

Abstract
The success of anticancer therapy is usually limited by the development of drug resistance. Such acquired resistance is driven, in part, by intratumoural heterogeneity — that is, the phenotypic diversity of cancer cells co-inhabiting a single tumour mass. The introduction of the cancer stem cell (CSC) concept, which posits the presence of minor subpopulations of CSCs that are uniquely capable of seeding new tumours, has provided a framework for understanding one dimension of intratumoural heterogeneity. This concept, taken together with the identification of the epithelial-to-mesenchymal transition (EMT) programme as a critical regulator of the CSC phenotype, offers an opportunity to investigate the nature of intratumoural heterogeneity and a possible mechanistic basis for anticancer drug resistance. In fact, accumulating evidence indicates that conventional therapies often fail to eradicate carcinoma cells that have entered the CSC state via activation of the EMT programme, thereby permitting CSC-mediated clinical relapse. In this Review, we summarize our current understanding of the link between the EMT programme and the CSC state, and also discuss how this knowledge can contribute to improvements in clinical practice.
The fact that most malignant tumours are composed of multiple phenotypically distinct subpopulations of neoplastic cells has been established by >60 years of research1-4. Nowadays, the phenotypic diversity of neoplastic cells within a tumour is considered a major driver of the development of resistance to therapy and, for this reason, is attracting increasing interest5. The biological heterogeneity arising through both genetic and non-genetic mechanisms contributes to the phenotypic differences between the distinct subpopulations of cancer cells residing within individual tumours6,7. Advances in genome-sequencing technologies are enabling rapid progress in delineating the types of genetic changes that underlie the phenotypic alteration and diversification of cancer cells8,9. In parallel, the ability of cancer cells to frequently interconvert between multiple alternative phenotypic states, doing so without concomitant mutational changes in their genomes, is increasingly recognized; however, the nature of the non-genetic — that is, epigenetic — regulatory mechanisms that contribute substantially to the behaviours of cancer cells and the tumours that they form remains largely unexplored, revealing a critical need to uncover these mechanisms10,11.
The introduction of cancer stem cell (CSC) concept, which postulates the presence of minor populations of CSCs that are uniquely capable of seeding new tumours, has provided new insights into how epigenetic regulatory mechanisms can contribute to the phenotypic diversity of distinct subpopulations of cancer cells within a tumour12,13. This concept is based on the notion that various phenotypically distinct cancer cells residing within the same tumour mass are organized in a hierarchy, resembling the stem-cell hierarchy of the corresponding non-neoplastic tissue. Indeed, the functional parallels between CSCs and non-neoplastic stem cells are considered to be extensive, including the unique ability to entirely regenerate complex neoplastic and non-neoplastic tissues, respectively, under appropriate conditions14. Accordingly, in tumours, the epigenetically defined state of CSCs should, in principle, enable the tumour cells to self-renew in order to generate new CSCs, and to spawn progeny that differentiate into less-tumorigenic and non-self-renewing offspring — that is, the non-CSCs that are thought, in most cases, to form the bulk of the tumour. Implicit in this paradigm is the important hypothesis that continued tumour growth, metastasis formation, and recurrence after therapy can largely be attributed to the relatively rare subpopulations of CSCs within individual tumours15,16. Notably, discrete subpopulations of CSCs have now been identified in many types of human cancer, demonstrating the wide applicability of the CSC concept17.
The penetration of the CSC concept has caused some to argue that the often limited efficacy of conventional anticancer therapies is attributable to the targeting of the bulk population of non-CSCs within individual tumours, while not eliminating the rare subpopulation of CSCs18-20. Indeed, accumulating evidence has shown that CSCs are more resistant than non-CSCs to various types of conventional therapies21-23. Moreover, given their tumour-initiating capacity, the surviving CSCs are qualified to serve as the precursors of new tumour masses, ultimately leading to clinical relapse. These observations highlight the critical importance of understanding the epigenetic mechanisms that distinguish CSCs from the bulk population of non-CSCs; insights into these mechanisms hold the promise of yielding novel therapeutic strategies designed to eradicate CSCs, with the possible benefit of achieving extended, if not permanent, clinical remissions.
The epigenetic changes that account for the phenotypic differences between CSCs and non-CSCs are only beginning to be uncovered. In the case of carcinomas, the difference between CSCs and non-CSCs is likely to be attributable largely to the cell-biological programme termed epithelial-to-mesenchymal transition (EMT)24,25. This programme imparts heritable phenotypic changes to carcinoma cells through epigenetic modifications without introducing new genetic alterations. Upon activation of the programme, carcinoma cells lose many of their epithelial characteristics, including the presence of epithelial cell junctions and apical–basal polarity, and instead acquire mesenchymal attributes, such as an elongated, fibroblast-like morphology as well as an increased capacity for migration and invasion26 (FIG. 1). In a number of carcinomas, only neoplastic cells within the CSC-enriched subpopulation exhibit aspects of EMT-programme activation27-30. Of note, in certain experimental models of carcinoma, forced induction of an EMT programme in epithelial tumour cells substantially increases their capacity for tumour initiation27,31,32. In addition, EMT-programme activation confers on these tumour cells resistance to many types of therapeutic agents — another important attribute of CSCs33,34.
Figure 1. Morphological and physiological changes associated with the epithelial-to-mesenchymal transition (EMT).
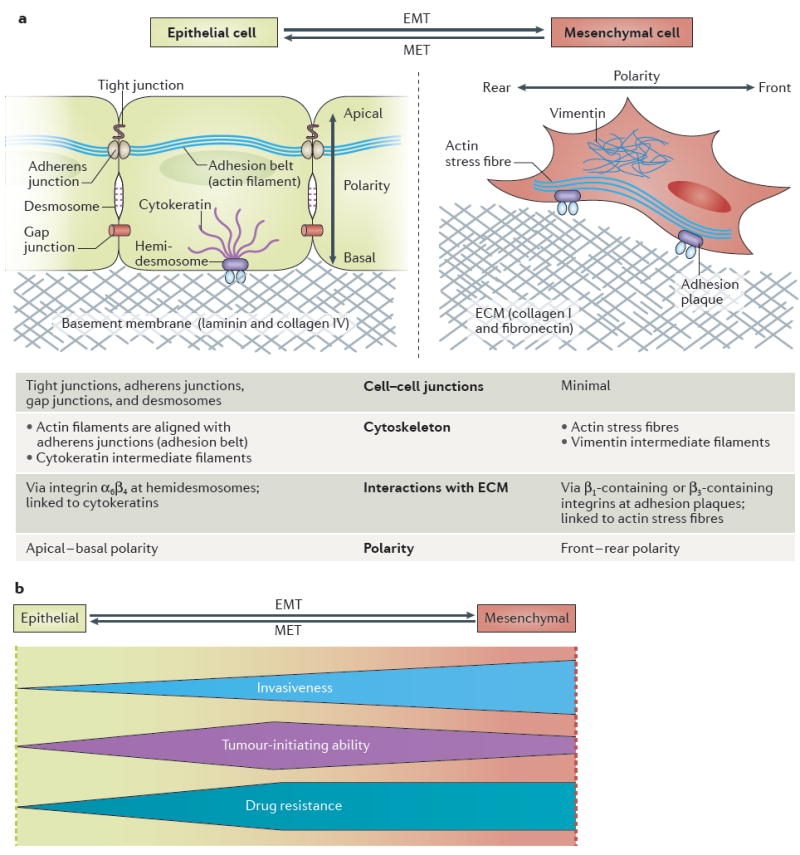
a ∣ A schematic overview of EMT-associated changes in cell physiology. Activation of the EMT programme induces profound changes in various aspects of cell morphology and physiology, most notably in cell cell junctions, cytoskeletal composition, cellular interactions with the extracellular matrix (ECM), and cell polarity. b ∣ Summary of the physiological outcomes of EMT in carcinoma: the profile of the shapes illustrates how the extent of invasiveness, the tumour-initiating ability, and degree of drug resistance change across the spectrum of EMT-programme activation. Carcinoma cells invade surrounding tissues either by individual-cell migration or multicellular migration (as cell cohorts). In general, migration of individual cells, which requires the strong activation of the EMT programme, results in faster tissue invasion than occurs by multicellular migration, the mode of migration that predominates when the EMT programme is only weakly activated189. The tumour-initiating ability of carcinoma cells is also affected by the level of EMT-programme activation, peaking at an intermediate level of EMT in these cells; extensive EMT activation is usually detrimental to tumour-initiating ability149. The drug resistance of carcinoma cells also seems to be maximal at an intermediate level of EMT-programme activation, but plateaus (rather than declines) with further activation of this programme149,184. MET, mesenchymal-to-epithelial transition.
Perhaps ironically, we still possess relatively few insights into why these two clinically important cell-biological programmes — the CSC phenotype and EMT — are so intimately interconnected. Herein, we summarize our current understanding regarding the causes and effects of EMT-programme activation in carcinoma cells, with a particular focus on how this programme is linked to the CSC state. We also discuss the opportunities and challenges in targeting the EMT programme and, by extension, CSCs as a strategy for carcinoma treatment.
Principles of the EMT programme
The roles of EMT and the reverse process, mesenchymalto-epithelial transition (MET), were initially studied in the context of embryonic morphogenesis35,36. The findings revealed that during gastrulation in amniotes, the epiblast cells in a specific area of the primitive ectoderm — the first embryonic epithelial tissue — undergo EMT, thereby differentiating into primary mesenchyme cells, and ingress from the ectodermal layer, ultimately giving rise to mesodermal and endodermal cell layers. As a further example, neural crest cells within the dorsal neural tube — composed of neuroepithelial cells — also undergo EMT, migrate to various sites in the developing embryo, and differentiate into a variety of distinct cell types, including melanocytes, peripheral neurons, glial cells, and connective-tissue cells. By contrast, during the embryonic development of a kidney, cells in the metanephric mesenchyme undergo MET in order to form the renal epithelium and, ultimately, generate nephrons37. These examples comprise only a few of the instances in which epithelial–mesenchymal interconversions have key roles in embryogenesis.
In addition, the EMT programme plays a central part in various pathological processes, including wound healing, tissue fibrosis, and carcinoma progression38,39. In all of these processes, activation of the EMT programme elicits changes in multiple fundamental aspects of cellular physiology that include: alterations in the cytoskeletal organization and associated changes in cell morphology from squamous, cuboidal, or columnar shapes to spindle-like, elongated forms (FIG. 1); dissolution of epithelial cell–cell junctions, including tight, adherens, and gap junctions; loss of apical–basal polarity and concomitant gain of front–rear polarity; acquisition of motility, as well as an ability to degrade and reorganize the extracellular matrix (ECM), enabling cell invasion; and reconfiguration of the expression patterns of at least 400 distinct genes40. Of note, both non-neoplastic and neoplastic epithelial cells can activate the multifaceted EMT programme to varying extents, often acquiring many, but not all, of the traits associated with the ‘canonical’ programme.
Various extracellular stimuli and corresponding intracellular signalling pathways have been implicated in the induction of EMT. The central role of TGFβ–SMAD signalling in multiple different models of EMT has been well established41,42. In addition, other signalling pathways, including the canonical or noncanonical Wnt, growth factor–receptor tyrosine kinase, and ECM–integrin signalling pathways, acting in various combinations, also seem to contribute critically to the induction of EMT in a variety of tissue contexts43. Importantly, the EMT process operating in carcinoma cells in vivo is often, and perhaps invariably, triggered by specific signals released by stromal cells into the tumour microenvironment, some of which are also involved in choreographing various inflammatory and hypoxic responses44.
In both non-neoplastic and neoplastic cells, these diverse signal-transduction pathways converge on the activation of a relatively small group of transcription factors that proceed to orchestrate the gene-expression changes associated with EMT45. These transcription factors, often referred to collectively as ‘EMT-inducing transcription factors’ (EMT-TFs), can typically be classified into one of three different protein families — namely, the Snail (including Snail and Slug; also known as SNAI1 and SNAI2, respectively), ZEB (including ZEB1 and ZEB2), and basic helix–loop–helix (including TWIST1, TWIST2, and TCF3) families45. In certain contexts, however, other transcription factors, such as PRRX1, YAP1/TAZ, and SOX4, also have critical roles in EMT. These master regulators of EMT programmes govern the transcription of EMT-associated genes via promoter activation or repression, often achieved through modification of chromatin structure45. The end results are suppression, to various extents, of genes associated with the epithelial phenotype, such as those encoding E-cadherin and cytokeratins, and upregulation of genes associated with the mesenchymal-cell phenotype, including those encoding N-cadherin, fibronectin, and vimentin45.
The EMT-TFs often act cooperatively to regulate the expression of common target genes and, in addition, frequently control the expression of one another39,45. For example, Snail is an upstream regulator that induces the expression of multiple other EMT-TFs, including SLUG, ZEB1, and TWIST1. On the other hand, TCF3 seems to function as a downstream effector whose expression is induced by various other EMT-TFs, such as Snail, Slug, and ZEB1 (REFS 40,46). Owing to these reciprocal interactions, experimental expression of one EMT-TF usually results in upregulation of certain other EMT-TFs, ultimately inducing, in many cases, the full spectrum of EMT-associated changes. Such coordinated, multifaceted shifts in cell phenotype can obscure the functional differences between the various individual EMT-TFs, which seem to be specialized for orchestrating distinct subprogrammes of the extensive canonical EMT programme. Moreover, the controls regulating the experimental expression of EMT-TFs rarely recapitulate those operating physiologically, resulting in phenotypic responses in cells that are not observed, for example, in spontaneously arising tumours. These disparities raise concerns regarding the interpretability of observations derived from experimental overexpression of an individual EMT-TF.
In addition to being regulated by signalling pathways, the expression and functions of EMT-TFs are controlled by other upstream regulatory mechanisms. In particular, studies have highlighted the important roles of noncoding microRNAs (miRNAs), which suppress the expression of specific proteins through a combination of mRNA destabilization and translational repression47. Among the best-characterized examples of miRNAs that regulate the EMT programme are members of the miR-200 family, which downregulate the expression of ZEB1 and ZEB2 proteins; intriguingly, the expression of miR-200 miRNAs is repressed, reciprocally, by ZEB1 and ZEB2, establishing a double-negative feedback loop48,49. Similarly, the expression of Snail is attenuated by members of the miR-34 family, which, in turn, is suppressed by Snail, forming another double-negative regulatory feedback loop between EMT-TFs and miRNAs50,51.
The expression levels of several EMT-TFs are also regulated at the level of post-translational protein stability: Snail is phosphorylated by GSK-3β at two consecutive serine-rich motifs, which targets this EMT-TF for ubiquitylation and subsequent proteasomal degradation52. By contrast, TWIST1 can be phosphorylated at Ser68 by ERK1/2, p38, and JNK, which protects this EMT-TF from ubiquitin-mediated degradation53. In addition, the Siah family of RING finger ubiquitin E3 ligases has a critical role in mediating the ubiquitylation and thus the subsequent proteasomal degradation of ZEB1 (REF. 54). Hence, EMT-TFs are controlled at multiple levels and by diverse upstream regulatory mechanisms, which cooperatively regulate activation of the EMT programme.
EMTs are known to have essential roles in embryogenesis, and the various EMT-TFs are specialized to orchestrate distinct steps of this process55. The associations of particular EMT-TFs with distinct embryogenic phases of development have been demonstrated most elegantly through gene-targeting studies in mice. Mice deficient in the Snai1 gene (encoding Snail) displayed the most severe phenotype observed to date in such studies: the embryos of these mice develop a morphologically abnormal mesodermal layer. More specifically, the cells within this layer fail to undergo EMT and retain their epithelial properties, resulting in the death of embryos at around embryonic day 8.5 (E8.5)56. Twist1-deficient and Zeb2-deficient mouse embryos both exhibit failure of neural tube closure and die at around E11.5 and E9.5, respectively57,58. Mice deficient for Snai2 (encoding Slug), Zeb1, Twist2, or Tcf3 can survive to term, but have various severe developmental defects59-63. Hence, different EMT-TFs enable distinct processes in embryonic morphogenesis, indicating that they control different forms of the EMT programme.
While the mechanisms underlying EMT-programme activation are becoming increasingly clear, those responsible for the induction of the reverse process — MET — remain enigmatic. The prevailing hypothesis is that MET results simply from the deactivation of a previously active EMT programme, caused by reduced or fully absent expression of EMT-TFs64,65; however, evidence also supports the presence of specific mechanisms that selectively activate the MET programme via pathways that do not directly involve the well-characterized EMT-TFs. For example, the reprogramming of mouse fibroblasts into pluripotent stem cells involves MET-programme activation that is mediated, in part, by the induction of epithelial gene expression by the transcription factor KLF4 (REF. 66). Moreover, during kidney development in mice, Wnt9b secreted by the ureteric epithelium triggers the secretion of two other signalling proteins, Wnt4 and Fgf8, from pre-tubular aggregates; the latter two factors then contribute critically to the transition of these initially mesenchymal aggregates into the renal epithelium, in part, through the activation of the Lhx1 transcription factor67.
EMT during carcinoma progression
In contrast to the general agreement on the contribution of EMTs to embryonic development, the involvement of this programme in carcinoma progression has been a matter of debate until very recently68. This controversy was largely attributable to scepticism raised by pathologists, who had not seen clear evidence of EMT in human carcinoma samples frequently enough to assert the wide involvement of the EMT programme in the pathogenesis of many cancer types69.
Indeed, the occurrence of a ‘complete EMT’, involving the loss of all epithelial traits and concomitant gain of the full spectrum of mesenchymal attributes (as seen in certain EMT processes during embryogenesis), is rare in human carcinomas. The exception to this rule derives from carcinosarcoma, a rare cancer type consisting of distinct epithelial (carcinomatous) and mesenchymal (sarcomatous) compartments that are derived from a common precursor-cell population70. The fact that the EMT programme in human carcinomas is usually activated only partially has probably obscured the detection of EMT markers in clinical carcinoma samples. The identification of EMT-programme components in islands of carcinoma cells has also been complicated by the presence of nearby stromal cells, which naturally display a diverse array of EMT-associated mesenchymal markers, confounding any clear association of EMT programmes specifically with the carcinoma cells.
Nonetheless, a rapidly growing number of studies are now adding to a large corpus of evidence for the activation of the EMT programme in, and its contributions to, the pathogenesis of human carcinomas. For more than 20 years, loss of expression of the adhesion protein E-cadherin in carcinoma cells has been recognized to be associated with metastasis and a poor prognosis71,72. More direct evidence of the relationship between the EMT programme and carcinoma progression was presented in the early 2000s, when the role of Snail as a driver of carcinoma progression was demonstrated. Specifically, expression of Snail in carcinoma cells was found to induce EMT in part via direct transcriptional repression of the gene encoding E-cadherin (CDH1)73. Moreover, Snail expression was detected in both invasive carcinoma cell lines and tumour specimens from patients with breast cancer, and the latter was found to correlate closely with histological dedifferentiation and the development of metastases74. A few years later, Twist1 was shown to be critical to the metastasis of mouse mammary carcinomas75. Results of a number of sub-sequent studies demonstrated correlations between the presence of EMT markers in many carcinoma types and poor patient prognoses, including, but not limited to, those with prostate, lung, pancreatic, liver, colorectal, or bladder carcinomas76-81.
The incomplete nature of the EMT programmes activated in human carcinomas — usually referred to as ‘partial EMT’ — could be inferred from early observations that loss of E-cadherin expression, a hallmark of invasiveness in carcinomas, is usually partial71,72. More recently, an RNA in situ hybridization analysis of circulating tumour cells (CTCs) from women with breast cancer revealed the presence of cancer cells that concomitantly express epithelial and mesenchymal transcripts, and thus exhibit partial EMT activation82. Remarkably, these partial EMT cells were observed in samples from patients harbouring any of the three major histological subtypes of invasive breast cancer (ER/PR-positive, HER2-positive, or triple-negative), but not in non-neoplastic breast tissues. This partial activation of the EMT programme results in extensive phenotypic diversity of the carcinoma cells within individual tumours.
In addition to being activated partially, the EMT programme in human carcinomas is often activated reversibly, permitting the cancer cells to revert via MET programmes to epithelial states during the course of carcinoma progression (FIG. 2). Thus, in many different types of carcinomas, cancer cells at the invasive front of the primary tumour frequently exhibit signs of EMT activation, including the reduced expression of E-cadherin, while cells that follow behind usually display many epithelial traits and maintain extensive cell–cell adhesions83,84. This observation, together with the dynamic interconversions between the ‘leaders’ and the ‘followers’ that have been shown to occur during the process of carcinoma invasion85, implies a plasticity of cell phenotypes and thus a reversibility of EMT programmes. Stated differently, carcinoma cells can flexibly activate EMT and MET programmes in the course of carcinoma invasion.
Figure 2. The patterns of epithelial-to-mesenchymal transition (EMT)-programme activation during carcinoma progression.
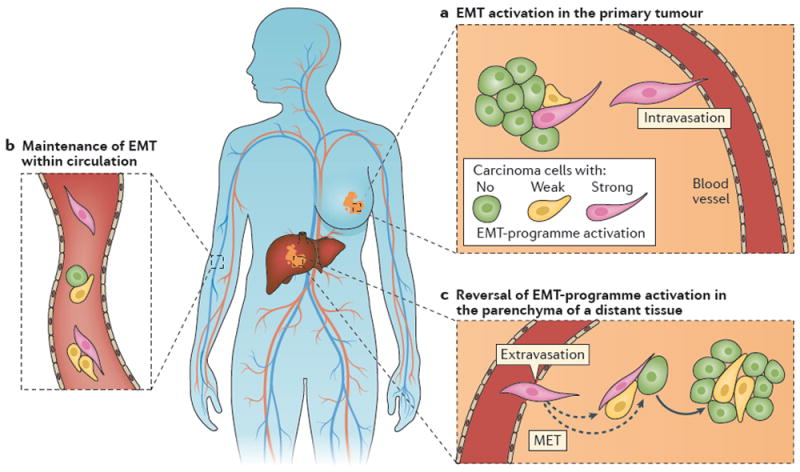
a ∣ In primary tumours, activation of the EMT programme enables particular carcinoma cells to invade the surrounding stroma; some of these cells eventually enter the systemic circulation. b ∣ Circulating tumour cells can demonstrate epithelial and/or mesenchymal traits. Indeed, cancer cells in the circulation, whether solitary or in clusters, frequently exhibit signs of at least partial EMT82. c ∣ Reversal of the EMT process — that is, activation of the mesenchymal-to-epithelial transition (MET) programme — following dissemination of carcinoma cells to distant tissues seems to be critical for the outgrowth of metastases from many types of carcinoma88,89.
The behaviour of disseminated carcinoma cells that seed metastatic colonies is of great interest: founders of metastatic colonies, which are likely to have undergone an EMT before leaving the primary tumour, usually produce progeny at distant sites that regain theirrin86,87 (FIG. 2). Moreover, studies in mouse models of breast or skin cancer have demonstrated that activation of the EMT programme in the primary tumour is crucial for the dissemination of tumour cells to the lungs, whereas the disseminated cells must subsequently undergo MET in order to efficiently form macroscopic metastases88,89. Gene-expression profiling of single cells from patient-derived xenograft (PDX) models of metastatic breast cancer has demonstrated that, before the formation of macroscopic metastases, disseminated tumour cells display stem-like transcriptional signatures, including the activation of the EMT programme; however, those in macroscopic colonies lose signs of EMT-programme activation and instead exhibit luminal-like, epithelial differentiation signatures90. Together, these observations indicate that the formation of macroscopic metastases usually involves inactivation of a previously active EMT programme in carcinoma cells.
How reversion of the EMT programme contributes mechanistically to the process of metastatic colonization remains to be resolved. In addition, restoration of the entire suite of epithelial traits is not necessary for the outgrowth of all types of metastases. Thus, in most cases of invasive lobular carcinoma of the breast, E-cadherin expression in the carcinoma cells is completely lost owing to CDH1 mutation91; however, these cancer cells frequently form macroscopic metastases in various organs. To summarize, although both clinical and pre-clinical observations clearly demonstrate that the EMT programme is regulated in a dynamic and reversible fashion during the progression of carcinomas, the functional significance of this reversibility awaits future study.
The timing of EMT-programme activation during the progression of carcinoma has been studied extensively using genetically-engineered mouse models, in which carcinoma cells were genetically labelled and thus could be distinguished from surrounding non-neoplastic cells. The findings revealed, intriguingly, that the EMT programme can be activated at pre-malignant stages of carcinoma development: in a breast cancer model driven by transgenic expression of a constitutively active mutant form of rat HER2, a high level of Twist1 expression in mammary epithelial cells was detected at the stage of atypical ductal hyperplasia (ADH)92. Similarly, in another mouse breast cancer model, with transgenic expression of polyomavirus middle-T antigen (PyMT), Snail expression in mammary epithelial cells was observed at the ADH stage93. Moreover, in a pancreatic cancer model based on elimination of p53 expression and concomitant expression of the constitutively active G12D-mutated KRas, the pancreatic epithelial cells began to express Zeb1 at the stage of pancreatic intraepithelial neoplasia94. These results are consistent with clinical observations that systemic dissemination of tumour cells can start early in the disease course of certain types of carcinomas, including those of the breast and prostate95.
Together, these findings suggest that activation of an EMT programme and the resulting dissemination of pre-neoplastic cells can occur early in the multistep process of carcinoma progression; however, several key questions regarding the activation of the EMT programme early in carcinoma progression remain unanswered. For instance, the types of genetic alterations that predispose pre-malignant cells to activate previously silent EMT programmes are unknown. Moreover, whether and how disseminated preneoplastic cells with EMT-programme activation contribute to the eventual formation of clinically detectable metastases is unclear96.
The activation of EMT programmes in carcinomas seems to be regulated not only temporally, but also spatially. As discussed, the dedifferentiation of epithelial tumour cells is particularly evident at the invasive fronts of carcinomas83,84, and data from genetically driven mouse models of carcinoma pathogenesis have reinforced the notion that the EMT programme is activated specifically at the invasive fronts of a tumour. In a breast cancer model with transgenic expression of the oncogenic transcription factor Myc, epithelium-derived cells with mesenchymal-like morphological features (spindle shape, elongated nuclei, and poorly defined cell borders) were enriched at the interface between tumour and stromal tissues97. Moreover, in a pancreatic cancer model, elevated expression of Zeb1 in the carcinoma cells was found to be evident particularly at the periphery of the tumour tissue, where abundant inflammatory cells accumulated94. The commonalities in both spatial and temporal patterns of EMT, observed among multiple carcinoma types in both humans and mice, indicate that EMT-programme activation is a highly regulated process, which is strongly influenced by contextual signals that individual carcinoma cells experience at various locations within a tumour.
Microenvironmental regulation of EMT
A large body of evidence points to the critical contributions made by resident and recruited non-neoplastic cell types within the tumour stroma to the regulation of carcinoma cell behaviours. These stromal cells can be categorized into three major classes: angiogenic vascular cells, infiltrating immune cells, and carcinoma-associated fibroblasts (CAFs)98.
The role of carcinoma-associated fibroblasts
CAFs have been reported to arise from multiple sources, including tissue-resident fibroblasts, mesenchymal stem cells, stellate cells, and endothelial cells; a distinct profile of secreted signalling molecules distinguishes CAFs from non-neoplastic tissue fibroblasts. At a biological level, CAFs display an ‘activated’ phenotype, reminiscent of that observed in cells actively participating in wound healing. Moreover, the classification of CAFs can substantially overlap with myofibroblasts that are abundant in the stroma of many high-grade carcinomas as well as in areas of active inflammation99,100. Although the precise molecular definition of this apparently heterogeneous collection of cells remains a matter of debate, a growing body of evidence supports the important role of CAFs in carcinoma progression101. Indeed, CAFs are particularly well characterized with respect to their involvement in EMT-programme activation in carcinoma cells (FIG. 3a).
Figure 3. The contribution of the tumour microenvironment to the activation of the epithelial-to-mesenchymal transition (EMT) programme.
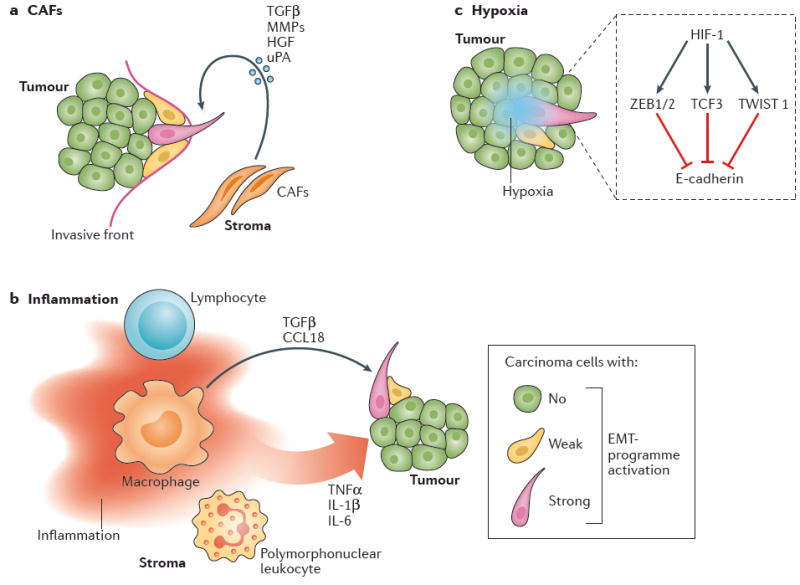
a ∣ Carcinoma-associated fibroblasts (CAFs) are frequently observed at the invasive front of tumours, and probably make an important contribution to the induction of EMT in nearby carcinoma cells by secreting various cytokines and enzymes. b ∣ The tumour microenvironment is often characterized by chronic inflammation; both soluble and cellular mediators of tumour-associated inflammation can contribute to the induction of an EMT programme in carcinoma cells. c ∣ Hypoxia is another common characteristic of the tumour microenvironment. HIF-1, the central mediator of the responses to tumour-associated hypoxia, has been demonstrated to trigger an EMT process, involving the prototypic transcription factors ZEB1/2, TCF-3, and TWIST1, in carcinoma cells. CCL18, C–C-motif chemokine 18; HGF, hepatocyte growth factor; HIF-1, hypoxia-inducible factor 1; MMP, matrix metalloproteinase; TGFβ, transforming growth factor β; TNFα, tumour necrosis factor α; uPA, urokinase plasminogen activator.
Intriguingly, the admixture of CAFs isolated from prostate cancer tissue enabled immortalized, but not transformed, human benign prostatic hyperplasia epithelial (BPH-1) cells to form tumours in athymic nude mice, whereas fibroblasts isolated from the corresponding non-neoplastic prostatic tissue did not102. Some of the resulting tumour cells had reduced E-cadherin expression and elevated vimentin expression, indicating the induction of EMT in BPH-1 cells by the co-inoculated CAFs, which might at least partially explain the increased tumour-initiating potential observed102,103.
Several subsequent reports identified potential mechanisms by which CAFs activate the EMT programme in nearby carcinoma cells. CAFs from human prostate cancers induced EMT in co-cultured PC-3 human prostate carcinoma cells via secretion of matrix metallo-proteinases (MMPs)104. In addition, the conditioned culture medium of CAFs isolated from invasive breast tumours induces EMT-like changes in multiple human breast cancer cell lines; this effect has largely been attributed to TGFβ secreted at high levels by CAFs105. Finally, CAFs have been shown to be enriched at the invasive fronts of multiple types of human carcinoma106-108, suggesting that signals arising from bidirectional interactions between carcinoma cell and CAFs account for the spatial patterns of EMT activation described previously.
The influence of inflammation
Several conditions specific to the tumour microenvironment seem to contribute to the activation of the EMT programme in carcinoma cells. Among these conditions, tissue inflammation comprises a pervasive and critical component of the tumour microenvironment. Diverse lines of evidence suggest that inflammation contributes to at least two stages of carcinoma pathogenesis. First, chronically inflamed tissues are fertile sites for the initiation of carcinomas. Thereafter, established carcinomas can undergo histopathological progression to states of high-grade malignancy, such as those associated with expression of EMT programmes, under the influence of inflammation-associated signals (FIG. 3b). Indeed, in this second stage involving established malignant tumours, inflammatory cells and soluble mediators of inflammation, such as cytokines and chemokines, are abundant within the tumour microenvironment. Thus, the RAS oncoproteins, products of the most frequently mutated oncogenes in human cancers, have been demonstrated to induce cancer cells to produce proinflammatory cytokines and chemokines, such as IL-8 and the CXCL1 (REFS 109,110). The resulting RAS-induced inflammatory tumour microenvironment contributes reciprocally to disease progression through several distinct mechanisms, including EMT-programme activation109,111.
Studies have highlighted the contribution of specific cytokines released by tumour-associated inflammatory cells to the activation of the EMT programme. TNFα treatment resulted in NF-κB-dependent stabilization of the otherwise unstable Snail protein in multiple human cancer cell lines, thereby activating EMT, which in turn increased tumour-cell invasiveness and dissemination to distant sites in preclinical models112. Other prototypic proinflammatory cytokines, including IL-1β and IL-6, have also been demonstrated to activate the EMT programme in carcinoma cells113,114. These observations provide a mechanistic link between tissue inflammation, heterotypic signalling, and activation of EMT programmes in carcinoma cells.
The contributions of cellular components of inflammation, such as macrophages, neutrophils, and myeloid-derived suppressor cells (MDSCs), to cancer progression have been studied extensively. Experiments in mice that lack expression of the key macrophage-lineage cytokine Csf1 and therefore lack functional macrophages have revealed that these cells are dispensable for the initiation and subsequent growth of PyMT-driven primary mammary carcinomas, but are critical to the formation of lung metastases from these tumours115. Moreover, close negative correlations between the abundance of tumour-associated macrophages (TAMs) and patient prognosis have been reported for multiple cancer types, including lung, breast, prostate, and ovarian cancers116.
Analyses of the mechanisms underlying promotion of carcinoma progression have demonstrated that TAMs, like CAFs, can contribute to the activation of EMT programmes in carcinoma cells. Thus, depletion of macrophages from mice bearing F9 teratocarcinoma allo-grafts results in the epithelial differentiation of tumour cells, thus indicating the critical role of macrophages in maintaining the mesenchymal properties of the tumour cells. This EMT effect is mediated, in part, by the secretion of TGFβ by macrophages117. Moreover, activated macrophages induce EMT in MCF7 human breast cancer cells following co-culture in vitro or co-implantation to form xenograft tumours in vivo; this effect is attributable, in part, to secretion of the chemokine CCL18 by macrophages118.
In addition, critical roles of neutrophils and MDSCs in the establishment, growth, and progression of various cancer types have been documented119,120. Intriguingly, such pro-tumour effects of these cells are also attributable, in part, to their capacity for activating EMT programmes within neighbouring cancer cells121,122. Hence, various secreted proteins and host cell types present in the inflammatory tumour microenvironment contribute, perhaps in a coordinated fashion, to the induction of EMT in carcinoma cells.
The effects of hypoxia
Hypoxia, which is often present in poorly vascularized regions of carcinomas, can also contribute to activation of the EMT programme (FIG. 3c). A direct connection between HIF-1 — a central transcriptional regulator of cellular responses to hypoxia — and the EMT of carcinoma cells has been demonstrated in the case of clear-cell renal-cell carcinoma (RCC), a disease in which frequent loss-of-function of the von Hippel-Lindau (VHL) tumour-suppressor protein results in the constitutive activation of HIF-1. In this context, HIF-1 represses the expression of E-cadherin indirectly by inducing the expression of ZEB1, ZEB2, and TCF3, thereby conferring mesenchymal attributes on the carcinoma cells123,124. In addition, multiple human carcinoma cell lines undergo EMT upon exposure to hypoxia in vitro, via a mechanism that is dependent, in part, on HIF-1 activation125-127. Notably, direct activation of the TWIST1 promoter by HIF-1 has also been demonstrated127. Hence, multiple distinct components of the tumour microenvironment and physiological conditions contribute to the activation of the EMT programme in carcinoma cells via diverse mechanisms. This complexity creates a serious challenge to the formulation of therapeutic strategies designed to effectively target the EMT programme.
The relationship between EMT and CSCs
The EMT programme has been portrayed for two decades as a major mechanism underlying carcinoma-cell invasion from the primary tumour mass into the surrounding stroma — the very first step of the complex series of events that lead to the formation of metastases128. This invasion step is followed by the entrance of cancer cells into the circulation (intravasation); transportation in the blood to the capillary beds of distant organs, where they become physically trapped; escape from capillary lumina to the tissue parenchyma (extravasation); and, ultimately, the outgrowth of these micrometastases into macroscopic metastases in the foreign tissue (colonization). These various processes, together, are often termed the ‘invasion-metastasis cascade’ (REF. 129). The scope of biological processes driven by the EMT programme, however, has been expanded far beyond the initial step of carcinoma-cell invasion, owing to the observation that experimental activation of EMT, via either the over-expression of Twist1 or Snail EMT-TFs, or treatment with TGFβ, confers many of the properties of CSCs on otherwise epithelial carcinoma cells27,31. These include CSC-specific cell-surface marker expression — that is, elevated and reduced expression of CD44 and CD24 glycoproteins, respectively, an increased ability to form spheres in a suspension culture, and an enhanced ability to seed tumours in mice. These observations, together with those from a series of follow-up studies, have now demonstrated that activation of the EMT programme in neoplastic cells is closely linked to entrance into the CSC state, across a wide variety of human carcinoma types, revealing multifaceted effects of the EMT programme in driving the malignant phenotype of carcinoma cells130 — and bringing this developmental programme to the heart of cancer biology (TABLE 1).
Table 1.
EMT–CSC connections in various carcinoma types
| Experimental system | Observations | Refs |
|---|---|---|
| Breast carcinoma | ||
| HMLER human mammary epithelial cells |
|
27,31 |
| Clinical samples (metaplastic or claudin-low subtypes) | The clinical samples of metaplastic and claudin-low subtypes of breast cancer simultaneously display a CSC-like profile of cell-surface markers (CD44high/CD24low) and signs of EMT-programme activation, which include reduced expression ofCDH1 mRNA | 190 |
| Clinical samples | The expression of miR-200c, an antagonist of the EMT programme, is reduced in the CSC fraction (CD44high/CD24low) compared with the non-CSC population of clinical breast cancer samples | 191 |
| Lung carcinoma | ||
| H1299 human non-small-cell lung cancer (NSCLC) cell line | The CD44high subpopulation of H1299 cells exhibits signs of EMT-programme activation (including increased expression of CDH2 and VIM mRNAs) and enhanced tumour-seeding ability in nude mice | 192 |
| LC31 human NSCLC cell line | TGFβ-induced EMT in LC31 cells confers elevated expression of the OCT4, NANOG, SOX2, KIT, and CD133 proteins, an increased potential for tumour sphere formation, and an enhanced tumour-seeding ability | 193 |
| Prostate carcinoma | ||
| PC3 human prostate cancer cell line | The induction of EMT by the ectopic expression of platelet-derived growth factor D (PDGF-D) in PC3 cells increases their in vitro tumour sphere formation and in vivo tumour-seeding abilities | 194 |
| Tumour cells from a Pten-null and KrasG12D-driven mouse prostate cancer model | Mesenchymal cell subpopulation (epithelial cell adhesion molecule (EpCAM)low/CD24low) of these tumour cells exhibits an enhanced ability to form tumour spheres, compared with that of a more epithelial cell subpopulation | 30 |
| Pancreatic carcinoma | ||
| Multiple human pancreatic cancer cell lines | The knockdown of zinc finger E-box-binding homeobox (ZEB1) expression in Panc1 and MiaPaCa2 human pancreatic cancer cell lines reduces their potentials forin vitro tumour sphere formation and in vivo tumour seeding | 32 |
| Clinical samples | Expression of the ZEB1 transcription factor is associated with tumour recurrence after surgery in pancreatic cancer clinical samples | 32 |
| Head and neck carcinoma | ||
| Clinical samples |
|
28 |
| Colon carcinoma | ||
| Clinical samples | The CSC subpopulations (CD133high/CD26high) of colon cancer clinical samples exhibit signs of EMT-programme activation, including increased expression of N-cadherin and vimentin, as well as reduced expression of E-cadherin | 29 |
The association between the EMT programme and the CSC state indicates that the activation of this programme in non-CSCs enables their conversion into CSCs. In addition, CSCs, by definition, should have the ability to differentiate into non-CSCs, triggered presumably by the activation of the MET programme. These assumptions, taken together with the aforementioned reversibility of EMT and MET in carcinoma cells, suggest the involvement of substantial phenotypic plasticity, enabling these cells to transition back-and-forth between the CSC and non-CSC states.
Indeed, tumour cells in the non-CSC subpopulation can spontaneously undergo EMT-like changes and acquire CSC-like cell-surface marker expression, as well as an enhanced capacity to seed tumours in mice131. A similar dedifferentiation of non-CSCs to CSCs has also been observed in multiple experimental systems132,133, indicating that the unidirectional stem-cell hierarchy operating in non-neoplastic tissues — whereby only stem cells can repopulate the entire cell population of a differentiated, functional adult tissue, but not the reverse — does not apply to neoplastic tissues. Moreover, given their observed plasticity, carcinoma cells might interconvert between multiple alternative states characterized by different degrees of mesenchymal features, one of which comprises CSCs. Such behaviour implies that EMT-induced non-CSC-to-CSC conversion should be considered simply as a phenotypic shift within a range of fluctuation, rather than true dedifferentiation. This plasticity of the CSC programme also points to the importance of further characterizing the differences between the stem-cell programmes of non-neoplastic tissues and those of neoplastic tissues, as this understanding will be critical to designing therapeutic strategies to selectively target CSCs without damaging non-neoplastic tissue stem cells.
EMT, CSCs, and metastasis
Given their unique property of being able to seed new tumours, CSCs — but not non-CSCs — should be capable of founding metastatic colonies after disseminating to foreign tissues134. This logic, when taken together with the aforementioned EMT–CSC connection, indicates that EMT-programme activation is necessary not only for the physical dissemination of carcinoma cells to distant tissues, but also for entrance into the CSC state that enables the disseminated cells to serve as founders of metastatic colonies and, thus, to succeed in colonizing foreign tissues. Indeed, activation of the EMT programme in carcinoma cells results in the formation of larger numbers of macroscopic lung metastases in mice, even when these cells are injected directly into the venous circulation, thereby bypassing the earlier steps of the invasion-metastasis cascade127,135,136. Moreover, as mentioned previously, disseminated carcinoma cells often exhibit signs of activation of the EMT programme before the formation of macrometastases90,137. These observations reinforce the notion that EMT-programme activation enables carcinoma cells to serve as founders of metastatic colonies.
More recently, while attempting to elucidate the molecular interactions of carcinoma cells infiltrating the lung parenchyma with ECM proteins, we and others discovered that CSC-like cells with high metastasis-seeding abilities develop mature adhesion plaques — macromolecular structures composed of clustered integrins — far more abundantly than do the related, poorly metastasis-seeding and thus non-CSC-like cells138,139. These mature adhesion plaques contribute functionally to the differing powers of metastasis seeding between these cell types, by enabling potent activation of focal adhesion kinase (FAK), the activity of which is critical to the initial proliferation of CSC-like cells within the lung tissue140,141. Further exploration revealed the mechanistic basis for the differing capabilities of CSC-like cells that have activated an EMT programme and non-CSC-like cells that have not done so to form mature adhesion plaques: this difference is attributable, in part, to the distinct potentials of these cell types, following their extravasation, to extend actin-rich, integrin-presenting protrusions141. These protrusions, which we named ‘filopodium-like protrusions’ (FLPs), are abundantly formed only by the EMT-activated cells; following formation of integrin–ECM connections, the FLPs contribute to the development of mature adhesion plaques, thereby enabling the potent activation of FAK signalling141. Intriguingly, in multiple carcinoma cell types, experimental activation of the EMT programme results in a remarkable increase in the ability of these cells to extend FLPs141. This ability, in turn, permits these EMT-activated cells to proliferate following extravasation, ultimately enabling them to seed metastases efficiently. This chain of causality provides one explanation for the link between the EMT programme and the enhanced metastasis-seeding ability of carcinoma cells (FIG. 4a).
Figure 4. The mechanistic link between the epithelial-to-mesenchymal transition (EMT) programme and cancer stem cell (CSC) status.
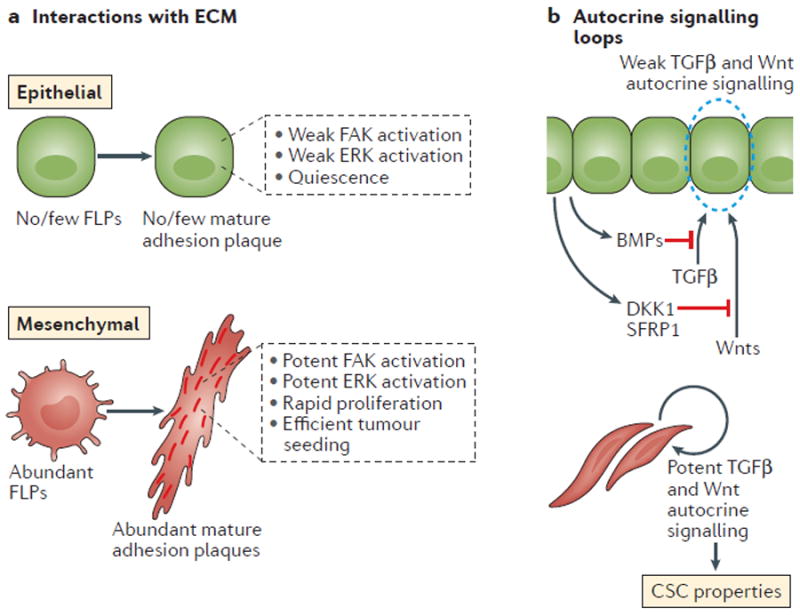
a ∣ The EMT programme enables carcinoma cells to interact productively with the surrounding extracellular matrix (ECM) proteins. Such changes in cell interactions with the ECM, in turn, reinforce the tumour-initiating ability of cancer cells with an active EMT programme. In particular, EMT enables the efficient development by carcinoma cells of integrin-containing mature adhesion plaques, and these plaques, once formed, trigger signalling pathways critical to the proliferation of cancer cells, such as those involving focal adhesion kinase (FAK) and extracellular signal-related kinase (ERK). b ∣ The activation of the EMT programme results in the establishment of several autocrine signalling loops, including the transforming growth factor β (TGFβ) and canonical and/or noncanonical Wnt pathways. These signalling loops contribute to the CSC properties of cells with an active EMT programme. BMPs, bone morphogenetic proteins; DKK1, Dickkopf-related protein 1; FLPs, filopodium-like protrusions; SFRP1, secreted Frizzled-related protein 1.
In truth, this depiction might oversimplify the complexity of the final, post-extravasation steps of metastasis formation: the EMT programme might enable physical dissemination of carcinoma cells to distant tissues and also confer on them an ability to initiate metastatic growths, but is unlikely to solve an additional and critical obstacle confronting these cells. This hurdle involves the adaptation of cells originating in one tissue to the microenvironment of a second, unfamiliar tissue. Most carcinoma cells are hypothesized to be poorly adapted to grow in foreign tissues, at least initially, and must contrive adaptive programmes in order to continue to expand. These adaptations are likely to be accompanied by, as mentioned, the reversion of the EMT programme (or activation of a MET programme) in carcinoma cells, and indeed robustly growing metastatic colonies are usually composed mostly of carcinoma cells that have regained their epithelial attributes86,87.
In contrast to the prevailing view that the EMT programme contributes critically to cancer metastasis, two reports have presented evidence of the contrary. In one of these studies, a linage-tracing approach was used to follow the fate of tumour cells that have expressed the EMT markers, S100A4 (also known as fibroblast-specific protein 1) and vimentin, and demonstrated that lung metastases in the PyMT-driven mouse mammary carcinoma model are formed predominantly by cells that have not previously expressed these markers142. In the other study, pancreas-specific deletion of either Snai1 or Twist1 was found to have no substantial effect on metastasis formation in a mouse model of pancreatic ductal adenocarcinoma143. These studies have raised questions regarding the ubiquitous requirement for EMT in cancer metastases, although the observations made therein need to be interpreted with caution. As discussed previously, the EMT programme is usually activated only partially in carcinoma cells, resulting in aggressive cells that might not be fully mesenchymal and, thus, might not express the two markers specific for highly mesenchymal cells, S100A4 and vimentin. Moreover, given their extensively overlapping functions, the effects of genetic elimination of a single EMT-TF are likely to be masked by the compensatory upregulation of others.
The molecular mechanism behind the EMT–CSC link
The connection between EMT and CSC status is now supported by substantial experimental evidence; however, the mechanistic link between these two phenotypes remains largely elusive. One of the potential explanations for this link is that alterations in the spectrum of secreted proteins (that is, the ‘secretome’) of carcinoma cells induced by the EMT programme establish autocrine signalling loops, which in turn are essential for the induction and maintenance of stem-cell properties. Indeed, results of a study analysing the secretome of transformed HMLER human mammary epithelial cells have revealed that the activation of the EMT programme induces autocrine signalling loops that are known to contribute critically to the ‘stemness’ of non-neoplastic and neoplastic cells; these include the TGFβ –SMAD and Wnt–β-catenin pathways144 (FIG. 4b). Consistent with this notion, the blockade of these auto-crine signalling loops is sufficient to prevent the acquisition of CSC properties by the HMLER cells following experimental activation of the EMT programme.
The EMT programme also seems to contribute to CSC phenotype through its effects on intracellular signalling pathways. For example, Snail has been reported to reduce the expression of the tumour-suppressor protein p53 in carcinoma cells via formation of a Snail–histone deacetylase 1 (HDAC1)–p53 ternary complex and subsequent deacetylation of p53, thereby promoting its protea-somal degradation145. Importantly, the tumour-initiating ability of neoplastic cells in the PyMT-driven mouse mammary tumour model is diminished by deletion of Snai1, and this ability can be restored through concomitant deletion of Tp53 (encoding p53)145. Hence, the EMT programme seems to confer CSC phenotypes on carcinoma cells through effects on both the extracellular and intracellular signalling machinery.
The importance of the EMT programme in driving the CSC phenotype itself requires further investigation. Indeed, whether activation of the EMT programme is necessary and/or sufficient for the acquisition of the CSC phenotype across various carcinoma types remains to be determined. In breast cancer, CSCs are demonstrated to be present in two alternative phenotypic states, namely, epithelial-like and mesenchymal-like, and cells in either state are capable of seeding tumours in mice146,147. Moreover, although the activation of the EMT programme markedly increases the tumour-initiating ability of carcinoma cells in many model systems, excessive activation of this programme is found to be detrimental for tumour initiation (FIG. 1b). This finding explains why increasing attention is being placed on the ‘partial EMT’ state, which seems to be critical for the maximal tumorigenic activity of CSCs148. More specifically, in a model of claudin-low breast cancer, in which most carcinoma cells display a highly mesenchymal phenotype, a subpopulation of cells with relatively epithelial characteristics exhibited greater tumorigenic activity than the bulk population of the cells149. Together, these observations indicate that the contribution of the EMT programme to the CSC phenotype is variable, most likely depending on cell type and/or coexisting genetic/epigenetic abnormalities. This variability underscores the complexity of the genetic/epigenetic mechanisms driving the CSC phenotype in carcinoma cells, an understanding of which will likely be critical to the future development of novel therapies directed against the (partially) mesenchymal cells in human carcinomas, including their subpopulations of CSCs.
EMT and therapeutic resistance
Soon after the introduction of the CSC concept, some researchers postulated that CSCs are more resistant than non-CSCs within the same tumours to elimination following conventional chemotherapy and radiotherapy12,150. This idea originated, at least in part, from early observations indicating that non-neoplastic tissue stem cells exhibit higher levels of resistance to chemotherapeutic agents than that of the differentiated cell types within the same tissues151. This resistance has been attributed to various mechanisms, including: elevated expression of antiapoptotic proteins; increased levels of ATP-binding cassette (ABC) transporters, transmembrane protein transporters that are known to mediate drug efflux and, thus, to confer multidrug resistance on cancer cells152-155; and the slow proliferation rate of stem cells156.
More recently, technical advances in the identification and isolation of CSC-enriched subpopulations of cells from various tumours types have made assessment of the relative sensitivity of CSCs and non-CSCs to therapeutic agents possible. These analyses have revealed, as anticipated, that chemotherapy and/or radiotherapy successfully eliminate the bulk population of non-CSCs, while leaving behind considerable numbers of CSCs in multiple cancer types, in both preclinical and clinical samples21,157,158. In addition, the survival of CSCs following certain kinds of molecularly targeted treatment and immunotherapy is greater than that of non-CSCs. For instance, the CSC-enriched subpopulation of chronic myeloid leukaemia (CML) cells was found to be far more resistant to treatment with the BCR–ABL1 kinase inhibitor imatinib than was the CSC-deprived subpopulation of the CML cells from the same patients159. In addition, in a mouse model of prostate cancer, therapeutic vaccination using a cDNA library derived from non-neoplastic prostate tissue results in the initial regression of the tumour followed by the recurrence of a more aggressive tumour composed of tumour cells with a much higher tumour-initiating ability than those of the original tumour160,161.
Nonetheless, further understanding of the connection between CSCs and therapeutic resistance had been hampered by the lack of reliable markers to identify cell populations highly enriched with CSCs, and by an insufficient understanding of the mechanisms that govern the phenotype of CSCs. In fact, the sets of markers used to identify cancer cell populations enriched for CSCs are being continuously updated, leaving room for further improvement. These difficulties greatly complicate attempts to predict the therapeutic resistance of ‘pure’ populations of CSCs. Nonetheless, the identification of the EMT programme as a common regulator of the CSC phenotype across various carcinoma types has provided us with an experimental opportunity to investigate, at the molecular level, how CSCs and therapeutic resistance are linked via EMT programmes (FIG. 5; TABLE 2).
Figure 5. The mechanism underlying epithelial-to-mesenchymal transition (EMT)-dependent acquisition of therapeutic resistance.
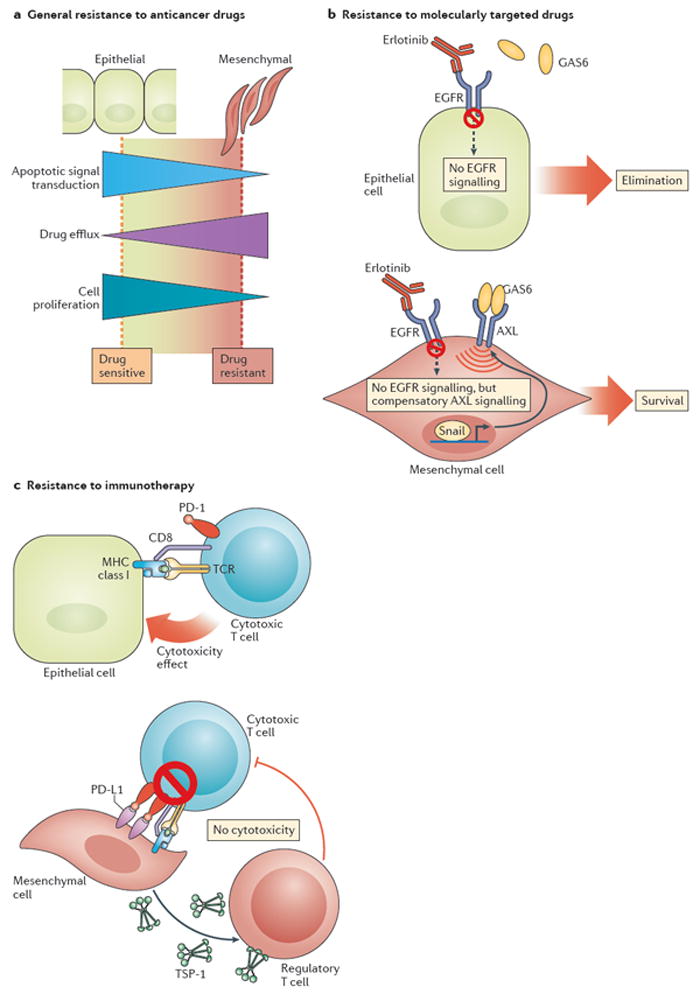
a ∣ EMT-associated downregulation of multiple apoptotic signalling pathways, enhanced drug efflux, and slow cell proliferation all contribute to enhance the general resistance of carcinoma cells to anticancer drugs. b ∣ In addition, the EMT-associated transcription factor Snail induces the expression of the AXL receptor tyrosine kinase on the surface of carcinoma cells. AXL signalling, triggered by the binding of its ligand growth arrest-specific protein 6 (GAS6), enables Snail-expressing carcinoma cells to override cytostatic effects of EGFR blockade with small-molecule inhibitors (such as erlotinib) or antagonistic monoclonal antibodies. c ∣ The EMT programme also activates several processes that enable carcinoma cells to evade the lethal effect of cytotoxic T cells. These changes include elevated expression of programmed cell death 1 ligand 1 (PD-L1), which binds to the programmed cell death protein 1 (PD-1) inhibitory immune-checkpoint receptor that is expressed by cytotoxic T cells and thereby diminishes their function; and increased secretion of thrombospondin-1 (TSP-1), which promotes the development of regulatory T cells within the tumour microenvironment that ultimately suppress the activity of cytotoxic T cells.
Table 2.
Therapy resistance conferred by epithelial-to-mesenchymal transition (EMT)
| Therapeutic agent | Observations | Refs |
|---|---|---|
| Inhibition of apoptotic signalling | ||
| Cisplatin | Slug blocks p53-mediated transcriptional induction of PUMA (also known as BBC3, encoding Bcl-2-binding component 3) expression by directly repressing the PUMA promoter region; multiple lung adenocarcinoma cell lines acquire cisplatin resistance through this mechanism | 195,196 |
| Tumour necrosis factor α (TNFα) treatment; γ-irradiation | Snail confers resistance against multiple apoptosis-inducing stimuli, in part by promoting AKT activation, upregulating the expression of the pro-survival protein Bcl-XL, and delaying cell-cycle progression | 197,198 |
| TNF-related apoptosis-inducing ligand (TRAIL) | EMT-programme activation diminishes E-cadherin-mediated clustering of the TRAIL receptors DR4 and DR5, thereby making carcinoma cells resistant to TRAIL-induced apoptosis | 199 |
| Enhancement of drug efflux | ||
| Doxorubicin | EMT-programme activation induces the expression of multiple members of the ATP-binding cassette (ABC) transporter family, thereby rendering these cells resistant to doxorubicin. | 200 |
| Protection against molecular targeted agents | ||
| EGFR inhibitors | The activation of EMT and subsequent expression of AXL receptor tyrosine kinase confer resistance to EGFR inhibitors onEGFR-mutant non-small-cell lung carcinoma (NSCLC) cells | 201,202 |
| EGFR inhibitors; PI3K inhibitors | An EMT-associated gene-expression signature predicts the resistance of NSCLC cells to EGFR inhibitors and PI3K inhibitors | 163 |
| Desensitization to immunotherapy | ||
| Dendritic cell (DC)-mediated immunotherapy (intratumoral injection of DCs pulsed with a tumour antigen) | Snail expression in melanoma cells contributes to resistance to DC-mediated and CTL-mediated immunotherapy via enhanced thrombospondin-1 expression and resultant induction of immunosuppressive regulatory T cells within the tumour tissue | 203 |
| Immune-checkpoint inhibition | Zinc finger E-box-binding homeobox (ZEB1)-mediated activation of EMT in NSCLC cells relieves miR-200-mediated repression of programmed cell death 1 ligand 1 (PD-L1) expression, a major inhibitory ligand for the programmed cell death protein 1 (PD-1) immune-checkpoint protein on CD8+ CTLs. This effect sensitizes these cells to immunotherapies targeting the PD-1 PD-L1 axis, while potentially conferring on them resistance to other strategies of activating antitumour immunity, such as the functional blockade of another immune-checkpoint protein, CTLA-4 | 204 |
Analyses of the relationships between gene-expression profiles of tumour samples and the clinical responses of the patients from which they were obtained have resulted in the identification of a strong correlation between an EMT-associated gene-expression signature and treatment resistance. For instance, an analysis of responses to chemotherapy in a large cohort of patients with breast cancer revealed a close association between therapeutic resistance and increased expression of genes that are usually expressed primarily by the stromal cells; this transcriptional upregulation seems to be caused, in part, by activation of the EMT programme within carcinoma cells162. In addition, findings of a study designed to interrelate gene-expression profiles and responses to EGFR or PI3K inhibitors in clinical samples and in various cell lines derived from patients with non-small-cell lung carcinoma (NSCLC) have also identified a 76-gene EMT signature as a robust predictor of clinical resistance to these agents163.
Collectively, these observations strongly support the notion that the EMT programme contributes critically to the development of resistance to various types of therapeutic agents in multiple cancer types; they also illustrate how the identification of the functional link between the EMT programme and the CSC phenotype has enabled us to understand at the molecular level the otherwise enigmatic phenomenon concerning the resistance of CSCs to multiple therapeutic modalities. Further study of the connections between the EMT programme, the CSC phenotype, and therapeutic resistance will certainly be required for the development of more effective treatment strategies to eradicate CSCs.
Targeting the EMT programme for therapy
The development of treatment strategies to directly target the EMT programme is both reasonable and promising, given the emerging importance of this programme in the malignant progression of carcinomas, as well as the frequent acquisition of therapeutic resistance by carcinoma cells. At least three different approaches might be pursued: preventing the induction of EMT; selectively targeting cells that have active components of an EMT programme; and/or reversing the process of EMT by forcing more-mesenchymal carcinoma cells to revert to an epithelial state via activation of the MET programme.
Prevention of EMT induction
EMTs can be prevented by interfering with the signalling processes that are critical to the activation and subsequent maintenance of EMT programmes. TGFβ signalling is among the best-characterized pathways involved in EMT induction; however, this cytokine has multifaceted effects on cancer cells and, therefore, attempts at the therapeutic inhibition of this pathway require caution. Specifically, although TGFβ potently promotes the progression of the tumours via mechanisms including the activation of the EMT programme and resulting invasion of carcinoma cells into the surrounding non-neoplastic tissue, it also negatively controls the initial stages of tumour formation through its antiproliferative effects164,165. Indeed, SKI and the SKI-like protein SnoN, endogenous inhibitors of TGFβ signalling, are known oncoproteins; increased expression of these proteins has been associated with the development of many types of human cancers, including melanoma and oesophageal cancer166. Hence, inhibition of TGFβ signalling might actually be counterproductive by promoting carcinoma-cell proliferation in the early stages of the disease. In addition, TGFβ signalling has critical roles in controlling the behaviours of stromal and immune cells, making it difficult to specifically target the process of TGFβ-mediated induction of EMT in the cancer cells. Nevertheless, inhibitors targeting this signalling pathway are currently undergoing both preclinical and clinical testing167,168; owing to the complex and multifaceted effects of TGFβ signalling, appropriate stratification of the patients and optimization of drug-administration protocols will be critical to the effective use of these inhibitors.
Other potential therapeutic targets for preventing the induction of EMT include hepatocyte growth factor (HGF)–HGF receptor (HGFR; a proto-oncogene also known, confusingly, as MET) signalling. Besides promoting cell proliferation, HGF–HGFR signalling also activates the EMT programme and induces cell motility169,170. This pathway contributes critically to cancer pathogenesis, as exemplified by its frequent activation, often by point mutation or amplification of the HGFR gene (MET), in many cancer types. Thus, HGF–HGFR signalling is a promising target of anticancer therapy, and substantial efforts have been made to develop antagonists of this pathway. In particular, a number of small-molecule inhibitors of the enzymatic activity of the HGFR tyrosine-kinase domain are being evaluated in clinical trials. Of note, two multikinase inhibitors with activity against this protein are already approved by the FDA: crizotinib, for the treatment of NSCLC; and cabozantinib, for the treatment of medullary thyroid cancer and RCC171. However, the extent to which HGFR inhibition contributes to the antitumour activity of these agents in these indications remains to be clarified.
In principle, blockade of EMT could also be accomplished by targeting the components of the tumour microenvironment that contribute to the activation of the EMT programme in carcinoma cells. Possible treatment strategies along this line include suppression of tumour-associated inflammation and hypoxia, as well as targeting of specific types of host cells, such as CAFs or TAMs172,173. As discussed, however, the manner in which each of these microenvironmental components triggers activation of the EMT programme in nearby carcinoma cells remains under investigation; a more detailed understanding of these mechanisms will be necessary in order to manipulate the microenvironmental factors in ways that will block EMT-programme activation.
Selective targeting of cells that have undergone EMT
Substantial efforts have been made to selectively target cancer cells that have undergone EMT, starting with attempts to therapeutically block the functions of classic EMT-associated markers. For instance, the natural compound withaferin A binds to, among other things, vimentin intermediate filaments, a standard marker of EMT, ultimately causing the degradation of these filaments174. Withaferin A treatment has been demonstrated to block the in vitro invasion and in vivo metastasis formation by breast cancer cells175, and is, therefore, an attractive target for further development as a therapeutic anticancer agent. In addition, treatment with monoclonal antibodies against the ectodomain of N-cadherin, another important marker of EMT, had the same effects on multiple prostate cancer cell lines176. The therapeutic index of both agents, however, remains to be examined in depth. Potential complications might plausibly arise from the fact that these mesenchymal markers are widely expressed by non-neoplastic mesenchymal cells, such as fibroblasts, resulting in on-target, off-tumour toxicities.
Defining and interfering with signalling pathways that contribute critically to the behaviours of carcinoma cells that have undergone EMT is another potential approach to anticancer therapy. One study along this line has identified AXL, a receptor tyrosine kinase activated by the GAS6 ligand, as a signalling protein whose expression and function are linked tightly with expression of an EMT signature163,177. The expression of AXL is strongly induced by the experimental activation of the EMT programme in the immortalized MCF10A human mammary epithelial cell line178. In addition, short hairpin RNA (shRNA)-mediated knockdown of AXL expression attenuates in vitro invasion as well as in vivo tumour initiation and metastasis formation by multiple mesenchymal-like breast cancer cell lines178. Similarly, in the aforementioned study that identified a strong correlation between the EMT-associated gene-expression signature and resistance to EGFR or PI3K inhibitors in NSCLC, the expression of an EMT-associated signature was closely accompanied by the elevated expression of AXL and its ligand, GAS6. Intriguingly, the combination of the EGFR inhibitor erlotinib and the AXL inhibitor SGI-7079 successfully overcame the resistance of mesenchymal-like NSCLC cells to erlotinib treatment in a mouse xenograft model163. Together, these experimental observations identify AXL as a critical mediator of EMT-associated phenotypes in cancer cells, including CSC-like properties, which suggests a strong potential of this receptor as a therapeutic target for the selective elimination of CSCs with an active EMT programme. The first AXL-specific small-molecule inhibitor, BGB324, entered clinical trials in 2013 (REF. 179). An initial clinical study in NSCLC has demonstrated that BGB324 can be safely administered to patients and a proportion of patients achieve durable disease stabilization for at least 6 months180.
High-throughput screening approaches have been used to identify therapeutic vulnerabilities in carcinoma cells that have an active EMT programme and thus have entered a CSC state. For example, a chemical compound screen of cells from the immortalized HMLE human mammary epithelial cell line, with or without prior experimental activation of the EMT programme, resulted in the identification of the potassium ionophore salinomycin as an agent with cytotoxic activity specifically in cells that have undergone EMT181. Findings of an analogous screen for kinase inhibitors demonstrated that inhibitors targeting PKCa selectively eliminate HMLE cells with an active EMT programme182. In both cases, the clinical utility of these and similar agents for targeting CSCs with active EMT programmes requires extensive testing and validation.
Reversing the process of EMT
From a therapeutic standpoint, reversing the process of EMT — inducing MET — would be beneficial, considering the critical contribution of the EMT programme to multiple malignant traits of cancer. By doing so, cancer cells in the CSC state with an active EMT programme will be forced to differentiate into non-CSCs and regain epithelial traits, thereby losing both enhanced tumorigenic activity and resistance to various therapeutic agents. This strategy is reminiscent of differentiation therapy used in patients with acute promyelocytic leukaemia (APL), whereby treatment with all-trans retinoic acid (ATRA) induces terminal differentiation of leukaemic promyelocytes, eventually leading to apoptotic death. ATRA is a component of the first-line treatment of APL and its use has dramatically improved the prognosis of patients with this disease183.
As reported in 2016, a screen for agents that reactivate the promoter of the E-cadherin gene (CDH1) in mesenchymal cells identified cholera toxin and forsko-lin, both of which increase intracellular levels of cyclic AMP and thereby enhance signalling through protein kinase A (PKA)184. Treatment of the spontaneously arising mesenchymal derivatives of HMLE cells with either of these two agents, or forced expression of a constitutively active PKA mutant, resulted in the induction of MET and associated reductions in invasiveness and sphere-forming potential184. Interestingly, this MET was accompanied by sensitization of these cells to therapeutic agents that included doxorubicin, paclitaxel, proteasome inhibitors, and EGFR inhibitors184. Additional studies will be needed to assess the general applicability and clinical utility of these findings. In particular, given the aforementioned requirement for reversion of the EMT programme at the last step of the invasion–metastasis cascade, therapeutic activation of the MET programme might adversely promote metastatic colony formation. Thus, the timing and strategy of MET-programme activation will need to be determined carefully, as a prerequisite to the success of this therapeutic approach. Nonetheless, such ‘differentiation therapy’ is an attractive avenue for the development of novel anticancer drugs.
Conclusions
Advances in basic and translational cancer biology have enabled us to understand, in unprecedented detail, the actions and the outcomes of cancer therapies. In particular, major conceptual advances, including the introduction of the CSC concept and elucidation of the connection between the EMT programme and the CSC state, have provided us with new insights into clinically significant problems, such as the development of resistance to therapy. Conventional therapeutics usually eliminate the bulk non-CSC population while sparing the minor subpopulations of CSCs, thereby leaving open the possibility of future local disease recurrence and/or the development of metastases17,185. Moreover, EMT seems to be a major strategy used by carcinoma cells to acquire a CSC phenotype, making this programme an attractive novel target for cancer therapy24. Indeed, therapeutic strategies based on this new knowledge are currently being designed and tested, and will probably result in improvements in cancer therapy in the near future186.
Nevertheless, the deduction that activation of the EMT programme in carcinoma cells generates CSCs has been challenged by more recent findings. Thus, while EMT-programme activation in the otherwise-epithelial carcinoma cells generally results in an enhanced potential for tumorigenesis, full activation of the entire EMT programme — that is, complete transition to a mesenchymal cell type — has been found to be detrimental to tumorigenic activity88,148,187. Moreover, under certain conditions, CSCs can undergo phenotypic drift, thus losing mesenchymal traits while maintaining their tumorigenic activity146,147. Such observations indicated that the EMT programme and the CSC phenotype are closely associated, but can actually be uncoupled from one another under certain conditions. Further investigation of the association and distinction between EMT and the CSC state will be required in order to exploit the EMT–CSC link for the improvement of therapeutic practice.
Another important challenge related to EMT–CSC connection derives from the phenotypic plasticity of non-CSCs. Unlike their counterparts in non-neoplastic tissues (that is, terminally differentiated cells), non-CSCs in the tumour tissue can spontaneously undergo EMT and dedifferentiate into new CSCs, thereby regenerating tumorigenic potential188. Hence, initial eradication of CSCs alone will probably not preclude disease recurrence; consideration of the simultaneous targeting of CSCs and non-CSCs, as well as a better understanding of the process of dedifferentiation of non-CSCs, will be critical to the development of effective anticancer therapies.
Research advances have provided solid evidence for the connection between the activation of the EMT programme and the development by carcinoma cells of resistance to therapeutics, not only in the experimental models, but also in clinical settings. Indeed, residual carcinoma cells surviving after various types of therapy, including chemotherapy, molecularly targeted therapy, and immunotherapy, commonly display signs of EMT activation. Thus, the efficiency of these therapeutic modalities in generating durable clinical responses might be improved substantially by targeting cancer cells that have activated portions of the EMT programme. However, the mechanisms accounting for the maintenance and/or induction of the EMT programme in these residual carcinoma cells remain largely obscure. EMT in carcinoma cells seems to be governed by highly complex and redundant mechanisms, posing a serious challenge to the selective and truly effective targeting of this programme. Overcoming these difficulties will enable us to design treatment strategies that have a minimal risk of developing resistance, ultimately paving the way to curative treatments of cancer. Finally, as is apparent from the present discussions, many of the biological traits of cancer cells are dictated by non-genetic mechanisms, revealing the limits of cancer genome sequencing in providing insights into many aspects of cancer cell biology and necessitating the integrated use of complementary approaches, such as epigenomics and transcriptomics.
Key points.
The cancer stem cell (CSC) concept posits that a subpopulation of neoplastic cells with stem-cell properties — particularly the capacity to self-renew and give rise to various more differentiated cell types — lies at the apex of a tumour cell hierarchy and serves as a critical driver of tumour progression
The phenotypic differences between CSCs and the bulk tumour cells that lack ‘stemness’ (that is, non-CSCs) seem to be attributable predominantly to epigenetic changes caused by the activation of a epithelial-to-mesenchymal transition (EMT) programme in the former
Thus, the CSC paradigm provides an explanation for how epigenetic mechanisms can drive the phenotypic diversity of neoplastic cells — an attribute critical for the development of resistance to therapy
Indeed, most conventional therapeutics are inefficient in eradicating carcinoma cells that have entered the CSC state via activation of the EMT programme, thereby permitting CSC-dependent disease relapse
Targeting the EMT programme in order to eliminate CSCs offers a promising avenue for the improvement of cancer therapy; however, the success of this approach will require an increase in our mechanistic understanding of the EMT–CSC link
Acknowledgments
We are grateful to Arthur W. Lambert (Whitehead Institute for Biomedical Research) for critically reading the draft of this article and providing invaluable comments. T.S. has received postdoctoral fellowships from the Human Frontier Science Program, the Japan Society for the Promotion of Science, and the Ludwig Fund for Cancer Research. R.A.W. is an American Cancer Society research professor and a Daniel K. Ludwig Foundation cancer research professor. The work of the authors has been funded by grants from the National Institutes of Health (P01 CA080111), the Breast Cancer Research Foundation, the Waxman Foundation, and the Virginia & Daniel K. Ludwig Fund for Cancer Research.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Levan A, Hauschka TS. Endomitotic reduplication mechanisms in ascites tumors of the mouse. J Natl Cancer Inst. 1953;14:1–43. [PubMed] [Google Scholar]
- 2.Makino S. Further evidence favoring the concept of the stem cell in ascites tumors of rats. Ann NY Acad Sci. 1956;63:818–830. doi: 10.1111/j.1749-6632.1956.tb50894.x. [DOI] [PubMed] [Google Scholar]
- 3.Prehn RT. Analysis of antigenic heterogeneity within individual 3-methylcholanthrene-induced mouse sarcomas. J Natl Cancer Inst. 1970;45:1039–1045. [PubMed] [Google Scholar]
- 4.Mitelman F. The chromosomes of fifty primary Rous rat sarcomas. Hereditas. 1971;69:155–186. doi: 10.1111/j.1601-5223.1971.tb02431.x. [DOI] [PubMed] [Google Scholar]
- 5.Alizadeh AA, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21:846–853. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 8.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 11.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 13.Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ. The origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5:899–904. doi: 10.1038/nrc1740. [DOI] [PubMed] [Google Scholar]
- 14.Avgustinova A, Benitah SA. Epigenetic control of adult stem cell function. Nat Rev Mol Cell Biol. 2016;17:643–658. doi: 10.1038/nrm.2016.76. [DOI] [PubMed] [Google Scholar]
- 15.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Migrating cancer stem cells — an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 16.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea — a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 17.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 19.Clarke MF, et al. Cancer stem cells — perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 20.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26:2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 22.Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 23.Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 25.Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15:338–344. doi: 10.1038/ncb2717. [DOI] [PubMed] [Google Scholar]
- 26.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 27.Mani SA, et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen YC, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. 2009;385:307–313. doi: 10.1016/j.bbrc.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 29.Pang R, et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell. 2010;6:603–615. doi: 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morel AP, et al. Generation of breast cancer stem cells through epithelial–mesenchymal transition. PLoS ONE. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellner U, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 33.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 35.Shook D, Keller R. Mechanisms, mechanics and function of epithelial–mesenchymal transitions in early development. Mech Dev. 2003;120:1351–1383. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn. 2005;233:706–720. doi: 10.1002/dvdy.20345. [DOI] [PubMed] [Google Scholar]
- 37.Dressler GR. The cellular basis of kidney development. Annu Rev Cell Dev Biol. 2006;22:509–529. doi: 10.1146/annurev.cellbio.22.010305.104340. [DOI] [PubMed] [Google Scholar]
- 38.Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 40.Taube JH, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zavadil J, Bottinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 42.Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 46.Hugo HJ, et al. Defining the E-cadherin repressor interactome in epithelial–mesenchymal transition: the PMC42 model as a case study. Cells Tissues Organs. 2011;193:23–40. doi: 10.1159/000320174. [DOI] [PubMed] [Google Scholar]
- 47.Diaz-Lopez A, Moreno-Bueno G, Cano A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag Res. 2014;6:205–216. doi: 10.2147/CMAR.S38156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burk U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 50.Kim NH, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial–mesenchymal transition. J Cell Biol. 2011;195:417–433. doi: 10.1083/jcb.201103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siemens H, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial–mesenchymal transitions. Cell Cycle. 2011;10:4256–4271. doi: 10.4161/cc.10.24.18552. [DOI] [PubMed] [Google Scholar]
- 52.Zhou BP, et al. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 53.Hong J, et al. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71:3980–3990. doi: 10.1158/0008-5472.CAN-10-2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen A, et al. The ubiquitin ligase Siah is a novel regulator of Zeb1 in breast cancer. Oncotarget. 2015;6:862–873. doi: 10.18632/oncotarget.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 56.Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial–mesenchymal transition. Mol Cell Biol. 2001;21:8184–8188. doi: 10.1128/MCB.21.23.8184-8188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen ZF, Behringer RR. Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- 58.Van de Putte T, et al. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease–mental retardation syndrome. Am J Hum Genet. 2003;72:465–470. doi: 10.1086/346092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang R, Lan Y, Norton CR, Sundberg JP, Gridley T. The Slug gene is not essential for mesoderm or neural crest development in mice. Dev Biol. 1998;198:277–285. [PubMed] [Google Scholar]
- 60.Higashi Y, et al. Impairment of T cell development in dEF1 mutant mice. J Exp Med. 1997;185:1467–1479. doi: 10.1084/jem.185.8.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sosic D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF-κB activity. Cell. 2003;112:169–180. doi: 10.1016/s0092-8674(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 62.Bain G, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 63.Zhuang Y, Soriano P, Weintraub H. The helix-loop-helix gene E2A is required for B cell formation. Cell. 1994;79:875–884. doi: 10.1016/0092-8674(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 64.Yao D, Dai C, Peng S. Mechanism of the mesenchymal–epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res. 2011;9:1608–1620. doi: 10.1158/1541-7786.MCR-10-0568. [DOI] [PubMed] [Google Scholar]
- 65.Gunasinghe NP, Wells A, Thompson EW, Hugo HJ. Mesenchymal–epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012;31:469–478. doi: 10.1007/s10555-012-9377-5. [DOI] [PubMed] [Google Scholar]
- 66.Li R, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 67.Little MH, McMahon AP. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4:a008300. doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ledford H. Cancer theory faces doubts. Nature. 2011;472:273. doi: 10.1038/472273a. [DOI] [PubMed] [Google Scholar]
- 69.Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- 70.Thompson L, Chang B, Barsky SH. Monoclonal origins of malignant mixed tumors (carcinosarcomas). Evidence for a divergent histogenesis. Am J Surg Pathol. 1996;20:277–285. doi: 10.1097/00000478-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 71.Mareel M, Vleminckx K, Vermeulen S, Bracke M, Van Roy F. E-Cadherin expression: a counterbalance for cancer cell invasion. Bull Cancer. 1992;79:347–355. [PubMed] [Google Scholar]
- 72.Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 73.Cano A, et al. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 74.Blanco MJ, et al. Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002;21:3241–3246. doi: 10.1038/sj.onc.1205416. [DOI] [PubMed] [Google Scholar]
- 75.Yang J, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 76.Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13:7003–7011. doi: 10.1158/1078-0432.CCR-07-1263. [DOI] [PubMed] [Google Scholar]
- 77.Soltermann A, et al. Prognostic significance of epithelial-mesenchymal and mesenchymal–epithelial transition protein expression in non-small cell lung cancer. Clin Cancer Res. 2008;14:7430–7437. doi: 10.1158/1078-0432.CCR-08-0935. [DOI] [PubMed] [Google Scholar]
- 78.Rasheed ZA, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–351. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee TK, et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial–mesenchymal transition. Clin Cancer Res. 2006;12:5369–5376. doi: 10.1158/1078-0432.CCR-05-2722. [DOI] [PubMed] [Google Scholar]
- 80.Spaderna S, et al. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. 2006;131:830–840. doi: 10.1053/j.gastro.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 81.Baumgart E, et al. Identification and prognostic significance of an epithelial–mesenchymal transition expression profile in human bladder tumors. Clin Cancer Res. 2007;13:1685–1694. doi: 10.1158/1078-0432.CCR-06-2330. [DOI] [PubMed] [Google Scholar]
- 82.Yu M, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinstein RS, Merk FB, Alroy J. The structure and function of intercellular junctions in cancer. Adv Cancer Res. 1976;23:23–89. doi: 10.1016/s0065-230x(08)60543-6. [DOI] [PubMed] [Google Scholar]
- 84.Gabbert H, Wagner R, Moll R, Gerharz CD. Tumor dedifferentiation: an important step in tumor invasion. Clin Exp Metastasis. 1985;3:257–279. doi: 10.1007/BF01585081. [DOI] [PubMed] [Google Scholar]
- 85.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mayer B, et al. E-Cadherin expression in primary and metastatic gastric cancer: down-regulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res. 1993;53:1690–1695. [PubMed] [Google Scholar]
- 87.Brabletz T, et al. Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA. 2001;98:10356–11036. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ocana OH, et al. Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 89.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial–mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lawson DA, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Berx G, et al. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene. 1996;13:1919–1925. [PubMed] [Google Scholar]
- 92.Husemann Y, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 93.Ye X, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–260. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rhim AD, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pantel K, Alix-Panabieres C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol. 2009;6:339–351. doi: 10.1038/nrclinonc.2009.44. [DOI] [PubMed] [Google Scholar]
- 96.Klein CA. Selection and adaptation during metastatic cancer progression. Nature. 2013;501:365–372. doi: 10.1038/nature12628. [DOI] [PubMed] [Google Scholar]
- 97.Trimboli AJ, et al. Direct evidence for epithelial–mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–945. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- 98.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 99.Mueller MM, Fusenig NE. Friends or foes — bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 100.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 101.Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–1523. doi: 10.1084/jem.20140692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hayward SW, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61:8135–8142. [PubMed] [Google Scholar]
- 104.Giannoni E, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial–mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–6956. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 105.Yu Y, et al. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br J Cancer. 2014;110:724–732. doi: 10.1038/bjc.2013.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nielsen BS, Sehested M, Timshel S, Pyke C, Dano K. Messenger RNA for urokinase plasminogen activator is expressed in myofibroblasts adjacent to cancer cells in human breast cancer. Lab Invest. 1996;74:168–177. [PubMed] [Google Scholar]
- 107.Martin M, Pujuguet P, Martin F. Role of stromal myofibroblasts infiltrating colon cancer in tumor invasion. Pathol Res Pract. 1996;192:712–717. doi: 10.1016/S0344-0338(96)80093-8. [DOI] [PubMed] [Google Scholar]
- 108.Nakayama H, et al. The role of myofibroblasts at the tumor border of invasive colorectal adenocarcinomas. Jpn J Clin Oncol. 1998;28:615–620. doi: 10.1093/jjco/28.10.615. [DOI] [PubMed] [Google Scholar]
- 109.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 110.Yang G, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci USA. 2006;103:16472–164773. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim H, Choi JA, Kim JH. Ras promotes transforming growth factor-β (TGF-β)-induced epithelial–mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J Biol Chem. 2014;289:22151–22160. doi: 10.1074/jbc.M114.556126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu Y, et al. Stabilization of snail by NFB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sullivan NJ, et al. Interleukin-6 induces an epithelial–mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–2947. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer. 2012;11:87. doi: 10.1186/1476-4598-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial–mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Su S, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25:605–620. doi: 10.1016/j.ccr.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 119.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016;37:41–52. doi: 10.1016/j.it.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Freisinger CM, Huttenlocher A. Live imaging and gene expression analysis in zebrafish identifies a link between neutrophils and epithelial to mesenchymal transition. PLoS ONE. 2014;9:e112183. doi: 10.1371/journal.pone.0112183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Toh B, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9:e1001162. doi: 10.1371/journal.pbio.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Krishnamachary B, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel–Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–2731. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 124.Esteban MA, et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–3575. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 125.Imai T, et al. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol. 2003;163:1437–1447. doi: 10.1016/S0002-9440(10)63501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lester RD, Jo M, Montel V, Takimoto S, Gonias SL. uPAR induces epithelial–mesenchymal transition in hypoxic breast cancer cells. J Cell Biol. 2007;178:425–436. doi: 10.1083/jcb.200701092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang MH, et al. Direct regulation of TWIST by HIF-1a promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 128.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial–mesenchymal transition? Cancer Res. 2005;65:5991–5995. doi: 10.1158/0008-5472.CAN-05-0616. [DOI] [PubMed] [Google Scholar]
- 129.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 130.Scheel C, Weinberg RA. Cancer stem cells and epithelial–mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22:396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chaffer CL, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gupta PB, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 133.Chaffer CL, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154:61–74. doi: 10.1016/j.cell.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012;198:281–293. doi: 10.1083/jcb.201202014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Waerner T, et al. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell. 2006;10:227–239. doi: 10.1016/j.ccr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 136.Onder TT, et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 137.Del Pozo Martin Y, et al. Mesenchymal cancer cell-stroma crosstalk promotes niche activation, epithelial reversion, and metastatic colonization. Cell Rep. 2015;13:2456–2469. doi: 10.1016/j.celrep.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Barkan D, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68:6241–6250. doi: 10.1158/0008-5472.CAN-07-6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shibue T, Weinberg RA. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci USA. 2009;106:10290–10295. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shibue T, Brooks MW, Inan MF, Reinhardt F, Weinberg RA. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012;2:706–721. doi: 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shibue T, Brooks MW, Weinberg RA. An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell. 2013;24:481–498. doi: 10.1016/j.ccr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fischer KR, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zheng X, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Scheel C, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ni T, et al. Snail1-dependent p53 repression regulates expansion and activity of tumour-initiating cells in breast cancer. Nat Cell Biol. 2016;18:1221–1232. doi: 10.1038/ncb3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim J, et al. Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proc Natl Acad Sci USA. 2012;109:6124–6129. doi: 10.1073/pnas.1203203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu S, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014;2:78–91. doi: 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jolly MK, et al. Coupling the modules of EMT and stemness: a tunable ‘stemness window’ model. Oncotarget. 2015;6:25161–25174. doi: 10.18632/oncotarget.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bierie B, et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci USA. 2017;114:E2337–E2346. doi: 10.1073/pnas.1618298114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–47. doi: 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 151.Lerner C, Harrison DE. 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp Hematol. 1990;18:114–118. [PubMed] [Google Scholar]
- 152.Bouwens L, De Blay E. Islet morphogenesis and stem cell markers in rat pancreas. J Histochem Cytochem. 1996;44:947–951. doi: 10.1177/44.9.8773559. [DOI] [PubMed] [Google Scholar]
- 153.Peters R, Leyvraz S, Perey L. Apoptotic regulation in primitive hematopoietic precursors. Blood. 1998;92:2041–2052. [PubMed] [Google Scholar]
- 154.Feuerhake F, Sigg W, Hofter EA, Dimpfl T, Welsch U. Immunohistochemical analysis of Bcl-2 andBax expression in relation to cell turnover and epithelial differentiation markers in the non-lactating human mammary gland epithelium. Cell Tissue Res. 2000;299:47–58. doi: 10.1007/s004419900127. [DOI] [PubMed] [Google Scholar]
- 155.Zhou S, et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001;7:1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 156.Potten CS, Loeffler M. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 157.Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS ONE. 2008;3:e3077. doi: 10.1371/journal.pone.0003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dallas NA, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Graham SM, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 160.Kottke T, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17:854–859. doi: 10.1038/nm.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Boisgerault N, et al. Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse. Mol Ther. 2013;21:1507–1516. doi: 10.1038/mt.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Farmer P, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 163.Byers LA, et al. An epithelial–mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bierie B, Moses HL. Tumour microenvironment: TGFβ : the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 165.Massague J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-β signaling. Cell Res. 2009;19:47–57. doi: 10.1038/cr.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Neuzillet C, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. doi: 10.1016/j.pharmthera.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 169.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 170.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 171.Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39:793–801. doi: 10.1016/j.ctrv.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 172.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7:513–520. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 173.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 174.Bargagna-Mohan P, et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol. 2007;14:623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Thaiparambil JT, et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int J Cancer. 2011;129:2744–2755. doi: 10.1002/ijc.25938. [DOI] [PubMed] [Google Scholar]
- 176.Tanaka H, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16:1414–1420. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hafizi S, Dahlback B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 2006;17:295–304. doi: 10.1016/j.cytogfr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 178.Gjerdrum C, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci USA. 2010;107:1124–1129. doi: 10.1073/pnas.0909333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013;31:775–776. doi: 10.1038/nbt0913-775a. [DOI] [PubMed] [Google Scholar]
- 180.Byers L, et al. A phase I/II and pharmacokinetic study of BGB324, a selective AXL inhibitor as monotherapy and in combination with erlotinib in patients with advanced non-small cell lung cancer (NSCLC) Eur J Cancer. 2017;69:S18–S19. [Google Scholar]
- 181.Gupta PB, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tam WL, et al. Protein kinase C a is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell. 2013;24:347–364. doi: 10.1016/j.ccr.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood. 2009;114:5126–5135. doi: 10.1182/blood-2009-07-216457. [DOI] [PubMed] [Google Scholar]
- 184.Pattabiraman DR, et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science. 2016;351:aad3680. doi: 10.1126/science.aad3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 186.Marcucci F, Stassi G, De Maria R. Epithelial–mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15:311–325. doi: 10.1038/nrd.2015.13. [DOI] [PubMed] [Google Scholar]
- 187.Schmidt JM, et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015;10:131–139. doi: 10.1016/j.celrep.2014.12.032. [DOI] [PubMed] [Google Scholar]
- 188.Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59:168–179. doi: 10.1373/clinchem.2012.184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015;36:13–22. doi: 10.1016/j.ceb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 190.Hennessy BT, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–4124. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Shimono Y, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Leung ELH, et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE. 2010;5:e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pirozzi G, et al. Epithelial to mesenchymal transition by TGFβ-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS ONE. 2011;6:e21548. doi: 10.1371/journal.pone.0021548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kong DJ, et al. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wu WS, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123:641–653. doi: 10.1016/j.cell.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 196.Wu DW, et al. FHIT loss confers cisplatin resistance in lung cancer via the AKT/NF-κB/Slug-mediated PUMA reduction. Oncogene. 2015;34:3882–3883. doi: 10.1038/onc.2015.203. [DOI] [PubMed] [Google Scholar]
- 197.Vega S, et al. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Escriva M, et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol. 2008;28:1528–1540. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lu M, et al. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol Cell. 2014;54:987–998. doi: 10.1016/j.molcel.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 200.Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011;2:e179. doi: 10.1038/cddis.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sequist LV, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang Z, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009;15:195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 204.Chen L, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. doi: 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]