

Abstract
The blockage of the hERG K+ channels is closely associated with lethal cardiac arrhythmia. The notorious ligand promiscuity of this channel earmarked hERG as one of the most important antitargets to be considered in early stages of drug development process. Herein we report on the development of an innovative and freely accessible web server for early identification of putative hERG blockers and non-blockers in chemical libraries. We have collected the largest publicly available curated hERG dataset of 5,984 compounds. We succeed in developing robust and externally predictive binary (CCR ≈0.8) and multiclass models (accuracy ≈0.7). These models are available as a web-service freely available for public at http://labmol.farma-cia.ufg.br/predherg/. Three following outcomes are available for the users: prediction by binary model, prediction by multi-class model, and the probability maps of atomic contribution. The Pred-hERG will be continuously updated and upgraded as new information became available.
Keywords: hERG, QSAR models, web-Server, Screening
Graphical Abstract
1 Introduction
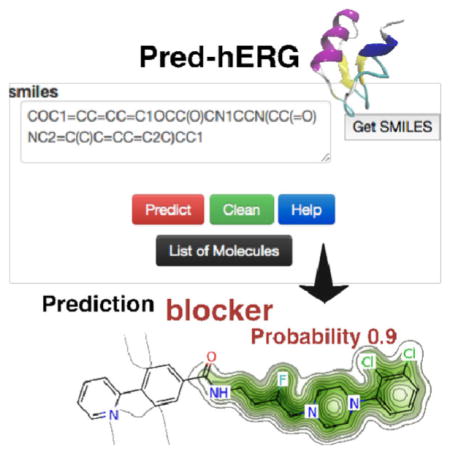
Several non-cardiovascular drugs (e.g., terfenadine,[1] cisapride,[2] sertindole[3]) have been withdrawn from the market due to their ability to inhibit the human ether-à-go-go related gene (hERG) K+ channels, which may lead to severe side effects such as heart arrhythmia and potentially death.[4] Moreover, the notorious ligand promiscuity of this channel[5] earmarked hERG as one of the most important antitargets to be considered in early stages of drug development process. Testing a new bioactive molecule for hERG safety is mandatory and required by the US FDA. Experimental evaluation of binding affinity to hERG K+ channel such as the “gold standard” patch-clamp electrophysiology,[6,7] the in vivo test on zebrafish,[8] etc., are laborious, expensive, and time consuming.[9] Therefore, there is a need in developing computational tools to reliably identify and filter out potential hERG blockers. Recently, we have built a series of QSAR (Quantitative Structure-Activity Relationship) models for hERG liability[10] using 4,833 diverse compounds. Herein, these models were retrained using a considerably larger dataset of 5,984 compounds and an improved validation protocol. Our Pred-hERG web-server (Figure 1) incorporates the new models and allows users for fast screening, even on large libraries of compounds. This service is freely available for public at http://labmol.farmacia.ufg.br/predherg/.
Figure 1.

General scheme for usage of Pred-hERG.
2 Pred-hERG Characteristics
2.1 Dataset Retrieval and Curation
We collected all available records related to the hERG channel from the ChEMBL[11] v.19 database (July, 2014). The original dataset consisted of 14,397 chemical records. Dataset curation (addition of explicit hydrogens, aromatization of functional groups, 2D structure cleaning, fragments removal, etc.) was performed using Indigo Open Source Standardizer following the workflow described by Fourches et al.[12] including the duplicate analysis. In addition, our previous experience showed higher chance of erroneous annotation for very active compounds (e.g., μM were confused with nM) and thus we flagged the compounds with activities lower than 0.3 μM. We also added to the flagged list compounds with activity higher than 300 μM. The flagged compounds comprehended around 10 % of the dataset and their chemical structures and activity values were validated manually by analyzing the original papers. Duplicates with confirmed conflicting annotations were removed. In the case of identical activities, only one record was kept. In total, 1311 compounds were annotated correctly, 140 were mis-annotated and corrected. One compound was absent in corresponding reference. Several compounds that had “less/greater than” operators were removed because they did not obey the threshold limit. Curated dataset consisted of 5,984 compounds including 2,191 non-blockers (activity ≥10 μM), 2,565 weak/moderate blockers (1 μM ≤activity ≤ 10 μM), and 1,228 strong blockers (≤1 μM). The dataset is available at http://labmol.farmacia.ufg.br/predhergmorein-formation/.
2.2 QSAR Modeling
Binary and multi-class QSAR models have been developed and validated according to the OECD principles, using two types of molecular descriptors and support vector machines (SVM)[13] modeling technique. Morgan fingerprints and Chemistry Development Kit (CDK)[14] descriptors were calculated using RDKit (http://www.rdkit.org) and PaDEL-Descriptor plugin for KNIME.[15] The models were built using the qsaR v.1.5 package (http://qsarr.r-forge.r-project.org/) and its integration workflow for KNIME v. 2.10.3. All these procedures were united in KSAR workflow. The 5-fold external cross-validation procedure was used to estimate the robustness of the developed models. Since there is no difference between random and rational selection of external folds for big datasets,[16] the compounds in the external folds were chosen randomly. The predicted probability maps revealing the predicted probability of atomic contributions for the structures became available in this version of Pred-hERG. The original code of probability maps[17] implemented in Python is also freely accessible for users. More detailed description of the QSAR modeling process is available in the Supporting Information.
2.3 Model Implementation and Usage
The Pred-hERG server employs many tools including Flask (http://flask.pocoo.org/), uWSGI (https://uwsgi-docs.read-thedocs.org/), nginx (http://nginx.org/), Python (https://www.python.org/), and JavaScript (http://www.ecma-inter-national.org/). Towards more user-friendly interface, Pred-hERG provides an interactive web interface, including the JSME, a free molecule editor in JavaScript,[18] written on JavaScript and support the latest versions of all most popular browsers. Users will not need any Java or Flash plugins to use it in their browser. Pred-hERG is implemented on Ubuntu Server.
The new version of the web server (Figure 1) has a simpler intuitive user interface. There are three possible ways of entering molecular structure information: (i) directly paste the SMILES string of the structure in the appropriate area and hit “Predict” button; (ii) draw the query molecule in the “molecular editor” box, click on the “Get SMILES” button to translate it to SMILES, and then hit “Predict” button. After the completion of the job, which takes less than one second for one molecule, the user will receive the outcome with the predicted probability maps in the page. If the user wants to predict more molecules, click on the “Clean” button and draw or paste and predict the next molecule. Alternatively, the user can also send a list of molecules by clicking on the “List of Molecules” button. In this case, is necessary to fill out the form with name and e-mail. Then, just paste the list of SMILES of the molecules in the box or upload a *.smi or *.sdf file with all the molecules, and submit the job to the server. The results will be sent by e-mail.
2.4 Outcome Interpretation
The user will receive three outcomes: (i) binary prediction (Consensus AD model); (ii) multiclass prediction (Consensus AD model); and (iii) probability maps extracted from the binary models using Morgan fingerprints. The results from each prediction are directly displayed in the website, along with the probability of the prediction for each class, which may influence the final decision of future use for that particular compound. For binary models, the probability of the compound to be hERG non-blocker or blocker is reported in parenthesis (in this order). Multiclass models have similar outcome, but the user will receive the probability of a compound to be a non-blocker, weak/moderate blocker, or strong blocker respectively. The predicted probability maps help to visualize the atomic contributions in a structure as predicted by the QSAR model. In the map, green atoms or fragments represent contribution towards blockage of hERG, while pink means that it contributes to decrease of hERG blockage, and gray means no contribution. Gray isolines delimit the region of split between the positive (green) and the negative (pink) contribution (see http://lab-mol.farmacia.ufg.br/predherg/predherghelp/ for an example).
3 Results and Discussion
As illustrated on Figure 2, the combination of different descriptors led to robust and predictive QSAR models, with correct classification rate (CCR) ranging between 0.83–0.84 and a coverage of 0.63–1.0 for binary models and accuracy ranging between 0.66–0.79 and a coverage of 0.58–0.81 for multiclass models. Consensus models were built by averaging the predicted values from each individual model as follows: Consensus is the average of Morgan and CDK models; Consensus AD is the average of the predictions made by Morgan and CDK models, but considering the applicability domain (AD); Consensus Rigor is the average of Morgan and CDK models, but only when both predictions were inside the AD. Divergent predictions between individual models were considered as inconclusive and were discarded. The Consensus model yielded the best performance (CCR =0.84 and accuracy =0.74 for binary and consensus classifiers, respectively); therefore, it has been selected to be the default model to be used in Pred-hERG. The complete statistical results of the developed models are available in the Supporting Information (Table S1 and S2).
Figure 2.

Evaluation of (a) binary QSAR models and (b) multiclass QSAR models for hERG liability implemented in Pred-hERG.
4 Conclusions
The Pred-hERG web server allows users to identify putative hERG blockers and non-blockers through a fast and user-friendly interface. No computational or programming skills are required from the user. Prediction time for a compound is less than one second. Three following outcomes are available for the users: prediction by binary model, prediction by multi-class model, and the probability maps of atomic contribution. The Pred-hERG will be continuously updated and upgraded as new information became available. This service is freely available for public at http://lab-mol.farmacia.ufg.br/predherg/.
Supplementary Material
Acknowledgments
The authors would like to thank ChemAxon for providing the academic license of their software. This work was supported by the State of Goias Research Foundation [FAPEG 201310267001095 to VMA]; the Coordination for the Improvement of Higher Education Personnel [CAPES fellowships to RCB and VMA]; the National Council for Scientific and Technological Development (CNPq); the National Institutes of Health [GM66940 and GM096967]; and the Environmental Protection Agency (Grant RD 83499901).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/minf.201500040.
References
- 1.Woosley RL. Annu Rev Pharmacol Toxicol. 1996;36:233–252. doi: 10.1146/annurev.pa.36.040196.001313. [DOI] [PubMed] [Google Scholar]
- 2.Rampe D, Roy ML, Dennis A, Brown AM. FEBS Lett. 1997;417:28–32. doi: 10.1016/s0014-5793(97)01249-0. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez PA, Pahissa J. Curr Drug Saf. 2010;5:97–104. doi: 10.2174/157488610789869265. [DOI] [PubMed] [Google Scholar]
- 4.Picard S, Goineau S, Guillaume P, Henry J, Hanouz JL, Rouet R. Cardiovasc Toxicol. 2011;11:285–307. doi: 10.1007/s12012-011-9133-z. [DOI] [PubMed] [Google Scholar]
- 5.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Proc Natl Acad Sci USA. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 7.Hancox JC, McPate MJ, El Harchi A, Hong Zhang Y. Pharmacol Ther. 2008;119:118–132. doi: 10.1016/j.pharmthera.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Wen D, Liu A, Chen F, Yang J, Dai R. J Appl Toxicol. 2012;32:834–842. doi: 10.1002/jat.2755. [DOI] [PubMed] [Google Scholar]
- 9.Heijman J, Voigt N, Carlsson LG, Dobrev D. Curr Opin Pharmacol. 2014;15C:16–21. doi: 10.1016/j.coph.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Tropsha A, Andrade CH. Curr Top Med Chem. 2014;14:1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fourches D, Muratov E, Tropsha A. J Chem Inf Model. 2010;50:1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vapnik V. The Nature of Statistical Learning Theory. Springer; New York: 2000. [Google Scholar]
- 14.Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. J Chem Inf Comput Sci. 2003;43:493–500. doi: 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazanetz MP, Marmon RJ, Reisser CBT, Morao I. Curr Top Med Chem. 2012;12:1965–1979. doi: 10.2174/156802612804910331. [DOI] [PubMed] [Google Scholar]
- 16.Martin TM, Harten P, Young DM, Muratov EN, Golbraikh A, Zhu H, Tropsha A. J Chem Inf Model. 2012;52:2570–2578. doi: 10.1021/ci300338w. [DOI] [PubMed] [Google Scholar]
- 17.Riniker S, Landrum GA. J Cheminform. 2013;5:43. doi: 10.1186/1758-2946-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bienfait B, Ertl P. J Cheminform. 2013;5 doi: 10.1186/1758-2946-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.