzed meta selective arene C-H borylation
3 mol% [Ir(COD)(OMe)]2, 6 mol% dtbpy, 1.5 eq. HBPin, hexanes, r.t., 20 h, no reaction
2 mol% 135, 2 mol% dmpe, 1.5 eq. HBPin, 150 ºC, 16 h, 97% yield
(Ind)Ir(COD) 135 has also been studied as a catalyst for hydrosilylation of terminal alkynes (Scheme 47).[97] Using 135 alone for the hydrosilylation of 1-octyne, a low 34% combined yield of hydrosilylated products was obtained, favoring β-(Z)-140. However, by adding biphosphinine 139, the yield can be increased to 96% and a switch in selectivity for β-(E)-140 is observed. The 135/139 catalyst system is complementary to [Ir(COD)2][BF4], which delivers high yields of β-(Z)-140. A broad substrate scope of aryl, alkyl, alkenyl, and silyl terminal alkynes were explored for the hydrosilylation, with yields ranging from 44% to 96%.
Scheme 47.
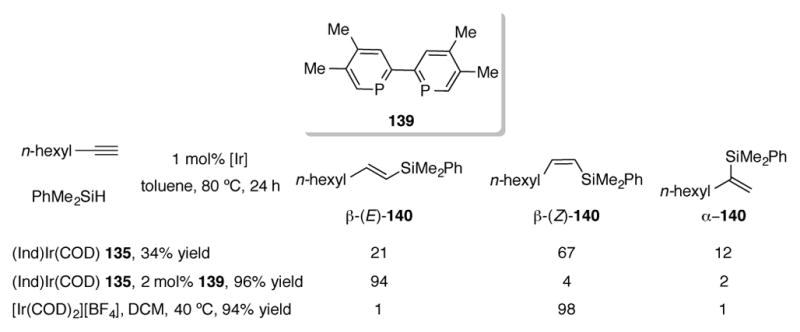
Hydrosilylation of terminal alkynes with (Ind)Ir(COD) 135
5. Nickel and Palladium
Compared to other d-block transition metals, the catalytic properties of indenylnickel and –palladium complexes in the context of organic synthesis have been underexplored. (1-MeInd)Ni(PPh3)Cl, which had previously been studied as a catalyst for dehydropolymerzation of phenylsilane,[98] can be used as a catalyst for the hydrosilylation of styrene with phenylsilane with 2 mol% catalyst loading.[99] Tethered amino complex 141 displays longer catalytic lifetimes and as little as 1 mol% of this complex can be used to give a quantitative yield of the α-silylated product 142 as the sole regioisomer (Scheme 48).[100] The tethered ligand prevents catalyst deactivation via decomposition to an unknown number of nickel complexes. Alternatively, bulky indenyl complexes [1,3-(TMS)2Ind]Ni(PPh3)Cl)[101] can be used instead of 141 to deliver 142 in an 82% yield.
Scheme 48.
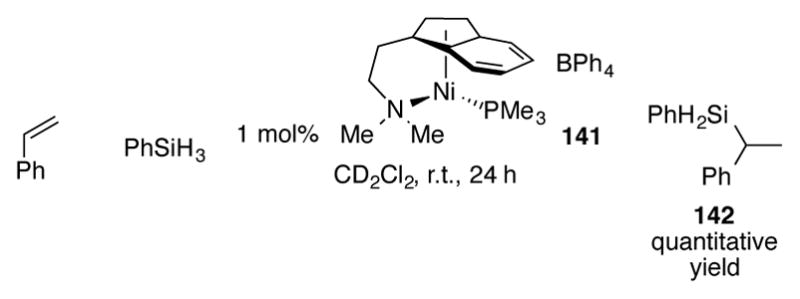
Hydrosilylation of styrene with indenylnickel complex 141
(Ind)Pd(PPh3)Cl can catalyze the hydrosilylation of styrene with trichlorosilane with 1 mol% catalyst loading to 92% conversion.[102] Like the nickel-catalyzed hydrosilylation described above, this reaction proceeds with complete α-regioselectivity. Phenylacetylene can also be hydrosilylated under these conditions, though a 1:1 mixture of regioisomers is observed.
There has been interest in using indenylpalladium complexes as pre-catalysts for cross-coupling reactions. (1-TMSInd)Pd(PPh3)Cl can catalyze the Heck coupling of PhX (X=Br, I) to styrene in nearly quantitative conversion (9:1 ratio (E):(Z) of stillbene).[100] The same complex can be used for the Buchwald-Hartwig coupling of aniline to o- and p-chlorotoluene in quantitative conversion.
More recently, (η3-1-t-Bu-Ind)LPdCl 143 (L=phosphine, NHC) has been shown to be exceptionally active catalysts for a wide range of cross-coupling reactions).[103] Hruszkewycz et al. noted that allylpalladium complexes 144, originally developed by the Nolan research group,[104] tended to form inactive PdI dimers during catalysis (Scheme 49a).[105] This would occur via comproportionation of the PdII precatalyst 144 with catalytically-active Pd0 generated under the reaction conditions. The sterically encumbering t-Butyl group of 143 prevents such dimers from forming, thus resulting in an especially active catalyst system. Scheme 49b shows a side-by-side comparison of IPr-143 with IPr-144 in room temperature Suzuki cross-coupling reactions (IPr=1,3-bis(2,6-diisopropylphenyl)-1,3-dihydro-2H-imidazol-2-ylidene). IPr-143 outperformed IPr-144 for all of the couplings tested, providing the desired biaryls in short reaction times and low catalyst loadings.
Scheme 49.
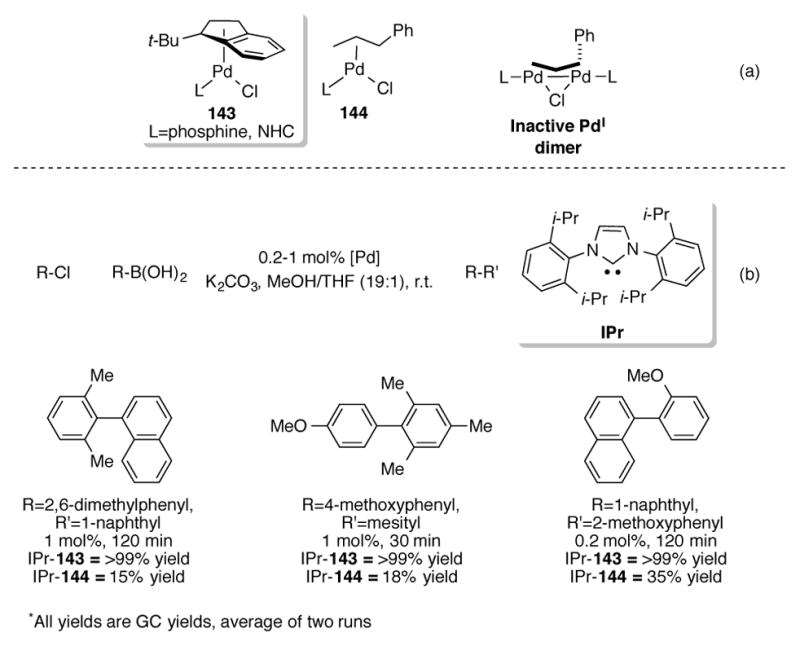
(a) Generation of inactive PdI dimers during catalysis (b) comparison of 143 and 144 for Suzuki cross-couplings
XPhos-143 can efficiently catalyze the Suzuki coupling of electron-rich heteroarylboronic acids and the α-arylation of aryl methyl ketones (Scheme 50a). RuPhos-143 is an effective catalyst for Buchwald-Hartwig amination reactions (Scheme 50b). Thus, (η3-1-t-Bu-Ind)LPdCl 143 complexes can be seen to be a versatile structural motif that can catalyze a diverse variety of reactions in high yield, mild reaction conditions, and low catalyst loadings.
Scheme 50.
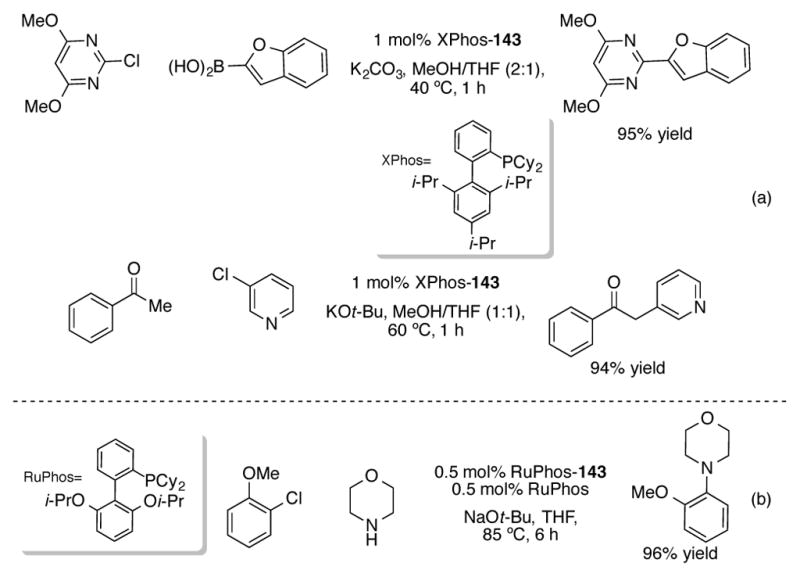
(a) Suzuki coupling of and α-arylation of ketones with XPhos-143 (b) Buchwald-Hartwig amination with RuPhos-143
6. Summary and Outlook
Since the discovery of the indenyl effect by Hart-Davis and Mawby in 1969, researchers have documented many cases in which the deliberate choice of an indenyl ligand had a consequence on catalysis. Indenylmetal complexes have been shown in many instances, but not all, to be superior catalysts to their cyclopentadienyl counterparts in terms of reactivity, selectivity, and stability. As can be seen from the examples presented in this review, it is difficult to predict a priori what impact an indenyl ligand will have on a catalytic reaction as this will be highly dependent on the metal, the reaction conditions, and the chemical transformation in question. For example, the positive effect of indenyl substitution in zirconium-catalyzed carboalumination appears to be purely steric in that it prevents substantial β-hydride elimination whereas in ruthenium-catalyzed redox isomerization, a true acceleration in reaction rate is observed.
Regardless, the indenyl ligand still remains an underexplored motif for transition metal catalysis, especially in the context of organic synthesis. The low usage of these indenyl complexes is likely due to their limited commercial availability. It is our hope that this review will inspire chemists to invent new and creative ways of designing and implementing these fascinating complexes.
Acknowledgments
We thank the NIH (GM-033049), National Science Foundation (CHE-1360634) for their generous support of our programs in catalysis.
Biographies
 Barry M. Trost was born in Philadelphia, PA in 1941 and studied at the University of Pennsylvania (BA, 1962). He obtained his PhD in 1965 at MIT. He moved to the University of Wisconsin where he was made Professor in 1969 and subsequently Vilas Research Professor in 1982. He moved to Stanford University in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. In addition to holding Visiting Professorships at several universities worldwide, he has been awarded numerous prizes worldwide. His interests span the entire field of organic synthesis, particularly in the development of novel methodology and strategy for total synthesis of bioactive complex molecules.
Barry M. Trost was born in Philadelphia, PA in 1941 and studied at the University of Pennsylvania (BA, 1962). He obtained his PhD in 1965 at MIT. He moved to the University of Wisconsin where he was made Professor in 1969 and subsequently Vilas Research Professor in 1982. He moved to Stanford University in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. In addition to holding Visiting Professorships at several universities worldwide, he has been awarded numerous prizes worldwide. His interests span the entire field of organic synthesis, particularly in the development of novel methodology and strategy for total synthesis of bioactive complex molecules.
 Michael Ryan was born in Whitefish Bay, WI and studied chemistry and physics at Boston College (BS, 2010). He completed his doctoral studies in 2016 at Stanford University in the laboratory of Professor Barry M. Trost on catalytic asymmetric cyclization reactions of ruthenium complexes. He is currently a postdoctoral associate at the University of Wisconsin-Madison in the laboratory of Professor Shannon S. Stahl working on copper-catalyzed aerobic oxidation reactions.
Michael Ryan was born in Whitefish Bay, WI and studied chemistry and physics at Boston College (BS, 2010). He completed his doctoral studies in 2016 at Stanford University in the laboratory of Professor Barry M. Trost on catalytic asymmetric cyclization reactions of ruthenium complexes. He is currently a postdoctoral associate at the University of Wisconsin-Madison in the laboratory of Professor Shannon S. Stahl working on copper-catalyzed aerobic oxidation reactions.
References
- 1.(a) Wilkinson G, Rosenblum M, Whiting MC, Woodward RB. J Am Chem Soc. 1952;74:2125. [Google Scholar]; (b) Pfab W, Fischer EOZ. anorg allg Chem. 1953;274:316. [Google Scholar]
- 2.For a review on the structure and bonding of the Cp ligand, see: Lauher JW, Hoffmann R. J Am Chem Soc. 1976;98:1729.
- 3.For example, the Cp ligand is a commonly used motif in Group 4 olefin polymerization catalysts: McKnight AL, Waymouth RM. Chem Rev. 1998;98:2587. doi: 10.1021/cr940442r.Hlatky GG. Coord Chem Rev. 1999;181:243.
- 4.Hart-Davis AJ, Mawby RJ. J Chem Soc A. 1969;(0):2403. [Google Scholar]
- 5.(a) Marder TB, Calabrese JC, Roe DC, Tulip TH. Organometallics. 1987;6:2012. [Google Scholar]; (b) Westcott SA, Kakkar AK, Stringer G, Taylor NJ, Marder TB. J Organomet Chem. 1990;394:777. [Google Scholar]
- 6.(a) Hart-Davis AJ, White C, Mawby RJ. Inorg Chim Act. 1970;4:441. [Google Scholar]; (b) Jones DJ, Mawby RJ. Inorg Chim Act. 1972;6:157. [Google Scholar]; (c) Turaki NN, Huggins JM, Lebioda L. Inorg Chem. 1988;27:424. [Google Scholar]
- 7.(a) Caddy P, Green M, O’Brien E, Smart LE, Woodward P. Angew Chem Int Ed Engl. 1977;16:648. [Google Scholar]; (b) Caddy P, Green M, O’Brien E, Smart LE, Woodward P. J Chem Soc, Dalton Trans. 1980;(6):962. [Google Scholar]
- 8.Rerek ME, Ji L-N, Basolo F. J Chem Soc, Chem Commun. 1983;(21):1208. [Google Scholar]
- 9.(a) Wild FRWP, Zsolnai L, Huttner G, Brintzinger HH. J Organomet Chem. 1982;232:233. [Google Scholar]; (b) Wild FRWP, Wasiucionek M, Huttner G, Brintzinger HH. J of Organomet Chem. 1985;288:63. [Google Scholar]
- 10.Brintzinger HH, Fischer D, Mülhaupt R, Rieger B, Waymouth RM. Angew Chem Int Ed Engl. 1995;34:1143. [Google Scholar]
- 11.Chen Z, Halterman RL. J Am Chem Soc. 1992;114:2276. [Google Scholar]
- 12.Akita M, Yasuda H, Nagasuna K, Nakamura A. Bull Chem Soc Jpn. 1983;56:554. [Google Scholar]
- 13.Colletti SL, Halterman RL. Tet Lett. 1992;33:1005. [Google Scholar]
- 14.Erker G, Aulbach M, Knickmeier M, Wingbermuehle D, Krueger C, Nolte M, Werner S. J Am Chem Soc. 1993;115:4590. [Google Scholar]
- 15.Kondakov DY, Negishi E. J Am Chem Soc. 1995;117:10771. [Google Scholar]
- 16.(a) Negishi E, Tan Z, Liang B, Novak T. PNAS. 2004;101:5782. doi: 10.1073/pnas.0307514101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Negishi E. Angew Chem Int Ed. 2011;50:6738. doi: 10.1002/anie.201101380. [DOI] [PubMed] [Google Scholar]; (c) Xu S, Negishi E. Acc Chem Res. 2016;49:2158. doi: 10.1021/acs.accounts.6b00338. [DOI] [PubMed] [Google Scholar]
- 17.For detailed mechanistic studies of this reaction see: Parfenova LV, Berestova TV, Tyumkina TV, Kovyazin PV, Khalilov LM, Whitby RJ, Dzhemilev UM. Tetrahedron: Asymmetry. 2010;21:299.
- 18.Kondakov DY, Negishi E. J Am Chem Soc. 1996;118:1577. [Google Scholar]
- 19.Liang B, Novak T, Tan Z, Negishi E. J Am Chem Soc. 2006;128:2770. doi: 10.1021/ja0530974. [DOI] [PubMed] [Google Scholar]
- 20.Tan Z, Liang B, Huo S, Shi J, Negishi E. Tetrahedron: Asymmetry. 2006;17:512. [Google Scholar]
- 21.Zhu G, Negishi E. Chem Eur J. 2008;14:311. doi: 10.1002/chem.200701512. [DOI] [PubMed] [Google Scholar]
- 22.Wipf P, Ribe S. Org Lett. 2000;2:1713. doi: 10.1021/ol005865w. [DOI] [PubMed] [Google Scholar]
- 23.Shaughnessy KH, Waymouth RM. Organometallics. 1998;17:5728. [Google Scholar]
- 24.(a) Yang X, Stern CL, Marks TJ. J Am Chem Soc. 1991;113:3623. [Google Scholar]; (b) Yang X, Stern CL, Marks TJ. J Am Chem Soc. 1994;116:10015. [Google Scholar]; (c) Wu Z, Jordan RF, Petersen JL. J Am Chem Soc. 1995;117(21):5867. [Google Scholar]; (d) Bochmann M, Sarsfield MJ. Organometallics. 1998;17:5908. [Google Scholar]
- 25.This catalyst system has also been used for standard ZACA processes: Petros RA, Camara JM, Norton JR. J Organomet Chem. 2007;692:4768.
- 26.Millward DB, Sammis G, Waymouth RM. J Org Chem. 2000;65:3902. doi: 10.1021/jo991429f. [DOI] [PubMed] [Google Scholar]
- 27.Huo S, Shi J, Negishi E. Angew Chem Int Ed. 2002;41:2141. [PubMed] [Google Scholar]
- 28.This fragment was also made via the method described in Scheme 3: Huo S, Negishi E. Org Lett. 2001;3:3253. doi: 10.1021/ol010142d.
- 29.(a) Novak T, Tan Z, Liang B, Negishi E. J Am Chem Soc. 2005;127:2838. doi: 10.1021/ja043534z. [DOI] [PubMed] [Google Scholar]; (b) Xu S, Lee CT, Wang G, Negishi E. Chem Asian J. 2013;8:1829. doi: 10.1002/asia.201300311. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xu S, Oda A, Kamada H, Negishi E. PNAS. 2014;111:8368. doi: 10.1073/pnas.1401187111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu S, Oda A, Negishi E. Chem Eur J. 2014;20:16060. doi: 10.1002/chem.201405053. [DOI] [PubMed] [Google Scholar]
- 30.Xu S, Oda A, Bobinski T, Li H, Matsueda Y, Negishi E. Angew Chem Int Ed. 2015;54:9319. doi: 10.1002/anie.201503818. [DOI] [PubMed] [Google Scholar]
- 31.6,7-dehydrostipiamide: Zeng X, Zeng F, Negishi E. Org Lett. 2004;6:3245. doi: 10.1021/ol048905v.Scyphostatin (fragment): Tan Z, Negishi E. Angew Chem Int Ed. 2004;43:2911. doi: 10.1002/anie.200353429.ionomycin (fragment): Cooksey JP, Kocienski PJ, Li Y, Schunk S, Snaddon TN. Org Biomol Chem. 4:2006. 3325. doi: 10.1039/b606262h.Spongidepsin: Zhu G, Negishi E. Org Lett. 2007;9:2771. doi: 10.1021/ol0707259.Yellow Scale Pheromone: Xu Z, Negishi E. Org Lett. 2008;10:4311. doi: 10.1021/ol8017566.(S,R,R,S,R,S)-4,6,8,10,16,18-hexamethyldocosane: Zhu G, Liang B, Negishi E. Org Lett. 2008;10:1099. doi: 10.1021/ol703056u.
- 32.Lipshutz BH, Butler T, Lower A. J Am Chem Soc. 2006;128:15396. doi: 10.1021/ja065769b.Similar results were observed with arylethynes: Wang G, Zhu G, Negishi E. J of Organomet Chem. 2007;692:4731. doi: 10.1016/j.jorganchem.2007.05.052.
- 33.For alkyne hydroamination reviews, see: Pohlki F, Doye S. Chem Soc Rev. 2003;32:104. doi: 10.1039/b200386b.Severin R, Doye S. Chem Soc Rev. 2007;36:1407. doi: 10.1039/b600981f.
- 34.(a) Pohlki F, Heutling A, Bytschkov I, Hotopp T, Doye S. Synlett. 2002;2002:0799. [Google Scholar]; (b) Heutling A, Pohlki F, Doye S. Chem Eur J. 2004;10:3059. doi: 10.1002/chem.200305771. [DOI] [PubMed] [Google Scholar]; (c) Heutling A, Severin R, Doye S. Synthesis. 2005;(7):1200. [Google Scholar]; (d) Marcseková K, Wegener B, Doye S. Eur J Org Chem. 2005;2005:4843. [Google Scholar]
- 35.Heutling A, Doye S. J Org Chem. 2002;67:1961. doi: 10.1021/jo016311p. However, high anti-Markovnikov selectivity (92:3) was observed when 4-methylaniline was used as the nucleophile. [DOI] [PubMed] [Google Scholar]
- 36.Buil ML, Esteruelas MA, López AM, Mateo AC, Oñate E. Organometallics. 2007;26:554. [Google Scholar]
- 37.Severin R, Reimer J, Doye S. Eur J Org Chem. 2010;2010:51. doi: 10.1021/jo100460v. [DOI] [PubMed] [Google Scholar]
- 38.Doye S, Severin R, Mujahidin D, Reimer J. Heterocycles. 2007;74:683. [Google Scholar]
- 39.Gräbe K, Zwafelink B, Doye S. Eur J Org Chem. 2009;2009:5565. [Google Scholar]
- 40.(a) Müller C, Loos C, Schulenberg N, Doye S. Eur J Org Chem. 2006;2006:2499. [Google Scholar]; (b) Müller C, Saak W, Doye S. Eur J Org Chem. 2008;2008:2731. [Google Scholar]
- 41.Bexrud JA, Beard JD, Leitch DC, Schafer LL. Org Lett. 2005;7:1959. doi: 10.1021/ol0503992. [DOI] [PubMed] [Google Scholar]
- 42.(a) Gagne MR, Stern CL, Marks TJ. J Am Chem Soc. 1992;114:275. [Google Scholar]; (b) Hong S, Marks TJ. Acc Chem Res. 2004;37:673. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 43.(a) Gilbert AT, Davis BL, Emge TJ, Broene RD. Organometallics. 1999;18:2125. [Google Scholar]; (b) Chai Z, Hua D, Li K, Zhou S, Chu J, Yang G. J Organomet Chem. 2014;768:136. [Google Scholar]
- 44.(a) Vitanova DV, Hampel F, Hultzsch KC. J Organomet Chem. 2007;692:4690. [Google Scholar]; (b) Xu X, Chen Y, Feng J, Zou G, Sun J. Organometallics. 2010;29:549. [Google Scholar]; (c) Chai Z, Hua D, Li K, Chu J, Yang G. Chem Commun. 2014;50:177. doi: 10.1039/c3cc47006g. [DOI] [PubMed] [Google Scholar]
- 45.Seyam AM, Stubbert BD, Jensen TR, O’Donnell JJ, III, Stern CL, Marks TJ. Inorg Chim Act. 2004;357:4029. [Google Scholar]
- 46.(a) Kubiak R, Prochnow I, Doye S. Angew Chem Int Ed. 2010;49:2626. doi: 10.1002/anie.200906557. [DOI] [PubMed] [Google Scholar]; (b) Preuß T, Saak W, Doye S. Chem Eur J. 2013;19:3833. doi: 10.1002/chem.201203693. [DOI] [PubMed] [Google Scholar]
- 47.(a) Herzon SB, Hartwig JF. J Am Chem Soc. 2007;129:6690. doi: 10.1021/ja0718366. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Herzon SB, Hartwig JF. J Am Chem Soc. 2008;130:14940. doi: 10.1021/ja806367e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trost BM, Kulawiec RJ. J Am Chem Soc. 1993;115:2027. [Google Scholar]
- 49.(a) Trost BM, Livingston RC. J Am Chem Soc. 1995;117:9586. [Google Scholar]; (b) Trost BM, Livingston RC. J Am Chem Soc. 2008;130:11970. doi: 10.1021/ja804105m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trost BM, Weiss AH. Org Lett. 2006;8:4461. doi: 10.1021/ol0615836. [DOI] [PubMed] [Google Scholar]
- 51.Trost BM, Breder A. Org Lett. 2011;13:398. doi: 10.1021/ol102706j. [DOI] [PubMed] [Google Scholar]
- 52.Trost BM, Breder A, Kai B. Org Lett. 2012;14:1708. doi: 10.1021/ol300278x. [DOI] [PubMed] [Google Scholar]
- 53.Trost BM, Xie J, Maulide N. J Am Chem Soc. 2008;130:17258. doi: 10.1021/ja807894t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koh JH, Jung HM, Kim M-J, Park J. Tet Lett. 1999;40:6281. [Google Scholar]
- 55.Bassetti M, Marini S, Tortorella F, Cadierno V, Díez J, Gamasa MP, Gimeno J. J Organomet Chem. 2000;593–594:292. [Google Scholar]
- 56.Yamamoto Y, Kitahara H, Ogawa R, Kawaguchi H, Tatsumi K, Itoh K. J Am Chem Soc. 2000;122:4310. [Google Scholar]
- 57.(a) Paih JL, Dérien S, Dixneuf PH. Chem Commun. 1999;(15):1437. [Google Scholar]; (b) Mauthner K, Soldouzi KM, Mereiter K, Schmid R, Kirchner K. Organometallics. 1999;18:4681. [Google Scholar]
- 58.Alvarez P, Gimeno J, Lastra E, García-Granda S, Van der Maelen JF, Bassetti M. Organometallics. 2001;20:3762. [Google Scholar]
- 59.Trost BM, Imi K, Indolese AF. J Am Chem Soc. 1993;115:8831. [Google Scholar]
- 60.Kündig EP, Saudan CM, Alezra V, Viton F, Bernardinelli G. Angew Chem Int Ed. 2001;40:4481. doi: 10.1002/1521-3773(20011203)40:23<4481::aid-anie4481>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 61.(a) Thamapipol S, Bernardinelli G, Besnard C, Kündig EP. Org Lett. 2010;12:5604. doi: 10.1021/ol1019103. [DOI] [PubMed] [Google Scholar]; (b) Thamapipol S, Kündig EP. Org Biomol Chem. 2011;9:7564. doi: 10.1039/c1ob06121f. [DOI] [PubMed] [Google Scholar]
- 62.Trost BM, Ryan MC, Maurer D. Org Lett. 2016;18:3166. doi: 10.1021/acs.orglett.6b01375. [DOI] [PubMed] [Google Scholar]
- 63.Manzini S, Fernández-Salas JA, Nolan SP. Acc Chem Res. 2014;47:3089. doi: 10.1021/ar500225j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manzini S, Blanco CAU, Nolan SP. Adv Synth Catal. 2012;354:3036. [Google Scholar]
- 65.Madrigal CA, García-Fernández A, Gimeno J, Lastra E. J Organomet Chem. 2008;693:2535. [Google Scholar]
- 66.Manzini S, Poater A, Nelson DJ, Cavallo L, Nolan SP. Chem Sci. 2013;5:180. [Google Scholar]
- 67.Manzini S, Nelson DJ, Nolan SP. ChemCatChem. 2013;5:2848. [Google Scholar]
- 68.Fernández-Salas JA, Manzini S, Nolan SP. Chem Eur J. 2014;20:13132. doi: 10.1002/chem.201404096. [DOI] [PubMed] [Google Scholar]
- 69.Fernández-Salas JA, Manzini S, Nolan SP. Adv Synth Catal. 2014;356:308. [Google Scholar]
- 70.Fernández-Salas JA, Manzini S, Piola L, Slawin AMZ, Nolan SP. Chem Commun. 2014;50:6782. doi: 10.1039/c4cc02096k. [DOI] [PubMed] [Google Scholar]
- 71.Piola L, Fernández-Salas JA, Manzini S, Nolan SP. Org Biomol Chem. 2014;12:8683. doi: 10.1039/c4ob01798f. [DOI] [PubMed] [Google Scholar]
- 72.Alkhaleeli DF, Baum KJ, Rabus JM, Bauer EB. Catal Commun. 2014;47:45. See also using IndRu tris(N-pyrrolyl)phosphine complexes: Stark MJ, Shaw MJ, Rath NP, Bauer EB. Eur J Inorg Chem. 2016:1093.
- 73.Lenze M, Sedinkin SL, Rath NP, Bauer EB. Tet Lett. 2010;51:2855. [Google Scholar]
- 74.Abdulla K, Booth BL, Stacey C. J Organomet Chem. 1985;293:103. [Google Scholar]
- 75.Borrini A, Diviersi P, Ingrosso G, Lucherini A, Serra G. J Mol Catal. 1985;30:181. [Google Scholar]
- 76.Orian L, Swart M, Bickelhaupt FM. ChemPhysChem. 2014;15:219. doi: 10.1002/cphc.201300934. [DOI] [PubMed] [Google Scholar]
- 77.Heller B, Oehme G. J Chem Soc, Chem Commun. 1995;(2):179. [Google Scholar]
- 78.(a) Gutnov A, Heller B, Fischer C, Drexler H-J, Spannenberg A, Sundermann B, Sundermann C. Angew Chem Int Ed. 2004;43:3795. doi: 10.1002/anie.200454164. [DOI] [PubMed] [Google Scholar]; (b) Hapke M, Kral K, Fischer C, Spannenberg A, Gutnov A, Redkin D, Heller B. J Org Chem. 2010;75:3993. doi: 10.1021/jo100122d. [DOI] [PubMed] [Google Scholar]
- 79.Heller B, Gutnov A, Fischer C, Drexler H-J, Spannenberg A, Redkin D, Sundermann C, Sundermann B. Chem Eur J. 2007;13:1117. doi: 10.1002/chem.200600826. [DOI] [PubMed] [Google Scholar]
- 80.Heller B, Hapke M, Fischer C, Andronova A, Starý I, Stará IG. J Organomet Chem. 2013;723:98. [Google Scholar]
- 81.Schumann H, Stenzel O, Dechert S, Girgsdies F, Blum J, Gelman D, Halterman RL. Eur J Inorg Chem. 2002;2002:211. [Google Scholar]
- 82.Lee BY, Chung YK, Jeong N, Lee Y, Hwang SH. J Am Chem Soc. 1994;116:8793. [Google Scholar]
- 83.Marder TB, Roe DC, Milstein D. Organometallics. 1988;7:1451. [Google Scholar]
- 84.Hauptman E, Sabo-Etienne S, White PS, Brookhart M, Garner JM, Fagan PJ, Calabrese JC. J Am Chem Soc. 1994;116:8038. [Google Scholar]
- 85.Smoleński P. J Organomet Chem. 2011;696:3867. [Google Scholar]
- 86.Garrett CE, Fu GC. J Org Chem. 1998;63:1370. [Google Scholar]
- 87.Brinkman JA, Nguyen TT, Sowa JR. Org Lett. 2000;2:981. doi: 10.1021/ol005653z. [DOI] [PubMed] [Google Scholar]
- 88.Iverson CN, Smith MR. J Am Chem Soc. 1999;121:7696. [Google Scholar]
- 89.Cho J-Y, Iverson CN, Smith MR. J Am Chem Soc. 2000;122:12868. [Google Scholar]
- 90.Chen H, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]
- 91.Cho J-Y, Tse MK, Holmes D, Maleczka RE, Smith MR. Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 92.Merola JS, Kacmarcik RT. Organometallics. 1989;8:778. [Google Scholar]
- 93.Norberg A, Smith MR, Maleczka RE. Synthesis. 2011;2011:857. doi: 10.1055/s-0030-1258443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.(a) Holmes D, Chotana GA, Maleczka RE, Smith MR. Org Lett. 2006;8:1407. doi: 10.1021/ol060205y. [DOI] [PubMed] [Google Scholar]; (b) Shi F, Smith MR, Maleczka RE. Org Lett. 2006;8:1411. doi: 10.1021/ol060207i. [DOI] [PubMed] [Google Scholar]
- 95.(a) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew Chem Int Ed. 2002;41:3056. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 96.Chotana GA, Kallepalli VA, Maleczka RE, Smith MR. Tetrahedron. 2008;64:6103. doi: 10.1016/j.tet.2008.02.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miyake Y, Isomura E, Iyoda M. Chem Lett. 2006;35:836. [Google Scholar]
- 98.Fontaine F-G, Kadkhodazadeh T, Zargarian D. Chem Commun. 1998:1253. [Google Scholar]
- 99.Fontaine F-G, Nguyen R-V, Zargarian D. Can J Chem. 2003;81:1299. [Google Scholar]
- 100.Sui-Seng C, Castonguay A, Chen Y, Gareau D, Groux LF, Zargarian D. Top Catal. 2006;37:81. [Google Scholar]
- 101.[1,3-(TMS)2Ind]Ni(PPh3)Me has also been tested in the Ni-catalyzed hydrosilylation of styrene with phenylsilane, though the reaction only proceeds to 79% conversion with 1 mol% catalyst, see: Yaofeng Chen Zargarian D. Can J Chem. 2009;87:280.
- 102.Sui-Seng C, Groux LF, Zargarian D. Organometallics. 2006;25:571. [Google Scholar]
- 103.Melvin PR, Nova A, Balcells D, Dai W, Hazari N, Hruszkewycz DP, Shah HP, Tudge MT. ACS Catal. 2015;5:3680. [Google Scholar]
- 104.Marion N, Navarro O, Mei J, Stevens ED, Scott NM, Nolan SP. J Am Chem Soc. 2006;128:4101. doi: 10.1021/ja057704z. and references therein. [DOI] [PubMed] [Google Scholar]
- 105.Hruszkewycz DP, Balcells D, Guard LM, Hazari N, Tilset M. J Am Chem Soc. 2014;136:7300. doi: 10.1021/ja412565c. [DOI] [PubMed] [Google Scholar]